Embed Size (px)
Citation preview

Mapping of Autosomal Recessive ChronicDistal Spinal Muscular Atrophy to
Chromosome 11q13Louis Viollet, MD,1,2 Annie Barois, MD,2 Jean G. Rebeiz, MD,3 Ziad Rifai, MD,4 Philippe Burlet,1
Mohammed Zarhrate,1 Elodie Vial,1 Michel Dessainte,5 Brigitte Estournet, MD,2 Bernard Kleinknecht, MD,6
John Pearn, MD,7 Raymond D. Adams, MD,8 Jon A. Urtizberea, MD,2 Didier P. Cros, MD,8
Kate Bushby, MD,9 Arnold Munnich, MD, PhD,1 and Suzie Lefebvre, PhD1
Distal spinal muscular atrophy is a heterogeneous group of neuromuscular disorders caused by progressive anterior horncell degeneration and characterized by progressive motor weakness and muscular atrophy, predominantly in the distalparts of the limbs. Here we report on chronic autosomal recessive distal spinal muscular atrophy in a large, inbred familywith onset at various ages. Because this condition had some of the same clinical features as spinal muscular atrophy withrespiratory distress, we tested the disease gene for linkage to chromosome 11q and mapped the disease locus to chro-mosome 11q13 in the genetic interval that included the spinal muscular atrophy with respiratory distress gene(D11S1889-D11S1321, Zmax � 4.59 at � � 0 at locus D11S4136). The sequencing of IGHMBP2, the human homo-logue of the mouse neuromuscular degeneration gene (nmd ) that accounts for spinal muscular atrophy with respiratorydistress, failed to detect any mutation in our chronic distal spinal muscular atrophy patients, suggesting that spinalmuscular atrophy with respiratory distress and chronic distal spinal muscular atrophy are caused by distinct geneslocated in the same chromosomal region. In addition, the high intrafamilial variability in age at onset raises the questionof whether nonallelic modifying genes could be involved in chronic distal spinal muscular atrophy.
Ann Neurol 2002;51:585–592
Spinal muscular atrophy (SMA) is a neuromusculardisorder characterized by progressive anterior horn celldegeneration, leading to motor weakness, muscular at-rophy, and denervation. Various SMA phenotypes havebeen reported; these differ according to the distributionof paralysis, age at onset, and mode of inheritance. Themajor clinical form is autosomal recessive proximalSMA, ascribed to deletions of the survival motor neu-ron gene (SMN1) in 95% of cases.1,2 Apart from thiswell-defined entity, SMA forms a clinically and genet-ically heterogeneous group of disorders. Among them,distal SMAs are characterized by the specific involve-ment of distal limb musculature. Distal SMAs are alsoknown as spinal-type Charcot-Marie-Tooth disease,3
distal hereditary motor neuropathy type II,4 and theneuronal motor neuropathy form of peroneal muscular
atrophy.5,6 Both autosomal recessive and dominantforms of distal SMA, with either adult or childhoodonset, have been reported.4,7–9 Dominant distal SMAsare genetically heterogeneous, and the disease-causinggenes have been mapped to chromosomes 2q14, 7p,7p15, 12q24, and 12q23-q24.10–14 However, a genefor very early and severe autosomal recessive distal spi-nal muscular atrophy with respiratory distress(SMARD1) has been mapped to chromosome 11q13-q21,15 and mutations in the gene encoding the immu-noglobulin �-binding protein 2 (IGHMBP2) havebeen reported in six SMARD1 families.16 This gene ishomologous to the mouse IGHMBP2 gene, which ac-counts for SMA in the neuromuscular degenerationmouse.17
Here we report on a form of chronic distal SMA
From the 1Unite de Recherches sur les Handicaps Genetiques del’Enfant, INSERM U 393, Institut Necker-Enfants Malades, Paris,France; 2Federation de Medecine de l’Enfant, Hopital RaymondPoincare, Garches, France; 3Department of Neurology and Neuro-pathology, American University Hospital, Beirut, Lebanon; 4De-partment of Neurology, Specialty Clinics Center, Beirut, Lebanon;5Genethon, Evry, France; 6Service d’Explorations FonctionnellesNeurologiques, Hopital Necker-Enfants-Malades, Paris, France;7Department of Paediatrics and Child Health, Royal Children’sHospital, Brisbane, Queensland, Australia; 8Department of Neurol-ogy, Massachusetts General Hospital, Boston, MA; and 9Institute ofHuman Genetics, University of Newcastle upon Tyne, Newcastleupon Tyne, United Kingdom.
Received May 23, 2001, and in revised form Jan 3, 2002. Acceptedfor publication Jan 4, 2002.
Published online Apr 23, 2002 in Wiley InterScience(www.interscience.wiley.com). DOI: 10.1002/ana.10182
Accession numbers and URLs for data in this article are listed in theAppendix.
