Embed Size (px)
Citation preview

Published: February 25, 2011
r 2011 American Chemical Society 4800 dx.doi.org/10.1021/jp111915y | J. Phys. Chem. C 2011, 115, 4800–4805
ARTICLE
pubs.acs.org/JPCC
Investigation of Ion Transport Traversing the “Ion Channels” byScanning Electrochemical Microscopy (SECM)Xiaoquan Lu,* Tianxia Wang, Xibing Zhou, Yao Li, Bowan Wu, and Xiuhui Liu
Key Laboratory of Bioelectrochemistry & Environmental Analysis of Gansu Province, College of Chemistry & Chemical Engineering,Northwest Normal University, Lanzhou, 730070, China
’ INTRODUCTION
The role of ion channels in cell physiology was regulated byprocesses occurring after protein biosynthesis, which are criticalfor both channel function and targeting of channels to appro-priate cell compartments.1-5 Hodgkin and Huxley first broughtforward the concept in 1952.6,7 Neher and Sakmann providedthe means for directly visualizing ion current through a singlechannel. Further literature relevant to this topic may be found inreviews and handbooks of the structure and function of voltage-gated ion channels in general.8-11 In 2003, Rod Mackinnonmade a groundbreaking work for discovering the structures ofpotassium and chloride channels.7 On the basis of main workabout ion channel engineering by the groups of Mutter,12,13
Montal,14 Armentrout,15-21 Schwarz,22-24 Kevan,25 ion-channelproteins and ion-channel-forming peptides have received in-creasing attention for sensing applications.
Ion channels are supramolecular structures embedded inbiological membranes. Ion channel-controlled transport throughbiological membrane is an extremely important aspect of theelectrophysiology of living cells. Different kinds of channels canopen and close synchronously, resulting in electric signals andresponses of the nervous system. The opened highly selective ionchannels allow some kinds of ions to enter along the electro-chemical gradient.26,27 Their activation, or opening, permits anion-specific flow of a few hundred to many thousands of ioniccharges to rush through the channel before its closure.28
With the development in the molecular biology and X-raydiffraction techniques, many experiments regarding the structur-al details of ion channels have been done.29 Many models weredeveloped to describe the ionic channels, with either theoreticalor experimental purposes. Simulation techniques were applied tothe study of these systems recently,29-31 such as anion-dopedcarbon nanotube,32 computer-aided design,33 and hydration
structure of the ions and nuclear magnetic resonance.34 Manybiological methods could also provide effective means to makeclear the structure of ion channels,1,6,26,35-38 but the details ofboth the process and the energy of ion through the ion channelscannot be obtained at the same time.
To address these issues, we have developed the L/L-AAOinterface. The L/L interface has been suggested as a simplemodel for biological and artificial membranes.39,40 More impor-tant is that in virtue of the interface not only can the life-likenessof ion channels be recurred easily but also both the process andthe energy of ion through the ion channels are obtainedcommendably simultaneously. Furthermore, the experimentalresults show the relationship between the rate constant and thedriving force. The constant of the ion controlled how the ionchannels open or closed. The driving force is an important factorof how fast the ion is through the ion channels.
To sumup the above arguments, studies of artificial ion channelsmust have receivedmuch attention because they both contribute tothe fundamental understanding of natural ion channels and maylead to applications such as drug discovery and sensors.4,41
’EXPERIMENTAL SECTION
Materials. Sodium chloride (NaCl, AR), Potassium chloride(KCl, AR), and 1,2-dichloroethane (DCE, 99.8%, AR) werepurchased from Beijing Chemical, China. Tetrabutylammoniumtetraphenylborate (TBATPB, 99%, Aldrich), tetrabutylammoniumchloride (TBACl, 99%, Aldrich), tetramethyammonium chloride(TMACl, 99%, Aldrich), and dibenzo-18-crown-6 (DB18C6, 98%,Aldrich) were used without purification. The AAO templates were
Received: December 15, 2010Revised: January 27, 2011
ABSTRACT: In the nervous system, ion channels are essentialfor all processes. The highly ordered porous anodic aluminumoxide (AAO) was modified at the interface between twoimmiscible electrolyte solutions (ITIES) to recover the processof ion transfer through the ion channels. The study on dynamicprocess of ion transfer through the ion channels obtained forDKþ
0 is 1.40� 10-8 cm2 s-1 andDNaþ0 is 2.74� 10-7 cm2 s-1.
