Embed Size (px)
DESCRIPTION
hiperplasia adrenal kongenital
Citation preview

MAKALAH
SISTEM ENDOKRIN
(MASALAH KELENJAR ADRENAL PADA WANITA)
`
Dosen Pembimbing :
Nur Hidaayah.S. Kep,. Ns, M. Kes
Disusun oleh :
1. Riyco Rachman P. 1130013085
2. Rizki Ramadhan 1130013086
3. Rochmatul Ummah 1130013087
4. Satria Achrudi A. 1130013088
5. Siti Maisaroh 1130013089
6. Sonya Dewi Finanti 1130013090
7. Baiq Paramita 1300120
PRODI S1 KEPERAWATAN
FAKULTAS KEPERAWATAN DAN KEBIDANAN
UNIVERSITAS NAHDLATUL ULAMA SURABAYA
2014-2015

KATA PENGANTAR
Puji dan syukur penulis panjatkan kehadirat Allah SWT, karena dengan
rahmat dan karunia-Nya kami masih diberi kesempatan untuk menyelesaikan
makalah ini. Tidak lupa kami ucapkan kepada dosen pembimbing dan teman-
teman yang telah memberikan dukungan dalam menyelesaikan makalah ini.
Penulis ucapkan banyak terima kasih kepada segala pihak yang telah
membantu dalam penulisan makalah ini. Semoga makalah ini bisa membantu bagi
siapa saja yang membutuhkan sedikit pengetahuan tentang salah satu materi
Sistem Pencernaan. Materi yang kami angkat dalam makalah ini adalah “SISTEM
ENDOKRIN (Masalah Kelenjar Adrenal Pada Wanita)”.
Namun demikian makalah ini masih jauh dari kesempurnaan, oleh sebab itu
kami sangat mengharapkan kritik dan saran yang membangun. Dan semoga
dengan selesainya makalah ini dapat bermanfaat bagi pembaca dan teman-teman.
Surabaya, Maret 2015
Penulis
ii

DAFTAR ISI
HALAMAN JUDUL
KATA PENGANTAR ............................................................................................
ii
DAFTAR ISI ............................................................................................................
iii
BAB I PENDAHULUAN ........................................................................................
1
A. Latar Belakang ..................................................................................................
1
B. Perumusan Masalah ..........................................................................................
2
C. Tujuan ...............................................................................................................
2
BAB II TINJAUAN TEORI....................................................................................
3
A. Definisi Congenital Adrenal Hyperplasia...........................................................
3
B. Etiologi................................................................................................................
4
C. Patofisiologi........................................................................................................
4
D. Aspek genetic pada CAH....................................................................................
7
E. Klasifikasi CAH..................................................................................................
9
F. Tanda dan Gejala ................................................................................................
14
G. Fungsi Repodruksi..............................................................................................
17
iii

H. Manifestasi Klinis...............................................................................................
18
I. Penatalaksanaan CAH ........................................................................................
19
J. Konsep Asuhan Keperawatan ............................................................................
20
BAB III PENUTUP..................................................................................................
25
A. Kesimpulan ........................................................................................................
25
B. Saran ...................................................................................................................
25
DAFTAR PUSTAKA
iv

BAB I
PENDAHULUAN
A. Latar Belakang
Di dalam dunia medis, tidak selalu mudah untuk menentukan sex dan
genderseseorang. Sex adalah sesuatu hal yang dapat membedakan apakah
seseorang itu pria ataupun wanita secara fisik, sedangkan gender adalah identitas
yang terdapat pada orang yang bersangkutan. Ketika genitalia luar seseorang tidak
dapat ditentukan secara pastiapakah pria ataupun wanita pada umumnya, maka hal
tersebut dikatakan Disorders ofSex Development (DSDs). DSDs adalah kondisi
congenital dimana perkembangankromosom, gonad atau anatomi seksual menjadi
tidak khas. DSDs meliputiperkembangan congenital dari genitalia yang ambigu
(misalkan pada 46,XX virilizingCongenital Adrenal Hyperplasia (CAH);
clitoromegaly; mikropenis); kelainanpemisahan congenital dari anatomi seksual
internal dan eksternal (misalkan padaComplete Androgen Insensitivity Syndrome
(CAIS); defisiensi enzim 5α-reduktase/ 5-AR); anomali kromosom seks (misalkan
pada Sindrom Turner, Sindrom Klinefelter, sexchromosome mosaicism); kelainan
perkembangan gonad (misalkan pada ovotestis). Jadiyang termasuk dalam
kategori DSDs adalah anomali dari seks kromosom, gonad, saluran reeproduksi
dan juga genitalia. DSDs dapat terjadi pada bayi dengan kariotipe 46,XXmaupun
46,XY.
Kasus paling sering dari pasien dengan 46,XX DSD adalah CAH, atau
sindromaadrenogenital. Kelainan ini terjadi sekitar 60% dari kasus interseksual
yang ada. CAHmerupakan sekelompok kelainan yang diturunkan secara
autosomal resesif danmenyebabkan defisiensi satu dari lima enzim yang
dibutuhkan dalam proses sintesishormon kortisol dan aldosteron dari kolesterol
pada korteks adrenal (steroidogenesis)sehingga produksi hormon steroid sex
(testosteron) menjadi berlebihan yang kemudianakan merubah perkembangan
karakteristik sexual wanita dengan kariotipe 46,XXmenjadi ke arah laki-laki
(maskulinisasi). Bentuk yang lebih berat dari CAH adalahmenurunnya produksi
hormon aldosteron dan terjadi salt-wasting. Lebih dari 90% kasusCAH
disebabkan karena defisiensi enzim 21-hidroksilase. Resiko mempunyai
1

anakdengan CAH tipe klasik adalah 1:16.000 bayi, untuk tipe non-klasik adalah
0,2% daripopulasi orang berkulit putih pada umumnya, namun lebih sering (1-
2%) pada populasietnik tertentu seperti Yahudi yang berasal dari Eropa Timur.
Kelainan ini dapatdiidentifikasi dengan melakukan pemeriksaan fisik maupun
pemeriksaan penunjang.Pemeriksaan fisik dilakukan terkait dengan tanda dan
gejala khas yang terkait dengankelainan tersebut.
Penelitian ini bertujuan untuk mengetahui profil karakteristik fisik pada
pasienCAH di Semarang. Dengan harapan, yang akan datang dalam
mengidentifikasi danmendiagnosis pasien CAH diantara kasus DSDs yang ada di
Indonesia, terutama denganpemeriksaan fisik, akan lebih sederhana dan akurat.
B. Rumusan Masalah
1. Bagaimana anatomi dan fisiologi mata?
2. Apa definisi dari fusi?
3. Apa definisi dari konvergensi dan divergensi?
4. Bagaimana epidemoligi strabismus?
5. Apa definisi strabismus?
6. Bagaimana etiologi strabismus?
7. Bagaimana patofisiologi dan WOC strabismus?
8. Bagaimana klasifikasi strabismus?
9. Bagaimana manifestasi klinis strabismus?
10. Bagaimana penatalaksanaan strabismus?
11. Apa saja pemeriksaan penunjang strabismus?
12. Bagaimana asuhan keperawatan strabismus?
C. Tujuan
1. Untuk mengetahui anatomi dan fisiologi mata.
2. Untuk menegetahui pengertian dari fusi.
3. Untuk menegetahui pengertiankonvergensi dan divergensi.
4. Untuk mengetahui epidemiologi strabismus.
5. Untuk mengetahui definisi strabismus.
6. Untuk mengetahui etiologi strabismus.
7. Untuk mengetahui patofisiologi dan WOC strabismus.
2

8. Untuk mengetahui klasifikasi strabismus.
9. Untuk mengetahui manifestasi strabismus.
10. Untuk mengetahui penatalaksanaan strabismus.
11. Untuk mengetahui pemeriksaan penunjang strabismus.
12. Untuk mengetahui asuhan keperawatan strabismus.
3

