Embed Size (px)
Citation preview

mmaMmmaam :||i|||i:||ip:l||||Ii|l||B;:i
Low endotoxin carbohydrates for the life sciences. Very low endotoxin carbohydrates are critical for many biotechnology and pharmaceutical processes. Fermentations, tissue culture work and certain critical pharmaceutical processes are among those that require such sugars. Our in-house technology and production know-how have led to the development of extremely low endotoxin levels in sugars such as maltose, sucrose, D-galactose and others. If your process requires low endotoxin carbohydrates or related compounds, put our products to work. Request information.
PFANSTIEHL LABORATORIES, INC. The source for carbohydrate chemistry 1219 Glen Rock Avenue / Waukegan, IL 60085-0439 1-708/623-0370 •Toll Free: 1-800/383-0126 • FAX:708/623-9173 International: Pfanstiehl (Europe) Ltd. +44 (0) 1606.331825 • FAX: +44 (0) 1606.331826 P7
MAKING THINGS WORK CIRCLE 88 ON READER SERVICE CARD
nusiin-Marketing A World Of Fine Chemicals5*
Custom and Toll Synthesis • Use of Corrosive, Flammable,
Toxic, and Carcinogenic Materials such as Acrylonitrile, Alkylamines, Bromine, Carbon Disulfide, Chlorosulfonic Acid, Dimethyl Sulfate, Epichlorohydrin, Hydrogen Fluoride, Hydrogen Sulfide, Nitric Acid, Nitromethane, Organometallics, Phosgene, Sodium Cyanide, Thionyl Chloride, and Thiophosgene
• Chiral Chemistry
• Equipment for Reactions at -90°C and 300°C
• High Pressure Reactions (1000 psi)
• Pilot Plant Reactors up to 2000 Liters; Production Plant Reactors up to 16,000 Liters
• FDA Inspected Facilities
AUSTIN CHEMICAL COMPANY, INC. 1565 Barclay Boulevard Buffalo Grove, IL 60089 (708)520-9600 FAX (708)520-9160
AUSTIN CHEMICAL COMPANY, INC. 24 South Holmdel Road
Holmdel, NJ 07733 (908)946-8667
FAX (908)946-8682
AUSTIN CHEMICAL COMPANY, INC. Liaison Office-Tianjin, China
CIRCLE 53 ON READER SERVICE CARD
SCIENCE/TECHNOLOGY
working to extend the range of combinatorial chemistry, in which large collections, or libraries, of distinct molecular entities are synthesized in parallel and then screened for potentially useful properties. His lab and others' labs have been involved in screening libraries consisting not only of antibodies and other biomolecules, but also polymers and small organic molecules. Schultz's goal, though, is to extend the combinatorial approach to the entire periodic table. This has led him and his coworkers to prepare and screen libraries of solid-state materials, such as high-temperature superconductors (C&EN, June 26, page 7).
In his lecture, a whirlwind survey of research from his lab, Schultz noted that high-temperature superconductors tend to have complex compositions and structures, and have been discovered largely by trial and error. The combinatorial approach is one way to carry out that discovery process more systematically and efficiently, he believes. And to be sure, a "huge amount of chemistry" is waiting to be discovered in unexplored regions of the solid-state realm, he said.
Schultz ended up taking his mostly biologically minded listeners down (what was for them) a surprising and unfamiliar avenue. But his take-home message fit in beautifully with the "two cultures" theme of the symposium: namely, that chemistry has a lot to learn from biology, and biology has a lot to learn from chemistry. •
New techniques enhance structure-activity work A group of industrial chemists and several academic collaborators are fomenting revolution in the normally sedate field of quantitative structure-activity relationships (QSAR). They are developing a series of QSAR techniques they hope will replace some traditional QSAR concepts that they believe are over the hill.
In QSAR, mathematical "descriptors" of molecular structure are used as a basis for predicting chemical and physical properties or biological activity. QSAR is widely used for drug discovery, among other applications. For example, it can be employed to predict partition coefficients, lipophilicities, or relative reactivi-
38 OCTOBER 9,1995 C&EN
Ή : H I ur,