The result offers tremendous advantages for linear relationshipbetween the half-wave potential and concentration of Kþ andNaþ. In addition, it will be helpful to understand both themechanism about the dynamics and thermodynamics processesof ion transfer through the ion channels and the role about ionic concentration in nerve conduction.

4801 dx.doi.org/10.1021/jp111915y |J. Phys. Chem. C 2011, 115, 4800–4805
The Journal of Physical Chemistry C ARTICLE
made according to the literature.42,43 The aqueous and organicphases were prepared with doubly distilled water (made by ourlaboratory) and DCE, respectively. DCE was washed several timeswith deionized water before use. Special precautions were taken fordealing with DCE and other hazardous chemicals.Apparatus and Electrochemical Measurements. A CHI-
900 electrochemical workstation (CH Instruments) was connectedto a personal computer, which was employed to record approachcurves and cyclic voltammograms. In this work, the nanopipetsemployed as the SECM tips were fabricated from borosilicate glasscapillaries (o.d. = 2.0 mm, i.d. = 1.16mm, L = 10 cm) using a laser-based puller (made by our laboratory). The pipets were filled withthe aqueous solutions from the back using a small syringe (5 μL).Before each experiment, the nanopipet was checked up by anOlympus optical microscope to ensure that there was no trappedbubble. As shown in Scheme 1, a three-electrode setup wasemployed with a nanopipet as the SECM tip, a Ag wire coatedwith AgTPBCl served as the reference electrode, and a Pt wireserved as the counter electrode. A silver wire coated with AgCl wasinserted into the pipet as the reference electrode in the aqueousphase. The tip was filled with x mM NaCl or x mM KCl aqueoussolution from the back by using a small syringe and biased at thepotential where the facilitated Naþ or Kþ transfer reaction wasdiffusion-controlled. The approach curves (iT-d curves) wereobtained by moving the tip toward the bottom L/L interface andrecording the tip current (iT) as the function of the tip interfaceseparation distance (d). The coordinate of the L/L interface (d0)was decided from the sharp increase in iT that occurredwhen the tiptouched the bottom DCE/W interface. Then, the droplet electro-des were for the cyclic voltammograms. A drop of a certain volumeof aqueous solution containing the KCl, FeCl3, and FeCl2 wastransferred to the surface of a freshly polished platinum diskelectrode (d = 2 mm) with a small syringe. The aqueous solutionspread spontaneously across the surface of this platinum electrodeand covered it completely. Then, the electrode was turned over andimmersed immediately in a DCE solution (of a certain volume)containingDB18C6 andTBATPB salt. All of the experiments werecarried out at room temperature (22 ( 2 �C).
’RESULTS AND DISCUSSION: TRANSMEMBRANEDELIVERY
System Setup. The transmembrane delivery of ions wascaused by the inhomogeneous concentrations between the
intracellular and extracellular ions and permeability of biomem-brane. Because ion channels are embedded in lecithin bilayer ofprotein molecules, the ions, especially the Kþ and Naþ passingthe biomembrane, must traverse the ion channels. Through thesechannels, cells could exchange the ions with outside. The modelin the discussion was used to embellish the L/L interface,which has the high similarity with biological membranes. AAOmembranes with nanochannels in the experiment have beensuccessfully fabricated via a two-step annodization method.According to the literature,42,43 the cylindrical uniformly sizedholes range from 20 to 40 nm in diameter, and the film thicknessvaries from 0.1 to 300 μm. The nanopores are so thick and fastthat the ions are just through the AAO pores, not the otheraperture. Before the experiment, the improvement of experi-mental device was that the AAO membrane could completelycover the measuring L/L interface. The transmembrane behaviorof ions gave rise to a concentration gradient between the in andout of cell, the same as the transmembrane voltage; therefore, theconcentration ofMþ in the tip (cMþ
w) should be higher than thatof DB18C6 in the DCE phase (cDB18C6
O ). In view of this, thebiomembrane could be simulated veritably and compactly bythe new system.Kþ and Naþ Transfer through the Ion Channels at the
Modified W/DCE Interface. Evaluation of Mechanism. Theways that a cell releases or takes up ions might be consideredchemically as follows: (1) active transport: ions pass through thebiomembrane directly; (2) passive transport: ions pass throughthe biomembrane facilitated by ionophores; and (3) ion path-way: certain ions pass the biomembrane through the ion channelsimmobilized in the biomembrane.44,45 Therefore, we could con-sider that ions traversing the ion channels belong to the third way.The following electrochemical cell(I) is employed for investi-
gation of alkali metal ion transfer at the modified W/DCEinterface facilitated by DB18C6.