BAB II
TINJAUAN TEORI
A. Definisi Congenital Adrenal Hyperplasia
CAH merupakan sekelompok kelainan yang diturunkan secara autosomal
resesif akibat adanya mutasi pada gen tersering CYP 21 dan menyebabkan
defisiensi satu dari lima ezim yang dibutuhkan dalam proses sintesis hormon
kortisol dan aldosteron darikolesterol pada korteks adrenal (steroidogenesis)
sehingga menyebabkan perubahanberupa produksi hormon steroid sex
(testosteron) menjadi berlebihan yang kemudianakan merubah perkembangan
karakteristik sexual wanita dengan kariotipe 46,XXmenjadi ke arah laki-laki
(maskulinisasi).
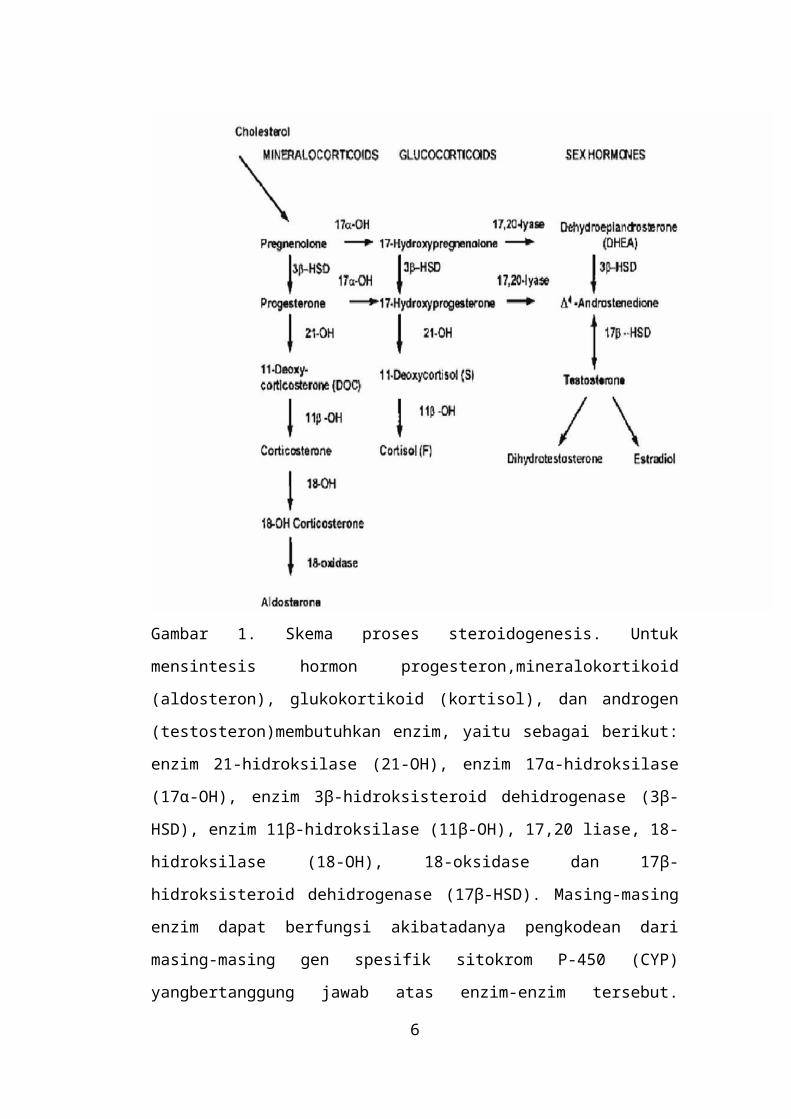
Gambar 1. Skema proses steroidogenesis. Untuk mensintesis hormon
progesteron,mineralokortikoid (aldosteron), glukokortikoid (kortisol), dan
4

androgen (testosteron)membutuhkan enzim, yaitu sebagai berikut: enzim 21-
hidroksilase (21-OH), enzim 17α-hidroksilase (17α-OH), enzim 3β-
hidroksisteroid dehidrogenase (3β-HSD), enzim 11β-hidroksilase (11β-OH),
17,20 liase, 18-hidroksilase (18-OH), 18-oksidase dan 17β-hidroksisteroid
dehidrogenase (17β-HSD). Masing-masing enzim dapat berfungsi akibatadanya
pengkodean dari masing-masing gen spesifik sitokrom P-450 (CYP)
yangbertanggung jawab atas enzim-enzim tersebut. Manakala terjadi mutasi
yangmenyebabkan salah satu enzim tidak dapat berperan dalam proses
steroidogenesis ini,maka akan terjadi akumulasi pada prekursor hormon tertentu
dan defisiensi maupunakumulasi pada hormon tertentu.
CAH merupakan penyakit yang diturunkan secara autosomal resesif.
Penyakit ini ditandai oleh defisiensi enzim yang terlibat dalam jalur
steroidogenesis pada kelenjar adrenal. Penyakit ini dapat terjadi pada wanita dan
laki-laki dan merupakan penyebab interseksual terbanyak pada individu dengan
46,XX ( Wilson, 2009)
Lebih dari 90% kasus CAH disebabkan karena defisiensi enzim 21-OH.
Ketikadefisiensi dari enzim 21-OH ini terjadi, maka progesteron dan 17-
hidroksiprogesteronakan terakumulasi, sedangkan jumlah 11-deoksikortikosteron
(DOC) dan 11-deoksikortisol akan menurun. Oleh karena jumlah 11-DOC dan 11-
deoksikortisolsedikit, hal ini menyebabkan produksi akhir dari dua prekursor
hormon tersebut, yaitualdosteron dan kortiol juga menurun. Selain itu, karena
adanya akumulasi dariprogesteron dan 17-hidroksiprogesteron akibat jalur
pembentukan aldosteron dankortisol yang terblok, maka akan semakin banyaklah
hormon-hormon tersebut diubahke jalur lain untuk menjadi androstenedion. Pada
akhirnya androstenedion ini akandiubah oleh enzim 17β-HSD menjadi testosteron
(androgen). Hal ini menyebabkanproduksi testosteron di perifer menjadi berlebih.
Testosteron dapat diaromatisasi menjadi estradiol akibat peran dari
enzimaromatase. Selain itu, testosteron juga dapat di konversi menjadi
dihidrotestosteronmelalui enzim 5-AR.
B. Etiologi
CAH dapat disebabkan karena hal-hal berikut ini:
1. Defisiensi enzim 21-hidroksilase
5

2. Defisiensi enzim 11β-hidroksilase
3. Defisiensi enzim 3β-hidroksisteroid dehidrogenase
4. Defisiensi enzim 17α-hidroksilase
5. Mutasi protein Steroidogenic acute regulatory (StAR)
C. Patofisiologi Congenital Adrenal Hyperplasia
Kelenjar adrenal mensintesis tiga kelas utama hormon, yaitu
mineralokortikoid,glukokortikoid dan androgen, misal: testosteron. Sintesis
hormon golonganmineralortikoid terjadi dalam zona glomerulosa korteks adrenal,
sedangkan hormone glukokortikoid dsintesis di zona fasikulata dan retikularis
korteks adrenal.
Ketiga hormon ini sangat penting bagi tubuh. Fungsi dari masing-masing
hormone tersebut adalah sebagai berikut:
1. Kortisol membantu tubuh dalam mengatasi stress ataupun tekanan seperti
pada kondisi luka maupun sakit
2. Aldosteron berperan dalam memastikan agar tubuh dapat menyimpan garam
dalam jumlah yang cukup, sedangkan
3. Testosteron terlibat dalam pembentukan sifat maskulin manusia, seperti
distribusi rambut pada tubuh dan perkembangan organ seks laki-laki. Baik
laki-laki maupun perempuan, keduanya memproduksi testosteron. Namun,
pada laki-laki produksi hormon ini jumlahnya lebih banyak
Hipofisis mengatur proses steroidogenesis di adrenal melaluiadrenocorticotropic
hormone (ACTH). ACTH menstimulasi sintesis steroid denganmeningkatkan
substrat utama dalam jalur steroidogenesis di adrenal. Proses tersebutdapat
terilustrasikan melalui Gambar 2.
6

