Embed Size (px)
Citation preview

Bio-engineering of stem/progenitor cells
Nichotherapy for stem cells:There goes the neighborhood
Jean-Pierre Levesque1)�, Ingrid G. Winkler2) and John E. J. Rasko3)4)�
Stem cells and their malignant counterparts require the
support of a specific microenvironment or ‘‘niche’’. While
various anti-cancer therapies have been broadly success-
ful, there are growing opportunities to target the environ-
ment in which these cells reside to further improve
therapeutic efficacy and outcome. This is particularly true
when the aim is to target normal or malignant stem cells.
The field aiming to target or use the niches that harbor,
protect, and support stem cells could be designated as
‘‘nichotherapy’’. In this essay, we provide a few examples
of nichotherapies. Some have been employed for deca-
des, such as hematopoietic stem cell mobilization,
whereas others are emerging, such as chemosensitization
of leukemia stem cells by targeting their niche.
Keywords:.bone marrow niche; granulocyte colony-stimulating
factor; leukemia; mobilization; Plerixafor; stem cell
Introduction
Adult stem cells require a specific microenvironment in theirtissue of residence to maintain their self-renewal capacity andprevent their differentiation into rapidly proliferating lineage-restricted progenitors. These stem cell-supportive micro-domains were conceived in the late 1970s under the genericterm of ‘‘niche’’ [1]. Thirty years later, many types of niches fordifferent adult stem cells (germinal and somatic) have beenidentified and characterized, providing important mechanisticinsights into the regulation of quiescence and self-renewal ofstem cells [2] (Fig. 1). Therapies that disrupt the interactionsbetween stem cells and their niches have been employed forover two decades to mobilize hematopoietic stem cells (HSCs)into the peripheral blood and facilitate their harvest for sub-sequent transplantation [3]. The importance of the micro-environment and niches is probably not limited to normalstem cells or to the bone marrow (BM). Indeed, several cancersappear to be hierarchically organized with an apical popu-lation of cancer-initiating cells that can self-renew and clo-nally reconstitute the tumor despite treatment [4]. Regardlessof whether cancer-sustaining cells are different to the originalcancer-initiating cells, the tumor microenvironment alsocritically regulates the behavior of cancer stem cells andultimately also that of their progeny and the overall tumor[5–8]. Alterations in BM micro-structure, such as the increasedreticulin observed in myelofibrosis, may be regarded as a formof ‘‘nichopathy’’. Furthermore, evidence is accumulating thatthis tumor microenvironment provides protection and resist-ance against therapies designed to destroy malignant cells [5,9–11]. Therapies that disrupt the interactions between cancerstem cells and the tumor environment to improve cancercytotoxic therapies are emerging.
Many anti-cancer treatments primarily target dividingcells. For instance, cytotoxic chemotherapies or targetedtherapies aim at directly killing the ‘‘malignant’’ cells.However, in some instances, treatments are known to targetthe environment in which these cells reside to further improvetherapeutic efficacy and outcome. In this essay, we review afew examples of the emerging concept of ‘‘nichotherapies’’that target interactions between stem cells and their nichesin the BM, such as (i) stem cell mobilization into the blood,
DOI 10.1002/bies.201200111
1) Stem Cell Biology Group, Biological Therapies Program, Mater MedicalResearch Institute, South Brisbane, Australia
2) Stem Cell and Cancer Group, Biological Therapies Program, MaterMedical Research Institute, South Brisbane, Australia
3) Gene and Stem Cell Therapy Program, Centenary Institute,Camperdown, Australia
4) Sydney Medical School, University of Sydney, and Cell and MolecularTherapies, Royal Prince Alfred Hospital, Camperdown, Australia
*Corresponding authors:Jean-Pierre LevesqueE-mail: [email protected] E. J. RaskoE-mail: [email protected]
Abbreviations:ALL, acute lymphoid leukemia; AML, acute myeloid leukemia; BM, bonemarrow; CML, chronic myeloid leukemia; CXCL12, chemokine C-X-C motifligand-12; CXCR4, chemokine C-X-C motif receptor-4; G-CSF, granulocytecolony-stimulating factor; HSC, hematopoietic stem cell; LSC, leukemia stemcell; MSC, mesenchymal stem cell; PlGF, placental growth factor; VCAM-1,vascular cell adhesion molecule-1.
