Embed Size (px)
Citation preview

COPHAR-1155; NO. OF PAGES 10
Mitosis-targeting therapies: a troubleshooting guideElena Domenech and Marcos Malumbres
Available online at www.sciencedirect.com
Several mitotic kinases and kinesins are currently considered
as cancer targets based on their critical role during the cell
division cycle and their significant level of expression in human
tumors. Yet, their use is limited by the lack of selectivity against
tumor cells, the low percentage of mitotic cells in many human
tumors, and dose-limiting side-effects. As a consequence,
initial clinical trials have shown limited responses. Despite
these drawbacks, inhibiting mitosis is a promising strategy that
deserves further development. Future advances will benefit
from more specific inhibitors with better pharmacodynamic
properties, a clear physiological characterization and cell-type-
specific requirements of old and new mitotic targets, and
rational strategies based on synthetic lethal interactions to
improve selectivity against tumor cells.
Addresses
Cell Division and Cancer Group, Spanish National Cancer Research
Centre (CNIO), Madrid, Spain
Corresponding author: Malumbres, Marcos ([email protected],
Current Opinion in Pharmacology 2013, 13:xx–yy
This review comes from a themed issue on Cancer
Edited by Massimo Santoro and Francesca Carlomagno
1471-4892/$ – see front matter, # 2013 Elsevier Ltd. All rights
reserved.
http://dx.doi.org/10.1016/j.coph.2013.03.011
IntroductionCell cycle deregulation is a common feature of human
cancers and multiple therapeutic strategies are aimed to
inhibit the cell division cycle in tumor cells. Current
efforts can be broadly divided into three different groups:
first, preventing the commitment to cell cycle entry
imposed by oncogenic signals [1]; second, abrogation of
the DNA damage checkpoint to increase lethality in
highly proliferating cells [2,3]; and third, taking advantage
of the special sensitivity of cells to abnormal chromosome
segregation during mitosis [4]. The two first approaches
are clearly based on the specific signals originated from
the oncogenic stress in tumor cells. This has been far
more difficult to achieve in targeted therapies directed
against mitosis given the conservation in the essential
mechanism that regulates chromosome segregation in
normal or tumor cells.
Classical antimitotic therapies have made use of spindle
poisons that bind tubulin subunits and either stabilize, for
Please cite this article in press as: Domenech E, Malumbres M. Mitosis-targeting therapies: a tro
www.sciencedirect.com
example, taxanes (paclitaxel, Nab-paclitaxel and doce-
taxel), or de-stabilize, for example, vinka-alkaloids (vinor-
elbine, vinblastine, vincristine) and microtubules. In all
these cases, these compounds prevent microtubule
dynamics, a process required for the generation of a
bipolar spindle and chromosome segregation during mito-
sis. Microtubules are also necessary for multiple cellular
functions during interphase and the use of these spindle
poisons is associated to side-effects such as neurotoxicity
that can lead to irreversible neuropathologies. It is
expected that dividing cells are more vulnerable to these
microtubule-disturbing agents due to the increased rate
or microtubule turnover during mitosis. In fact, micro-
tubule poisons can be considered as one of the major
additions to the clinic in the last decades. However,
whether mitotic inhibition explains the clinical success
of these drugs is now under debate [5,6�].
The success of spindle poisons and the susceptibility of
cells to mitotic arrest have led in the last years to a search
for improved therapeutic strategies based on specific
mitotic targets (Figure 1). Several mitotic kinases have
been evaluated including members of the Polo-like
kinase (Plk) and Aurora families. Mitotic targets also
include kinesins, a class of motor proteins with ATPase
activity that move along microtubule filaments and are
required for proper microtubule function. Abrogation of
the mitotic checkpoint has been also considered to
specifically generate lethal aberrations in chromosomally
unstable tumor cells. Preventing mitotic exit has been
recently proposed as a complementary alternative since
cells cannot survive for long time in mitosis and they
eventually die if components of the mitotic exit pathway,
such as the E3 ubiquitin ligase APC/C (Anaphase Pro-
moting Complex/Cyclosome) are inhibited. In this review
we will discuss the recent data generated from these
studies as well as proposals that need to be evaluated
in the near future.
Mitotic kinases and kinesinsMultiple enzymatic activities are required for mitosis.
These include centrosomal and mitotic kinases required
for the maintenance of the mitotic state, generation of
the mitotic spindle, and proper attachment of chromo-
somes to microtubules for segregation [7]. The cyclin-
dependent kinase Cdk1 is a major driver of mitotic entry
and progression. Its activity is essential for mitotic entry
and lack of Cdk1 activity prevents cell proliferation in
every cell type tested [8]. Inhibition of this kinase is
thought to result in high levels of toxicity in normal cells
and it is not frequently considered as an interesting
target for cancer therapy [9]. However, some recent
ubleshooting guide, Curr Opin Pharmacol (2013), http://dx.doi.org/10.1016/j.coph.2013.03.011
Current Opinion in Pharmacology 2013, 13:1–10

2 Cancer
COPHAR-1155; NO. OF PAGES 10
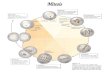
Figure 1
TARGETING MITOSIS
MICROTUBULEPOISONS
Vinka-AlkaloidsTaxanes
MITOTICKINASES
INHIBITORSPlk1
Aurora A
KINESINSINHIBITORS
Eg5CENP-E
SACDEPENDENT
ARREST
MITOTIC SLIPPAGE
CELL DEATH CELL CYCLEPROGRESSION
ARREST ININTERPHASE
INHIBITMITOTIC
EXIT
APC/CINHIBITORS
MITOTICCHECKPOINTABROGATION
Mps1 Aurora BINHIBITORS MITOTIC CELL
DEATH
Current Opinion in Pharmacology
Schematic representation of current therapeutic approaches targeting
mitosis. Most antimitotic drugs including microtubule poisons trigger a
mitotic arrest that depends on the Spindle Assembly Checkpoint (SAC).
This arrest is transient and cells exit from mitosis (mitotic slippage) with
different fates: proliferation, arrest or death. Inhibitors of the SAC
(checkpoint abrogation) may have deleterious effects by triggering rapid
mitotic exit with the subsequent chromosome aberrations. Preventing
mitotic exit, on the other hand, invariably results in cell death.
results suggest that specific combinations that may
change this view (see below).
Most current efforts to inhibit mitotic kinases in cancer
have focused on Plk1 and Aurora (A and B) kinases [10].
These three proteins are overexpressed in multiple
tumors and are essential components of cell division since
their inactivation results in lack of chromosome segre-
gation, polyploidy and, eventually, cell death. At least in
the case of Plk1, it seems that proliferation of primary
Please cite this article in press as: Domenech E, Malumbres M. Mitosis-targeting therapies: a tro
Current Opinion in Pharmacology 2013, 13:1–10
cells exhibit a lower dependency on this kinase whereas
tumor cells are more sensitive to loss of Plk1 activity [11].
Multiple small-molecule inhibitors have been developed
in the last years and some of them have been tested in
clinical trials (Table 1). Dose-escalation clinical trials with
the most advanced Plk1 compounds, BI6727 or
ON01910.Na have been published in 2012 [12,13].
Reversible hematological toxicity was the main side-
effect and some limited but encouraging antitumor
activity was observed promoting Phase II studies in
monotherapy (Table 1) and combination (Table 2) in
solid and hematopoietic tumors. Aurora kinase inhibitors
display similar toxicity and have also shown preliminary
activity (a few partial responses and a significant effect in
stable disease) in hematological and solid tumors [14–19].
Mps1 inhibitors have been only evaluated in preclinical
assays (Table 1). Mps1 is required for the Spindle Assem-
bly Checkpoint (SAC) and its inhibition results in defec-
tive chromosome segregation, mitotic catastrophe and
frequent cell death in a variety of tumor cell lines, with
certain preference for p53 mutant cells [20–22].