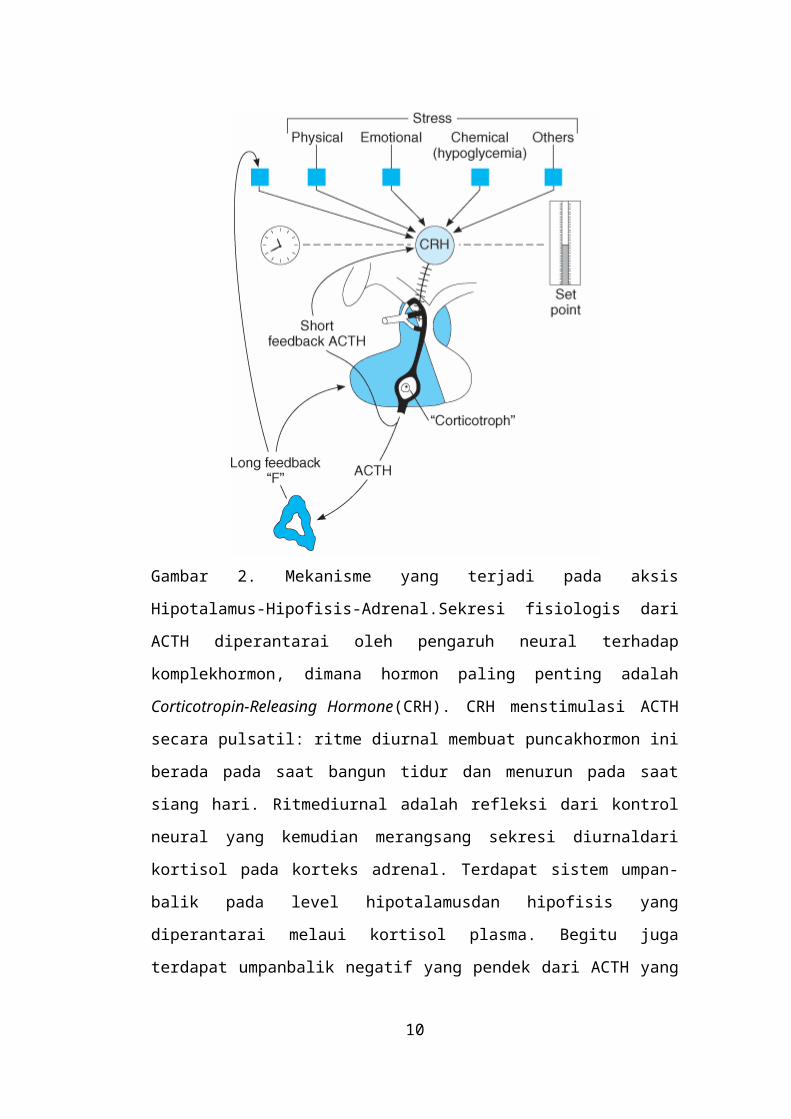
Gambar 2. Mekanisme yang terjadi pada aksis Hipotalamus-Hipofisis-
Adrenal.Sekresi fisiologis dari ACTH diperantarai oleh pengaruh neural terhadap
komplekhormon, dimana hormon paling penting adalah Corticotropin-Releasing
Hormone(CRH). CRH menstimulasi ACTH secara pulsatil: ritme diurnal
membuat puncakhormon ini berada pada saat bangun tidur dan menurun pada saat
siang hari. Ritmediurnal adalah refleksi dari kontrol neural yang kemudian
merangsang sekresi diurnaldari kortisol pada korteks adrenal. Terdapat sistem
umpan-balik pada level hipotalamusdan hipofisis yang diperantarai melaui
kortisol plasma. Begitu juga terdapat umpanbalik negatif yang pendek dari ACTH
yang mempengarui sekresi dari CRH. Jadi,kondisi apapun yang dapat
menurunkan sekresi kortisol akan mengakibatkanmeningkatnya sekresi ACTH.
Dengan ini, kortisol memberikan efek umpan-baliknegatif terhadap sekresi
ACTH.
Kebanyakan CAH yang memiliki defek pada suatu enzim yang memblok
sintesiskortisol akan mengganggu kontrol umpan-balik sekresi ACTH melalui
kortisol. SekresiACTH kemudian menjadi berlebihan yang selanjutnya akan
7
memicu terjadinyahiperplasia adrenocortical. Hal ini menyebabkan stimulasi
sintesis produk-produk dariadrenal berlebihan, dan dengan adanya defisiensi salah
satu enzim dari jalursteroidogenesis akan menyebabkan akumulasi dari molekul
prekursor jalur tersebut.Prekursor-prekursor tersebut akan teralihkan ke jalur lain
yaitu jalur androgen, sehinggamenyebabkan level androgen menjadi tinggi.
Gejala-gejala klinik yang timbul dari berbagai jenis CAH tergantung dari
hormone apa yang diproduksi secara berlebihan atau hormon apa yang defisiensi.
Perbandinganfenotip dari masing-masing jenis CAH dapet dilihat dalam tabel 1.
CAH dapatdisebabkan karena hal-hal berikut ini:
1) Defisiensi enzim 21-OH
Defisiensi enzim ini terjadi paling sering, lebih dari 90-95% dari seluruh
kasus CAH. Enzim 21-OH adalah enzim yang terlibat dalam konversi
kolesterol menjadi kortisol dan aldosteron, tapi tidak dalam konversi
menjadi testosteron. Pada defisiensi enzim 21-OH, jalur aldosteron dan
kortisol dihambat, sedangkan jalur androgen yang tidak dipengaruhi oleh
enzim 21-OH menjadi terstimulasi secara berlebihan. Virilisasi pada kasus
defisiensi enzim 21-OH terjadi karena sekresi yang berlebihan dari
androgen adrenal.
2) Defisiensi 11β-OH
Defisiensi enzim terjadi sekitar 5-8% dari kasus CAH. Pada proses
steroidogenesis, hal tersebut juga mengakibatkan turunnya sintesis kortisol
yang kemudian mengakibatkan overproduksi dari prekursor kortisol dan
steroid seks seperti yang terjadi pada kasus defisiensi enzim 21-OH,
sehingga defisiensi enzim 11β-OH memiliki gambaran klinik berupa
virilisasi yang mirip dengan kelainan pada kasus defisiensi enzim 21-OH.
Temuan tambahan pada banyak kasus defisiensi enzim 11β-OH, namun
tidak semua, adalah adanya hipertensi. Hipertensi ini mungkin berasal dari
akumulasi berlebihan prekursor aldosteron, 11-DOC, yaitu steroid yang
memiliki aktifitas menyimpan garam.
3) Defisiensi enzim 3β-HSD
Defisiensi enzim 3β-HSD merupakan penyebab kedua terbesar dari CAH,
yaitu sekitar 10% dari kasus. Tidak seperti CAH karena defisiensi enzim
8

21-OH maupun 11β-OH yang hanya mempengaruhi fungsi adrenal, pada
defisiensi enzim 3β-HSD akan berakibat pada kelenjar adrenal maupun
fungsi gonad. Bayi yang baru lahir dengan defisiensi enzim 3β-HSD
memiliki gejala dari defisiensi kortisol dan aldosteron. Pada anak
perempuan dapat memiliki perkembangan seksual yang normal maupun
virilisasi ringan yang kebanyakan terdeteksi pada masa pubertas. Oleh
karena hiperandrogenisme, maka dapat terjadi anovulasi kronik bahkan
amenore primer.
4) Defisiensi enzim 17α-OH
Defisiensi enzim-enzim ini juga dapat menimbulkan kelainan-kelainan
pada proses steroidogenesis di adrenal dan di gonad. Uniknya terjadi
kompensasi dari sekresi ACTH yang memacu produksi berlebih dari
mineralokortikoid, sehingga menyebabkan hipertensi dan hipokalemia.
Wanita dengan defisiensi enzim 17α-OH akan mengalami sexual
infantilism dan hypergonadotropic hypogonadism. Hipergonadotropisme
terjadi karena defisiensi estrogen.
5) Defisiensi Steroidogenic Acute Regulatory (StAR) protein
Protein StAR adalah fosfoprotein mitokondria yang bertanggung jawab
mengangkut kolesterol dari luar ke dalam membran interna mitokondria
yang kemudian diubah menjadi pregnenolon oleh P450cc. Kehilangan
enzim ini menyebabkan gangguan pada steroidogenesis di adrenal maupun
gonad. Kerusakan overium dapat terjadi setelah masa pubertas aibat adanya
kerusakan sel-sel ovarium.
Tabel 1. CAH berdasarkan masing-masing defisensi enzim pada steroidogenesis.
Perbedaan gen-gen yang terlibat dalam masing-masing penyebab dari CAH,
lokasi darimasing-masing gen tersebut, beberapa manifestasi klinis yang dapat
timbul pada masingmasingdefisiensi enzim pada CAH, insidensi, besar hormon
korteks adrenal, kadarmetabolit yang dapat meningkat, tekanan darah, level
natrium dan level kalium.
9

D. Aspek genetik pada Congenital Adrenal Hyperplasia
Semua tipe CAH diturunkan melaui cara autosomal resesif yang
disebabkankarena perubahan pada sepasang gen. Seseorang dengan CAH
mengalami perubahandalam masalah penyalinan dari gen yang bertanggung jawab
untuk memproduksi enzimyang terlibat dalam pemecahan kolesterol. Seseorang
akan terkena CAH akibatditurunkannya salah satu gen yang telah berubah dari ibu
dan satu gen lain, diturunkandari ayah, yang juga telah berubah dan kemudian
akan menjadi bakal pasangan gen tersebut.
Sintesis enzim yang terlibat dalam sintesis kortisol dan aldosteron
merupakanprotein sitokrom P450 (CYP). CYP21 adalah gen yang mengkode
enzim 21-OH;CYP11B1 mengkode enzim 11β-OH; dan CYP17 mengkode enzim
17α-OH. Hal inidapat dilihat pada tabel 1.
Berdasarkan studi dari genetik molekuler, gen yang mengkode sitokrom
P450spesifik untuk enzim 21-OH (P450c21) terletak di kompleks HLA
polimorfik padakromosom 6p21.3, yaitu CYP21 yang letaknya bersama dengan
pseudogen atauhomolog inaktif darinya yaitu CYP21P. Kedua gen ini
bertanggung jawab untukmenyebabkan terjadinya defisiensi enzim 21-OH. Oleh
karena CYP21 dan CYP21Pmemiliki 98% kemiripan dalam urutan nukleotidanya,
maka diketahui terdapat banyakmutasi yang menyebabkan produk dari gen
menjadi terinaktivasi. Hal ini termasukdelesi 8-bp pada exon 3, frame shift
10

