Embed Size (px)
DESCRIPTION
The catalytic performance of mono- and bimetallic Pd (0.6, 1.0 wt.%)–Pt (0.3 wt.%) catalysts supported on ZrO2 (70, 85 wt.%)–Al2O3(15, 0 wt.%)–WOx (15 wt.%) prepared by sol–gel was studied in the hydroisomerization of n-hexane.
Citation preview

www.elsevier.com/locate/apcata
Applied Catalysis A: General 290 (2005) 97–109
Isomerization of n-hexane over mono- and bimetallic Pd–Pt catalysts
supported on ZrO2–Al2O3–WOx prepared by sol–gel
A. Barrera a, J.A. Montoya a,*, M. Viniegra b, J. Navarrete a, G. Espinosa a,A. Vargas a, P. del Angel a, G. Perez b
a Instituto Mexicano del Petroleo, Ingenierıa Molecular, Eje Central Lazaro Cardenas 152, C.P. 07730, Mexico, D.F., Mexicob Division de CBI, Universidad Autonoma Metropolitana-Iztapalapa, 09340 Mexico, D.F., Mexico
Received 23 November 2004; received in revised form 16 May 2005; accepted 18 May 2005
Available online 27 June 2005
Abstract
The catalytic performance of mono- and bimetallic Pd (0.6, 1.0 wt.%)–Pt (0.3 wt.%) catalysts supported on ZrO2 (70, 85 wt.%)–Al2O3
(15, 0 wt.%)–WOx (15 wt.%) prepared by sol–gel was studied in the hydroisomerization of n-hexane. The catalysts were characterized by N2
physisorption, XRD, TPR, XPS, Raman, NMR, and FT-IR of adsorbed pyridine. The preparation of ZrW and ZrAlW mixed oxides by sol–gel
favored the high dispersion of WOx and the stabilization of zirconia in the tetragonal phase. The Al incorporation avoided the formation of
monoclinic-WO3 bulk phase. The catalysts increased their SBET for about 15% promoted by Al2O3 addition. Various oxidation states of WOx
species coexist on the surface of the catalysts after calcination. The structure of the highly dispersed surface WOx species is constituted mainly
of isolated monotungstate and two-dimensional mono-oxotungstate species in tetrahedral coordination. The activity of Pd/ZrW catalysts in
the hydroisomerization of n-hexane is promoted both with the addition of Al to the ZrW mixed oxide and the addition of Pt to Pd/ZrAlW
catalysts. The improvement in the activity of Pd/ZrAlW catalysts is ascribed to a moderated acid strength and acidity, which can be correlated
to the coexistence of W6+ and reduced-state WOx species (either W4+ or W0). The addition of Pt to the Pd/ZrAlW catalyst does not modify
significantly its acidic character. Selectivity results showed that the catalyst produced 2MP, 3MP and the high octane 2,3-dimethylbutane (2,3-
DMB) and 2,2-dimethylbutane (2,2-DMB) isomers.
# 2005 Elsevier B.V. All rights reserved.
Keywords: Bimetallic Pd–Pt catalysts; ZrO2–Al2O3–WO3 ternary oxide; Sol–gel method; Structure; Reducibility; XPS; Raman; NMR; Acidity;
Isomerization of n-hexane
1. Introduction
In recent years the catalytic processes to promote the
skeletal isomerization of light n-alkanes such as n-hexane
(n-C6H14) have played an important role in order to increase
the octane number of gasoline [1,2]. The industrial catalysts
used for the isomerization of alkanes are those based in Pt
supported on chlorinated alumina and Pt or Pd supported on
mordenite [1,2]. However, the former catalyst results in
serious environmental problems related to the recovery of
corrosive acids and it is sensitive to moisture and sulfur
impurities, while Pt or Pd supported on mordenite is less
* Corresponding author. Tel.: +52 55 91 75 8375; fax: +52 55 95 75 6239.
E-mail address: [email protected] (J.A. Montoya).
0926-860X/$ – see front matter # 2005 Elsevier B.V. All rights reserved.
doi:10.1016/j.apcata.2005.05.011
active and works well at high temperatures with low yield of
branched isomers [3,4]. Arata et al. [5] have found that the
catalysts based on sulfated zirconia are highly active in the
hydroisomerization of alkanes at low temperatures, however
the inconvenient with these materials is that they are quickly
deactivated. It has also been reported that Pt catalysts
supported on zirconia promoted with tungsten are active in
the hydroisomerization of light n-alkanes at moderated
pressure and temperature [6–11]. However, the improve-
ment of their acid properties and hydrogenation capability is
highly desired in order to improve their catalytic properties
in the hydroisomerization of alkanes. On this respect, it is
known that the acidity of tungstated zirconia can be
increased by the addition of certain promoters as for
example Al [12–14]. Other noble metals than Pt, as for

A. Barrera et al. / Applied Catalysis A: General 290 (2005) 97–10998
example Pd with high hydrogenation capability, have been
scarcely studied [15,16]. An important aspect to consider is
the method of preparation of the catalytic materials. The
majority of Pt catalysts supported on zirconia promoted with
tungsten are prepared mainly by sequential impregnation of
zirconia with tungsten and Pt solutions to give Pt/WOx/ZrO2
systems [7–10,17]. However, the activity and selectivity of
the catalyst could depend of the form of interaction between
species which is a function of the method of preparation
used.
In this work, we have studied the activity and selectivity
in the hydroisomerization of n-hexane over mono- and
bimetallic Pd–Pt catalysts supported on ZrO2–Al2O3–WOx
oxides prepared by sol–gel via the hydrolysis of alkoxides
assisted by the aqueous ammonia metatungstate solution.
The activity test was carried out by applying high-
throughput reaction techniques. We have selected the more
active catalysts and tried to establish correlations between
their physicochemical properties and their catalytic perfor-
mance in the hydroisomerization of n-hexane.
2. Experimental
2.1. Catalysts preparation
70 wt.% ZrO2–15 wt.% Al2O3–15 wt.% WO3 (ZrAlW)
ternary oxides were prepared by the sol–gel method from
organic precursors with the hydrolysis process assisted by
the addition of the aqueous ammonia metatungstate solution
as follows: required volumes of zirconium(IV) propoxide,
Zr(O-Prop)4 (Aldrich), and aluminum-tri-sec-butoxide,
Al(O-sec-Bu)3 (Aldrich), were dissolved in a glass flask
containing 1-propanol (Fermont), and hexylene glycol, 2-
methyl-2,4-pentanediol (Aldrich), as a complexing agent
[18]. The solution was heated with continuous stirring to
70 8C and kept there for 1 h. Then, the required volume of
the aqueous ammonia metatungstate (AMT) solution
(NH4)6H2W12O40�nH2O (Aldrich) was added to the homo-
geneous alkoxide solution in the glass flask to perform the
hydrolysis. The gels obtained were aged in the glass flask
with the remaining AMT solution at 40 8C for 2 h and then at
room temperature for 12 h. After that, the gels were dried in
an oven at 100 8C for 12 h followed by calcination at 800 8Cfor 4 h. 85 wt.% ZrO2–15 wt.% WO3 (ZrW) binary oxides
were prepared by the same method except for the addition of
the alumina precursor.
The ZrW and ZrAlW oxides calcined at 800 8C were
impregnated with solutions of varying concentrations of
PdCl2 (Aldrich) to give catalysts with 0.3, 0.6, and 1.0 wt.%
of Pd. Some samples were coimpregnated with a
H2PtCl6�xH2O solution (Aldrich) to give bimetallic catalysts
with Pd (0.6 wt.%)–Pt (0.3 wt.%), and Pd (1.0 wt.%)–Pt
(0.3 wt.%). In order to study the effect of Pt, the ZrAlW
oxide was also impregnated with H2PtCl6�xH2O to give
catalysts with 0.3 and 0.9 wt.% Pt. After impregnation, the
samples were dried in two stages, firstly at 60 8C for 4 h and
then at 100 8C for 12 h. Finally the samples were calcined at
600 8C for 3 h.
2.2. Catalyst characterization
The surface area of Pd, Pt and Pd–Pt catalysts after
calcination at 600 8C for 3 h was determined by nitrogen
physisorption in an Autosorb gas sorption system (from
Quantachrome) on the basis of the BET equation from the
nitrogen isotherms. Before the measurements, the samples
were flushed with Ar at 200 8C for 2 h.
X-ray diffraction patterns of the catalysts calcined at
600 8C for 3 h were obtained in a Bruker-Axs D8 Discover
with GADDS (General Area Detector Diffraction Systems,
two-dimensional (2D) detector) diffractometer fitted with a
Cu anode tube (40 kV, 40 mA) by using the combinatorial
approach for both measurements and patterns evaluation
with the sample holder designed for 48 samples in one plate.
In this equipment the area detector is situated at 15 cm from
the sample and the step size is 0.058 with a scan time of
300 s.
Temperature-programmed reduction (TPR) profiles of
the catalysts calcined at 600 8C for 3 h were obtained under
hydrogen flow (5% H2/Ar) by using a thermodesorption
apparatus, Multipulse RIG model (from ISRI), equipped
with a thermal conductivity detector (TCD). Samples of
100 mg and a gas flow rate of 30 cm3 min�1 were used in the
experiments. The TPR profiles were registered by heating
the samples from 25 to 1000 8C at a rate of 10 8C min�1 and
the rate of hydrogen consumption was monitored by TCD.
The amount of hydrogen consumed was obtained by the
integration of the TPR profiles peaks with the standard
procedure included in the software of the Multipulse RIG
unit. This procedure presents splitting for unresolved peaks
with a vertical line dropped to the base line. Hydrogen pulses
were used to calibrate the TPR signal with the same flow
used in the TPR experiments.
