Embed Size (px)
Citation preview

169
Abstract Dominant optic atrophy (DOA) and Charcot-Marie-Tooth type 2A disease (CMT2A) are inherited neurological disorders primarily affecting retinal ganglion cells and peripheral nerves, and secondarily, other neuronal cells with high ener-getic requirements. The discovery of OPA1 mutations in DOA patients and MFN2 mutations in CMT2A patients, has defined a new subset of mitochondrial diseases related to mitochondrial dynamics. OPA1 and MFN2 are both dynamin GTPases that promote the fusion of the mitochondrial network. Since diseases related to OPA1 and MFN2 mutations show some clinical similarities, their pathophysiologi-cal mechanisms are believed to share some common mitochondrial dysfunctions. These mechanisms are closely interdependent and involve mitochondrial fusion, motility along the axons, energy production and metabolism, maintenance of the organelle and its genome, as well as susceptibility to apoptosis. This chapter presents recent findings on: (1) the clinical diversity of OPA1- and MFN2-related diseases; (2) the spectrum of mutations in the OPA1 and MFN2 genes and their consequences at the protein level; and (3) the pathophysiology of DOA and CMT2A as character-ized in cellular and mouse models. Finally, we discuss the therapeutic perspectives opened up by the current knowledge of these diseases.
G. Lenaers (), C. Delettre, E. Sarzi, and C. Hamel Inserm U1051, Institut des Neurosciences de Montpellier, Universités de Montpellier I et II, Montpellier, France e-mail: [email protected]
D. Bonneau, P. Amati-Bonneau, V. Procaccio, and P. Reynier UMR CNRS 6214 - Inserm U771, Angers, France and Département de Biochimie et Génétique, Centre Hospitalier Universitaire, Angers, France
D. Miléa UMR CNRS 6214 - Inserm U771, Angers, France and Département d’Ophtalmologie, Centre Hospitalier Universitaire, Angers, France
C. Verny UMR CNRS 6214 - Inserm U771, Angers, France and Département de Neurologie, Centre Hospitalier Universitaire, Angers, France
Chapter 6Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
Guy Lenaers, Dominique Bonneau, Cécile Delettre, Patrizia Amati-Bonneau, Emmanuelle Sarzi, Dan Miléa, Christophe Verny, Vincent Procaccio, Christian Hamel, and Pascal Reynier
B. Lu (ed.), Mitochondrial Dynamics and Neurodegeneration, DOI 10.1007/978-94-007-1291-1_6, © Springer Science+Business Media B.V. 2011

170 G. Lenaers et al.
Keywords Dominant optic atrophy • Charcot-Marie-Tooth disease • Mitochondria • MFN2 • OPA1
Abbreviations
aa amino acidADP adenosine-5’-diphosphateANT adenine nucleotide translocatorATP adenosine-5’-triphosphateCMT disease Charcot-Marie-Tooth diseaseCPEO chronic progressive external ophthalmoplegiaDOA dominant optic atrophyDRG dorsal root ganglionENU N-ethyl-N-nitrosureaGTP guanosine triphosphateGTPase guanosine triphosphate phosphorylaseHR heptad repeat domainIMM inner mitochondrial membrane.LHON Leber’s hereditary optic neuropathyMEF mouse embryonic fibroblastsMFN2 Mitofusin 2mtDNA mitochondrial DNAOMA1 overlapping activity with M-AAA protease 1OMM outer mitochondrial membraneOPA1 optic atrophy 1OXPHOS oxidative phosphorylationPhb1/2 prohibitins 1 and 2RGC retinal ganglion cellTM transmembrane domainVEP visual evoked potentialYME1L YME1-like protein 1
6.1 OPA1-Associated Diseases
6.1.1 Historical Description of Dominant Optic Atrophy (DOA)
In 1959, the Danish ophthalmologist Poul Kjer described an infantile optic atrophy with a dominant mode of inheritance identified in 19 families (Kjer 1959). Dominant optic atrophy (DOA; MIM #165500), also called Kjer disease, is classi-cally described as a progressive, isolated optic neuropathy, developing insidiously

1716 Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
over the first two decades of life. Rarely, the clinical presentation of the disease is atypical including acute, severe visual loss, reversible visual loss, or late occurrence in the life course. Bilateral visual loss is generally moderate, but, in some cases, leading to legal blindness (Votruba et al. 1998). The loss of the central visual field, due to centrocecal, central or paracentral scotoma, is commonly bilateral and sometimes progressive, associated with difficulty in discriminating colors in the blue-yellow axis, corresponding to dyschromatopsia of the tritanopic type. Ophthalmoscopic examination discloses temporal or diffuse pallor of the optic nerve heads, sometimes associated with optic disc excavation. The rare histopatho-logical postmortem studies published several years ago described a selective loss of retinal ganglion cells (RGC) that form the optic nerve (Johnston et al. 1979; Kjer et al. 1983). More recently, in vivo optical coherence tomography confirmed the thinning of the optic fiber layer around the optic disc in patients, whereas the other retinal layers remain unaltered (Milea et al. 2010). From a genetic point of view, DOA has incomplete penetrance with highly heterogeneous clinical expressivity even within families (Hoyt 1980; Votruba et al. 1998).
The classical clinical presentation of DOA is distinct from that of Leber’s hereditary optic neuropathy (LHON), which is characterized by an acute and sequential onset of visual loss in both eyes occurring at an older age, typically 18–35 years (Carelli et al. 2004). LHON, which is exclusively transmitted mater-nally through mitochondrial DNA (mtDNA) mutations, predominantly affects males, whereas DOA affects males and females equally.
6.1.2 The Discovery of OPA1
The locus for the main DOA-causing gene, optic atrophy 1 (OPA1, MIM *605290), was mapped to chromosome 3q28-29 (Eiberg et al. 1994; Lunkes et al. 1995). Subsequently, DOA was found to be genetically heterogeneous as two further loci were identified, i.e. OPA4 (MIM %605293) and OPA5 (MIM %610708), mapping to chromosomes 18q12.2-q12.3 (Kerrison et al. 1998) and 22q12.1-q13.1 (Barbet et al. 2005) respectively. In 2000, studies on mitochondrial dynamics in yeast, led to the identification of the human ortholog gene of the MGM1/Msp1 genes of Saccharomyces cerevisiae and Schizosaccharomyces pombe. This gene, encoding a mitochondrial GTPase of the dynamin family was localized on chromosome 3 at the morbid locus linked to DOA. Gene sequencing in six families suffering from DOA unraveled pathogenic mutations, indicating that the ortholog of the MGM1/Msp1 genes was indeed OPA1, the gene responsible for DOA (Delettre et al. 2000). Meanwhile, another team from Germany and the United Kingdom reached the same conclusion using positional cloning (Alexander et al. 2000). These two articles published in Nature Genetics in 2000, marked the beginning of the explo-ration of a new subtype of mitochondrial disorders involving mitochondrial network dynamics.

172 G. Lenaers et al.
6.1.3 Revisiting Clinical Phenotypes of DOA on the Basis of Current Knowledge of the OPA1 Gene
Since 2000, the systematic sequencing of OPA1 in patients with different forms of optic atrophy has led to a better understanding of the pathophysiology of DOA and to refining the clinical spectrum of the disease, which has turned out to be larger than expected. Indeed, OPA1 mutations are responsible for many different pheno-types ranging from mild disorders affecting only the RGCs, as in DOA, to severe and complex phenotypes, named “DOA+”, which are multi-systemic diseases rather similar to mitochondrial diseases due to oxidative phosphorylation (OXPHOS) defects. The routine screening of patients with isolated optic nerve atrophies, which could not be classified as DOA on the basis of genetic or clinical parameters, has led to the discovery of novel OPA1 mutations and phenotype-genotype correlations. Moreover, the identification of multi-systemic forms of DOA has revealed that RGCs, although highly sensitive to these diseases, are not the only cells affected by OPA1 dysfunction.
The molecular screening of 980 patients with sporadic and familial optic neuropa-thies showed that 295 patients (30.1%) carried an OPA1 mutation, 131 patients (13.4%) a LHON-related mtDNA mutation and 14 patients (1.4%) an OPA3 mutation responsible for a rare form of DOA associated with cataract (Ferre et al. 2009; Reynier et al. 2004). In addition, this study showed the high prevalence of pathogenic OPA1 mutations in 157 of the 392 (40%) patients with apparent sporadic optic atrophy, demonstrating the frequent absence of the dominant inheritance considered to be characteristic of this disease.
DOA is classically associated with a progressive, irreversible loss of vision. We have reported several cases of DOA presenting with a large clinical heterogeneity, including cases of spontaneous recovery of vision (Cornille et al. 2008), or with late-onset, LHON-like acute optic atrophy in a 62-year-old woman carrying an OPA1 mutation (Nochez et al. 2009). Several LHON patients referred to our laboratory for mtDNA-mutation screening were actually found to harbor an OPA1 mutation, suggesting a degree of clinical overlap between the two disorders.
A recent study of 104 patients, from 45 unrelated families, sums up the new clinical forms of DOA described over the past 10 years (Yu-Wai-Man et al. 2010a). Nearly 20% of patients with DOA have extra-ocular impairment associated with optic neuropathy; these clinical forms being referred to as “DOA+” (Table 6.1). The commonest form of this impairment is bilateral, progressive sensorineural deafness beginning in adolescence (Amati-Bonneau et al. 2005). The association of DOA with deafness was initially reported in patients carrying the same OPA1 p.R445H missense mutation (Amati-bonneau et al. 2003, 2005; Li et al. 2005; Payne et al. 2004; Shimizu et al. 2003) and other OPA1 mutations were later reported (Ke et al. 2006; Puomila et al. 2005; Yu-Wai-Man et al. 2010b).
Another “DOA+” clinical phenotype associates ataxia, myopathy, peripheral neuropathy, and chronic progressive external ophthalmoplegia (Amati-Bonneau et al. 2008; Hudson et al. 2008). Finally, more rarely, other disorders mimicking