Address correspondence to Dr Viollet, Unite de Recherches sur lesHandicaps Genetiques de l’Enfant, INSERM U 393, InstitutNecker-Enfants Malades, 149 Rue de Sevres, 75743 Paris Cedex 15,France. E-mail: [email protected]
© 2002 Wiley-Liss, Inc. 585
with a broad range of ages at onset (from infancy toearly adulthood) in a large, consanguineous family andprovide evidence of mapping of the disease gene to thegenetic interval encompassing the SMARD1 gene onchromosome 11q13.
Subjects and MethodsThe subjects belonged to a very large, inbred Lebanese familywith three major branches. All 5 affected children have atleast 1 common ancestor. Seventeen members participated inthe study, and informed consent was obtained from each in-dividual. For Individual II3, clinical and histological datawere obtained from reports by Dr Ziad Rifai and ProfessorRobert Griggs, who examined the patient at Strong Memo-rial Hospital of the University of Rochester (Rochester, NewYork). Medical data were completed by our own physicalexamination and by electrophysiological studies, which wereperformed at Necker-Enfants Malades Hospital (Paris,France). For Individuals II7, II8, II10, and II11, clinical, his-tological, and neurophysiological data were collected with thehelp of Professor Jean Rebeiz in Beirut, Lebanon. These datawere completed by clinical and electrophysiological reportsby Professor Raymond D. Adams of the Department of
Neurology at Massachusetts General Hospital (Boston, MA).Concerning Individual II7, data were also obtained from aclinical report by Pearn and Hudgson that was based on aprevious clinical and genetic study of 12 distal SMA cases(Case 4).18 These data were completed by respiratory inves-tigations in Garches, France, and Beirut, Lebanon.
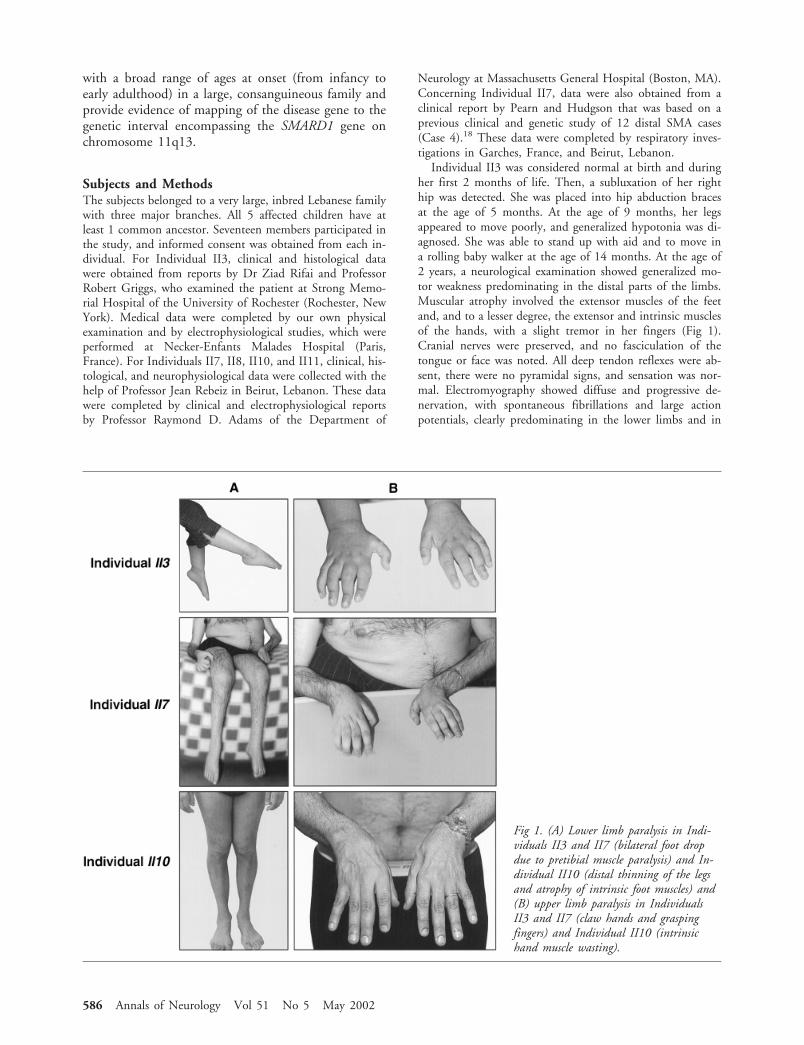
Individual II3 was considered normal at birth and duringher first 2 months of life. Then, a subluxation of her righthip was detected. She was placed into hip abduction bracesat the age of 5 months. At the age of 9 months, her legsappeared to move poorly, and generalized hypotonia was di-agnosed. She was able to stand up with aid and to move ina rolling baby walker at the age of 14 months. At the age of2 years, a neurological examination showed generalized mo-tor weakness predominating in the distal parts of the limbs.Muscular atrophy involved the extensor muscles of the feetand, and to a lesser degree, the extensor and intrinsic musclesof the hands, with a slight tremor in her fingers (Fig 1).Cranial nerves were preserved, and no fasciculation of thetongue or face was noted. All deep tendon reflexes were ab-sent, there were no pyramidal signs, and sensation was nor-mal. Electromyography showed diffuse and progressive de-nervation, with spontaneous fibrillations and large actionpotentials, clearly predominating in the lower limbs and in
Fig 1. (A) Lower limb paralysis in Indi-viduals II3 and II7 (bilateral foot dropdue to pretibial muscle paralysis) and In-dividual II10 (distal thinning of the legsand atrophy of intrinsic foot muscles) and(B) upper limb paralysis in IndividualsII3 and II7 (claw hands and graspingfingers) and Individual II10 (intrinsichand muscle wasting).