XPS spectra of mono- and bimetallic Pd–Pt catalysts after
calcination were recorded on a VG ESCALAB 250
apparatus after excitation with a monochromatic Al Ka
radiation (hn = 1486.6 eV). Samples were embedded onto
Indium foils which were pressed manually and placed to a
holder. Calibration of the energy position of an XPS peak
was performed by using the binding energy of carbon 1s
peak at 284.8 eV. The unresolved XP spectra for the W 4f
transition were deconvoluted with three doublets by using a
standard Gaussian function. A binding energy splitting of
2.1 � 0.1 eV was adopted for the W 4f7/2–W 4f5/2
separation. The binding energies values were assigned to
the corresponding oxidation state according to literature
data.
Raman spectra of the mono- and bimetallic Pd–Pt
catalysts were recorded in the 100–1400 cm�1 wavenumber
range using a ThermoNicolet Raman apparatus (Almega
model) equipped with a Nd:YVO4DPSS laser source. The

A. Barrera et al. / Applied Catalysis A: General 290 (2005) 97–109 99
excitation line of the laser was 532 nm and the laser power
was of 25 mW.27Al NMR–MAS spectra of the catalysts calcined at
600 8C for 3 h were recorded at 23 kHz with a Bruker 400
spectrometer and using AlCl3 0.1 M solution as the standard
reference. Scan conditions were 0.35 ms pulse with a delay
time of 1.0 s. The unresolved 27Al NMR spectra were
deconvoluted in three peaks by using a standard Lorentzian
function.
FT-IR of adsorbed pyridine experiments were performed
in order to determine the type and amount of surface acid
sites of the ZrW mixed oxide and catalysts with 1.0 wt.% of
palladium. They were carried out by using a Fourier
transform infrared (FT-IR) Perkin-Elmer spectrometer
Model 170-SX. Pretreatment of the samples prior to the
adsorption of pyridine consisted in outgassing followed by
heating to 500 8C at 20 8C min�1 and cooling to room
temperature. After the pretreatment, the samples were
exposed to saturated pyridine vapor for 20 min. IR spectra
were recorded after desorption at 50, 100, 200, 300, and
400 8C.
2.3. Catalytic activity test
Hydroisomerization of n-hexane (n-C6H14) over the Pd,
Pt and Pd–Pt catalysts calcined at 600 8C for 3 h was
performed in a Multi-Channel Fixed Bed Reactor (Symyx).
This equipment is appropriate to evaluate the activity and
selectivity of 48 samples in parallel by applying high-
throughput testing techniques with a matrix arrangement of
6 columns � 8 rows. Briefly, this system consists of six
reactor heads each one containing eight wells for steel
micro-reactors of approximately 4 mm inner diameter and
47 mm length. The six reactor heads are connected
independently to six chromatographs (Agilent, 6850 Series)
equipped with a SPB-1 capillary column (Supelco) and a
flame ionization detector (FID) to do the reaction product
analysis. For the catalytic evaluation 100 mg of catalyst was
diluted with 200 mg of inert silicon carbide by mixing them
and then packed within the micro-reactors and fixed into the
well reactor heads. Pretreatment of the catalysts was carried
out in situ prior to the activity test, and consisted in a drying-
reduction program; drying the samples at 260 8C for 2 h in
helium (200 cm3 min�1) followed by reduction in hydrogen
flow (200 cm3 min�1) at 350 8C for 3 h.
Table 1
Specific surface area and H2 uptake from the TPR profiles of mono- and bime
calcination of the supports at 800 8C/4 h
Catalysts SBET (m2 g�1)
Pd (0.6 wt.%)/ZrW 69
Pd (0.6 wt.%)/ZrAlW 89
Pd (0.6 wt.%)–Pt (0.3 wt.%)/ZrAlW 80
Pt (0.3 wt.%)/ZrAlW 85
Pd (1.0 wt.%)/ZrW 67
Pd (1.0 wt.%)/ZrAlW 78
Pd (1.0 wt.%)–Pt (0.3 wt.%)/ZrAlW 87
The experimental conditions in the hydroisomerization of
n-hexane were as follows: reaction pressure of 0.689 MPa,
reaction temperature of 220, 240 and 260 8C, the total feed
composition was a mixture of H2 = 0.269 mol/h,
He = 0.135 mol/h, and a liquid flow of n-hexane = 0.184 -
mol/h, giving a WHSV = 3.695 for every single well. The
H2/n-C6 molar ratio was 1.469, which is very close to that
used in the industrial process.
The catalytic evaluation procedure is fully automatized,
and for every run the pressure, reactor and accessory
temperature and reactant flows are set and controlled in real
time. When the experimental conditions are reached the first
row of six reaction products are analyzed simultaneously in
the six GC’s, the time for analysis method is 10.7 min, and
after the next injection proceeds, and so on until the eight
rows are analyzed. The 48 chromatograms are saved in a
database. In this way every six catalysts row are evaluated
simultaneously and at the same reaction time but the next
one has 10.7 min more of the reaction time. Therefore, the
total time for one run experiment is about 90 min. To
estimate if the deactivation process is important the catalysts
are evaluated in a different well position. In this particular
type of catalyst it was found that deactivation by carbon
deposition was not important at least during the run
experiment.
3. Results
3.1. BET surface area
The BET surface area (SBET) of the Pd, Pt, and bimetallic
Pd–Pt catalysts supported on ZrW and ZrAlW prepared by
sol–gel via the hydrolysis assisted by the AMT solution is
shown in Table 1. Their SBET were in the range 67–
89 m2 g�1 with the lowest values observed in the catalysts
supported on ZrW (alumina free). The addition of 15 wt.%
of alumina increased the SBET of the catalysts for about 15%.
3.2. X-ray diffraction
The XRD patterns of the Pd and bimetallic Pd–Pt
catalysts supported on ZrAlW after calcination at 600 8C are
shown in Fig. 1. Pd, Pt or Pd–Pt phases were not observed.
For the catalysts supported on ZrW (0 wt.% of Al2O3),
tallic Pd–Pt/ZrO2–Al2O3–WOx catalysts calcined at 600 8C/3 h; previous
H2 uptake of the TPR peak at T = 80 8C (mmol g�1)
11.28
31.17
35.97
–
69.00
38.60
27.60
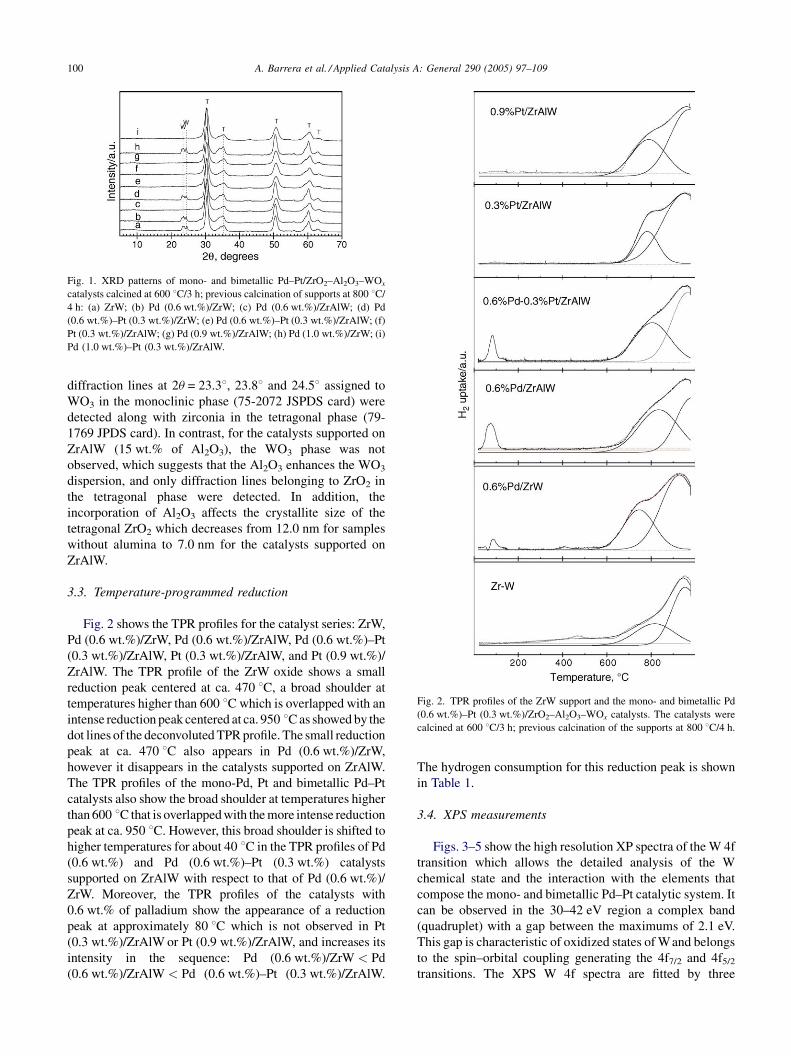
A. Barrera et al. / Applied Catalysis A: General 290 (2005) 97–109100
Fig. 1. XRD patterns of mono- and bimetallic Pd–Pt/ZrO2–Al2O3–WOx
catalysts calcined at 600 8C/3 h; previous calcination of supports at 800 8C/
4 h: (a) ZrW; (b) Pd (0.6 wt.%)/ZrW; (c) Pd (0.6 wt.%)/ZrAlW; (d) Pd
(0.6 wt.%)–Pt (0.3 wt.%)/ZrW; (e) Pd (0.6 wt.%)–Pt (0.3 wt.%)/ZrAlW; (f)
Pt (0.3 wt.%)/ZrAlW; (g) Pd (0.9 wt.%)/ZrAlW; (h) Pd (1.0 wt.%)/ZrW; (i)
Pd (1.0 wt.%)–Pt (0.3 wt.%)/ZrAlW.
