Embed Size (px)
DESCRIPTION
testo di ricerca sulla DAAO e sulla sua estrazione
Citation preview

Laboratorio di Metodologie Biochimiche Turno D
Gruppo 2 Edoardo Sirtori
Matricola: 722062
Giorno 1
Purificazione e caratterizzazione della D-AMINOACIDO OSSIDASI da Rhodotorula gracilis in E.coli.
Nella prima giornata di laboratorio ci siamo dedicati all’estrazione della proteina RgDAAO che si accumula in forma solubile nel citoplasma delle cellule di E.coli. Dopo una breve spiegazione teorica sulle tecniche e a ciò di cui ci saremmo occupati, nella prima giornata, abbiamo iniziato la nostra esperienza di laboratorio calcolando quanto tampone di rottura avremmo dovuto inserire all’interno del nostro pellet, il campione di pellet era già stato precedentemente misurato facendo la differenza tra peso netto e tara; in questo caso: 12,9(tara) - 17,9 = 4,5 grammi di pellet ora sapendo quanti grammi di pellet abbiamo all’interno della nostra falcon dobbiamo calcolare il quantitativo di tampone di rottura che dobbiamo inserire per rompere le cellule, sapendo che per ogni grammo di pellet dobbiamo aggiungere 3 / 4 mL di tampone di rottura. quindi facendo una moltiplicazione andiamo a calcolare il quantitativo di tampone da aggiungere: 4,5 (g di pellet) x 3,5 (mL di Tampone per grammo) = 15,75 mL di Tampone di rottura. Una volta aggiunto il tampone di rottura aiutandosi con un puntale abbiamo rotto manualmente il pellet per far si che le cellule di grandi dimensioni vengano rotte. Prima di poter purificare il nostro enzima dobbiamo rompere le pareti cellulari, questo processo richiede l’utilizzo di un sonicatore. Il nostro campione ha eseguito un totale di 5 giri di sonicazione ovvero 30’’ di sonicazione e poi un minuto di riposo nel ghiaccio. una volta terminato questo processo abbiamo lasciato il campione all’assistente che lo avrebbe posto dentro una centrifuga per centrifugare le cellule a 18500 rpm a 4°c per un totale di 60 minuti. Mentre il nostro campione si trovava nella centrifuga abbiamo calcolato la Retta di Taratura mediante il metodo del Reattivo del Biureto della BSA (albumina di siero bovino). il reattivo del Biureto è una soluzione che va a reagire in presenza di ioni Cu2+ e Biureto, le proteine reagiscono a questa soluzione poiché i legami peptidici presentano dei complessi simili a quelli del biureto. Questo metodo viene utilizzato per osservare la concentrazione di proteine presenti in soluzione indipendentemente da quale essa sia poiché il seguente sistema si basa sulla loro concentrazione, questo sistema possiede una scarsa sensibilità per cui possiamo utilizzarlo per osservare le concentrazioni di proteine comprese in un range che va da 0,5 e 2,5 mg/mL. Per questo metodo abbiamo preparato 7 cuvette di plastica da 1mL, che successivamente le andremo a posizionare dentro lo spettrofotometro per calcolarne l’Assorbanza. All’interno delle nostre cuvette andremo a inserire:
- X Volume di soluzione contenente BSA - X di H2O - 0.5 mL di reattivo del Biureto per poter costruire una retta di taratura prendiamo diverse concentrazioni di BSA comprese nel range di osservazione del Biureto. Le concentrazioni prese in esame sono:
1) 0,5 % 2) 0,9% 3) 1,3% 4) 1,7% 5) 2,1% 6) 2,5%
�1

Il totale di soluzione da inserire nelle cuvette deve essere pari a 1mL, quindi dovremo calcolare quanti mL di H2O andremo ad aggiungere. Per poter trovare il volume di BSA da inserire all’interno delle cuvette ci avvaliamo della formula: Vi x Ci = Vf x Cf
Vi = al volume di H2O da aggiungere Ci = concentrazione del biureto sempre a 0,5 mg/mL Vf = volume della cuvetta Cf = concentrazione della proteina BSA scelte da noi ora sapendo ciò possiamo applicare la formula per poter trovare il volume di BSA, che in questo caso diventa: Vi = Vf x Cf / Ci
Applicando la formula trovo che:
1) X = 1mL x 0,5 mg/mL / 0,5 mg/mL = 0,1 mL di BSA 2) X = 1mL x 0,9 mg/mL / 0,5 mg/mL = 0,18 mL di BSA 3) X = 1mL x 1,3 mg/mL / 0,5 mg/mL = 0,26 mL di BSA 4) X = 1mL x 1,7 mg/mL / 0,5 mg/mL = 0,34 mL di BSA 5) X = 1mL x 2,1 mg/mL / 0,5 mg/mL = 0,42 mL di BSA 6) X = 1mL x 2,5 mg/mL / 0,5 mg/mL = 0,5 mL di BSA
Una volta trovato quanti mL di BSA devo inserire nella provetta eseguo un semplice sottrazione per calcolare gli mL di H2O da aggiungere, quindi nelle cuvette andremo ad aggiungere:
1) 0,5 mL - 0,1 mL = 0,4 mL di H2O 2) 0,5 mL - 0,18 mL = 0,32 mL di H2O 3) 0,5 mL - 0,26 mL = 0,24 mL di H2O 4) 0,5 mL - 0,34 mL = 0,16 mL di H2O 5) 0,5 mL - 0,42 mL = 0,08 mL di H2O 6) 0,5 mL - 0,5 mL = 0 mL di H2O
Ora avendo trovato tutti i dati possiamo procedere con la preparazione delle cuvette.
