Embed Size (px)
Citation preview

Leaders
Pulmonary preinvasive neoplasia
K M Kerr
AbstractAdvances in molecular biology have in-creased our knowledge of the biology ofpreneoplastic lesions in the human lung.The recently published WHO lung tumourclassification defines three separate le-sions that are regarded as preinvasiveneoplasia. These are (1) squamous dyspla-sia and carcinoma in situ (SD/CIS), (2)atypical adenomatous hyperplasia (AAH),and (3) diVuse idiopathic pulmonary neu-roendocrine cell hyperplasia (DIP-NECH). SD/CIS is graded in four stages(mild, moderate, severe, and CIS), basedupon the distribution of atypical cells andmitotic figures. Most airways showingSD/CIS demonstrate a range of grades;many epithelia are hard to assess and thereproducibility of this complex systemremains to be established. Detailed crite-ria are, however, welcome and provide anobjective framework on which to comparevarious molecular changes. Alterations ingene expression and chromosome struc-ture known to be associated with malig-nant transformation can be demonstratedin CIS, less so in dysplasias, but also inmorphologically normal epithelium. Thechanges might be sequential, and theirfrequency and number increase with aty-pia. Less is known of the “risk of progres-sion” of SD/CIS to invasive “central”bronchial carcinoma. It may take betweenone and 10 years for invasion to occur, yetthe lesion(s) may be reversible if carcino-gen exposure ceases.
AAH may be an important precursorlesion for peripheral “parenchymal” ad-enocarcinoma of the lung: the “adenoma”in an adenoma–carcinoma sequence.There is good morphological evidencethat AAH may progress from low to highgrade to bronchioloalveolar carcinoma(BAC; a non-invasive lesion by defini-tion). Invasion then develops within BACand peripheral lung adenocarcinomaevolves. The molecular events associatedwith this progression are not well under-stood and studies are hampered by a lackof clear criteria to distinguish high gradeAAH from BAC. Nonetheless, as withSD/CIS, the patterns of expression oftumour associated genes are consistentwith neoplastic progression. We have littleidea of the incidence of AAH in the
normal or “smoking” populations. It isfound more frequently in cancer bearinglungs, especially in those with adenocarci-noma, and is more common in women. Nodata are available on the risk of progres-sion of AAH. DIPNECH is an exception-ally rare lesion associated with thedevelopment of multiple carcinoid tu-mours. Almost nothing is known of itsbiology.
Knowledge of these lesions will becrucial in the design and understanding oflung cancer screening programmes,where it is likely that the morphologicaland, more importantly perhaps, the mo-lecular characteristics of these lesions willprovide useful targets for detection andpossibly even treatment.(J Clin Pathol 2001;54:257–271)
Keywords: lung cancer; preneoplasia; carcinogenesis
With recent advances in molecular biology, theadvent of microdissection techniques, and agreater understanding of the genomic changesinvolved in malignant transformation, there hasbeen a resurgence of interest in the genesis ofprimary lung cancer.
Lung cancer is the most common fatalmalignancy world wide and global case num-bers are set to rise as the smoking epidemictakes hold in Asia, particularly in China. Recentreports confirm that this disease remains verycommon in the UK and, in Scotland in particu-lar, it is the major cause of death from cancer inboth sexes.1 However, the origins of thiscommon disease are not well understood. Theprogression of disease through squamousmetaplasia and carcinoma in situ in the bronchihas been long recognised,2 3 but it is onlyrecently that some of the molecular and geneticchanges associated with the morphologicaltransformation have been elucidated.
But how many carcinomas of the lung arisein this way? It seems likely that the vast major-ity of squamous cell cancers and probably mostothers that arise in the central airways do so.However, peripheral lung cancers, predomi-nantly adenocarcinoma, might arise through adiVerent mechanism and a diVerent series ofprogenitor or preinvasive lesions.
The concept of “scar cancer” is longestablished, based upon observations that adultlungs often show peripheral scars (old tubercu-losis, infarcts, and other inflammatory foci)
J Clin Pathol 2001;54:257–271 257
Department ofPathology, AberdeenRoyal Infirmary andAberdeen UniversityMedical School,Foresterhill, AberdeenAB25 2ZD, UKK M Kerr
Correspondence to:Dr [email protected]
Accepted for publication20 September 2000
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from

and peripheral lung tumours frequently pos-sess central scarring, which is believed, bysome, to have induced the tumour. Morerecent studies have suggested that this is notthe way in which most peripheral cancers arise.On the basis of morphological and radiologicalobservations, survival data, and studies of col-lagen types in scars,4–6 the prevailing view isthat, in most peripheral tumours (largelyadenocarcinoma) with central scarring, thefibrosis postdates the development of thetu-mour. Shimosato presented a critique of thescar cancer hypothesis containing the followingpoints4:• Most adenocarcinomas arise in areas of lung
that show no evidence of fibrosis.• The central fibrotic scar is usually smaller
than the tumour and might also be present inmetastases from the same tumour, includinglymph node deposits.5
• Metastatic lung tumours can have centralscars.
• Psammoma bodies present in the centralscar point to the earlier presence of tumour.
• Radiological evidence of cicatrisation is usu-ally seen after the tumour becomes visible onthe chest radiograph.
• The relatively poor prognosis of tumourswith central scars might imply that the lesionhas a longer natural history.Nonetheless, several diVuse fibrosing
conditions are undoubtedly associated with anincreased incidence of lung cancer andprobably an excess of adenocarcinoma.Although this association with diVusefibrosis probably accounts for only a smallnumber of cases overall, various studies onpostmortem and surgically resected materialfound lung cancer in between 10% and 20% oflungs showing diVuse pulmonary fibrosis orhoneycomb lung.7 The association is madebetween tumour development and the hyper-plasia of alveolar lining cells, which may be
atypical, seen in these conditions. Why pa-tients with systemic sclerosis should beparticularly at risk is not clear. Other causes ofdiVuse lung fibrosis include asbestos and silicaand these factors are also linked to thedevelopment of lung cancer. It seems, how-ever, that with asbestos, fibrogenesis andcarcinogenesis might have independentmechanisms.8 The same may be true for silicaexposure. Fibrosis associated with silica expo-sure (silicosis) appears to be associated with anexcess risk of lung cancer but, for exposure tosilica without fibrosis, the evidence is tenuous.9
The topic of scar cancer has been reviewedelsewhere.10
More recently, considerable attention hasbeen paid to atypical adenomatous hyperplasia(AAH) as a preinvasive lesion, as the adenomain a putative “adenoma–carcinoma” sequencein the lung periphery,11 12 leading to the devel-opment of bronchioloalveolar carcinoma andinvasive peripheral adenocarcinoma of thelung. Even less is understood regarding theorigins of pulmonary neuroendocrine tu-mours, although there is some evidence thatsome carcinoid tumours might be related tofocal bronchial neuroendocrine cell hyperpla-sia.
In this review, I will describe the recognisedpulmonary preneoplastic lesions, bronchialsquamous dysplasia/carcinoma in situ (CIS),AAH, and neuroendocrine cell hyperplasia,and their morphology. I will then consider theprogression of squamous dysplasia/CIS toinvasive disease, including what is understoodof the molecular basis of this progression.Finally, I will discuss what is known of theprobable progression of AAH to adenocarci-noma. Nothing is known about the molecularevents that might occur in neuroendocrine cellhyperplasia.
WHO classification of preinvasive lesionsin the lungThe recently published third edition of theWorld Health Organisation (WHO) lungtumour classification13 lists three main forms ofpreinvasive lesion in the lung: (1) squamousdysplasia and CIS, (2) AAH, and (3) diVuseidiopathic pulmonary neuroendocrine cell hy-perplasia (DIPNECH). The morphology ofthese are described in turn.
SQUAMOUS DYSPLASIA/CIS
Many reports refer to the grading of dysplasiaof squamous epithelium in the bronchi asbeing similar to that in other sites such as thecervix or oesophagus. Some authors refer tothe usual mild, moderate, and severe atypias,as well as CIS, whereas others tend to lumpthe grades together, or refer instead to lowgrade and high grade preinvasive disease, thelatter category including CIS. The WHOclassification provides criteria for the distinc-tion of mild, moderate, and severe dysplasiaand CIS. These are summarised below andpresented in an abbreviated diagrammaticform in fig 1.
Figure 1 A diagrammatic summary of the WHO criteria13 for bronchial squamousdysplasia/carcinoma in situ (CIS). The distribution of various features is related to thelower, middle, and upper thirds of the multilayered “squamous” epithelium.
Increasing epithelial thickness
Basalzoneexpansion/crowding
Verticalnuclei
Verticalnuclei
Mitoses
Mitoses
Mitoses
Chaos
Increasing cell size and pleomorphism
Loss of progression to maturation
Basalzoneexpansion/crowding
Basalzoneexpansion/crowding
Verticalnuclei
Milddysplasia
Moderatedysplasia
Severedysplasia
CIS
258 Kerr
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from

Mild dysplasiaMild dysplasia is present when the architec-tural and cytological disturbance is minimal(fig 2A). Basilar zone expansion with cellularcrowding is limited to the lower third of theepithelium, as are vertically oriented nuclei.Mitoses are absent. This would occur in anepithelium that is clearly squamous and is dis-tinct, in this classification, from basal cellhyperplasia. The latter is present when thebasal epithelial cell layer is more than threecells thick and may well be a precursor of(immature) squamous metaplasia.
Moderate dysplasiaModerate dysplasia exhibits more cytologicalirregularity but still has finely granular chroma-tin. Maturation of cells from the base to theluminal surface shows only partial progression,but there is still clear flattening of thesuperficial cells (fig 2B). The basilar zone nowoccupies two thirds of the epithelium, as dovertically oriented nuclei, and mitoses arepresent in the lower third.
Severe dysplasiaSevere dysplasia exhibits considerable cellularpleomorphism, coarse, uneven chromatin, andlittle cell maturation, but still shows superficialcell flattening. Basilar zone crowding extendswell into the upper third, vertical nuclei andmitotic figures are present in the lower twothirds (fig 2C), and the prickle cell layer hasalmost disappeared.
Carcinoma in situCIS may or may not be associated with epithe-lial thickening. Cytological aberration is ex-treme, mitoses occur at all levels, and matura-tion is absent such that, if the epithelium wereinverted, it would not look diVerent (fig 3).Although this is the usual form of CIS, with aflat topography, a more unusual form existswhere the in situ lesion develops into anexophytic, polypoid, or papillary growth thatcan cause mechanical or functional airwayobstruction, despite the absence of mucosalinvasion.
