Embed Size (px)
Citation preview

1
Méthodes d’études en histologie BUTS de ce chapitre :
- Connaître le circuit de prise en charge d’un prélèvement histologique humain. - Connaître en détail les techniques histologiques et cytologiques de routine. - Connaître les techniques spécialisées et leurs principes généraux.
I- Généralités et définitions. « Histologie » signifie étymologiquement « science des tissus ». Le concept de tissu, de derme, a été inauguré fin XVIIème / début XVIIIème par Xavier Bichat, sans microscope ; ce concept a été élaboré grâce à ses travaux de dissection anatomique.
Xavier Bichat (1771-1802) L'histologie s'est constituée vers le milieu du XIXème siècle à partir de la conjonction de deux événements, en particulier grâce à l'avènement de la théorie cellulaire (1838), établie à partir des travaux de Théodore Schwann et de Schleiden.
Theodor Schwann (1810-1882) Cette théorie postule que tout organisme est constitué de cellules, et est complétée par Virchow en 1858, lorsque ce dernier affirme que toute cellule provient d'une autre cellule : l'unité élémentaire de la vie est donc la cellule. D'autre part, les protozoaires sont constitués d'une seule cellule ; d'autres organismes sont au contraire constitués de plusieurs cellules : ce sont les métazoaires. A partir du début du XIXème siècle, et surtout vers 1840, le microscope optique est perfectionné et l'optique bénéficie de grandes améliorations. Des lentilles beaucoup plus performantes sont en effet

2
fabriquées, ce qui permet d'obtenir des images beaucoup plus proches de la réalité. C'est à cette époque que les techniques histologiques se mettent en place. Un tissu est constitué de cellules et des substances qu'elles synthétisent ; ces substances constituent la matrice extracellulaire ou MEC. Bien que plus complexe, l'examen histologique est aujourd'hui souvent assimilé à l'examen microscopique. L'histologie s'est rapidement intéressée à la morphologie des cellules (l'histochimie), mais aussi à leur fonctionnement et à la conception des tissus (histophysiologie). Au début des années 1960, la création du microscope électronique permet d'améliorer le grandissement et introduit une nouvelle façon de voir les cellules et les tissus. Aujourd'hui, l'histologie a pour but de mettre en évidence au sein de la cellule, in situ (contrairement à la biochimie), les protéines, l'ADN et l'ARNm. On distingue quatre familles de tissus, qui sont étudiés dans le cadre de l'histologie générale : Les épithéliums. Les tissus conjonctifs. Les tissus musculaires. Les tissus nerveux. Ces tissus sont caractérisés par la composition moléculaire de leur MEC, la nature de leur cellule et la proportion de celles-ci par rapport à la MEC. L'histologie spéciale s'intéresse à l'étude de la structure des organes en étudiant l'anatomie microscopique d'une part et l'histophysiologie d'autre part. Les organes sont regroupés en systèmes ou en appareils ; ceux-ci ont chacun une ou plusieurs fonctions bien définies et assurent le fonctionnement de l'organisme. L'anatomie pathologique, quant à elle, étudie les tissus pathologiques. Le diagnostic de nombreuses pathologies repose sur l'examen microscopique de petits échantillons tissulaires appelés biopsies. II- Les préparations tissulaires.
1) Microscopie optique.
Le prélèvement En histologie humaine, le prélèvement peut se faire sur un cadavre (le plus rapidement possible après le décès), sur pièces opératoires (rapidement ou bien les pièces doivent être fixées rapidement) ou encore de façon globale, sans recoupes (= biopsie). On utilise des instruments bien tranchants, afin de ne pas écraser les tissus et donc d'éviter la formation d'artefacts (le Scalpel). La fixation
(Source Wikipédia) Le but de la fixation est de conserver les structures. En effet, le prélèvement des tissus provoque leur mort : les cellules déversent leurs enzymes, ce qui provoque une autodigestion du tissu. De plus, à l'air ambiant, les prélèvements peuvent être contaminés par des bactéries, ce qui entraîne une putréfaction des tissus. Intérêts de la fixation : - immobilisation des constituants tissulaires/cellulaires ; - prévient l’autolyse cellulaire ; - prévient de la putréfaction bactérienne post-mortem ; - permet la technique histologique et les colorations ultérieures.