N.B. la settima cuvetta è detta bianco poiché dentro ad essa andrò ad inserire 0,5 mL di Biureto e 0,5 mL di H2O. questo mi servirà una volta trovata l’assorbanza per eliminare l’assorbanza dovuta dalle proteine presenti.
in questa tabella troviamo i valori riscontrati:
�2
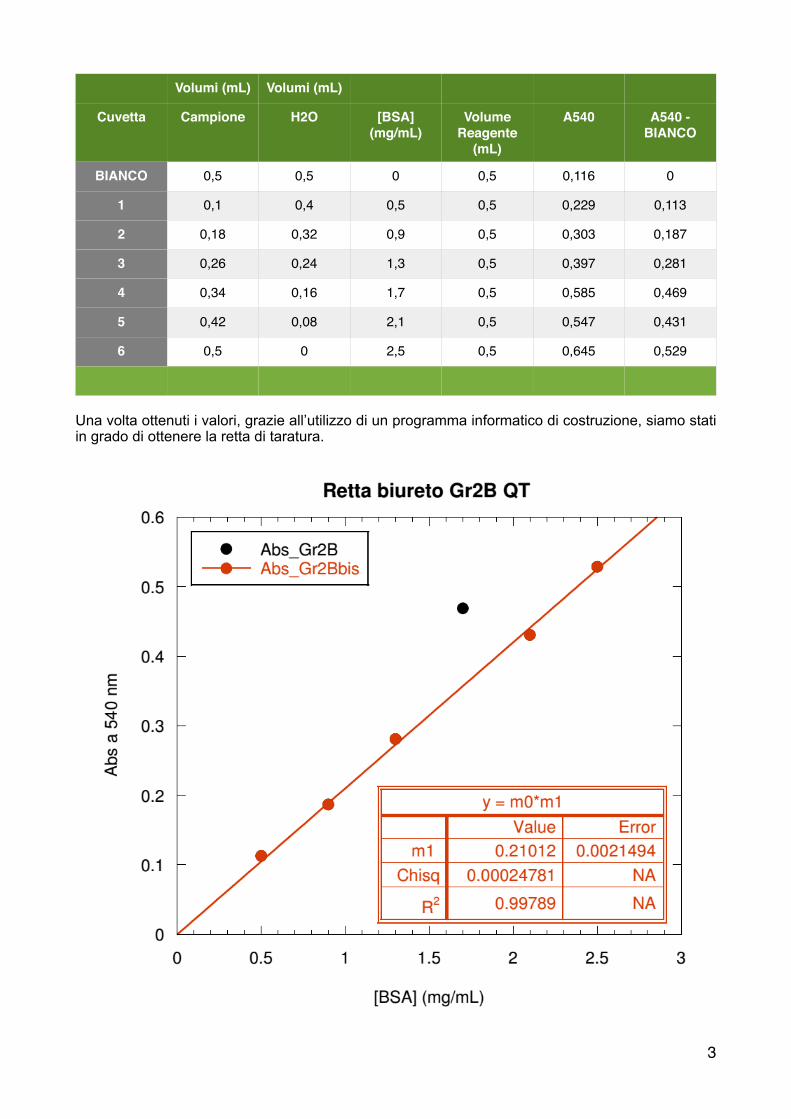
Una volta ottenuti i valori, grazie all’utilizzo di un programma informatico di costruzione, siamo stati in grado di ottenere la retta di taratura.
���
Volumi (mL) Volumi (mL)
Cuvetta Campione H2O [BSA](mg/mL)
Volume Reagente
(mL)
A540 A540 - BIANCO
BIANCO 0,5 0,5 0 0,5 0,116 0
1 0,1 0,4 0,5 0,5 0,229 0,113
2 0,18 0,32 0,9 0,5 0,303 0,187
3 0,26 0,24 1,3 0,5 0,397 0,281
4 0,34 0,16 1,7 0,5 0,585 0,469
5 0,42 0,08 2,1 0,5 0,547 0,431
6 0,5 0 2,5 0,5 0,645 0,529
�3

Y= 0,210x è l’equazione della retta di calibrazione ottenuta interpolando i punti ricavati sperimentalmente. Ponendo sulle ordinate di questa retta il valore di assorbanza di un campione a concentrazione incognita, trattato nelle stesse condizioni di dosaggio, sarà possibile leggere sulle ascisse la corrispondente quantità di sostanza presente nel campione in esame. Dal valore del coefficiente di regressione R² si può dedurre che la retta ha un buon fitting in quanto il valore è molto vicino a 1.
Una volta ottenuta la retta troviamo la Y ovvero la pendenza della retta che è uguale a y= 0,210x. alla fine della giornata, una volta ottenuta la retta abbiamo preso 0,5 mL di estratto grezzo e lo abbiamo posto all’interno di una eppendorf, che avremmo utilizzato per successive analisi.
�4

Giorno 2
Purificazione mediante cromatografia di affinità per metallo chelato in FPLC. Cambio del tampone mediante gel-filtrazione in PD10.
Nella seconda giornata di laboratorio abbiamo purificato il nostro estratto grezzo mediante la cromatografia mediante sistema FPLC (Fast Protein Liquid Chromatography). inizialmente abbiamo unito i campioni dei prime 3 gruppi, dopo di che siamo andati ad aggiungere alla soluzione di estratto grezzo (EG) NaCl al tampone per evitare le interazioni ioniche che si possono andare a generare tra le proteine e la resina. per sapere la quantità di NaCl da aggiungere utilizziamo la seguente formula:
Vi x Ci = Vf x Cf
dove Vi: volume iniziale di soluzione d concentrazione Ci da aggiungere (NaCl) Ci: concentrazione della soluzione iniziale da aggiungere Vf: volume finale Cf: concentrazione finale desiderata
ma poiché FD = Ci/ Cf Vf = VEG + Vi
sostituendo alla fine ottengo la seguente formula: Vi = VEG / (FD - 1) dove
VEG: volume della soluzione iniziale (estratto grezzo) FD: fattore di diluizione, pari a Ci/Cf quindi ottengo che :
Vi = 48 /(5-1) = 48/4 = 12 mL di NaCl
il volume finale ottenuto è di 60 mL.
Una volta preparato il campione lo andiamo a inserire all’interno della colonna, precedentemente preparata col nichel, all’interno del Super Loop (valvola di iniezione da 50 mL), eseguiamo due caricamenti per purificare l’interno campione. una volta caricato il campione. seguiamo la sua corsa grazie al computer che ci mostra l’assorbanza. lentamente osserviamo come alla resina si lega la proteina poiché essa si colora. una volta che abbiamo caricato tutto l’EG incolonna eseguiamo due eluizione: la prima a 5% per staccare dalla resina i fattori contaminanti che si possono essere legati ad essa, dopo di che faremo la seconda eluizione al 100% che stacca la proteina dalla e la andiamo a raccogliere in provetta, alla fine da 60 mL di EG otteniamo 4,5 mL di EP (enzima puro), che vengono divisi tra i tre gruppi per un totale di 1,5 mL di EP a gruppo. una volta terminato la purificazione, siamo tornati nel laboratorio dove gli altri 3 gruppi avevano preparato la cromatografia su gel-filtrazione, questo tipo di cromatografia serve a cambiare il tampone al nostro EP. Cartogramma dei gruppi 1-2-3.