mutation pada exon 7, dan nonsense mutation pada exon 8. Lokasi dari gen-gen
tresebut dapat dilihat pada gambar 3.
Gambar 3. Regio kromosom 6p21.3 yang mengandung gen-gen yang
bertanggungjawab pada enzim 21-OH. 5 CYP21 dan CYP21P adalah gen aktif
dan pseudogen darienzim 21-OH. C4A dan C4B mengkode komponen keempat
dari komplemen serum. RP1mengkode protein inti untuk fungsi yang masih
belum diketahui, sedangkan RP2berhubungan dengan pseudogen. TNXB
mengkode tenascin-X, sedangkan TNX Aberhubungan dengan pseudogen. Daerah
yang digambarkan dengan regio 30-kb adalahdaerah yang terdelesi sekitar 20%
dari kromosom defisiensi 21-OH.
Mutasi lain pada CYP21P dapat mempengaruhi proses splicing dari
messenger
RNA (mRNA) atau mempengaruhi urutan dari asam amino. Namun, mutasi
yangpaling sering menyebabkan defisiensi enzim 21-OH adalah dua tipe
rekombinasidiantara gen CYP21 dengan CYP21P. Sekitar 75% terjadi delesi pada
pseudogen yangkemudian ditransfer ke CYP21 selama mitosis melalui proses
yang dinamakan “geneconversion”. Sekitar 20% diantaranya terjadi rekombinasi
meiotik yang menghilangkan30-kb segmen gen yang meliputi ujung 3’ dari
CYP21P, semua komplemen gen dariC4B yang berdekatan dan ujung 5’ dari
CYP21, yang pada akhirnya menghasilkannonfunctional chimeric pseudogene.
Lebih dari 60 mutasi lain yang dapat terjaditerhitung sebesar 5% dari kasus.
Gambar skematis dari salah satu mutasi ini, dapat dilihat dari gambar 4.
11
Gambar 4. Gen-gen dari enzim 21-OH mengalami unequal crossover selama
proses meiosis. Gen-gen tersebut kemudian menghasilan anak kromosom yang
memiliki tigaalel CYP21 atau satu gen CYP21 yang tak berfungsi sebagai akibat
dari proses delesiyang besar.
CYP 21 adalah salah satu gen manusia yang paling polimorfik. Pada
sperma,kemungkinan terjadi rekombinasi spontan antara gen CYP21 dengan
CYP21P adalah 1:1000 sampai 1:100.000 sel.
Ketika gen CYP21 berubah, akan menyebabkan situasi dimana enzim 21-
OHmenjadi tidak dapat diproduksi, atau diproduksi dalam jumlah yang sedikit.
Banyak genyang dapat mengurangi level dari enzim 21-OH. Jumlah enzim 21-OH
yang diproduksi,bergantung pada jenis dan kombinasi dari perubahan gen CYP21
dan sebagian hal inijuga menentukan derajat beratnya penyakit CAH akibat
defisiensi enzim 21-OH.
Karekteristik kombinasi dari alel HLA atau haplotipe HLA berhubungan
denganberbedanya jenis dari defisiensi enzim 21-OH. Genotipe dari defisiensi
enzim 21-OHtipe klasik berasal dari adanya dua alel yang mengalami kerusakan
berat. Defisiensienzim 21-OH tipe non-klasik merupakan hasil dari adanya dua
alel dari defisiensienzim 21-OH yang mengalami kerusakan ringan atau satu alel
namun mengalamikerusakan berat dan satu alael lainnya mengalami kerusakan
ringan.
Dua puluh lima persen alel dari classic type defisiensi enzim 21-OH
terjadikarena delesi dari CYP21; sedangkan 75% sisanya disebabkan mutasi kecil
padaCYP21 yang mana beberapa diantaranya adalah mutasi titik de novo,
yangmenghasilkan substitusi asam amino yang menyebabkan terganggunya
sintesis protein.Pada nonclassic type defisiensi enzim 21-OH, merupakan kejadian
yang timbulkarena substitusi ringan dari asam amino yang berlangsung lama pada
gen yangmengkode enzim 21-OH.
12

Orang tua yang memiliki anak dengan CAH disebut sebagai carrier karena
salah satudari mereka memiliki satu gen CYP21 yang telah mengalami perubahan
dan satu gen lainyang tidak mengalami perubahan. Carrier biasanya tidak
memiliki gejala karena merekamasih memiliki satu gen yang tidak mengalami
perubahan yang dapat memproduksienzim 21-OH dalam jumlah yang cukup
untuk mencegah timbulnya CAH. Anak yanglahir dari orang tua yang keduanya
adalah carrier untuk tipe CAH yang sama akanmemiliki peluang 25% untuk
terkena CAH, 50% peluang untuk menjadi seorang carrierdan juga 25% peluang
untuk tidak menjadi carrier maupun anak yang mengidap CAH.
E. Klasifikasi klinis Congenital Adrenal Hyperplasia
Dua tipe fenotipe mayor yang diketahui dari defisiensi enzim 21-
hidroksilase,yaitu: classic type dan non classic type (onset lambat). Classic type
dibagi lagi menjadiclassic simple virilizing type dan classic salt-wasting type.
Dalam non classic type,pasien mengalami defek biokimiawi namun hanya sedikit
tanda jelas dari hiperandrogenisme yang tampak. Berikut di bawah ini Tabel 2
yang mendeskripsikansecara ringkas perbedaan fenotip dari masing-masing tipe
CAH karena defisiensi enzim21-OH.
Tabel 2. Perbandingan fenotip berbagai tipe CAH karena defisiensi enzim 21-OH.
13

1. Classic type dari CAH
Abnormalitas biokimiawi maupun klinis akan muncul baik pada
masaprenatal maupun postnatal. Progesteron, 17-OH-progesteron,
androstenedion,dan testosteron disekresikan dalam jumlah besar dalam rahim ibu
sebagaiakibat dari meningkatnya stimulasi ACTH karena defisiensi enzim 21-
OHyang mengganggu sintesis kortisol. Kemudian, ekskresi metabolit dari
steroidsteroidtersebut juga meningkat di urin. Abnormalitas pada sekresi
kortisoljuga dihubungkan dengan adanya perubahan pada sekresi hormon
darihipofisis seperti GH dan TSH.
Bayi yang secara genetik adalah perempuan, lalu mengalami defisiensienzim
21-OH congenital, hal ini berarti androgen yang diproduksi di kelenjaradrenalnya
berada dalam jumlah yang sangat besar akibat sekresi dari korteksadrenal yang
mengalami hiperplasia. Genitalia eksterna pada seseorang yanggenetiknya adalah
perempuan, memiliki keambiguitasan mulai dari levelringan sampai berat. Hal ini
14

terjadi akaibat adanya virilisasi. Genitalia internaseperti uterus dan tuba fallopi,
tidak dipengaruhi oleh tingginya kadarandrogen.Hal ini juga terjadi pada bayi
laki-laki. Bayi laki–laki yang mengalamidefisiensi enzim 21-OH, saat dilahirkan
tidak menunjukkan bahwa genitalnyamengalami abnormalitas. Pada masa
postnatal, anak laki-laki dan perempuanyang tidak ditangani, produksi
androgennya yang massif tetap berlanjut, lalumenyebabkan:
a. Pertumbuhan yang cepat
b. Mempercepat pematangan epifisial,
c. Pembesaran progresif dari penis dan klitoris
d. Rambut pada muka, ketiak dan pubis yang muncul lebih dini
e. Berjerawat
f. Tanpa pengobatan, dapat menyebabkan penutupan epifisial dini
sehinggamenyebabkan perawakannya menjadi pendek.
Pasien dengan classic type dari CAH karena defisiensi enzim 21-
OHmengalami disfungsi adrenomedular yang ditandai dengan
menurunnyaproduksi epinefrin, metanefrin dan normetanefrin; serta mengalami
perubahanstruktur yang besar dari medula adrenal yang ditandai dengan
adanyadisplasia, berkurangnya ekspresi dari enzim tirosin hidroksilase, dan
adanyadeplesi dari vesikel sekretori yang berisi epinefrin. Pasien dengan fenotip
yanglebih berat, yaitu adanya salt-wasting dan riwayat adanya krisis
adrenalternyata produksi dari epinefrin dan metanefrin oleh kelenjar
adrenalnyasangat rendah.
Berikut ini adalah pembagian lebih spesifik dari classic type CAH:
a). Classic Simple Virilizing Type dari CAH
Congenital Adrenal Hyperplasia akibat defisiensi enzim 21-OH tipeclassic
simple virilizing terjadi jika enzim 21-OH diproduksi dalamjumlah yang sedikit.
Pada tipe ini, enzim masih dapat berperan untukmencegah terjadinya level
garam yang rendah pada tubuh, jugamencegah krisis adrenal.
Perempuan akan lahir dengan maskulinisasi ringan pada genitaliaeksterna,
seperti pembesaran clitoris, fusi sebagian dari labiaperkembangan dari sinus
urogenitalia, sehingga akan menampilkankeambiguan seksual dan bahkan
15

