Embed Size (px)
Citation preview

Neuropathology introduction, Part IIBrain biopsy

• Introduction to brain biopsies:– What to look for– Procedures– How to handle– Intraoperative consultation
• Examples on common brain tumors and mimickers

Brain biopsies
• Look for:– Age, sex and main complaints– Family history (present in 16% of patients with
brain tumors)– Previous results– Prior treatment– Purpose of procedure– Imaging features (Location)

Procedures• Biopsy : to determine diagnosis or theraputic resection• Stereotactic needle biopsy:
• through small hole in skull• tissue may be non diagnostic (necrotic, may have missed lesion)• important for pathologist to indicate if lesional tissue is obtained• perform cytology by touching slide to gauze• report presence of large vessel to neurosurgeon
• Post-therapy biopsy• Therapeutic resection: if tumor was not previously completely excised;
may be:• gross total resection (100% removal of known mass)• radical subtotal (95-99% removal)• subtotal (75-95% removal)• partial (10-75% removal)
• compare to prior slides to determine treatment response (fibrosis, necrosis)

What to do with the specimen• Assess the volume• Give priorities according to your differential
diagnoses:• Air dried (can be used for FISH)• Smear (H&E)• Flow cytometry• molecular genetics • Cytogenetics• Paraffin• EM• Culture and sensitivity

Gross pathologyExamples:• Infarcts: section perpendicular to surface of brain, submit sections of
arachnoid, gray and white matter; save gray matter for EM if considering CADASIL or mitochondrial disease
• Hematopoietic lesions: also B5, cell culture media (for flow cytometry), touch preparations, flash frozen tissue for molecular analysis
• Infectious disorders: recommended that surgeon obtain cultures from sterile operating field
• Toxic-metabolic disorders: recommend fixation of gray and white matter in formalin, glutaraldehyde, frozen tissue bank
• Semiliquid or gelatinous masses: usually tumors, including oligodendroglioma, lymphoma, pituitary adenoma
• Vascular malformation: vessels larger than usual aggregate of arachnoid vessels, often associated with hemorrhage, may involve meninges or parenchyma

Margins of resection
• usually impossible to assess due to piecemeal nature of resection or infiltrative growth pattern
• may be able to determine for some tumors like childhood pilocytic astrocytoma of cerebellum, cerebral dysembryoplastic neuroepithelial tumor or meningioma

Staging
• No TNM classification exists for CNS tumors for these reasons:– For T component, histology and location are more
important than tumor size– For N component, brain and spinal cord have no
lymphatics, so lymph nodes cannot be staged– For M component, most patients die before metastatic
disease is identified (except for CSF seeding)– Generally favorable prognostic factors: younger age,
total resection vs. subtotal resection

What to assess?
• Neoplasm: yes or no? Yes - hypercellular, composed of atypical fibrillar cells; if no, may be infectious, inflammatory, toxic-metabolic, traumatic, vascular, development or degenerative
• Primary or metastatic neoplasm?• Glial, neuronal and other primary tumor
considerations• Correlate pathologic diagnosis with age, sex,
location and imaging characteristics

Intraoperative consultation
• Frozen section and smear:– determine if lesional tissue is present– determine if adequate sampling– provide preliminary information to assist
neurosurgeon– perform special techniques (culture, B5 for
lymphoma, touch preparations)

Intraoperative consultation
• For some tumors, attempts at total resection are made (meningioma, schwannoma, solitary metastases, cysts, ependymoma, hemangioblastoma, cerebellar pilocytic astrocytoma, craniopharyngioma), so intraoperative consultation may be helpful

• Recommended to assess undefined lesions by both frozen section and cytologic preparation
• Cytologic preparation: • adds fine nuclear detail• reveals glial-type processes or epithelioid features of
carcinomas• shows discohesiveness associated with pituitary
adenoma, oligodendroglioma, medulloblastoma or lymphoma
• may be more accurate than frozen section


Brain tumors
• Peaks in childhood, then declines to age 25 years, then increases with age
• Childhood tumors: – 33% in anterior fossa– 67% in posterior fossa
• Commonest:– astrocytoma–medulloblastoma/PNET– ependymoma