�5
���
Mentre gli altri gruppi purificavano la proteina, ci siamo dedicati a calcolare la concentrazione delle proteine con il metodo del Reattivo del Biureto, usando il nostro EG. Questa volta andiamo a osservare l’assorbanza dei campioni a diverse quantità di EG portati a 0,5 mL con H2O.
In questa tabella troviamo i valori riscontrati:
Una volta che abbiamo trovato i seguenti fattori, calcoliamo calcolo le concentrazioni EG in ogni cuvetta:
Y = m x X dove: Y: Assorbanza m: pendenza della retta di taratura X: mg/mL di proteine in cuvetta per calcolarla facciamo : X = m / Y
0,152 / 0,210 = 0,72 mg/mL 0,239 / 0,210 = 1,14 mg/mL 0,352 / 0,210 = 1,68 mg/mL
Volume Volume Volume
Cuvetta EG (mL) H2O (mL) Reagente (mL) A540 A540 -Bianco
Bianco 0 0,5 0,5 0,116 0
1 0,20 0,48 0,5 0,268 0,152
2 0,40 0,46 0,5 0,355 0,239
3 0,60 0,44 0,5 0,468 0,352
�6

Si calcolare ora il fattore di diluizione considerando il volume di EG di ogni cuvetta. Fattore Diluizione = 1000 µL/ EG µm
1000 / 20 = 50 1000 / 40 = 25 1000 / 60 = 16,7
Moltiplicando il fattore di diluizione per la concentrazione di EG all’interno di ogni cuvetta si ricava la concentrazione di EG iniziale tenendo conto delle diluizioni fatte.
0,72 x 50 = 36 mg/mL 1,14 x 25 = 28,5 mg/mL 1,68 x 16,7 = 28,1 mg/mL Si calcola la concentrazione media 36 + 28,5 + 28,1 = 92,6 / 3 = 30,86 mg/mL
A questo punto, con i dati fin ora raccolti, è possibile calcolare la quantità di proteine nell’estratto grezzo: 30,86 x 17,5 = 540,05 mg
Si calcola l’efficienza di rottura: mg totali / g di cellule = efficenza di rottura 540,05 / 4,5 = 120
Una volta ottenuta l’efficenza di rottura, possiamo eseguire la Gel-filtrazione con PD10. Questa tecnica viene utilizzata perché durante la purificazione con FPLC è stato aggiunto dell’imidazolo che non è stabile, quindi prendiamo la PD10 e la calibriamo con il tampone di Storage in cui l’enzima è stabile. carichiamo fino ad un massimo di 2,5 mL ed aspettiamo l’eluizione del campione, possiamo seguirne la corsa dato che la proteina DAAO è di colore giallo. Quando la proteina si trova presso la bocca di uscita della PD10 la raccolgo usando una provetta graduata. la frazione cha abbiamo raccolto è il nostro Campione di Enzima Puro (EP).
PD10
�7

Giornata 3
Determinazione dell’attività enzimatica nell’estratto grezzo e in quello puro. Determinazione spettro di assorbimento e concentrazione di His - RgDAAO nel campione puro.
Si determina l’attività enzimatica nel campione EG e nell’enzima purificato (campione EP) tramite dosaggio continuo allo spettrofotometro. Il dosaggio dell’attività D-aminoacido ossidasi si effettuerà in maniera indiretta, cioè mediante l’utilizzo accoppiato dell’enzima perossidasi da rafano (HRP) e del suo substrato cromogenico, o-dianisidina (o-DNS). La perossidasi riduce l’H₂O₂, uno dei prodotti della reazione catalizzata dalla DAAO, ad acqua e ossida o-DNS portando allo sviluppo di una colorazione rosso-bruna, con un massimo di assorbimento a 440 nm. Considerando la velocità di comparsa della colorazione è possibile monitorare la produzione di H₂O₂ e a sua volta questa è proporzionale l’attività della DAAO per il substrato in esame, la D-alanina. Prima di proseguire con la preparazione delle cuvette è necessario diluire opportunamente i campioni con il Tampone di Storage, in quanto si devono utilizzare concentrazioni di substrato che garantiscono un andamento rettilineo iniziale, poiché Vo deve essere simile a Vmax.Prima di inserire le cuvette nello spettrofotometro, per la lettura dell’assorbanza, si aggiunge in ogni cuvetta 10 µL di HRP, nelle cuvette per l ‘estratto grezzo 10 µL di campione EG 40X, e nelle cuvette per l’enzima purificato 10 µL del campione EP, per un volume finale di 1000 µL. Subito dopo l‘aggiunta del campione viene registrata per alcuni minuti l’assorbanza della miscela di reazione (EG o EP). Dalla parte iniziale del tracciato, regione rettilinea, si ricava il valore della velocità iniziale di reazione, proporzionale all’attività della DAAO, espressa come ΔA/min. Da tale valore, conoscendo il coefficiente di estinzione dell’o-DNS, pari a 13 mM⁻¹cm⁻¹, si ottengono le unità enzimatiche per unità di volume (U/mL).