menyulitkan dalam penentuan jenis kelamin bayi ini saat lahir. Jarang diagnosis
dari keadaan ini tidak dibuatsaat periode neonatal.
Bila ditangani dengan adekuat, periode menstruasi dapat normalsetelah
menarche dan kehamilan mungkin terjadi. Secara keseluruhan,tingkat fertilitas
dilaporkan rendah, yang dikatakan akibat introitusvaginae yang inadekuat
sehingga menyebabkan ketidakpuasan saatcoitus, kemudian meningkatnya level
androgen menimbulkan disfungsi ovarium.
Pada laki-laki yang tidak mendapatkan penanganan, akan memilikigenitalia
normal, namun mungkin mengalami pubertas dini juga.
Classic simple virilizing type CAH akibat defisiensi enzim 21-OH,dapat
menyebabkan pertumbuhan linear yang cepat pada masa kanak–kanak
disebabkan karena banyaknya androgen yang muncul lebihdini, namun pada
saat dewasa jika tetap tidak ditangani, nantinya akanterlihat pendek, baik pada
laki-laki maupun pada wanita.
b). Classic Salt-Wasting Type dari CAH
Tipe ini adalah tipe yang paling berat dari CAH akibat defisiensienzim 21-
OH. Sekitar 75% dari kasus classic type CAH akibat defisiensienzim 21-OH,
terjadi pembuangan garam dan juga hipotensi,dikarenakan hiponatremia,
hiperkalemia, natriuresis yang tidak sesuai,dan rendahnya aldosteron pada
serum dan urin bersamaan dengantingginya aktivitas plasma renin, pada
akhirnya dapat melanjut menjadikrisis adrenal.
Classic simple virilizing type CAH akibat defisiensi enzim 21-OH,jika tidak
ditangani, akan menyebabkan kehilangan garam yang akanmemacu terjadinya
krisis adrenal. Krisis adrenal merupakan keadaanyang mengancam kehidupan,
ditandai dengan adanya dehidrasi berat,tekanan darah yang sangat rendah,
melemahnya otot jantung, danmuntah.
Hal ini terjadi karena enzim 21-OH diproduksi dalam jumlah yangsangat
sedikit atau tidak sama sekali. Pembuangan garam terjadi akibatsekresi
inadekuat dari steroid yang bertanggung jawab untuk menahangaram, terutama
aldosteron. Selain itu, hormon prekursor dari enzim 21-OH dapat berperan
16

sebagai antagonis dari mineralokorticoid pada bayiyang tubulus renalisnya
masih imatur.CAH tipe ini, jika tidak tertangani, juga dapat
menyebabkanpertumbuhan yang cepat di masa kanak-kanak namun
berperawakanpendek di usia dewasa.
Meningkatnya angka keselamatan dari Classic simple virilizing typeCAH
akibat defisiensi enzim 21-OH terjadi akibat adanya suplemen
darimineralokortikoid eksogen. Telah diketahui bahwa defek dari
biosintesisaldosteron yang nyata terjadi pada masa kecil, akan
mengalamiperbaikan sejalan dengan usia dan bahkan perbaikan spontan
secaraparsial dapat terjadi pada usia dewasa. Variasi dalam
kemampuanmemproduksi mineralokortikoid dapat disebabkan oleh enzim
adrenallain yeng sejalan dengan aktivitas enzim 21-OH. Oleh karena itu,
sangatdiperlukan evaluasi dari kadar sodium dan mineralokortikoid
denganmengukur aktivitas plasma renin pada pasien yang pada saat lahir
telahdiketahui mengalami pembuangan garam.
Meskipun telah diklaim bahwa pembuangan garam ini berhubungandengan
beratnya virilisasi, oleh karena itu, sangat penting untukmengenali virilisme
yang sama seperti pada simple virilizing type. Jadi,walaupun tingkat virilisasi
pada bayi dengan defisiensi enzim 21-OHdalam derajat ringan, tetap harus
diobservasi tanda-tanda yang potensialuntuk mengancam kehidupan dari krisis
adrenal dalam minggu-minggupertama kehidupan.
Anak perempuan yang tidak tertangani mungkin mengalamikesalahan
disangka sebagai anak laki-laki sejak lahir karena keduanyaterlihat sama-sama
memiliki genitalia eksternal yang maskulin. Namun,organ-organ seksual
internal mereka normal. Anak laki-laki yang tidaktertangani akan memiliki
genitalia eksterna yang terlihat normal, namunia akan mengalami pubertas dini.
Ciri-ciri dari pubertas tersebut sepertiadanya rambut pubis, pembesaran phallus,
suara yang dalam dan berat,peningkatan kekuatan otot, dapat terjadi jauh
sebelum waktu pubertasnormal, atau bahkan dapat muncul pada usia dua sampai
tigatahun.
Anak laki-laki yang tidak terdeteksi pada newborn screening, beradapada
resiko yang tinggi untuk krisis adrenal akibat pembuangan garam,karena
17

genitalia eksternanya yang tampak normal tidak membuat dokterwaspada akan
kondisinya kemudian mengalami krisis kehilangan garamyang tidak terprediksi.
Sebaliknya, pada anak perempuan biasanyaterdiagnosis dan tertangani secara
dini karena dokter telah terperingatioleh genitalia eksternalnya yang ambigu.
2. Nonclassic type dari CAH
Non classic type dari CAH adalah bentuk yang paling ringan dari CAH akibat
defisiensi enzim 21-OH. Level enzim 21-OH disini menurun ringan. Gejala
kliniknya bervariasi dan dapat muncul di usia berapapun.Pria dan wanita dengan
tipe ini terlihat normal disaat lahir dan tidakmenderita kekurangan garam. Tipe ini
dapat menyebabkan perkembangan rambut pubis prematur pada anak-anak,
bahkan hal ini dapat ditemukan pada pasien berumur enam bulan. Meningkatnya
androgen yang diproduksi oleh kelenjar adrenal membuat penutupan dari lempeng
epifisial lebih dini. Hal ini wajar, namun tidak selalu ditemukan, bahwa anak
dengan kelainan inimemiliki penuaan umur tulang dan peningkatan pertumbuhan
linear dengan sangat cepat, lalu biasanya berperawakan lebih pendek dari tinggi
yang dapat diperkirakan berdasarkan tinggi midparental dan dari persentil
pertumbuhan linear.
Pada wanita muda dapat terjadi kebotakan dengan pola seperti pada laki-laki
dan juga jerawat karena androgen yang menjadi salah satu tanda. Menarche dapat
terjadi secara normal ataupun tertunda, amenore sekunder maupun siklus
menstruasi yang irregular serta infertile anovulatoir sering terjadi. Fenomena ini
mungkin terjadi akibat hormon steroid seks dari adrenal yang berada dalam
jumlah besar, mengganggu siklus pelepasan gonadotropin dan/atau adanya efek
langsung dari androgen adrenal terhadap ovarium, yang pada akhirnya akan
membuat terbentuknya kista pada ovarium yang kemudian disini dapat diproduksi
androgen. Oleh karena itu, CAH yang memiliki onset lambat, diketahui menjadi
penyebab sekunder dari Polycistic OvarySyndrome (PCOS). Hirsutisme dan
oligomenore dapat terjadi dan bervariasi tergantung dari grup etniknya.
Pada anak laki-laki, biasanya tidak menimbulkan gejala walaupun tidak
ditangani, namun pada beberapa diantaranya, tanda-tanda fisik yang dapat
ditemukan adalah pertumbuhan janggut dini, jerawat, pertumbuhan linear cepat,
rambut pubis, pembesaran phallus, dan biasanya testis yang kecil. Pada pria
18

dewasa, tanda-tanda dari androgen yang besar sulit untuk ditemukan, tapi secara
teoritis dapat memiliki manifestasi berupa perawakan pendek, dan/atau
mengurangi kesuburan (subfertil) dan oligospermia akibat hormon steroid seks
adrenal yang memicu supresi dari aksis hipotalamus-hipofisis-gonad.
Adanya defisiensi enzim 21-OH dapat ditemukan dengan tidak sengaja pada
evaluasi pemeriksaan massa pada adrenal. Meningkatnya insidensi tumor adrenal
dapat ditemukan baik pada pasien pria maupun wanita dengan CAH homozigot
dan juga heterozigot.7 Penurunan sintesis dari kortisol tidak signifikan pada
pasien dengan non classic type CAH akibat defisiensi enzim 21-OH.
F. Tanda dan Gejala
Pasien yang diduga untuk mengidap CAH adalah dengan tanda dan gejala
sebagaiberikut:
a) Bayi perempuan yang lahir mengalami virilisasi prenatal dan genitalia
eksternanya ambigu, atau yang menjadi tervirilisasi di saat postnatal pada
anak lakilaki maupunperempuan, atau yang mengalami pubertas prekoks
ataupun adrenarche.
b) Laki-laki yang mengalami virilisasi di masa kanak-kanak, misalkan
pubertaspseudoprekoks
c) Bayi laki-laki atau perempuan dengan insufisiensi adrenal dengan atau tanpa
krisisakibat kehilangan garam di empat minggu pertama kehidupan.
1. Genitalia eksterna yang rancu
Perempuan dengan classic type defisiensi enzim 21-OH akan
terpaparandrogen adrenal sistemik dalam jumlah yang tinggi semenjak minggu
ketujuhkehamilan. Hal ini menyebabkan bayi perempuan yang secara genetic
mengandung kromosom XX biasanya menghasilkan genitalia yang tidak khas.Di
dalam pelvis, tidak akan ada perkembangan dari ductus Wolfii, namunstruktur dari
ductus Mülleri akan berkembang normal, yaitu ovarium, uterus,tuba fallopi,
vagina bagian atas dan struktur lain yang dibentuk dari ductusmulleri akan
terbentuk dengan baik karena tidak terpapar oleh antimullerianhormone (AMH)
dan juga struktur-struktur tersebut tidak dipengaruhi olehhormon steroid seks
(testosteron).
19

