Embed Size (px)
Citation preview

Mitochondrial Membrane ProteinAssociated Neurodegenration: A
Novel Variant of NeurodegenerationWith Brain Iron Accumulation
Eva C. Schulte, MD,1,2,3 Malte C. Claussen, MD,1
Angela Jochim, MD,1 Tobias Haack, MD,2,3
Monika Hartig, MD,2,3 Maja Hempel, MD,2,3
Holger Prokisch, PhD,2,3 Ursula Haun-Junger, MD,4
Juliane Winkelmann, MD,1,2,3 Bernhard Hemmer, MD,1
Annette F€orschler, MD,5 and Rudiger Ilg, MD1*
1Neurologische Klinik und Poliklinik, Klinikum rechts der Isar,
Technische Universit€at Munchen, Munich, Germany; 2Institut fur
Humangenetik, Helmholtz Zentrum Munchen, Munich, Germany;3Institut fur Humangenetik, Klinikum recht der Isar, Technische
Universit€at Munchen, Munich, Germany; 4Sozialp€adiatrisches
Zentrum, Klinikum Bremen-Mitte, Bremen, Germany; 5Abteilung fur
Neuroradiologie, Klinikum rechts der Isar, Technische Universit€at
Munchen, Munich, Germany
ABSTRACTBackground: Recently, mutations in an open-readingframe on chromosome 19 (C19orf12) were identified asa novel genetic factor in neurodegeneration with brainiron accumulation (NBIA). Because of the mitochondriallocalization of the derived protein, this variant is referredto as mitochondrial membrane protein-associated neu-rodegeneration with brain iron accumulation (MPAN).Methods/Results: We describe the clinical phenotypeand MRI of 3 newly identified individuals with MPAN dueto either previously reported or novel homozygous orcompound heterozygous genetic alterations in C19orf12.Conclusions: MPAN is characterized by a juvenile-onset, slowly progressive phenotype with predominantlower limb spasticity, generalized dystonia, and cogni-tive impairment. Typical additional features include axo-nal motor neuropathy and atrophy of the optic nerve.MRI showed iron deposition in the globus pallidus andsubstantia nigra without the eye-of-the-tiger sign,which is typical for PKAN, the most frequent form ofNBIA. VC 2012 Movement Disorder Society
Key Words: neurodegeneration with brain iron accu-mulation (NBIA); genetics; MRI; mitochondrial membraneprotein-associated neurodegeneration (MPAN); C19orf12
We describe 3 cases of mitochondrial membraneprotein-associated neurodegeneration (MPAN), anovel form of neurodegeneration with brain iron accu-mulation (NBIA, formerly known as Hallervorden–
Spatz syndrome). NBIA subsumes a group of heredi-tary neurodegenerative disorders characterized by ironaccumulation primarily in the basal ganglia, presentingwith a variety of neurologic changes with prominentprogressive motor and behavioral symptoms.1,2
Although very rare (1–3 cases/1 million population),significant progress has been made in stratifying thisheterogeneous group into genetic and phenotypic enti-ties. Accounting for up to 50% of cases, pantothenatekinase–associated neurodegeneration (PKAN), attrib-uted to autosomal recessive mutations in the PANK2gene, is the most common form.3,4 Other NBIA var-iants include infantile and noninfantile neuraxonaldystrophy (collectively also termed PLA2G6-associ-ated neurodegeneration [PLAN]),5,6 static encephalop-athy of childhood with neurodegeneration inadulthood, fatty acid hydroxylase-associated neurode-generation, Kufor–Rakeb syndrome, and Woodhouse–Sakati syndrome.2,5,6 Recently, an open-reading frameon chromosome 19, C19orf1, was identified as a novelgenetic factor in NBIA.7 In a cohort of 52 NBIApatients from Poland, deletions and missense muta-tions were found in 19, rendering C19orf12 the sec-ond most common genetic locus affected in NBIA.7
Both genetically and clinically, NBIA ascribed to var-iants in C19orf12 represents a distinct group that hasbeen termed MPAN.7 Based on this discovery, weidentified 3 previously unreported individuals withMPAN who had so far been classified as idiopathicforms of NBIA. We describe the phenotype and MRIthat, according to the current state of knowledge, isbelieved to be characteristic of this subgroup of NBIA.