DILUIZIONE EG 1/40 calcolo dell’attività enzimatica mediante le formule:
Estratto Grezzo lettura assorbanza a 40X
È necessario tener conto delle diluizioni per considerare le unità presen2 nel volume di campione aggiunto in cuve6a e per risalire alle unità nel campione originale
cuvetta 𝚫A/min
1 0,1084
2 0,1202
3 0,131
�8

si calcola la media usando la Formula:
Lettura assorbanza del campione EP 100X
calcolo la media:
Per ricavare la concentrazione di proteina si osserva lo spe6ro nell’UV visibile in un intervallo di lunghezza d’onda tra 342-‐800 nm, conoscendo ques2 da2 si u2lizza la legge di Lambert-‐Beer per ricavare la concentrazione di estra6o puro:
Cuvetta 𝚫A/min
1 0,2422
2 0,222
3 0,2000
�9
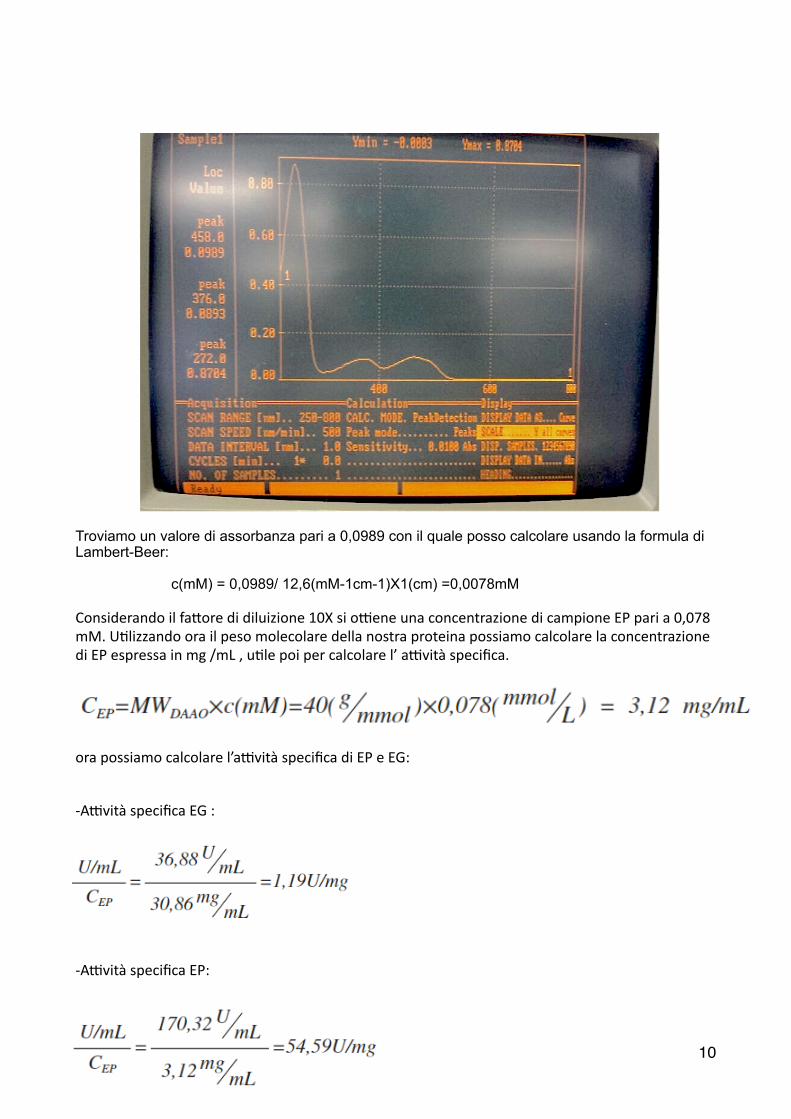
Troviamo un valore di assorbanza pari a 0,0989 con il quale posso calcolare usando la formula di Lambert-Beer:
c(mM) = 0,0989/ 12,6(mM-1cm-1)X1(cm) =0,0078mM
Considerando il fa6ore di diluizione 10X si oLene una concentrazione di campione EP pari a 0,078 mM. U2lizzando ora il peso molecolare della nostra proteina possiamo calcolare la concentrazione di EP espressa in mg /mL , u2le poi per calcolare l’ aLvità specifica.
ora possiamo calcolare l’aLvità specifica di EP e EG:
-‐ALvità specifica EG :
-‐ALvità specifica EP:
�10

a questo punto possiamo calcolare il fa6ore di purificazione, facendo un rapporto tra aLvità di EP e aLvità di EG:
�11
Giornata 4
DETERMINAZIONE DEI PARAMETRI CINETICI DELLA DAAO
Per determinare i parametri cine2ci di un enzima bisogna misurare la V₀ a diverse concentrazione di substrato, in modo da poter costruire sperimentalmente la curva di Michaelis-‐Menten. La velocità di reazione viene misurata con lo spe6rofotometro, a diverse concentrazioni di D-‐alanina in presenza di una concentrazione costante di enzima.