Bioessays 35: 183–190,� 2012 WILEY Periodicals, Inc. www.bioessays-journal.com 183
Revie
wessays

(ii) sensitization of leukemia cells to chemotherapy by dis-lodging them from their protective niches, and (iii) redirectionof endogenous or transplanted stem cells to specific sitesto increase tissue regeneration. Moreover there is a burgeon-ing literature concerning methods to facilitate ex vivo expan-sion of BM stem and progenitor cells, many of which seekto reproduce cellular and molecular components of theniche [12]. Since much of the knowledge concerning theniche is based on research involving HSC transplantationand HSC niches, our focus and examples in this paperare predominately drawn from this field. However, as aware-ness of other important niches, such as those in the gastro-intestinal tract and solid organs, expands so will the scope ofnichotherapies.
HSC niche in the BM
Normally HSCs are located within specific regions in the BMthat orchestrate their behavior. The complex interplay ofcells, molecules, biophysical states, and biomechanical forcesnot only regulates HSC quiescence and mobilization, butalso modulates the extended microenvironment comprisingdaughter cells and lineage-restricted progenitors (Fig. 1).A variety of different cell types have been identified as con-tributing to the HSC niche, including various mesenchymalstem (or stromal) cell (MSC) populations such as CD146þ
perivascular MSCs [13], nestin-positive MSCs [14], leptinreceptor-positive perivascular MSCs [15], so-called chemokineC-X-C motif ligand-12 (CXCL12) abundant reticular (CAR)cells as well as progeny cells derived from MSCs, suchas osteoprogenitors or osteoblast-lineage cells [16–18].Endothelial cells also form an additional or overlappingHSC niche [15, 19]. These niches appear to be further regulatedby local Schwann cells ensheathing sympathetic nerves [20] aswell as a specialized population of macrophages such as
osteoblast-supportive macrophages called ‘‘osteomacs’’ [21]or macrophages expressing a-smooth muscle actin [22]. Anarea of current controversy surrounds the idea as to whetherdistinct niches maintain distinct HSC populations or fates [23].At present, the so-called ‘‘endosteal’’ or ‘‘osteoblastic’’ nicheis thought to maintain HSCs in a quiescent state, whereas the‘‘vascular’’ niche promotes HSC proliferation and differen-tiation [24–26]. However, the ‘‘endosteal’’ region (bone inter-face) is richly vascularized [27–29], and the ‘‘vascular’’ nicheitself may include perivascular MSCs [13, 30]. Furthermore, itis clear that the most potent and quiescent HSCs, with greatestself-renewal potential, preferentially reside in poorly perfusedregions of the BM. Conversely, more proliferative but lesspotent HSC reside closer to the blood flow [31]. However,vascular niches may not be uniform in function. BM sinusesare flaccid with very low blood velocity, suggesting poor localperfusion [32]. Such vascular niches may harbor quiescentHSCs. This model fits with the observation that HSC nichesare often hypoxic [33, 34] and that the oxygen-labile tran-scription factor HIF-1a regulates HSC quiescence and self-renewal once stabilized in a hypoxic microenvironment [35].In addition, only about 20% of BM endothelial cells expressthe endothelial cell-specific adhesion molecule E-selectin,directly promoting HSC proliferation [26]. Absence or blockadeof this adhesion molecule at the vascular niche increases HSCquiescence and HSC self-renewal potential and can protectendogenous HSC from chemotherapy-induced damage in vivo,facilitating more rapid blood recovery post-chemotherapy [26].Therefore, although the respective contribution of nichecellular and biophysical components is complex and notcompletely understood, there remains great potential for suchtherapies targeting the niche to improve treatment outcome instem cells.
First nichotherapies targeted chemotacticinteractions to mobilize HSCs fortransplantation
HSC transplantation has been in clinical practice for over 50years; however, the means by which HSCs are harvested hasconsiderably changed since its early days. Rather than harvestHSCs for transplantation from a few hundred BM aspirationswith the donor under general anesthesia, cells are nowcollected as peripheral blood stem cells by leukapheresisfollowing their mobilization. The mobilization of HSCs intothe peripheral blood may be considered the first example ofnichotherapy (Fig. 2A). Systemic administration of granulo-cyte colony-stimulating factor (G-CSF) or other cytokinesdramatically alters the BM microenvironment and HSCniches via mechanisms involving specialized niche-supportivemacrophages [21, 36, 37], granulocytes [38–41], and adrener-gic nerves [42]. HSC niche function is rapidly impaired follow-ing G-CSF administration with a down-regulation of factorsthat retain HSCs in their niches, such as CXCL12, vascular celladhesion molecule-1 (VCAM-1) and Kit ligand [43]. In addition,a counter-gradient of sphingosine-1-phosphate created by thecomplement-mediated attack of erythrocytes, may serve toattract HSCs towards the blood [44, 45].