MPANAll 3 individuals visited our department for routine
treatment reevaluation of a slowly progressive psycho-motor disorder. As required by clinical regulations, allprovided written informed consent for genetic testing.
------------------------------------------------------------Additional Supporting Information may be found in the online version ofthis article.
Eva C. Schulte and Malte C. Claussen contributed equally to the article.
*Correspondence to: Rudiger Ilg, Neurologische Klinik und Poliklinik,Klinikum rechts der Isar, Technische Universit€at Munchen, Ismaningerstr.22, 81675 Munich, Germany; [email protected]
Funding agencies: This study was financed exclusively by in-housefunding from the Neurologische Klinik & Poliklinik, Klinikum rechts derIsar, Technische Universit€at Munchen, Munich, Germany.Relevant conflicts of interest/financial disclosures: Holger Prokischreceives research support from the Impulse and Networking Fund of theHelmholtz Association within the framework of the Helmholtz Alliance forMental Health in an Ageing Society (HA-215) and the German FederalMinistry of Education and Research (BMBF)–funded German Center forDiabetes Research (DZD e.V.), Systems Biology of Metabotypes grant(SysMBo 0315494A), and the German Network for MitochondrialDisorders (mitoNET 01GM0867). Rudiger Ilg is supported by the medicalfaculty of the Technische Universit€at Munchen (KKF).Full financial disclosures and author roles may be found in the onlineversion of this article.
Received: 25 January 2012; Revised: 10 September 2012;Accepted: 25 September 2012Published online 19 November 2012 in Wiley Online Library(wileyonlinelibrary.com). DOI: 10.1002/mds.25256
224 Movement Disorders, Vol. 28, No. 2, 2013
S C H U L T E E T A L .

The 3 individuals presented with progressive psycho-motor deterioration (gait impairment, clumsiness, dys-arthria, and progressive cognitive deficits). Initialsymptoms consisted of general clumsiness along withretarded motor and scholastic development. Duringthe course of the disease, gait disturbance from severespasticity of the lower limbs became most prominent.Pyramidal signs (ie, upgoing plantars) were found inall 3 individuals. In contrast to PKAN, generalizeddystonia was less pronounced and parkinsonism,which can occur in PKAN, was not present. Diseaseprogression in 2 individuals was comparatively slow,with preserved ambulation at 10 and 25 years afterdisease onset, whereas the third individual was wheel-chair bound by age 13. Two individuals showedimpulsive behavior. Retinitis pigmentosa, frequentlyobserved in PKAN, was not found in any of our individ-uals. Instead, bilateral atrophy of the optic nerve waspresent in 2 of the 3 cases (Table 1). Similar to a signifi-cant part of the Polish MPAN cohort,7 in 2 of the cases,nerve conduction studies revealed prominent general-ized axonal motor neuropathy, a feature not typicallyseen in PKAN. Additional clinical features in singleindividuals included low-frequency bilateral actiontremor of the hands and congenital strabism. Increasedserum creatine kinase (CK) levels have been reported inindividuals with MPAN.8 In our cases, only MPAN_2
had an elevated serum CK (584 U/L; normal < 174 U/L). We also noted slightly increased serum lactate dehy-drogenase levels in all 3 individuals (MPAN_1, 369 U/L; MPAN_2, 270 U/L; and MPAN_3, 289 U/L; normal< 244 U/L), whereas all other routine blood tests wereunremarkable. Electroencephalography was normal inall cases. MRI showed prominent T2 hypointensity ofthe globus pallidus highly suggestive of iron deposition6
in all 3 MPAN subjects. In contrast to the PKAN sub-type, where the so-called eye-of-the-tiger sign, a centralT2 hyperintense area within the T2 hypointense (iron-rich) globus pallidus, is nearly pathognomonic, the eye-of-the-tiger sign was not seen in any of our individuals.In contrast to other subtypes of NBIA, all individualsshowed T2 hypointensity of the substantia nigra (Fig. 1)and additional T1 hyperintensity of the caudate nucleusand putamen. In the most severely affected individual,additional frontotemporal atrophy was present.