Brain tumors
• Adults: metastases are the commonest than primary brain tumors (usually not biopsied)
• Of biopsied tumors, 67% arise in anterior fossa– Glioma– meningioma– metastases– pituitary adenoma
• 33% in posterior fossa – schwannoma

Brain tumors• Most common spinal cord tumors:– Schwannoma– meningioma – Ependymoma
• Metastasis of primary CNS tumors outside CNS is rare:– usually along brain and spinal cord via subarachnoid
space– surgery related

Neurofibromatosis type 1• One of the most common genetic disorders• Autosomal dominant• Caused by mutations of a gene (17q) that encodes a protein called
neurofibromin• Neurofibromin is involved in control of cell proliferation and acts as
a tumor suppressor.• Patients with NF1 have a variety of tumors, including bilateral optic
nerve astrocytomas, plexiform neurofibromas and malignant peripheral nerve tumors of peripheral nerves.
• NF1 also causes café au lait spots of the skin, axillary and inguinal freckles, dysplasia of the sphenoid wing and other skeletal abnormalities, fibromuscular dysplasia of arteries and other lesions.

Bilateral acoustic neurofibromatosis (NF2)
• Autosomal dominant condition• Acoustic and spinal schwannomas,
meningiomas, ependymomas• Inactivation of the NF2 gene on chromosome
22q• This gene encodes a structural protein,
schwannomin or merlin, which has tumor suppressor activity.

Von Hippel-Lindau Disease
• Autosomal dominant • Hemangioblastomas of the cerebellum and
retina, cysts of the liver and pancreas, pheochromocytomas, and tumors of the kidneys
• It is linked to VHL, a tumor suppressor gene on chromosome 3p.
• The product of this gene is involved in mRNA transcription.

Some common mutations• LOW-GRADE ASTROCYTOMA:
TP53 mutation • ANAPLASIC ASTROCYTOMA-GLIOBLASTOMA
EGFR amplificationp16 alteration (chromosome 9p loss) PTEN alteration (chromosome 10q loss)
• OLIGODENDROGLIOMA: 1p and 19q loss • MEDULLOBLASTOMA: LOH17p • ATYPICAL TERATOID-RHABDOID TUMOR : Loss of 22q • MENINGIOMA: Loss of 22q • SCHWANNOMA: Loss of 22q, mutations of the NF2 gene

Brain tumors
• Gliomas:• astrocytomas, oligodendrogliomas,
and ependymomas
• Neuronal tumors• Poorly differentiated neoplasms• Meningiomas

• TUMORS OF CRANIAL AND SPINAL NERVES Schwannoma Neurofibroma
• MESENCHYMAL TUMORS Sarcoma Hemangioblastoma
• CEREBRAL LYMPHOMAS• GERM CELL TUMORS
Teratoma Craniopharyngioma
• TUMORS OF THE PITUITARY GLAND Pituitary adenoma
• METASTATIC TUMOR
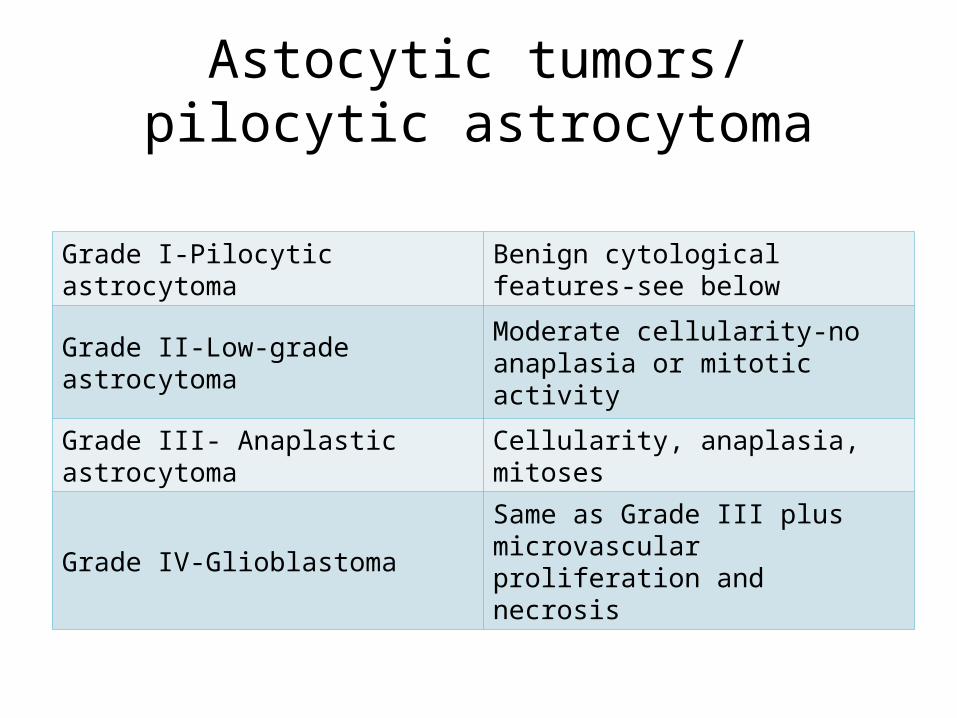
Astocytic tumors/ pilocytic astrocytoma
Grade I-Pilocytic astrocytoma Benign cytological features-see below
Grade II-Low-grade astrocytoma Moderate cellularity-no anaplasia or mitotic activity
Grade III- Anaplastic astrocytoma Cellularity, anaplasia, mitoses
Grade IV-Glioblastoma Same as Grade III plus microvascular proliferation and necrosis