Figure 1. Known components of the HSC niche. Components ofthe extracellular matrix (ECM) including collagen, fibronectin, andlaminin contribute to the physical microenvironment comprisingdiverse biomechanical interactions [53].
J.-P. Levesque et al. Bio-engineering of stem/progenitor cells....
184 Bioessays 35: 183–190,� 2012 WILEY Periodicals, Inc.
Revie
wessays
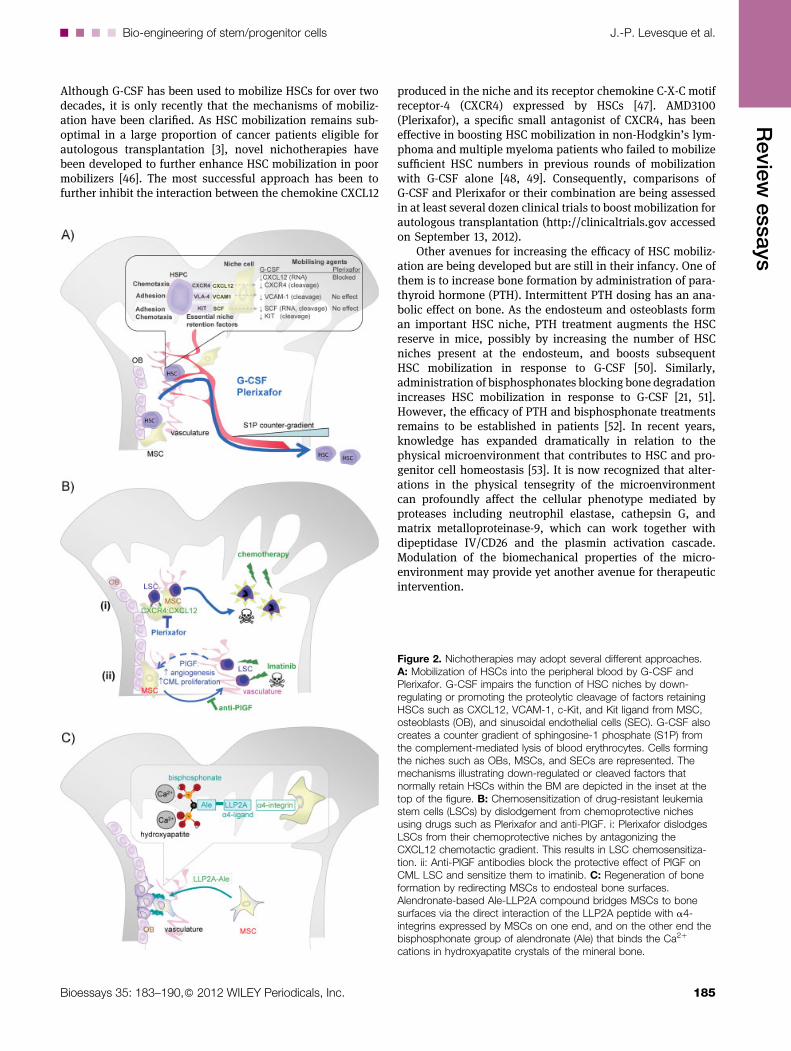
Although G-CSF has been used to mobilize HSCs for over twodecades, it is only recently that the mechanisms of mobiliz-ation have been clarified. As HSC mobilization remains sub-optimal in a large proportion of cancer patients eligible forautologous transplantation [3], novel nichotherapies havebeen developed to further enhance HSC mobilization in poormobilizers [46]. The most successful approach has been tofurther inhibit the interaction between the chemokine CXCL12
produced in the niche and its receptor chemokine C-X-C motifreceptor-4 (CXCR4) expressed by HSCs [47]. AMD3100(Plerixafor), a specific small antagonist of CXCR4, has beeneffective in boosting HSC mobilization in non-Hodgkin’s lym-phoma and multiple myeloma patients who failed to mobilizesufficient HSC numbers in previous rounds of mobilizationwith G-CSF alone [48, 49]. Consequently, comparisons ofG-CSF and Plerixafor or their combination are being assessedin at least several dozen clinical trials to boost mobilization forautologous transplantation (http://clinicaltrials.gov accessedon September 13, 2012).