Per o6enere la concentrazione finale di D-‐alanina(mM) uso la formula:
Vi x Ci = Vf x Cf che diventa —> Cf = Vi X Ci / Vf
quindi sos2tuisco e calcolo:
1) 10µL X 25 mM/1000 µL = 0,25mM
2) 20µL X 25mM/1000 µL =0,50mM
3) 50µL X 25mM/1000 µL =1,25mM
4) 100µL X 25mM/1000 µL =2,50mM
5) 200µL X 25mM/1000 µL =5mM
6) 400µL X 25mM/1000 µL =10mM
Dosaggio Dosaggio Dosaggio Dosaggio Dosaggio Dosaggio
1 2 3 4 5 6
[D-Ala] (mM) 0,25 0,50 1,25 2,50 5 10
1/[D-Ala] (mM-1)
4 1,96 0,80 0,40 0,20 0,10
V0 (𝚫Abs440/
min)
0,0544 0,0998 0,161 0,1234 0,1452 0,1658
1/v0 (𝚫Abs440/
min)-1
18,38 10,02 6,21 8,10 6,88 6,03
�12
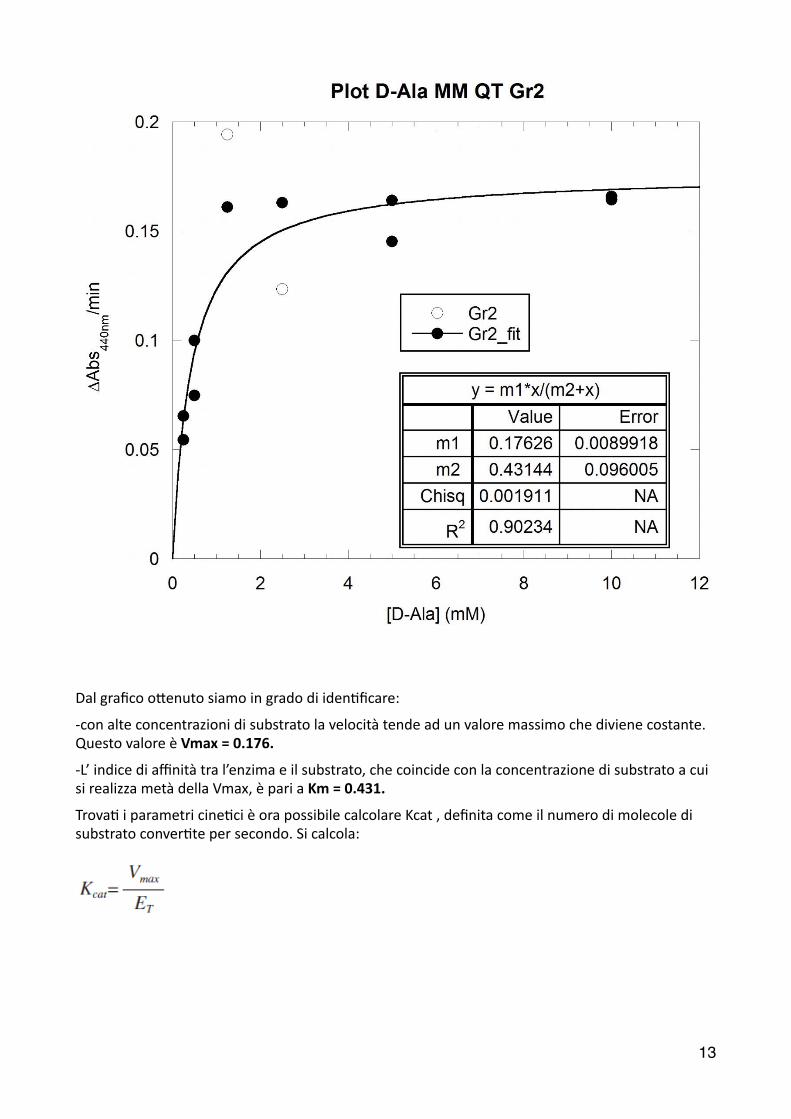
���
Dal grafico o6enuto siamo in grado di iden2ficare:
-‐con alte concentrazioni di substrato la velocità tende ad un valore massimo che diviene costante. Questo valore è Vmax = 0.176.
-‐L’ indice di affinità tra l’enzima e il substrato, che coincide con la concentrazione di substrato a cui si realizza metà della Vmax, è pari a Km = 0.431.
Trova2 i parametri cine2ci è ora possibile calcolare Kcat , definita come il numero di molecole di substrato conver2te per secondo. Si calcola:
�13

Tenendo conto che la Vmax ricavata è espressa come ΔAbs/min abbiamo potuto calcolare Kcat facendo:
Per determinare i parametri cine2ci è u2le u2lizzare, oltre alla curva di Michaelis-‐Menten , la linearizzazione dei doppi reciproci, che rappresenta una delle tecniche più pra2che. Si riportano in grafico i valori di 1/[D-‐Ala] in ascissa e quelli di 1/V₀ in ordinata.
�14
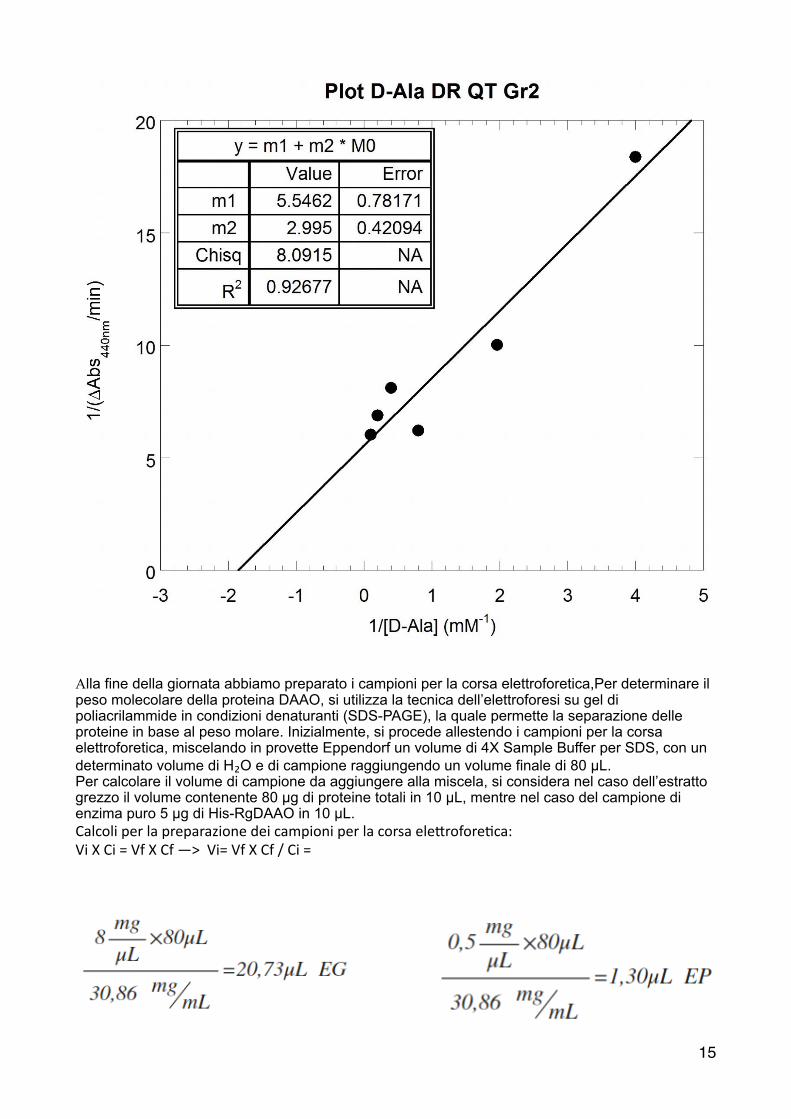
!