1736 Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
hereditary spastic paraparesis (Yu-Wai-Man et al. 2010b), peripheral neuropathy (Spinazzi et al. 2008), or multiple sclerosis have been reported (Verny et al. 2008). Interestingly, in a few cases, the clinical presentations of OPA1 mutations excluded optic nerve involvement (Milone et al. 2009; Yu-Wai-Man et al. 2010b), showing that some OPA1-associated diseases tend towards phenotypes far removed from the initial description of DOA.
The “DOA+” multi-systemic syndrome is described as beginning with optic atrophy during childhood and then extending to chronic progressive external ophthalmoplegia (CPEO), ataxia, sensorineural deafness, sensory-motor neuropathy and myopathy in adult life (Amati-Bonneau et al. 2008; Hudson et al. 2008; Stewart et al. 2008). The muscle abnormalities observed in patients with “DOA+” were similar to those found in mitochondrial myopathies, with the presence of cytochrome c oxidase-negative fibers and ragged red fibers. In fact, these patients harbored multiple mtDNA deletions in skeletal muscle, suggesting that mitochondrial fusion and the OPA1 protein, in particular, play a key role in mtDNA maintenance.
OPA1 mutations are by far the commonest genetic causes of DOA. However, the rate of detection of OPA1 mutations in DOA families is highly variable, rang-ing from 32% to 89% (Cohn et al. 2007; Delettre et al. 2001; Marchbank et al. 2002; Pesch et al. 2001; Thiselton et al. 2002; Toomes et al. 2001). OPA1-related DOA has incomplete penetrance, and several carriers of OPA1 mutations remain asymptomatic. Initially, the penetrance of the disease was estimated to be as high as 98% (Kivlin et al. 1983), but the identification of OPA1 mutations led to a re-evaluation of this figure, with results ranging from 43% to 90% in more recent studies (Cohn et al. 2007; Puomila et al. 2005; Toomes et al. 2001).
Table 6.1 Clinical and pathophysiological similarities and differences between OPA1- and MFN2-related diseases. CPEO: Chronic progressive external ophthalmoplegia
DOA/OPA1 Clinical phenotypes CMT2A/MFN2
+ Optic neuropathy +/−+/− Peripheral neuropathy ++/− Sensorineural deafness +/−+/− Encephalopathy +/−+/− Myopathy and CPEO −+/− Multiple sclerosis-like −+/− Tremor +/−
Pathophysiological conditions+ Mitochondrial morphological anomalies ++ Energy production defect +? Axonal transport alteration ++ Alteration of neuron connectivity ?+ Increased susceptibility to apoptosis −+ Mitochondrial DNA instability −+/− Haploinsufficiency −+/− Dominant negative effect +

174 G. Lenaers et al.
6.1.4 Epidemiology of DOA
DOA and LHON are the two commonest forms of hereditary optic atrophy. The prevalence of DOA is estimated at about 1/50,000 in the general population (Lyle 1990), but the actual prevalence was found to be much higher in Denmark (1/12,000) (Kjer et al. 1996; Thiselton et al. 2001). The discovery of OPA1 also helped to show that the higher prevalence of DOA observed in the Danish popula-tion was due to a founder effect involving the c.2826delT mutation, hypothesized to have been introduced at least 69 generations ago, much before the Viking era (Thiselton et al. 2001). Recently, the prevalence of DOA due to OPA1 mutations has been re-evaluated to over 1/35,000 in a large cohort of patients of the north of England (Yu-Wai-Man et al. 2010a).
6.2 MFN2-Associated Charcot-Marie-Tooth Diseases
6.2.1 Clinical Description and Classification of Charcot-Marie-Tooth Diseases
Charcot-Marie-Tooth disease (CMT) owes its name to three neurologists who first described the disorder in 1886 (Charcot and Marie 1886; Tooth 1886). CMT is now known to include a heterogeneous group of hereditary chronic neuropathies, affect-ing sensory and motor neurons (Skre 1974), characterized by motor weakness and muscle atrophy affecting the limbs, reduction or abolition of tendon reflexes, and distal sensory loss, often associated with orthopedic deformities, such as hollow feet. The disease progresses slowly, gradually leading to impaired mobility, and often to wheelchair dependency. The CMT group of diseases is clinically heteroge-neous, even within the same family.
Several forms of CMT have been described on the basis of electrophysiological criteria (nerve conduction velocities), modes of inheritance, and the molecular genetic classification. The electrophysiology helps to distinguish the axonal forms from the demyelinating forms of CMT since the latter are specifically associated with motor nerve conduction velocities lower than 38 m/s. The axonal forms correspond to predominantly axonal lesions that do not affect the integrity of the myelin sheath. The modes of inheritance may be autosomal dominant, X-linked or autosomal recessive. The most common forms of CMT are the autosomal dominant demyelinating form (CMT1), the axonal form (CMT2), and the X-linked forms (CMTX). Autosomal recessive forms of CMT are rarer but more severe, and may be either axonal (AR-CMT2) or demyelinating (CMT4). So far, more than 30 genes have been linked to the different forms of CMT. Among these genes, four encode mitochondrial proteins, two of which are involved in mitochondrial dynamics: mitofusin 2 (MFN2) and the ganglioside-induced differentiation-associated protein 1 (GDAP1).

1756 Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
6.2.2 The Discovery of MFN2
In 2004, the MFN2 gene was linked with axonal, autosomal dominant Charcot-Marie-Tooth disease (CMT type 2A; MIM #118210), (Zuchner et al. 2006). Subsequently, it was found that MFN2 mutations occurred in 9–33% of CMT2 patients (Calvo et al. 2009; Kijima et al. 2005; Verhoeven et al. 2006). On the basis of clinical criteria, the disease penetrance was estimated at 74–100% (Lawson et al. 2005).
6.2.3 “CMT2A+” Phenotypes
Like DOA linked to OPA1 mutations, CMT due to MFN2 mutations may be associ-ated with other neurological disorders, as in the case of hereditary motor and sensory neuropathy type VI, which includes peripheral and optic neuropathies (Zuchner et al. 2006). Patients with this disorder suffer from severe peripheral neuropathy, often with a subacute onset of optic atrophy followed by slow recovery of visual acuity. This observation, illustrating the similarities between the peripheral neuropa-thy found in some patients carrying OPA1 mutations and the optic neuropathy reported in some patients with MFN2 mutations, suggests the operation of common pathophysiological mechanisms. Hearing loss, found in patients carrying OPA1 mutations, was also detected in some patients carrying MFN2 mutations (Calvo et al. 2009; Verhoeven et al. 2006). A more complex form of CMT, associating peripheral neuropathy with cognitive impairment and defective vision was also reported in a family carrying a missense MFN2 mutation (Del Bo et al. 2008), with one member of the family additionally presenting spastic paraplegia. This observation, as well as the presence of pyramidal signs in patients, (Calvo et al. 2009; Zhu et al. 2005), demonstrates the possible involvement of the central nervous system in the pathophysiology of MFN2-related CMT. Recently the clinical spectrum of MFN2 mutations was further extended to the CMT1 or demyelinating forms of the disease, as well as to intermediate forms of the disease (Braathen et al. 2010).
6.3 Pathophysiology of OPA1- and MFN2-Associated Diseases
The diversity and variable severity of the phenotypes associated with OPA1 and MFN2 mutations indicate the operation of complex pathogenic mechanisms, linking neuronal and mitochondrial pathophysiology. Considering the energy requirement of RGCs and the complexity of axonal transport, the specific structure of the RGCs and the length of the peripheral nerve are likely to be critical pathophysiological parameters. Moreover, mitochondrial involvement is highly complex since it concerns several essential, interdependent functions, such as energy production,

176 G. Lenaers et al.
apoptosis, oxidative stress, maintenance and expression of mtDNA, mitophagy, and cell signaling.
6.3.1 Mutation Spectrum of the OPA1 Gene
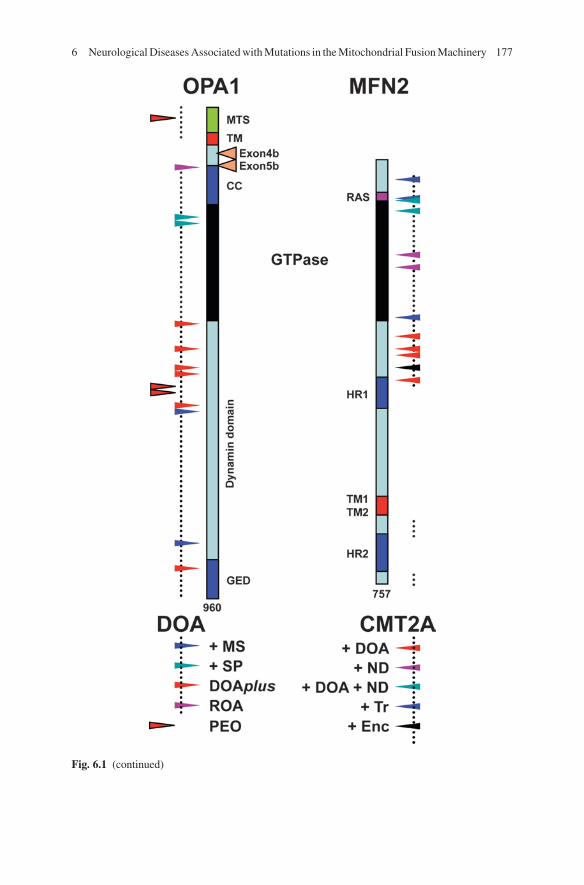
The OPA1 gene, located on chromosome 3q28-29, spans approximately 100 kb and is composed of 31 exons, including three that undergo alternative splicing (4, 4b and 5b) corresponding to a total of eight protein isoforms (Delettre et al. 2001). The main isoform, excluding exon 4b et 5b, encoded by a 2,880 nucleotide-long open reading frame, is a 960 amino acid protein ubiquitously expressed and located in the inter-membrane space, associated with the inner mitochondrial membrane (IMM). The main protein domains are the N-terminal mitochondrial targeting sequence (aa 1–88), a transmembrane domain (aa 97–115), the GTPase domain (aa 260–567), the dynamin central region (aa 568–901), the coiled-coil domain (aa 209–260), and the GTPase putative effector domain (aa 902–960) (Fig. 6.1).
We have developed a locus specific database called eOPA1 (http://lbbma.univ-angers.fr/eOPA1/) as the main depository of OPA1 sequence variations (Ferre et al. 2005, 2009). As of October 2010, 224 pathogenic mutations have been published in this database. These mutations span all the exons, with the exception of exons 4, 4b and 5, in which no mutations have so far been reported. The exons encoding the GTPase domain and the C-terminal coiled-coil domain are more frequently mutated. Heterozygous mutations are mostly family specific, but the mutation c.2708_2711delTTAG in exon 27, is present in about 17% of cases.
A total of 41% of the mutations results in premature termination of translation, 25% are missense mutations, 25% are in frame splice variants, and 6% are micro-deletions or duplications. In addition, large genomic rearrangements of OPA1 gene have been revealed by fluorescence in situ hybridization (FISH) or multiplex ligation-dependent probe amplification (MLPA); (Fuhrmann et al. 2009, 2010; Toomes et al. 2001; Yu-Wai-Man et al. 2010b).
OPA1 mutations leading to splicing defects and premature stop codons induce non-sense mRNA decay, and the reduction of mRNA levels (Schimpf et al. 2008). These observations concord with the low mRNA levels found in fibroblasts from OPA1 patients and mouse models, with a decrease of up to 50% in OPA1 transcripts; in one patient the entire OPA1 gene was found to be deleted, suggesting that haplo-insufficiency is the main pathophysiological mechanism in “pure” DOA (Alavi et al. 2007; Davies et al. 2008; Olichon et al. 2007b). Consequently, pheno-type-genotype correlations have been difficult to establish for most OPA1 mutations leading to DOA, suggesting that other gene modifiers, such as those involved in mRNA decay, may modulate the severity of the disease.
Nevertheless, in syndromic forms of DOA, missense mutations have been systematically identified downstream to, or within, the GTPase domain, with one exception situated in the GED C-terminal domain (Amati-Bonneau et al. 2008; Hudson et al. 2008). In these cases, the OPA1 protein levels were always normal,
1776 Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
Fig. 6.1 (continued)