There can be considerable overlap betweenthese four categories and in any particular casea range of grades may be seen. These lesionsare infrequently encountered by the surgicalhistopathologist in endoscopic biopsy material.They do, however, shed cytologically atypicalcells into sputum and bronchial brushings andwashings, although here their importance canbe diYcult to determine. Bronchial squamousdysplasia/CIS is relatively frequent in lungresection specimens from cigarette smokerswith lung cancer but, in this context, reportingand classification of the lesions is less impor-tant, outside of any academic considerations.Given that, on conventional bronchoscopy,most of these preinvasive lesions are not visible,
Figure 2 (A) Mild dysplasia: basal zone crowding andminimal cytological atypia. (B) Moderate dysplasia:vertically orientated nuclei occupy the lower two thirds ofthe epithelium whereas maturation is still clearly visibletowards the surface. (C) Severe dysplasia: greatercytological aberration, mitoses are clearly present in thelower two thirds of the epithelium, and there is virtually nomaturation. Note the sharp transition from normalrespiratory epithelium on the left.
Figure 3 Carcinoma in situ. Full thickness severecytological atypia with a chaotic appearance.
Pulmonary preinvasive neoplasia 259
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from
their appearance in bronchial biopsy specimensis fortuitous. If fluorescence bronchoscopytechniques, such as LIFE (lung imagingfluorescence endoscopy), become more widelyused, the situation might change.14 15 Suchtechniques greatly increase the sensitivity ofbronchoscopy in finding areas of abnormalepithelium, based on their tendency to showless autofluorescence than normal mucosa.Unfortunately, the specificity of this techniqueis not high and perhaps only one third of allbiopsies taken from areas signalled as “abnor-mal” during LIFE show any histologicalabnormality. However, histological normalitydoes not guarantee the absence of genomicalterations in bronchial epithelial cells (seebelow).
Until these WHO criteria are widely read,tried, and subjected to tests of reproducibility,we will not know how useful this classificationis. The authors acknowledge that lack ofexperience and ill defined boundaries betweencategories will lead to “problems with repro-ducibility” and that “the clinical significance ofthe grading system remains to be established”.It is perhaps surprising, especially if you are a“lumper” rather than a “splitter”, that fourseparate categories, including a distinctionbetween severe dysplasia and CIS, weredefined, rather than two (low versus high gradedysplasia), which seems to be the currenttrend. Nonetheless, we now have criteria forthese four grades. Time will tell us whether ornot this was the correct choice.
Another factor also needs to be taken intoaccount. This process is one that does not nec-essarily aVect the full thickness of the epithe-lium. The process of squamous metaplasia/dysplasia appears to start in a zone of
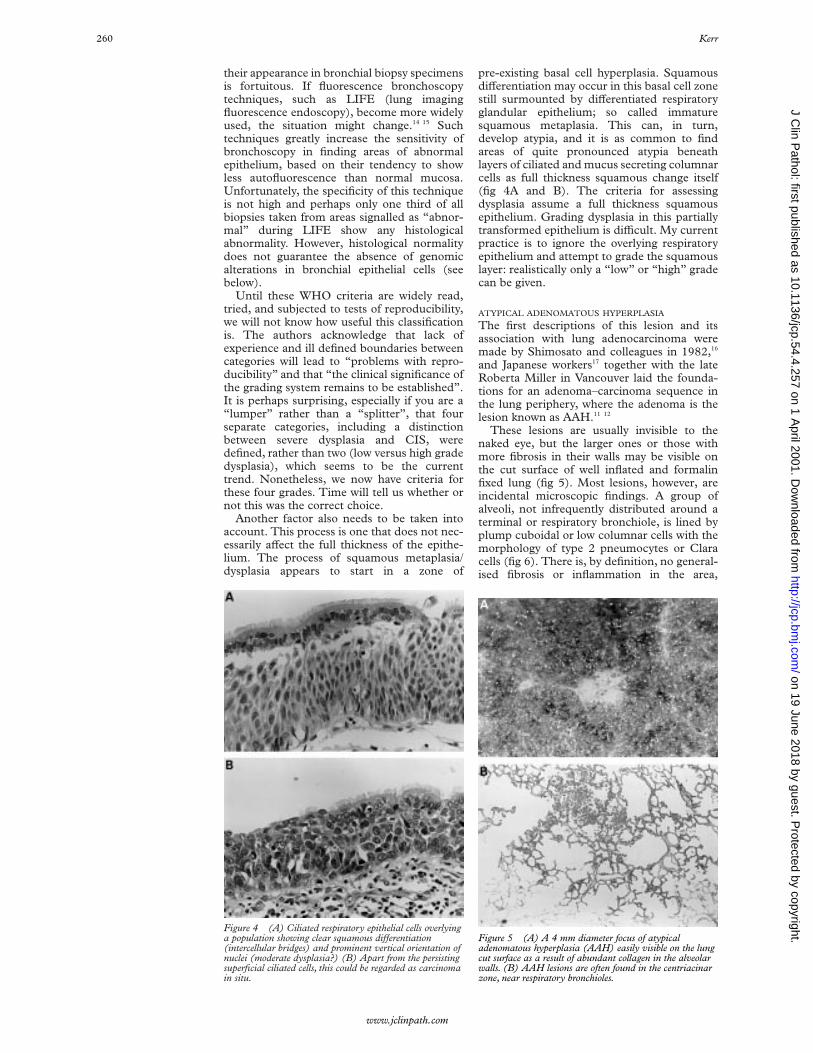
pre-existing basal cell hyperplasia. SquamousdiVerentiation may occur in this basal cell zonestill surmounted by diVerentiated respiratoryglandular epithelium; so called immaturesquamous metaplasia. This can, in turn,develop atypia, and it is as common to findareas of quite pronounced atypia beneathlayers of ciliated and mucus secreting columnarcells as full thickness squamous change itself(fig 4A and B). The criteria for assessingdysplasia assume a full thickness squamousepithelium. Grading dysplasia in this partiallytransformed epithelium is diYcult. My currentpractice is to ignore the overlying respiratoryepithelium and attempt to grade the squamouslayer: realistically only a “low” or “high” gradecan be given.
ATYPICAL ADENOMATOUS HYPERPLASIA
The first descriptions of this lesion and itsassociation with lung adenocarcinoma weremade by Shimosato and colleagues in 1982,16
and Japanese workers17 together with the lateRoberta Miller in Vancouver laid the founda-tions for an adenoma–carcinoma sequence inthe lung periphery, where the adenoma is thelesion known as AAH.11 12
These lesions are usually invisible to thenaked eye, but the larger ones or those withmore fibrosis in their walls may be visible onthe cut surface of well inflated and formalinfixed lung (fig 5). Most lesions, however, areincidental microscopic findings. A group ofalveoli, not infrequently distributed around aterminal or respiratory bronchiole, is lined byplump cuboidal or low columnar cells with themorphology of type 2 pneumocytes or Claracells (fig 6). There is, by definition, no general-ised fibrosis or inflammation in the area,
Figure 4 (A) Ciliated respiratory epithelial cells overlyinga population showing clear squamous diVerentiation(intercellular bridges) and prominent vertical orientation ofnuclei (moderate dysplasia?) (B) Apart from the persistingsuperficial ciliated cells, this could be regarded as carcinomain situ.
Figure 5 (A) A 4 mm diameter focus of atypicaladenomatous hyperplasia (AAH) easily visible on the lungcut surface as a result of abundant collagen in the alveolarwalls. (B) AAH lesions are often found in the centriacinarzone, near respiratory bronchioles.
260 Kerr
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from
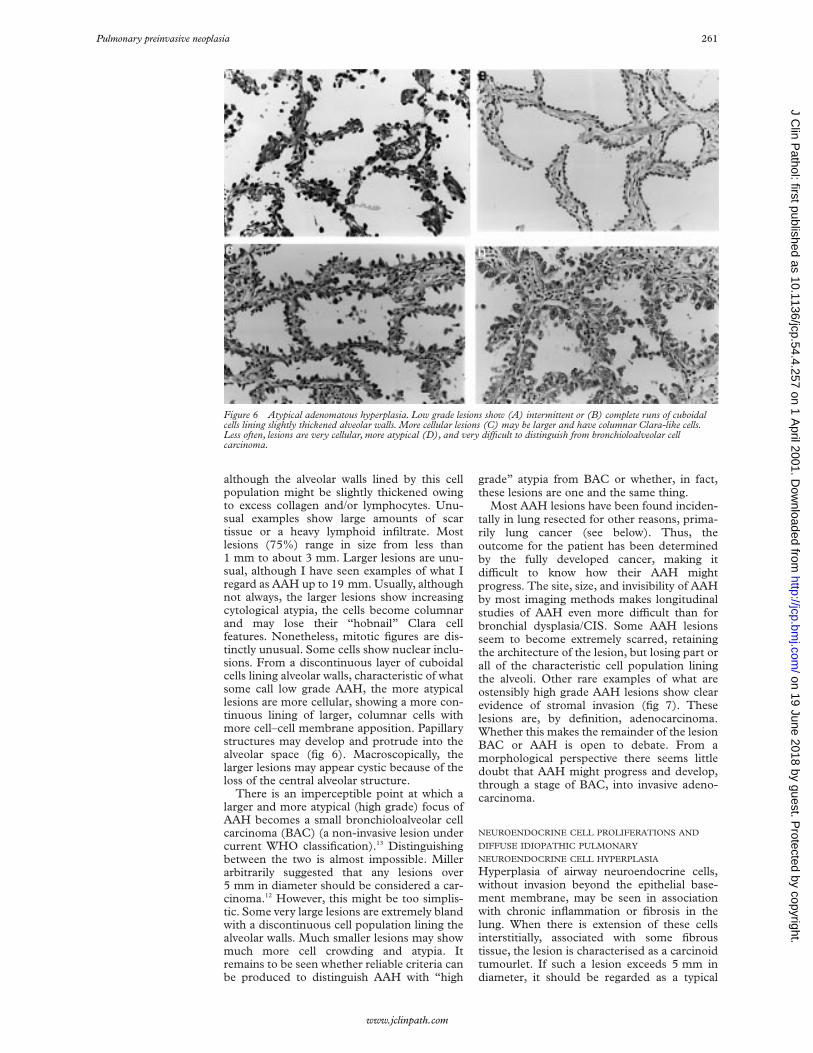
although the alveolar walls lined by this cellpopulation might be slightly thickened owingto excess collagen and/or lymphocytes. Unu-sual examples show large amounts of scartissue or a heavy lymphoid infiltrate. Mostlesions (75%) range in size from less than1 mm to about 3 mm. Larger lesions are unu-sual, although I have seen examples of what Iregard as AAH up to 19 mm. Usually, althoughnot always, the larger lesions show increasingcytological atypia, the cells become columnarand may lose their “hobnail” Clara cellfeatures. Nonetheless, mitotic figures are dis-tinctly unusual. Some cells show nuclear inclu-sions. From a discontinuous layer of cuboidalcells lining alveolar walls, characteristic of whatsome call low grade AAH, the more atypicallesions are more cellular, showing a more con-tinuous lining of larger, columnar cells withmore cell–cell membrane apposition. Papillarystructures may develop and protrude into thealveolar space (fig 6). Macroscopically, thelarger lesions may appear cystic because of theloss of the central alveolar structure.