Several members of the kinesin superfamily such as Eg5
or Cenp-E (Figure 1 and Table 1) have also emerged as
putative anticancer targets [23]. In the first clinical trials
with Eg5 or Cenp-E inhibitors, tolerability was accepta-
ble with neutropenia as the major dose-limiting effect.
However, the evidence for clinical efficacy was reduced
with very limited objective responses and these com-
pounds were mostly ineffective in patients [24–31].
Targeting mitosis in combination therapiesCombination strategies are frequently tested to improve
efficacy while maintaining acceptable overall toxicity.
Taxanes, for instance, are used in combination with
platinum analogs for lung cancer and with anthracyclines
for breast cancer, and new microtubule poisons in de-
velopment are evaluated in combination with these cyto-
toxic agents [32]. In a similar effort, Plk1 and Aurora
inhibitors have been combined with a variety of che-
motherapeutic agents including conventional cytotoxic
agents, antimicrotubule poisons or other targeted thera-
pies (Table 2). Combination of Aurora-A or Plk1 inhibi-
tors with genotoxic agents (e.g. DNA synthesis inhibitors)
may provide additional benefits given the role of these
kinases in the recovery process that occurs following
DNA damage [10]. In fact, combination of Plk1 or Aurora
inhibitors with radiotherapy or cytotoxic compounds such
as cisplatin, cyclophosphamide or doxorubicine has
resulted in improved efficacy in preclinical assays
[33,34�,35–37]. Multiple clinical studies are now in pro-
gress to evaluate the combination of Plk1 or Aurora
inhibitors with cytotoxic compounds such as DNA syn-
thesis inhibitors (Table 2).
Since Aurora kinases are required for the mitotic arrest in
the presence of some microtubule poisons, inactivation of
ubleshooting guide, Curr Opin Pharmacol (2013), http://dx.doi.org/10.1016/j.coph.2013.03.011
www.sciencedirect.com

Targeting mitosis: a troubleshooting guide Domenech and Malumbres 3
COPHAR-1155; NO. OF PAGES 10
Please cite this article in press as: Domenech E, Malumbres M. Mitosis-targeting therapies: a troubleshooting guide, Curr Opin Pharmacol (2013), http://dx.doi.org/10.1016/j.coph.2013.03.011
Table 1
Mitotic drugs: recent preclinical and clinical studies as monotherapy
Target Inhibitor Tumor types Description Referencesa
Plk1 BI2536 Hematopoietic and solid
tumor cell lines
Antiproliferative and apoptotic in
cultured cells and xenografts
[60,61]
Phase I/II in solid tumors and
relapsed hematopoietic
malignancies
Partial antitumor activity. Reversible
neutropenia
NCT00706498, NCT00710710,
NCT00412880, NCT00376623,
NCT00243087, NCT00701766 [62,63]
BI6727 Phase I advanced solid
tumors
Some partial responses and stable
disease. Most trials ongoing
NCT01145885, NCT01121406,
NCT01023958, NCT01348347,
NCT01662505, NCT00969553 [12]
GSK461364 Tumor cell lines and xenograft
models
Mitosis arrest and growth inhibition.
Uses pH3 as a pharmacodynamic
biomarker
[64]
Phase I in advanced solid
tumors and non-Hodgkins
lymphoma
Prolonged stable disease in 15% of
the patients
NCT00536835 [65]
CBB2001 HeLa cells and xenografts Cell cycle arrest and inhibition of
tumor growth in xenografts
[66]
ON01910.Na Pancreatic cancer cell lines Tumor-specific apoptosis. Cyclin B1
proposed as a biomarker
[67]
Phase I/II hematopoietic and
advanced solid cancer
Propose AKT2 phosphorylation and
cyclin D1 expression as biomarkers.
Mostly ongoing/no results published
NCT00533416, NCT00854646,
NCT01048619, NCT00854945,
NCT01167166, NCT00861510,
NCT01326377, NCT00867061,
NCT00906334, NCT01241500,
NCT01538537, NCT01538563 [13,68�]
Aurora AZD1152
(AurkB-specific)
Solid tumor cell lines Growth inhibition and apoptosis in
vivo and in vitro
[69]
Phase I/II study AML Overall hematologic response rate
27%. One out of the four entered
complete remission
NCT00497991, NCT01019161,
NCT00497731, NCT00497679 [70–72]
Phase I trials patients with
advanced solid malignancies
No objective tumor responses
observed, but stable disease in 23%
of the patients
[19]
MLN8237
(AurkA-specific)
Phase I refractory ovarian,
fallopian tube or primary
peritoneal carcinoma
Modest response as a single agent,
durable disease
[17,18]
Phase I advanced solid
tumors and phase I/II AML
Good tolerability and favorable
pharmacokinetics, produces
neutropenia and stomatitis. Mostly
ongoing/unpublished
NCT00962091, NCT00500903,
NCT00651664, NCT00830518,
NCT00530699 [15,16]
MLN8054
(AurkA-specific)
Phase I advanced solid tumor Dose-escalating study.
Recommended dose for phase II
clinical trials was not established
NCT00249301, NCT00652158 [73]
VX-680 (MK-0457,
pan Aurk inhibitor)
Hepatoblastoma and CLL cell
lines
Inhibition cell proliferation and
induction of apoptosis
[74–76]
Phase I/II Advanced
hematopoietic and solid tumors
Ongoing/no results published NCT00104351, NCT00099346,
NCT00290550, NCT00405054,
NCT00500006, NCT00111683
AMG 900
(pan-Aurora
inhibitor)
Solid tumor cell lines Inhibition of proliferation including
paclitaxel-resistant cells
[77]
Phase I acute leukemias and
solid tumor
Ongoing/no results published NCT01380756, NCT00858377
Mps1 NMS-P715 Cultured cells Aneuploidy and cell death in a
variety of tumor cell lines. Inhibits
tumor growth
[20]
MPI-0479605 Colon carcinoma cell lines Aberrant mitosis and growth arrest.
Cells undergo mitotic catastrophe
and/or apoptosis
[21,78]
Reversine Colon carcinoma cell lines Low doses selectively kill p53-null
cells
[22]
Cenp-E GSK923295 Mesenchymal xenographs Showed significant antitumor
activity in xenograft tumor models
[79]
Phase I in refractory cancer Limited side-effects. One patient
with a durable partial response
NCT00504790 [26]
www.sciencedirect.com Current Opinion in Pharmacology 2013, 13:1–10

4 Cancer
COPHAR-1155; NO. OF PAGES 10
Table 1 (Continued )
Target Inhibitor Tumor types Description Referencesa
Eg5 AZD4877 Phase I solid tumor, phase II
advanced bladder cancer
Acceptable safety profile. But little
evidence of clinical efficacy
NCT00661609, NCT00661609
[25,27,28,80]
Phase I/II AML Study terminated due to lack of
efficacy
NCT00486265 [24]
SB-715992 Phase I metastatic cancer and
tumors resistant to taxanes
and/or platinum
The most common toxicities were
nausea, diarrhea, fatigue and
neutropenia. Some partial
responses. Mostly ongoing/
unpublished data
NCT00354250, NCT00363272,
NCT00085813, NCT00095953,
NCT00095992, NCT00095628,
NCT00103311, NCT00097409,
NCT00096499, NCT00089973,
NCT00607841 [81–83]
SB-743921 Lung cancer patient explants Complete tumor regression in
xenografts
[84]
Phase I lymphoma, solid tumors Partial responses NCT00343564, NCT00136513 [29]
ARRY-520 Solid and hematopoietic cells Mitotic defects, arrest and
apoptosis. Good responses
particularly in hematological tumors
[85–87,88�]
Phase I solid advanced
cancer, advanced myeloid
leukemia
Relative lack of clinical activity in
solid tumors
NCT00462358, NCT00637052 [31]
ARQ621 Phase I metastatic solid, h
ematologic
Ongoing/no results published NCT00825487
LY2523355 Phase I Solid and
hematopoietic tumors
Ongoing/no results published NCT01059643, NCT01214629,
NCT01358019, NCT01214642,
NCT01214655, NCT01025284
MK0731 Phase I Solid tumors Lengthening of stable disease in
patients with taxane resistant
tumors
NCT00104364 [30,89]
Abbreviations: AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; pH3, phospho-histone H3.a Clinical trials are indicated using the ClinicalTrials.gov identifier (http://www.clinicaltrials.gov/show/NCTXXXXXXXX).
these kinases may result in deleterious effects [38],
similar to those obtained after checkpoint abrogation in
the absence of Mps1 [39]. Combination of Aurora kinase
inhibition with classical microtubule agents displayed
promising efficacy in specific preclinical assays (Table
2). A few clinical trials are now exploring the advantages
of combining Aurora or Eg5 inhibitors with taxanes for the
treatment of solid tumors (Table 2). Additional trials in
combination with a variety of targeted therapies, such as
proteasome, HDAC or tyrosine kinase inhibitors are also
in progress (Table 2).