Fig. 2. TPR profiles of the ZrW support and the mono- and bimetallic Pd
(0.6 wt.%)–Pt (0.3 wt.%)/ZrO2–Al2O3–WOx catalysts. The catalysts were
calcined at 600 8C/3 h; previous calcination of the supports at 800 8C/4 h.
diffraction lines at 2u = 23.38, 23.88 and 24.58 assigned to
WO3 in the monoclinic phase (75-2072 JSPDS card) were
detected along with zirconia in the tetragonal phase (79-
1769 JPDS card). In contrast, for the catalysts supported on
ZrAlW (15 wt.% of Al2O3), the WO3 phase was not
observed, which suggests that the Al2O3 enhances the WO3
dispersion, and only diffraction lines belonging to ZrO2 in
the tetragonal phase were detected. In addition, the
incorporation of Al2O3 affects the crystallite size of the
tetragonal ZrO2 which decreases from 12.0 nm for samples
without alumina to 7.0 nm for the catalysts supported on
ZrAlW.
3.3. Temperature-programmed reduction
Fig. 2 shows the TPR profiles for the catalyst series: ZrW,
Pd (0.6 wt.%)/ZrW, Pd (0.6 wt.%)/ZrAlW, Pd (0.6 wt.%)–Pt
(0.3 wt.%)/ZrAlW, Pt (0.3 wt.%)/ZrAlW, and Pt (0.9 wt.%)/
ZrAlW. The TPR profile of the ZrW oxide shows a small
reduction peak centered at ca. 470 8C, a broad shoulder at
temperatures higher than 600 8C which is overlapped with an
intense reduction peak centered at ca. 950 8C as showed by the
dot lines of the deconvoluted TPR profile. The small reduction
peak at ca. 470 8C also appears in Pd (0.6 wt.%)/ZrW,
however it disappears in the catalysts supported on ZrAlW.
The TPR profiles of the mono-Pd, Pt and bimetallic Pd–Pt
catalysts also show the broad shoulder at temperatures higher
than 600 8C that is overlapped with the more intense reduction
peak at ca. 950 8C. However, this broad shoulder is shifted to
higher temperatures for about 40 8C in the TPR profiles of Pd
(0.6 wt.%) and Pd (0.6 wt.%)–Pt (0.3 wt.%) catalysts
supported on ZrAlW with respect to that of Pd (0.6 wt.%)/
ZrW. Moreover, the TPR profiles of the catalysts with
0.6 wt.% of palladium show the appearance of a reduction
peak at approximately 80 8C which is not observed in Pt
(0.3 wt.%)/ZrAlW or Pt (0.9 wt.%)/ZrAlW, and increases its
intensity in the sequence: Pd (0.6 wt.%)/ZrW < Pd
(0.6 wt.%)/ZrAlW < Pd (0.6 wt.%)–Pt (0.3 wt.%)/ZrAlW.
The hydrogen consumption for this reduction peak is shown
in Table 1.
3.4. XPS measurements
Figs. 3–5 show the high resolution XP spectra of the W 4f
transition which allows the detailed analysis of the W
chemical state and the interaction with the elements that
compose the mono- and bimetallic Pd–Pt catalytic system. It
can be observed in the 30–42 eV region a complex band
(quadruplet) with a gap between the maximums of 2.1 eV.
This gap is characteristic of oxidized states of Wand belongs
to the spin–orbital coupling generating the 4f7/2 and 4f5/2
transitions. The XPS W 4f spectra are fitted by three
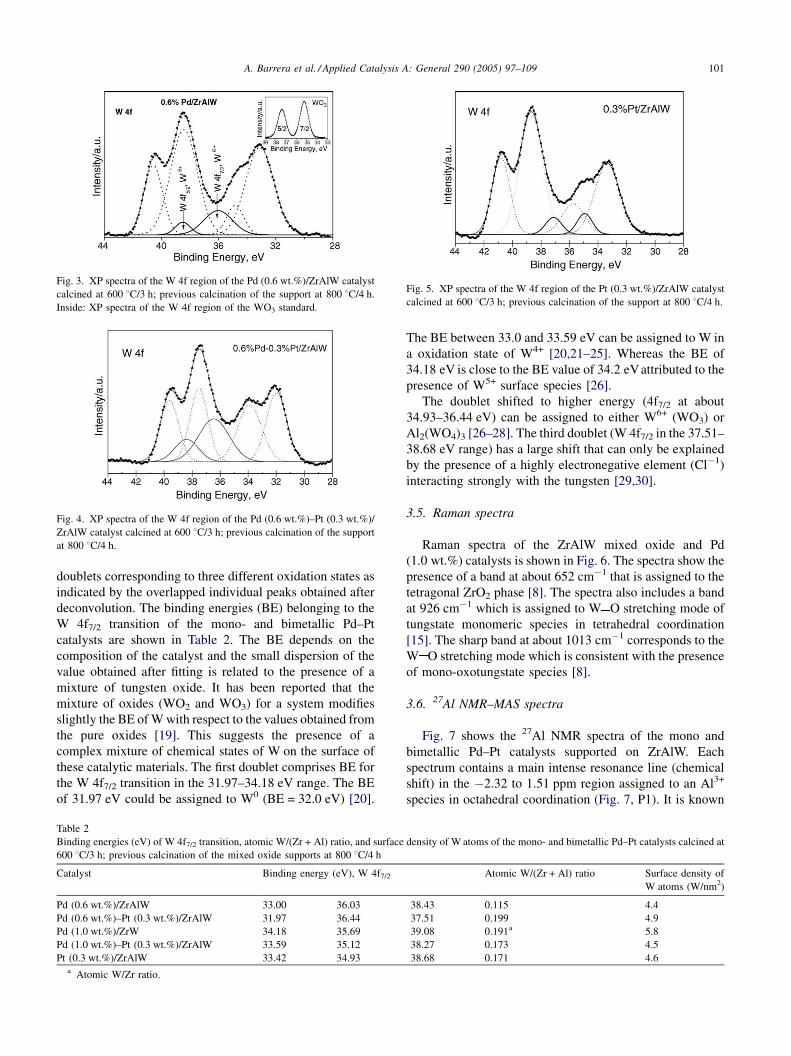
A. Barrera et al. / Applied Catalysis A: General 290 (2005) 97–109 101
Fig. 3. XP spectra of the W 4f region of the Pd (0.6 wt.%)/ZrAlW catalyst
calcined at 600 8C/3 h; previous calcination of the support at 800 8C/4 h.
Inside: XP spectra of the W 4f region of the WO3 standard.
Fig. 4. XP spectra of the W 4f region of the Pd (0.6 wt.%)–Pt (0.3 wt.%)/
ZrAlW catalyst calcined at 600 8C/3 h; previous calcination of the support
at 800 8C/4 h.
Fig. 5. XP spectra of the W 4f region of the Pt (0.3 wt.%)/ZrAlW catalyst
calcined at 600 8C/3 h; previous calcination of the support at 800 8C/4 h.
doublets corresponding to three different oxidation states as
indicated by the overlapped individual peaks obtained after
deconvolution. The binding energies (BE) belonging to the
W 4f7/2 transition of the mono- and bimetallic Pd–Pt
catalysts are shown in Table 2. The BE depends on the
composition of the catalyst and the small dispersion of the
value obtained after fitting is related to the presence of a
mixture of tungsten oxide. It has been reported that the
mixture of oxides (WO2 and WO3) for a system modifies
slightly the BE of W with respect to the values obtained from
the pure oxides [19]. This suggests the presence of a
complex mixture of chemical states of W on the surface of
these catalytic materials. The first doublet comprises BE for
the W 4f7/2 transition in the 31.97–34.18 eV range. The BE
of 31.97 eV could be assigned to W0 (BE = 32.0 eV) [20].
Table 2
Binding energies (eV) of W 4f7/2 transition, atomic W/(Zr + Al) ratio, and surface
600 8C/3 h; previous calcination of the mixed oxide supports at 800 8C/4 h
Catalyst Binding energy (eV), W 4f7/2
Pd (0.6 wt.%)/ZrAlW 33.00 36.03
Pd (0.6 wt.%)–Pt (0.3 wt.%)/ZrAlW 31.97 36.44
Pd (1.0 wt.%)/ZrW 34.18 35.69
Pd (1.0 wt.%)–Pt (0.3 wt.%)/ZrAlW 33.59 35.12
Pt (0.3 wt.%)/ZrAlW 33.42 34.93a Atomic W/Zr ratio.
The BE between 33.0 and 33.59 eV can be assigned to W in
a oxidation state of W4+ [20,21–25]. Whereas the BE of
34.18 eV is close to the BE value of 34.2 eVattributed to the
presence of W5+ surface species [26].
The doublet shifted to higher energy (4f7/2 at about
34.93–36.44 eV) can be assigned to either W6+ (WO3) or
Al2(WO4)3 [26–28]. The third doublet (W 4f7/2 in the 37.51–
38.68 eV range) has a large shift that can only be explained
by the presence of a highly electronegative element (Cl�1)
interacting strongly with the tungsten [29,30].
3.5. Raman spectra
Raman spectra of the ZrAlW mixed oxide and Pd
(1.0 wt.%) catalysts is shown in Fig. 6. The spectra show the
presence of a band at about 652 cm�1 that is assigned to the
tetragonal ZrO2 phase [8]. The spectra also includes a band
at 926 cm�1 which is assigned to W O stretching mode of
tungstate monomeric species in tetrahedral coordination
[15]. The sharp band at about 1013 cm�1 corresponds to the
W O stretching mode which is consistent with the presence
of mono-oxotungstate species [8].