Genetic evaluation of the coding regions of C19orf12 inthe affected individuals only was performed by Sangersequencing. One individual (MPAN_3) was homozygousfor an 11-bp deletion (c.204_214del [p.Gly69ArgfsX10]),which is predicted to result in a truncated protein andwhich is the most frequent variant in C19orf12 observedin MPAN to date.7 MPAN_1 also carried this deletion on1 allele and a missense variant (c.32 C>T [p.Thr11Met])on the other. The p.Thr11Met missense variant has
Table 1. Summary of clinical, genetic, and MRI features observed in 3 individuals suffering from MPAN
MPAN_1 MPAN_2 MPAN_3
Sex F M MAge of onset 7 3 6Age at presentation 34 19 18C19orf12 mutation c.32 C>T (p.Thr11Met) c.53 A>G (p.Asp18Gly) c.204_214del (p.Gly69ArgfsX10)
c.204_214del (p.Gly69ArgfsX10) c.395 T>A (p.Leu132Gln) c.204_214del (p.Gly69ArgfsX10)Pyramidal signs þ þþ þþSpasticity þ þþ þOromandibular dystonia � þ �Generalized dystonia þ þ þParkinsonism � � �Dysarthria No spontaneous speech þ þOptic atrophy � þþ (þ)Eye movements Unremarkable Not assessable Saccadic pursuitMotor axonal neuropathy þþ þþ �Psychiatric signs þ þþ �Cognitive impairment þþ þþ þWheelchair bound (age) 13Additional clinical features Low-frequency hand tremor None StrabismSerum creatine kinase (U/L) 115 (normal < 174) 589 (normal < 174) 143 (normal < 174)MRI findings Bilateral T2 hypointensity
in GP and SNBilateral T2 hypointensityin GP and SN
Bilateral T2 hypointensity in GP & SN,No eye-of-the-tiger sign
No eye-of-the-tiger sign No eye-of-the-tiger sign T1 hyperintensity in CN and PutT1 hyperintensity inCN and Put
T1 hyperintensity inCN and Put
Family history 1 Unaffected sister No siblings 2 Unaffected brothersNo neurologic/psychiatricdiseases
No neurologic/psychiatricdiseases
No neurologic/psychiatric diseases
Ethnicity German from Russia Turkish Polish
GP, globus pallidus; SN, substantia nigra; CN, caudate nucleus; Put, putamen.