Targeting mitosis: troubles and proposalsIn a general overview, the first generation of clinical trials
for mitotic targeted therapies has not reached the expec-
tations raised from preclinical models, at least for solid
tumors. In general, the current generation of targeted
mitotic inhibitors displays less anticancer activity than
paclitaxel although neurotoxicity has been eliminated as
expected. Major side-effects in patients include hemato-
logical alterations and neutropenia usually sets the dose
limit. It has been argued that mitotic rates in human
tumors are much more lower than in preclinical models,
suggesting that the duration of exposure limits the effect
of these targeted antimitotic drugs [5,6�,40]. Considering
these limitations, it is critical to evaluate how antimitotic
therapies can be improved in the clinic. In the following
sections we will discuss some of these issues and some
outlines to take into consideration in the future.
Please cite this article in press as: Domenech E, Malumbres M. Mitosis-targeting therapies: a tro
Current Opinion in Pharmacology 2013, 13:1–10
Improving inhibitors and targets
The selection of mitotic targets has been driven by the
pioneer identification of major mitotic regulators in
screenings in yeast or Drosophila [41,42]. These screens
identified essential, well-conserved regulators such as
Cdk1, Aurora A/B or Plk1 and these are the targets that
first attracted the interest of pharmaceutical companies
and reached clinical studies. The essential role of these
kinases in the cell cycle also determines the toxicity
expected from their inhibition in vivo. The focus on
Plk1 and Aurora kinases, and the lack of interest in
Cdk1 due to its essential requirements for mitotic entry,
is somehow surprising given that Aurora-A, Aurora-B or
Plk1 are also essential for mammalian development and
cell proliferation [7]. One of the reasons for losing the
interest in Cdk1 was perhaps the presence of other family
members, such as Cdk4/6, that are not essential for
normal tissue homeostasis but specifically required for
proliferation under specific oncogenic signals [1,7,9]. It
can be predicted that a similar situation may apply for
other mitotic regulators. Aurora C for instance is dispen-
sable for mammalian development although it is specifi-
cally required during specific stages and tissues and
mutated in a few tumors [43�]. The example is not as
clear for Plk as the other family members (Plk2–Plk5) are
thought to function as tumor suppressors [44]. The poten-
tial as cancer targets of kinesins [23] or many other kinases
such as the Nek family [45], Greatwall [46] or haspin [47]
remains to be explored.
ubleshooting guide, Curr Opin Pharmacol (2013), http://dx.doi.org/10.1016/j.coph.2013.03.011
www.sciencedirect.com

Targeting mitosis: a troubleshooting guide Domenech and Malumbres 5
COPHAR-1155; NO. OF PAGES 10
Table 2
Therapeutic treatments using mitotic inhibitors in combination with additional compounds
Target Combination Model Description Referencesa
Plk1 BI2536, BI6727, ON01910
and DNA damage agents
(doxorubicine, cisplatin,
cyclophosphamide,
irinotecan, etc.)
Breast cancer cell lines, phase
I/II solid and hematopoietic
tumors
Impaired tumor growth in
xenografts. Combinations
lead to a faster complete
response and prevent relapse.
Clinical trials ongoing
NCT00804856,
NCT00824408,
NCT00969761,
NCT01125891,
NCT01165905,
NCT00861783,
NCT00861328
[34,90�,91]
BI2536 and bortezomib
(proteasome inhibitor)
Tumor cell lines Enhanced antiproliferative
and apoptotic effects
[92]
BI6727, BIBF1120 (VEGFR
inhibitor) and BIBW2992
(EGFR inhibitor)
Phase I advanced solid
tumors
Ongoing/no results published NCT01022853,
NCT01206816
Aurora kinases AZD1152, PHA-680632,
MLN8237 and DNA damage
agents (platinum,
gemcitabine, radiation, etc.)
Solid tumor cell lines. Phase I
in AML
Synergistic effects in
proliferation and apoptosis.
Enhance the effectiveness of
the single therapy.
Radiosensitizer specially in
p53-deficient cells
NCT00952588,
NCT00926731
[36,37,93–95]
VX-680, VE-465, AT9283,
CYC3, MLN8237 and
microtubule agents
Tumor cell lines. Phase I in
adenocarcinoma
Synergistic or additive effect
in different cell lines
NCT01677559
[96–99]
MLN8237 and bortezomid Phase I lymphoma and
multiple myeloma
Ongoing/no results published NCT01695941,
NCT01034553
MLN8237 and vorinostat
(HDAC inhibitor)
Tumor cell lines. Phase I in
lymphoma
Tumor growth inhibition in
xenografts but additive
cytotoxicity in leukemias
NCT01567709
[100,101]
MLN8237 and rapamycin
(mTOR inhibitor)
Uterine leiomyosarcoma cell
lines
Synergistic antiproliferative
effect (only when
preadministered)
[102]
MLN8237 and erlotinib (EGFR
inhibitor)
Phase I NSCLC Ongoing/no results published NCT01471964
Kinesins SB-715992 and carboplatin or
capecitabine
Phase I solid tumors Ongoing/no results published NCT00136578,
NCT00119171
SB-715992 and docetaxel Phase I solid tumors Ongoing/no results published NCT00169520
ARRY-520, carfilzomib,
bortezomid and
dexamethalone
Phase I relapsed or refractory
multiple myeloma
Ongoing/no results published NCT01248923,
NCT01372540
Abbreviations: AML, acute myeloid leukemia; NSCLC, non-small cell lung cancer.a Clinical trials are indicated using the ClinicalTrials.gov identifier (http://www.clinicaltrials.gov/show/NCTXXXXXXXX).
Another major problem with these major targets is that
our knowledge about their function mostly comes from
studies in yeast, flies, frogs or a reduced number of
mammalian cell lines. Clinical studies frequently report
differential levels of expression of some of these proteins
in cancer but the relevance of that information is not
really clear, and the tumor-associated expression of these
proteins may simply correlate with cell proliferation [48].
In addition, the general concepts of oncogene and tumor
suppressors are insufficient to classify these proteins [48].
An interesting example is Plk1, a protein frequently
overexpressed in human tumors and with predictive value
in specific malignancies. On the basis of that, Plk1 is
frequently considered as an oncogene. Yet, the only
mutations found in human tumors are loss-of-function
mutations and Plk1-heterozygous mice display increased
susceptibility to spontaneous tumor formation, suggesting
Please cite this article in press as: Domenech E, Malumbres M. Mitosis-targeting therapies: a tro
www.sciencedirect.com
that Plk1 may function as a tumor suppressor similar to the
rest of the Plk family [44]. Whereas these data do not
preclude the possible use of Plk1 inhibitors in cancer
treatment, it is clear that we are far from understanding
the physiological relevance of these proteins in specific
adult tissues, a must to properly select biomarkers and to
improve their clinical use in patients.