178 G. Lenaers et al.
suggesting a mutant dominant negative effect. Interestingly, the R445H mutation affecting a highly conserved amino acid in the GTPase domain is responsible for the clinical presentation of most of these “DOA+” patients, ranging from DOA and deafness to severe multi-systemic disorders.
6.3.2 Mutation Spectrum of the MFN2 Gene
The MFN2 gene, located on chromosome 1p36.2, comprises 19 exons (Zuchner et al. 2004). The main transcript, which is 4,685 nucleotides long, including an open reading frame containing 2,271 nucleotides, encodes a protein composed of 757 amino acids with a Ras domain (aa 84–99), a GTPase domain (aa 100–260), two transmembrane domains (aa 628–647), two hydrophobic heptad repeat domains (HR1 and HR2, aa 405–425 and 702–742) allowing coiled-coil interaction and fusion between adjacent mitochondria.
CMT2A is inherited as a dominant trait caused by heterozygous mutations in the MFN2 gene; however, in some rare cases, compound heterozygous or homozygous mutations have been found (Calvo et al. 2009; Nicholson et al. 2008). To date, more than 70 mutations have been identified (see http://www.molgen.ua.ac.be/CMTMutations/Mutations/Mutations.cfm) (Calvo et al. 2009), the large majority of these (>90%) being missense mutations, whereas only a few truncating mutations have been reported; these later mutations target either the last 10 amino acids of the C-terminal domain, the last 340 amino acids including the two TM and the HR2 domains, or result from splicing defects. So far, no duplication or deletion in the MFN2 gene has been reported. The distribution of mutations in the MFN2 protein is not homogeneous, indeed most of the mutations (>85%) affect the half of the N-terminal domain of the protein including the GTPase domain, and the two surrounding domains (aa 1–99 and 260–405), the structure and function of which remain unknown. Conversely, from the first coiled-coil HR1 domain to the carboxy-end of the protein, only a few mutations have been identified, none of which affect the transmembrane TM1 and TM2 domains (Cartoni and Martinou 2009). This suggests that the dysfunc-tional element of the mutated MFN2 protein is strictly located on the external side of the outer mitochondrial membrane (OMM), i.e. towards the cytoplasm.
Fig. 6.1 (continued) Mutation spectrum in OPA1 and MFN2 proteins and clinical presentations of syndromic DOA and CMT2A. OPA1 (left) and MFN2 (right) proteins are schematically represented. MTS: mitochondrial targeting sequence; TM: transmembrane domain; CC/HR: coiled coil domains/heptad repeats; RAS: Ras binding domain; GTPase: guanosine tri-phosphate hydrolase domain; and GED: GTPase effector domain. In addition, for OPA1, the domains corresponding to the alternate spliced exons 4b and 5b are shown. The domains mutated in OPA1 and MFN2 in “pure” DOA (left) and CMT2A (right) patients are indicated by dotted lines. Mutations responsible for peculiar syndromic phenotypes are represented by arrowheads of different colors. DOA: Dominant optic atrophy; CMT2A: Charcot Marie Tooth type 2A; +MS: plus multiple sclerosis; +SP: plus spastic paraplegia; ROA: recessive optic atrophy; PEO: progressive external ophthalmoplegia (without DOA); +ND: plus neurosensorial deafness; +Tr: plus tremor; and + Enc: plus encephalopathy

1796 Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
On the basis of the accumulated clinical and genetic data collected over the past few years, it now appears possible to evaluate genotype-phenotype correlations, although this should be done with caution since the severity of the disease remains highly variable between individuals sharing the same molecular defect. Thus, patients presenting a CMT2A syndrome associated with optic atrophy carry mutations in the domain (aa 260–405) located between the GTPase and the HR1 domains. Similarly, CMT2A patients with neurosensorial hearing loss carry muta-tions in the GTPase domain, while the two most common mutations at position R94Q (Ras domain) and R104W (GTPase domain) are responsible for CMT2A plus optic atrophy and hearing loss. In addition, CMT2A patients with tremor carry MFN2 mutations in the amino-terminal domain upstream of the GTPase domain. Finally, one of the two splicing mutations in MFN2, which induce erroneous splicing of intron 13 leading to truncated forms of the MFN2 protein, is associated with a subacute and severe fatal encephalopathy that follows shortly the onset of the polyneuropathy (Boaretto et al. 2010) (Fig. 6.1).
Taken together, these findings have three important consequences on the pathophysiological hypothesis concerning the relationship between CMT2A and the mutant MFN2 protein. Firstly, the different amino-terminal domains of MFN2 have distinct functions. Secondly, MFN2 must be anchored to the outer mitochon-drial membrane by the two TM domains and exerts its pathological effect by inter-acting with partners thanks to the integrity of the HR domains. Thirdly, CMT2A is caused by a dominant negative pathophysiological mechanism depending on the mutant MFN2 protein. This idea is supported by the fact that the amount of MFN2 remains unchanged in fibroblasts from CMT2A patients (Amiott et al. 2008). Moreover, the heterozygous MFN2+/− mouse does not present any symptom of the disease (Chen et al. 2003), thus ruling out the hypothesis that haplo-insufficiency might be the main causative mechanism of CMT2A.
6.3.3 Consequences on Mitochondrial Network Morphology
The dynamin OPA1, and the mitofusins MFN1 and MFN2, are the only actors known to be involved in the fusion of the mitochondrial network (Song et al. 2009; Chen and Chan 2010). The mitofusins are responsible for tethering adjacent mitochondria and promoting the fusion of the OMMs in which they are embedded (Rojo et al. 2002). OPA1, which is closely associated with the IMM in the intermembrane space and cristae (Olichon et al. 2002), is directly involved in the fusion of the IMM (Song et al. 2009). We may therefore expect OPA1 and MFN2 mutations to lead to various defects in mitochondrial network structure and function in DOA and CMT2A.
Primary cultures of skin fibroblasts from DOA and CMT2A patients have been studied to investigate the pathophysiology of these diseases. Simultaneously, mutated alleles of OPA1 and MFN2 were expressed in common cell lines, such as HeLa and MEF cells, to study the consequences of the mutations in a homoge-neous genetic background; RNA-interference experiments targeting the OPA1 and

180 G. Lenaers et al.
MFN2 genes were also carried out. Similar gene overexpression and silencing experiments were performed in other cell types, such as purified neurons from the retina, the cerebellum, or the spinal cord. Finally, murine models have recently allowed the investigation of the consequences of OPA1 and MFN2 mutations on animal physiology.
The convergence of the results obtained with these different models has helped to unravel the pathophysiology of DOA and CMT2A. In fibroblasts from patients carrying OPA1 mutations leading to haplo-insufficiency, there were practically no detectable defects in mitochondrial morphology under normal growth conditions (Olichon et al. 2007b). Nevertheless, when galactose was substituted for glucose in the cell culture medium to force the cells to use mitochondrial OXPHOS, OPA1 fibroblasts showed a fragmented mitochondrial network compared to controls (Zanna et al. 2008). This phenotype with highly punctuated mitochondria was also prominent in fibroblasts with OPA1 dominant negative mutations found in DOA+, independently of the media used, suggesting that missense OPA1 alleles have a more severe effect on fusion than the 50% reduction in OPA1 abundance. (Amati-Bonneau et al. 2005).
These findings were confirmed in HeLa cells in which the expression of different missense OPA1 mutations induced severe fragmentation, whereas that of truncative OPA1 mutations had no effect on the mitochondrial network. In cells carrying truncative OPA1 mutations, the level of OPA1 expression was markedly reduced, suggesting that the truncated protein is unstable, thus reinforcing the hypothesis of haplo-insufficiency in “pure” DOA (Olichon et al. 2007b). In addition, some elegant recent research has demonstrated that OPA1 associates with cardiolipins, which are negatively charged phospholipids enriched in the IMM, to stimulate GTP hydrolysis and membrane tubulation. Interestingly, different missense OPA1 mutations induce specific defects in membrane scaffolding, thus revealing for the first time the pos-sible consequences of these mutations on the disorganization of the IMM (Ban et al. 2010). This is of further relevance to the apoptotic process since the release of cyto-chrome c during programmed cell death depends on major structural changes of the IMM, associated with the processing and release of OPA1 in the cytoplasm. These findings are concordant with the results of silencing specific OPA1 isoforms, which induce independent effects on mitochondrial dynamics and apoptosis, suggesting that these processes operate independently in mitochondria (Olichon et al. 2003, 2007a). Similar experiments on mouse RGCs suggest that OPA1 is required for the mitochondrial fusion and necessary to protect these cells from apoptotic insults (Kamei et al. 2005) and for the distribution of mitochondria in the somas and den-drites of the RGCs (Williams et al. 2010).
The investigation of the dysfunction of MFN2 in fibroblasts from CMT2A patients revealed limited alteration of the mitochondrial network, suggesting that the MFN2 defect in fibroblast cells either has mild consequences on the equilibrium between fusion and fission, or is readily compensated by other processes. It is thus possible that the role of MFN2 in fibroblasts is secondary to that of MFN1, and that the eightfold higher GTPase activity of MFN1 compared to that of MFN2 is responsible for most of the membrane tethering and fusion activity in fibroblasts

1816 Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
(Amiott et al. 2008; Ishihara et al. 2004). More importantly, the absence of any major effect on the structure of the mitochondrial network may be related to the lack of susceptibility to apoptosis found in MFN2 fibroblasts (Guillet et al. 2010). Similarly, since MFN2 is responsible for tethering the endoplasmic reticulum to the mitochondrial network, the absence of network modification suggests that the physical relationship between the ER and the mitochondrial network, as well as the calcium uptake by mitochondria, remain unaffected in MFN2 mutated cells (de Brito and Scorrano 2008). Conversely, the overexpression of MFN2 alleles either with trun-cated TM or HR1/2 domains, or mutated in the GTPase domain, lead to a dominant negative effect on fusion, leaving fragmented mitochondria that remain merely apposed but unfused (Eura et al. 2003; Honda et al. 2005). Interestingly, the overex-pression of wild-type MFN2 induces mitochondrial fragmentation and dysfunction with concomitant cell death, illustrating the need for a balance between mitochon-drial fusion and fission (Huang et al. 2007).
Similarly, the expression of MFN2-mutated proteins in neurons induces the clustering of fragmented mitochondria in the soma, by slowing down the physio-logical anterograde and retrograde mitochondrial transport in axons, without affecting the transport of other cargoes (Baloh et al. 2007; Misko et al. 2010). These phenomena clearly play a role in the peculiar cell structure of RGCs and on the axonal length of neurons in patients affected by CMT2A.
6.3.4 Consequences on Mitochondrial Metabolism
Cells carrying OPA1 mutations present a significant deficit of mitochondrial respiration. The study of fibroblasts from DOA patients reveals a coupling defect associated with the reduction of membrane potential and respiratory complex activities (Chevrollier et al. 2008). The reduction of the quantity of OPA1 protein in these fibroblasts can alter the structure of the mitochondrial cristae and reduce the impermeability of the inner membrane, allowing a mild dissipation of the proton gradient (Olichon et al. 2003, 2007a). This observation is in agreement with the finding of a partial uncoupling of the respiration in OPA1-mutated fibroblasts, correlated with the severity of the disease, (Chevrollier et al. 2008). Moreover, the calcium clearance was found to be impaired in RGCs silenced for OPA1, underscoring the importance of membrane potential in importing cytoplasmic calcium into the matrix (Dayanithi et al. 2010). Similarly, mild alteration of the structure of the IMM was found to disrupt the organization between the respiratory complexes, explaining the reduction of complex IV activity observed in DOA fibroblasts, that is compensated in basic growth condition to allowing normal ATP production (Chevrollier et al. 2008). However, when galactose was used instead of glucose, forcing cells to rely on OXPHOS alone, OPA1-mutated fibroblasts presented impaired complex I-driven production of ATP compared to control cells (Zanna et al. 2008). A similar alteration of ATP production was reported in DOA patients, with an in vivo study of the calf muscle by phosphorus magnetic resonance spectroscopy showing that the de novo synthesis of phosphocreatine was delayed after exercise