Other avenues for increasing the efficacy of HSC mobiliz-ation are being developed but are still in their infancy. One ofthem is to increase bone formation by administration of para-thyroid hormone (PTH). Intermittent PTH dosing has an ana-bolic effect on bone. As the endosteum and osteoblasts forman important HSC niche, PTH treatment augments the HSCreserve in mice, possibly by increasing the number of HSCniches present at the endosteum, and boosts subsequentHSC mobilization in response to G-CSF [50]. Similarly,administration of bisphosphonates blocking bone degradationincreases HSC mobilization in response to G-CSF [21, 51].However, the efficacy of PTH and bisphosphonate treatmentsremains to be established in patients [52]. In recent years,knowledge has expanded dramatically in relation to thephysical microenvironment that contributes to HSC and pro-genitor cell homeostasis [53]. It is now recognized that alter-ations in the physical tensegrity of the microenvironmentcan profoundly affect the cellular phenotype mediated byproteases including neutrophil elastase, cathepsin G, andmatrix metalloproteinase-9, which can work together withdipeptidase IV/CD26 and the plasmin activation cascade.Modulation of the biomechanical properties of the micro-environment may provide yet another avenue for therapeuticintervention.
Figure 2. Nichotherapies may adopt several different approaches.A: Mobilization of HSCs into the peripheral blood by G-CSF andPlerixafor. G-CSF impairs the function of HSC niches by down-regulating or promoting the proteolytic cleavage of factors retainingHSCs such as CXCL12, VCAM-1, c-Kit, and Kit ligand from MSC,osteoblasts (OB), and sinusoidal endothelial cells (SEC). G-CSF alsocreates a counter gradient of sphingosine-1 phosphate (S1P) fromthe complement-mediated lysis of blood erythrocytes. Cells formingthe niches such as OBs, MSCs, and SECs are represented. Themechanisms illustrating down-regulated or cleaved factors thatnormally retain HSCs within the BM are depicted in the inset at thetop of the figure. B: Chemosensitization of drug-resistant leukemiastem cells (LSCs) by dislodgement from chemoprotective nichesusing drugs such as Plerixafor and anti-PIGF. i: Plerixafor dislodgesLSCs from their chemoprotective niches by antagonizing theCXCL12 chemotactic gradient. This results in LSC chemosensitiza-tion. ii: Anti-PlGF antibodies block the protective effect of PlGF onCML LSC and sensitize them to imatinib. C: Regeneration of boneformation by redirecting MSCs to endosteal bone surfaces.Alendronate-based Ale-LLP2A compound bridges MSCs to bonesurfaces via the direct interaction of the LLP2A peptide with a4-integrins expressed by MSCs on one end, and on the other end thebisphosphonate group of alendronate (Ale) that binds the Ca2þ
cations in hydroxyapatite crystals of the mineral bone.
....Bio-engineering of stem/progenitor cells J.-P. Levesque et al.
Bioessays 35: 183–190,� 2012 WILEY Periodicals, Inc. 185
Revie
wessays

Dislodging leukemia cells from theirprotective niche to enhancechemotherapies
Myeloid leukemias, including both acute myeloid leukemia(AML) and chronic myeloid leukemia (CML), are hierarchicallyorganized with an apex comprising phenotypically identifi-able leukemia-initiating cells (a subset of leukemia stem cells:LSCs) that self-renew and can clonally re-initiate the leukemia.These leukemia-initiating cells generate the progenitors thatproduce the bulk of leukemia cells [4, 54]. Although theexistence of AML stem cells is mainly supported by xenotrans-plantation experiments into immunodeficient mice [54, 55],direct evidence of a rare CML stem cell population resistant toprolonged tyrosine kinase inhibitor treatment and persistingin patients after years of treatment has been reported [56]. Acommon trait between AML and CML stem cells is their resist-ance to treatment. AML is a heterogeneous disease caused by adiverse collection of genetic lesions (>100) [57]. Due to thisdiversity of causative genetic lesions, conventional cytotoxictreatment using cytarabine combined with an anthracycline(doxorubicin or daunorubicin) has remained the standardtreatment since the early 1980s [58, 59].