There is an imperceptible point at which alarger and more atypical (high grade) focus ofAAH becomes a small bronchioloalveolar cellcarcinoma (BAC) (a non-invasive lesion undercurrent WHO classification).13 Distinguishingbetween the two is almost impossible. Millerarbitrarily suggested that any lesions over5 mm in diameter should be considered a car-cinoma.12 However, this might be too simplis-tic. Some very large lesions are extremely blandwith a discontinuous cell population lining thealveolar walls. Much smaller lesions may showmuch more cell crowding and atypia. Itremains to be seen whether reliable criteria canbe produced to distinguish AAH with “high
grade” atypia from BAC or whether, in fact,these lesions are one and the same thing.
Most AAH lesions have been found inciden-tally in lung resected for other reasons, prima-rily lung cancer (see below). Thus, theoutcome for the patient has been determinedby the fully developed cancer, making itdiYcult to know how their AAH mightprogress. The site, size, and invisibility of AAHby most imaging methods makes longitudinalstudies of AAH even more diYcult than forbronchial dysplasia/CIS. Some AAH lesionsseem to become extremely scarred, retainingthe architecture of the lesion, but losing part orall of the characteristic cell population liningthe alveoli. Other rare examples of what areostensibly high grade AAH lesions show clearevidence of stromal invasion (fig 7). Theselesions are, by definition, adenocarcinoma.Whether this makes the remainder of the lesionBAC or AAH is open to debate. From amorphological perspective there seems littledoubt that AAH might progress and develop,through a stage of BAC, into invasive adeno-carcinoma.
NEUROENDOCRINE CELL PROLIFERATIONS AND
DIFFUSE IDIOPATHIC PULMONARY
NEUROENDOCRINE CELL HYPERPLASIA
Hyperplasia of airway neuroendocrine cells,without invasion beyond the epithelial base-ment membrane, may be seen in associationwith chronic inflammation or fibrosis in thelung. When there is extension of these cellsinterstitially, associated with some fibroustissue, the lesion is characterised as a carcinoidtumourlet. If such a lesion exceeds 5 mm indiameter, it should be regarded as a typical
Figure 6 Atypical adenomatous hyperplasia. Low grade lesions show (A) intermittent or (B) complete runs of cuboidalcells lining slightly thickened alveolar walls. More cellular lesions (C) may be larger and have columnar Clara-like cells.Less often, lesions are very cellular, more atypical (D), and very diYcult to distinguish from bronchioloalveolar cellcarcinoma.
Pulmonary preinvasive neoplasia 261
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from

carcinoid tumour. Such a proliferation of neu-roendocrine cells, in association with chronicinflammation, is not regarded as a preinvasivelesion.
Hyperplasia of neuroendocrine cells is alsoseen in the airway mucosa in lungs bearingtypical (usually peripheral) carcinoid tu-mours.18 This process may also occur in lungsshowing higher grade neuroendocrine malig-nancy (fig 8). The relation between the hyper-plasia and the tumour, if any, is not currentlyunderstood.
In 1992 Aguayo et al described six unusualpatients who had diVuse hyperplasia of airwayneuroendocrine cells and evidence of airwaysobstruction.19 These patients also had multiplepulmonary carcinoid tumourlets. In these andsubsequent cases the airway fibrosis, perhapsbecause of the eVects of some paracrine secre-tion, was presumed to be the cause of thewidespread small airways obstruction. As wellas multiple carcinoid tumourlets, some of thesepatients also had one or more carcinoid
tumours. Such cases are rare, so experience ofthis lesion is limited. In the above context,however, it is not unreasonable to suggest thatthe widespread neuroendocrine cell hyperpla-sia (DIPNECH) might be a preinvasive lesion,which may give rise to typical carcinoidtumours. What relation this lesion or a similarmore localised process might have to mostinvasive neuroendocrine tumours—from carci-noids (typical and atypical) to the high gradelarge cell and small cell neuroendocrinecarcinomas—is not known. However, it istempting to speculate that such focal hyperpla-sia might provide a basis upon which neoplas-tic transformation is more likely to occur (seebelow).
SQUAMOUS DYSPLASIA/CIS AND ITS PROGRESSION
Little is known about the rate and risks of pro-gression of squamous dysplasia to CIS andultimately invasive disease.
There is a large amount of animal experi-mental work20–22 using a variety of carcinogensand subjects, including the infamous smokingbeagles. These data suggest that the earliestchange in the epithelium, in keeping with simi-lar observations in epithelia at other sites, ishyperplasia in the basal layer of cells. Such ahyperplasia in the stem cell compartment ofthe epithelium is one of the earliest stepsrequired in currently accepted theories of mul-tistep carcinogenesis.23 As a result of chronicirritation/stimulation, this expanded cell com-partment may diVerentiate towards a pheno-type better adapted to the prevailing environ-ment (cigarette smoke—for example) andsquamous metaplasia occurs. Thus, the earliestchanges represent a relatively acute response toinjury. Over time, a more subacute preneoplas-tic form of nuclear “injury” takes place which,
Figure 7 (A) This 1.7 cm lesion with a rather solid and cystic centre has alveolar spaces clearly visible in the periphery.(B) Whole mount of the same lesion. Closer examination (C) shows a periphery typical of atypical adenomatoushyperplasia but the central, solid zone shows (D) papillary bronchioloalveolar-like areas and clear evidence of stromalinvasion (arrows).
Figure 8 A hyperplastic focus of neuroendocrine cells inthe airway epithelium, found in the same lobe as anatypical carcinoid tumour (immunoperoxidase withanti-chromogranin).
262 Kerr
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from

in turn, leads to the expression of the evolvingabnormal genotype as an abnormal phenotype,namely dysplasia and carcinoma in situ.
Experimental studies suggest that all of thesechanges, up to and including CIS, might bereversible if the stimulus/carcinogen is re-moved.21 22 At some point, however, the process(and lesion) become irreversible and we have,by definition, a neoplasm.24 There is alsoevidence that preinvasive dysplasias may “waxand wane” depending on the dose/duration ofexposure to particular carcinogens. Removal ofthe carcinogens might cause the visible lesionto regress (phenotypic regression); however,the genomic damage (see later) might not dis-appear and may persist in microscopicallyapparently normal respiratory epithelium. Inmore prolonged experiments there is alsoevidence that the dose and type of carcinogenmight determine the type of cancer that devel-ops.21 22 This is an interesting parallel with therecently observed increase in small cell carci-noma and adenocarcinoma and the fall insquamous lung cancers in the Western worldthat might, at least in part, be related tochanges in smoking habits or in the tobacco oradditives used in cigarettes.
Clinical studies of bronchial preinvasivelesions are limited and often involve a popula-tion who are “at risk” or the subject of a lungcancer screening programme. Bronchial hyper-plasia and squamous metaplasia are commonin the airways of cigarette smokers,25 whereasdysplasias and carcinoma in situ are particu-larly found in cancer bearing lungs. Auerbachet al found dysplasia in 40% of heavysmokers.2 3 In another study, one third ofpatients who had atypical cells in sputum wenton to develop lung carcinoma.26 In a study ofuranium miners, who were subjected torepeated sputum examination, increasinglysevere cytological abnormality could be ob-served in squamous cells with, in some, thedevelopment of invasive carcinoma over aperiod of one to 10 years.27 Up to 20% ofworker cohorts exposed to chromates ormustard gas have also been shown to developpreinvasive bronchial changes and to have anincreased risk of developing invasive cancer.28
In studies of populations screened usingsputum cytology, some individuals will showatypical (squamous) cells. Most of thesepatients, not including an at risk occupationalgroup, will be cigarette smokers. In such a situ-ation the presence of atypical cells in thesputum usually leads to bronchoscopy, andsome studies have shown that it may take six to36 months for a bronchoscopically visible(standard technique) lesion to appear, if one isnot present at the initial examination.29–32
In those patients where atypical sputumcytology leads to bronchoscopy and a lesion isseen (if not at the initial investigation, then atsubsequent examination and perhaps afterselective bronchial brushings are made in anattempt to localise the source of the abnormalcells to a particular airway), biopsy of thevisible lesions yields a range of histologicalabnormalities. All of these patients would havea normal chest radiograph so that, should
malignant cells be detected, these lesions areclassified as so called radiographically occultsquamous carcinoma of the lung.29–32 In areview of several reported series of such“occult” carcinomas, Carter and Patchefskyfound that 25% of the patients had CIS, 42%had early invasive disease confined to themucosa, whereas 33% showed a small, radio-graphically invisible cancer that extendedbeyond the bronchial cartilage.28
It is generally accepted that a tumour has tobe at least 1 cm in diameter and unobscured byrib, vascular, or mediastinal shadows to be reli-ably detectable on a plain chest radiograph. At1 cm diameter, a tumour has alreadyundergone around 30 doublings of its cellpopulation, something that possibly took sev-eral years. Thus, a radiologically visible lungtumour is usually already relatively late in itsnatural history, having been invasive formonths or years before detection, and mayalready have metastasised. Hence, the failure ofmost radiograph based screening programmesto improve the cure rate for treating lung can-cer. If fluorescence bronchoscopy becomeswidely used it is possible that many more earlylesions will be found. Even if patients withoccult lung cancer are treated by surgery or,more recently, by photodynamic treatmentusing laser bronchoscopy, the apparent “cure”rates of 80–95% are marred by the finding thatup to 25% of patients develop a second lungcancer, sometimes again occult.
Nagamoto et al have made an extensive studyof a series of occult lung cancers, includingCIS, found through the Miyagi programme ofmass screening between 1982 and 1991.30–32
They distinguished between two patterns ofoccult carcinoma. Most cases (86%) were clas-sified as the “creeping type” where thedominant growth pattern was a horizontalspread within the mucosa, sometimes over sev-eral centimetres. The remainder seemed tohave a predominantly transmural rather thanlongitudinal growth pattern (penetrating type).These latter lesions were thought to be morerapidly growing and more likely to becomeadvanced in a short time. However, even thecreeping type of lesion showing minimalinvasion did have a chance of metastasis,31
although only when the extent of the lesion waslongitudinally > 20 mm, when 24% showednodal spread. No lesion < 20 mm in extent wasfound to spread. In a study of 19 cases ofisolated CIS discovered during the samescreening study,32 and treated by surgicalresection, no patient had lymph node metas-tases. Follow up of these patients showed anexcellent prognosis. Six of the patients showeda polypoid or micronodular protuberance inthe mucosa on bronchoscopy. Such an exo-phytic and exuberant thickening of the in situlesion is unusual, but was also described bySpencer et al.33 These lesions may havebronchoscopic, cytological, and even biopsyappearances that are highly suggestive ofmalignancy. I have seen at least two such caseswhere, after lobectomy, the preoperative diag-nosis of squamous carcinoma required revisionto in situ disease. In neither was there evidence
Pulmonary preinvasive neoplasia 263
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from

of nodal metastatic disease, yet in one case inparticular, there was pronounced nodal en-largement, presumably secondary to the con-siderable “obstructive” pneumonia in thesegments distal to the airway containing thepolypoid CIS. Even if such lesions are too smallto cause mechanical obstruction, functionalobstruction might result from the loss ofmucociliary function if there is extensivereplacement of normal respiratory mucosa by(atypical) squamous epithelium.