182 G. Lenaers et al.
(Lodi et al. 2004, 2010). Taken together, these observations support the hypothesis of an energetic defect as one of the principal causes of DOA. Hence, a reduction in respi-ration coupling may progressively impair neuronal functions, particularly in the transmission of action potentials and the transport of cargoes along the axons.
Although the MFN2 mutations identified in CMT2A affect the cytoplasmic domains of the protein, it is now well established that they are responsible for severe defects within mitochondria, and not only in the process of oxidative respira-tion. MFN2 mutations reduce mitochondrial membrane potential and induce partial uncoupling of respiration, reducing ATP production. This process appears to be compensated by a higher respiratory rate, although in normal conditions of cell culture, i.e. in the presence of glucose, respiratory complex activities were found to be normal in fibroblasts from CMT2A patients (Loiseau et al. 2007). The mito-chondrial phenotype and energetic metabolism of such fibroblasts cultured in a galactose medium have not yet been investigated, but MFN2 mutations may induce subtle defects under restrictive oxidative metabolism.
The effect of silencing MFN2, studied in L6E9 myotubes, revealed increased complex II activity and a shift of mitochondrial substrate consumption with a reduced input of pyruvate, glucose, and fatty acids into mitochondrial fuelling. The amounts of the respiratory complexes present, except for complex IV, appear to be linked to the integrity of the first 600 amino acids of MFN2, independently of the fusion activity or attachment of the protein to the OMM (Pich et al. 2005). In addition, the level of expression of ANT3, an adenine nucleotide translocase embedded in the IMM, depends on the integrity of MFN2, suggesting that in parallel to its fusion activity, MFN2 could modulate the mitochondrial ATP/ADP exchanges (Guillet et al. 2010). These findings suggest that MFN2 has an indirect regulatory function in modulating gene expression, uncoupled from its fusion activity in the mitochondrial network. This suggests that MFN2 contributes to a regulatory cross-talk pathway linking mitochondrial dynamics to nuclear gene expression.
Studies on dorsal root ganglion cells (DRG), expressing the mutated forms of MFN2, have revealed normal ATP levels and oxidative respiration, although the mitochondrial network appeared to be affected with fragmented and aggregated mitochondria surrounding the nucleus (Baloh et al. 2007). These results further illustrate the dichotomy of MFN2 functions, making it critical to choose appro-priate cellular models for studying the pathophysiological mechanisms of MFN2 mutations.
6.3.5 Consequences on the Integrity of MtDNA
It has been recently demonstrated that mitochondrial fusion plays an important role in the maintenance of the integrity of the mitochondrial genome. Indeed, studies of muscle biopsies from patients with syndromic DOA revealed the presence of mtDNA deletions; however, such deletions were not found in fibroblasts from these

1836 Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
patients (Amati-Bonneau et al. 2008; Hudson et al. 2008). The characterization of the deletion boundaries showed that the deletions were similar to those observed in other disorders associated with mtDNA deletions, suggesting a common mecha-nism underlying the instability of mtDNA. Another study found similar mtDNA deletions in patients with non-syndromic DOA, suggesting that there may be no correlation between the clinical presentation of DOA and the presence of mtDNA deletions (Yu-Wai-Man et al. 2010c).
One study indicated that lymphocytes from OPA1 patients contained smaller amounts of mtDNA than controls (Kim et al. 2005). However, this finding was not confirmed in any other studies involving fibroblasts from OPA1 patients or OPA1 mouse models. In contrast, a recent study revealed that the amount of mtDNA increased two to fourfold in skeletal fibers with even higher levels of amplification in “DOA+” samples (Yu-Wai-Man et al. 2010c). More importantly, in addition to the increased mtDNA copy number, the ratio of wild-type mitochondrial genome versus mutant genome decreased, supporting the hypothesis that the inhibition of mitochondrial dynamics may somehow favor mutant over wild-type mtDNA proliferation (Malena et al. 2009).
Few studies have addressed the consequences of MFN2 mutations on mtDNA integrity. To date, two reports on fibroblasts from CMT2A patients have suggested that mtDNA content and integrity are unaffected by pathological MFN2 alleles (Amiott et al. 2008; Guillet et al. 2010); however, as in the case of fibroblasts from OPA1 patients, the fibroblasts from CMT2A patients may not offer the best approach for the investigation of mtDNA integrity. Since MFN2 is not uniformly expressed in all tissues, we cannot rule out the contribution of neuronal mtDNA deletions to the pathophysiological mechanism of CMT2A, particularly in syndro-mic cases. Indeed, the deletion of both mitofusins in mouse muscle has drastic effects on the quantitative and qualitative integrity of mtDNA, highlighting the importance of mitofusins in mtDNA maintenance (Chen et al. 2010).
6.3.6 Mouse Models of DOA and CMT2A
The development and investigation of animal models of DOA and CMT2A are of critical importance to gain deep insights in the pathophysiological mechanisms of these diseases. Indeed, in vitro primary cultures of RGCs or peripheral neurons fail to reproduce several of the main constraints imposed on these cells under physio-logical conditions. For example, in the eye the RGC axons are unmyelinated and are constantly solicited for the transduction of visual information. Moreover, these axons are subjected to the assaults of the intra-ocular pressure and exposure to daylight, which generate oxidative stress. Similarly, the peripheral neurons have the longest axons in the body. This means that axonal transport, including that of mitochondria, as well as the energy distribution must be maintained at a high level of efficiency to avoid jeopardizing cell survival. Thus, only animal models of DOA and CMT2A can satisfactorily reproduce these neuronal specificities, allowing the chronological

184 G. Lenaers et al.
follow-up of the impact of the disease on mitochondrial pathophysiology, cell survival and neuronal functionality.
Two OPA1 mouse models have been created from an N-ethyl-N-nitrosurea (ENU) mutagenized library, each encoding a protein with a truncation in the GTPase domain. Molecular analysis has revealed that the mutated allele is counter-selected so that haplo-insufficiency is the princeps mechanism driven by these mutations (Alavi et al. 2007; Davies et al. 2007). Visual assessment tests were performed in order to charac-terize the clinical phenotype of these animals, but no significant alteration of the electroretinogram was observed, whatever the age of the animals. In contrast, a reduc-tion of amplitude of the scotopic, but not the photopic, visual evoked potential (VEP) was detected in the older animals, reflecting an altered transduction of visual informa-tion from the retina to the brain (Heiduschka et al. 2010). This reduction of the VEP followed a decrease in visual acuity detected in 12-month-old mice by means of an optokinetic drum. The absence of any delay in the VEP suggests that RGC dysfunction runs parallel to the phenomenon of apoptosis, which progresses from the soma to the synapse. Histological examination of the retina revealed a decrease in the dendritic length of the RGC-ON subpopulation, but not that of the RGC-OFF subpopulation (Williams et al. 2010). Since the RGC-ON neurons contact bipolar cells in sublamina b of the inferior parietal lobule, the decreased dendritic length may reflect retarded dendritic outgrowth due to defective mitochondrial transport. Moreover, autophago-somes have been found in the RGC layer of DOA mouse models, attesting the autophagy of RGCs and the clearance of apoptotic by-products (White et al. 2009).
In the optic nerve of older DOA mouse models, some abnormal myelination was observed beyond the lamina cribrosa, with an increase in the number of microglial cells together with the loss of the largest axons. In parallel, alterations of the IMM structure were observed in the OPA1(+/−) animals with greater numbers of cristae, forming vesicule-like structures. However, no abnormalities were reported in the mitochondrial network or abundance of the organelles, and no mtDNA deletions were found, but whether the mitochondrial activity is altered in these models has yet to be determined.
Surprisingly, some secondary clinical, neurological and neuromuscular symptoms were observed, although in a very mild presentation. Thus, locomotor activity was reduced and some tremor was observed in the older mouse models, but no hearing loss was detected. Similarly, although food intake was normal during life, lower weight and less body fat were found in old OPA1 mice, possibly reflecting the lasting uncoupling of mitochondrial respiratory activity (Alavi et al. 2009).
No MFN2 knock-in animal models have yet been developed. However, two transgenic mouse strains, expressing a pathological MFN2 allele only in neurons, have been developed using neuron-specific promoters. One of these mouse strains has a mutation in the GTPase domain (T105 M) (Detmer et al. 2008), and the other has a mutation upstream of the GTPase domain (R94Q) (Cartoni et al. 2010), cor-responding to two commonest mutations found in CMT2A patients. Both mouse models present locomotor impairment and hindleg gait defects, mimicking the major syndrome of the disease. Nevertheless, the phenotype and its severity result from a subtle but significant difference in the level of expression of the pathological

1856 Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
alleles. Indeed, the expression of the T105M allele induces a dose-dependent gradient of mitochondria, with several aggregates around the nucleus and clusters in the proximal axonal region, but few in the distal region. Conversely, the expression of R94Q leads to an increase in the number of mitochondria, particularly in the distal part of axons of small diameter, thus reproducing some of the findings in CMT2A patients and suggesting that the underlying pathophysiological mechanisms may be similar. In parallel, the T105M expression induced a reduction of the number of motoneuron axons, whereas the number of small axons increased in the R94Q mouse model (Cartoni et al. 2010; Detmer et al. 2008). Thus, both models mimic the clinical presentation of CMT2A but possibly with different pathophysiological mechanisms.
6.4 Therapeutic Strategies for DOA and CMT2A
DOA and CMT2A remain orphan pathologies with no currently available treatment. As in the case of several other genetic diseases, the two therapeutic approaches that may be envisaged are based on gene therapy and pharmacological therapy. The direct involvement of OPA1 in the function and regulation of mitochondrial net-works in patients with DOA, and that of MFN2 in patients with CMT2A, suggests some promising therapeutic strategies.
6.4.1 Gene Therapy
The theory of gene therapy is fairly straightforward. It states that if a pathological condition is due to the presence of reduced quantities of a certain protein then the normal protein content may be restored by using an appropriate vector carrying the transgene encoding the protein. Could this theory have a practical application in diseases such as DOA and CMT2A?
In most cases of DOA, the disease is caused by OPA1 haplo-insufficiency. Moreover, RGC somas, which form the innermost layer of the retina facing the vitreous humor, are readily accessible to micro-injections into the vitreous humor of the eye globe. Thus, microinjections of a vector with an affinity for RGCs and encoding the OPA1 gene should enhance the protein content and restore its full activity in the mitochondrial network of the target cells. However, this attractive strategy remains conditioned by the possibility of controlling the ectopic pro-duction of OPA1 since overexpression of this protein might be more deleterious than beneficial. In addition, it would be of capital importance to choose the appropriate OPA1 isoform to be expressed, since all eight isoforms of OPA1 are expressed in the RGCs. The tools required to address these problems have now been assembled and the first pre-clinical trials on mouse models should soon be under way.