Alla fine della giornata abbiamo preparato i campioni per la corsa elettroforetica,Per determinare il peso molecolare della proteina DAAO, si utilizza la tecnica dell’elettroforesi su gel di poliacrilammide in condizioni denaturanti (SDS-PAGE), la quale permette la separazione delle proteine in base al peso molare. Inizialmente, si procede allestendo i campioni per la corsa elettroforetica, miscelando in provette Eppendorf un volume di 4X Sample Buffer per SDS, con un determinato volume di H₂O e di campione raggiungendo un volume finale di 80 µL. Per calcolare il volume di campione da aggiungere alla miscela, si considera nel caso dell’estratto grezzo il volume contenente 80 µg di proteine totali in 10 µL, mentre nel caso del campione di enzima puro 5 µg di His-RgDAAO in 10 µL. Calcoli per la preparazione dei campioni per la corsa ele6rofore2ca: Vi X Ci = Vf X Cf —> Vi= Vf X Cf / Ci =
�15
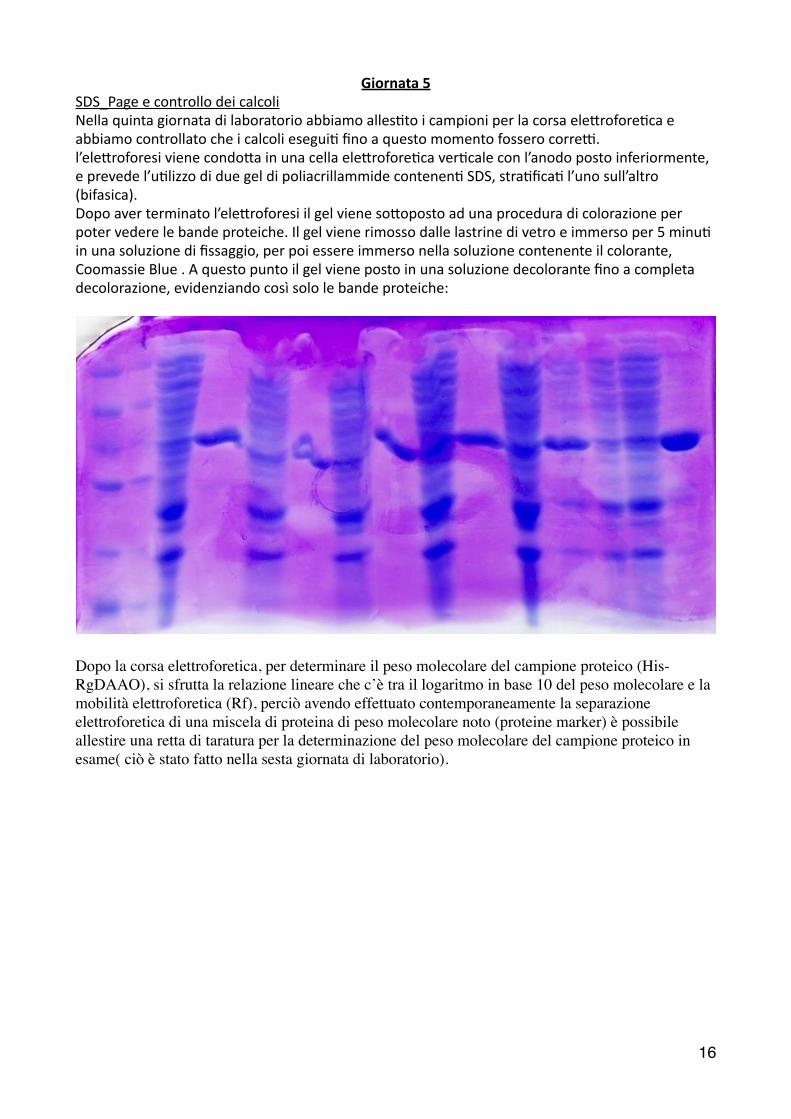
Giornata 5 SDS_Page e controllo dei calcoli Nella quinta giornata di laboratorio abbiamo alles2to i campioni per la corsa ele6rofore2ca e abbiamo controllato che i calcoli esegui2 fino a questo momento fossero correL. l’ele6roforesi viene condo6a in una cella ele6rofore2ca ver2cale con l’anodo posto inferiormente, e prevede l’u2lizzo di due gel di poliacrillammide contenen2 SDS, stra2fica2 l’uno sull’altro (bifasica). Dopo aver terminato l’ele6roforesi il gel viene so6oposto ad una procedura di colorazione per poter vedere le bande proteiche. Il gel viene rimosso dalle lastrine di vetro e immerso per 5 minu2 in una soluzione di fissaggio, per poi essere immerso nella soluzione contenente il colorante, Coomassie Blue . A questo punto il gel viene posto in una soluzione decolorante fino a completa decolorazione, evidenziando così solo le bande proteiche:
���
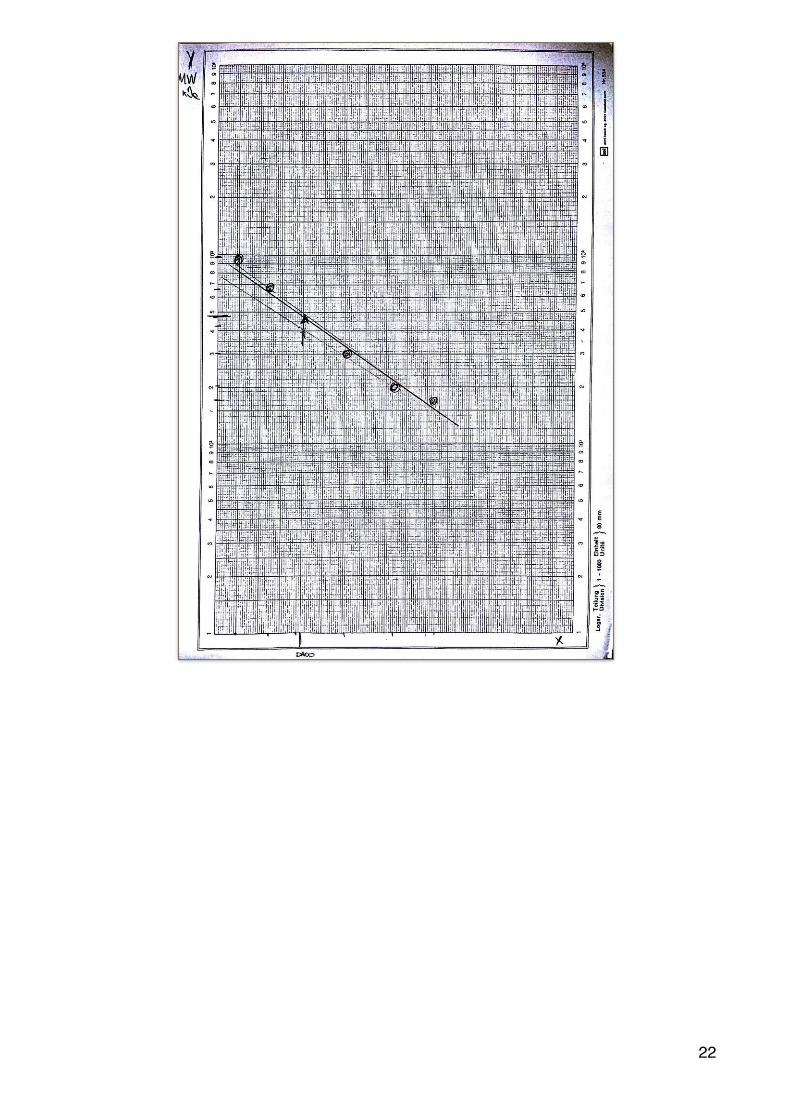
Dopo la corsa elettroforetica, per determinare il peso molecolare del campione proteico (His-RgDAAO), si sfrutta la relazione lineare che c’è tra il logaritmo in base 10 del peso molecolare e la mobilità elettroforetica (Rf), perciò avendo effettuato contemporaneamente la separazione elettroforetica di una miscela di proteina di peso molecolare noto (proteine marker) è possibile allestire una retta di taratura per la determinazione del peso molecolare del campione proteico in esame( ciò è stato fatto nella sesta giornata di laboratorio).