Synthetic lethal interactions
As indicated above, the essential role of these molecules
determines undesired effects in patients. However, their
partial inhibition may synergize with specific oncogenic
alterations providing certain selectivity against tumor
cells. This has been elegantly proposed for Cdk1.
Cdk1 is required for proper BRCA1 activity and partial
inhibition of this kinase impairs repair by homologous
recombination. As a consequence, Cdk1-inhibited tumor
ubleshooting guide, Curr Opin Pharmacol (2013), http://dx.doi.org/10.1016/j.coph.2013.03.011
Current Opinion in Pharmacology 2013, 13:1–10

6 Cancer
COPHAR-1155; NO. OF PAGES 10
cells are hypersensitive to inhibition of poly(ADP-ribose)
polymerase (PARP) whereas normal cells are not sensi-
tized [49��]. Partial inhibition of Cdk1 therefore
represents a plausible strategy for expanding the utility
of PARP inhibitors to BRCA-proficient cancers.
Additional uses have been proposed for Cdk1 inhibitors
in the treatment of Myc-induced tumors [50], or to
promote differentiation in leukemias, especially those
resistant to Flt3 inhibitory therapies [51�] or in megakar-
yocytic leukemia [52��].
Similar proposals start to emerge for other mitotic reg-
ulators. In addition to the interactions between Plk1 and
the DNA damage response, Plk1 inhibition preferentially
reduces survival of cells with mutant p53 or Ras onco-
genes [53,54]. A more recent screen in lung cancer cells
has shown that treatment with retinoids can sensitize cells
to Plk1 inhibitors [55]. A parallel screen for sensitizers for
Aurora kinase inhibitors found a significant effect after
downregulation of platelet-derived growth factor receptor
(PDGFR), which has been shown to be overexpressed in
pancreatic cancer cells and tumor tissues [56]. In fact,
tyrosine kinase inhibitors enhance the antiproliferative
effect of Plk1 or Aurora kinase inhibitor (Table 2). Many
more possibilities will arise in the future although the
relevance of these combinations has not been analyzed in
the clinic yet.
Inhibiting mitotic progression and mitotic exit: a deadly
combination
One of the mechanisms that explain resistance to mitotic
drugs is mitotic slippage. In the presence of antimitotic
drugs such as microtubule poisons or Plk1 inhibitors, the
activity of the SAC prevents the degradation of cyclin B1,
inactivation of Cdk1 and mitotic exit. However, this arrest
is transient as a consequence of the slow but maintained
degradation of cyclin B1 even in the presence of an active
checkpoint. This arrest is likely insufficient for an effi-
cient therapeutic response [5] and cells that exit mitosis
can remain viable (Figure 1). Cell fate during or after
mitotic arrest is dictated by multiple networks but pre-
venting mitotic exit invariably results in cell death. It has
been therefore proposed that blocking mitotic exit may
have stronger effects than targeting the spindle or mitotic
entry [57]. In fact, elimination of Cdc20, the APC/C
cofactor required for cyclin B1 degradation and mitotic
exit, is highly efficient in killing cells in mitosis and
preventing tumor growth in vivo [58]. A small-molecule
inhibitor of the APC/C is now available for preclinical
studies [59]. These data suggest that preventing mitotic
slippage or modulating the cell death pathways that
trigger cell death in mitosis should enhance the efficacy
of other antimitotic compounds [4].
ConclusionsThe data published in the last few years support the
interest in antimitotic therapies but also display the
Please cite this article in press as: Domenech E, Malumbres M. Mitosis-targeting therapies: a tro
Current Opinion in Pharmacology 2013, 13:1–10
limited effect of strategies currently in clinical trials.
Overall, monotherapy studies suggest that some Plk1
or Aurora kinase inhibitors are reasonably well tolerated
although antitumor responses in advanced cancers seem
to be modest. The availability of newer and improved
compounds will improve the current situation although it
is expected that the toxicity derived from the essential
role of these major kinases will limit their clinical success.
The development of successful antimitotic therapies
requires a better knowledge of the functional and phys-
iological relevance of current and new mitotic targets, and
subsequent therapeutic proposals based on synthetic
lethal interactions with oncogenic mutations present in
tumor cells. Patient stratification based on the functional
relevance of these proteins in specific cell types and
under specific oncogenic backgrounds will be the key
for the success of antimitotic therapies in the future.
AcknowledgementsE.D. is supported by the Fondo de Investigaciones Sanitarias. The CellDivision and Cancer group of the CNIO is supported by grants from theMinisterio de Economıa y Competitividad (MINECO; SAF2012-38215),Fundacion Ramon Areces, the OncoCycle Programme (S2010/BMD-2470)from the Comunidad de Madrid, and the European Union SeventhFramework Programme (MitoSys project; HEALTH-F5-2010-241548).
References and recommended readingPapers of particular interest, published within the period of review,have been highlighted as:
� of special interest�� of outstanding interest
1. Malumbres M: Cell cycle-based therapies move forward.Cancer Cell 2012, 22:419-420.
2. Toledo LI, Murga M, Fernandez-Capetillo O: Targeting ATR andChk1 kinases for cancer treatment: a new model for new (andold) drugs. Mol Oncol 2011, 5:368-373.
3. Bucher N, Britten CD: G2 checkpoint abrogation andcheckpoint kinase-1 targeting in the treatment of cancer. Br JCancer 2008, 98:523-528.
4. Manchado E, Guillamot M, Malumbres M: Killing cells bytargeting mitosis. Cell Death Differ 2012, 19:369-377.
5. Komlodi-Pasztor E, Sackett DL, Fojo AT: Inhibitors targetingmitosis: tales of how great drugs against a promising targetwere brought down by a flawed rationale. Clin Cancer Res 2012,18:51-63.
6.�
Mitchison TJ: The proliferation rate paradox in antimitoticchemotherapy. Mol Biol Cell 2012, 23:1-6.
These two articles [5,6�] discuss the relevance of mitotic index andduration of mitosis in the efficacy of cancer therapies directed againstmitosis in human tumors.
7. Malumbres M: Physiological relevance of cell cycle kinases.Physiol Rev 2011, 91:973-1007.
8. Malumbres M, Barbacid M: Cell cycle, CDKs and cancer: achanging paradigm. Nat Rev Cancer 2009, 9:153-166.
9. Malumbres M, Pevarello P, Barbacid M, Bischoff JR: CDKinhibitors in cancer therapy: what is next? Trends PharmacolSci 2008, 29:16-21.
10. Lens SM, Voest EE, Medema RH: Shared and separate functionsof polo-like kinases and aurora kinases in cancer. Nat RevCancer 2010, 10:825-841.
11. Raab M, Kappel S, Kramer A, Sanhaji M, Matthess Y, Kurunci-Csacsko E, Calzada-Wack J, Rathkolb B, Rozman J, Adler T et al.:
ubleshooting guide, Curr Opin Pharmacol (2013), http://dx.doi.org/10.1016/j.coph.2013.03.011
www.sciencedirect.com

Targeting mitosis: a troubleshooting guide Domenech and Malumbres 7
COPHAR-1155; NO. OF PAGES 10
Toxicity modelling of Plk1-targeted therapies in geneticallyengineered mice and cultured primary mammalian cells. NatCommun 2011, 2:395.
12. Schoffski P, Awada A, Dumez H, Gil T, Bartholomeus S, Wolter P,Taton M, Fritsch H, Glomb P, Munzert G: A phase I, dose-escalation study of the novel Polo-like kinase inhibitorvolasertib (BI 6727) in patients with advanced solid tumours.Eur J Cancer 2012, 48:179-186.