3.6. 27Al NMR–MAS spectra
Fig. 7 shows the 27Al NMR spectra of the mono and
bimetallic Pd–Pt catalysts supported on ZrAlW. Each
spectrum contains a main intense resonance line (chemical
shift) in the �2.32 to 1.51 ppm region assigned to an Al3+
species in octahedral coordination (Fig. 7, P1). It is known
density of W atoms of the mono- and bimetallic Pd–Pt catalysts calcined at
Atomic W/(Zr + Al) ratio Surface density of
W atoms (W/nm2)
38.43 0.115 4.4
37.51 0.199 4.9
39.08 0.191a 5.8
38.27 0.173 4.5
38.68 0.171 4.6
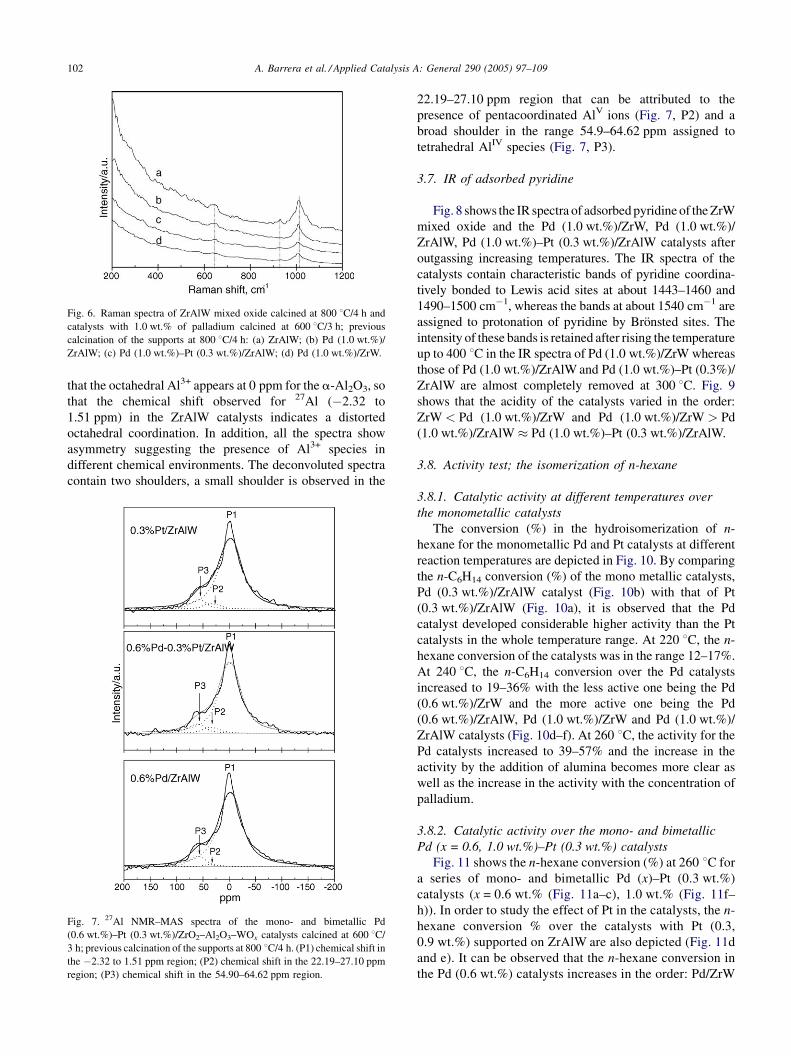
A. Barrera et al. / Applied Catalysis A: General 290 (2005) 97–109102
Fig. 6. Raman spectra of ZrAlW mixed oxide calcined at 800 8C/4 h and
catalysts with 1.0 wt.% of palladium calcined at 600 8C/3 h; previous
calcination of the supports at 800 8C/4 h: (a) ZrAlW; (b) Pd (1.0 wt.%)/
ZrAlW; (c) Pd (1.0 wt.%)–Pt (0.3 wt.%)/ZrAlW; (d) Pd (1.0 wt.%)/ZrW.
that the octahedral Al3+ appears at 0 ppm for the a-Al2O3, so
that the chemical shift observed for 27Al (�2.32 to
1.51 ppm) in the ZrAlW catalysts indicates a distorted
octahedral coordination. In addition, all the spectra show
asymmetry suggesting the presence of Al3+ species in
different chemical environments. The deconvoluted spectra
contain two shoulders, a small shoulder is observed in the
Fig. 7. 27Al NMR–MAS spectra of the mono- and bimetallic Pd
(0.6 wt.%)–Pt (0.3 wt.%)/ZrO2–Al2O3–WOx catalysts calcined at 600 8C/
3 h; previous calcination of the supports at 800 8C/4 h. (P1) chemical shift in
the �2.32 to 1.51 ppm region; (P2) chemical shift in the 22.19–27.10 ppm
region; (P3) chemical shift in the 54.90–64.62 ppm region.
22.19–27.10 ppm region that can be attributed to the
presence of pentacoordinated AlV ions (Fig. 7, P2) and a
broad shoulder in the range 54.9–64.62 ppm assigned to
tetrahedral AlIV species (Fig. 7, P3).
3.7. IR of adsorbed pyridine
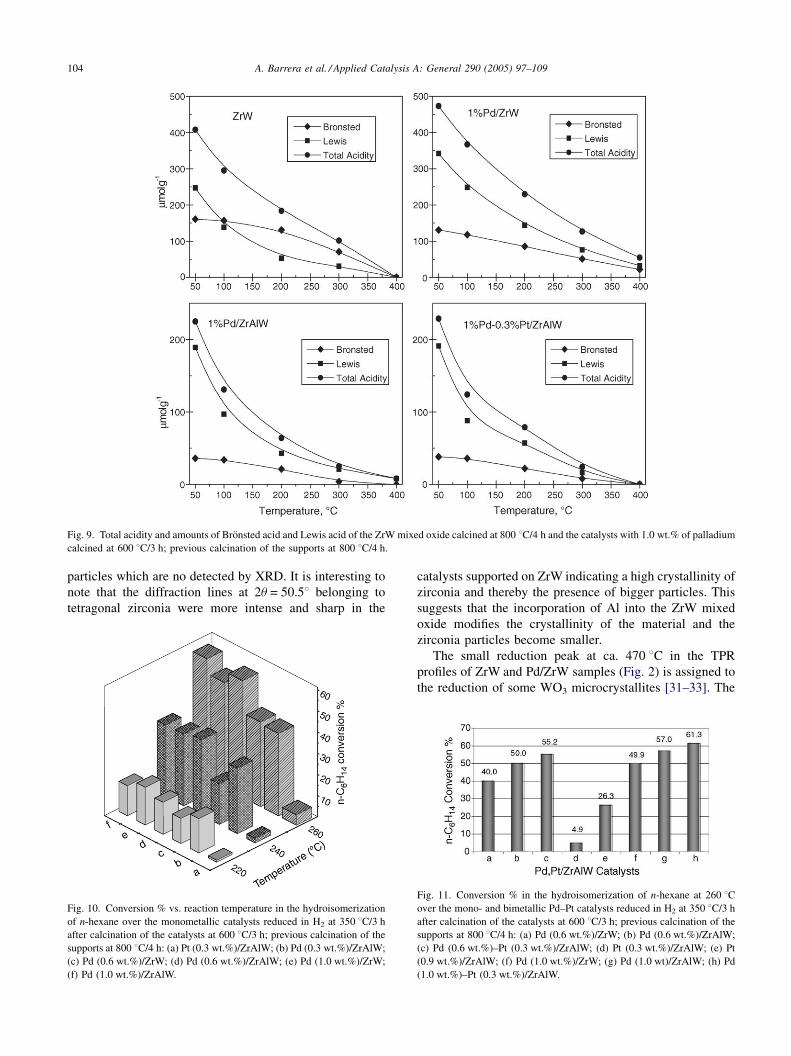
Fig. 8 shows the IR spectra of adsorbed pyridine of the ZrW
mixed oxide and the Pd (1.0 wt.%)/ZrW, Pd (1.0 wt.%)/
ZrAlW, Pd (1.0 wt.%)–Pt (0.3 wt.%)/ZrAlW catalysts after
outgassing increasing temperatures. The IR spectra of the
catalysts contain characteristic bands of pyridine coordina-
tively bonded to Lewis acid sites at about 1443–1460 and
1490–1500 cm�1, whereas the bands at about 1540 cm�1 are
assigned to protonation of pyridine by Bronsted sites. The
intensity of these bands is retained after rising the temperature
up to 400 8C in the IR spectra of Pd (1.0 wt.%)/ZrW whereas
those of Pd (1.0 wt.%)/ZrAlW and Pd (1.0 wt.%)–Pt (0.3%)/
ZrAlW are almost completely removed at 300 8C. Fig. 9
shows that the acidity of the catalysts varied in the order:
ZrW < Pd (1.0 wt.%)/ZrW and Pd (1.0 wt.%)/ZrW > Pd
(1.0 wt.%)/ZrAlW � Pd (1.0 wt.%)–Pt (0.3 wt.%)/ZrAlW.
3.8. Activity test; the isomerization of n-hexane
3.8.1. Catalytic activity at different temperatures over
the monometallic catalysts
The conversion (%) in the hydroisomerization of n-
hexane for the monometallic Pd and Pt catalysts at different
reaction temperatures are depicted in Fig. 10. By comparing
the n-C6H14 conversion (%) of the mono metallic catalysts,
Pd (0.3 wt.%)/ZrAlW catalyst (Fig. 10b) with that of Pt
(0.3 wt.%)/ZrAlW (Fig. 10a), it is observed that the Pd
catalyst developed considerable higher activity than the Pt
catalysts in the whole temperature range. At 220 8C, the n-
hexane conversion of the catalysts was in the range 12–17%.
At 240 8C, the n-C6H14 conversion over the Pd catalysts
increased to 19–36% with the less active one being the Pd
(0.6 wt.%)/ZrW and the more active one being the Pd
(0.6 wt.%)/ZrAlW, Pd (1.0 wt.%)/ZrW and Pd (1.0 wt.%)/
ZrAlW catalysts (Fig. 10d–f). At 260 8C, the activity for the
Pd catalysts increased to 39–57% and the increase in the
activity by the addition of alumina becomes more clear as
well as the increase in the activity with the concentration of
palladium.