586 Annals of Neurology Vol 51 No 5 May 2002

the anterior parts of the leg muscles. Motor nerve conduc-tion velocity was normal in the distal nerves (45m/sec in theright deep peroneal nerve). Distal sensory action potentialsand sensory nerve conduction velocity were normal (20�Vand 43m/sec, respectively, in the right sural nerve). A quad-riceps muscle biopsy showed large, fiber grouping consistentwith SMA. When she was 6 years of age, lower limb weak-ness was stable, but she had bilateral hip subluxation andgenu recurvatum. She was still able to stand up alone butcould not walk without aid. Slight dorsal kyphoscoliosis andmarked lumbar hyperlordosis were noted. She required in-tensive physiotherapy and wore a trunk orthosis. Respiratoryfunction was normal (respiratory vital capacity was 106% ofthe normal value), but a chest X-ray showed an unusuallyhigh right hemidiaphragm (Fig 2). Hypomobility of the righthemidiaphragm was confirmed by chest fluoroscopy. At the
age of 8 years, she was a very alert child, following a normalcurriculum at school.
Individual II7, a boy, experienced his first symptoms atthe age of 6 months, with bilateral floppiness of the feet andpoor spontaneous mobility contrasting with otherwise nor-mal motor development. He acquired the ability to sit un-aided, to walk with assistance at the age of 2 years, and towalk independently at the age of 4 years. When the patientwas 2 years of age, a diagnosis of SMA was made on thebasis of electrophysiological evidence of denervation withnormal nerve conduction velocities and histopathological ev-idence of muscle denervation with large-group atrophy and anormal sural nerve biopsy. He retained walking ability untilthe age of 12 years and then became wheelchair-bound aftera knee fracture. A neurological examination showed severefoot muscle involvement with muscular atrophy and second-ary hand paralysis, which worsened by the age of 10 years(see Fig 1). Trunk muscles were also involved, and trunkweakness progressed, causing hyperlordosis and severe ky-phoscoliosis. Facial weakness was never mentioned, and in-telligence was normal. Deep tendon reflexes were either di-minished or abolished, and cutaneous plantar reflexes werenormal. Neither palpable thickening of peripheral nerves norany sensory defect was present. When he was 30 years of age,a measurement of respiratory function showed a severe de-crease of vital capacity to 34% of the normal value. A chestX-ray taken with the patient in a sitting position showed anelevation of both hemidiaphragms, strongly suggesting bilat-eral diaphragmatic paralysis (see Fig 2). At this time, the pa-tient used a computer keyboard to write because of handparalysis, and he had experienced a generalized and progres-sive deterioration of muscle strength.
Individual II8, the younger brother of II7, did well duringinfancy and was able to walk at the age of 1 year. When hewas 2 years of age, a foot paralysis similar to that in hisbrother occurred. A muscle biopsy at the age of 4 yearsshowed neurogenic atrophy. Despite a distribution of muscleweakness similar to his brother’s, the course of the diseasewas less severe, and he could walk unaided until the age of16 years. By the age of 12 years, his hand muscle strengthwas flagging. At the age of 28 years, he was unable to standup from a sitting position any longer, but he could still walkwith aid. His hands and feet were weak, as were his shoul-ders, but he was still able to write with a pencil. The diag-nosis of SMA was clinically obvious. A respiratory investiga-tion was not undertaken for this patient.
Individual II10 was seen at the age of 43 years. He hadhad normal motor development during infancy and child-hood, and he had practiced several sports, including water,sky, and scuba diving, until the age of 26 years. The patientcomplained about a slight weakness of the lower extremities,especially in the pretibial muscle group, from the age of 23years. He experienced a progressive worsening of his condi-tion, and 3 years later, a neurological examination showed amuscular atrophy of the lower extremities, with spindly legs,slight foot drop contracture, and inability to walk on theheels (see Fig 1A). Muscle strength testing showed weaknessof the dorsiflexor muscles of the toes and feet (3/5 on theMedical Research Council scale), even though the thighmuscles were almost normal (4.5/5) and the trunk muscleswere strong. In the upper limbs, the forearm and hand mus-
Fig 2. Frontal chest X-rays in prone position. For IndividualII3, the white arrow indicates the abnormal elevation of theright hemidiaphragm due to unilateral diaphragmatic paraly-sis. For Individual II7, white arrows indicate the elevation ofboth hemidiaphragms during inspiration due to bilateral dia-phragmatic insufficiency. For Individual II10, there is normaldescent for both hemidiaphragms during inspiration, givingevidence of normal diaphragmatic function.