rsfAgjAgClj5 mM TBACl
organic phase TBAþ ISErsf
j5 mM TBATPBCl þ 0:5 mM DB18C6j1, 2 - DEC
rsfjxMMCl þ 10 mM TBAClj
H2OrsfAgAgCl
Re fcellðIÞ
where M is Naþ or Kþ.The first process is the alkali metal ion (Mþ) transfer
facilitated by DB18C6 from water to DCE at the tip, and itsmechanism could be considered chemically as a transfer by
Scheme 1. (A) Ions through the Ion Channels; (B) Simple Model for Single Ion through the Ion Channel; (C) Model for a Three-Electrode to Investigation of Ion Transport Traversing the “Ion Channels” by (SECM); (D) Scanning Electron Micrographs of AAO
4802 dx.doi.org/10.1021/jp111915y |J. Phys. Chem. C 2011, 115, 4800–4805
The Journal of Physical Chemistry C ARTICLE
interfacial complexation (TIC) process45-47
MþðwÞ þDB18C6ðDCEÞ sfkb ½M-DB18C6�þðDCEÞ ðat the tipÞ
The second process is transfer by interfacial dissociation (TID)at the bottom W/DCE interface.
½M -DB18C6�þðDCEÞ sfkfMþðwÞ þDB18C6ðDCEÞ ðat the ITIESÞ
When the tip approaches the ITIES, Mþ is released from thecomplex and transferred through the AAO pores into theaqueous solution, and DB18C6 is regenerated to its neutral formby interfacial dissociation and diffuses back to the tip to producethe enhancement.Dynamics and Thermodynamics of Ions Transport. A
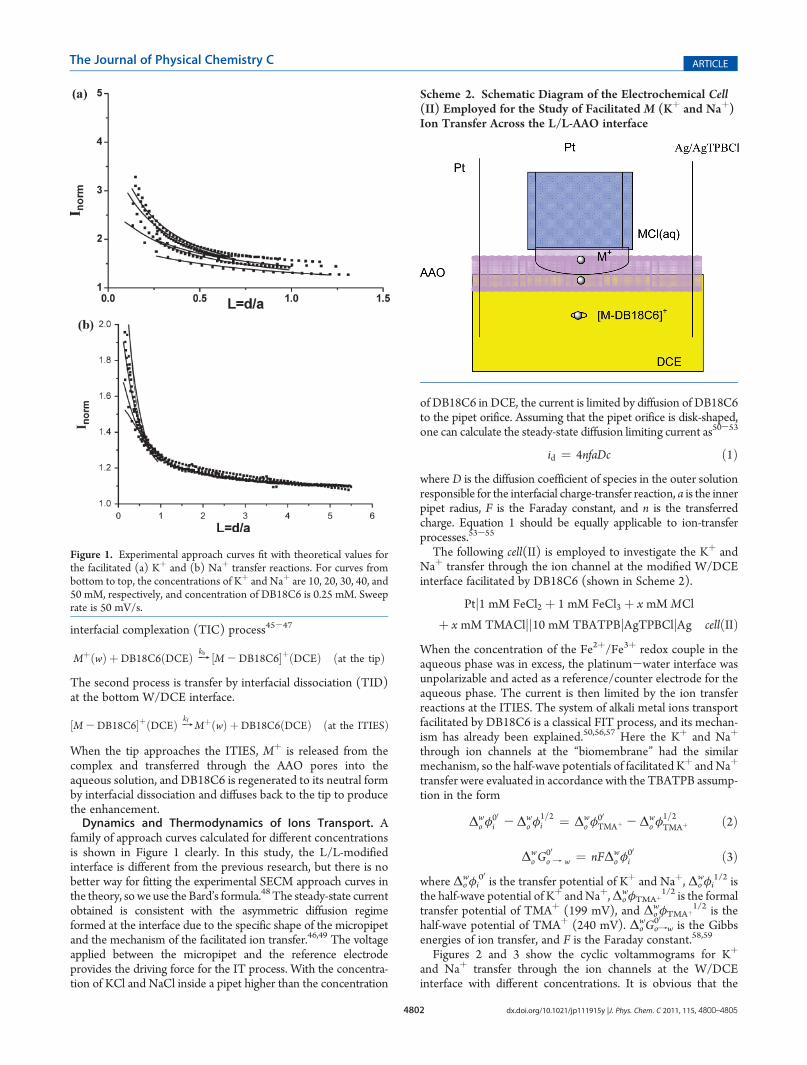
family of approach curves calculated for different concentrationsis shown in Figure 1 clearly. In this study, the L/L-modifiedinterface is different from the previous research, but there is nobetter way for fitting the experimental SECM approach curves inthe theory, so we use the Bard’s formula.48 The steady-state currentobtained is consistent with the asymmetric diffusion regimeformed at the interface due to the specific shape of the micropipetand the mechanism of the facilitated ion transfer.46,49 The voltageapplied between the micropipet and the reference electrodeprovides the driving force for the IT process. With the concentra-tion of KCl and NaCl inside a pipet higher than the concentration
of DB18C6 in DCE, the current is limited by diffusion of DB18C6to the pipet orifice. Assuming that the pipet orifice is disk-shaped,one can calculate the steady-state diffusion limiting current as50-53