3.8.2. Catalytic activity over the mono- and bimetallic
Pd (x = 0.6, 1.0 wt.%)–Pt (0.3 wt.%) catalysts
Fig. 11 shows the n-hexane conversion (%) at 260 8C for
a series of mono- and bimetallic Pd (x)–Pt (0.3 wt.%)
catalysts (x = 0.6 wt.% (Fig. 11a–c), 1.0 wt.% (Fig. 11f–
h)). In order to study the effect of Pt in the catalysts, the n-
hexane conversion % over the catalysts with Pt (0.3,
0.9 wt.%) supported on ZrAlW are also depicted (Fig. 11d
and e). It can be observed that the n-hexane conversion in
the Pd (0.6 wt.%) catalysts increases in the order: Pd/ZrW
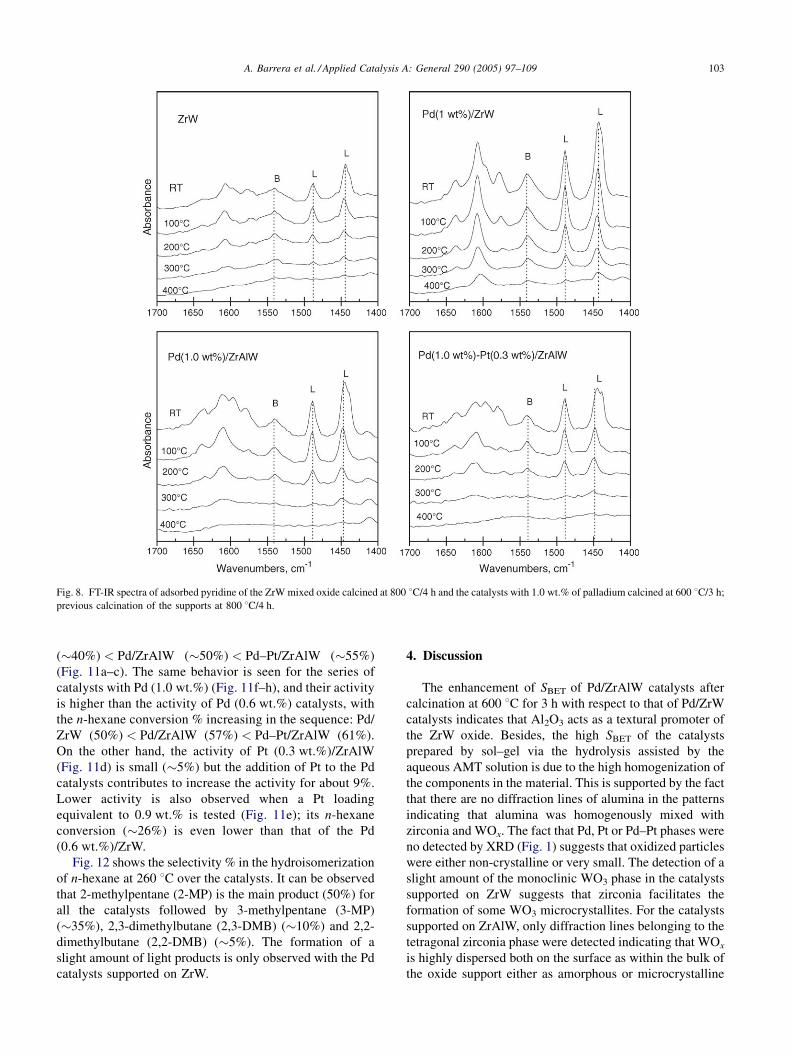
A. Barrera et al. / Applied Catalysis A: General 290 (2005) 97–109 103
Fig. 8. FT-IR spectra of adsorbed pyridine of the ZrW mixed oxide calcined at 800 8C/4 h and the catalysts with 1.0 wt.% of palladium calcined at 600 8C/3 h;
previous calcination of the supports at 800 8C/4 h.
(�40%) < Pd/ZrAlW (�50%) < Pd–Pt/ZrAlW (�55%)
(Fig. 11a–c). The same behavior is seen for the series of
catalysts with Pd (1.0 wt.%) (Fig. 11f–h), and their activity
is higher than the activity of Pd (0.6 wt.%) catalysts, with
the n-hexane conversion % increasing in the sequence: Pd/
ZrW (50%) < Pd/ZrAlW (57%) < Pd–Pt/ZrAlW (61%).
On the other hand, the activity of Pt (0.3 wt.%)/ZrAlW
(Fig. 11d) is small (�5%) but the addition of Pt to the Pd
catalysts contributes to increase the activity for about 9%.
Lower activity is also observed when a Pt loading
equivalent to 0.9 wt.% is tested (Fig. 11e); its n-hexane
conversion (�26%) is even lower than that of the Pd
(0.6 wt.%)/ZrW.
Fig. 12 shows the selectivity % in the hydroisomerization
of n-hexane at 260 8C over the catalysts. It can be observed
that 2-methylpentane (2-MP) is the main product (50%) for
all the catalysts followed by 3-methylpentane (3-MP)
(�35%), 2,3-dimethylbutane (2,3-DMB) (�10%) and 2,2-
dimethylbutane (2,2-DMB) (�5%). The formation of a
slight amount of light products is only observed with the Pd
catalysts supported on ZrW.
4. Discussion
The enhancement of SBET of Pd/ZrAlW catalysts after
calcination at 600 8C for 3 h with respect to that of Pd/ZrW
catalysts indicates that Al2O3 acts as a textural promoter of
the ZrW oxide. Besides, the high SBET of the catalysts
prepared by sol–gel via the hydrolysis assisted by the
aqueous AMT solution is due to the high homogenization of
the components in the material. This is supported by the fact
that there are no diffraction lines of alumina in the patterns
indicating that alumina was homogenously mixed with
zirconia and WOx. The fact that Pd, Pt or Pd–Pt phases were
no detected by XRD (Fig. 1) suggests that oxidized particles
were either non-crystalline or very small. The detection of a
slight amount of the monoclinic WO3 phase in the catalysts
supported on ZrW suggests that zirconia facilitates the
formation of some WO3 microcrystallites. For the catalysts
supported on ZrAlW, only diffraction lines belonging to the
tetragonal zirconia phase were detected indicating that WOx
is highly dispersed both on the surface as within the bulk of
the oxide support either as amorphous or microcrystalline
A. Barrera et al. / Applied Catalysis A: General 290 (2005) 97–109104
Fig. 9. Total acidity and amounts of Bronsted acid and Lewis acid of the ZrW mixed oxide calcined at 800 8C/4 h and the catalysts with 1.0 wt.% of palladium
calcined at 600 8C/3 h; previous calcination of the supports at 800 8C/4 h.
particles which are no detected by XRD. It is interesting to
note that the diffraction lines at 2u = 50.58 belonging to
tetragonal zirconia were more intense and sharp in the
Fig. 10. Conversion % vs. reaction temperature in the hydroisomerization
of n-hexane over the monometallic catalysts reduced in H2 at 350 8C/3 h
after calcination of the catalysts at 600 8C/3 h; previous calcination of the
supports at 800 8C/4 h: (a) Pt (0.3 wt.%)/ZrAlW; (b) Pd (0.3 wt.%)/ZrAlW;
(c) Pd (0.6 wt.%)/ZrW; (d) Pd (0.6 wt.%)/ZrAlW; (e) Pd (1.0 wt.%)/ZrW;
(f) Pd (1.0 wt.%)/ZrAlW.
catalysts supported on ZrW indicating a high crystallinity of
zirconia and thereby the presence of bigger particles. This
suggests that the incorporation of Al into the ZrW mixed
oxide modifies the crystallinity of the material and the
zirconia particles become smaller.
The small reduction peak at ca. 470 8C in the TPR
profiles of ZrW and Pd/ZrW samples (Fig. 2) is assigned to
the reduction of some WO3 microcrystallites [31–33]. The
Fig. 11. Conversion % in the hydroisomerization of n-hexane at 260 8Cover the mono- and bimetallic Pd–Pt catalysts reduced in H2 at 350 8C/3 h
after calcination of the catalysts at 600 8C/3 h; previous calcination of the
supports at 800 8C/4 h: (a) Pd (0.6 wt.%)/ZrW; (b) Pd (0.6 wt.%)/ZrAlW;
(c) Pd (0.6 wt.%)–Pt (0.3 wt.%)/ZrAlW; (d) Pt (0.3 wt.%)/ZrAlW; (e) Pt
(0.9 wt.%)/ZrAlW; (f) Pd (1.0 wt.%)/ZrW; (g) Pd (1.0 wt)/ZrAlW; (h) Pd
(1.0 wt.%)–Pt (0.3 wt.%)/ZrAlW.