186 G. Lenaers et al.
In contrast to the therapeutic strategy outlined for DOA, CMT2A offers a much greater challenge. Since the disease is caused by the dominant negative effect of the mutated MFN2 protein on the wild-type protein, it is far from obvious that the pathological phenotype would be rescued by the ectopic expression of MFN2. Indeed, fibroblast studies have shown that the overexpression of MFN2 does not in itself complement the mutated MFN2, whereas that of MFN1 does so by forming hetero-oligomeric complexes with the mutated MFN2 protein (Detmer and Chan 2007). The specific effects of the overexpression of MFN1 and MFN2 in peripheral nerves expressing mutated MFN2 have yet to be investigated. Another important aspect that needs to be considered is the delivery of the therapeutic vectors to the soma of the peripheral neurons affected in CMT2A. It would obviously be difficult to design a practical strategy that would allow the exhaustive transduction of a vector to neurons located all along the spinal cord and in the dorsal root ganglia. Furthermore, the current mouse models of CMT2A were obtained by transgenesis of a mutated allele, rather than by knocking-in the MFN2 gene. For all these reasons, we are still far from the point of testing the benefits of gene therapy for CMT2A patients.
6.4.2 Pharmacological Therapy
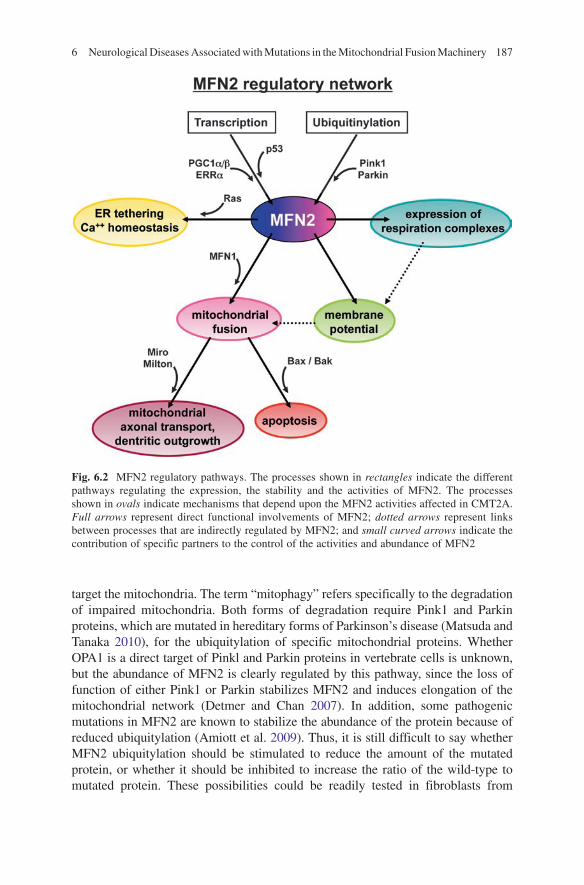
Pharmacological therapy offers an interesting alternative to gene therapy. Although no pharmacological therapy could be expected to correct gene mutations, it may prove effective in dealing with various mechanisms affected by these genetic diseases. In particular, it would be important to elucidate the factors involved in the production and function of OPA1 in DOA, and MFN2 in CMT2A, so as to deter-mine how their activities could be appropriately modulated by drugs. The pathways regulating the production and function of MFN2 (Fig. 6.2) and OPA1 (Fig. 6.3) suggest the eventuality of novel therapeutic approaches to the treatment of CMT2A and DOA.
Whereas little is known about the regulation of the expression of the OPA1 gene, three pathways have been identified in skeletal muscle for the regulation of the MFN2 gene. They involve PGC1a and PGC1b, which are PPARg co-activators, the estrogen-related receptor a, which acts in synergy with PPARg and the anti-tumoral protein p53; all of these induce the transcription of MFN2 by binding to its promoter (Wang et al. 2010; Zorzano 2009). It may therefore be hypothesized that drugs activating these pathways, such as glitazones, resveratrol and estrogen mimetics, could increase the abundance of MFN2 in peripheral neurons. However, it is not clear whether the increase of mutated as well as wild-type MFN2 proteins would exert a positive or negative effect on the basic functions of the protein. It further remains to be demonstrated that these pathways are active in peripheral neurons, and that they can be stimulated through oral treatment.
A second pathway regulates the turnover of OPA1 and MFN2 according to their rate of degradation. The degradation may target a particular protein, or globally
1876 Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
Fig. 6.2 MFN2 regulatory pathways. The processes shown in rectangles indicate the different pathways regulating the expression, the stability and the activities of MFN2. The processes shown in ovals indicate mechanisms that depend upon the MFN2 activities affected in CMT2A. Full arrows represent direct functional involvements of MFN2; dotted arrows represent links between processes that are indirectly regulated by MFN2; and small curved arrows indicate the contribution of specific partners to the control of the activities and abundance of MFN2
target the mitochondria. The term “mitophagy” refers specifically to the degradation of impaired mitochondria. Both forms of degradation require Pink1 and Parkin proteins, which are mutated in hereditary forms of Parkinson’s disease (Matsuda and Tanaka 2010), for the ubiquitylation of specific mitochondrial proteins. Whether OPA1 is a direct target of Pinkl and Parkin proteins in vertebrate cells is unknown, but the abundance of MFN2 is clearly regulated by this pathway, since the loss of function of either Pink1 or Parkin stabilizes MFN2 and induces elongation of the mitochondrial network (Detmer and Chan 2007). In addition, some pathogenic mutations in MFN2 are known to stabilize the abundance of the protein because of reduced ubiquitylation (Amiott et al. 2009). Thus, it is still difficult to say whether MFN2 ubiquitylation should be stimulated to reduce the amount of the mutated protein, or whether it should be inhibited to increase the ratio of the wild-type to mutated protein. These possibilities could be readily tested in fibroblasts from

188 G. Lenaers et al.
CMT2A patients by means of drugs or siRNA targeting the Parkin E3-ubiquitin ligase, and evaluating MFN2 turnover, the mitochondrial network structure and respiration parameters.
Mitophagy follows a quality-control process, called “kiss-and-run”, which evaluates the functionality of mitochondria by assessing their membrane potential (Twig et al. 2008). OPA1 is required for this process, and its downregulation in DOA could affect its sensor activity, leading to the progressive accumulation of impaired mitochondria (Liu et al. 2009). Thus, stabilizing the amount of OPA1 or increasing its expression could favor the maintenance of efficient mitochondria and sustain the perennity of their distribution.
A third pathway controlling the activities of OPA1 and MFN2 corresponds to the post-translational modifications of these proteins. It is not yet clear whether MFN2 undergoes other modifications than the ubiquitinylation that controls its turn-over. But the involvement of OPA1 in mitochondrial fusion is clearly regulated by an amino-terminal cleavage that in physiological conditions affects about 50% of all isoforms, except for those, including the alternate-spliced exon 4b, which are 100%
Fig. 6.3 OPA1 regulatory pathways. The processes shown in rectangles indicate the different pathways regulating the expression, the stability and the activities of OPA1. The processes shown in ovals indicate mechanisms that depend upon the OPA1 activities affected in DOA. Full arrows represent direct functional involvements of OPA1; dotted arrows represent links between pro-cesses that are indirectly regulated by OPA1; and small curved arrows indicate the contribution of specific partners to the control of the activities and abundance of OPA1

1896 Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
cleaved (Song et al. 2007). Under conditions of stress, all OPA1 isoforms undergo cleavage (Baricault et al. 2007; Guillery et al. 2008) leading to a markedly frag-mented mitochondrial network and increasing susceptibility to apoptosis (Griparic et al. 2007). Thus, the reduction of OPA1 cleavage, through the modulation of the inner membrane proteases, YME1L (Song et al. 2007) and OMA1 (Ehses et al. 2009; Head et al. 2009), involved in this proteolytic process, should offer an interesting approach towards increasing the resistance to apopotic stimuli. This could further be promoted by stimulating the activity of the Phb1 and Phb2 prohibitins, which control the proteases involved in OPA1 processing (Merkwirth et al. 2008).
The three pathways described above sustain OPA1 and MFN2 activity by increas-ing their active amounts in mitochondria. However, the drugs capable of acting on these pathways are far from being adequately characterized. Other strategies for the treatment of DOA and CMT2A diseases may rely on products acting on the down-stream mitochondrial activities affected by OPA1 and MFN2 mutations. Thus, patients may expect to benefit from strategies, compatible with chronic treatment, that would stimulate mitochondrial fusion, foster mitochondrial transport, restore membrane potential, boost mitochondrial respiration and ATP production, promote calcium homeostasis, and inhibit the apoptotic process. Drugs targeting these pro-cesses remain to be characterized, but the methodology that would enable their identification is now available. High-throughput drug screening on fibroblasts from DOA and CMT2A patients would help to determine the most potent drugs for these diseases. Further testing would allow identification of the precise targets and path-ways stimulated or inhibited by these drugs. Furthermore, the drugs identified as potentially active against DOA and CMT2A would probably also prove useful for the treatment of other mitochondrial diseases.
To conclude this section on therapeutic strategies, it should be mentioned that prosthetic therapy using a cochlear implant has been successfully used to treat neurosensorial deafness in syndromic DOA patients with the OPA1-R445H muta-tion, leading to marked auditive improvement (Huang et al. 2009). This prosthetic approach, unfortunately not applicable in the optic and peripheral neuropathies in cases of DOA and CMT2A, could nevertheless be used to treat the neurosensorial deafness of syndromic patients with either disease.
Acknowledgements We are grateful to Kanaya Malkani for critical reading and comments on the manuscript. Our work is supported by INSERM, the University Hospital of Angers (PHRC 04–12), the University of Angers, France; and by grants from the following patients’ associations: “Association contre les Maladies Mitochondriales (AMMi)”, “Ouvrir les Yeux (OLY)”, “Retina France” and “Union Nationale des Aveugles et Déficients Visuels (UNADEV)”.
References
Alavi, M. V., Bette, S., Schimpf, S., Schuettauf, F., Schraermeyer, U., Wehrl, H. F., Ruttiger, L., Beck, S. C., Tonagel, F., Pichler, B. J., Knipper, M., Peters, T., Laufs, J. & Wissinger, B. (2007) A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain, 130, 1029–42.