MOLECULAR BIOLOGICAL AND GENETIC STUDIES
ON SQUAMOUS DYSPLASIA/CIS
Extensive work has been carried out on a rangeof preinvasive bronchial lesions. This hasgenerally involved either immunohistochemis-try, or microdissection of lesional tissuefollowed by DNA extraction, PCR amplifica-tion, and experiments to seek gene mutation orloss of heterozygosity (LOH) of variouschromosomal regions.
Cell proliferation and related markersAs might be expected from morphologicalstudies, hyperproliferation, as demonstrated bythe use of anti-proliferative cell nuclear antigen(PCNA) antibodies, seems to be an early eventin the transforming bronchial epithelium. Theproliferative component was shown to risefrom around 25% in apparently normalepithelium, through 35–40% in low and highgrade dysplasias, to 85–90% in invasivesquamous carcinomas, both early and ad-vanced.34 The distribution of positively stainingnuclei mirrored the change in epithelial cyto-architecture from the base to the more super-ficial layers as atypia increased.35
In a study of Rb, p16, and cyclin D1 expres-sion in bronchial preinvasive lesions, Brambillaet al found that although Rb loss was not seenin dysplasia/CIS, loss of p16 expression wasfound in moderate dysplasia (12%) and CIS(30%), exclusively in lesions found in cancerbearing lungs, and not in lesions from patientswho did not have cancer.36 The same authorsfound cyclin D1 overexpression in earlierlesions: 6% of hyperplasia/metaplasia, 17% ofmoderate dysplasia, 46% of moderate dyspla-sia, and 38% of CIS lesions. It was also overex-pressed in preinvasive lesions in 14% of thosewho did not have cancer.
DNA ploidyStudies of the DNA content of cell nuclei, as arough measure of chromosomal gain or loss,have been used to measure the degree of“nuclear aberration” in a malignant cell popu-lation. Hirano et al demonstrated aneuploidy inall of the invasive squamous carcinomas testedbut not in normal bronchial mucosa.34 Of thepreinvasive lesions, 8% of the low grade casesand 33% of the high grade cases wereaneuploid. These authors suggest that thedevelopment of aneuploidy is a relatively earlyevent in the progression to malignancy and alsoshowed that the development of aneuploidydepended on the pre-existence of a hyperprolif-erative state in the bronchial epithelium. Thesegraded changes in the nuclei of preneoplastic
lesions have also been shown by others, both inretrospective analysis of biopsy material fromhuman patients and in dogs experimentallyexposed to various carcinogens.21
bcl-2 expressionThe bcl-2 protein is thought to inhibitprogrammed cell death. Therefore, (over)ex-pression in tumours might give a cell popula-tion some survival advantage. Bcl-2 protein hasbeen shown to be overexpressed in lung cancer(25% of squamous carcinomas, 10% of adeno-carcinomas, 64% of small cell carcinomas)37–39
and in preinvasive bronchial lesions.40 Kata-bami et al showed that 33% of bronchialdysplasias expressed Bcl-2 protein, with a weakassociation with an increasing degree ofdysplasia.41 Brambilla et al found bcl-2 overex-pression in association with bax downregula-tion, but this did not correlate with p53status.42
p53 expressionVarious studies have found an increase instainable p53 protein with increasingly severebronchial squamous dysplasia (table 1). Sev-eral studies describe localisation of p53 proteinin nuclei in the zone of the epithelium, whichshowed loss of nuclear polarity.43 44 In a largeseries, Bennett et al also found p53 protein in6.7% of squamous metaplasias but none innormal epithelium.44 Katabami et al found thatin dysplasia in patients with pneumoconiosis,low grade dysplasia (mild/moderate) had agreater tendency to p53 positivity (40%) thanin those without pneumoconiosis.41 Brambillaand co-workers found p53 expression in allcases of squamous dysplasia/CIS from cancerbearing lungs, but similar lesions from lungswith no cancer showed no staining, implyingthat the stabilisation of p53 in the preinvasivelesion had a high predictive value for invasion.42
The demonstration of stainable p53 proteinmight infer gene alteration, but the detection ofgene mutation might be more important. In astudy that included 13 examples of bronchialsquamous metaplasia and 22 squamous dys-plasias (ungraded), only two of the dysplasiasshowed evidence of p53 gene mutation.45
Earlier, smaller studies also found evidence ofp53 gene mutation, sometimes diVerent fromany concurrent tumour, sometimes the samemutation as found in adjacent tumour.46–48
The general conclusion is that even in thevery early stages of malignant transformation,in the least dysplastic of the preinvasive lesions,abnormal expression of p53 occurs. Thissuggests that p53 mutation might be arelatively early event in central bronchialcarcinogenesis.
Table 1 Stainable p53 protein in bronchial preinvasivedisease
Author (reference) Low gradedysplasias
High grade dysplasias(including CIS)
Nuorva et al (43) 17% 78%Bennett et al (44) 28.2% 59.1%Hirano et al (34) 0% 6%Katabami et al (41) 20% 100%Brambilla et al (42) 23% 59%
264 Kerr
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from

TelomeraseTelomerase is activated and expressed in mosthuman cancers and it may be onemechanism—by preventing progressive tel-omere shortening, and therefore cellularsenescence—that confers immortality totumour cell populations. Most adult somaticcells have inactive telomerase, but at somestage in the carcinogenic process it becomesreactivated. Yashima et al,49 using in situhybridisation, demonstrated much greater tel-omerase positivity in both hyperplastic anddysplastic bronchial epithelium (70–80%)when compared with normal controls (20%),and CIS and invasive disease were positive in95–100% of cases. However, preinvasive le-sions, including CIS, had enzyme activity thatwas only three to fourfold higher than normal,whereas in invasive disease, telomerase activitywas 40 times greater. Once more, even at theearliest stages, telomerase dysregulation ap-pears to occur as part of the multistage patho-genesis of lung cancer.
K-ras mutationK-ras is more commonly found mutated inadenocarcinoma than in squamous carcinoma.Nonetheless, mutation of K-ras might be ofimportance in the development of at least somecentral “bronchogenic” carcinomas arisingfrom the hyperplasia–dysplasia–CIS sequencein the bronchial/bronchiolar epithelium. In astudy concentrating primarily on squamousdysplasias in six patients whose tumours (fiveadenocarcinomas, one squamous carcinoma)all showed a K-ras mutation, Sugio et al wereunable to demonstrate K-ras mutation in anynormal or hyperplastic epithelium and foundonly one mutated preinvasive lesion out of the12 studied.50 This suggests that K-ras mutationmight either be a very late or relativelyunimportant step in central bronchial carcino-genesis. This is also the conclusion of anotherstudy, which found no evidence of K-ras muta-tion in bronchial adenocarcinoma, a tumourthat might arise from bronchial epithelialdysplasia, whereas 35% of peripheral adeno-carcinomas, which arise by a diVerent mech-anism (see below), showed K-ras mutation.51
Human papillomavirus (HPV)HPV-6 DNA was found in one of 10 squamousmetaplastic lesions by Béjui-Thivolet et al,52
whereas 18% of squamous carcinomas con-tained HPV DNA from several subtypes,predominantly HPV-18. No cases of preinva-sive disease with dysplasia were studied andother work suggests that HPV has no impor-tant role to play in the genesis of bronchogenicsquamous carcinoma.53
The FHIT (fragile histidine triad) tumoursuppressor geneFHIT is a tumour suppressor gene at 3p14.2,which spans the FRA3B fragile site. It appearsto be a frequently deleted gene in commonhuman carcinomas, including lung. It has beensuggested that the FHIT protein might play arole in cell death through apoptosis and/oraVect cell proliferation, but its true function is
not known. However, its role as a tumour sup-pressor gene is reflected in the fact that mostprimary lung cancers show loss of FHITprotein expression as well as aberrant RNA andaltered FHIT genome DNA.54 55
Sozzi et al also found that FHIT proteinexpression is lost in 73% of non-small cell lungcancers and in 93% of precancerous lesions.56
Tumours from smokers are more likely to loseFHIT expression (75%) than those from non-smokers (39%).57 This change is independentof p53 status and seemed to be more of a fea-ture in squamous carcinoma (87%) than inadenocarcinoma (57%). In the preinvasivelesions studied, the loss of FHIT was very fre-quent and appeared to increase as the grade ofdysplasia increased. Moderate dysplasiashowed 60% loss, whereas all cases of severedysplasia and CIS studied showed loss ofFHIT protein. Fong et al also examined prein-vasive bronchial lesions, but demonstrated that3p14.2 LOH (implying FHIT deletion) onlyoccurred at the CIS stage and not before, sug-gesting that FHIT loss might be a relatively lateevent in central bronchial carcinogenesis.58
Genomic alterations and LOH studiesSeveral chromosomal sites appear to beinvolved frequently in lung cancer. The mostcommon and earliest is loss of genome at 3p. Ininvasive disease there are at least five loci(3p12–13, 3p14.2, 3p21, 3p21.3, and 3p25)that are lost frequently. In preneoplastic lesionsthe identifiable losses tend to be more local-ised, selective, and appear relatively early.Kohno et al demonstrated that 7.7% ofsquamous metaplasias and 9.1% of squamousdysplasias showed LOH at 3p.45 Wistuba et alfound 3p LOH more extensively, frequentlyand earlier, such that, in invasive squamouscarcinoma and CIS, most of 3p was deleted.59
In normal or mildly abnormal epithelium, 3ploss was much less and more focal, whereas inthe squamous dysplasias the degree of 3p losswas intermediate. This reflected some of thesame group’s earlier work, using diVerent tech-niques, where a study of 3p14, 3p21.3, and3p25 again showed early loss, with 76% ofhyperplasias, 86% of dysplasias, and all exam-ples of CIS being aVected.60
The 3p loss in the early lesions did notappear to occur at random, with even the nor-mal epithelium showing 3p21, 3p22–24, and3p25 losses, whereas 3p14.2 and 3p14–21losses began to appear in the hyperplastic/metaplastic lesions. These results indicate thatloss of 3p14.2 occurs much earlier in theproposed sequence of events than has beensuggested by Fong et al.58
Some 9p21 losses were described by Wis-tuba et al.59 These occurred in early disease andwere almost invariably present in dysplasia,CIS, and invasive tumours. LOH at 17p13(p53) and 13q (Rb) seemed to be a slightlylater phenomenon associated with some dys-plasias, but almost all CIS/invasive disease.Similarly, Kohno and colleagues45 found a 9pdeletion in 9% of dysplasias and 17p LOH in36.4%, whereas neither was shown insquamous metaplasia.