�16

Giornata 6
Determinazione dell’attività cinetica con substrati alternativi Controllo western-blot e SDS-Page
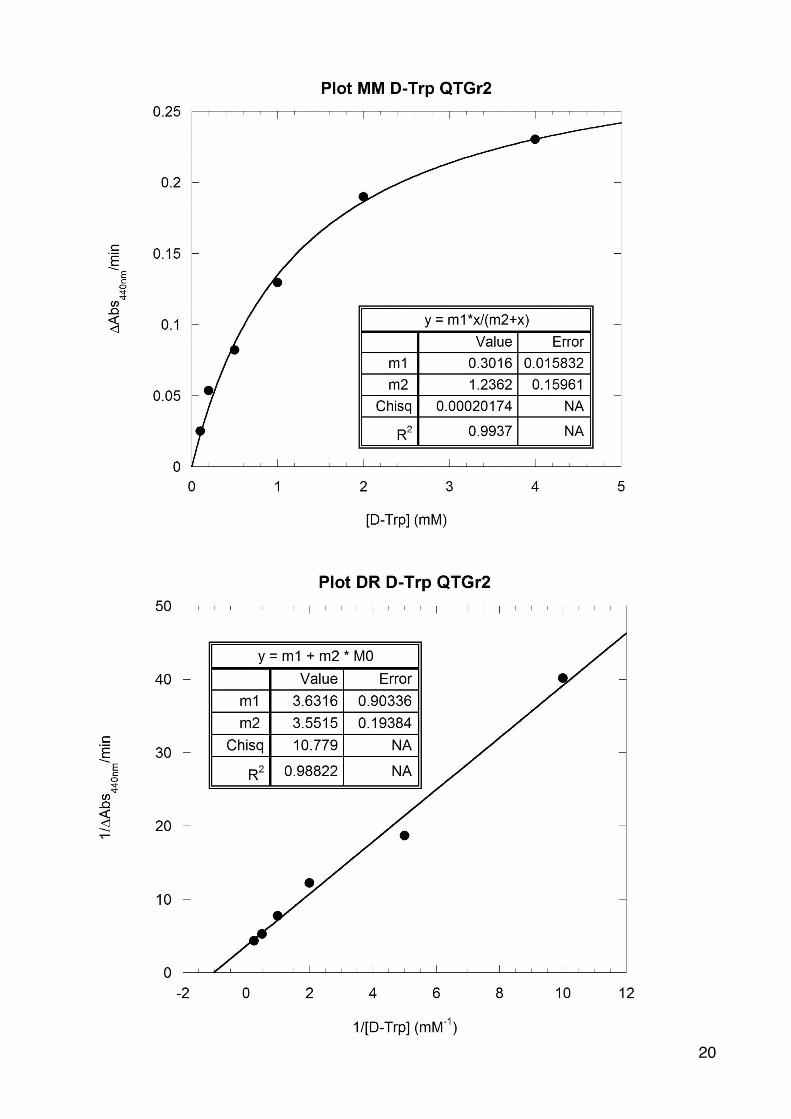
La DAAO è un enzima che agisce specificatamente su un buon numero di D-aminoacidi, pertanto in questa giornata abbiamo determinato l’attività enzimatica dell’enzima puro (EP) utilizzando diversi D-aminoacidi come substrato. in nostro gruppo ha usato la D-metionina e il D-triptofano.
Calcolo della concentrazione finale di substrato con D-‐Me2onina
Analogamente a quanto già effe6uato per la D-‐Ala, prima di alles2re le cuve6e è necessario diluire il campione EP di un Fd 100X (con due diluizioni da 10X). Anche in questo caso si determinano i parametri cine2ci u2lizzando la curva di Michaelis-‐Menten e il metodo di linearizzazione dei doppi reciproci.