Le fixateur le plus commun en microscopie optique (MO) et le plus utilisé dans le monde est le formol à 4% (formaldéhyde à 10%). Son principe repose sur le fait qu'il réagit avec les groupements aminés des protéines. La durée de

3
fixation est variable et la quantité de fixateur utilisée doit être au moins dix fois plus importante que le volume de tissu à fixer : quelques heures suffisent donc pour fixer les petits fragments. Le fixateur souvent utilisé en France est le liquide de Bouin (acide picrique+formol+acide acétique+eau). Il présente le désavantage de provoquer un phénomène d'auto-fluorescence cellulaire pouvant gêner les observations au microscope photonique à fluorescence. Il dégrade aussi rapidement les acides nucléiques. Rôle du fixateur : - précipitation, polymérisation, coagulation des protéines ; - mise en place des liaisons covalentes ; - tue les cellules ; - blocage des réactions enzymatiques. L’inclusion
(Source Wikipédia) Pour que la lumière puisse passer à travers le tissu à examiner, celui-ci doit être très fin. Or les tissus sont mous, il faut donc leur donner une consistance solide. C'est le principe de l'inclusion : l'inclusion en paraffine consiste à infiltrer et à enrober les tissus à examiner avec de la paraffine. L'inclusion est précédée de deux étapes essentielles. Il faut tout d'abord procéder à la déshydratation : on passe les tissus dans des bains d'alcool de degré croissant (70°, 80°, 90°, 95°, 99°, puis enfin 100°). L’intérêt de la déshydratation est d’éliminer le fixateur. L'alcool (éthanol) est ensuite remplacé par un solvant miscible à la paraffine : il s'agit soit de xylène, soit de toluène (hydrocarbures). Ces substances éliminent l’éthanol. Au fur et à mesure de leur infiltration par le solvant, les tissus ont tendance à s'éclaircir : cette étape est donc parfois appelée éclaircissement ou clarification. Une fois totalement imprégné, le tissu est placé dans de la paraffine fondue (portée à 56/58°C) ; la chaleur provoque l'évaporation du solvant (et sa dissolution dans la paraffine) : les espaces ainsi libérés sont remplis par la paraffine. Puis la paraffine est placée dans de petits moules, à température ambiante, ce qui provoque son durcissement et donc la rigidification des fragments tissulaires prélevés. On procède alors au démoulage : on obtient des fragments tissulaires inclus dans un bloc de paraffine. La microtomie
(Source Wikipédia) On isole ensuite des coupes dans le bloc de paraffine. On utilise pour cela un microtome, qui fait avancer le bloc sur un rasoir : le bloc avance d'environ 2 à 3 um à chaque fois (en MO classique, les coupes mesurent environ 5 um). L'ensemble des tranches vont former un ruban dans lequel on retrouve des coupes sériées de prélèvement tissulaire. Etalement et collage des coupes sur des lames de verre On chauffe sur une plaque chauffante la paraffine et ainsi la paraffine colle à la lame.

4
Coloration des lames
(Source Wikipédia)
Les tissus de l'organisme ne sont pas spontanément colorés, ce qui rend les observations difficiles. Les colorants utilisés en histologie sont plus ou moins sélectifs ; la plupart sont des composants acides ou basiques en milieu aqueux qui forment des sels avec les radicaux ionisés des tissus. Des composants acides sont utilisés pour les zones tissulaires basophiles, et des composants basiques sont utilisés pour les zones tissulaires acidophiles .
La coloration la plus utilisée est HES : hématéine/éosine/safran. L'hématéine est une substance plutôt basique, qui colore les noyaux en violet donc colore les acides nucléiques. L'éosine est une substance plutôt acide, qui colore plutôt les cytoplasmes (en rose) donc colore les protéines. Enfin, le safran colore les fibres de collagène en jaune.