Pulmonary preinvasive neoplasia 265
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from
Thus, very early in the evolution of preinva-sive disease, localised specific LOH of 3p andoften 9p occurs, even in phenotypically normalor hyperplastic epithelium. As the lesionadvances and becomes dysplastic, more 3pdeletions, including 3p14.2, begin to appearand 17p LOH also becomes a feature. By thetime the stage of severe dysplasia/CIS isreached, there is extensive loss of 3p andpersistent loss of the 9p and 17p regions.
In a study of the next stage—early invasivesquamous carcinoma (usually radiographicallyoccult carcinoma)—Endo et al showed fre-quent LOH, with losses at 3p21 (53%), 5p21(44%), and 17p13 (61%) being particularlyprominent.61 Another study has shown that 5q(APC-MCC) loss might be an important eventin central bronchial carcinogenesis, but not inevents occurring in the periphery of the lung.62
What is not known is the relative “cause andeVect” relation between these various geneticabnormalities. It is tempting to speculate that,given the relation between many of these find-ings and the grades of atypia (table 2), thegenomic lesions or abnormal gene expressionmight in some way be responsible for thechange in morphology. Of course the morphol-ogy of the epithelium is a mirror of genomicchange, but whether or not it is a reflection ofthese particular changes is not known. What isknown is that some of these genomic changesmight be widespread in phenotypically normalepithelium, that the phenotype/histology is, insome (perhaps many) cases reversible, butwhen this occurs, some of the genomicalterations persist, perhaps accounting for thecontinuing high risk of developing lung cancermany years after the cessation of smoking.Brambilla and colleagues suggest that stablep53 overexpression, p16 loss and, perhaps,cyclin D1 overexpression in preinvasive bron-chial disease might be related to a greater like-lihood of progression to invasion.36 42 Theseinteresting findings require further attention.
So far, this discussion has primarily consid-ered the evolution of squamous carcinomafrom in situ disease in the central airwayepithelium. It seems likely that some of the rarecentral bronchial adenocarcinomas might arisefrom the same preneoplastic lesion. As forsmall cell carcinoma, another predominantly
central tumour, its progenitor lesion is essen-tially unknown. In a few cases, squamousdysplasia/CIS can be seen in the adjacentairway mucosa, but this is hardly proof of anorigin. There has been a suggestion, based onthe frequent presence of pronounced genomicabnormalities in morphologically normal mu-cosa near small cell tumours, that this tumourmight arise almost de novo from the bronchialmucosa, without going through a stage of amorphologically recognisable preinvasive le-sion.63 There are no detailed molecular biologi-cal studies on neuroendocrine cell hyperplasia.
AAH: a preinvasive lesion that progressesto adenocarcinomaThere is an increasing body of evidence to sup-port the hypothesis that AAH is a preinvasivelesion and indeed can be regarded as theadenoma in an adenoma–carcinoma sequencein the lung periphery.11 This evidence comesfrom morphological observations, the associ-ation of AAH with malignancy, morphometricand cytofluorimetric data, and immunohisto-chemical and molecular biological studies,which are reviewed below.
MORPHOLOGICAL EVIDENCE
Most foci of atypical adenomatous hyperplasia,as described in detail earlier, look very diVerentfrom lung adenocarcinoma, including BAC.However, as lesions of AAH become moreatypical and larger there are no morphologicalfeatures that reliably separate AAH fromBAC.63 Indeed, the largest and most atypicalAAH and BAC lesions might be the samething.64 Within the scope of the current WHOdefinition13 of BAC as a non-invasive tumourgrowing by replacement of alveolar lining cells,this is easy to understand. Furthermore, mostpeople who have studied AAH will have seenexamples of (usually larger) lesions that have,for the most part, histological features of AAHbut with varying grades of atypia and which, infoci, may well demonstrate invasion. The onlyreasonable conclusion, therefore, is that pro-gression to invasion has occurred in part of thepre-existing AAH. Careful examination oflarger well established invasive adenocarcino-mas, lesions that might contain zones of BAC-like tumour, may show an AAH-like zonearound the margin. Here I make the distinction(or try to) between lepidic spread of tumourand the presence of alveoli lined by cells thatare much less pleomorphic than those in thetumour. Although absolute proof that thesezones represent an edge of residual “adenoma”is lacking, these observations suggest such apossibility.
ASSOCIATION OF AAH WITH MALIGNANCY
Most AAH lesions are very small, cause nosymptoms, and are radiologically invisible.Thus, they are found incidentally, usually inlung surgically resected for another reason.One postmortem study prospectively soughtAAH and found only two patients with AAH in100 consecutive cases.65 Given the frequency ofpulmonary pathology at necropsy, such mate-rial is less than ideal for finding AAH lesions.
Table 2 The various changes occurring with progression from normal bronchial mucosa toinvasive carcinoma in central bronchial carcinogenesis
NormalSquamousmetaplasia
Low gradedysplasia
High gradedysplasia
Carcinomain situ
Invasivecarcinoma
Hyperproliferation + ++ ++ ++ +++ +++3p LOH + + ++ ++ +++ +++9p LOH + + ++ ++ +++ +++p53 overexpression + + ++ ++ +++ +++Rb expression ++ ++ ++ ++ ++ ++Cyclin D1 overexpression + + ++ ++Telomerase overexpression + + + + +++bcl-2 overexpression + + ++ ++Aneuploidy + ++ ++ +++p53 mutation + ++p16 loss + ++FHIT loss + ++ +++13q and 17p LOH + +++ +++5p and 5q LOH +
Normal refers to morphologically normal epithelium in cigarette smokers. Data summarisedfrom discussion in text and references.LOH, loss of heterozygosity.
266 Kerr
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from

Atypical adenomatous hyperplasia is mostcommonly found in lung resected for primarylung cancer, but has been found, less fre-quently, in lung resected for benign tumours ormetastatic disease. The presence of coincidentpathology such as pneumonia, obstructivepneumonitis, or fibrosis make AAH almostimpossible to find. Careful examination of wellinflated and formalin fixed lung (I prefer 1 cmthick parasagittal slices of resection specimensand constantly wash the cut surface of the lungwith water) will improve pick up rates. Optingnot to look for lesions macroscopically, relyinginstead on random block taking for histologicalidentification will reduce success. Clearly,however, more blocks will help to identify morelesions. I sample all likely foci and take up to sixrandom parenchymal blocks. Whatever thetechnique, determined by interest or prevailinglaboratory policy, the number of AAH lesionswill always be underestimated.
Overall figures for AAH incidence quotedrange from 9.3% to 21.4% of cases resectedfrom primary lung cancer, whereas lungsremoved for benign or metastatic disease yieldAAH in between 4.4% and 9.6% of cases.66–70
Furthermore, it is pertinent to note that AAHis more often found, and more often in largenumbers, in patients who have had lungresected for adenocarcinoma or large cellundiVerentiated carcinoma than for squamouscarcinoma. This does appear to be a trueassociation, although the presence of distantobstructive pneumonitis associated with a cen-tral bronchial tumour will make lesions morediYcult to find. Published data show that AAHis present in up to 34.5% and 25%, respec-tively, of lungs resected for adenocarcinoma orlarge cell undiVerentiated carcinoma.69 Thehighest published figure in squamous carci-noma is 11.1% of cases.67 Nakanishi describedAAH in 6.9% of squamous cancer bearinglungs yet found lesions in 34.5% of those withadenocarcinoma.69 In our own series of 554lungs resected for primary lung cancer, wefound AAH in 12% of cases, whereas it waspresent in 23.2% of lungs resected foradenocarcinoma and 12.5% of large cell undif-ferentiated cancer resections, but in only 3.3%of lungs with squamous carcinoma.70
AAH: CASES WITH MULTIPLE LESIONS
Some patients reported in the literature havevery large numbers of AAH lesions. To someextent, the number of lesions found will beproportional to the enthusiasm of the patholo-gist for finding them, the amount of lung tissueblocked, the presence of other pathologyobscuring the lesions, the state of lung fixationand expansion, and so on. Nonetheless, it istempting to believe that those patients withlarge numbers of AAH lesions might in someway be diVerent to those with relatively few.Not infrequently, those patients with largenumbers of AAH lesions also have multiplesynchronous primary carcinomas, sometimes“adenocarcinoma in AAH”. Those patientswith many AAH lesions almost invariably haveadenocarcinoma as their associated malig-nancy.
The patient described by Nadav and col-leagues71 had three synchronous primary BAC-type adenocarcinomas (but a single AAHlesion) associated with Li-Fraumeni syndrome,whereas two of Weng and colleague’s patientshad multiple AAH and two synchronousprimary lung cancers.66 Suzuki et al describe apatient with bilateral stage I BAC lesions andalso 12 AAH lesions.72 Patients with LOH inthe tuberous sclerosis gene associated regionsin adenocarcinoma may have multiple AAHlesions.73 Anami et al also described a patientwho underwent left upper lobectomy; theoperative specimen yielded two papillary ad-enocarcinomas and 161 AAH lesions.74
In our series of 70 patients with AAH, eightpatients had between six and eight AAHlesions, whereas four had 12, 19, 34, and 42lesions. Of these patients all had adenocarci-noma, except one who had a mixed carcinoma,including adenocarcinoma. Ten of the 70patients had multiple synchronous primaryadenocarcinomas (up to six in each resectionspecimen) and many had multiple AAHlesions.70
It is not known whether patients with such apropensity to develop large numbers of AAHlesions and cancers have a particular constitu-tive genetic lesion underlying this change. Thepatient described above with Li-Fraumeni syn-drome had only one AAH lesion. Nonetheless,this will be an interesting area of future study.
AAH: NUCLEAR SIZE, DNA CONTENT, AND CELL
PROLIFERATION
In 1986 Kodama et al morphometricallymeasured mean nuclear area (MNA) anddemonstrated an increase in mean values fromlow, through high grade AAH, to adenocarci-noma.75 Also of interest was their finding of twocell populations in two lesions. In each of theselesions (one was (apparently) an AAH, theother an adenocarcinoma), one cell populationhad a scatter of MNA typical of adenocarci-noma, the other was characteristic of AAH.This is of interest given the morphologicalobservations referred to above. A similarprogression of increasing MNA from AAHthrough BAC to “overt BAC” (adenocarci-noma with bronchioloalveolar features) wasdescribed by Kitamura et al.76
Nuclear DNA content was measuredcytofluorimetrically in a range of AAH lesionsand small adenocarcinomas by Nakayama etal.77 Using this measure, small solitary AAHlesions showed less DNA than areas of AAH inconjunction with adenocarcinoma, whereas thedata for this latter group were indistinguishablefrom those for adenocarcinoma. They alsofound aneuploid stem cell lines in 53.8% ofAAH foci and 76.9% of adenocarcinomas.These authors conclude that AAH was eitheran adenoma or a well diVerentiated adenocar-cinoma.
In a more sophisticated morphometric studyusing 12 variate cluster analysis, Mori and col-leagues78 found that lesions classified morpho-logically as AAH could be separated into twogroups, one of which showed characteristics
Pulmonary preinvasive neoplasia 267
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from

identical to adenocarcinomas of type II pneu-mocytes. These AAH lesions tended to bemore atypical than those without anadenocarcinoma-like data pattern.