�17
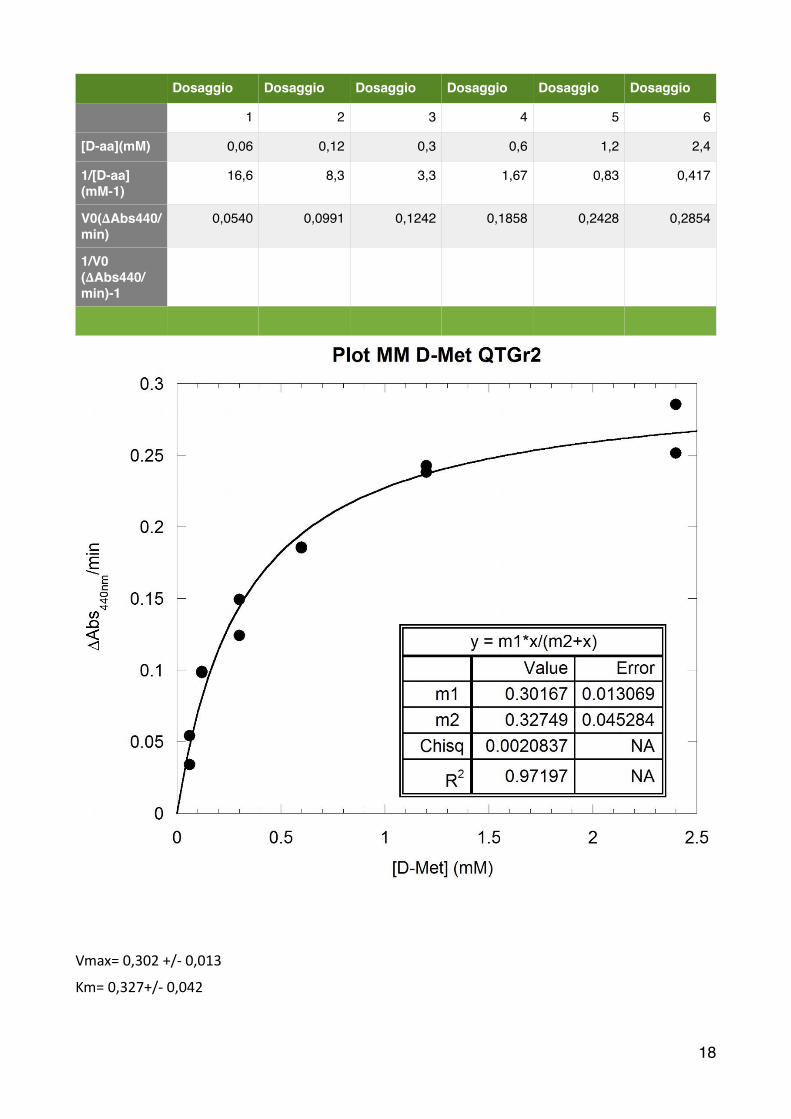
�
Vmax= 0,302 +/-‐ 0,013
Km= 0,327+/-‐ 0,042
Dosaggio Dosaggio Dosaggio Dosaggio Dosaggio Dosaggio
1 2 3 4 5 6
[D-aa](mM) 0,06 0,12 0,3 0,6 1,2 2,4
1/[D-aa](mM-1)
16,6 8,3 3,3 1,67 0,83 0,417
V0(𝚫Abs440/min)
0,0540 0,0991 0,1242 0,1858 0,2428 0,2854
1/V0(𝚫Abs440/min)-1
�18
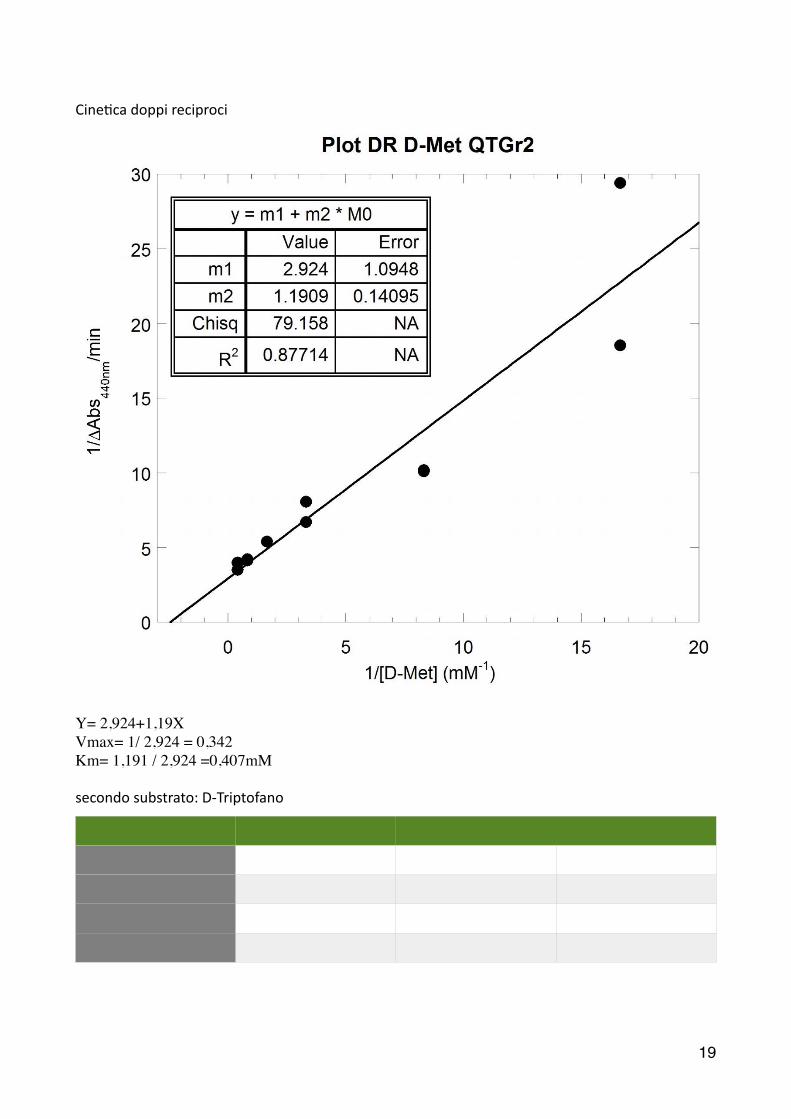
Cine2ca doppi reciproci
���
Y= 2,924+1,19XVmax= 1/ 2,924 = 0,342Km= 1,191 / 2,924 =0,407mM
secondo substrato: D-‐Triptofano
�19
�20

alla fine della ultima giornata di laboratorio abbiamo costruito la retta dell’enzima DAAO, usando un righello abbiamo misurato la lunghezza delle bande, avendo come controllo delle proteine a peso noto(vedi immagine sopra per i calcoli). una volta trovati tutti i dati, li abbiamo posizionati per costruire la retta. (vedi immagine sotto)
�21
�22





