Namun, tingginya level testosteron dalamdarah dapat memperbesar phallus,
vagina gagal terbentuk pada perineum(introitus vagina menutup secara komplet
maupun parsial), sinus urogenitalterletak pada pemisahan vagina dan uretra,
batang maupun ujung dari phallusterlihat seperti milik laki-laki. Testosteron dapat
membuat fusi labia mayorsecara parsial dan membuat kulit dari labia menjadi tipis
dan memiliki ruggaeseperti pada scrotum, tapi tidak terdapat gonad (testis) yang
dapat di palpasi.Jadi, tergantung dari beratnya hiperandrogenisme, bayi wanita
dapat terpengaru secara ringan yang biasanya genitalianya menjadi tidak khas,
ataumenjadi virilisasi yang berat dan akan terlihat seperti laki-laki.
Anak laki-laki justru terjadi sebaliknya. Anak laki-laki dengan classic
typedefisiensi enzim 21-OH tidak memiliki tanda yang khas atas penyakit
inikecuali hiperpigmentasi yang bervariasi dan tidak begitu kentara dan
adanyapembesaran penis.
Ada beberapa sistem untuk menilai derajat genitalia yang ambigu.
Derajatdari maskulinisasi yang rendah seperti yang terdapat pada
AndrogenInsensitivity Syndrome (AIS) dinilai dengan Quigley score, ataupun
derajatdari maskulinisasi dengan Prader Stage seperti yang diterapkan pada
virilisasi di kasus CAH. Berikut pada Gambar 5 ilustrasi dari Prader stage.
Prader 0 : Genitalia eksterna wanita normal
Prader 1 : Genitalia eksterna wanita dengan clitoromegaly
Prader 2 : Clitoromegaly dengan fusi labia parsial membentuk sinusurogenital
berbentuk corong
20

Prader 3 : Peningkatan pembesaran phallus. Fusi labioscrotal lengkap membentuk
sinus urogenital dengan satu lubang
Prader 4 : Fusi scrotum lengkap dengan pintu urogenital di dasar atau di batang
phallus
Prader 5 : Genitalia eksterna pria normal
Pada bayi dengan Prader derajat 4: lebih terlihat seperti laki-lakidibanding
wanita dengan scrotum yang kosong ukuran phallus seperti penis yang normal
tapi tergantung bebas dalam perineum karena adanya tarikan darichordae yang
mengarahkannya ke arah umbilicus. Ostium urethra / vaginayang kecil pada basis
atau pada batang dari phallus akan dipertimbangkansebagai hypospadia pada laki-
laki.
Bayi dengan derajat 5: Genitalia eksterna bayi-bayi ini tidak terlihatambigu,
tapi biasanya disimpulkan sebagai laki-laki biasa dengan undescencustestis. Pada
banyak kasus, diagnosis dari CAH tidak terbentuk sampaiditemukan adanya
pembuangan garam yang berkembang pada mingguberikutnya.Oleh karena itu,
dalam pemeriksaan fisik genitalia eksterna selainmenentukan apakah gonad yang
21

dapat dipalpasi dan derajat virilisasi menurutPrader scale, perlu juga mengukur
panjang phallus. Normal penis bayi barulahir dengan masa gestasi normal adalah
sekitar 3cm (diukur dari tuberkulumpubis dampai dengan ujung penis). Jika
mikropenis dapat kurang dari 2,0-2,5cm, meskipun ukuran ini bervariasi
tergantung dari etniknya.
Selain itu, chordae juga harus diperhatikan, karena chordae
dapatmemperkecil panjang phallus dari ukuran yang sebenarnya. Ada
tidaknyahipospadia, posisi dari meatus urethra, derajat fusi lipatan labioscrotal
danada atau tidaknya introitus vaginae juga harus dipastikan.
Pada pemeriksaan fisik, biasanya hiperpigmentasi sering ditemukan didaerah
genitalia dan papilla mammae. Hal ini dikarenakan rendahnya enzimyang
berperan dalam sintesis kortisol sehingga terjadi umpan balik negative yang
membuat ACTH meningkat selanjutnya mempengaruhi pigmentasi kulit.Waktu
dari gestasi bayi juga harus dicari, karena pada anak wanita yang lahirpreterm,
memiliki clitoris dan labia minora yang lebih prominent disbanding anak laki-
laki; sedangkan pada anak laki-laki, testis biasanya mulai turun kearah scrotum
saat usia gestasi sekitar 34 minggu.
2. Virilisasi postnatal
Pasien yang tidak tertangani secara dini maupun yang mendapatkan
penanganan namun tidak adekuat, akan mendapatkan paparan jangka panjang dari
hormon seks (testosteron) dalam jumlah yang besar. Hal ini menyebabkan rambut
pubis dan rambut aksila dapat tumbuh dini. Pembesaran klitoris dapat terjadi dan
terus berlanjut pada perempuan sehingga menyerupai penis. Pada laki laki, penis
akan membesar walaupun testisnya kecil, karena androgenyang ada berasal dari
adrenal. Paparan lama terhadap androgen akan memicu aksis hipotalamus-
hipofisis gonad sehingga menyebabkan pubertas prekoks.
3. Salt-wasting
Tujuh puluh lima persen pasien dengan classic type dari CAH
dengandefisiensi enzim 21-OH mengalami gangguan berat dalam meng-
hidroksilasiprogesteron dan sehingga sintesis aldosteron menjadi tidak
adekuat.Meningkatnya level prekursor dari 21-OH, yaitu progesteron dan 17-
22

hidroksiprogesteron, dapat berperan sebagai antagonis mineralokortikoid, yang
pada
akhirnya memperburuk efek defisiensi aldosteron. Karena aldosteron
mengatur homeostasis dari natrium, maka ekskresi natrium dari ginjal pada pasien
yang tidak tertangani akan meningkat dan dapat menyebabkan hipovolemia
sertahipereninemia. Pasien ini juga tidak dapat mengekskresi kalium dengan
efisien sehingga menyebankan hiperkalemia, khususnya pada bayi. Defisinsi
kortisol dapat merusak fungsi jantung, merusak respon vaskular terhadap
katekolamin,glomerular filtration rate (GFR), dan meningkatkan sekresi
antidiuretikhormon (ADH).
Jadi, defisiensi kortisol dan aldosteron bersama-sama menyebabkan dehidrasi
akibat hiponatremia dan pada pasien yang tidak tertanganidengan adekuat. Selain
itu, karena perkembangan dari medula adrenalbergantung pada glukokortikoid,
maka pasien dengan salt-wasting type dariCAH akibat defisiensi enzim 21-OH
juga dapat mengalami defisiensikatekolamin, yang berpotensi untuk menyebabkan
syok eksaserbasi.
Pasien dengan salt-wasting type diidentifikasi melalui pengukuranelektrolit
serum, aldosteron dan renin plasma, yaitu hiperkalemia, rendahnyalevel
aldosteron, hipereninemia.
4. Pertumbuhan linear
Congenital Adrenal Hyperplasia dapat mempengaruhi pertumbuhan
linear,walaupun dengan pengawasan terapi yang ketat. Sebuah meta-analisis
datadari 18 senter pasien menunjukkanbahwa tinggi orang dewasa pada
pasiendengan classic type dari CAH sekitar 1,4 SD dibawah rata-rata
populasi.Penanganan yang tidak adekuat maupun penanganan yang berlebihan
tetapdapat membuat pasien memiliki resiko berperawan pendek, karena penyebab
utamanya adalah penutupan lempeng epifisial dini yang dipicu oleh
jumlahhormon steroid seks yang tinggi dan pada akhirnya
menyebabkanterhambatnya sumbu pertumbuhan yang dipicu oleh glukokortikoid.
Walaupun begitu, rangsangan hormon steroid seks yang tinggi pada masa kanak-
kanak menyebabkan anak-anak laki-laki maupun perempuan terlihat lebih tinggi
dibandingkan anak lain yang seusianya.
23