A N O V E L V A R I A N T O F N B I A
Movement Disorders, Vol. 28, No. 2, 2013 225
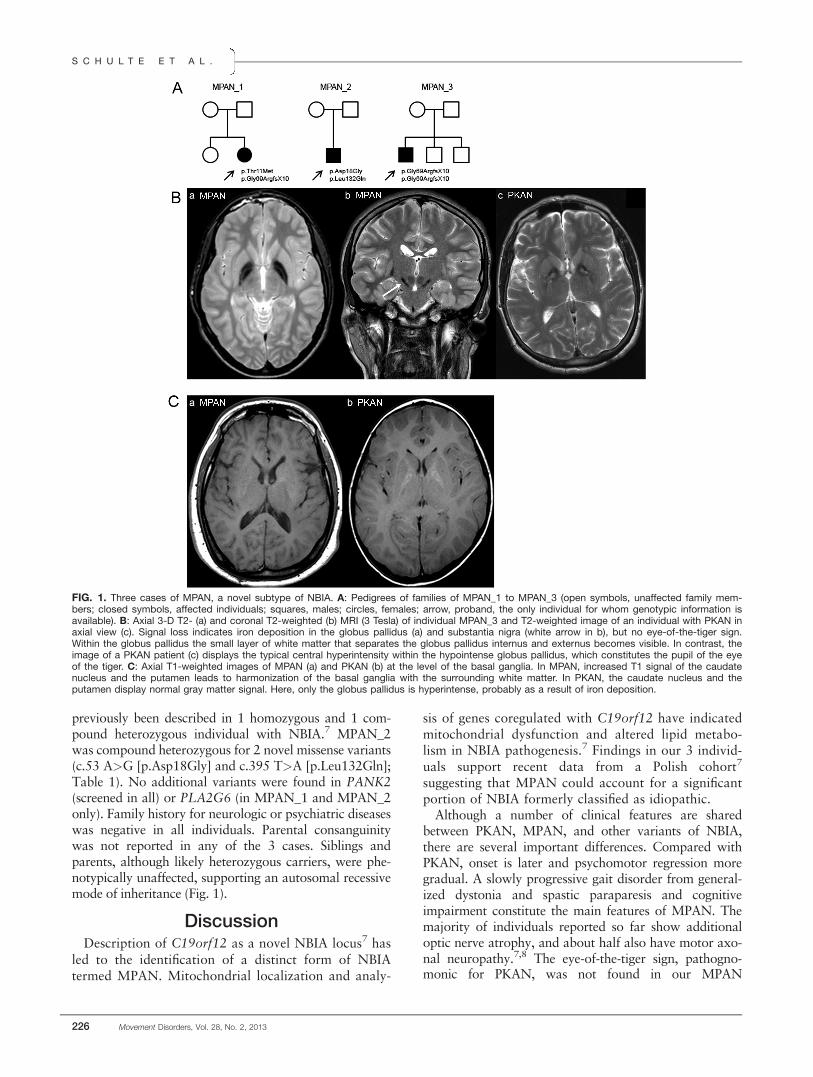
previously been described in 1 homozygous and 1 com-pound heterozygous individual with NBIA.7 MPAN_2was compound heterozygous for 2 novel missense variants(c.53 A>G [p.Asp18Gly] and c.395 T>A [p.Leu132Gln];Table 1). No additional variants were found in PANK2(screened in all) or PLA2G6 (in MPAN_1 and MPAN_2only). Family history for neurologic or psychiatric diseaseswas negative in all individuals. Parental consanguinitywas not reported in any of the 3 cases. Siblings andparents, although likely heterozygous carriers, were phe-notypically unaffected, supporting an autosomal recessivemode of inheritance (Fig. 1).
DiscussionDescription of C19orf12 as a novel NBIA locus7 has
led to the identification of a distinct form of NBIAtermed MPAN. Mitochondrial localization and analy-
sis of genes coregulated with C19orf12 have indicatedmitochondrial dysfunction and altered lipid metabo-lism in NBIA pathogenesis.7 Findings in our 3 individ-uals support recent data from a Polish cohort7
suggesting that MPAN could account for a significantportion of NBIA formerly classified as idiopathic.
Although a number of clinical features are sharedbetween PKAN, MPAN, and other variants of NBIA,there are several important differences. Compared withPKAN, onset is later and psychomotor regression moregradual. A slowly progressive gait disorder from general-ized dystonia and spastic paraparesis and cognitiveimpairment constitute the main features of MPAN. Themajority of individuals reported so far show additionaloptic nerve atrophy, and about half also have motor axo-nal neuropathy.7,8 The eye-of-the-tiger sign, pathogno-monic for PKAN, was not found in our MPAN
FIG. 1. Three cases of MPAN, a novel subtype of NBIA. A: Pedigrees of families of MPAN_1 to MPAN_3 (open symbols, unaffected family mem-bers; closed symbols, affected individuals; squares, males; circles, females; arrow, proband, the only individual for whom genotypic information isavailable). B: Axial 3-D T2- (a) and coronal T2-weighted (b) MRI (3 Tesla) of individual MPAN_3 and T2-weighted image of an individual with PKAN inaxial view (c). Signal loss indicates iron deposition in the globus pallidus (a) and substantia nigra (white arrow in b), but no eye-of-the-tiger sign.Within the globus pallidus the small layer of white matter that separates the globus pallidus internus and externus becomes visible. In contrast, theimage of a PKAN patient (c) displays the typical central hyperintensity within the hypointense globus pallidus, which constitutes the pupil of the eyeof the tiger. C: Axial T1-weighted images of MPAN (a) and PKAN (b) at the level of the basal ganglia. In MPAN, increased T1 signal of the caudatenucleus and the putamen leads to harmonization of the basal ganglia with the surrounding white matter. In PKAN, the caudate nucleus and theputamen display normal gray matter signal. Here, only the globus pallidus is hyperintense, probably as a result of iron deposition.