A. Barrera et al. / Applied Catalysis A: General 290 (2005) 97–109 105
Fig. 12. Selectivity % in the hydroisomerization of n-hexane at 260 8C over
the mono- and bimetallic Pd–Pt catalysts reduced in H2 at 350 8C/3 h after
calcination of the catalysts at 600 8C/3 h; previous calcination of the
supports at 800 8C/4 h: (a) Pt (0.3 wt.%)/ZrAlW; (b) Pd (0.3 wt.%)/ZrAlW;
(c) Pd (0.6 wt.%)/ZrW; (d) Pd (0.6 wt.%)/ZrAlW; (e) Pd (0.6 wt.%)–Pt
(0.3 wt.%)/ZrAlW; (f) Pd (1.0 wt.%)/ZrW; (g) Pd (1.0 wt.%)/ZrAlW; (h) Pd
(1.0 wt.%)–Pt (0.3 wt.%)/ZrAlW.
disappearance of this reduction peak in the TPR profiles of
the catalysts supported on ZrAlW suggests that the addition
of Al to the ZrW mixed oxide support prevent the formation
of WO3 microcrystallites. The broad shoulder observed
between 600 and 900 8C in the TPR profiles is assigned to
the reduction of dimeric tungstate species in tetrahedral
coordination [31,34], whereas the intense reduction peak
observed at temperatures higher than 800 8C corresponds to
the reduction of isolated monomeric tungstate species in
tetrahedral coordination which are strongly bound to ZrO2
and Al2O3 [31,34]. No reduction peak attributed to the
presence of octahedral tungstate species was detected in the
TPR profiles. Therefore, only monomeric and dimeric
tungstate species in tetrahedral coordination are present in
the catalysts as inferred by the inflection point separating the
overlapped reduction peaks. The shifting in the reduction
temperature for about 40 8C higher in the catalysts supported
on ZrAlW indicates that Al3+ interacts strongly with the
tetrahedrally coordinated tungstate species retarding their
reduction. Since no reduction peaks are observed at
temperatures lower than 200 8C in the Pt/ZrAlW catalysts,
it is deduced that platinum oxide species reduce at room
temperature or lower and that there is a negligible interaction
with the tungstate species. However, the reduction peak at
ca. 80 8C in the TPR profiles of the samples is observed only
in the catalysts containing Pd, therefore this peak must be
assigned to the reduction of PdO species. This reduction
peak is observed for about 45 8C higher than that reported
for the reduction of PdO supported on zirconia [35]
suggesting the existence of a strong interaction between PdO
and the surface WOx species of the mixed oxide supports.
The variation of PdO reducibility at ca. 80 8C (Table 1) in the
sequence Pd (0.6 wt.%)/ZrW < Pd (0.6 wt.%)/ZrAlW < Pd
(0.6 wt.%)–Pt (0.3 wt.%)/ZrAlW, and Pd (1.0 wt.%)/
ZrW > Pd (1.0 wt.%)/ZrAlW > Pd (1.0 wt.%)–Pt (0.3
wt.%)/ZrAlW could be related to the interaction extent
between PdO and the surface WOx species since the atomic
W/(Zr + Al) ratio (Table 2) in the catalysts varied in the
same way as the H2 uptake, i.e. the atomic ratio of tungsten
increases in the Pd (0.6 wt.%) catalysts whereas it decreases
in the Pd (1.0 wt.%) catalysts. This observation suggests that
the variation in the reducibility of PdO could depend on both
the load of palladium as well as the amount of surface WOx
species interacting with PdO. The hydrogen uptake for this
reduction peak corresponds to less than 100% of reduction
of PdO species. Thereby, it is assumed that an important
amount of PdO species are already reduced at room
temperature or lower, which is evidenced by the small
negative peak at ca. 70 8C in the TPR profile of Pd
(0.6 wt.%)/ZrW indicating the decomposition of palladium
hydride which is formed upon absorption of hydrogen on
Pd0 [36]. This observation agrees with the references that
report that a high amount of metallic palladium is already
present in Pd/WO3 and Pd/WO3/Al2O3 samples before any
hydrogen treatment which was ascribed to an autoreduction
of the metallic precursor containing chlorine [37].
The presence of the surface WOx species is corroborated
from the W 4f XP spectra of the catalysts calcined at 600 8C/
3 h (Figs. 3–5). However, a striking result is the coexistence
of various oxidation states as observed after deconvolution
of the XP spectra. The lower BE of 31.97 eVobserved in the
first component of the W 4f XP spectra of the bimetallic Pd
(0.6 wt.%)–Pt (0.3 wt.%)/ZrAlW catalyst (Table 2) is close
to the BE value of 32.0 eV assigned to the presence of W0
[20]. However, this BE could correspond to the so-called b-
phase (W3O) which is metastable and it is almost impossible
to distinguish from metallic tungsten by XPS [37]. The
maximum BE value for the first component of W 4f XP
spectra is observed in the Pd (1.0 wt.%)/ZrW catalyst and
corresponds to the presence of W5+ species [20–24,26].
Whereas, the BE between 33.0 and 33.59 eVobserved in the
remainder catalysts are in the range corresponding to W4+
[20–24,26].
Concerning the presence of reduced-state WOx species in
the calcined samples, it has been suggested that a
condensation phenomenon of surface monomeric tungstate
species leading to polymeric species could explain the
formation of W5+ in WO3/ZrO2 [26]. It is also known that the
reduction of W6+–W4+ can occur by injection of trapped
electrons into the WO3 lattice [39]. In our catalysts a high
amount of Pd0 and Pt0 is already present before any
hydrogen treatment, then, an electronic transfer from the
noble metal to the surface WOx species could occur due to
interaction effects [40]. Moreover, the reduction of WOx
species could be facilitated by the delocalization of the
negative charge onto the WOx network [41], promoted in our
opinion by the attractive potential of coordinative unsatu-
rated Zr4+ and Al3+ ions (Lewis acid sites) of the Zr–O–W
and Zr–O–Al–O–W mixed oxides.

A. Barrera et al. / Applied Catalysis A: General 290 (2005) 97–109106
The BE in the range 36.03 and 36.44 eV observed in the
second component of the W 4f XP spectra (Table 2) are close
to the values assigned to hexavalent tungsten compounds
(W6+) corresponding to either WO3 or Al2(WO4)3 species
[26–28]. Since Al3+ interacts strongly with the tetrahedrally
coordinated tungstate species in Pd (0.6 wt.%) catalysts
supported on ZrAlW, as inferred from their TPR profiles
(Fig. 2), it is assumed that these BE must correspond to the
presence of Al2(WO4)3 species. Whereas, the BE of
35.69 eV observed in the W 4f XP spectra of Pd
(1.0 wt.%)/ZrW is close to the BE value of 35.7 eV assigned
to W6+ species [11,27], confirming the formation of some
WO3 microcrystallites on the surface of Pd/ZrW catalysts.
The BE of 34.93 and 35.12 eV observed in the W 4f XP
spectra of Pt (0.3 wt.%)/ZrAlW and Pd (1.0 wt.%)–Pt
(0.3 wt.%)/ZrAlW catalysts are slightly lower than the BE
value of 35.2 eV (W6+) of the standard WO3.
The BE (37.51–38.68 eV) observed in the third
component of the XP spectra are higher than those reported
of WO2Cl2 (37.1 eV) or WOCl4 (37.4 eV) [30]. These high
BE are due to the presence of tungsten compounds having
highly electronegative ligands [38]. It is known that residual
chlorine is remaining when WO3 is impregnated with
metallic precursors containing chlorine even after calcina-
tion of the catalysts at 700 8C, the relative amount of which
depends on the WO3 loading [23]. Since the ZrWand ZrAlW
mixed oxide supports were impregnated from PdCl2 and
H2PtCl6�xH2O, then residual chlorine could be located on
tungsten oxide to form WxOyCl compounds [37].
Regarding the structure of the surface WOx species,
Raman spectra show that isolated monomeric tungstate
species in tetrahedral coordination ([WO4] units) are present
in the ZrAlW mixed oxide and Pd (1.0 wt.%) catalysts as
inferred by the band at about 926 cm�1 (W O stretching
mode [15]) (Fig. 6). The existence of two-dimensional
mono-oxotungstate species is also deduced from the band at
1013 cm�1 (W O stretching mode) [8,41–43]. According to
Iglesia et al. [41], these terminal W O bonds are common in
well-dispersed WOx domains which are observed at tungsten
surface coverage lower than the theoretical monolayer
(�7 W atoms nm�2). Besides, no Raman bands at about
300, 600, 807, and 830 cm�1 characteristics of W–O–W
linkages [8,15,31,41–43] or 270, 715, and 800 cm�1
ascribed to bulk WO3 [8,15,43] are present in the catalysts.
Table 327Al NMR–MAS spectra of mono- and bimetallic Pd–Pt catalysts calcined at 60
Catalysts Population %
P1, octahedral (�2.32 to 1.51 ppm)
Pd (0.6 wt.%)/ZrW –
Pd (0.6 wt.%)/ZrAlW 90.47
Pd (0.6 wt.%)–Pt (0.3 wt.%)/ZrAlW 86.71
Pt (0.3 wt.%)/ZrAlW 88.28
Pd (1.0 wt.%)/ZrW –
Pd (l.0 wt.%)/ZrAlW 64.10
Pd (1.0 wt.%)–Pt (0.3 wt.%)/ZrAlW 70.39
This observation suggests the lack of W–O–W connectivity
and confirms that the WOx species are highly dispersed on
the surface of the mixed oxides. In our catalysts, the
theoretical WOx surface coverage (Table 2) for 15 wt.% of
tungsten oxide is slightly below of that predicted for a two-
dimensional polytungstate monolayer (�7 W atoms nm�2)
[41], which is due to the high dispersion of WOx species as
on the surface as within the bulk of the mixed oxide
supports. It is well known that the formation of octahedrally
coordinated tungstate species requires WOx surface cov-
erages higher than a monolayer [26]. Then, it is suggested
that the structure of the highly dispersed WOx species in
these catalysts consists mainly of oxotungstate species in
which the tungsten is in tetrahedral coordination form. This
is corroborated from their TPR profiles because there is no
reduction peaks assigned to octahedrally coordinated WOx
species, and monomeric and dimeric WOx species in
tetrahedral coordination are predominant in the catalysts
(Fig. 2). Besides, the reduction of a slight amount of WO3
microcrystallites is observed only in the Pd/ZrW catalysts
having the higher WOx surface coverage (Fig. 2, Table 2).
Therefore, we can conclude on the basis of TPR and Raman
experiments that the structure of the surface WOx species in
the catalysts consists of oxotungstate species which are
constituted mainly of monomeric and dimeric species in
tetrahedral coordination.