Argyrophilic nucleolar organiser region(AgNOR) counts were shown to increase fromlow grade to high grade AAH and to BAC byNakanishi et al.79 They also examined DNAaneuploidy and found none in low grade AAH,but detected aneuploidy in 25% of high gradeAAH and 34.6% of BAC lesions. There are fewstudies of cell proliferation in AAH but oursand others, using anti-PCNA markers, foundthat the growth fraction in AAH is very low.80–82
All of these findings provide more objectiveevidence for the morphological observations ofincreasing atypia in AAH and the transition toadenocarcinoma.
AAH: IMMUNOHISTOCHEMISTRY
Several antigens have been shown to beexpressed, or overexpressed, in AAH whencompared with normal peripheral lung epithe-lium. Many authors have shown the expressionof carcinoembryonic antigen (CEA), with atendency for more to be expressed in larger ormore atypical lesions.67 69 76 83 CEA is almostuniversally expressed in lung adenocarcinoma.
Mori et al,84 in a re-examination of casessubjected to detailed morphological analysis,studied low and high grade AAH and BACwith a range of antibodies, including anti-CEA.Other antigens stained for were surfactant apo-protein A (SAP), urine protein 1 (UP1), andcytochrome p450. These authors reinforcedthe conclusion of their earlier work—that highgrade AAH lesions are closely related toBAC—by finding that both high grade AAHsand BACs had identical staining profiles (highCEA, p450 (including CYP1A1), and SAP andlow UP1), whereas low grade AAH lesionsshowed the reverse. A later study by Kitamuraand colleagues85 also used antibodies to UP1and SAP, but found that UP1 was notexpressed in AAH lesions but was found inBAC and was widespread in “overt BAC”(adenocarcinoma with bronchioloalveolar fea-tures). SAP staining was found in all lesions butappeared to decrease as the lesions advanced.
Blood group antigens expressed in normalalveolar epithelium and low grade AAH arealso lost as lesions progress to high grade atypiaand BAC.69
AAH: ONCOGENES AND TUMOUR SUPPRESSOR
GENES
One of the earliest studies of oncogene expres-sion in AAH lesions stained for p53 proteinand c-erbB2, the product of the neu onco-gene.86 Only 7% of AAH lesions expressedc-erbB2, and these were universally high gradelesions. Expression of p53 protein was found in58% of all lesions, with higher grade AAHlesions showing more staining. Other studieshave found either similar staining,87 or muchless stainable p53 protein in 10% of AAHlesions,76 although the relation with AAH gradewas preserved. In a small series of patients withcancer, Katabami and colleagues41 found that
although 25% of AAH lesions in patients with-out pneumoconiosis had raised p53, 36.4% ofAAH lesions in patients with pneumoconiosisshowed excess staining.
Slebos et al found stainable p53 in none oftheir low grade AAH lesions, 9% of high gradeAAH, and in 50% of their early BAC samples.88
One high grade AAH lesion showing p53 pro-tein overexpression had a mutation in the p53gene, whereas three others showed no muta-tion. Kohno and colleagues45 also found p53gene mutation to be an infrequent event,occurring in only one lesion of 20 examined,and concluded that p53 mutation may be arelatively late event in peripheral adenocarcino-genesis, when compared with events leading tothe development of bronchial squamous carci-noma.
In a study comparing p53 expression withthe expression of p53 inducible cyclin depend-ent kinase inhibitor p21Waf1/Cip1, Hayashi et alfound that, although reactive lesions showed acorrelation between p53 and p21 expression,this was lost in both AAH and carcinomas ofvarious stages.80 They suggested that p21expression might become a p53 independentevent during neoplastic transformation, even atthe very early AAH stage.
Cell cycle regulators were studied by Kura-sono et al.81 They found that, whereas cyclinD1 appeared to be overexpressed in most AAHfoci, but less so in BAC, the loss of Rb and p16expression was rare or not seen at all in AAH,and was relatively infrequent in adenocarci-noma. They suggested that although cyclin D1overexpression might occur early in adenocar-cinoma tumorigenesis, it did not appear to berequired to maintain the malignant phenotype.
Four studies have reported the presence ofK-ras mutation (point mutation at codon 12)in AAH. This is an important finding becauseK-ras mutations are frequent in human pulmo-nary adenocarcinoma. Such mutations werefirst described by Ohshima et al in 1994,89 whofound mutation in AAH in two of six patientsstudied. Sagawa et al found only one AAHlesion with a K-ras mutation in the 39 lesionsexamined.90 Westra et al found that 39% ofAAH lesions had the mutation, which wasevenly distributed among diVerent grades ofAAH and similar in frequency to that found inadenocarcinoma.91 Furthermore, these authorsshowed that the AAH lesion often showed dif-ferent base changes at codon 12 of the K-rasgene when compared with those found in theconcurrent adenocarcinoma from the samepatient. This is consistent with the notion thatAAH represents “field cancerisation” in thelung periphery. In our own work51 K-ras muta-tion was found in fewer AAH lesions (15% ofthose studied) but was seen in 35% of periph-eral parenchymal type adenocarcinomas (ad-enocarcinoma with bronchioloalveolar fea-tures), the type of adenocarcinoma arisingfrom AAH, although never being found inadenocarcinomas of bronchial origin.
268 Kerr
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from

AAH: LOSS OF HETEROZYGOSITY AND OTHER
GENOMIC ALTERATIONS
Much work, already detailed above, has beencarried out on the preinvasive lesions thatoccur in the central bronchial epithelium.Much less is known of AAH lesions. Kohno etal studied 28 AAH lesions, eight low grade and20 high grade.45 No LOH was identified in lowgrade lesions but two high grade lesionsshowed 3p LOH and one showed 9p loss.Although no AAH lesions were studied bySuzuki et al,73 they showed that lungs with anadenocarcinoma, which demonstrated LOH inthe tuberous sclerosis gene associated regions(TSC1 and TSC2–9q and 16p, respectively),were more likely also to have multiple AAHlesions. Nadav et al also described an interest-ing patient with Li-Fraumeni syndrome whohad multiple synchronous lung cancer andAAH lesions.71 Anami et al studied a singleunusual patient who had two synchronous lungadenocarcinomas and 161 AAH lesions.74 SixAAH lesions greater than 3 mm were exam-ined and 17q LOH was shown in two, 9q LOHin one, whereas another demonstrated micros-atellite instability at D17S791. Kitaguchi et alstudied 31 AAH lesions and showed 3p, 9p,and 17p loss in 19%, 13%, and 5%, respec-tively.92 All lesions with genetic alterations werehigher grade. The 3p LOH is clearly of interest,even though to date very few lesions have beenstudied, given the finding that 43% of BAClesions show evidence of LOH within theFHIT gene at region 3p14.2.93
AAH lesions have been shown to bemonoclonal proliferations by analysis based onthe X-linked polymorphic human androgenreceptor gene.94 Of interest also, in this paper,was the presence of identical monoclonality intwo cases of BAC and the contiguous AAH.This finding supports the earlier contentionthat morphological observations fit with thehypothesis that the AAH-like edge, which issometimes observed around a peripheral ad-enocarcinoma (with or without bronchioloal-veolar features), is evidence of the pre-existingadenoma from which the invasive carcinomaarose.
AAH: FATE OF THE LESION
Despite the increasing amount of data, summa-rised above and in table 3, which support a
progression towards malignancy at a morpho-logical and molecular level, there are no data tosuggest the risk of progression of AAH to inva-sive carcinoma. The almost impossible task ofidentifying these lesions before resection rulesout any longitudinal study. It remains to beseen whether or not state of the art spiral com-puted tomography (CT) scanning can oVersome chance of detecting the largest examplesof AAH.95 96
In 1997 Suzuki et al described a follow upstudy where patients, whose resected cancerswere associated with AAH, were comparedwith those who did not have AAH.97 The pres-ence of AAH in the lungs did not appear toaVect the survival of the patients adversely.Conversely, in another study98 there is evidencethat patients with stage IA disease and AAHhad a better postoperative survival than thosewithout AAH. Our data mirror those of Suzukiet al.70
Morphological observations of our owncases suggest that a small proportion of AAHlesions might undergo sclerosis and possibleregression, leaving behind a scar in the lung,which retains the topography of an AAHlesion. Sometimes this process aVects only partof the lesion, and some or all of the patient’slesions may show this change. What relationthis has with the heavy infiltration of AAH bylymphocytes occasionally seen in some patientsis not known.
Until it becomes possible to make longitudi-nal observations of lesions, or an animal modelis found, evidence that AAH does progressthrough BAC to invasive adenocarcinoma willlargely be based on observational and circum-stantial evidence, as described in this review.
Pulmonary preinvasive lesions: relevanceto clinical practiceApart from academic interest in the process ofcarcinogenesis, a knowledge of pulmonary pre-invasive lesions also has relevance to severalclinical situations. If progress is ever to be madewith screening for lung cancer, then moreneeds to be understood about its progenitorlesions. It is clear that detecting pulmonaryneoplasia at a stage when invasion is alreadypresent is too late. To date, screening haslargely been based upon examining sputumspecimens for atypical cells, or mass radio-graphy campaigns. Morphology on sputumalone is not good at diVerentiating invasivefrom preinvasive bronchial disease, and itremains to be seen whether any other specificmarker can be found that can reliably identifyexfoliated neoplastic cells. However, fluores-cence bronchoscopy (LIFE) might increase thedetection of squamous dysplasia/CIS, a lesionmore or less invisible using standard visualisa-tion methods. Histopathologists may face hav-ing to deal with large numbers of biopsies frombronchi examined using the LIFE technique.The experience of Lam et al would suggest thatamong the frequent non-atypical biopsy speci-mens taken during LIFE procedures, a varietyof dysplasias will be diagnosed.14 What then?We have so little idea of how these progress andcurrently have such limited therapeutic options
Table 3 The various morphological, morphometric, immunohistochemical, and molecularbiological findings in AAH and adenocarcinoma
Low grade AAH High grade AAHAdenocarcinoma(including BAC)
Cellularity and atypia + ++ +++Mean nuclear area + ++ +++Nuclear DNA content + ++ ++Aneuploidy + ++ ++AgNOR count + ++ +++CEA, p450, ApoA expression + ++ ++p53 expression + ++ +++K-ras mutation −/+ + ++C-erb-B2 expression + ++Loss of Rb and p16 +3p, 7p, 17p/q LOH + ++Blood group antigen expression +++ + +Cyclin D1 overexpression +++ ++ +
AAH, atypical adenomatous hyperplasia; AgNOR, silver stained nucleolar organising region;BAC, bronchioloalveolar cell carcinoma; CEA, carcinoembryonic antigen; LOH, loss ofheterozygosity.