id ¼ 4nfaDc ð1Þwhere D is the diffusion coefficient of species in the outer solutionresponsible for the interfacial charge-transfer reaction, a is the innerpipet radius, F is the Faraday constant, and n is the transferredcharge. Equation 1 should be equally applicable to ion-transferprocesses.53-55
The following cell(II) is employed to investigate the Kþ andNaþ transfer through the ion channel at the modified W/DCEinterface facilitated by DB18C6 (shown in Scheme 2).
Ptj1 mM FeCl2 þ 1 mM FeCl3 þ x mM MCl
þ x mM TMACljj10 mM TBATPBjAgTPBCljAg cellðIIÞWhen the concentration of the Fe2þ/Fe3þ redox couple in theaqueous phase was in excess, the platinum-water interface wasunpolarizable and acted as a reference/counter electrode for theaqueous phase. The current is then limited by the ion transferreactions at the ITIES. The system of alkali metal ions transportfacilitated by DB18C6 is a classical FIT process, and its mechan-ism has already been explained.50,56,57 Here the Kþ and Naþ
through ion channels at the “biomembrane” had the similarmechanism, so the half-wave potentials of facilitated Kþ and Naþ
transfer were evaluated in accordance with the TBATPB assump-tion in the form
Δwo φ
00i -Δw
o φ1=2i ¼ Δw
o φ00TMAþ -Δw
o φ1=2TMAþ ð2Þ
Δwo G
00o f w ¼ nFΔw
o φ00i ð3Þ
where Δowφi00 is the transfer potential of Kþ and Naþ, Δo
wφi1/2 is
the half-wave potential of Kþ andNaþ,ΔowφTMAþ
1/2 is the formaltransfer potential of TMAþ (199 mV), and Δo
wφTMAþ
1/2 is thehalf-wave potential of TMAþ (240 mV). Δo
wGofw00 is the Gibbs
energies of ion transfer, and F is the Faraday constant.58,59
Figures 2 and 3 show the cyclic voltammograms for Kþ
and Naþ transfer through the ion channels at the W/DCEinterface with different concentrations. It is obvious that the
Figure 1. Experimental approach curves fit with theoretical values forthe facilitated (a) Kþ and (b) Naþ transfer reactions. For curves frombottom to top, the concentrations of Kþ and Naþ are 10, 20, 30, 40, and50 mM, respectively, and concentration of DB18C6 is 0.25 mM. Sweeprate is 50 mV/s.