Low grade astrocytoma• Most frequent in children and young adults• Most LGAs are poorly demarcated • Some astrocytomas involve a large part of the
brain or the entire CNS in a diffuse fashion (gliomatosis cerebri).
• Histologically, the tumor cells can be stellate, spindle-shaped with fiber like processes, or plump with a large eosinophilic cytoplasmic mass (gemistocytic astrocytomas)

GLIOBLASTOMA MULTIFORME-WHO GRADE IV
• The most malignant glioma. • It occurs most frequently in middle aged adults. • GBM arises most commonly de novo (primary
GBM). • Some GBMs arise by malignant transformation of
low-grade astrocytomas (secondary GBM). • Imaging shows a the contrast-enhancing
properties of GBM. Contrast enhancing should not be equated with malignancy. Pilocytic astrocytoma also enhances.

GBM
• On naked eye examination , poorly defined mass with variegated (multiform) appearance due to necrosis and hemorrhage.
• Also, large MS lesions, especially Schilder’s disease, may involve both hemispheres and be confused with GBM.
• Microscopically, GBM shows high cellularity, cellular and nuclear anaplasia which is the basis of the designation "multiforme", mitoses, microvascular proliferation, and necrosis.

GBM• Losses of chromosome 10 involving the tumor
suppressor PTEN and other chromosomal loci convert low-grade astrocytoma to anaplastic astrocytoma and GBM.
• Overexpression of the Epidermal Growth Factor Receptor (EGFR) gene on 7p characterizes GBMs that arise de novo (primary GBMs) and provides a potential target for EGFR inhibitors.
• The status of expression of these genes and others that interact with them determines the response of GBM to tyrosine kinase inhibitors and temozolomide, which are used in GBM chemotherapy.

PILOCYTIC ASTROCYTOMA-WHO GRADE I
• astrocytoma of children and young adults. • Most PAs arise in the cerebellum and
hypothalamus.
• Grossly, circumscribed and often cystic. Histologically, they are sparsely cellular tumors without anaplasia or mitoses. They show a biphasic pattern, consisting of cellular and fibrillary perivascular areas, alternating with loose microcystic zones.

Pilocytic astrocytoma
• The tumor cells often contain Rosenthal fibers and granular eosinophilic droplets.
• The word pilocytic (hair cell) refers to the fiber-like appearance of the tumor cells and their fibrillary stroma, but large parts of these tumors, especially in the loose areas, do not fit this description.
• PAs are highly vascular and enhance with contrast injection.