13. Seetharam M, Fan AC, Tran M, Xu L, Renschler JP, Felsher DW,Sridhar K, Wilhelm F, Greenberg PL: Treatment of higher riskmyelodysplastic syndrome patients unresponsive tohypomethylating agents with ON 01910.Na. Leuk Res 2012,36:98-103.
14. Arkenau HT, Plummer R, Molife LR, Olmos D, Yap TA, Squires M,Lewis S, Lock V, Yule M, Lyons J et al.: A phase I dose escalationstudy of AT9283, a small molecule inhibitor of aurora kinases,in patients with advanced solid malignancies. Ann Oncol 2012,23:1307-1313.
15. Cervantes A, Elez E, Roda D, Ecsedy J, Macarulla T,Venkatakrishnan K, Rosello S, Andreu J, Jung J, Sanchis-Garcia JM et al.: Phase I pharmacokinetic/pharmacodynamicstudy of MLN8237, an investigational, oral, selective aurora akinase inhibitor, in patients with advanced solid tumors. ClinCancer Res 2012, 18:4764-4774.
16. Dees EC, Cohen RB, von Mehren M, Stinchcombe TE, Liu H,Venkatakrishnan K, Manfredi M, Fingert H, Burris HA 3rd,Infante JR: Phase I study of aurora A kinase inhibitor MLN8237in advanced solid tumors: safety, pharmacokinetics,pharmacodynamics, and bioavailability of two oralformulations. Clin Cancer Res 2012, 18:4775-4784.
17. Matulonis UA, Sharma S, Ghamande S, Gordon MS, Del Prete SA,Ray-Coquard I, Kutarska E, Liu H, Fingert H, Zhou X et al.: Phase IIstudy of MLN8237 (alisertib), an investigational Aurora Akinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primaryperitoneal carcinoma. Gynecol Oncol 2012, 127:63-69.
18. Mosse YP, Lipsitz E, Fox E, Teachey DT, Maris JM, Weigel B,Adamson PC, Ingle MA, Ahern CH, Blaney SM: Pediatric phase Itrial and pharmacokinetic study of MLN8237, aninvestigational oral selective small-molecule inhibitor ofAurora kinase A: a Children’s Oncology Group Phase IConsortium study. Clin Cancer Res 2012, 18:6058-6064.
19. Schwartz GK, Carvajal RD, Midgley R, Rodig SJ, Stockman PK,Ataman O, Wilson D, Das S, Shapiro GI: Phase I study of barasertib(AZD1152), a selective inhibitor of Aurora B kinase, in patientswith advanced solid tumors. Invest New Drugs 2013, 31:370-380.
20. Colombo R, Caldarelli M, Mennecozzi M, Giorgini ML, Sola F,Cappella P, Perrera C, Depaolini SR, Rusconi L, Cucchi U et al.:Targeting the mitotic checkpoint for cancer therapy withNMS-P715, an inhibitor of MPS1 kinase. Cancer Res 2010,70:10255-10264.
21. Tardif KD, Rogers A, Cassiano J, Roth BL, Cimbora DM,McKinnon R, Peterson A, Douce TB, Robinson R, Dorweiler I et al.:Characterization of the cellular and antitumor effects of MPI-0479605, a small-molecule inhibitor of the mitotic kinaseMps1. Mol Cancer Ther 2011, 10:2267-2275.
22. Jemaa M, Galluzzi L, Kepp O, Boileve A, Lissa D, Senovilla L,Harper F, Pierron G, Berardinelli F, Antoccia A et al.: Preferentialkilling of p53-deficient cancer cells by reversine. Cell Cycle2012, 11:2149-2158.
23. Rath O, Kozielski F: Kinesins and cancer. Nat Rev Cancer 2012,12:527-539.
24. Kantarjian HM, Padmanabhan S, Stock W, Tallman MS, Curt GA,Li J, Osmukhina A, Wu K, Huszar D, Borthukar G et al.: Phase I/IImulticenter study to assess the safety, tolerability,pharmacokinetics and pharmacodynamics of AZD4877 inpatients with refractory acute myeloid leukemia. Invest NewDrugs 2012, 30:1107-1115.
25. Esaki T, Seto T, Ariyama H, Arita S, Fujimoto C, Tsukasa K,Kometani T, Nosaki K, Hirai F, Yagawa K: Phase I study to assessthe safety, tolerability and pharmacokinetics of AZD4877 inJapanese patients with solid tumors. Arch Drug Inf 2011, 4:23-31.
Please cite this article in press as: Domenech E, Malumbres M. Mitosis-targeting therapies: a tro
www.sciencedirect.com
26. Chung V, Heath EI, Schelman WR, Johnson BM, Kirby LC,Lynch KM, Botbyl JD, Lampkin TA, Holen KD: First-time-in-human study of GSK923295, a novel antimitotic inhibitor ofcentromere-associated protein E (CENP-E), in patients withrefractory cancer. Cancer Chemother Pharmacol 2012, 69:733-741.
27. Jones R, Vuky J, Elliott T, Mead G, Arranz JA, Chester J,Chowdhury S, Dudek AZ, Muller-Mattheis V, Grimm MO et al.:Phase II study to assess the efficacy, safety and tolerability ofthe mitotic spindle kinesin inhibitor AZD4877 in patients withrecurrent advanced urothelial cancer. Invest New Drugs 2013http://dx.doi.org/10.1007/s10637-013-9926-y.
28. Infante JR, Kurzrock R, Spratlin J, Burris HA, Eckhardt SG, Li J,Wu K, Skolnik JM, Hylander-Gans L, Osmukhina A et al.: A Phase Istudy to assess the safety, tolerability, and pharmacokineticsof AZD4877, an intravenous Eg5 inhibitor in patients withadvanced solid tumors. Cancer Chemother Pharmacol 2012,69:165-172.
29. Holen KD, Belani CP, Wilding G, Ramalingam S, Volkman JL,Ramanathan RK, Vasist LS, Bowen CJ, Hodge JP, Dar MM et al.: Afirst in human study of SB-743921, a kinesin spindle proteininhibitor, to determine pharmacokinetics, biologic effects andestablish a recommended phase II dose. Cancer ChemotherPharmacol 2011, 67:447-454.
30. Holen K, DiPaola R, Liu G, Tan AR, Wilding G, Hsu K,Agrawal N, Chen C, Xue L, Rosenberg E et al.: A phase I trialof MK-0731, a kinesin spindle protein (KSP) inhibitor, inpatients with solid tumors. Invest New Drugs 2012, 30:1088-1095.
31. Khoury HJ, Garcia-Manero G, Borthakur G, Kadia T, Foudray MC,Arellano M, Langston A, Bethelmie-Bryan B, Rush S, Litwiler Ket al.: A phase 1 dose-escalation study of ARRY-520, a kinesinspindle protein inhibitor, in patients with advanced myeloidleukemias. Cancer 2012, 118:3556-3564.
32. Cortes J, Vidal M: Beyond taxanes: the next generation ofmicrotubule-targeting agents. Breast Cancer Res Treat 2012,133:821-830.
33. Tyagi S, Bhui K, Singh R, Singh M, Raisuddin S, Shukla Y: Polo-like kinase1 (Plk1) knockdown enhances cisplatinchemosensitivity via up-regulation of p73alpha in p53 mutanthuman epidermoid squamous carcinoma cells. BiochemPharmacol 2010, 80:1326-1334.
34.�
Maire V, Nemati F, Richardson M, Vincent-Salomon A, Tesson B,Rigaill G, Gravier E, Marty-Prouvost B, De Koning L, Lang G et al.:Polo-like kinase 1: a potential therapeutic option incombination with conventional chemotherapy for themanagement of patients with triple-negative breast cancer.Cancer Res 2013, 73:813-823.
Plk1 is overexpressed in triple-negative breast tumors and its inhibitionleads to impaired tumor growth. Combination of BI-2536 with DNAdamaging agents results in complete responses and lack of relapses,which is the major risk associated to these tumors.