Pulmonary preinvasive neoplasia 269
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from

that these diagnoses may be of limited clinicalvalue. Their presence might help to persuade apatient to stop smoking, and lesions may betreatable by photodynamic therapy, but the realhope is that some form of chemopreventiveintervention can be oVered, with surveillanceusing fluorescence bronchoscopy. Will this beyet another contribution to the ever increasingnumbers of biopsy specimens pouring throughthe doors of histopathology departments?
The lung periphery is much harder to “getat”. Unless a very specific marker comes tolight, I doubt whether bronchoalveolar lavagehas anything to oVer in terms of detectingAAH. Given the size and architecture of mostlesions, they are invisible on plain chest x rayfilms or CT scans using standard resolution.Even high resolution CT would be unlikely toproduce a specific enough appearance to bediagnostic. Nonetheless, the work of Eguchiand colleagues95 and Sone and colleagues96 is ofinterest because leading edge radiology mightprovide a means of detecting relatively largelesions that could be excised, perhaps usingstereotactic needle localisation. This again,opens the door to a potential screeningsituation. This prospect, in turn, raises theissue of the diagnosis of AAH and its diVeren-tiation from BAC, as currently defined by theWHO. Using existing criteria, distinguishing ahigh grade AAH from BAC is very diYcult,certainly using morphology alone. As yet, noreliable adjunct marker or group of markershas been found to help. In any case, it may bepointless to try to separate what is essentiallythe same disease, pulmonary alveolar intraepi-thelial neoplasia or adenocarcinoma in situ.
Thanks to Dr A Chapman, Mr S Pritchard, and Professor JSimpson for their review and comments on this manuscript.
1 Sharp L, Brewster D. The epidemiology of lung cancer inScotland: a review of trends in incidence, survival and mor-tality and prospects for prevention. Health Bull (Edinb)1999;57:318–11.
2 Auerbach O, Hammond EC, Garfinkel L. Changes in bron-chial epithelium in relation to smoking. N Engl J Med1979;300:381–6.
3 Auerbach O, Stout AP, Hammond EC, et al. Changes inbronchial epithelium in relation to sex, age, residence,smoking and pneumonia. N Engl J Med 1962;267:111–19.
4 Shimosato Y, Hashimoto T, Kodama T, et al. Prognosticimplications of fibrotic focus (scar) in small peripheral lungcancers. Am J Surg Pathol 1980;4:365–73.
5 Madri JA, Carter D. Scar cancer of the lung: origin and sig-nificance. Hum Pathol 1984;15:625–31.
6 Barsky SH, Huang SJ, Bhuta S. The extracellular matrix ofpulmonary scar carcinomas is suggestive of a desmoplasticorigin. Am J Pathol 1986;124:412–19.
7 Fraire AE, Greenberg SD. Carcinoma and diVuse interstitialfibrosis of lung. Cancer 1973;31:1078–86.
8 Henderson DW, de Klerk NH, Hammar SP, et al. Asbestosand lung cancer: is it attributable to asbestosis or to asbes-tos fiber burden? In: Corrin B, ed. Pathology of lung tumours.Edinburgh: Churchill Livingstone, 1997:83–118.
9 Weill H, McDonald JC. Exposure to crystalline silica andrisk of lung cancer: the epidemiological evidence. Thorax1995;51:97–102.
10 Kerr KM. Adenomatous hyperplasia and the origin ofperipheral adenocarcinoma of the lung. In: Corrin B, ed.Pathology of lung tumours. Edinburgh: Churchill Living-stone, 1997:119–34.
11 Miller RR, Nelems B, Evans KG, et al. Glandular neoplasiaof the lung. A proposed analogy to colonic tumours. Cancer1988;61:1009–14.
12 Miller RR. Bronchioloalveolar cell adenomas. Am J SurgPathol 1990;14:904–12.
13 Travis WD, Colby TV, Corrin B, et al, eds. Histological typingof lung and pleural tumours. WHO international histologicalclassification of tumours, 3rd ed. Berlin: Springer, 1999.
14 Lam S, MacAulay C, Hung J, et al. Detection of dysplasiaand carcinoma in situ by a lung imaging fluorescence(LIFE) device. J Thorac Cardiovasc Surg 1993;105:1035–40.
15 George PJM. Fluorescence bronchoscopy for the earlydetection of lung cancer. Thorax 1999;54:180–3.
16 Shimosato Y, Kodama T, Kameya T. Morphogenesis ofperipheral type adenocarcinoma of the lung. In: ShimosatoY, Melamed MR, Nettesheim P, eds. Morphogenesis of lungcancer, Vol. 1. Boca Raton, FL: CRC Press, 1982:65–90.
17 Kodama T, Biyajima S, Watanabe S, et al. Morphometricstudy of adenocarcinomas and hyperplastic epitheliallesions in the peripheral lung. Am J Clin Pathol 1986;85:146–51.
18 Miller R, Muller NL. Neuroendocrine cell hyperplasia andobliterative bronchiolitis in patients with peripheral carci-noid tumours. Am J Surg Pathol 1995;19:653–8.
19 Aguayo SM, Miller YE, Waldron JA, et al. Idiopathic diVusehyperplasia of pulmonary neuroendocrine cells and airwaydisease. N Engl J Med 1992;327:1285–8.
20 Melamed MR, Zaman MB. Pathogenesis of epidermoidcarcinoma of lung. In: Shimosato Y, Melamed MR,Nettesheim P, eds. Morphogenesis of lung cancer, Vol. 1. BocaRaton, FL: CRC Press, 1982:37–64.
21 Nasiell M, Auer G, Kato H. Cytological studies in man andanimals on the development of bronchogenic carcinoma.In: McDowell EM, ed. Lung carcinomas. Edinburgh:Churchill Livingstone, 1987:207–42.
22 Nettesheim P, Klein-Szanto AJP, Yarita T. Experimentalmodels for the study of morphogenesis of lung cancer. In:Shimosato Y, Melamed MR, Nettesheim P, eds. Morpho-genesis of lung cancer, Vol. 2. Boca Raton, FL: CRC Press,1982:131–66.
23 Cho KR, Vogelstein B. Genetic alterations in the adenoma–carcinoma sequence. Cancer 1992;70:1727–31.
24 Willis RA. In: Willis RA, ed. The pathology of tumours, 4th ed.London: Butterworths, 1967:1.
25 Auerbach O. Pathogenesis of lung cancer. Cancer 1961;7:11–21.
26 Suprun H, Hjerpe A, Nasiell M, et al. A correlative cytologicstudy of the incidence of pulmonary cancer and other lungdiseases associated with squamous metaplasia of the bron-chial epithelium. In: Niebergs HE, ed. Prevention and detec-tion of cancer. Part 2. Detection. New York: Marcel Dekkar,1980:1303–20.
27 Saccomanno G, Archer VE, Auerbach O, et al. Developmentof carcinoma of the lung as reflected in exfoliated cells.Cancer 1974;33:256–70.
28 Carter D, Patchefsky AS. Keratinising lesions. In: Tumoursand tumour like lesions of the lung. Philadelphia: Saunders,1998:120–47.
29 Tao LC, Chamberlain DW, Delarue NC, et al. Cytologicdiagnosis of radiographically occult squamous cell carci-noma of the lung. Cancer 1982;50:1580–6.
30 Nagamoto N, Saito Y, Suda H, et al. Relationship betweenlength of longitudinal extension and maximal depth oftramsmural invasion in roentgenographically occultsquamous cell carcinoma of the bronchus (nonpolypoidtype). Am J Surg Pathol 1989;13:11–20.
31 Nagamoto N, Saito Y, Ohta S, et al. Relationship betweenlymph node metastasis to primary tumour size and micro-scopic appearance of roentgenographically occult lungcancer. Am J Surg Pathol 1989;13:1009–13.
32 Nagamoto N, Saito Y ,Sato M, et al. Clinicopathologicalanalysis of 19 cases of isolated carcinoma in situ of thebronchus. Am J Surg Pathol 1993;17:1234–43.
33 Spencer H, Dail DH, Arneaud J. Non-invasive bronchialepithelial papillary tumors. Cancer 1980;45:1486–97.
34 Hirano T, Franzen B, Kato H, et al. Genesis of squamouscell lung carcinoma. Sequential changes of proliferation,DNA ploidy and p53 expression. Am J Pathol 1994;144:296–302.
35 Pendelton N, Dixon GR, Burnett HE, et al. Expression ofproliferating cell nuclear antigen (PCNA) in dysplasia ofthe bronchial epithelium. J Pathol 1993;170:169–72.
36 Brambilla E, Gazzeri S, Moro D, et al. Alterations of Rbpathway (Rb–p16INK4–cyclin D1) in preinvasive bron-chial lesions. Clin Cancer Res 1999;5:243–50.
37 Pezzella F, Turley H, Kuzu I, et al. bcl-2 protein in non-smallcell lung carcinoma. N Engl J Med 1993;329:690–4.
38 Brambilla E, Negoescu A, Gazzeri S, et al. Apoptosis-relatedfactors p53, bcl-2 and bax in neuroendocrine lungtumours. Am J Pathol 1996;149:1941–52.
39 Kennedy MM, Lamb D, King G, et al. Cell proliferation,cell loss and expression of bcl-2 and p53 in human pulmo-nary neoplasms. Br J Cancer 1997;75:545–7.
40 Walker C, Robertson L, Myskow M, et al. Expression ofthe bcl-2 protein in normal and dysplastic bronchialepithelium and in lung carcinomas. Br J Cancer 1995;72:164–9.
41 Katabami M, Dosaka-Akita H, Honma K, et al. p53 andbcl-2 expression in pneumoconiosis-related pre-cancerouslesions and lung cancers: frequent and preferential p53expression in pneumoconiotic bronchiolar dysplasias. Int JCancer 1998;75:504–11.
42 Brambilla E, Gazzeri S, Lantuejoul S, et al. p53 mutantimmunophenotype and deregulation of p53 transcriptionpathway (bcl2, bax and waf1) in precursor bronchiallesions of lung cancer. Clin Cancer Res 1998;4:1609–18.
43 Nuorva K, Soini Y, Kamel D, et al. Concurrent p53 expres-sion in bronchial dysplasias and squamous cell lung carci-nomas. Am J Pathol 1993;142:725–32.
44 Bennett WP, Colby TV, Travis WD, et al. p53 protein accu-mulates frequently in early bronchial neoplasia. Cancer Res1993;53: 4817–22.
45 Kohno H, Hiroshima K, Toyozaki T, et al. p53 mutation andallelic loss of chromosome 3p, 9p of preneoplastic lesions inpatients with non-small cell lung carcinoma. Cancer1999;85:341–7.
270 Kerr
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from

46 Sundaresan V, Ganly P, Hasleton P, et al. p53 and chromo-some 3 abnormalities, characteristic of malignant lungtumours, are detectable in preinvasive lesions of the bron-chus. Oncogene 1992;7:1989–97.
47 Sozzi G, Miozzo M, Donghi R, et al. Deletions of 17p andp53 mutations in preneoplastic lesions of the lung. CancerRes 1992;52:6079–82.