S C H U L T E E T A L .
226 Movement Disorders, Vol. 28, No. 2, 2013

individuals and was only described in 1 individual fromthe recently published Polish cohort.7 Instead, all 3 indi-viduals showed additional prominent nigral T2 hypoin-tensity as a sign of iron deposition also in the substantianigra (Fig. 1). This pattern is similar to subjects withPLAN,6 who also show optic atrophy in some cases.9 Incontrast to PLAN, our subjects showed no cerebellar atro-phy but additional T1 hyperintensity of the caudate nu-cleus and putamen. Depending on the tissueconcentration, iron can either be hyper- or isointense inT1.6 The combination of T1 hyperintensity and normalgray matter signal in T2 suggests either a discrepancybetween paramagnetic effects on T1 and T2 as reportedfor manganese or could be a result of a low iron concen-tration, which has not yet led to signal extinction in theT2 sequence.10
Further instrumental diagnostic and genetic examina-tion of the parents would be interesting to evaluatewhether a heterozygous carrier status coincides withmild disease features such as asymptomatic polyneurop-athy, signs of subclinical brain iron deposition, ordecreased visual acuity. From a genetics perspective, it isnoteworthy that MPAN_3, who is of Polish origin, washomozygous for C19orf12 p.Gly69ArgfsX10, the origi-nal and most common genetic variant identified in thepreviously reported Polish NBIA cohort. It may be possi-ble that this variant arose in Eastern Europe, whereas inother regions such as, for example, Turkey, from whereboth parents of MPAN_2 originate, other genetic var-iants of C19orf12 may have arisen independently. Thisconception is further supported by a recent report of 2Turkish brothers with an NBIA phenotype similar to the1 of our 3 individuals (spasticity, dystonia, optic atrophy,and peripheral neuropathy) who harbor a novel homozy-gous missense mutation (c.362 T>A, p.Leu121Gln) inC19orf12.8 Interestingly, the genetic variant found inMPAN_2 (p.Leu132Gln) is close to the p.Leu121Glnmutation and a previously identified missense mutation(p.Lys142Gln), but not in the predicted transmembranedomain where previously reported MPAN mutations arefound. Clinically, MPAN_2 was most severely affectedwith the earliest disease onset and the most rapid pro-gression. One possible explanation is that the variantsidentified in MPAN_2 harbor the most deleterious effecton MPAN-relevant C19orf12 function or the presence ofadditional modifying genetic factors.
So far, the clinical management of NBIA has been lim-ited to symptomatic treatment of motor symptomsincluding bilateral pallidal deep brain stimulation incases with severe generalized dystonia.11 In PKAN, theCoA precursors calcium panthothenate and panthetineare discussed as potential therapeutic agents,12 andresults of a small phase II pilot study indicate possiblereduction of pallidal iron deposition by the iron-chelat-ing agent deferiprone. The clinical benefit here is unclear,however.13 Moreover, it remains to be elucidatedwhether PKAN and MPAN share a common diseasemechanism or merely have enhanced iron deposition inthe brain as a unifying feature.