ties with biological receptors to support drug-design efforts. But Jack W. Frazer of Tuolumne, Calif., team leader of a scientific computing group that consults for Eastman Kodak, believes QSAR techniques up to now "have not realized their true potential//
Several years ago, Frazer's group was asked to bring new insights and computer technology to the drug discovery process at Kodak's Sterling Winthrop pharmaceuticals division (which has since been sold). Frazer says that when he evaluated QSAR as part of the drug-modeling process, "I decided some major changes would be required/'
For example, 'Traditional QSAR uses only one or two linear models to capture a ligand's noncovalent bonding interactions with a protein. . . . You can't capture such a complex thing with one or two simple linear models," says Frazer. And with traditional techniques, "QSAR practitioners never knew the quality of any prediction. When they made a prediction for an
unknown, they didn't know if it was good, bad, or indifferent."
So Frazer and coworkers began developing a series of techniques that Frazer calls "a new paradigm" in QSAR. The techniques include a matrix of models to replace the traditional one or two linear QSAR models, new descriptors that relate properties to local substructure parameters, and equations to assess the quality of predictions. When Frazer presented some of these concepts at a recent Gordon Research Conference on QSAR, he says, "A lot of people liked them but others said they plain didn't understand them.... I've come to the conclusion it's because the paradigm is so different from what they've been used to."
Frazer explains, "I classify a data set into subsets that are dominated by common noncovalent bonding interactions. Then, I construct a matrix of several hundred models using a special variable selection routine that puts me in the right part of descriptor space to make good predictions of the unknown's property values. This is much
different from what QSAR modelers are currently doing."
Frazer says his group also has developed "a new cross-validation that moves you to the part of structure and descriptor space where the linearities more accurately point to the property value of interest. And we qualify every prediction. We haven't got the mathematics refined yet that will tell us the exact correctness of predictions. But we can classify predictions as being in three classes—within the range of measurement noise, quite a ways beyond that range, or just indeterminate."
The matrix of models technique is also capable of modest extrapolation to properties beyond the range of those in the original data set. For instance, Frazer and coworkers have used a data set on cholecystokinin inhibitors to demonstrate the ability of the technique to accurately predict new compounds with greatly improved property values. And Frazer believes new local surface descriptors the team is developing will further improve the technique's capabilities.
More than just lithium. More than just organics.
• Standard lithium-and silicon-based products
• Unique lithium and other organometallic reagents
• Synthetic intermediates based on organolithiums and other organometallics
• Safe handling and analytical support
• Process development assistance
Available Worldwide:
U.S.: Fax:
1-800-362-2548 704-868-5370
Europe: +44(0)51-334-8085 Fax: +44(0)51-334-8501
Japan: 81-06-399-2331 Fax: 81-06-399-2345
Rely On The Industry Leader With More Than Thirty Years Of Experience
ISO 9002 certified -FMC
FMC Corporation, Lithium Division, 449 North Cox Road, Gastonia, North Carolina 28054
CIRCLE 28 ON READER SERVICE CARD
OCTOBER 9,1995 C&EN 39
FMC

SCIENCE/TECHNOLOGY
Chemistry professor Curtis M. Bren-eman at Rensselaer Polytechnic Institute, Troy, N.Y., who collaborates with Frazer's group, explains that traditional QSAR is generally good at predicting interpolated properties, but not extrapolated properties. "Extrapolation is something you don't do because the model blows up/ ' he says.
But Frazer's matrix of models technique "is capable of extrapolation," he says, "and that's a really powerful thing. The way it does it is by not building just one of these models but by building sets of models and by analyzing how well these different sets predict properties."
The ability to extrapolate is important, says Breneman, because drug researchers often want to calculate properties outside existing data sets. "They pretty much know what the activities or properties of molecules in their data set are," says Breneman, "but they want the home run, the drug that's going to really be good. In order to do that you have to be able to extrapolate, to go outside your data set with your predictions."
In addition, he says, by using a matrix of models "you get more information than you do from a single model, so you have a better idea about the quality of the prediction you've made."
According to chemistry professor Peter A. Politzer of the University of New Orleans, another academic collaborator of Frazer's, "A novel aspect of Frazer's approach is that it will permit the design of new molecules with predicted and desired properties, rather than trying to predict the properties of already-designed molecules." Such an application can be viewed as inverse QSAR, says Politzer. "You decide what properties are wanted, and then the method should give you molecules with those desired properties."
Another tool that enables modelers to propose sets of molecules with tailored properties is the transferable atom equivalent (TAE) modeling technique, developed by Breneman and coworkers as part of the QSAR project. TAE-generated van der Waals surface properties of molecules can be used to produce high-quality QSAR indexes.
TAE modeling calculates electron densities and other properties of molecules of any size, including proteins. The technique assembles densities in atomic chunks, based on the "theory of atoms in molecules" developed by
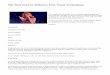
Oxazole Tetrahydropyridine
Pyrrolidine Thiophene
Molecular surface ionization potential (encoded by different colors) is one of many parameters that can be estimated with the transferable atom equivalent (TAE) technique developed by Breneman and coworkers. Surface ionization potential affects the intermolecular interactions of molecules. Numerical data obtained from such TAE-generated plots can be used as descriptors in QSAR models.
chemistry professor Richard F. W. Ba-der of McMaster University, Hamilton, Ontario. "We've extended Bader's method to be able to actually reconstruct molecules from these atomic chunks, which we call transferable atoms," says Breneman.
Breneman points out that TAE is philosophically similar to MEDLA (molecular electron density Lego assembler), a technique developed by Paul G. Mezey and a coworker at the departments of chemistry and of mathematics and statistics of the University of Saskatchewan, Saskatoon (C&EN, Aug. 14, page 29). But unlike MEDLA, TAE can be used to obtain energetics data that can otherwise be calculated only by high-level quantum chemical calculations.
Mezey comments that he sees "the main advantage of Frazer's matrix of models method in the fact that it is multidimensional. Clearly, complex drug interactions cannot be modeled by one or two parameters. A logical
step is then to model molecules by a complete shape description, replacing quantitative structure-activity relationships, QSAR, with quantitative shape-activity relations, QShAR."
He adds, "In our laboratory we have developed shape group methods for QShAR studies in drug design and toxi-cological risk assessment, using quantum chemical shape descriptors of electron density." Mezey has written a book on this topic—"Shape in Chemistry: An Introduction to Molecular Shape and Topology," VCH Publishers, New York, 1993.
According to Breneman, the rationale for supplementing older QSAR techniques with the matrix of models technique and other new QSAR-related methods is, "Let's take what we have here and do a paradigm shift. Let's throw out a lot of old thoughts about things we can't do and try some new techniques that are more related to the actual structure of molecules."
Stu Borman
40 OCTOBER 9,1995 C&EN