35. Tao Y, Zhang P, Girdler F, Frascogna V, Castedo M, Bourhis J,Kroemer G, Deutsch E: Enhancement of radiation response inp53-deficient cancer cells by the Aurora-B kinase inhibitorAZD1152. Oncogene 2008, 27:3244-3255.
36. Zhang L, Zhang S: ZM447439, the Aurora kinase B inhibitor,suppresses the growth of cervical cancer SiHa cells andenhances the chemosensitivity to cisplatin. J Obstet GynaecolRes 2011, 37:591-600.
37. Ma Y, Weimer J, Fredrik R, Adam-Klages S, Sebens S, Caliebe A,Hilpert F, Eckmann-Scholz C, Arnold N, Schem C: Aurora kinaseinhibitor AZD1152 has an additional effect of platinum on asequential application at the human ovarian cancer cell lineSKOV3. Arch Gynecol Obstet 2013 http://dx.doi.org/10.1007/s00404-013-2719-x.
38. Sehdev V, Katsha A, Ecsedy J, Zaika A, Belkhiri A, El-Rifai W: Thecombination of alisertib, an investigational Aurora kinase Ainhibitor, and docetaxel promotes cell death and reducestumor growth in preclinical cell models of uppergastrointestinal adenocarcinomas. Cancer 2013, 119:904-914.
ubleshooting guide, Curr Opin Pharmacol (2013), http://dx.doi.org/10.1016/j.coph.2013.03.011
Current Opinion in Pharmacology 2013, 13:1–10

8 Cancer
COPHAR-1155; NO. OF PAGES 10
39. Janssen A, Kops GJ, Medema RH: Elevating the frequency ofchromosome mis-segregation as a strategy to kill tumor cells.Proc Natl Acad Sci U S A 2009, 106:19108-19113.
40. Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo AT: Mitosis is nota key target of microtubule agents in patient tumors. Nat RevClin Oncol 2011, 8:244-250.
41. Archambault V, Glover DM: Polo-like kinases: conservation anddivergence in their functions and regulation. Nat Rev Mol CellBiol 2009, 10:265-275.
42. Nurse P: A long twentieth century of the cell cycle and beyond.Cell 2000, 100:71-78.
43.�
Fernandez-Miranda G, Trakala M, Martin J, Escobar B,Gonzalez A, Ghyselinck NB, Ortega S, Canamero M, Perez deCastro I, Malumbres M: Genetic disruption of aurora B uncoversan essential role for aurora C during early mammaliandevelopment. Development 2011, 138:2661-2672.
Aurora C is an Aurora kinase usually not considered as a cancer targetdue to its restricted pattern of expression. This manuscript reports thefirst essential role of Aurora C in mammals and its function in embryoniccells, a cell-type that may resemble certain type of tumors.
44. de Carcer G, Manning G, Malumbres M: From Plk1 to Plk5:functional evolution of polo-like kinases. Cell Cycle 2011,10:2255-2262.
45. Fry AM, O’Regan L, Sabir SR, Bayliss R: Cell cycle regulation bythe NEK family of protein kinases. J Cell Sci 2012,125:4423-4433.
46. Glover DM: The overlooked greatwall: a new perspective onmitotic control. Open Biol 2012, 2:120023.
47. Higgins JM: Haspin: a newly discovered regulator of mitoticchromosome behavior. Chromosoma 2010, 119:137-147.
48. Perez de Castro I, de Carcer G, Malumbres M: A census ofmitotic cancer genes: new insights into tumor cell biology andcancer therapy. Carcinogenesis 2007, 28:899-912.
49.��
Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ,Moreau LA, Unitt C, Bronson RT, Thomas HD et al.: CompromisedCDK1 activity sensitizes BRCA-proficient cancers to PARPinhibition. Nat Med 2011, 17:875-882.
This manuscript renews the interest in Cdk1 as a cancer target byshowing that Cdk1 inhibition results in impaired BRCA1 activity andsensitizes cells to PARP inhibitors, highlighting the relevance of thesearch for synthetic lethal interactions for tumor treatment.
50. Goga A, Yang D, Tward AD, Morgan DO, Bishop JM: Inhibition ofCDK1 as a potential therapy for tumors over-expressing MYC.Nat Med 2007, 13:820-827.
51.�
Radomska HS, Alberich-Jorda M, Will B, Gonzalez D, Delwel R,Tenen DG: Targeting CDK1 promotes FLT3-activated acutemyeloid leukemia differentiation through C/EBPalpha. J ClinInvest 2012, 122:2955-2966.
This manuscript reports the control of the transcription factor C/EBPaby Cdk1. Cdk1 inhibition relieves the differentiation block in cellswith mutated Flt3, suggesting possible uses for Cdk1 inhibition in thetreatment of Flt3-mutant leukemias, especially those resistant to Flt3inhibitors.
52.��
Wen Q, Goldenson B, Silver SJ, Schenone M, Dancik V, Huang Z,Wang LZ, Lewis TA, An WF, Li X et al.: Identification of regulatorsof polyploidization presents therapeutic targets for treatmentof AMKL. Cell 2012, 150:575-589.
This article shows an elegant exercise to analyze the relevance ofinhibition of mitosis in cells that can undergo special cell cycles suchas megakaryocytes. The inhibition of mitotic kinases such as Auroraprevents growth of acute megakaryocytic leukemia cells by inhibitingtheir mitotic cell cycles. In the presence of these inhibitors, these tumorcells differentiate toward the megakaryocytic lineage and becomepolyploid.
53. Guan R, Tapang P, Leverson JD, Albert D, Giranda VL, Luo Y:Small interfering RNA-mediated Polo-like kinase 1 depletionpreferentially reduces the survival of p53-defective,oncogenic transformed cells and inhibits tumor growth inanimals. Cancer Res 2005, 65:2698-2704.
54. Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR,Westbrook TF, Wong KK, Elledge SJ: A genome-wide RNAi
Please cite this article in press as: Domenech E, Malumbres M. Mitosis-targeting therapies: a tro
Current Opinion in Pharmacology 2013, 13:1–10
screen identifies multiple synthetic lethal interactions with theRas oncogene. Cell 2009, 137:835-848.
55. Liu-Sullivan N, Zhang J, Bakleh A, Marchica J, Li J, Siolas D,Laquerre S, Degenhardt YY, Wooster R, Chang K et al.: PooledshRNA screen for sensitizers to inhibition of the mitoticregulator polo-like kinase (PLK1). Oncotarget 2011,2:1254-1264.
56. Xie L, Kassner M, Munoz RM, Que QQ, Kiefer J, Zhao Y,Mousses S, Yin HH, Von Hoff DD, Han H: Kinome-wide siRNAscreening identifies molecular targets mediating thesensitivity of pancreatic cancer cells to Aurora kinaseinhibitors. Biochem Pharmacol 2012, 83:452-461.
57. Huang HC, Shi J, Orth JD, Mitchison TJ: Evidence that mitoticexit is a better cancer therapeutic target than spindleassembly. Cancer Cell 2009, 16:347-358.
58. Manchado E, Guillamot M, de Carcer G, Eguren M, Trickey M,Garcia-Higuera I, Moreno S, Yamano H, Canamero M,Malumbres M: Targeting mitotic exit leads to tumor regressionin vivo: modulation by Cdk1, Mastl, and the PP2A/B55alpha,delta phosphatase. Cancer Cell 2010, 18:641-654.
59. Zeng X, Sigoillot F, Gaur S, Choi S, Pfaff KL, Oh DC, Hathaway N,Dimova N, Cuny GD, King RW: Pharmacologic inhibition of theanaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage.Cancer Cell 2010, 18:382-395.