Scheme 2. Schematic Diagram of the Electrochemical Cell(II) Employed for the Study of Facilitated M (Kþ and Naþ)Ion Transfer Across the L/L-AAO interface

4803 dx.doi.org/10.1021/jp111915y |J. Phys. Chem. C 2011, 115, 4800–4805
The Journal of Physical Chemistry C ARTICLE
half-wave potentials for ion transfer shift negatively withincreasing concentration of alkali metal ion. At half-wavepotentials of ∼0.45 V, the voltammogram is very well-shapedwith a peak.The relationship between the anodic peak currents and the
root of sweep rates obeys the Randles/Sevcik equation60-63
ip ¼ 0:4463� 10-3n3=2F3=2AðRTÞ-1=2D1=2MþcMþv1=2 ð4Þ
where ip is the anodic peak current of the anodic polarization,A isthe area of the micro-L/L interface, cMþ is the metal ion diffusioncoefficient in aqueous solution, and the other parameters havebeen defined as above.The kinetic parameters can be found from the literature for
given values ΔE1/4 andΔE3/4; the standard rate constant (κ0) is
given by
k0 ¼ λD0
að5Þ
where κ0, D�, and R have been defined in the context. This
method64 has been successfully taken to evaluate the standardrate constant of Kþ and Naþ transport across bionic membrane.As shown in Figure 4, Δo
wφi1/2 linearly depends on the
logarithm of the concentration of Naþ (log Kr), Kr is ratio ofconcentration of Naþ and DB18C6, and the slope was found tobe-0.07465. Kþ is the same as Naþ, and the slope was found tobe-0.13793. The result offers tremendous advantages for linearrelationship between the half-wave potential and concentrationof Kþ and Naþ. In other words, the half-wave potential increaseswith the concentrations of alkali metals ion increasing.Effect of AAOon the Ion Transfer at the BionicMembrane.
Figures 2 and 3 present steady-state voltammograms for thetransfer of Kþ and Naþ through the ion channel at the W/DCEinterface facilitated by DB18C6 with different concentrations. Asdescribed by Stewart et. al,65,66 simple IT reactions at a micro-pipet are characterized by an asymmetric diffusion regime. It isobvious that the peak currents for facilitated ion transfer increaseand the peak currents for Kþ transfer increase with increasingconcentrations of alkali metals ion. (See the inset in Figure 3.)Table 1 shows the kinetic rate constants of the FIT at thedifferent potentials added externally under various concentra-tions of the redox couple in the water droplet. These results are in
Figure 2. Cyclic voltammograms of the transfer of Kþ facilitated byDB18C6 through ion channels on the Pt electrode. The concentrationsof Kþ are 10, 20, 30, 40, and 50 mM, respectively, and the concentrationof DB18C6 is 0.25 mM. Sweep rate is 50 mV/s. Inset: Concentration ofKþ is 10 mM; the DCE contains TBATPBCl but without DB18C6.
Figure 3. Cyclic voltammograms of the transfer of Naþ facilitated byDB18C6 through ion channels on the Pt electrode. The concentrationsof Naþ are 10, 20, 30, 40, and 50 mM, respectively, and the concentra-tion of DB18C6 is 0.25 mM. Sweep rate is 50 mV/s. Inset: Cyclicvoltammogram obtained with TMAþ ion transfer across the AAO-W/DCE interface.
Figure 4. Relationship between the half-wave potential of the FIT of theMþ system and the logarithm of Kr (Kr = CMþ/cDB18C6; CMþ isconcentration of the Kþ or Naþ ion in the aqueous phase and cDB18C6is concentration of the DB18C6 in the DCE).
Table 1. Kinetic Parameters for Facilitated Transfer of Kþ
and Naþ with DB18C6 through the Ion Channels at theBionic Membrane
ion Kr Δowφ1/2 (V) Δo
wφi00 (V) Δ0
wGtr,i00 ,wfo (kJ 3mol-1) Kf (cm 3 s
-1)
Na 20 0.464 0.251 24.2 0.049
40 0.446 0.232 22.4 0.082
80 0.431 0.217 20.9 0.122
120 0.428 0.215 20.7 0.152
160 0.408 0.194 18.8 0.167
K 20 0.509 0.295 28.5 0.002
40 0.474 0.260 25.1 0.003
80 0.444 0.230 22.3 0.006
120 0.428 0.214 20.7 0.007
160 0.413 0.199 19.3 0.008

4804 dx.doi.org/10.1021/jp111915y |J. Phys. Chem. C 2011, 115, 4800–4805
The Journal of Physical Chemistry C ARTICLE
good agreement with the theoretical simulation developed byStewart et al. The value obtained forDKþ
0 is 1.40� 10-8 cm2 s-1
andDNaþ0 is 2.74� 10-7 cm2 s-1, which is smaller than the value
of 1.5 � 10-5 cm2 s-1 from the previous research.60,67
From the experimental results, we can conclude that theAAO plays a block effect. To investigate the effect of the AAOon the rate constant, we further researched its structure.The serrated structures initiate to form at the bottom ofnanochannels during the growth of the oxide membranes. Theperiodic serrated channels are aligned along the same directionwith an inclination angle of 20-30� to the stem channel. Thecurrent density is concentrated around the bubble42,68-71 so thation transport is suppressed through the bubble region. There-fore, it should be rather easy to understand why the AAO has ablock on the electron transfer. The first process at the tip is thealkali ion (Mþ) transfer facilitated by DB18C6 from water toDCE; the second process is the interfacial dissociation reaction atthe bottom W/DCE interface. When the tip approaches thesubstrate (aqueous) phase,Mþ is released from the complex andtransferred to the aqueous solution, and DB18C6 is regeneratedto its neutral form by interfacial dissociation and diffuses back tothe tip to produce the current enhancement. Here the kineticparameters for facilitated transfer of Kþ are a little smaller thanNaþ, which may be caused by the radii of Kþ (1.38 Å) larger thanthe Naþ (1.02 Å) . This indicated that the resistance of Kþ islarger than that of Naþ when they through the ion channels. TheL/L interface modified by the AAO is not quite flat, which leadsto the experimental approach curves not being smooth.