Cependant, pour que l'on puisse utiliser une coloration, la paraffine doit être éliminée. On procède donc au déparaffinage, qui consiste à passer les lames dans des bains de toluène ou de xylène afin de dissoudre la paraffine. On effectue ensuite une réhydratation : l'alcool se mélange avec l'eau et le toluène, on passe les lames dans des bains d'alcool de degré décroissant (de 100° à 70°, voire 50/40°). On peut également avoir recours à une coloration histochimique, appelée PAS (Periodic Acid Schiff), qui met en évidence le glycogène, les glycoprotéines et à peu près tout ce qui contient des sucres. Les techniques d'immunomarquage sont également assimilées à des techniques de coloration ; on utilise des anticorps couplés à des substances que l'on peut révéler en microscopie (enzymes, substances fluorescentes, ... ) (voir plus loin). L'hybridation in situ, quant à elle, consiste à mettre en évidence des séquences spécifiques d'ADN en utilisant une sonde marquée. Le montage des lames
(Source Wikipédia) Les lames sont montées pour préserver les colorations (plus difficile avec les marquages, pour lesquels on utilise généralement des photos). Les lames sont déshydratées grâce à des bains en toluène, puis on colle des lamelles de verre par-dessus (grâce à des résines synthétiques) afin de préserver les préparations. Les lames ainsi montées peuvent être conservées pendant plusieurs dizaines voire plusieurs centaines d'années. Ces techniques sont cependant mises en défaut pour un certain nombre d'analyse, car l'alcool dissout les graisses.
2) Microscopie électronique. La microscopie électronique (ME) repose exactement sur les mêmes principes que la MO mais un faisceau d'électrons remplace le faisceau de photons et les prélèvements s'effectuent sous vide ; on utilise d'autre part des lentilles magnétiques.
5
La préparation des tissus comporte les mêmes grandes étapes qu'en MO, mais avec des adaptations dues à la contrainte de rapidité qui est encore plus importante pour la fixation. Le fixateur utilisé est le glutaraldéhyde (plusieurs bains, 1heure au total), et l'on effectue généralement une post-fixation grâce à l'acide osmique (tetroxyde d’osmium) afin d'obtenir de meilleures observations. La