60. Lee C, Fotovati A, Triscott J, Chen J, Venugopal C, Singhal A,Dunham C, Kerr JM, Verreault M, Yip S et al.: Polo-like kinase 1inhibition kills glioblastoma multiforme brain tumor cells inpart through loss of SOX2 and delays tumor progression inmice. Stem Cells 2012, 30:1064-1075.
61. Wagenblast J, Hirth D, Thron L, Arnoldner C, Diensthuber M,Stover T, Hambek M: Effects of the Polo-like-kinase-1-inhibitorBI2536 in squamous cell carcinoma cell lines of the head andneck. Oncol Lett 2012, 4:175-177.
62. Mross K, Frost A, Steinbild S, Hedbom S, Rentschler J, Kaiser R,Rouyrre N, Trommeshauser D, Hoesl CE, Munzert G: Phase I doseescalation and pharmacokinetic study of BI 2536, a novelPolo-like kinase 1 inhibitor, in patients with advanced solidtumors. J Clin Oncol 2008, 26:5511-5517.
63. Hofheinz RD, Al-Batran SE, Hochhaus A, Jager E, Reichardt VL,Fritsch H, Trommeshauser D, Munzert G: An open-label, phase Istudy of the polo-like kinase-1 inhibitor, BI 2536, in patientswith advanced solid tumors. Clin Cancer Res 2010,16:4666-4674.
64. Gilmartin AG, Bleam MR, Richter MC, Erskine SG, Kruger RG,Madden L, Hassler DF, Smith GK, Gontarek RR, Courtney MPet al.: Distinct concentration-dependent effects of the polo-like kinase 1-specific inhibitor GSK461364A, includingdifferential effect on apoptosis. Cancer Res 2009,69:6969-6977.
65. Olmos D, Barker D, Sharma R, Brunetto AT, Yap TA,Taegtmeyer AB, Barriuso J, Medani H, Degenhardt YY, Allred AJet al.: Phase I study of GSK461364, a specific and competitivePolo-like kinase 1 inhibitor, in patients with advanced solidmalignancies. Clin Cancer Res 2011, 17:3420-3430.
66. Lan R, Lin G, Yin F, Xu J, Zhang X, Wang J, Wang Y, Gong J,Ding YH, Yang Z et al.: Dissecting the phenotypes of Plk1inhibition in cancer cells using novel kinase inhibitorychemical CBB2001. Lab Invest 2012, 92:1503-1514.
67. Jimeno A, Chan A, Cusatis G, Zhang X, Wheelhouse J, Solomon A,Chan F, Zhao M, Cosenza SC, Ramana Reddy MV et al.:Evaluation of the novel mitotic modulator ON 01910.Na inpancreatic cancer and preclinical development of an ex vivopredictive assay. Oncogene 2009, 28:610-618.
68.�
Olnes MJ, Shenoy A, Weinstein B, Pfannes L, Loeliger K, Tucker Z,Tian X, Kwak M, Wilhelm F, Yong AS et al.: Directed therapy forpatients with myelodysplastic syndromes (MDS) bysuppression of cyclin D1 with ON 01910.Na. Leuk Res 2012,36:982-989.
Lack of biomarkers is a relevant drawback in current antimitotic therapies.In this article, cyclin D1 is proposed as a biomarker for Plk1 inhibition inmyelodysplastic syndromes.
ubleshooting guide, Curr Opin Pharmacol (2013), http://dx.doi.org/10.1016/j.coph.2013.03.011
www.sciencedirect.com

Targeting mitosis: a troubleshooting guide Domenech and Malumbres 9
COPHAR-1155; NO. OF PAGES 10
69. Diaz RJ, Golbourn B, Shekarforoush M, Smith CA, Rutka JT:Aurora kinase B/C inhibition impairs malignant glioma growthin vivo. J Neurooncol 2012, 108:349-360.
70. Tsuboi K, Yokozawa T, Sakura T, Watanabe T, Fujisawa S,Yamauchi T, Uike N, Ando K, Kihara R, Tobinai K et al.: A Phase Istudy to assess the safety, pharmacokinetics and efficacy ofbarasertib (AZD1152), an Aurora B kinase inhibitor, inJapanese patients with advanced acute myeloid leukemia.Leuk Res 2011, 35:1384-1389.
71. Dennis M, Davies M, Oliver S, D’Souza R, Pike L, Stockman P:Phase I study of the Aurora B kinase inhibitor barasertib(AZD1152) to assess the pharmacokinetics, metabolism andexcretion in patients with acute myeloid leukemia. CancerChemother Pharmacol 2012, 70:461-469.
72. Lowenberg B, Muus P, Ossenkoppele G, Rousselot P, Cahn JY,Ifrah N, Martinelli G, Amadori S, Berman E, Sonneveld P et al.:Phase 1/2 study to assess the safety, efficacy, andpharmacokinetics of barasertib (AZD1152) in patients withadvanced acute myeloid leukemia. Blood 2011, 118:6030-6036.
73. Dees EC, Infante JR, Cohen RB, O’Neil BH, Jones S, vonMehren M, Danaee H, Lee Y, Ecsedy J, Manfredi M et al.: Phase 1study of MLN8054, a selective inhibitor of Aurora A kinase inpatients with advanced solid tumors. Cancer ChemotherPharmacol 2011, 67:945-954.
74. Dewerth A, Wonner T, Lieber J, Ellerkamp V, Warmann SW,Fuchs J, Armeanu-Ebinger S: In vitro evaluation of the Aurorakinase inhibitor VX-680 for Hepatoblastoma. Pediatr Surg Int2012, 28:579-589.
75. de Paula Careta F, Gobessi S, Panepucci RA, Bojnik E, Morato deOliveira F, Mazza Matos D, Falcao RP, Laurenti L, Zago MA,Efremov DG: The Aurora A and B kinases are up-regulated inbone marrow-derived chronic lymphocytic leukemia cells andrepresent potential therapeutic targets. Haematologica 2012,97:1246-1254.
76. Chowdhury A, Chowdhury S, Tsai MY: A novel Aurora kinase Ainhibitor MK-8745 predicts TPX2 as a therapeutic biomarker innon-Hodgkin lymphoma cell lines. Leuk Lymphoma 2012,53:462-471.
77. Payton M, Bush TL, Chung G, Ziegler B, Eden P, McElroy P,Ross S, Cee VJ, Deak HL, Hodous BL et al.: Preclinical evaluationof AMG 900, a novel potent and highly selective pan-aurorakinase inhibitor with activity in taxane-resistant tumor celllines. Cancer Res 2010, 70:9846-9854.
78. Vijay Kumar D, Hoarau C, Bursavich M, Slattum P, Gerrish D,Yager K, Saunders M, Shenderovich M, Roth BL, McKinnon Ret al.: Lead optimization of purine based orally bioavailableMps1 (TTK) inhibitors. Bioorg Med Chem Lett 2012,22:4377-4385.
79. Lock RB, Carol H, Morton CL, Keir ST, Reynolds CP, Kang MH,Maris JM, Wozniak AW, Gorlick R, Kolb EA et al.: Initial testing ofthe CENP-E inhibitor GSK923295A by the pediatric preclinicaltesting program. Pediatr Blood Cancer 2012, 58:916-923.
80. Gerecitano JF, Stephenson JJ, Lewis NL, Osmukhina A, Li J, Wu K,You Z, Huszar D, Skolnik JM, Schwartz GK: A Phase I trial of thekinesin spindle protein (Eg5) inhibitor AZD4877 in patients withsolid and lymphoid malignancies. Invest New Drugs 2013,31:355-362.
81. Burris HA, 3rd, Jones SF, Williams DD, Kathman SJ, Hodge JP,Pandite L, Ho PT, Boerner SA, Lorusso P: A phase I study ofispinesib, a kinesin spindle protein inhibitor, administeredweekly for three consecutive weeks of a 28-day cycle inpatients with solid tumors. Invest New Drugs 2011, 29:467-472.