190 G. Lenaers et al.
Alavi, M. V., Fuhrmann, N., Nguyen, H. P., Yu-Wai-man, P., Heiduschka, P., Chinnery, P. F. & Wissinger, B. (2009) Subtle neurological and metabolic abnormalities in an Opa1 mouse model of autosomal dominant optic atrophy. Exp Neurol, 220, 404–9.
Alexander, C., Votruba, M., Pesch, U. E., Thiselton, D. L., Mayer, S., Moore, A., Rodriguez, M., Kellner, U., Leo-Kottler, B., Auburger, G., Bhattacharya, S. S. & Wissinger, B. (2000) OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet, 26, 211–5.
Amati-bonneau, P., Guichet, A., Olichon, A., Chevrollier, A., Viala, F., Miot, S., Ayuso, C., Odent, S., Arrouet, C., Verny, C., Calmels, M. N., Simard, G., Belenguer, P., Wang, J., Puel, J. L., Hamel, C., Malthiery, Y., Bonneau, D., Lenaers, G. & Reynier, P. (2005) OPA1 R445H muta-tion in optic atrophy associated with sensorineural deafness. Ann Neurol, 58, 958–63.
Amati-bonneau, P., Odent, S., Derrien, C., Pasquier, L., Malthiery, Y., Reynier, P. & Bonneau, D. (2003) The association of autosomal dominant optic atrophy and moderate deafness may be due to the R445H mutation in the OPA1 gene. Am J Ophthalmol, 136, 1170–1.
Amati-bonneau, P., Valentino, M. L., Reynier, P., Gallardo, M. E., Bornstein, B., Boissiere, A., Campos, Y., Rivera, H., De la Aleja, J. G., Carroccia, R., Iommarini, L., Labauge, P., Figarella-branger, D., Marcorelles, P., Furby, A., Beauvais, K., Letournel, F., Liguori, R., La Morgia, C., Montagna, P., Liguori, M., Zanna, C., Rugolo, M., Cossarizza, A., Wissinger, B., Verny, C., Schwarzenbacher, R., Martin, M. A., Arenas, J., Ayuso, C., Garesse, R., Lenaers, G., Bonneau, D. & Carelli, V. (2008) OPA1 mutations induce mitochondrial DNA instability and optic atro-phy ‘plus’ phenotypes. Brain, 131, 338–51.
Amiott, E. A., Cohen, M. M., Saint-georges, Y., Weissman, A. M. & Shaw, J. M. (2009) A mutation associated with CMT2A neuropathy causes defects in Fzo1 GTP hydrolysis, ubiquitylation, and protein turnover. Mol Biol Cell, 20, 5026–35.
Amiott, E. A., Lott, P., Soto, J., Kang, P. B., Mccaffery, J. M., Dimauro, S., Abel, E. D., Flanigan, K. M., Lawson, V. H. & Shaw, J. M. (2008) Mitochondrial fusion and function in Charcot-Marie-Tooth type 2A patient fibroblasts with mitofusin 2 mutations. Exp Neurol, 211, 115–27.
Baloh, R. H., Schmidt, R. E., Pestronk, A. & Milbrandt, J. (2007) Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci, 27, 422–30.
Ban, T., Heymann, J. A., Song, Z., Hinshaw, J. E. & Chan, D. C. (2010) OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum Mol Genet, 19, 2113–22.
Barbet, F., Hakiki, S., Orssaud, C., Gerber, S., Perrault, I., Hanein, S., Ducroq, D., Dufier, J. L., Munnich, A., Kaplan, J. & Rozet, J. M. (2005) A third locus for dominant optic atrophy on chromosome 22q. J Med Genet, 42, e1.
Baricault, L., Segui, B., Guegand, L., Olichon, A., Valette, A., Larminat, F. & Lenaers, G. (2007) OPA1 cleavage depends on decreased mitochondrial ATP level and bivalent metals. Exp Cell Res, 313, 3800–8.
Boaretto, F., Vettori, A., Casarin, A., Vazza, G., Muglia, M., Rossetto, M. G., Cavallaro, T., Rizzuto, N., Carelli, V., Salviati, L., Mostacciuolo, M. L. & Martinuzzi, A. (2010) Severe CMT type 2 with fatal encephalopathy associated with a novel MFN2 splicing mutation. Neurology, 74, 1919–21.
Braathen, G. J., Sand, J. C., Lobato, A., Hoyer, H. & Russell, M. B. (2010) MFN2 point mutations occur in 3.4% of Charcot-Marie-Tooth families. An investigation of 232 Norwegian CMT families. BMC Med Genet, 11, 48.
Calvo, J., Funalot, B., Ouvrier, R. A., Lazaro, L., Toutain, A., de Mas, P., Bouche, P., Gilbert-dussardier, B., Arne-bes, M. C., Carriere, J. P., Journel, H., Minot-myhie, M. C., Guillou, C., Ghorab, K., Magy, L., Sturtz, F., Vallat, J. M. & Magdelaine, C. (2009) Genotype-phenotype correlations in Charcot-Marie-Tooth disease type 2 caused by mitofusin 2 mutations. Arch Neurol, 66, 1511–6.
Carelli, V., Ross-cisneros, F. N. & Sadun, A. A. (2004) Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res, 23, 53–89.

1916 Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
Cartoni, R., Arnaud, E., Medard, J. J., Poirot, O., Courvoisier, D. S., Chrast, R. & Martinou, J. C. (2010) Expression of mitofusin 2(R94Q) in a transgenic mouse leads to Charcot-Marie-Tooth neuropathy type 2A. Brain, 133, 1460–9.
Cartoni, R. & Martinou, J. C. (2009) Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp Neurol, 218, 268–73.
Charcot, J. & Marie, P. (1886) Sur une forme particulière d’atrophie musculaire progressive sou-vent familiale débutant par les pieds et les jambes et atteignant plus tard les mains. Revue de Médecine, 6, 97–138.
Chen, H. & Chan, D. C. (2010) Physiological functions of mitochondrial fusion. Ann N Y Acad Sci, 1201, 21–5.
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E. & Chan, D. C. (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol, 160, 189–200.
Chen, H., Vermulst, M., Wang, Y. E., Chomyn, A., Prolla, T. A., Mccaffery, J. M. & Chan, D. C. (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell, 141, 280–9.
Chevrollier, A., Guillet, V., Loiseau, D., Gueguen, N., de Crescenzo, M. A., Verny, C., Eng, M. F., Dollfus, H., Odent, S., Milea, D., Goizet, C., Amati-bonneau, P., Procaccio, V., Bonneau, D. & Reynier, P. (2008) Hereditary optic neuropathies share a common mitochondrial coupling defect. Ann Neurol, 63, 794–8.
Cohn, A. C., Toomes, C., Potter, C., Towns, K. V., Hewitt, A. W., Inglehearn, C. F., Craig, J. E. & Mackey, D. A. (2007) Autosomal dominant optic atrophy: penetrance and expressivity in patients with OPA1 mutations. Am J Ophthalmol, 143, 656–62.
Cornille, K., Milea, D., Amati-bonneau, P., Procaccio, V., Zazoun, L., Guillet, V., El Achouri, G., Delettre, C., Gueguen, N., Loiseau, D., Muller, A., Ferre, M., Chevrollier, A., Wallace, D. C., Bonneau, D., Hamel, C., Reynier, P. & Lenaers, G. (2008) Reversible optic neuropathy with OPA1 exon 5b mutation. Ann Neurol, 63, 667–71.
Davies, V. J., Hollins, A. J., Piechota, M. J., Yip, W., Davies, J. R., White, K. E., Nicols, P. P., Boulton, M. E. & Votruba, M. (2007) Opa1 deficiency in a mouse model of autosomal domi-nant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual func-tion. Hum Mol Genet, 16, 1307–18.
Davies, V. J., Powell, K. A., White, K. E., Yip, W., Hogan, V., Hollins, A. J., Davies, J. R., Piechota, M., Brownstein, D. G., Moat, S. J., Nichols, P. P., Wride, M. A., Boulton, M. E. & Votruba, M. (2008) A missense mutation in the murine Opa3 gene models human Costeff syndrome. Brain, 131, 368–80.
Dayanithi, G., Chen-kuo-chang, M., Viero, C., Hamel, C., Muller, A. & Lenaers, G. (2010) Characterization of Ca2+ signalling in postnatal mouse retinal ganglion cells: involvement of OPA1 in Ca2+ clearance. Ophthalmic Genet, 31, 53–65.
de Brito, O. M. & Scorrano, L. (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature, 456, 605–10.
Del Bo, R., Moggio, M., Rango, M., Bonato, S., D’angelo, M. G., Ghezzi, S., Airoldi, G., Bassi, M. T., Guglieri, M., Napoli, L., Lamperti, C., Corti, S., Federico, A., Bresolin, N. & Comi, G. P. (2008) Mutated mitofusin 2 presents with intrafamilial variability and brain mitochondrial dysfunction. Neurology, 71, 1959–66.
Delettre, C., Griffoin, J. M., Kaplan, J., Dollfus, H., Lorenz, B., Faivre, L., Lenaers, G., Belenguer, P. & Hamel, C. P. (2001) Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet, 109, 584–91.
Delettre, C., Lenaers, G., Griffoin, J. M., Gigarel, N., Lorenzo, C., Belenguer, P., Pelloquin, L., Grosgeorge, J., Turc-carel, C., Perret, E., Astarie-dequeker, C., Lasquellec, L., Arnaud, B., Ducommun, B., Kaplan, J. & Hamel, C. P. (2000) Nuclear gene OPA1, encoding a mitochon-drial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet, 26, 207–10.
Detmer, S. A. & Chan, D. C. (2007) Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol, 176, 405–14.