déshydratation est effectuée grâce à de l'oxyde de propylène. Pour l'inclusion, on utilise des résines synthétiques d'époxy. Celles-ci présentent les avantages suivants : - stable au bombardement électronique, - peu de rétrécissement, - durcissement uniforme. Les désavantages sont que ces résines sont très visqueuses et présente une compatibilité limitée avec l’éthanol. Les coupes sont réalisées sur un ultra-microtome avec rasoir de verre ou de diamant : on obtient des coupes d'environ 80 nm d'épaisseur. Ces coupes sont ensuite déposées sur de petites grilles de cuivre mesurant quelques centimètres de diamètre, et l'observation se fait entre les barreaux. Il n'existe pas de techniques de coloration en ME, mais des techniques de contraste des tissus. On utilise fréquemment l'acétate d'uranyle (qui contraste les nucléoles) et le citrate de plomb, qui contraste les membranes (l'acétate d'uranyle et citrate de plomb sont des sels de métaux lourds). Remarque : La cryohistologie repose sur l'utilisation d'un microtome à congélation (environ -18°C/-25°C) ; utilisée en particulier pour les lipides, elle permet une observation rapide et possède des applications très utiles. Elle est notamment utilisée pour des examens extemporanés (au bloc opératoire), associée à des techniques de coloration rapides mais moins efficaces.
Revenons en détail sur les colorations.
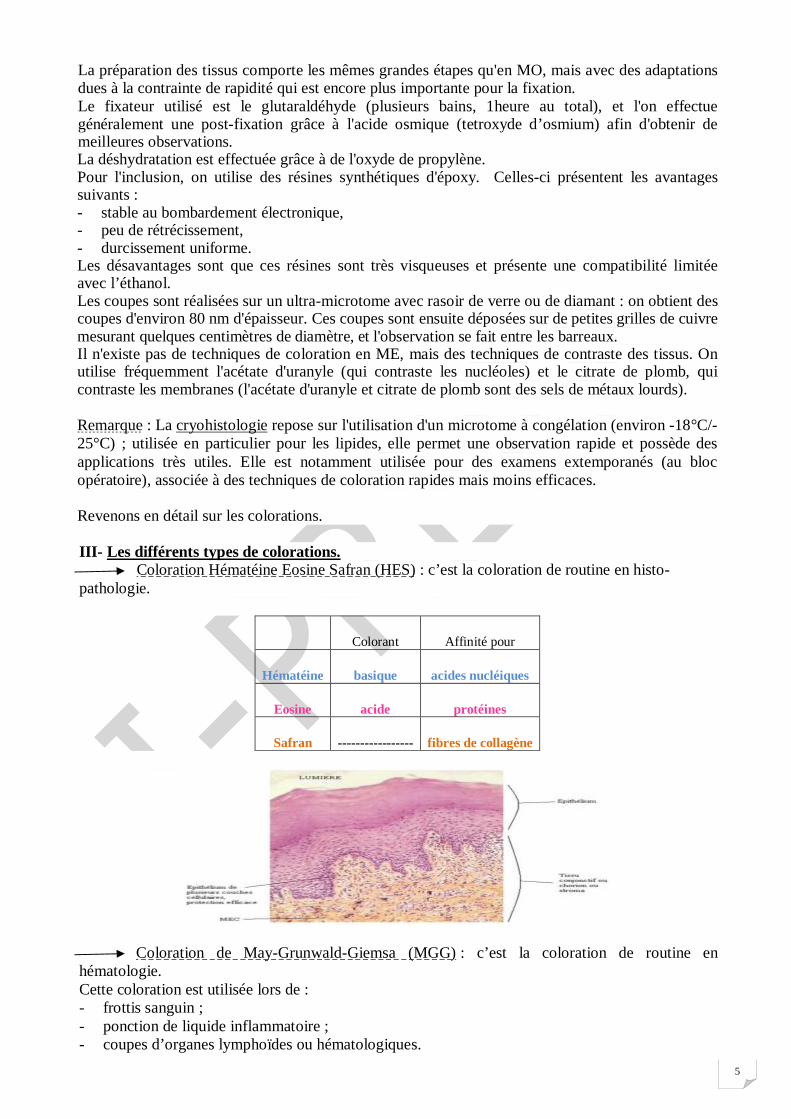
III- Les différents types de colorations. Coloration Hématéine Eosine Safran (HES) : c’est la coloration de routine en histo-pathologie.
Colorant Affinité pour
Hématéine basique acides nucléiques
Eosine acide protéines
Safran ----------------- fibres de collagène
Coloration de May-Grunwald-Giemsa (MGG) : c’est la coloration de routine en hématologie. Cette coloration est utilisée lors de : - frottis sanguin ; - ponction de liquide inflammatoire ; - coupes d’organes lymphoïdes ou hématologiques.
6
MGG Hématologie (coloration)
basophile bleu éosinophile orange neutrophile beige-rose azurophile pourpre
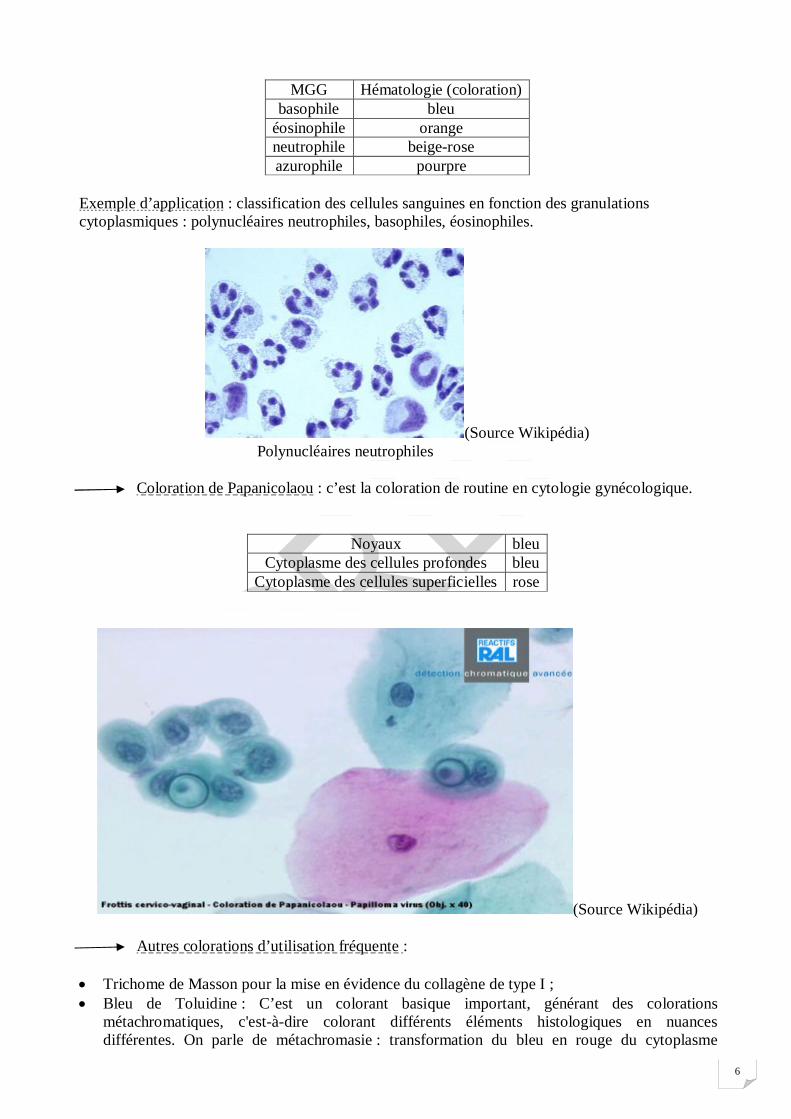
Exemple d’application : classification des cellules sanguines en fonction des granulations cytoplasmiques : polynucléaires neutrophiles, basophiles, éosinophiles.