’CONCLUSIONS
In this Article, the new method is described for investigatingion transport traversing the “ion channels” by SECM, which hasthe same trend of change on relationship between the half-wavepotential and concentration with traditional L/L interface. Usingthis method, the lively process of ions traversing the bionicmembrane can be monitored simply. The dynamics and thermo-dynamics processes have been determined using SECM, and theresult indicates a tremendous advantage for a linear relationshipbetween the half-wave potential and concentration of Kþ andNaþ. Moreover, the L/L-AAO interface not only has theadvantages of traditional biomimetics but also has better researchof process of ions traversing the bionic membrane. The L/L-AAO interface could provide a simple and feasible method tostudy the ion transfer reaction for the future, which contributes tothe biochemical applications such as drug discovery and sensors.
’AUTHOR INFORMATION
Corresponding Author*Tel:þ86-931-7971276. Fax:þ86-931-7971323. E-mail: [email protected].
’ACKNOWLEDGMENT
This work was supported by the Natural Science Foundationof China (nos. 20775060, 20927004, 20875077) and the KeyLaboratory of Polymer Materials of Gansu Province.
’REFERENCES
(1) Schmalhofer, W. A.; Sanchez, M.; Dai, G.; Dewan, A.; Secades,L.; Hanner, M.; Knaus, H. G.; McManus, O. B.; Kohler, M.; Kaczorowski,G. J.; Garcia, M. L. Biochemistry 2005, 44, 10135–10144.
(2) Sumikama, T.; Saito, S.; Ohmine, I. J. Phys. Chem. B 2006,110, 20671–20677.
(3) Umezawa, Y.; Aoki, H. Anal. Chem. 2004, 76, 320–326.(4) Woolf, T. B.; Roux, B. Annu. Rev. Biophys. Biomol. Struct. 1994,
23, 11631–11635.(5) Valiyaveetil, F. I.; Zhou, Y.; MacKinnon, R. Biochemistry 2002,
41, 10771–10777.(6) Lehmann-horn, F.; Jurkat-rott, K. Physiol. Rev. 1999, 79, 1317–
1372.(7) Hebert, S. C.; Giebisch, G. J. Am. Soc. Nephrol. 2004,
15, 1096–1097.(8) Patlak, J. Physiol. Rev. 1991, 71, 1047–1080.(9) Martinac, B. J. Cell Sci. 2004, 117, 2449–2460.(10) Catterall, A. W. Trends Neurosci. 1993, 16, 500–506.(11) Rue, C.; Armentrout, P. B. J. Phys. Chem. A 2002,
106, 9788–9797.(12) Mutter, M.; Altmann, K. H.; Tuchscherer, G.; Vuilleumier, S.
Tetrahedron 1988, 44, 771–785.(13) Tuchscherer, G.; Steiner, V.; Altmann, K. H.; Mutter, M.
Methods Mol. Biol. 1994, 36, 261–285.(14) Oiki, S.; Danho, W.; Montal, M. Proc. Natl. Acad. Sci. U.S.A.