The incorporation of Al within the Zr–O–W mixed oxide
framework prepared by sol–gel is suggested from the 27Al
NMR spectra of the mono- and bimetallic Pd–Pt catalysts
supported on ZrAlW (Fig. 7, Table 3) by the resonance line
observed in the 54.9–64.6 ppm region (P3) attributed to AlIV
species in tetrahedral coordination [12,44–46]. The higher
amount of tetrahedral AlIV species in the catalysts with
1.0 wt.% of palladium could be due to a high redispersion of
Al atoms caused by a higher redissolution of alumina
aggregates or to a higher transformation of the octahedral
AlVI species during calcination. The presence of bulk
alumina aggregates is inferred from the main intense
resonance line in the �1.7 to 1.1 ppm region (P1) of the 27Al
NMR spectra which is associated to AlVI species in an
octahedral coordination and represent AlVI�O� units [44].
The transformation of AlVI species into AlIV species is
corroborated by the presence of unsaturated aluminum ions
in octahedral position as deduced by the resonance line
0 8C/3 h: population % of each deconvoluted spectral component
P2, pentahedral (26.3–35.17 ppm) P3, tetrahedral (57.13–64.62 ppm)
– –
1.28 8.23
5.18 8.18
4.22 7.49
– –
9.84 26.00
9.17 20.40

A. Barrera et al. / Applied Catalysis A: General 290 (2005) 97–109 107
observed in the 22.2–27.1 ppm region (P2) which is close to
the chemical shift produced by five oxygen atoms
surrounding an aluminum nucleus (AlV pentacoordinated
species) [45].
The activity of palladium catalysts in the hydroisome-
rization of n-hexane is promoted by both, the addition of Al
to the ZrW oxide support and the addition of Pt to the
catalyst. The addition of Al to the ZrW oxide improves the n-
C6H14 conversion in the palladium catalysts for about 25%,
whereas the addition of Pt to the catalysts contributes to
increase the activity for about 10% (Fig. 11). The lower
activity of the palladium catalysts supported on ZrW is
correlated with the presence of some surface WO3
microcrystallites as deduced from the BE of 35.69 eV
assigned to W6+ species (Table 2). However, these surface
W6+ species coexist with W5+ species (BE of 34.18 eV). The
presence of surface W5+ in Pd/ZrW catalysts could be
related to the formation of the nonstoichiometric W20O58
(WO2.59) phase which is constituted of W6+ and W5+ cations
[26,37,47]. Moreover, the existence of cracking leading to a
slight amount of light products as well as skeletal
isomerization to form 2MP, 3MP, 2,2DMB and 2,3DMB
in the Pd/ZrW catalysts (Fig. 12c and f) suggests the
participation of strong acid sites. This is corroborated by the
intensity of the bands at 1445, 1489 cm�1 (Lewis sites), and
1540 cm�1 (Bronsted sites) that is retained up to 400 8C in
the FT-IR spectra of adsorbed pyridine of the Pd (1.0 wt.%)/
ZrW catalyst, whereas those of ZrW, Pd (1.0 wt.%)/ZrAlW
and Pd (1.0 wt.%)–Pt (0.3 wt.%)/ZrAlW are completely
removed at this temperature (Fig. 8), inferring therefore that
the acid strength of this catalyst was stronger. Besides, the
acidity of the Pd (1.0 wt.%)/ZrW catalyst was enhanced with
respect to that of the ZrW mixed oxide due to the increase in
the number of Lewis acidic sites (Fig. 9) although the
number of Bronsted acid sites was keep nearly constant.
Therefore, we assume that the strong acidic sites of the Pd/
ZrW catalysts would be generated by the coexistence of the
surface W6+ and W5+ species in intimate contact with
palladium.
Nevertheless, a striking result in our work is that the
improvement in the activity of the palladium catalysts
promoted by Al is correlated with a lower acid strength and
acidity than those of Pd/ZrW as observed from the
disappearance of the bands at 1448, 1490 cm�1 (Lewis
sites), and 1540 cm�1 (Bronsted sites) at 300 8C in the FT-IR
spectra of the Pd (1.0 wt.%)/ZrAlW catalyst (Fig. 8) and by
the decrease in the total amount of Bronsted and Lewis sites
of this catalyst (Fig. 9). The lower acid strength and the
decrease in the amount of acid sites of ZrAlW with respect to
that of ZrW is also confirmed from its FT-IR spectra of
adsorbed pyridine (figure not shown). The lower acidic
character of Pd/ZrAlW suggests that the nature of the
surface WOx species must be different than that in Pd/ZrW
catalysts. This is corroborated from the W 4f XP spectra of
the Pd (0.6 wt.%)/ZrAlW catalyst where W6+ cations (BE of
36.03 eV) corresponding to Al2(WO4)3 species coexist with
W4+ cations (BE of 33.0 eV) attributed to WO2 species and
from its Raman spectra showing a higher amount of mono-
oxotungstate species in tetrahedral coordination (Fig. 6).
Therefore, we suggest from the above-mentioned
considerations that the improvement in the activity of Pd/
ZrAlW catalysts with respect to that of Pd/ZrW is associated
to a lower acid strength and acidity of the catalyst, which can
be correlated to the coexistence between surface W6+
species and reduced-state WOx species (either W4+ or W0)
forming tetrahedrally coordinated oxotungstate species.
Another possibility of the decrease in the acidity of Pd/
ZrAlW catalysts could be due to the generation of Lewis
basic sites (coordinative unsaturated oxide ions of alumina)
during the calcination of the ZrAlW supports at 800 8C. It is
well known that the generation of Lewis basic sites in
alumina occurs easily by dehydration when a proton
bounded to an oxygen atom bridging octahedral and
tetrahedral aluminum atoms is combined with a basic
hydroxyl of an aluminum atom in octahedral coordination
[48]. Therefore, by the regular dehydration process of
alumina both AlV pentacoordinated atoms (Lewis acid sites)
and coordinative unsaturated (cus) oxygen atoms (Lewis
basic sites) are generated. In our catalysts a higher
population of AlV species (Lewis acid sites) is observed
in the 27Al NMR spectra of the Pd (1.0 wt.%)/ZrAlW and Pd
(1.0 wt.%)–Pt (0.3 wt.%)/ZrAlW (Table 3), which are the
more active catalysts. These catalysts have a lower acid
strength and amount of acid sites than that of Pd (1.0 wt.%)/
ZrW, therefore we infer that these catalysts should contain
also a high amount of cus oxide ions of alumina (Lewis basic
sites). Thereby, we believe that the lower acid strength and
the decrease in the amount of acid sites in Pd/ZrAlW could
be related to both the coexistence W6+ and reduced-state
WOx species (W4+ or W0) forming tetrahedrally coordinated
oxotungstate species and the presence of Lewis basic sites of
alumina. This assumption suggests a participation of both
Lewis acid sites (reduced-state WOx species and cus AlV
atoms) as well as Lewis basic sites (cus oxide ions of
alumina) on the skeletal isomerization of n-hexane which
must influence the catalytic selectivity. The lower acidic
character could explain the absence of cracking in the mono-
and bimetallic palladium catalysts supported on ZrAlW.
The coexistence between different oxidation states of
tungsten is also observed in the Pd (0.6 wt.%)–Pt (0.3 wt.%)/
ZrAlW catalyst where the surface W6+ cations (BE of
36.44 eV) assigned to Al2(WO4)3 species coexist with the
highly reduced tungsten oxide phase, W3O (BE of 31.97 eV)
which could be formed from the W20O58 phase [37,47]. The
same observation occurs with the most active Pd (1.0 wt.%)–
Pt (0.3 wt.%)/ZrAlW catalyst, where W6+ cations (BE of
35.12 eV) coexist with W4+ species (BE of 33.59 eV)
corresponding to WO2. Because the acidity of this catalyst
was similar to that of the Pd (1.0 wt.%)/ZrAlW, it is deduced
that neither the presence of a slightly lower amount of
pentacoordinate AlV species nor the addition of Pt to the Pd/
ZrAlW catalyst modifies significantly its acidic character.

A. Barrera et al. / Applied Catalysis A: General 290 (2005) 97–109108
Moreover, the improvement in the activity of the bimetallic
Pd–Pt catalysts is not only due to a higher load of metal as
confirmed by comparing the activity of the Pd (0.6 wt.%)–Pt
(0.3 wt.%)/ZrAlW with that of Pt (0.9 wt.%)/ZrAlW having
the same load of noble metal (Fig. 11c and e), since the n-
hexane conversion of the last catalyst was nearly the half of
the former. Therefore, we presume that the improvement in
the activity of the bimetallic Pd–Pt/ZrAlW catalysts could
be due to a modification of the intrinsic properties of surface
palladium atoms induced by the neighboring platinum
atoms.
Although, the properties responsible for the acidic
character in the palladium catalysts are also observed in
the Pt (0.3 wt.%)/ZrAlW, such as for instance the coex-
istence between W4+ species (BE of 33.42 eV) and W6+
species (BE of 34.93 eV) and an important population % of
pentacoordinated AlV species, however its lower activity
compared with that of the mono- and bimetallic Pd–Pt/
ZrAlW catalysts (Figs. 10 and 11) can be correlated to a
negligible interaction between Pt and WOx species as
inferred from its TPR profile. Another possibility would be
the sintering of platinum upon calcination at 600 8C, since it
is well known that platinum is less resistant to thermal
sintering than palladium [49].
The higher n-hexane conversion of the catalysts with
1.0 wt.% of palladium compared with that of the 0.6 wt.% of
Pd catalysts having the same composition of the mixed oxide
support could be due to a higher density of accessible
metallic sites to dissociate hydrogen required in the
dehydrogenation and rehydrogenation steps in the hydro-
isomerization of n-hexane. The mono- and bimetallic Pd–Pt/
ZrAlW catalysts are complex systems due to many factors
involved in their catalytic performance. In order to elucidate
the role of palladium in the Pd/ZrAlW catalysts as well as
the promotion effect of Pt on the improvement in the
catalytic activity in the hydroisomerization of n-hexane
further experiments must be performed.
5. Conclusions
(1) Addition of Al2O3 to the ZrW mixed oxides prepared by
sol–gel via the hydrolysis assisted by the aqueous
ammonia metatungstate solution generates solids with
higher specific surface area.