Viollet et al: Autosomal Recessive Chronic Distal SMA Mapping 587

cles were rather thin, and abduction, adduction, and exten-sion of fingers were evaluated at 3.5 of 5 (see Fig 1B). Theshoulder muscles were normal. Cranial musculature was en-tirely normal, and no fasciculation was detected. There wereno sensory changes. All reflexes were present, especially anklejerks. Plantar reflexes were flexor. A muscle biopsy showeddenervation atrophy. Electromyography confirmed the de-nervation with normal motor nerve conduction velocity(45m/sec in the right tibial nerve), normal distal sensory ac-tion potentials, and normal sensory nerve conduction veloc-ity (45m/sec in the right sural nerve), supporting the diag-nosis of distal SMA. At the age of 43 years, the patient’sphysical examination was largely unchanged, but he had de-veloped a limping gait and felt tired after walking a longdistance or climbing stairs. Respiratory function was normal,with full respiratory capacity (115% of normal values). A flu-oroscopic examination showed normal position for both he-midiaphragms (see Fig 2).
Individual II11 underwent her first neurological examina-tion at the age of 19 years for lower back pain. At that time,a physical examination showed definite atrophy of her feetand, to a lesser extent, of her hands. SMA was suspected andconfirmed when she was 26 years of age, both by musclebiopsy showing denervation atrophy and by electromyogra-phy. At the age of 38 years, she was able to write and wasnot limited in her ability to walk or climb stairs, but she hada waddling gait. Respiratory function was normal, with re-spiratory capacity at 119% of the normal value. No dia-phragmatic paralysis was detected on chest fluoroscopy.
MethodsDNA was extracted from blood leukocytes by sodium dode-cyl sulfate lysis, proteinase K digestion, phenol/chloroformextraction, ethanol precipitation, and Tris–ethylenediami-netetraacetic acid resuspension. SMN exon 7 deletion wastested as described previously.19
For genotyping, genomic DNA (200ng) was amplifiedwith 0.5U of Thermus aquaticus polymerase (Gibco BRL) ina buffer containing 1.5mM MgCl2, 20�M deoxynucleotide,and 20�M primers, in a final volume of 20�l. Amplificationconditions were 94°C for 10 minutes followed by 30 cyclesat 94°C for 1 minute, at 55°C for 1 minute, at 72°C for 1minute, and at 72°C for a final extension of 10 minutes.Polymerase chain reaction products (3�l) were mixed with3�l of formamide in a loading buffer (xylene cyanol/bromo-phenol blue/ethylenediaminetetraacetic acid), incubated at94°C for 5 minutes, and electrophoresed at 14,000V for 4hours on a 6% polyacrylamide, 7.5M urea denaturing gel.Gels were blotted onto charged nylon membranes (HybondN�; Amersham Pharmacia, Piscataway, NJ). Membraneswere hybridized with a deoxycytidine triphosphate oligonu-cleotide labeled by chemoluminescence according to themanufacturer’s instructions (enhanced-chemoluminescencedirect nucleic acid labeling and detection systems; AmershamLife Science) and exposed to X-ray films.
For genotype analysis, the order of markers in theSMARD1 region was taken from the radiation hybrid map ofthe Marshfield Medical Research Foundation Center forMedical Genetics, and haplotypes were assigned manually.For statistical analyses, two-parameter linkage analyses were
performed with the M-LINK and LINKMAP options of ver-sion 5.1 of the LINKAGE program in each of the threebranches of the family.20 Allele frequencies were obtainedfrom the Centre d’Etudes du Polymorphisme Humain geno-type database.
For IGHMBP2 gene mutation screening, all 15 exonswere amplified from the genomic DNA of Individual II7 andof at least one control individual, with the intronic primersused by Grohmann and colleagues.16 DNA (200ng) was am-plified with 0.5U of Thermus aquaticus polymerase (GibcoBRL) in a buffer containing 1.5mM MgCl2, 20�M de-oxynucleotide, and 20�M primers in a final volume of 20�l.Amplification conditions were 94°C for 10 minutes followedby 30 cycles at 94°C for 1 minute, at 55°C for 1 minute, at72°C for 1 minute, and at 72°C for a final extension of 10minutes.