(Source Wikipédia) Polynucléaires neutrophiles Coloration de Papanicolaou : c’est la coloration de routine en cytologie gynécologique.
Noyaux bleu Cytoplasme des cellules profondes bleu
Cytoplasme des cellules superficielles rose
(Source Wikipédia) Autres colorations d’utilisation fréquente : · Trichome de Masson pour la mise en évidence du collagène de type I ; · Bleu de Toluidine : C’est un colorant basique important, générant des colorations
métachromatiques, c'est-à-dire colorant différents éléments histologiques en nuances différentes. On parle de métachromasie : transformation du bleu en rouge du cytoplasme

7
(mastocytes, polynucléaires basophiles).
Colorations spéciales : mise en évidence d’un composé donné non identifié sur l’HES. · Orcéine : coloration des fibres élastiques. · Réactif de Schiff à l’acide périodique :étude des polysaccharides et des mucines (coloration
pourpre/magenta). · Bleu alcian : mise en évidence de certaines mucines (famille de grandes protéines, fortement
glycosylées entrant dans la composition de nombreux mucus) et de certains glycosaminoglycannes.
Colorations spéciales : détection de l’accumulation de certaines substances : · Détection de métaux comme le fer : coloration de Perls (hémochromatose : maladie
hématologique se caractérisant par une anomalie héréditaire de l'utilisation du fer par l'organisme).
· Détection du cuivre (maladie de Wilson). · Détection du calcium (méthode de Von Kossa).
Colorations spéciales : détection d’agents infectieux : · Détection de Bactéries : coloration de Gram, coloration de Ziehl, coloration de Whartin-Starry · Détection de Champignons : coloration de Grocott.
Colorations spéciales nécessitant un conditionnement particulier : · Coloration de cellules encore vivantes (non fixées). On peut prendre l’exemple du Bleu de
Crézyl qui permet la coloration du RER des réticulocytes ( cellule précédant le stade de globule rouge)
· Mise en évidence d’inclusions lipidiques : coloration par diffusion d’huiles ( Soudan IV, rouge ; Noir Soudan, noir et O red Oil, rouge).Cette coloration ne peut se faire uniquement à partir de tissus/ cellules frais ou congelées car les inclusions lipidiques disparaissent après traitement par le toluène. Discutons de la partie observation avec les différents types de microscopes.
IV- La microscopie
a) Le microscope optique. Le microscope repose sur trois systèmes de lentilles. Le premier condense la lumière sur l’objet à examiner, tandis que le second constitue l’objectif : les lentilles élargissent l’image et la projettent vers le troisième système de lentilles qui lui constitue l’oculaire. Les préparations histologiques sont généralement colorées et observées par trans-illumination. Le grandissement du microscope est égal au produit du grandissement de l’objectif par le grandissement de l’oculaire. Les objets de taille inférieure à environ 0,1-0,2 µm ne sont pas distinguables : cela constitue la limite de la microscopie optique.

8
(Source Wikipédia)
b) Le Microscope à contraste de phase.
C'est le signe d'une différence d'indice de réfraction, densité d'un objet ou encore la vitesse de traversée de l'objet par la lumière.
La lumière va se déplacer différemment dans les différentes structures. La lumière va plus vite dans l'air que dans l'eau que dans le verre. Le microscope à contraste de phase est très utilisé en bactériologie grâce à sa particularité de révélation des objets peu opaques, notamment dans le domaine dentaire dans la recherche en parodontologie, recherche de la flore bactérienne de la gencive dite maladie parodontale ou parodontie.
c) Le Microscope a Fluorescence.