1988, 85, 2393–2397.(15) More, M. B.; Ray, D.; Armentrout, P. B. J. Am. Chem. Soc. 1999,
121, 417–423.(16) Ye, S. J.; Clark, A. A.; Armentrout, P. B. J. Phys. Chem. B 2008,
112, 10291–10302.(17) Armentrout, P. B.; Ervin, K. M.; Rodgers, M. T. J. Phys. Chem. A
2008, 112, 10071–10085.(18) Gengeliczki, Z.; Sztaray, B.; Baer, T.; Iceman, C.; Armentrout,
P. B. J. Am. Chem. Soc. 2005, 127, 9393–9402.(19) Chen, Y. M.; Clemmer, D. E.; Armentrout, P. B. J. Phys. Chem.
1994, 98, 11490–11498.(20) Ye, S. J.; Armentrout, P. B. J. Phys. Chem. A 2008, 112, 3587–
3596.(21) Clemmer, D. E.; Sunderlin, L. S.; Armentrout, P. B. Inorg. Chem.
2004, 43, 1976–1985.(22) Roithova, J.; Schroder, D.; Schwarz, H. J. Phys. Chem. A 2004,
108, 5060–5068.(23) Schalley, C. A.; Wesendrup, R.; Schroder, D.; Schwarz, H.
Organometallics 1996, 15, 678–683.(24) Schroder, D.; Siilzle, D.; Dutuit, M.; Baer, T.; Schwarz, H. J. Am.
Chem. Soc. 1994, 116, 6395–6400.(25) Ranjit, K. T.; Bae, J. Y.; Chang, Z.; Kevan, L. J. Phys. Chem. B
2002, 106, 583–588.(26) Matzner, O.; Devor, M. Brain Res. 1992, 597, 92–98.(27) Xu, Y. D.; Bakker, E. Langmuir 2009, 25, 568–573.(28) Sansom, M. S. P.; Shrivastava, I. H.; Ranatunga, K. M.; Smith,
G. R. Trends Biochem. Sci. 2000, 25, 368–374.(29) Jordan, P. C. J. Phys. Chem. 1987, 91, 6582–6591.(30) Demi, T. J. Chem. Phys. 1991, 95, 9242–9247.(31) Sørensen, T. S. J. Chem. Soc., Faraday Trans. 1992, 88, 571–589.(32) Dehez, F.; Tarek, M.; Chipot, C. J. Phys. Chem. B 2007,
111, 10633–10635.(33) Pandeya, S.; Bortei, D. A.; White, M. H. Comput. Methods
Programs Biomed. 2007, 85, 1–7.(34) Yu, L. P.; Sun, C. H.; Song, D. Y.; Shen, J. W.; Xu, N.;
Gunasekera, A.; Hajduk, P. J.; Olejniczak, E. T. Biochemistry 2005,44, 15834–15841.
(35) Guidoni, L.; Torre, V.; Carloni, P. Biochemistry 1999,38, 8599–8604.
(36) Luzhkov, V. B.; Qvist, J. A. Biochim. Biophys. Acta 2005,1747, 109–120.
(37) Narahashi, T. J. Pharmacol. Exp. Ther. 2000, 294, 1–26.(38) Noda, M.; Ikeda, T.; Kayano, T.; Suzuki, H.; Takeshima, H.;
Kurasaki, M.; Takahashi, H.; Numa, S. Nature 1986, 320, 188–192.(39) Ohkouchi, T.; Kakutani, T.; Senda, M. Bioelectrochem. Bioenerg.
1991, 25, 71–80.

4805 dx.doi.org/10.1021/jp111915y |J. Phys. Chem. C 2011, 115, 4800–4805
The Journal of Physical Chemistry C ARTICLE
(40) Ding, Z. F.; Quinn, B. M.; Bard, A. J. J. Phys. Chem. B 2001,105, 6367–6374.(41) Sansom, M. S. Curr. Opin. Struct. Biol. 1998, 8, 237–244.(42) Li, D. D.; Zhao, L; Jiang, C. H.; Lu, J. G. Nano Lett. 2010,
10, 2766–2771.(43) Yuan, J. H.; He, F. Y.; Sun, D. C.; Xia, X. H. Chem. Mater. 2004,
16, 1841–1844.(44) Zhan, D. P.; Mao, S. N.; Zhao, Q.; Chen, Z.; Hu, H.; Jing, P.;
Zhang, M. Q.; Zhu, Z.W.; Shao, Y. H.Anal. Chem. 2004, 76, 4128–4136.(45) Girault, H. H.; Bockris, J. O. M.; Conway, B. E.; White, R. E. In
Modern Aspects of Electrochemistry; Plenum: New York, 1993; Vol. 25, p1.(46) Shao, Y. H.; Osborne, M. D.; Girault, H. H. J. Electroanal. Chem.