For direct sequencing, amplification products were loadedonto a 1% agarose gel with a low melting temperature, pu-rified by phenol/chloroform extraction, and recovered byethanol precipitation. Bidirectional sequencing of the puri-fied fragments was performed with the intronic primers usedfor polymerase chain reaction amplification and the Big DyeTerminator Cycle Sequencing kit (Applied Biosystems, Fos-ter City, CA) on an automatic fluorometric ABIPRISMDNA sequencer 377 (Applied Biosystems). Data were col-lected and analyzed with ABI DNA sequencing analysis soft-ware, version 3.4.1.
ResultsThe analysis of four microsatellite DNA markers flank-ing the survival motor neuron gene on chromosome5q13 (D5S435, D5F149S1/S2 [C212], D5F150S1/S2[C272], and D5S351)2 showed that at least 1 affectedindividual and 1 unaffected individual shared the samehaplotype at the SMN1 locus, supporting the viewthat, in this family, autosomal recessive chronic distalSMA was not linked to chromosome 5q13. In addi-tion, the analysis of the survival motor neuron geneshowed that none of the affected individuals carried ahomozygous SMN1 exon 7 deletion (data not shown).
Sixteen hypervariable microsatellite markers from theGenethon final linkage map (D11S4076 toD11S917)21 were used to scan the SMARD1 region onchromosome 11q13-q21.15 Haplotype analyses showedthat all affected siblings were homozygous for a coseg-regating segment of seven markers in the 11q13 region,consistent with parental consanguinity. A recombina-tion event in Individual II5 placed the disease locusdistal to marker D11S1889. The crossovers in Individ-uals II7, II8, II10, and II11 placed the disease locusproximal to D11S1321 (Fig 3). Pairwise linkage be-tween the disease locus and polymorphic markersof chromosome 11q13 (D11S4113, D11S4136,D11S4196, D11S1314, and D11S4207) gave positivevalues in all three branches of the family (data notshown). Maximum LOD score values were obtained forprobe AFMb032zg5 at the D11S4136 locus (Zmax �
588 Annals of Neurology Vol 51 No 5 May 2002
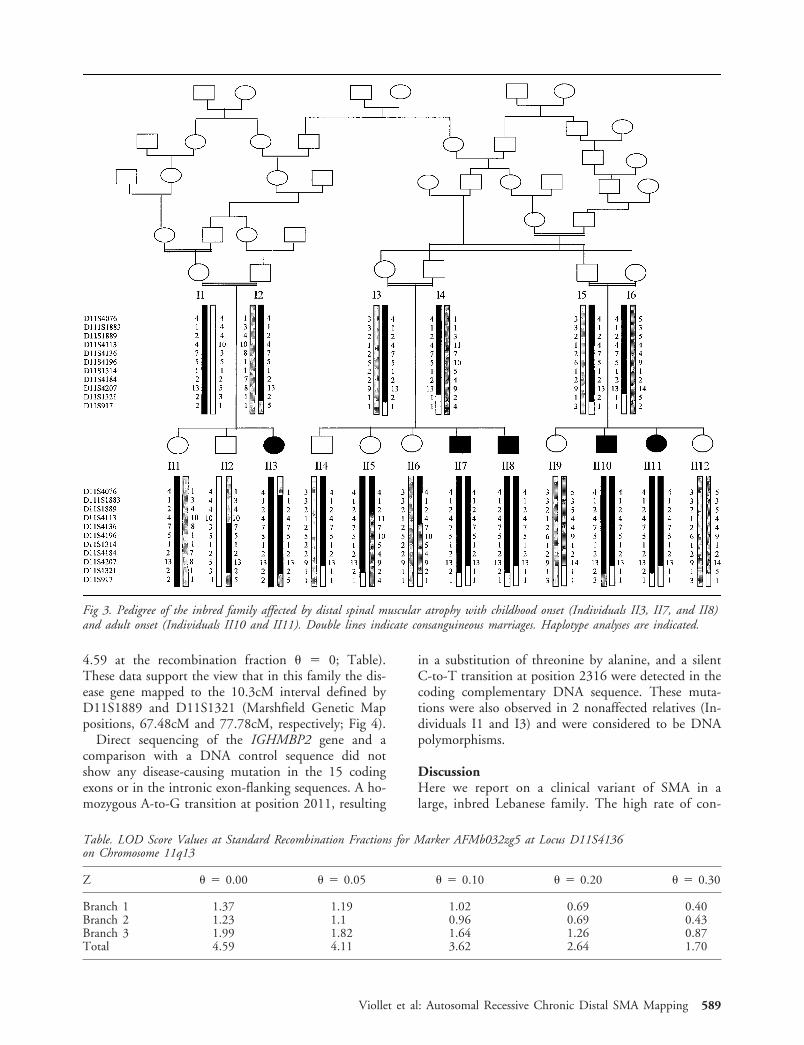
4.59 at the recombination fraction � � 0; Table).These data support the view that in this family the dis-ease gene mapped to the 10.3cM interval defined byD11S1889 and D11S1321 (Marshfield Genetic Mappositions, 67.48cM and 77.78cM, respectively; Fig 4).