La fluorescence est la propriété que possèdent certains corps d'émettre de la lumière après avoir absorbé des photons de plus haute énergie. La microscopie en fluorescence repose sur la formation d'une image par détection de cette lumière émise.
De nombreuses techniques de marquage peuvent être utilisées :
- Le marquage simple se fait par affinité entre un fluorochrome et la molécule à marquer. - L'immunomarquage direct ou indirect fait intervenir un anticorps marqué.
Application : dénaturation de l'ADN marqué par des fluorochromes.

9
(Source Wikipédia)
d) Le microscope électronique. La microscopie électronique à transmission est une technique de microscopie où un faisceau d'électrons est « transmis » à travers un échantillon très mince. Les effets d'interaction entre les électrons et l'échantillon donnent naissance à une image, dont la résolution peut atteindre 0,8 Angströms. Les images obtenues ne sont généralement pas explicites, et doivent être interprétées à l'aide d'un support théorique. L'intérêt principal de ce microscope est de pouvoir combiner cette grande résolution avec les informations de l'espace de Fourier, c'est-à-dire la diffraction. Il est aussi possible d’étudier la composition chimique de l’échantillon en étudiant le rayonnement X provoqué par le faisceau électronique. Contrairement aux microscopes optiques, la résolution n’est pas limitée par la longueur d’onde des électrons, mais par les aberrations dues aux lentilles magnétiques.
(Source science-et-vie.net)
V- Immunohistochimie (IHC) et immunocytochimie. L'immunohistochimie (ou « IHC ») est le nom d'une méthode de localisation de protéines dans les cellules d'une coupe de tissu, par la détection d'antigènes au moyen d'anticorps. L'immunohistochimie exploite le fait qu'un anticorps se lie spécifiquement à des antigènes dans les tissus biologiques. Les anticorps peuvent être d'origine polyclonale ou monoclonale. a) Intérêt et définitions.
L’intérêt de cette technique est de repérer des antigènes (protéines le plus souvent) d’intérêt au niveau cellulaire ou extracellulaire à l’aide d’anticorps spécifiques.
Un antigène est une molécule capable d’activer le système immunitaire et de conduire à la production d’anticorps.
Un anticorps est une glycoprotéine, sécrétée par les plasmocytes, capable de se lier spécifiquement à l’antigène.

10
(Source svt.edunet) On appelle épitope le site de l’antigène où se fixe l’anticorps.
(Source encyclopedie-enligne)
b) Principes IHC et retour sur les définitions. Ø exploiter la réponse immunitaire de diverses espèces pour produire des anticorps spécifiques.
Ø réaliser la liaison antigène-anticorps spécifique. Ø détecter (visualiser) le complexe antigène-anticorps.
Un antigène possède en général plusieurs épitopes (voir schéma ci-dessus). En fonction de la configuration de l’antigène, un épitope peut ne pas être accessible à l’anticorps. Plusieurs anticorps peuvent se lier à un même épitope avec des affinités différentes. Un même épitope peut parfois être présent sur plusieurs antigènes.
La principale difficulté de l’IHC est de disposer d’un bon anticorps spécifique et pouvant atteindre son épitope sur coupe On distingue 2 types d’anticorps : les anticorps polyclonaux et les anticorps monoclonaux. Les anticorps spécifiques peuvent être fabriqués en injectant à plusieurs reprises un échantillon de l'antigène (protéine à détecter) à un animal (le plus souvent, lapin ou chèvre), et en recueillant ensuite le sérum riche en anticorps (antisérum). Cet antisérum contient différents anticorps dits polyclonaux, produits par différents plasmocytes, reconnaissant divers antigènes de la protéine d'intérêt. Avantages : simple, peu couteux. Inconvénients : - risques de réactions croisées (signal non spécifique) ; - quantités limitées de sérum . Un anticorps monoclonal correspond à une population d'anticorps identiques dirigés contre le

11
même site antigénique d'une protéine. Ces anticorps sont produits en grande quantité en culture par un clone de lymphocytes B selon la technique des hybridomes. Après avoir immunisé une souris contre un antigène donné, on prélève dans sa rate des lymphocytes B. Ceux-ci sont fusionnés avec des plasmocytes tumoraux immortalisés. Après sélection, les hybridomes ainsi obtenus sont une source permanente et stable d'un seul type d'anticorps monoclonal. La spécificité des anticorps monoclonaux est supérieure à celle des sérums polyclonaux, mais leur sensibilité peut être inférieure.