(2) Z
irconia in the tetragonal phase predominates in themixed oxides and mono- and bimetallic Pd–Pt catalysts,
whereas the addition of Al2O3 to ZrW modifies its
crystallinity and avoids the formation of WO3 bulk
nanocrystals. In these materials, the WOx species are
found highly dispersed.
(3) T
here is a coexistence of various oxidation states oftungsten on the surface of the mono- and bimetallic Pd–
Pt catalysts after calcination. The presence of reduced-
state WOx species could be ascribed to either an
electronic transfer from the noble metal to the surface
WOx species or the delocalization of the negative charge
onto the WOx network.
(4) T
he structure of the highly dispersed WOx species inthese materials consists of oxotungstate species which
are constituted mainly of monomeric and dimeric
species in tetrahedral coordination.
(5) T
he activity of Pd/ZrW catalysts in the hydroisomeriza-tion of n-hexane is promoted by both the addition of Al
to the ZrW mixed oxide and the addition of Pt to the Pd/
ZrAlW catalysts.
(6) T
he improvement in the activity of Pd/ZrAlW catalystsis associated to a lower acid strength and acidity of the
catalyst, which can be related to the coexistence of
surface W6+ and reduced-state WOx species (either W4+
or W0) forming tetrahedrally coordinated oxotungstate
species. The Lewis basic sites of alumina should not be
discarded.
(7) T
he lower acid strength and the amount of acid sites ofthe mono-Pd and bimetallic Pd–Pt catalysts supported
on ZrAlW suggests a participation of both Lewis acid
sites (reduced-state WOx species and cus AlV atoms) as
well as Lewis basic sites (cus oxide ions of alumina) on
the skeletal isomerization of n-hexane.
(8) T
he addition of Pt to the Pd/ZrAlW catalyst does notmodify significantly its acidic character. Therefore, the
improvement in the activity in the bimetallic Pd–Pt/
ZrAlW catalysts could be due to a modification of the
intrinsic properties of surface palladium atoms induced
by the neighboring platinum atoms.
Acknowledgements
The authors gratefully acknowledge the technical
assistance of Mr. Carlos Franco in the development of the
RMN experiments. A. Barrera thanks to the Instituto
Mexicano del Petroleo for the scholarship granted during his
post-doctoral stay.
References
[1] A. Corma, J.M. Serra, A. Chica, Catal. Today 81 (2003) 405.
[2] Y. Ono, Catal. Today 81 (2003) 3.
[3] Chong-J. Cao, S. Han, Chang-L. Chen, Nan-P. Xu, Chuny-Y. Mou,
Catal. Commun. 4 (2003) 511.
[4] S.T. Sie, in: G. Ertl, H. Knozinger, J. Weitkamp (Eds.), Handbook of
Heterogeneous Catalysis, vol. 4, Wiley/VCH, Weinheim, 1997, p.
1998.
[5] M. Hino, S. Kobayashi, K. Arata, J. Am. Chem. Soc. 101 (1979) 6439.
[6] J.G. Santiesteban, D.C. Calabro, W.S. Borghard, C.D. Chang, J.C.
Vartuli, Y.P. Tsao, M.A. Natal-Santiago, R. Bastian, J. Catal. 183
(1999) 314.
[7] S.R. Vaudagna, R.A. Comelli, N.S. Fıgoliı, Appl. Catal. 164 (1997) 265.
[8] S.R. Vaudagna, S.A. Canavese, R.A. Comelli, N.S. Figoliı, Appl.
Catal. 168 (1998) 93.
[9] A.V. Ivanov, T.V. Vasina, O.V. Masloboishchikova, E.G. Khelkovs-
kaya-Sergeeva, L.M. Kustov, J.I. Houzvicka, Catal. Today 73 (2002)
95.

A. Barrera et al. / Applied Catalysis A: General 290 (2005) 97–109 109
[10] E. Iglesia, D.G. Barton, S.L. Soled, S. Miseo, J.E. Baumgartner, W.E.
Gates, G.A. Fuentes, G.D. Meitzner, Stud. Surf. Sci. Catal. 101 (1996)
533.
[11] M. Arribas, F. Marquez, A. Martınez, J. Catal. 190 (2000) 314.
[12] S.-T. Wong, T. Li, S. Cheng, J.-F. Lee, C.-Y. Mou, J. Catal. 215 (2003) 45.
[13] C.-L. Chen, T. Li, S. Cheng, N.-P. Xu, C.-Y. Mou, Catal. Lett. 78
(2002) 223.
[14] W. Hua, Y. Xia, Y. Yue, Z. Gao, J. Catal. 196 (2000) 104.
[15] C. Bigey, L. Hilaire, G. Maire, J. Catal. 198 (2001) 208.
[16] Y. Ono, in: J.M. Thomas, K.I. Zamaraev (Eds.), Perspectives in
Catalysis, Blackwell, London, 1992, p. 533.
[17] D. Barton, Stuart L. Soled, G.D. Meitzner, G.A. Fuentes, E. Iglesia, J.
Catal. 181 (1997) 57.
[18] K. Masuda, M. Kawai, K. Kuno, N. Kachi, F. Mizukami, in: V.G.
Poncelet, P.A. Jacobs, P. Grange (Eds.), Preparation of Catalysts, Stud.
Surf. Sci. and Catal., vol. 63, Elsevier, Amsterdam, 1991, p. 229.
[19] S. Kumar, D.R. Chopra, G.L. Smith, J. Vac. Sci. Technol. B 10 (1992)
1218.
[20] M.A. Arribas, F. Marquez, A. Martınez, J. Catal. 190 (2000) 309.
[21] L. Salvati, L.E. Makovsky, J.M. Stencel, F.R. Brown, D.M. Hercules,
J. Phys. Chem. 85 (1981) 3700.
[22] M. Occhiuzzi, D. Cordischi, D. Gazzoli, M. Valigi, P.C. Heydorn,
Appl. Catal. A: Gen. 269 (2004) 169.
[23] J.R. Regalbuto, T.H. Fleish, E.E. Wolf, J. Catal. 107 (1987) 114.
[24] J.F. Fiedor, A. Proctor, M. Houalla, D.M. Hercules, Surf. Interf. Anal.
23 (1995) 204.
[25] M. Regragui, M. Addou, A. Outzourhit, J.C. Bernede, E.E. Idrissi,
Thin Solid Films 358 (2000) 40.
[26] F. Di Gregorio, V. Keller, J. Catal. 225 (2004) 45.
[27] H.K. Plummer, S. Shinozaki Jr., K.H. Adams, H.S. Gandhi, J. Mol.
Catal. 20 (1983) 251.
[28] W. Grunert, E.S. Shpiro, R. Feldhaus, K. Anders, G.V. Antoshin,
K.H.M. Minachev, J. Catal. 107 (1987) 522.
[29] G.E. McGuire, G.K. Schweitzer, T.A. Carlson, Inorg. Chem. 12 (1973)
2451.
[30] P.G. Gassman, D.W. Maomber, S.M. Willging, J. Am. Chem. Soc. 107
(1985) 2380.
[31] V.M. Benitez, C.A. Querini, N.S. Fıgoli, Appl. Catal. A: Gen. 252
(2003) 427.
[32] I. Wachs, C. Chersich, J. Hardengerg, Appl. Catal. 13 (1985) 335.
[33] V. Benitez, N.S. Fıgoli, Catal. Commun. 3 (2002) 487.
[34] J. Horsley, I. Wachs, J. Brown, G. Via, F. Hardcastle, J. Phys. Chem. 91
(1987) 4014.
[35] K. Fuimoto, F.H. Ribeiro, M. Avalos-Borja, E. Iglesia, J. Catal. 179
(1998) 431.
[36] X.L. Seoane, N.S. Fıgoli, P.C. L’Argentiere, J.A. Gonzalez, A. Arcoya,
Catal. Lett. 47 (1997) 213.
[37] C. Bigey, L. Hilaire, G. Maire, J. Catal. 184 (1999) 406.
[38] M. Sun, T. Burgi, R. Cattaneo, R. Prins, J. Catal. 197 (2001) 172.
[39] R.J. Colton, A.M. Guman, J.W. Rabalais, J. Appl. Phys. 49 (1)
(1978).
[40] P.C. L’Argentiere, N.S. Fıgoli, React. Kinet. Catal. Lett. 64 (2) (1998)
221.
[41] E. Iglesia, Structure and catalytic function of oxide nanostructures, in:
Proceedings of the XIX Simposio Iberoamericano de Catalisis, Mer-
ida, Yucatan, Mexico, September 5–11, 2004.
[42] D.G. Barton, M. Shtein, R.D. Wilson, S.L. Soled, E. Iglesia, J. Phys.
Chem. B 103 (1999) 630.
[43] S. Kuba, P. Lukinskas, R.K. Grasselli, B.C. Gates, H. Knozinger, J.
Catal. 216 (2003) 253.
[44] M.L. Occelli, S. Biz, A. Auroux, P.S. Iyer, Appl. Catal. A: Gen. 179
(1999) 117.
[45] J.A. Wang, X. Bokhimi, A. Morales, O. Novaro, T. Lopez, R. Gomez,
J. Phys. Chem. B 103 (2) (1999) 299.
[46] E. Zhao, S.E. Hardcastle, G. Pacheco, A. Garcia, A.L. Blumenfeld, J.J.
Friapiat, Micropor. Mesopor. Mater. 31 (1999) 9.
[47] C. Bigey, G. Maire, J. Catal. 196 (2000) 224.
[48] H. Knozinger, P. Ratnasamy, Catal. Rev. 17 (1978) 31.
[49] J.C. Summers, D.R. Monroe, Ind. Eng. Chem. Prod. Res. Dev. 20
(1981) 23.





