Direct sequencing of the IGHMBP2 gene and acomparison with a DNA control sequence did notshow any disease-causing mutation in the 15 codingexons or in the intronic exon-flanking sequences. A ho-mozygous A-to-G transition at position 2011, resulting
in a substitution of threonine by alanine, and a silentC-to-T transition at position 2316 were detected in thecoding complementary DNA sequence. These muta-tions were also observed in 2 nonaffected relatives (In-dividuals I1 and I3) and were considered to be DNApolymorphisms.
DiscussionHere we report on a clinical variant of SMA in alarge, inbred Lebanese family. The high rate of con-
Table. LOD Score Values at Standard Recombination Fractions for Marker AFMb032zg5 at Locus D11S4136on Chromosome 11q13
Z � � 0.00 � � 0.05 � � 0.10 � � 0.20 � � 0.30
Branch 1 1.37 1.19 1.02 0.69 0.40Branch 2 1.23 1.1 0.96 0.69 0.43Branch 3 1.99 1.82 1.64 1.26 0.87Total 4.59 4.11 3.62 2.64 1.70
Fig 3. Pedigree of the inbred family affected by distal spinal muscular atrophy with childhood onset (Individuals II3, II7, and II8)and adult onset (Individuals II10 and II11). Double lines indicate consanguineous marriages. Haplotype analyses are indicated.
Viollet et al: Autosomal Recessive Chronic Distal SMA Mapping 589

sanguinity, the involvement of both genders, and theabsence of any identified neurological disorder in theparents suggest an autosomal recessive mode of inher-itance. Affected individuals consistently presentedwith bilateral muscle weakness and atrophy, predom-inantly in the distal parts of the limbs. Progressive,symmetrical muscular wasting, normal sensation, andthe absence of central nervous system involvementsupported the diagnosis of distal SMA. A conclusivediagnosis was based on electrophysiological and histo-logical studies, which showed severe denervation andabsence of sensory track involvement.9 These criteriaand the absence of SMN1 gene deletion were consis-tent with a diagnosis of distal hereditary motor neu-ropathy or spinal-type Charcot Marie Tooth disease,according to the Second European NeuromuscularConsortium.22
Multiple cases of chronic distal SMA, segregating oneither an autosomal dominant or autosomal recessivemode of inheritance, have been reported.3,5,7,18,23 Al-though their prevalence remains largely unknown,Pearn and Hudgson estimated that distal SMAs ac-counted for 10% of all SMA cases in a population sur-vey of northeastern England.18 Their study included 1of our patients (Individual II3) among 12 cases of dis-tal SMA in 8 kindred. In all their cases, as well as inour patient, affected individuals presented with anidentical pattern of muscle wasting beginning in thelower limbs and associated with proximal weakness.During the natural course of the disease, there was aprogressive worsening during childhood and a relativestabilization into adulthood. In our adult-onset cases, avery slow, progressive worsening of muscle weaknesswas observed. The slow progression of the disease wasalso mentioned by Gardner-Medwin and colleagues ina case report of SMA with childhood and adolescenceonset and benign evolution (Case 17).24 As observed inour pedigree, Pearn and Hudgson noted different ages
at onset. For this reason, they suggested the existenceof at least two separate genes for the early-onset andjuvenile-onset forms of autosomal recessive distal SMA.In fact, our pedigree supports the view that the samedisease gene could account for either childhood-onsetor adult-onset chronic distal SMA and raises the ques-tion of whether nonallelic modifying genes could alsobe involved.
Diaphragmatic paralysis was noted in 2 of our pa-tients who presented with childhood-onset chronic dis-tal SMA. Diagnosis was suspected by paradoxical chestmotion and was diagnosed by chest X-ray and fluoros-copy, showing elevation and hypomobility of the he-midiaphragms. At variance with proximal SMA pa-tients, no chest atrophy was noted in our distal SMApatients, suggesting a relative sparing of intercostalmuscles in comparison with diaphragmatic muscle. Di-aphragmatic dysfunction is not a rare clinical feature inmotor neuron disorders and could be misdiagnosed ifthe paralysis is slight and unilateral (as in IndividualII3). Conversely, diaphragmatic dysfunction could beresponsible for restrictive respiratory insufficiency inthe most severe cases, as observed in IndividualII7.25,26 Unfortunately, in the cases of childhood onsetdistal SMA reported by Pearn and Hudgson, respira-tory function and diaphragmatic mobility was not in-vestigated. In addition, the long-term follow-up of thepatients was not reported, and no late respiratory in-sufficiency was documented.