Avantages : un seul anticorps par clone (limite les réactions croisées) ; Inconvénients : - long, coûteux, résultats incertains
c) Technique de l’immunohistochimie L'immunofluorescence directe sur coupe en congélation est une méthode très spécifique qui évite toutes les réactions non spécifiques dues à l'accrochage de l'Ac au récepteur du Fc des immunoglobulines, à condition d'utiliser l'Ac à la dilution optimale. Mais c'est une méthode peu sensible (pas d'amplification du signal) et qui nécessite d'utiliser des Ac couplés à un chromogène fluorescent (Fluorescéine, Rhodamine, Rouge Texas….). Intérêt : permet facilement les doubles marquages et évite les problèmes liés aux peroxydases endogènes. Le marquage est labile dans le temps : des milieux de montages spéciaux permettent d'éliminer l'effacement du marquage. Pour la révélation de l’immunohistochimie indirecte, les enzymes utilisées sont la peroxydase ou la phosphatase alcaline et les chromogènes sont le plus souvent la diaminobenzidine (réaction de couleur brune) ou l'aminoethylcarbazole (réaction de couleur rouge). Il existe dans la méthode immunoenzymatique indirecte un risque de marquage non spécifique par fixation de l'Ac secondaire sur le récepteur du Fc des immunoglobulines : ces sites de fixation non spécifique doivent donc être bloqués au cours de la technique. L’intensité du signal obtenu après marquage d’une réaction antigène-anticorps dépend du nombre de molécules colorées visibles. Plusieurs mécanismes d’amplification sont possibles, parmi lesquelles les méthodes à trois couches, d’autant que l’Ac intermédiaire divalent peut aussi servir à attacher un complexe réactif : peroxydase-antiperoxydase, avidine-biotine péroxydase (ou phosphatase alcaline) et streptavidine-biotine-peroxydase (voir schémas ci-dessous). On peut multiplier le nombre de molécules réactives en les attachant à un polymère (voir schéma ci-dessous )ou bien l'amplification peut être générée par la création d’un complexe qui multiplie les molécules d’enzyme attachées de façon non covalente à l’anticorps (tyramide). L’amplification peut être apportée par l’augmentation du temps d’incubation et aussi par le prétraitement des coupes déparaffinées par la chaleur ou des enzymes.
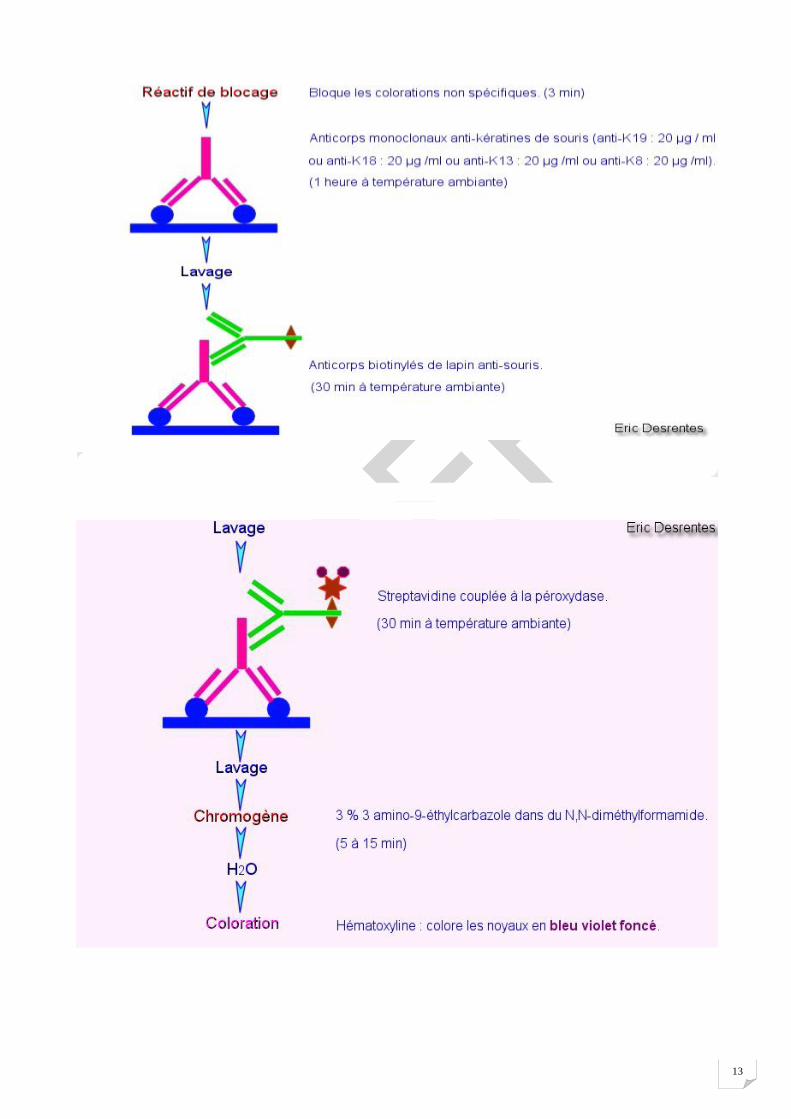
Méthode immunoenzymatique de type streptavidine-biotine-peroxydase : 1) l'anticorps primaire (bleu) se fixe à l'antigène ; 2) l'anticorps secondaire (vert) se fixe à l'anticorps primaire et porte une molécule de biotine (flèche rouge) ; 3) la biotine fixe un complexe streptavidine-peroxydase (croix)