192 G. Lenaers et al.
Detmer, S. A., Vande Velde, C., Cleveland, D. W. & Chan, D. C. (2008) Hindlimb gait defects due to motor axon loss and reduced distal muscles in a transgenic mouse model of Charcot-Marie-Tooth type 2A. Hum Mol Genet, 17, 367–75.
Ehses, S., Raschke, I., Mancuso, G., Bernacchia, A., Geimer, S., Tondera, D., Martinou, J. C., Westermann, B., Rugarli, E. I. & Langer, T. (2009) Regulation of OPA1 processing and mito-chondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol, 187, 1023–36.
Eiberg, H., Kjer, B., Kjer, P. & Rosenberg, T. (1994) Dominant optic atrophy (OPA1) mapped to chromosome 3q region. I. Linkage analysis. Hum Mol Genet, 3, 977–80.
Eura, Y., Ishihara, N., Yokota, S. & Mihara, K. (2003) Two mitofusin proteins, mammalian homo-logues of FZO, with distinct functions are both required for mitochondrial fusion. J Biochem, 134, 333–44.
Ferre, M., Amati-bonneau, P., Tourmen, Y., Malthiery, Y. & Reynier, P. (2005) eOPA1: an online database for OPA1 mutations. Hum Mutat, 25, 423–8.
Ferre, M., Bonneau, D., Milea, D., Chevrollier, A., Verny, C., Dollfus, H., Ayuso, C., Defoort, S., Vignal, C., Zanlonghi, X., Charlin, J. F., Kaplan, J., Odent, S., Hamel, C. P., Procaccio, V., Reynier, P. & Amati-bonneau, P. (2009) Molecular screening of 980 cases of suspected heredi-tary optic neuropathy with a report on 77 Novel OPA1 mutations. Hum Mutat, 30, E692–E705.
Fuhrmann, N., Alavi, M. V., Bitoun, P., Woernle, S., Auburger, G., Leo-kottler, B., Yu-wai-man, P., Chinnery, P. & Wissinger, B. (2009) Genomic rearrangements in OPA1 are frequent in patients with autosomal dominant optic atrophy. J Med Genet, 46, 136–44.
Fuhrmann, N., Schimpf, S., Kamenisch, Y., Leo-kottler, B., Alexander, C., Auburger, G., Zrenner, E., Wissinger, B. & Alavi, M. V. (2010) Solving a 50 year mystery of a missing OPA1 muta-tion: more insights from the first family diagnosed with autosomal dominant optic atrophy. Mol Neurodegener, 5, 25.
Griparic, L., Kanazawa, T. & Van der Bliek, A. M. (2007) Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol, 178, 757–64.
Guillery, O., Malka, F., Landes, T., Guillou, E., Blackstone, C., Lombes, A., Belenguer, P., Arnoult, D. & Rojo, M. (2008) Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol Cell, 100, 315–25.
Guillet, V., Gueguen, N., Verny, C., Ferre, M., Homedan, C., Loiseau, D., Procaccio, V., Amati-bonneau, P., Bonneau, D., Reynier, P. & Chevrollier, A. (2010) Adenine nucleotide translocase is involved in a mitochondrial coupling defect in MFN2-related Charcot-Marie-Tooth type 2A disease. Neurogenetics, 11, 127–33.
Head, B., Griparic, L., Amiri, M., Gandre-babbe, S. & Van der Bliek, A. M. (2009) Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol, 187, 959–66.
Heiduschka, P., Schnichels, S., Fuhrmann, N., Hofmeister, S., Schraermeyer, U., Wissinger, B. & Alavi, M. V. (2010) Electrophysiological and histologic assessment of retinal ganglion cell fate in a mouse model for OPA1-associated autosomal dominant optic atrophy. Invest Ophthalmol Vis Sci, 51, 1424–31.
Honda, S., Aihara, T., Hontani, M., Okubo, K. & Hirose, S. (2005) Mutational analysis of action of mitochondrial fusion factor mitofusin-2. J Cell Sci, 118, 3153–61.
Hoyt, C. S. (1980) Autosomal dominant optic atrophy. A spectrum of disability. Ophthalmology, 87, 245–51.
Huang, P., Yu, T. & Yoon, Y. (2007) Mitochondrial clustering induced by overexpression of the mitochondrial fusion protein Mfn2 causes mitochondrial dysfunction and cell death. Eur J Cell Biol, 86, 289–302.
Huang, T., Santarelli, R. & Starr, A. (2009) Mutation of OPA1 gene causes deafness by affecting function of auditory nerve terminals. Brain Res, 1300, 97–104.
Hudson, G., Amati-bonneau, P., Blakely, E. L., Stewart, J. D., He, L., Schaefer, A. M., Griffiths, P. G., Ahlqvist, K., Suomalainen, A., Reynier, P., Mcfarland, R., Turnbull, D. M., Chinnery, P. F. & Taylor, R. W. (2008) Mutation of OPA1 causes dominant optic atrophy with external oph-thalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain, 131, 329–37.

1936 Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
Ishihara, N., Eura, Y. & Mihara, K. (2004) Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci, 117, 6535–46.
Johnston, P. B., Gaster, R. N., Smith, V. C. & Tripathi, R. C. (1979) A clinicopathologic study of autosomal dominant optic atrophy. Am J Ophthalmol, 88, 868–75.
Kamei, S., Chen-kuo-chang, M., Cazevieille, C., Lenaers, G., Olichon, A., Belenguer, P., Roussignol, G., Renard, N., Eybalin, M., Michelin, A., Delettre, C., Brabet, P. & Hamel, C. P. (2005) Expression of the Opa1 mitochondrial protein in retinal ganglion cells: its downregulation causes aggregation of the mitochondrial network. Invest Ophthalmol Vis Sci, 46, 4288–94.
Ke, T., Nie, S. W., Yang, Q. B., Liu, J. P., Zhou, L. N., Ren, X., Liu, J. Y., Wang, Q. & Liu, M. G. (2006) The G401D mutation of OPA1 causes autosomal dominant optic atrophy and hearing loss in a Chinese family. Zhonghua Yi Xue Yi Chuan Xue Za Zhi, 23, 481–5.
Kerrison, J. B., Koenekoop, R. K., Arnould, V. J., Zee, D. & Maumenee, I. H. (1998) Clinical features of autosomal dominant congenital nystagmus linked to chromosome 6p12. Am J Ophthalmol, 125, 64–70.
Kijima, K., Numakura, C., Izumino, H., Umetsu, K., Nezu, A., Shiiki, T., Ogawa, M., Ishizaki, Y., Kitamura, T., Shozawa, Y. & Hayasaka, K. (2005) Mitochondrial GTPase mitofusin 2 muta-tion in Charcot-Marie-Tooth neuropathy type 2A. Hum Genet, 116, 23–7.
Kim, J. Y., Hwang, J. M., Ko, H. S., Seong, M. W., Park, B. J. & Park, S. S. (2005) Mitochondrial DNA content is decreased in autosomal dominant optic atrophy. Neurology, 64, 966–72.
Kivlin, J. D., Lovrien, E. W., Bishop, D. T. & Maumenee, I. H. (1983) Linkage analysis in domi-nant optic atrophy. Am J Hum Genet, 35, 1190–5.
Kjer, B., Eiberg, H., Kjer, P. & Rosenberg, T. (1996) Dominant optic atrophy mapped to chromo-some 3q region. II. Clinical and epidemiological aspects. Acta Ophthalmol Scand, 74, 3–7.
Kjer, P. (1959) Infantile optic atrophy with dominant mode of inheritance: a clinical and genetic study of 19 Danish families. Acta Ophthalmol Suppl, 164, 1–147.
Kjer, P., Jensen, O. A. & Klinken, L. (1983) Histopathology of eye, optic nerve and brain in a case of dominant optic atrophy. Acta Ophthalmol (Copenh), 61, 300–12.
Lawson, V. H., Graham, B. V. & Flanigan, K. M. (2005) Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology, 65, 197–204.
Li, C., Kosmorsky, G., Zhang, K., Katz, B. J., Ge, J. & Traboulsi, E. I. (2005) Optic atrophy and sensorineural hearing loss in a family caused by an R445H OPA1 mutation. Am J Med Genet A, 138A, 208–11.
Liu, X., Weaver, D., Shirihai, O. & Hajnoczky, G. (2009) Mitochondrial ‘kiss-and-run’: interplay between mitochondrial motility and fusion-fission dynamics. Embo J, 28, 3074–89.
Lodi, R., Tonon, C., Valentino, M. L., Iotti, S., Clementi, V., Malucelli, E., Barboni, P., Longanesi, L., Schimpf, S., Wissinger, B., Baruzzi, A., Barbiroli, B. & Carelli, V. (2004) Deficit of in vivo mito-chondrial ATP production in OPA1-related dominant optic atrophy. Ann Neurol, 56, 719–23.
Lodi, R., Tonon, C., Valentino, M. L., Manners, D., Testa, C., Malucelli, E., La Morgia, C., Barboni, P., Carbonelli, M., Schimpf, S., Wissinger, B., Zeviani, M., Baruzzi, A., Liguori, R., Barbiroli, B. & Carelli, V. (2010) Defective Mitochondrial Adenosine Triphosphate Production in Skeletal Muscle From Patients With Dominant Optic Atrophy Due to OPA1 Mutations. Arch Neurol.
Loiseau, D., Chevrollier, A., Verny, C., Guillet, V., Gueguen, N., Pou de Crescenzo, M. A., Ferre, M., Malinge, M. C., Guichet, A., Nicolas, G., Amati-bonneau, P., Malthiery, Y., Bonneau, D. & Reynier, P. (2007) Mitochondrial coupling defect in Charcot-Marie-Tooth type 2A disease. Ann Neurol, 61, 315–23.
Lunkes, A., Hartung, U., Magarino, C., Rodriguez, M., Palmero, A., Rodriguez, L., Heredero, L., Weissenbach, J., Weber, J. & Auburger, G. (1995) Refinement of the OPA1 gene locus on chromosome 3q28-q29 to a region of 2-8 cM, in one Cuban pedigree with autosomal dominant optic atrophy type Kjer. Am J Hum Genet, 57, 968–70.
Lyle, W. (1990) Genetic Risk. University of Waterloo Press.Malena, A., Loro, E., Di Re, M., Holt, I. J. & Vergani, L. (2009) Inhibition of mitochondrial
fission favours mutant over wild-type mitochondrial DNA. Hum Mol Genet, 18, 3407–16.Marchbank, N. J., Craig, J. E., Leek, J. P., Toohey, M., Churchill, A. J., Markham, A. F.,
Mackey, D. A., Toomes, C. & Inglehearn, C. F. (2002) Deletion of the OPA1 gene in a

194 G. Lenaers et al.
dominant optic atrophy family: evidence that haploinsufficiency is the cause of disease. J Med Genet, 39, e47.
Matsuda, N. & Tanaka, K. (2010) Uncovering the roles of PINK1 and parkin in mitophagy. Autophagy, 6, 952–4.
Merkwirth, C., Dargazanli, S., Tatsuta, T., Geimer, S., Lower, B., Wunderlich, F. T., Von Kleist-retzow, J. C., Waisman, A., Westermann, B. & Langer, T. (2008) Prohibitins control cell pro-liferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev, 22, 476–88.
Milea, D., Sander, B., Wegener, M., Jensen, H., Kjer, B., Jorgensen, T. M., Lund-andersen, H. & Larsen, M. (2010) Axonal loss occurs early in dominant optic atrophy. Acta Ophthalmol, 88, 342–6.
Milone, M., Younge, B. R., Wang, J., Zhang, S. & Wong, L. J. (2009) Mitochondrial disorder with OPA1 mutation lacking optic atrophy. Mitochondrion, 9, 279–81.
Misko, A., Jiang, S., Wegorzewska, I., Milbrandt, J. & Baloh, R. H. (2010) Mitofusin 2 is neces-sary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci, 30, 4232–40.
Nicholson, G. A., Magdelaine, C., Zhu, D., Grew, S., Ryan, M. M., Sturtz, F., Vallat, J. M. & Ouvrier, R. A. (2008) Severe early-onset axonal neuropathy with homozygous and compound heterozygous MFN2 mutations. Neurology, 70, 1678–81.
Nochez, Y., Arsene, S., Gueguen, N., Chevrollier, A., Ferre, M., Guillet, V., Desquiret, V., Toutain, A., Bonneau, D., Procaccio, V., Amati-bonneau, P., Pisella, P. J. & Reynier, P. (2009) Acute and late-onset optic atrophy due to a novel OPA1 mutation leading to a mitochondrial coupling defect. Mol Vis, 15, 598–608.
Olichon, A., Baricault, L., Gas, N., Guillou, E., Valette, A., Belenguer, P. & Lenaers, G. (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem, 278, 7743–6.
Olichon, A., Elachouri, G., Baricault, L., Delettre, C., Belenguer, P. & Lenaers, G. (2007a) OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell Death Differ, 14, 682–92.
Olichon, A., Emorine, L. J., Descoins, E., Pelloquin, L., Brichese, L., Gas, N., Guillou, E., Delettre, C., Valette, A., Hamel, C. P., Ducommun, B., Lenaers, G. & Belenguer, P. (2002) The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett, 523, 171–6.
Olichon, A., Landes, T., Arnaune-pelloquin, L., Emorine, L. J., Mils, V., Guichet, A., Delettre, C., Hamel, C., Amati-bonneau, P., Bonneau, D., Reynier, P., Lenaers, G. & Belenguer, P. (2007b) Effects of OPA1 mutations on mitochondrial morphology and apoptosis: relevance to ADOA pathogenesis. J Cell Physiol, 211, 423–30.
Payne, M., Yang, Z., Katz, B. J., Warner, J. E., Weight, C. J., Zhao, Y., Pearson, E. D., Treft, R. L., Hillman, T., Kennedy, R. J., Meire, F. M. & Zhang, K. (2004) Dominant optic atrophy, sensorineural hearing loss, ptosis, and ophthalmoplegia: a syndrome caused by a missense mutation in OPA1. Am J Ophthalmol, 138, 749–55.
Pesch, U. E., Leo-kottler, B., Mayer, S., Jurklies, B., Kellner, U., Apfelstedt-sylla, E., Zrenner, E., Alexander, C. & Wissinger, B. (2001) OPA1 mutations in patients with autosomal dominant optic atrophy and evidence for semi-dominant inheritance. Hum Mol Genet, 10, 1359–68.
Pich, S., Bach, D., Briones, P., Liesa, M., Camps, M., Testar, X., Palacin, M. & Zorzano, A. (2005) The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet, 14, 1405–15.
Puomila, A., Huoponen, K., Mantyjarvi, M., Hamalainen, P., Paananen, R., Sankila, E. M., Savontaus, M. L., Somer, M. & Nikoskelainen, E. (2005) Dominant optic atrophy: correlation between clinical and molecular genetic studies. Acta Ophthalmol Scand, 83, 337–46.
Reynier, P., Amati-bonneau, P., Verny, C., Olichon, A., Simard, G., Guichet, A., Bonnemains, C., Malecaze, F., Malinge, M. C., Pelletier, J. B., Calvas, P., Dollfus, H., Belenguer, P., Malthiery, Y., Lenaers, G. & Bonneau, D. (2004) OPA3 gene mutations responsible for autosomal domi-nant optic atrophy and cataract. J Med Genet, 41, e110.