OLIGODENDROGLIOMA • Usually affects the cerebral hemispheres of middle-aged adults.• They are insidious, slow-growing• Microscopically, the tumor cells are uniform and have round central
nuclei surrounded by a clear space or halo (unstained cytoplasm) which is an artifact of processing.
• They infiltrate the cortex diffusely. • Oligodendrogliomas are traversed by delicate capillaries and have a
tendency to calcify• Some oligodendrogliomas have an astrocytic component. Such
mixed tumors are called oligoastrocytomas.• Oligodendrogliomas are among the most chemosensitive solid
tumors. They show losses of chromosomes 1p and 19q which correlate with increased sensitivity to PVC and temozolomide chemotherapy and longer survival.

EPENDYMOMA• Ependymomas are predominantly tumors of children and
adolescents. • They arise most frequently in the fourth ventricle and
cause hydrocephalus by blocking CSF flow. • However, they may occur anywhere in relation to the
ventricular system or central canal and are the most common primary intra-axial tumors in the spinal cord and filum terminale.
• Ependymomas are well demarcated from the surrounding brain and spinal cord and grow in an exophytic fashion, protruding into and out of the fourth ventricle. Spinal ependymomas are circumscribed intra-axial masses.

• An anaplastic version of ependymoma, called ependymoblastoma, is seen infrequently in young children.
• Most ependymomas are histologically and biologically low-grade, but surgical resection of fourth-ventricle ependymomas is difficult.

MEDULLOBLASTOMA
• Medulloblastoma is the second most frequent BT in children after pilocytic astrocytoma.
• Most medulloblastomas occur in the first decade of life. There is a second peak in the early 20s
• The term primitive neuroectodermal tumor (PNET), which has been applied to medulloblastoma and other "small blue cell tumors" of the brain, reflects the embryonal nature and undifferentiated appearance of these tumors.

MEDULLOBLASTOMA• Medulloblastomas are tumors of the cerebellum,
arising usually in the midline, especially in the posterior vermis.
• A few of them, usually in older patients, arise in the cerebellar hemispheres.
• On MRI imaging, they are mostly compact, isointense and show contrast enhancing.
• On gross examination, medulloblastomas are soft, pink-red, and well demarcated.
• They can block the fourth ventricle and the aqueduct, causing hydrocephalus.

MEDULLOBLASTOMA
• Microscopically, they are highly cellular and are composed of diffuse masses of small, undifferentiated oval or round cells.
• Some medulloblastomas show neuronal, glial and other differentiation.
• Neuronal differentiation is manifested by neuropil and rosette formation.
• Rosettes are groups of tumor cells arranged in a circle around a fibrillary center.
• Infrequent mature neurons may also be found in medulloblastomas.
• Glial differentiation in some tumors is reflected by GFAP-positive cells.
• More unusual lines of differentiation result in formation of striated muscle cells (medullomyoblastoma) and melanin-producing cells.

Medulloblastoma
• Medulloblastoma is a highly malignant tumor. It infiltrates and destroys brain tissue and tends to seed the subarachnoid space and spread along the walls of the ventricles.
• Treatment combines resection, to reduce the tumor mass and decompress the fourth ventricle, shunting of the lateral ventricles, radiation of the tumor bed and the entire neuraxis and intrathecal chemotherapy.
• Medulloblastoma is sensitive to radiation.

MENINGIOMA • Meningiomas arise from arachnoidal cells.• They constitute about 20% of BT and • affect mostly adults, women almost twice as
frequently as men. • They may be located anywhere in the brain or spinal
cord• Meningiomas are circumscribed; they may be attached
to the dura, though they do not arise from the dura per se.
• Usually, they displace brain tissue without invading it. Some meningiomas grow flat on the surface of the brain.

• Microscopically, they have a variety of appearances• Classified into several histological types, most of which
have no prognostic significance• Meningothelial meningiomas are composed of diffuse
masses of arachnoidal-like cells• Transitional meningiomas, tumor cells are arranged in
whorls with hyalinized and calcified• Fibroblastic meningiomas are composed of fascicles of
fiber-like cells with abundant interstitial collagen• Many meningiomas are histologically mixed.• Meningiomas tend to infiltrate overlying bone, even
muscle. This peculiar phenomenon does not indicate malignancy.

• Malignant meningiomas are relatively infrequent.• They display overt histological anaplasia and
increased mitoses and invade the brain.• About 10% of meningiomas display histological
features that are intermediate between benign and malignant meningiomas, such as increased cellularity, mitotic activity, a diffuse patternless cellular growth, and necrosis.
• These atypical meningiomas grow more rapidly and are more prone to recur after surgical resection.