Méthode immunoenzymatique avec amplification du signal par un polymère : 1) l'anticorps primaire (bleu) se fixe à l'antigène ; 2) l'anticorps secondaire (vert) se fixe à l'anticorps primaire et est couplé à un polymère inerte (ligne noire) qui porte plusieurs molécules d'anticorps secondaire et de peroxydase

12
L'utilisation de tissus congelés ou de tissus fixés et inclus en paraffine comporte des avantages et inconvénients : · congélation : nécessité de disposer de tissus frais, d'un système de stockage (au moins à - 80 °C), de pratiquer des coupes au cryostat ; morphologie moins bien conservée que sur des coupes fixées et incluses dans la paraffine. L'intérêt majeur est de pouvoir visualiser des épitopes détruits par des procédés de fixation et d'inclusion. Néanmoins, les indications de l'immunohistochimie sur coupes à congélation tendent à diminuer, d'une part en raison de la fabrication de nouveaux Ac reconnaissant des épitopes résistants à la fixation, d'autres part grâce aux techniques de démasquage antigénique (micro-ondes, autocuiseur, bain-marie) et aux systèmes de révélation ultrasensibles. · tissus fixés inclus en paraffine : les avantages sont l'utilisation a posteriori quand le tissu a été fixé et la possibilité d'études rétrospectives (sur blocs ou sur lames) ainsi qu'une morphologie bien conservée. Mais certains épitopes sont détruits : pour y remédier, nécessité d'une bonne fixation (ni trop courte, ni trop longue). Le choix du fixateur est important (en règle préférer le formol tamponné), car certains épitopes sont détruits par certains fixateurs (alcooliques surtout) et l'éventuelle décalcification. Eviter les températures excessives au moment de l'inclusion et du séchage des lames. Nécessité pour de nombreux épitopes d'un démasquage antigénique (prétraitement qui permet de rompre les liaisons moléculaires créées par le fixateur et modifiant la configuration spatiale des épitopes et leur accessibilité aux antigènes) : digestion enzymatique ou plus souvent chauffage des coupes dans un tampon au four à micro-ondes, autocuiseur, ou bain-marie.
Exemple : Un premier anticorps de souris anti-kératines se fixe spécifiquement sur les kératines. La présence de cet anticorps est révélé par la fixation d'un anticorps secondaire biotynilé de lapin anti-souris (kit DAKO LSAB kit, Peroxidase universale K680). Cette méthode utilise la haute affinité de la streptavidine pour la biotine. La streptavidine est couplée à l'enzyme péroxydase et va se fixer sur l'anticorps secondaire biotynilé. L'activité de la péroxydase induira la coloration du chromogène.
13

14





