48 Vahakangas KH, Samet JM, Metcalf RA, et al. Mutations ofp53 and ras genes in radon-associated lung cancer fromuranium miners. Lancet 1992;339:576–80.
49 Yashima K, Litzky LA, Kaiser L, et al. Telomeraseexpression in respiratory epithelium during the multistagepathogenesis of lung carcinomas. Cancer Res 1997;57:2373–7.
50 Sugio K, Kishimoto Y, Virmani AK, et al. K-ras mutationsare a relatively late event in the pathogenesis of lung carci-nomas. Cancer Res 1994;54:5811–15.
51 Cooper CA, Carey FA, Bubb VJ, et al. The pattern of K-rasmutation in pulmonary adenocarcinoma defines a newpathway of tumour development in the human lung. JPathol 1997;181:401–4.
52 Bejui-Thivolet F, Liagre N, Chignol MC, et al. Detection ofhuman papillomavirus DNA in squamous bronchial meta-plasia and squamous cell carcinomas of the lung by in situhybridisation using biotinylated probes in paraYn-embedded specimens. Hum Pathol 1990;21:111–16.
53 Carey FA, Salter DM, Kerr KM, et al. An investigation intothe role of human papilloma virus in endobronchial papil-lary squamous tumours. Respir Med 1990;84:445–7.
54 Ong ST, Fong KM, Bader SA, et al. Precise localisation ofthe FHIT gene to the common fragile site at 3p14.2(FRA3B) and characterisation of homozygous deletionswithin FRA3B that aVect FHIT transcription in tumourcell lines. Genes Chromosomes Cancer 1997;20:16–23.
55 Sozzi G, Tornielli S, Tagliabue E, et al. Absence of Fhit pro-tein in primary lung tumours and cell lines with FHIT geneabnormalities. Cancer Res 1997;57:5207–12.
56 Sozzi G, Pastorino U, Moiraghi L, et al. Loss of FHIT func-tion in lung cancer and preinvasive bronchial lesions. Can-cer Res 1998;58:5032–7.
57 Sozzi G, Sard L, De Gregorio L, et al. Association betweencigarette smoking and FHIT gene alterations in lungcancer. Cancer Res 1997;57:2121–3.
58 Fong KM, Biesterveld EJ, Virmani A, et al. FHIT andFRA3B 3p14.2 allele loss are common in lung cancer andpreneoplastic bronchial lesions and are associated withcancer-related FHIT cDNA splicing aberrations. CancerRes 1997;57:2256–67.
59 Wistuba II, Behrens C, Milchgrub S, et al. Sequentialmolecular abnormalities are involved in the multistagedevelopment of squamous cell lung carcinoma. Oncogene1999;18:643–50.
60 Hung J, Kishimoto Y, Sugio K, et al. Allele-specific chromo-some 3p deletions occur at an early stage in the pathogen-esis of lung carcinoma. JAMA 1995;273:558–63.
61 Endo C, Sagawa M, Sato M, et al. Sequential loss of hetero-zygosity in the progression of squamous cell carcinoma ofthe lung. Br J Cancer 1998;78:612–15.
62 Carey FA, Bubb VJ, Carter R, et al. Loss of heterozygosity atchromosome 5q is seen in proximal but not in parenchymalpulmonary carcinomas. J Pathol 1994;173:151A.
63 Colby TV, Wistuba II, Gazdar A. Precursors to pulmonaryneoplasia. Adv Anat Pathol 1998;5:205–15.
64 Kitamura H, Kameda Y, Ito T, et al. Atypical adenomatoushyperplasia of the lung. Implications for the pathogenesis ofperipheral lung adenocarcinoma. Am J Clin Pathol1999;111:610–22.
65 Sterner DJ, Masuko M, Roggli VL, et al. Prevalence of pul-monary atypical alveolar cell hyperplasia in an autopsypopulation: a study of 100 cases. Mod Pathol 1997;10:469–73.
66 Weng S-Y, Tsuchiya E, Kasuga T, et al. Incidence of atypicalbronchioloalveolar cell hyperplasia of the lung: relation tohistological subtypes of lung cancer. Virchows Archiv APathol Anat Histopathol 1992;420:463–71.
67 Morinaga S, Shimosato Y. Pathology of the microadenocar-cinoma in the periphery of the lung [in Japanese]. Pathologyand Clinical Medicine 1987;5:74–80.
68 Kodama T, Nishiyama H, Nishiwaki Y, et al. Histopathologi-cal study of adenocarcinoma and hyperplastic epitheliallesions of the lung. Lung Cancer 1988;8:325–33.
69 Nakanishi K. Alveolar epithelial hyperplasia and adenocar-cinoma of the lung. Arch Pathol Lab Med 1990;114:363–8.
70 Chapman AD, Kerr KM. The association between atypicaladenomatous hyperplasia and primary lung cancer. Br JCancer 2000;83:632–6.
71 Nadav Y, Pastorino U, Nicholson AG. Multiple synchro-nous lung cancers and atypical adenomatous hyperplasia inLi-Fraumeni syndrome. Histopathology 1998;33:52–4.
72 Suzuki K, Takahashi K, Yoshida J, et al. Synchronous doubleprimary lung carcinomas associated with multiple atypicaladenomatous hyperplasia. Lung Cancer 1998;19:131–9.
73 Suzuki K, Ogura T, Yokose T, et al. Loss of heterozygosity inthe tuberous sclerosis gene associated regions in adenocar-cinoma of the lung accompanied by multiple atypicaladenomatous hyperplasia. Int J Cancer 1998;79:384–9.
74 Anami Y, Matsuno Y, Yamada T, et al. A case of double pri-mary adenocarcinoma of the lung with multiple atypicaladenomatous hyperplasia. Pathol Int 1998;48:634–40.
75 Kodama T, Biyajima S, Watanabe S, et al. Morphometricstudy of adenocarcinomas and hyperplastic epitheliallesions in the peripheral lung. Am J Clin Pathol 1986;85:146–51.
76 Kitamura H, Kameda Y, Nakamura N, et al. Atypicaladenomatous hyperplasia and bronchoalveolar lung carci-noma. Analysis by morphometry and the expressions ofp53 and carcinoembryonic antigen. Am J Surg Pathol 1996;20:553–62.
77 Nakayama H, Noguchi M, Tsuchiya R, et al. Clonal growthof atypical adenomatous hyperplasia of the lung:cytofluorometric analysis of nuclear DNA content. ModPathol 1990;3:314–20.
78 Mori M, Chiba R, Takahashi T. Atypical adenomatoushyperplasia of the lung and its diVerentiation from adeno-carcinoma. Characterisation of atypical cells by morphom-etry and multivariate cluster analysis. Cancer 1993;72:2331–40.
79 Nakanishi K, Hiroi S, Kawai T, et al. Argyrophilicnucleolar-organiser region counts and DNA status inbronchioloalveolar epithelial hyperplasia and adenocarci-noma of the lung. Hum Pathol 1998;29:235–9.
80 Hayashi H, Miyamoto H, Ito T, et al. Analysis ofp21waf1/cip1 expression in normal, premalignant andmalignant cells during the development of human lungadenocarcinoma. Am J Pathol 1997;151:461–70.
81 Kurasono Y, Ito T, Kameda Y, et al. Expression of cyclin D1,retinoblastoma gene protein and p16 MTS1 protein inatypical adenomatous hyperplasia and adenocarcinoma ofthe lung. An immunohistochemical analysis. Virchows Arch1998;432:207–15.
82 Carey FA, Wallace WAH, Fergusson RJ, et al. Alveolaratypical hyperplasia in association with primary pulmonaryadenocarcinoma: a clinicopathological study of 10 cases.Thorax 1992;47:1041–3.
83 Rao SK, Fraire AE. Alveolar cell hyperplasia in associationwith adenocarcinoma of lung. Mod Pathol 1995;9:99–108.
84 Mori M, Tezuka F, Chiba R, et al. Atypical adenomatoushyperplasia and adenocarcinoma of the human lung.Cancer 1996;77:665–74.
85 Kitamura H, Kameda Y, Ito T, et al. CytodiVerentiation ofatypical adenomatous hyperplasia and bronchioloalveolarlung adenocarcinoma: immunohistochemical and ul-trastructural studies. Virchows Arch 1997;431:415–24.
86 Kerr KM, Carey F, King G, et al. Atypical alveolarhyperplasia: relationship with pulmonary adenocarcinoma,p53 and c-erbB2 expression. J Pathol 1994;174:249–56.
87 Pueblitz S, Hieger LR. Expression of p53 and CEA in atypi-cal adenomatous hyperplasia of the lung [letter]. Am J SurgPathol 1997;21:867–9.
88 Slebos RJC, Baas IO, Clement MJ, et al. p53 alterations inatypical alveolar hyperplasia of the human lung. HumPathol 1998;29:801–8.
89 Ohshima S, Shimizu Y, Takahama M. Detection of c-Ki-rasgene mutation in paraYn sections of adenocarcinoma andatypical bronchioloalveolar cell hyperplasia of human lung.Virchows Arch 1994;424:129–34.
90 Sagawa M, Saito Y, Fujimura S, et al. K-ras point mutationoccurs in the early stage of carcinogenesis in lung cancer.Br J Cancer 1998;77:720–3.
91 Westra WH, Baas IO, Hruban RH, et al. K-ras oncogeneactivation in atypical alveolar hyperplasias of the humanlung. Cancer Res 1996;56:2224–8.
92 Kitaguchi S, Takeshima Y, Nishisaka T, et al. Genomicinstability and K-ras mutation in atypical adenomatoushyperplasia of the lung. Lung Cancer 1997;18(suppl1):175–6.
93 Marchetti A, Pellegrini S, Bertacca G, et al. FHIT and p53gene abnormalities in bronchioloalveolar carcinomas. Cor-relations with clinicopathological data and K-ras muta-tions. J Pathol 1998;184:240–6.
94 Niho S, Yokose T, Suzuki K, et al. Monoclonality of atypicaladenomatous hyperplasia of the lung. Am J Pathol1999;154:249–54.
95 Eguchi K, Kaneko M, Kobayashi T, et al. High-resolutioncomputed tomography of bronchioloalveolar cell carci-noma. Lung Cancer 1997;18(suppl 2):16.
96 Sone S, Takashima S, Li F, et al. Mass screening for lungcancer with mobile spiral computed tomography scanner.Lancet 1998;351:1242–5.
97 Suzuki K, Nagai K, Yoshida J, et al. The prognosis ofresected lung carcinoma associated with atypical adenoma-tous hyperplasia. Cancer 1997;79:1521–6.
98 Takigawa N, Segawa Y, Nakata M, et al. Clinicalinvestigation of atypical adenomatous hyperplasia of thelung. Lung Cancer 1999;25:115–21.
Pulmonary preinvasive neoplasia 271
www.jclinpath.com
on 19 June 2018 by guest. Protected by copyright.
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.54.4.257 on 1 A
pril 2001. Dow
nloaded from





