In contrast to dominant distal SMA, only two auto-somal recessive distal SMA genes have been mapped upto now. Recently, a gene for a novel form of autosomalrecessive distal hereditary motor neuropathy with child-hood onset and upper motor neuron involvement(dHMN-J) was mapped to chromosome 9p21.1-p12 ina Jordanian population.27 However, no pyramidal fea-ture was elicited in our patients, suggesting that distalSMA in our pedigree was a distinct entity. Grohmann
Fig 4. Genetic map of chronic distal spinal muscular atrophy (SMA) and spinal muscular atrophy with respiratory distress (SMARD1)gene regions on chromosome 11q13-q21. Filled bars indicate the disease-associated haplotypes reported in the chronic distal SMA familyand in the SMARD1 families reported by Grohmann and colleagues. The position of the IGHMBP2 gene is estimated from an inte-grated map at the database of the Human Genome Project Working Draft at the University of California at Santa Cruz.
590 Annals of Neurology Vol 51 No 5 May 2002

and colleagues assigned a gene for an early-onset formof SMA (SMARD1) to chromosome 11q13-q21.15 Inthis condition, the patients had severe respiratory dis-tress due to bilateral diaphragmatic paralysis, requiringrespiratory assistance in the first weeks of life. A gen-eralized muscle weakness was noted, predominating inthe distal musculature of the limbs.28–30 These featuresprompted us to test the SMARD1 locus in our pedi-gree. Here we report on the mapping of a chronic dis-tal SMA gene to the 11q13 region in the 10.3cM re-gion encompassing IGHMBP2, the human homologueof the mouse nmd gene (see Fig 4). IGHMBP2 hasbeen shown to account for SMA in mice and forSMARD1 in six unrelated families.16,17 For these rea-sons, we tested the IGHMBP2 gene in the family westudied. The absence of detectable mutations in ourpatients supports the view that chronic distal SMA andSMARD1 are distinct entities mapping to chromosome11q13. Because of the substantial size of our geneticinterval, further work will be necessary to refine themapping of the chronic distal SMA locus, and se-quence analysis of the other candidate genes located inthe 11q13 region will hopefully allow identification ofthe gene causing chronic distal SMA.
This work was supported by the Association Francaise contre lesMyopathies (8644, L.V.).
We thank the following people for their assistance with this study:the patients and their family for their participation and their effortsto help us; Dr Carsten Bonneman (Division of Neurology, Chil-dren’s Hospital of Philadelphia, Philadelphia, PA), Dr SusanaQuijano-Roy (Clinical Neurophysiology Department, Children’sHospital, Boston, MA), Dr Robert C. Griggs (Department of Neu-rology, Strong Memorial Hospital, University of Rochester, Roch-ester, NY), and Dr Andre Megarbane (Unite de Genetique Medi-cale, Universite Saint Joseph, Beirut, Lebanon), who helped usgather medical data; and Dr Josseline Kaplan (INSERM U393, Ho-pital Necker-Enfants Malades, Paris, France) for her help in linkageanalyses.
References1. Melki J, Lefebvre S, Burglen L, et al. De novo and inherited
deletions in the region of the spinal muscular atrophy gene.Science 1994;264:1474–1477.
2. Lefebvre S, Burglen L, Reboullet S, et al. Identification andcharacterization of a spinal muscular atrophy-determining gene.Cell 1995;80:155–165.
3. Dyck PJ, Lambert EH. Lower motor and primary sensory neu-ron diseases with peroneal muscular atrophy. I. Neurologic, ge-netic and electrophysiologic findings in hereditary polyneurop-athies. Arch Neurol 1968;28:603–618.
4. Harding AE. Inherited neuronal atrophy and degeneration pre-dominantly of lower motor neurons. In: Dyck PJ, Thomas PK,Griffin JW, et al, eds. Peripheral neuropathy. 3rd ed.Philadelphia: W. B. Saunders, 1993:1051–1064.
5. Davis CJF, Bradley WG, Madrid R. The peroneal muscularatrophy syndrome. J Genet Hum 1978;26:311–349.
6. Buchthal F, Behse F. Peroneal muscular atrophy (PMA) andrelated disorders. I. Clinical manifestations as related to biopsyfindings, nerve conduction and electromyography. Brain 1977;100:41–66.
7. Meadows JC, Marsden CD, Harriman DGF. Chronic spinalmuscular atrophy in adults. 2. Other forms. J Neurol Sci 1969;9:551–566.
8. Martin-Sneesens L. Formes a evolution tres prolongee del’amyotrophie spinale de Werdnig-Hoffmann. J Genet Hum1962;11:251–269.
9. Emery AEH. The nosology of spinal muscular atrophies. J MedGenet 1971;8:481–495.
10. McEntagart M, Norton N, Williams H, et al. Localization ofthe gene for distal hereditary motor neuronopathy VII(dHMN-VII) to chromosome 2q14. Am J Hum Genet 2001;68:1270–1276.
11. Christodoulou K, Kyriakides T, Hristova AH, et al. Mappingof a distal form of a distal spinal muscular atrophy with upperlimb predominance to chromosome 7p. Hum Mol Genet 1995;4:1629–1632.