1956 Neurological Diseases Associated with Mutations in the Mitochondrial Fusion Machinery
Rojo, M., Legros, F., Chateau, D. & Lombes, A. (2002) Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci, 115, 1663–74.
Schimpf, S., Fuhrmann, N., Schaich, S. & Wissinger, B. (2008) Comprehensive cDNA study and quantitative transcript analysis of mutant OPA1 transcripts containing premature termination codons. Hum Mutat, 29, 106–12.
Shimizu, S., Mori, N., Kishi, M., Sugata, H., Tsuda, A. & Kubota, N. (2003) A novel mutation in the OPA1 gene in a Japanese patient with optic atrophy. Am J Ophthalmol, 135, 256–7.
Skre, H. (1974) Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin Genet, 6, 98–118.Song, Z., Chen, H., Fiket, M., Alexander, C. & Chan, D. C. (2007) OPA1 processing controls
mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol, 178, 749–55.
Song, Z., Ghochani, M., Mccaffery, J. M., Frey, T. G. & Chan, D. C. (2009) Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell, 20, 3525–32.
Spinazzi, M., Cazzola, S., Bortolozzi, M., Baracca, A., Loro, E., Casarin, A., Solaini, G., Sgarbi, G., Casalena, G., Cenacchi, G., Malena, A., Frezza, C., Carrara, F., Angelini, C., Scorrano, L., Salviati, L. & Vergani, L. (2008) A NOVEL DELETION IN THE GTPase DOMAIN OF OPA1 CAUSES DEFECTS IN MITOCHONDRIAL MORPHOLOGY AND DISTRIBUTION, BUT NOT IN FUNCTION. Hum Mol Genet.
Stewart, J. D., Hudson, G., Yu-wai-man, P., Blakeley, E. L., He, L., Horvath, R., Maddison, P., Wright, A., Griffiths, P. G., Turnbull, D. M., Taylor, R. W. & Chinnery, P. F. (2008) OPA1 in multiple mitochondrial DNA deletion disorders. Neurology, 71, 1829–31.
Thiselton, D. L., Alexander, C., Morris, A., Brooks, S., Rosenberg, T., Eiberg, H., Kjer, B., Kjer, P., Bhattacharya, S. S. & Votruba, M. (2001) A frameshift mutation in exon 28 of the OPA1 gene explains the high prevalence of dominant optic atrophy in the Danish population: evi-dence for a founder effect. Hum Genet, 109, 498–502.
Thiselton, D. L., Alexander, C., Taanman, J. W., Brooks, S., Rosenberg, T., Eiberg, H., Andreasson, S., van Regemorter, N., Munier, F. L., Moore, A. T., Bhattacharya, S. S. & Votruba, M. (2002) A comprehensive survey of mutations in the OPA1 gene in patients with autosomal dominant optic atrophy. Invest Ophthalmol Vis Sci, 43, 1715–24.
Toomes, C., Marchbank, N. J., Mackey, D. A., Craig, J. E., Newbury-ecob, R. A., Bennett, C. P., Vize, C. J., Desai, S. P., Black, G. C., Patel, N., Teimory, M., Markham, A. F., Inglehearn, C. F. & Churchill, A. J. (2001) Spectrum, frequency and penetrance of OPA1 mutations in domi-nant optic atrophy. Hum Mol Genet, 10, 1369–78.
Tooth, H. (1886) The peroneal type of progressive muscular atrophy. HK Lewis, London.Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., Stiles, L., Haigh,
S. E., Katz, S., Las, G., Alroy, J., Wu, M., Py, B. F., Yuan, J., Deeney, J. T., Corkey, B. E. & Shirihai, O. S. (2008) Fission and selective fusion govern mitochondrial segregation and elimi-nation by autophagy. Embo J, 27, 433–46.
Verhoeven, K., Claeys, K. G., Zuchner, S., Schroder, J. M., Weis, J., Ceuterick, C., Jordanova, A., Nelis, E., de Vriendt, E., van Hul, M., Seeman, P., Mazanec, R., Saifi, G. M., Szigeti, K., Mancias, P., Butler, I. J., Kochanski, A., Ryniewicz, B., de Bleecker, J., Van Den Bergh, P., Verellen, C., Van Coster, R., Goemans, N., Auer-grumbach, M., Robberecht, W., Milic Rasic, V., Nevo, Y., Tournev, I., Guergueltcheva, V., Roelens, F., Vieregge, P., Vinci, P., Moreno, M. T., Christen, H. J., Shy, M. E., Lupski, J. R., Vance, J. M., De Jonghe, P. & Timmerman, V. (2006) MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain, 129, 2093–102.
Verny, C., Loiseau, D., Scherer, C., Lejeune, P., Chevrollier, A., Gueguen, N., Guillet, V., Dubas, F., Reynier, P., Amati-Bonneau, P. & Bonneau, D. (2008) Multiple sclerosis-like disorder in OPA1-related autosomal dominant optic atrophy. Neurology, 70, 1152–3.
Votruba, M., Moore, A. T. & Bhattacharya, S. S. (1998) Clinical features, molecular genetics, and pathophysiology of dominant optic atrophy. J Med Genet, 35, 793–800.
Wang, W., Cheng, X., Lu, J., Wei, J., Fu, G., Zhu, F., Jia, C., Zhou, L., Xie, H. & Zheng, S. (2010) Mitofusin-2 is a novel direct target of p53. Biochem Biophys Res Commun.

196 G. Lenaers et al.
White, K. E., Davies, V., Hogan, V., Piechota, M., Nichols, P., Turnbull, D. M. & Votruba, M. (2009) OPA1 deficiency is associated with increased autophagy in retinal ganglion cells in a murine model of dominant optic atrophy. Invest Ophthalmol Vis Sci.
Williams, P. A., Morgan, J. E. & Votruba, M. (2010) Opa1 deficiency in a mouse model of domi-nant optic atrophy leads to retinal ganglion cell dendropathy. Brain.
Yu-wai-man, P., Griffiths, P. G., Burke, A., Sellar, P. W., Clarke, M. P., Gnanaraj, L., Ah-kine, D., Hudson, G., Czermin, B., Taylor, R. W., Horvath, R. & Chinnery, P. F. (2010a) The prevalence and natural history of dominant optic atrophy due to OPA1 mutations. Ophthalmology, 117, 1538–46, 1546 e1.
Yu-wai-man, P., Griffiths, P. G., Gorman, G. S., Lourenco, C. M., Wright, A. F., Auer-grumbach, M., Toscano, A., Musumeci, O., Valentino, M. L., Caporali, L., Lamperti, C., Tallaksen, C. M., Duffey, P., Miller, J., Whittaker, R. G., Baker, M. R., Jackson, M. J., Clarke, M. P., Dhillon, B., Czermin, B., Stewart, J. D., Hudson, G., Reynier, P., Bonneau, D., Marques, W., Jr., Lenaers, G., Mcfarland, R., Taylor, R. W., Turnbull, D. M., Votruba, M., Zeviani, M., Carelli, V., Bindoff, L. A., Horvath, R., Amati-bonneau, P. & Chinnery, P. F. (2010b) Multi-system neu-rological disease is common in patients with OPA1 mutations. Brain, 133, 771–86.
Yu-wai-man, P., Sitarz, K. S., Samuels, D. C., Griffiths, P. G., Reeve, A. K., Bindoff, L. A., Horvath, R. & Chinnery, P. F. (2010c) OPA1 mutations cause cytochrome c oxidase deficiency due to loss of wild-type mtDNA molecules. Hum Mol Genet, 19, 3043–52.
Zanna, C., Ghelli, A., Porcelli, A. M., Karbowski, M., Youle, R. J., Schimpf, S., Wissinger, B., Pinti, M., Cossarizza, A., Vidoni, S., Valentino, M. L., Rugolo, M. & Carelli, V. (2008) OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mito-chondrial fusion. Brain, 131, 352–67.
Zhu, D., Kennerson, M. L., Walizada, G., Zuchner, S., Vance, J. M. & Nicholson, G. A. (2005) Charcot-Marie-Tooth with pyramidal signs is genetically heterogeneous: families with and without MFN2 mutations. Neurology, 65, 496–7.
Zorzano, A. (2009) Regulation of mitofusin-2 expression in skeletal muscle. Appl Physiol Nutr Metab, 34, 433–9.
Zuchner, S., de Jonghe, P., Jordanova, A., Claeys, K. G., Guergueltcheva, V., Cherninkova, S., Hamilton, S. R., Van Stavern, G., Krajewski, K. M., Stajich, J., Tournev, I., Verhoeven, K., Langerhorst, C. T., De Visser, M., Baas, F., Bird, T., Timmerman, V., Shy, M. & Vance, J. M. (2006) Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol, 59, 276–81.
Zuchner, S., Mersiyanova, I. V., Muglia, M., Bissar-tadmouri, N., Rochelle, J., Dadali, E. L., Zappia, M., Nelis, E., Patitucci, A., Senderek, J., Parman, Y., Evgrafov, O., Jonghe, P. D., Takahashi, Y., Tsuji, S., Pericak-vance, M. A., Quattrone, A., Battaloglu, E., Polyakov, A. V., Timmerman, V., Schroder, J. M. & Vance, J. M. (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet, 36, 449–51.





