At present, it is important to realize that iron depo-sition in the basal ganglia and the substantia nigrawithout the characteristic eye-of-the-tiger sign along-side relatively late psychomotor regression with promi-nent spasticity of the lower limbs, atrophy of the opticnerve, and sometimes severe motor axonal neuropathyshould raise suspicion for MPAN, and C19orf12should be assessed for genetic alterations.
Video. The MPAN phenotype. Videos of MPAN_2showing generalized action-induced dystonia predomi-nantly of the upper extremities, together with retro- andtorticollis and oromandibular dystonia, spasticity of thelower limbs with upgoing plantar reflexes (Babinskisign)—note that at the time of videotaping, spasticity ofthe lower extremities was relatively mild under therapywith baclofen 62.5 mg/day)—and interdigital muscular at-rophy caused by axonal motor polyneuropathy.
Acknowledgments: We are greatly indebted to the 3 individuals whoparticipated in this study and their families. We thank Angelika Klucken andher colleagues at Hoffnungsbaum e.V. for the good collaboration and theirgreat commitment in supporting individuals with NBIA.
References1. Hayflick SJ, Westaway SK, Levinson B, et al. Genetic, clinical, and
radiographic delineation of Hallervorden-Spatz syndrome. N EnglJ Med 2003;348:33–40.
2. Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineationof neurodegeneration with brain iron accumulation. J Med Genet2009;46:73–80.
3. Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, HayflickSJ. A novel pantothenate kinase gene (PANK2) is defective in Haller-vorden-Spatz syndrome. Nat Genet 2001;28:345–349.
4. Hartig MB, Hortnagel K, Garavaglia B, et al. Genotypic and pheno-typic spectrum of PANK2 mutations in patients with neurodegenera-tion with brain iron accumulation. Ann Neurol 2006;59:248–256.
5. Schneider S A, Hardy J, Bhatia K P. Syndromes of neurodegenera-tion with brain iron accumulation (NBIA): An update on clinicalpresentations, histological and genetic underpinnings, and treat-ment considerations. Mov Disord 2012;27:42–53.
6. Kruer MC, Boddaert N, Schneider SA, et al. Neuroimaging fea-tures of neurodegeneration with brain iron accumulation. AJNRAm J Neuroradiol 2012;33:407–414.
7. Hartig MB, Iuso A, Haack T, et al. Absence of an orphan mito-chondrial protein (C19orf12) causes a distinct clinical subtype ofneurodegeneration with brain iron accumulation (NBIA). Am JHum Genet 2011;89:543–550.
8. Horvath R, Holinski-Feder E, Neeve VCM, et al. A new phenotypeof brain iron accumulation with dystonie, optic atrophy, and pe-ripheral neuropathy. Mov Disord 2012;27:789–793.
9. Morgan NV, Westaway SK, Morton JE, et al. PLA2G6, encodinga phospholipase, A2, is mutated in neurodegenerative disorderswith high brain iron. Nat Genet 2006;38:752–754.
10. Ginat DT, Meyers SP. Intracranial lesions with high signal inten-sity on T1-weighted MR images: differential diagnosis. Radio-graphics 2012;32:499–516.
11. Timmermann L, Pauls KA, Wieland K, et al. Dystonia in neurode-generation with brain iron accumulation: outcome of bilateral pal-lidal stimulation. Brain 2010;133:701–712.
12. Rana A, Seinen E, Siudeja K, et al. Pantethine rescues a Drosophilamodel for pantothenate kinase-associated neurodegeneration. ProcNatl Acad Sci U S A 2010;107:6988–6993.
13. Zorzi G, Zibordi F, Chiapparini L, et al. Iron-related MRI imagesin patients with pantothenate kinase-associated neurodegeneration(PKAN) treated with deferiprone: results of a phase II pilot trial.Mov Disord 2011;26:1756–1759.
A N O V E L V A R I A N T O F N B I A
Movement Disorders, Vol. 28, No. 2, 2013 227





