1991, 318, 101–109.(47) Zhang, J.; Unwin, P. R. Langmuir. 2002, 18, 2313–2318.(48) Wei, C.; Bard, A. J.; Mirkin, M. V. J. Phys. Chem. 1995,
99, 16033–16042.(49) Shao, Y. H.; Mirkin, M. V. Anal. Chem. 1997, 69, 1627–1634.(50) Beattie, P. D.; Delay, A.; Girault, H. H. J. Electroanal. Chem.
1995, 380, 167–175.(51) Tsionsky, M.; Zhou, J.; Amemiya, S.; Fan, F. R. F.; Bard, A. J.;
Dryfe, R. A. W. Anal. Chem. 1999, 71, 4300–4305.(52) Solomont, T.; Bard, A. J. J. Phys. Chem. 1995, 99, 17487–17489.(53) Su, B.; Zhang, S.; Yuan, Y. H.; Guo, J.; Gan, L.; Shao, Y. Anal.
Chem. 2001, 74, 373–378.(54) Shao, Y. H.; Mirkin, M. V. Anal. Chem. 1998, 70, 3155–3161.(55) Li, F.; Chen, Y.; Sun, P.; Zhang, M.; Gao, Z.; Zhan, D. P.; Shao,
Y. H. J. Phys. Chem. B 2004, 108, 3295–3302.(56) Lee, H. J.; Beriet, C.; Girault, H. H. J. Electroanal. Chem. 1998,
453, 211–219.(57) Shao, Y. H.; Mirkin, M. V. J. Phys. Chem. B 1998,
102, 9915–9921.(58) Yuan, Y.; Gao, Z.; Guo, J.; Shao, Y. H. J. Electroanal. Chem.
2002, 526, 85–91.(59) Chen, Y.; Gao, Z.; Li, F.; Ge, L.; Zhang, M.; Zhan, D. P.; Shao,
Y. H. Anal. Chem. 2003, 75, 6593–6601.(60) Zhan, D. P.; Yuan, Y.; Xiao, Y.; Wua, B.; Shao, Y. H. Electrochim.
Acta 2002, 47, 4477–4483.(61) Reymond, F. D.; Lagger, G.; Carrupt, P. A.; Girault, H. H.
J. Electroanal. Chem. 1998, 451, 59–76.(62) Reymond, F.; Carrupt, P. A.; Girault, H. H. J. Electroanal. Chem.
1998, 449, 49–65.(63) Tomaszewski, L.; Ding, Z. F.; Fermı’n, D.; Cacote, H. M.;
Pereira, C.; Silva, F.; Girault, H. H. J. Electroanal. Chem. 1998,453, 171–177.(64) Shao, Y. H.; Mirkin, M. V. J. Am. Chem. Soc. 1997,
119, 8103–8104.(65) Stewart, A. A.; Taylor, G.; Girault, H. H.; McAleer, J.
J. Electroanal. Chem. 1990, 296, 491–515.(66) Stewart, A. A.; Shao, Y. H.; Pereira, C. M.; Girault, H. H.
J. Electroanal. Chem. 1991, 305, 135–139.(67) Samec, Z.; Mare�cek, V.; Colombini, M. P. J. Electroanal. Chem.
1998, 257, 147–154.(68) Li, D. D.; Jiang, C. H.; Jiang, J. H.; Lu, J. G. Chem. Mater. 2009,
21, 253–258.(69) Bronstein, L. M.; Chernyshov, D. M.; Karlinsey, R.; Zwanziger,
J. W.; Matveeva, V. G.; Sulman, E.M.; Demidenko, G. N.; Hentze, H.-P.;Antonietti, M. Chem. Mater. 2003, 15, 2623–2631.(70) Lau, K. H. A.; Tan, L. S.; Tamada, K.; Sander, M. S.; Knoll, W.
J. Phys. Chem. B 2004, 108, 10812–10818.(71) Moon, J. M.; Wei, A. J. Phys. Chem. B 2005, 109, 23336–23341.





