82. Souid AK, Dubowy RL, Ingle AM, Conlan MG, Sun J, Blaney SM,Adamson PC: A pediatric phase I trial and pharmacokineticstudy of ispinesib: a Children’s Oncology Group phase Iconsortium study. Pediatr Blood Cancer 2010, 55:1323-1328.
83. Gomez HL, Philco M, Pimentel P, Kiyan M, Monsalvo ML,Conlan MG, Saikali KG, Chen MM, Seroogy JJ, Wolff AA et al.:Phase I dose-escalation and pharmacokinetic study ofispinesib, a kinesin spindle protein inhibitor, administered ondays 1 and 15 of a 28-day schedule in patients with no prior
Please cite this article in press as: Domenech E, Malumbres M. Mitosis-targeting therapies: a tro
www.sciencedirect.com
treatment for advanced breast cancer. Anticancer Drugs 2012,23:335-341.
84. Good JA, Wang F, Rath O, Kaan HY, Talapatra SK, Podgorski D,Mackay SP, Kozielski F: Optimized S-trityl-L-cysteine basedinhibitors of kinesin spindle protein with potent in vivoantitumor activity in lung cancer xenograft models. J MedChem 2013, 56:1878-1893.
85. Carter BZ, Mak DH, Woessner R, Gross S, Schober WD, Estrov Z,Kantarjian H, Andreeff M: Inhibition of KSP by ARRY-520induces cell cycle block and cell death via the mitochondrialpathway in AML cells. Leukemia 2009, 23:1755-1762.
86. Kim KH, Xie Y, Tytler EM, Woessner R, Mor G, Alvero AB: KSPinhibitor ARRY-520 as a substitute for Paclitaxel in Type Iovarian cancer cells. J Transl Med 2009, 7:63.
87. Woessner R, Tunquist B, Lemieux C, Chlipala E, Jackinsky S, DewolfW Jr, Voegtli W, Cox A, Rana S, Lee P et al.: ARRY-520, a novel KSPinhibitor with potent activity in hematological and taxane-resistant tumor models. Anticancer Res 2009, 29:4373-4380.
88.�
Tunquist BJ, Woessner RD, Walker DH: Mcl-1 stabilitydetermines mitotic cell fate of human multiple myeloma tumorcells treated with the kinesin spindle protein inhibitor ARRY-520. Mol Cancer Ther 2010, 9:2046-2056.
Surprisingly, not many studies have analyzed the combination betweenthe inhibition of mitosis and promotion of apoptosis. In this article,downregulation of the antiapoptotic protein Mcl1 is shown to increasecell death in response to Eg5 inhibitors.
89. Cox CD, Garbaccio RM: Discovery of allosteric inhibitors ofkinesin spindle protein (KSP) for the treatment of taxane-refractory cancer: MK-0731 and analogs. Anticancer AgentsMed Chem 2010, 10:697-712.
90.�
Grinshtein N, Datti A, Fujitani M, Uehling D, Prakesch M, Isaac M,Irwin MS, Wrana JL, Al-Awar R, Kaplan DR: Small moleculekinase inhibitor screen identifies polo-like kinase 1 as a targetfor neuroblastoma tumor-initiating cells. Cancer Res 2011,71:1385-1395.
Low dosis of Plk1 inhibitors are cytotoxic for tumor-initiating cellswhereas much higher dosis are required to inhibit normal neural stemcells.
91. Ma WW, Messersmith WA, Dy GK, Weekes CD, Whitworth A,Ren C, Maniar M, Wilhelm F, Eckhardt SG, Adjei AA et al.: Phase Istudy of Rigosertib, an inhibitor of the phosphatidylinositol 3-kinase and Polo-like kinase 1 pathways, combined withgemcitabine in patients with solid tumors and pancreaticcancer. Clin Cancer Res 2012, 18:2048-2055.
92. Leinung M, Hirth D, Tahtali A, Diensthuber M, Stover T,Wagenblast J: Fighting cancer from different signallingpathways: effects of the proteasome inhibitor Bortezomib incombination with the polo-like-kinase-1-inhibitor BI2536 inSCCHN. Oncol Lett 2012, 4:1305-1308.
93. Tao Y, Zhang P, Frascogna V, Lecluse Y, Auperin A, Bourhis J,Deutsch E: Enhancement of radiation response by inhibition ofAurora-A kinase using siRNA or a selective Aurora kinaseinhibitor PHA680632 in p53-deficient cancer cells. Br J Cancer2007, 97:1664-1672.
94. Niermann KJ, Moretti L, Giacalone NJ, Sun Y, Schleicher SM,Kopsombut P, Mitchell LR, Kim KW, Lu B: Enhancedradiosensitivity of androgen-resistant prostate cancer:AZD1152-mediated Aurora kinase B inhibition. Radiat Res2011, 175:444-451.
95. Zhou N, Singh K, Mir MC, Parker Y, Lindner DJ, Dreicer R,Ecsedy JA, Teh BT, Zhang Z, Almasan A et al.: The investigationalAurora kinase A inhibitor MLN8237 induces defects in cellviability and cell cycle progression in bladder cancer cells invitro and in vivo. Clin Cancer Res 2013, 19:1717-1728.
96. Curry J, Angove H, Fazal L, Lyons J, Reule M, Thompson N,Wallis N: Aurora B kinase inhibition in mitosis: strategies foroptimising the use of aurora kinase inhibitors such as AT9283.Cell Cycle 2009, 8:1921-1929.
97. Yoshida K, Nagai T, Ohmine K, Uesawa M, Sripayap P, Ishida Y,Ozawa K: Vincristine potentiates the anti-proliferative effect ofan aurora kinase inhibitor, VE-465, in myeloid leukema cells.Biochem Pharmacol 2011, 82:1884-1890.
ubleshooting guide, Curr Opin Pharmacol (2013), http://dx.doi.org/10.1016/j.coph.2013.03.011
Current Opinion in Pharmacology 2013, 13:1–10

10 Cancer
COPHAR-1155; NO. OF PAGES 10
98. Lin Y, Richards FM, Krippendorff BF, Bramhall JL, Harrington JA,Bapiro TE, Robertson A, Zheleva D, Jodrell DI: Paclitaxel andCYC3, an aurora kinase A inhibitor, synergise in pancreaticcancer cells but not bone marrow precursor cells. Br J Cancer2012, 107:1692-1701.
99. Jeet V, Russell PJ, Verma ND, Khatri A: Targeting aurora kinases:a novel approach to curb the growth & chemoresistance ofandrogen refractory prostate cancer. Curr Cancer Drug Targets2012, 12:144-163.
100. Fiskus W, Hembruff SL, Rao R, Sharma P, Balusu R,Venkannagari S, Smith JE, Peth K, Peiper SC, Bhalla KN: Co-treatment with vorinostat synergistically enhances activity
Please cite this article in press as: Domenech E, Malumbres M. Mitosis-targeting therapies: a tro
Current Opinion in Pharmacology 2013, 13:1–10
of Aurora kinase inhibitor against human breast cancer cells.Breast Cancer Res Treat 2012, 135:433-444.
101. Muscal JA, Scorsone KA, Zhang L, Ecsedy JA, Berg SL: Additiveeffects of vorinostat and MLN8237 in pediatric leukemia,medulloblastoma, and neuroblastoma cell lines. Invest NewDrugs 2013, 31:39-45.
102. Brewer Savannah KJ, Demicco EG, Lusby K, Ghadimi MP,Belousov R, Young E, Zhang Y, Huang KL, Lazar AJ, Hunt KK et al.:Dual targeting of mTOR and aurora-A kinase for the treatmentof uterine Leiomyosarcoma. Clin Cancer Res 2012, 18:4633-4645.
ubleshooting guide, Curr Opin Pharmacol (2013), http://dx.doi.org/10.1016/j.coph.2013.03.011
www.sciencedirect.com