SCHWANNOMA
• Schwannomas arise most often in cranial and spinal nerve roots and peripheral nerves
• Ninety percent of schwannomas arise in the 8th nerve root (acoustic Schwannoma, cerebellopontine angle tumor)
• Microscopically, they consist of fascicles of spindle cells that are arranged in palisades. Less frequently they form a loose reticular pattern.
• They are benign, slow-growing tumors and cause symptoms by compression

NEUROFIBROMA
• Neurofibromas are peripheral nerve tumors composed of a mixture of Schwann cells and fibroblasts..
• They cause a fusiform enlargement of the nerve in which they arise.
• Microscopically, their cells are loosely arranged in a wavy pattern.
• Multiple neurofibromas that involve long segments of peripheral nerves (plexiform-from a Greek word that means braid- neurofibromas) are characteristic of NF1.

CRANIOPHARYNGIOMA• Children and adolescents, suprasellar• Thought to arise from epithelial remnants of Rathke's pouch that
are trapped in the pituitary stalk.• Grossly, they show a mixture of solid and cystic areas.• Microscopically, they are composed of sheets of squamous
epithelial cells and keratin, set in a loose connective tissue stroma. • Islands of keratin often calcify. • Water accumulating in the central portion of the epithelial islands
causes them to loosen, creating an appearance that resembles adamantinoma.
• Cholesterol crystals, formed from break down of keratin, float in the greasy fluid that fills the cysts giving it an iridescent appearance
• Cysts, calcification, and the suprasellar location are the criteria for the radiological diagnosis of craniopharyngiomas.
• Other common suprasellar tumors are pilocytic astrocytoma and germ cell tumors.

HEMANGIOBLASTOMA• Hemangioblastomas are sporadic or familial. • The latter are associated with the von Hippel Lindau
disease.• They occur in young to middle-aged adults. Typically,
they are found in the cerebellum as a mural nodule within a cyst.
• In von Hippel Lindau disease, there are multiple hemangioblastomas involving the retina, spinal cord, and brain.
• Hemangioblastoma is a benign tumor which consists of numerous delicate capillaries set in a background of clear foamy cells.

CEREBRAL LYMPHOMA • Primary cerebral lymphomas are thought to arise from indigenous
brain histiocytes (microglia) or from rare lymphocytes that are normally present in the meninges and around vessels.
• Most often, they affect immunosuppressed• The brain, especially subarachnoid space, is also a frequent site of
metastasis of systemic lymphoma and leukemia. • Cerebral lymphomas are single or multiple, poorly defined tumors
with necrosis, similar to glioblastoma.• Microscopically, most of them are large, B-cell lymphomas. The
tumor cells form dense perivascular sheaths or diffuse masses. • Meningeal spread is very common and some cerebral lymphomas
arise in the subarachnoid space. • Cerebral lymphomas, like their extracerebral high-grade
counterparts, are highly malignant.

METASTATIC TUMORS
• Men:• the most common primary is carcinoma of the lung,
• Women:• carcinoma of the breast
• The tumor with the highest rate of metastasis is melanoma.
• Metastatic tumors are frequently multiple. Meningeal carcinomatosis (diffuse spread of tumor in the subarachnoid space)

Demyelinative diseases
• Loss of myelin with variable loss of axons. • In contrast: infarcts, contusions, encephalitis,
and other conditions destroy myelin and axons equally.
• Autoimmune inflammatory diseases. • There are also virus-induced demyelinative
diseases, such as progressive multifocal leukoencephalopathy.

MULTIPLE SCLEROSIS• It is characterized by multiple lesions (plaques) involving the
brain and spinal cord and has an unpredictable clinical course.• MS affects one in every 500 persons, women twice as frequently
as men.• It is more common in young adults• Causes a variety of neurological deficits (visual or sensory loss,
paralysis, ataxia, brainstem signs, psychiatric, dementia). • Many MS cases evolve over a long period (20-30 years) with
remissions and exacerbations. • Some cases have an acute, fulminant, even fatal course, and
others go into a relentlessly progressive phase after a period of remissions and exacerbations.