G. Fungsi reproduksi
Pada anak perempuan dengan bentuk apapun dari defisiensi enzim 21-OH,
akan mengalami gangguan dari reproduksi, seperti oligomenore atau amenore,
yang dapat berkembang di usia dewasa. Masalah kesuburan berhubungandengan
penyesuaian dari segi psikososial. Wanita dengan classic salt-wastingtype ataupun
simple virilizing type dari CAH yang lahir pada tahun 1940anmaupun 1950an
memiliki kecenderungan untuk menjalani hubunganheteroseksual, terutama jika
introitus vaginae inadekuat atau level androgensecara kronik terus meningkat.
Paparan prenatal terhadap androgen selanjutnyadapat mempengaruhi perilaku seks
seseorang. Telah diketahui bahwa,kebanyakan wanita yang dilaporkan berperilaku
lebih ke arah laki-laki selamamasa kanak-kanak dalam hal pemilihan mainan,
permainan dan agresifitas.Namun, kebanyakan wanita menjadi heteroseksual dan
identitas seksualmereka hampir selalu wanita. Jika wanita seperti ini diterapi,
mereka dapathamil dan melahirkan, kebanyakan dengan cara sectio caesaria.
Sekitar 80%wanita dengan simple virilizing type dari CAH dan sekitar 60% dari
classicsalt-wasting type CAH adalah fertil.
Laki-laki memiliki masalah reproduksi yang relatif lebih
sedikitdibandingkan wanita khususnya dalam fungsi gonad. Kebanyakan jumlah
spermanya normal dan subur.
H. Manifestasi klinis
1. Salt losing
1. Hiponatremia
2. Gagal tumbuh
3. Dehidrasi
4. Hiperkalemia
5. Krisis adrenal:
6. bayi tidak mau minum, muntah, diare, BB turun drastis, dehidrasi,
hiperkalemia, hiponatremia, asidosis
a. Ambigous Genitalia
24

1. Pseudohermafoditisme dengan klitoromegali
2. Fusi partial komplet lipatan labioskrotal
3. Gradasi dengan skala Prader
4. Biasanya ada korelasi antara gambaran genitalia dengan ada/tidaknya
salt losing atau kadar hiponatremia
b. Postnatal virilization
1. Laki-laki:
a. Terdiagnosa usia 3-7 tahun
b. isoseksual prekok
c. Usia tulang maju
d. Karakterisktik prapubertas prekok
2. Remaja dan wanita dewasa:
a. Klitoromegali, virilisasi, hirsutisme, menstruasi iregular,
infertilisasi,jerawat
b. Cryptic
c. Pertumbuhan Linear
1. Percepatan laju pertumbuhan
2. Umur tulang maju
3. Mempercepat penutupan epifisis
4. Tinggi dewasa pendek
5. Efek androgen mengurangi tinggi potensi dewasa
6. Efek glukokortikoid
2. Tipe Non klasik
a. Pubertas prekoks, usia tulang maju, pertumbuhan yang pesat
Perempuan:
ovarium polikistik, hirsutisme, menstruasi tidak teratur, perawakan
pendek, fertilitas menurun
b. Heterozigot
Kelebihan androgen walaupun ringan
I. Penatalaksanaan
1. Glukokortikoid
25

Semua pasien defisiensi 21-hydroxylase klasik dan non klasik diobati dengan
glukokortikoid. Pemberian terapi ini menekan sekresi CRH dari hipotalamus dan
ACTH dari hipofisis yang berlebihan dan mengurangi kadar steroid seks. Pada
anak dipilih hidrokortison dengan dosis 10-20 mg/M2/hari dibagi dalam dua atau
tiga kali sehari. Dosis suprafisiologis ini (pada keadaan fisiologis sekresi kortisol
pada anak dan remaja 6-7 mg/M2/hari) dibutuhkan untuk menekan androgen
adrenal secara adekuat dan meminimalkan kemungkinan terjadinya insufisiensi
adrenal.
Pada remaja dan dewasa dapat diberikan terapi prednison dosis rendah (5-7,5
mg/hari dibagi dalam 2 kali pemberian) atau deksametason dosis rendah (dosis
total sebesar 1,25-1,5 mg diberikan dosis tunggal atau berbagi dalam dua kali
pemberian). Pasien harus dimonitor secara cermat adanya tanda-tanda sindroma
cushing iatrogenik seperti kenaikan berat badan yang cepat, striae dan osteopenia.
2. Mineralokortikoid
Bayi dengan defisiensi 21-hydroxylase tipe salt wasting membutuhkan
pemberian mineralokortikoid (fludrokortison, biasanya 0,1-0,2 mg dapat sampai
0,4 mg/hari) dengan suplemen natrium klorida (1 sampai 2 gram per hari, tiap
gram natrium klorida mengandung 17 mEq natrium).
3. Farmakologis
4. Adrenalektomi
Bilamana terapi hormonal tidak adekuat atau tidak berkesinambungan pada
perempuan yang virilisasinya terus melanjut dan adanya gangguan pertumbuhan
liniar, adrenalektomi melalui laparoskopi merupakan salah satu alternatif untuk
mengurangi terapi glukokortikoid. Dengan pertimbangan karena penyakit addison
lebih mudah diatasi dengan pemberian glukokortikoid dan mineralokortikoid
dosis rendah dibandingkan adanya kelenjar adrenal yang mensekresi steroid seks
berlebihan.
5. Terapi gen
6. Pembedahan Korektif
7. Konseling Psikologi
Orang tua harus ditawarkan uinggi anak perempuan ntuk konseling psikologi
segera setelah anak didiagnosis HAK ditegakkan. Selanjutnya, dilakukan
26

penilaian pada keluarga secara berkala seperti pada penyakit lain, ini sangat
berguna untuk memprediksi masalah di masa mendatang. Karena anak tersebut
akan berkembang menjadi dewasa maka mereka harus secara berkala
mendapatkan informasi mengenai keadaan mereka oleh orang tuanya dan dokter
yang bersangkutan sesuai dengan usia anak tersebut. Bila dilakukan psikoterapi
maka pelaksana terapi medis dan psikolog harus saling berkomunikasi sehingga
keduanya memahami keadaan pasien dan keluarganya.
Meskipun perkembangan psikoseksual pada perempuan dengan HAK klasik
masih belum dipahami secara baik namun konseling harus segera dilakukan
mengingat ada kecenderungan tinggi anak perempuan yang menderita HAK akan
muncul perilaku tomboy dan cenderung memiliki kesukaan pada permainan yang
bersifat maskulin.
8. Penatalaksaan pubertas dini
Diagnosis pasti pubertas dini membutuhkan uji stimulasi GnRH. Kadar LH
dan FSH yang diukur sebelum pemberian GnRH secara bolus dan 30 menit
sesudahnya akan menunjukkan peningkatan kadar LH lebih besar daripada FSH.
Keadaan ini membutuhkan terapi supresi dengan pemberian analog GnRH.
Tujuan terapi adalah untuk menekan gonadotropin hipofisis, maka terjadi supresi
produksi steroid seks gonad, disamping itu untuk menambah tinggi badan saat
dewasa dengan mencegah fusi epifisis secara dini.
J. Konsep Asuhan keperawatan
1. PENGKAJIAN
Anamnesa :
Keluhan,riwayat kesehatan mencakup informasi tentang tingkat aktivitas
kliendan kemampuan untuk melakukan aktivitas rutin dan perawatan diri.
Review Of Sistem:
B1 : Asidosis, sianosis, RR meningkat
B2 : Hipotensi, nadi irreguler
B3 : Penurunan kesadaran
B4 : Mual, muntah, diare
B5 : Oliguria, diuresis
27

B6 : akral dingin dan pucat, memar, edema
Psikososial : fungsi mental, suasana hati, tingkat depresi. Keluarga klien
merupakan sumber terbaik untuk mendapatkan informasi tentang perubahan
ini.
2. DIAGNOSA KEPERAWATAN
1) Gangguan keseimbangan elektrolit b/d kekurangan natrium dan
kelebihan kalium
2) Kekurangan volume cairan tubuh b/d intake dan output yg tidak
seimbang
3) Perubahan nutrisi kurang dari kebutuhan tubuh b/d intake tidak
adekuat
4) Intoleransi aktivitas b/d kelemahan
5) Gangguan gambaran diri b/d perubahan dalam perubahan fungsi dan
karakteristik tubuh
6) Resti infeksi b/d kelemahan
3. INTERVENSI
1. Gangguan Keseimbangan Elektrolit berhubungan dengan kekurangan
natrium dan kelebihan kalium
Kriteria hasil :
a) Pengeluaran urin adekuat (1 cc/kg BB/jam)
b) TTV (Dalam Batas Normal)
c) Turgor kulit elastis
d) Pengisian kapiler naik kurang dari 3 detik
e) Membran mukosa lembab
f) Warna kulit tidak pucat
g) BB ideal (TB 100) – 10% (TB – 100) CARI DULUUUU
h) Hasil lab
Ht : W = 37 – 47 %
L = 42 – 52 %
Ureum = 15 – 40 mg/dl
Natrium = 135 – 145 mEq/L
28