12. Sambuughin N, Sivakumar K, Selenge B, et al. Autosomaldominant distal spinal muscular atrophy type V (dSMA-V) andCharcot-Marie-Tooth type 2D (CMT2D) segregate within asingle large kindred and map to a refined region on chromo-some 7p15. J Neurol Sci 1998;26:23–28.
13. Timmerman V, De Jonghe P, Simokovic S, et al. Distal hered-itary motor neuropathy type II (distal HMN II): mapping of alocus to chromosome 12q24. Hum Mol Genet 1996;5:1065–1069.
14. Van der Vleuten AJW, Van Ravenswaaij-Arts CMA, FrijnsCJM, et al. Localisation of the gene for a dominant congenitalspinal muscular atrophy predominantly affecting the lowerlimbs to chromosome 12q23–q24. Eur J Hum Genet 1998;6:376–382.
15. Grohmann K, Wienker TF, Saar K, et al. Diaphragmatic spinalmuscular atrophy with respiratory distress is heterogeneous, andone form is linked to chromosome 11q13–q21. Am J HumGenet 1999;65:1459–1462.
16. Grohmann K, Schuelke M, Diers A, et al. Mutations in thegene encoding immunoglobulin �-binding protein 2 cause spi-nal muscular atrophy with respiratory distress type 1. NatGenet 2001;29:75–77.
17. Cox GA, Mahaffey CL, Frankel WN. Identification of themouse neuromuscular degeneration gene and mapping of a sec-ond site suppressor allele. Neuron 1998;21:1327–1337.
18. Pearn J, Hudgson P. Distal spinal muscular atrophy: a clinicaland genetic study of 8 kindred. J Neurol Sci 1979;43:183–191.
19. Van der Steege G, Grootscholten PM, Van de Vlies P, et al.PCR-based DNA test to confirm clinical diagnosis of autoso-mal recessive spinal muscular atrophy. Lancet 1995;345:985–986.
20. Lathrop GM, Lalouel JM. Easy calculations of LOD scores andgenetic risks on small computers. Am J Hum Genet 1984;36:460–465.
21. Schuler GD, Boguski MS, Stewart EA, et al. A gene map of thehuman genome. Science 1996;274:540–546.
22. De Jonghe P, Timmerman V, Van Broeckhoven C. Secondworkshop of the European Neuromuscular Consortium, Sep-tember 26–28, 1997. Neuromusc Disord 1998;8:426–431.
23. Harding AE, Thomas K. Hereditary distal spinal muscular at-rophy. A report on 34 cases and a review of the literature.J Neurol Sci 1980;45:337–348.
24. Gardner-Medwin D, Hudgson P, Walton JN. Benign spinalmuscular atrophy arising in childhood and adolescence. J Neu-rol Sci 1967;5:121–158.
Viollet et al: Autosomal Recessive Chronic Distal SMA Mapping 591

25. Parhad IM, Clark AW, Barron KD, Staunton SB. Diaphragmaticparalysis in motor neuron disease. Neurology 1978;28:18–22.
26. McCredie M, Lovejoy FW, Kaltreider NL. Pulmonary functionin diaphragmatic paralyis. Thorax 1962;17:213–217.
27. Christodoulou K, Zamba E, Tsingis M, et al. A novel form ofdistal hereditary motor neuropathy maps to chromosome9p21.1-p12. Ann Neurol 2000;48:877–884.
28. Bove KE, Iannaccone ST. Atypical infantile spinomuscular at-rophy presenting as acute diaphragmatic paralysis. PediatrPathol 1988;8:95–107.
29. Bertini E, Gadisseux JL, Palmieri G, et al. Distal infantile spinalmuscular atrophy associated with paralysis of the diaphragm: avariant of infantile spinal muscular atrophy. Am J Med Genet1989;33:328–335.
30. Novelli G, Capon F, Tamisari L, et al. Neonatal spinal muscu-lar atrophy with diaphragmatic paralysis is unlinked to 5q11.2-q13. J Med Genet 1995;32:216–219.
Appendix
Accession numbers and URLs for data in this article are asfollows: Centre d’Etudes du Polymorphisme Humain, forallele frequencies, http://www.cephb.fr/cgi-bin/wdb/ceph/systeme/form; Genethon, for genetic markers, ftp://ftp.genethon.fr/pub/Gmap/Nature-1995/data/; Human GenomeProject Working Draft at the University of Californiaat Santa Cruz, for an integrated physical map, http://genome.ucsc.edu; Marshfield Medical Research FoundationCenter for Medical Genetics, for genetic markers, http://research.marshfieldclinic.org/genetics/; and Online Mende-lian Inheritance in Man, for SMA1 [MIM 253300] andSMARD1 [MIM 604320], http://www.ncbi.nlm.nih.gov/Omim.
592 Annals of Neurology Vol 51 No 5 May 2002





