Predisposing factors
• Genetic factors: • relatives of patients is 7 times higher than in the
general population. • Environmental factors:• higher incidence in high latitude zones. Prevalence
in the northern US is 4-6 times higher than in the South.
• Viruses, particularly measles and HTLV-1, have been suspected (but not proven) to be involved in the pathogenesis of MS.

MS• The evolution of the MS plaque is as follows:
• acute phase, activated mononuclear cells destroy myelin and to a variable degree, oligodendroctes.
• With time, gliosis develops, and plaques reach a burned-out stage consisting of demyelinated axons traversing glial scar tissue.
• In more advanced lesions, remyelination is ineffective because gliosis creates a barrier between the myelin producing cells and their axonal targets.
• The pathological process may be arrested at any time, sometimes after partial demyelination.
• In fulminant MS cases, large lesions with diffuse activity develop and expand inexorably. Although myelin is preferentially affected, axonal loss is significant, and necrosis and cavitation may develop, especially in severe, acute lesions.

MS• Because of the predilection of plaques for the optic
nerves, most MS patients present with visual loss (optic neuritis).
• Spinal lesions cause paralysis and sensory loss (transverse myelitis).
• Usually, these patients have plaques elsewhere in the brain or develop them later.
• These other plaques may be clinically silent, whereas the optic and spinal lesions always cause symptoms.

MS
• Biopsy diagnosis of acute MS (especially with stereotactic needle biopsies) may be tricky because cellularity and reactive astrocytes in the lesions may be misinterpreted as a neoplasm.

MS plaques• Usually multiple• Long-standing plaques are firm (sclerosis)
because of gliosis• Plaques are randomly distributed• Predilection for the periventricular white matter,
optic nerves, and spinal cord but spare no part of the CNS
• They may involve gray matter such as cerebral cortex, deep nuclei, and brainstem. In these locations, they involve myelinated axons while sparing the neuronal bodies.

Neuromyelitis optica-NMO (Devic's disease)
• A special monophasic or relapsing inflammatory demyelinative disease that involves the optic nerves and spinal cord
• IgG (NMO IgG) and complement are deposited in the lesions along the blood-brain barrier (BBB).
• The IgG represents antibodies to the water channel protein Aquaporin-4 (AQP4), which is present in astrocytic processes along the BBB.
• A commercially available test, which detects NMO IgG in serum, distinguishes NMO from other inflammatory demyelinative diseases
• Acute MS and NMO cause extensive axonal damage.

ACUTE DISSEMINATED ENCEPHALOMYELITIS
• An acute demyelinating disease, most commonly affects children. • Commonly follows a viral infection.• After an initial phase characterized by malaise, fever, headache, stiff neck and
drowsiness, multifocal deficits (hemiplegia, ataxia, sensory abnormalities, visual loss) appear within hours or a few days.
• MRI :• Shows multiple bilateral hyperintense lesions in the centrum semiovale and deep
nuclei. • Some large confluent lesions resemble brain tumors because of perilesional edema
and mass effect. • Microscopic:
• Perivenous inflammation (T- lymphocytes and macrophages), demyelination, and axonal loss
• ADEM is not an infection. It is thought to be a T-cell mediated immune reaction triggered by the preceding viral infection or vaccination.
• Similarity between viral and myelin proteins probably accounts for some ADEM cases.

CENTRAL PONTINE MYELINOLYSIS
• degeneration of a symmetrical midline patch of the basis pontis.
• Neurons of the nuclei pontis are relatively spared. • No inflammation is seen. • Initially was thought to be a complication of
alcoholism. • It occurs:• in hyponatremic patients when hyponatremia is
corrected rapidly• in patients with severe hyperosmolality that was not
preceded by hyponatremia.

• Pathologyoutlines.com• www.neuropathologyweb.org• Lester, Manual of surgical pathology













![PET Molecular Imaging Directed Biopsy: A Review · of pediatric brain tumor patients. Lopez et al. [18] correlated the histopatho - logic criteria of biopsy sites with PET uptake](https://img.pdfslide.net/doc/110x75/5ede100ead6a402d666955bf/pet-molecular-imaging-directed-biopsy-a-review-of-pediatric-brain-tumor-patients.jpg)