Calium = 3,3 – 5,0 mEq/L
Kretanium = 0,6 – 1,2 mg/dl
Intervensi
1) Pantau TTV, catat perubahan tekanan darah pada perubahan posisi,
kekuatan dari nadi perifer
Rasional : Hipotensi pastoral merupakan bagian dari hipovolemia
akibat kekurangan aldosteron.
2) Ukur dan timbang BB klien
Rasional : Peningkatan BB yang cepat disebabkan oleh adanya
retensi cairan dan natrium yang berhubungan dengan pengobatan
steroids
3) Kaji pasien mengenai rasa haus, kelelahan, nadi cepat, pengisian
kapiler memanjang, turgor kulit jelek, membran mukosa kering, catat
warna kulit dan temperaturnya
Rasional :Mengidentifikasi adanya hipotermia dan
mempengaruhikebutuhanvolume pengganti
4) Periksa adanya status mental dan sensori
Rasional : Dehidrasi berat menurunkan curah jantung, berat dan
perfusi jaringan terutama jaringan otak
5) Auskultasi bising usus ( peristaltik khusus) catat dan laporan adanya
mual muntah dan diare
Rasional :Kerusakan fungsi saluran cerna meningkatkan kehilangan
cairan dan elektrolit.
Kolaborasi
6) Berikan cairan, antara lain :
a) Cairan Na Cl 0,9 %
Rasional : Kebutuhan cairan pengganti 4 – 6 liter, dengan
pemberian cairan NaCl 0,9 % melalui IV 500 – 1000 ml/jam,
dapat mengatasi kekurangan natrium yang sudah terjadi
b) Larutan glukosa
Rasional : Dapat menghilangkan hipovolemia
29

7) Berikan obat sesuai dosis
a) Kartison (ortone) / hidrokartison (cortef) 100 mg intravena setiap
6 jam untuk 24 jam, Mineral kartikoid, flu dokortisan, deoksikortis
25 – 30 mg/hr peroral
Rasional : Dosis hidrokortisol yang tinggi mengakibatkan retensi
garam berlebihan yang mengakibatkan gangguan tekanan darah dan
gangguan elektrolit
8) Pasang / pertahankan kateter urin dan selang NGT sesuai indikasi
Rasional : Menfasilitasi pengukuran haluaran dengan akurat baik urin
maupun lambung, berikan dekompresi lambung dan membatasi
muntah
9) Pantau hasil laborat
a) Hematokrit ( Ht)
Rasional :Peningkatan kadar Ht darah merupakan indikasi
terjadinya hemokonsentrasi yang akan kembali normal sesuai
dengan terjadinya dehidrasi pada tubuh
b) Ureum atau kreatin
Rasional peningkatan kadar ureum dan kreatinin darah merupakan
indikasi terjadinya kerusakan tingkat sel karena dehidrasi / tanda
serangan gagal jantung
c) Natrium
Rasional :Hiponatremia merupakan indikasi kehilangan melalui
urin yang berlebihan katena gangguan reabsorbsi pada tubulus
ginjal
d) Kalium
Rasional : Penurunan kadar aldusteron mengakibatkan penurunan
natrium dan air sementara itu kalium tertahan sehingga
2. Kekurangan volume cairan tubuh b/d intake dan output yg tidak
seimbang
Tujuan: Kebutuhan cairan terpenuhi
Kriteria Hasil
30

a). TTV dalam batas normal
b). Mukosa bibir lembab
c). Mata tidak cowong
d). Turgor baik
e). Produksi urin 1 cc/kg BB/jam
Intervensi
1. Jelaskan pada klien tentang akibat dari kurang cairan dan elektrolit.
Rasional : Klien mengerti dan kooperative dengan perawat
2. Lakukan obs.TTV Klien.
Rasional : deteksi terus menerus keadaan pasien.
3. Lakukan obs. tanda-tanda dehidrasi.
Rasional : mengetahui derajat dehidrasi klien
4. Lakukan obs. intake dan out put.
Rasional : menghindari defisit dan overload
5. Lakukan kolaborasi dengan dokter dalam pemberian cairan
perinfus.
Rasional : membantu menambah intake cairan
3. Perubahan nutrisi, kurang dari kebutuhan tubuh b.d masukan nutrisi
yang tidak adekuat.
Tujuan: kebutuhan nutrisi terpenuhi dalam waktu 3 hari
Kriteria Hasil:
a) BB naik
b) Hb > 12 gr/dl
c) Alb 3,5 gr/dl
d) Menunjukkan pernbaikan nafsu makan
e) Mual muntah tidak ada
Intervensi
a. Beri penjelasan terhadap pentingnya nutrisi bagi tubuh dan proses
penyembuhan
Rsional : Pengetahuan yang meningkat dapat meningkatkan
perilaku hidup sehat
b. Berikan makanan yang menarik dan merangsang selera makan
31

Rasional : Untuk meningkatkan selera makan sehingga
meningkatkan intake bagi tubuh
c. Berikan makanan dalam porsi kecil tapi sering
Rasional : Makanan dalam porsi besar lebih sulit dikonsumsi
pasien saat anorexia
d. Berikan diit tktp rendah lemak
Rasional : Meningkatkan asupan gizi yang adekuat mempercepat
proses penyembuhan
e. Timbang berat badan tiap 2-3 hari
Rasional : Megetahui perkembangan tubuh
f. Kolaborasi dengan dokter dalam pemberian nutrisi parenteral dan
robaransia
Rasional : Dibutuhkan bila intake tidak mencukupi dan efek
farmakologis roboransia untuk meningkatkan nafsu makan.
4. Intoleransi aktivitas b/d kelemahan
Tujuan : meningkatkan toleransi aktivitas
Kriteria hasil:
Klien menentukan dan melakukan aktivitas sesuai kemampuan
Intervensi :
1. Kaji tingkat kelelahan, kemampuan untuk melakukan ADL
Rasional : memberikan informasi tentang energi cadangan dan
respon untuk beraktifitas
2. Berikan periode istirahat dan tidur yang cukup
Rasional : meningkatkan istirahat dan menghemat energi
3. Instruksikan tentang tehnik menghemat tenaga, misal:
menggunakan kursi saat mandi, sisir rambut.
Rasional : Mencegah terjadinya kelelahan
4. Beri dorongan untuk melakukan aktifitas/ perawatan diri secara
bertahap jika dapat ditoleransi.
Rasional : Membantu penyesuaian tubuh terhadap perubahan
aktivitas
5. Beri bantuan sesuai dengan kebutuhan.
Rasional : Aktivitas mandiri membantu dalam perubah
32

BAB III
PENUTUP
A. Kesimpulan
CAH merupakan sekelompok kelainan yang diturunkan secara autosomal
resesifakibat adanya mutasi pada gen tersering CYP 21 dan menyebabkan
defisiensi satu dari lima ezim yang dibutuhkan dalam proses sintesis hormon
kortisol dan aldosteron darikolesterol pada korteks adrenal (steroidogenesis)
sehingga menyebabkan perubahanberupa produksi hormon steroid sex
(testosteron) menjadi berlebihan yang kemudianakan merubah perkembangan
karakteristik sexual wanita dengan kariotipe 46,XXmenjadi ke arah laki-laki
(maskulinisasi). Yang disebabkan karena hal-hal berikut ini:
1. Defisiensi enzim 21-hidroksilase
2. Defisiensi enzim 11β-hidroksilase
3. Defisiensi enzim 3β-hidroksisteroid dehidrogenase
4. Defisiensi enzim 17α-hidroksilase
5. Mutasi protein Steroidogenic acute regulatory (StAR)
Klasifikasi klinis Congenital Adrenal Hyperplasia ada dua tipe fenotipe mayor
yang diketahui dari defisiensi enzim 21-hidroksilase,yaitu: classic type dan non
classic type (onset lambat). Classic type dibagi lagi menjadiclassic simple
virilizing type dan classic salt-wasting type. Dalam non classic type,pasien
mengalami defek biokimiawi namun hanya sedikit tanda jelas dari
hiperandrogenisme yang tampak.
Dengan fungsi reproduksiPada anak perempuan dengan bentuk apapun dari
defisiensi enzim 21-OH, akan mengalami gangguan dari reproduksi, seperti
oligomenore atau amenore, yang dapat berkembang di usia dewasa. Masalah
kesuburan berhubungandengan penyesuaian dari segi psikososial. Wanita dengan
classic salt-wastingtype ataupun simple virilizing type dari CAH yang lahir pada
tahun 1940anmaupun 1950an memiliki kecenderungan untuk menjalani
33

hubunganheteroseksual, terutama jika introitus vaginae inadekuat atau level
androgensecara kronik terus meningkat.
B. Saran
Kami berharap semoga makalah ini dapat memberikan pengetahuan lebih
luas lagi mengenai penyakit dalam sistem Endokrin.
34

DAFTAR PUSTAKA
Kamus Kedokteran Dorland. Edisi 29. Jakarta: EGC; 2005. Seks; p.1978.
Speiser PW, White PC. Congenital Adrenal Hyperplasia (Review). N Eng J Med
2003;349:776-88
Hutcheson J, Snyder III HM. Ambiguous Genitalia and Intersexuality.
Pennsylvania.
2006. Available from: URL:
http://emedicine.medscape.com/article/1015520overview
35






