In the absence of allogenic transplantation to induce agraft-versus-leukemia response, survival of AML patients inresponse to chemotherapy treatment alone is very poor withapproximately 25–30% survival after three years [59, 60].While 60% of adult patients respond to initial cytotoxicchemotherapy, 60–70% of these responders relapse. Oncerelapsed, the outcome in adult AML is very poor with<10% survival beyond two years. Experiments in the mouseindicate that the high incidence of relapse could be due toa population of leukemia-initiating cells that resist initialtreatment and re-initiate the leukemia when chemotherapyis stopped. Indeed, treatment of immunodeficient micetransplanted with primary human AML cells with cytarabinekills the bulk of AML cells in the BM except in the trabecularendosteal region and along endothelial sinusoids, whereresidual CD34þ leukemia cells survive [9, 61]. This suggeststhat the endosteal and perivascular regions of the BMprovide niches that support and protect LSCs, similar tonormal HSCs, which preferentially reside in these areas ofthe BM [14, 28].
The observations mentioned above provide a rational foradjuvant nichotherapy to dislodge AML stem cells from theirprotective BM niches in order to sensitize LSCs to chemo-therapy. Proof-of-principle was provided in mice usingthe CXCR4 inhibitors AMD3100/Plerixafor and AMD3465,which both block the chemotactic interactions betweenniche stromal cells that express CXCL12 and its receptorCXCR4 on LSCs and HSCs. Treatment of mice withPlerixafor or AMD3465 increased the response of acute pro-myelocytic leukemia to cytarabine [62] and of AML withfetal liver kinase-3 (Flt3) mutations to the Flt3 tyrosinekinase inhibitor sorafenib [63], resulting in decreased leukemiaburden and increased mouse survival [62, 63] (Fig. 2B). Theseencouraging results have prompted the initiation of abouta dozen clinical trials to test the efficacy of Plerixafor inincreasing the sensitivity of malignant cells to chemotherapy
treatment (http://clinicaltrials.gov, accessed on September 13,2012).
The relatively rapid embracing of this form of nichotherapyaimed at sensitizing hematological neoplasms to chemother-apy has expanded to chronic lymphoid leukemia (two trials)and myelodysplastic syndromes (two trials). Considering thevariable resistance of CML-initiating cells to prolonged tyro-sine kinase inhibition, CML may become the next target.Following a similar rationale, it has been recently reportedthat stromal cells from the BM of CML patients produce abnor-mally large amounts of placental growth factor (PlGF), amember of the vascular endothelial growth factor family.PlGF produced in the leukemic BM simulates BM angiogenesisand CML cell proliferation [64]. Interestingly, treatment withan anti-PlGF antibody prolonged survival of mice harboringimatinib-resistant or -sensitive CML cells [64]. Therefore,targeting PlGF produced by BM stromal cells in response toCML cells may enhance the efficacy of tyrosine kinase inhibi-tors such as imatinib (Fig. 2B).
Although no trials targeting B-cell acute lymphoblasticleukemia (B-ALL) have been registered, there is a soundrationale to combine Plerixafor with chemotherapy treatmentfor B-ALL. As ALL cells are highly dependent on L-asparaginefor their survival and proliferation, cytotoxic cocktails ofteninclude asparaginase, an enzyme that degrades asparagine.However, during ALL, BM MSCs express very high levels ofasparagine synthetase [10]. This local production of aspargineby these MSCs may thus protect nearby ALL cells from therapy[10]. As B-ALL cells express high levels of CXCR4, co-admin-istration with Plerixafor may potentially enhance the cytotoxiceffect of asparaginase by dislodging ALL cells from ‘‘protec-tive’’ MSCs. Although it is too early to evaluate the efficacy ofthese newer nichotherapies to treat leukemia, we look forwardto the results of well-designed clinical trials.
Potential nichotherapies for solid tumors
By analogy with hematological malignancies, one can imaginethat similar nichotherapies could be developed for metastasiz-ing cancers that are often chemoresistant. Two prominentcandidates are advanced prostate and breast cancer, whichboth metastasize to the bone and BM. Advanced stages ofthese diseases are incurable with current therapies eitherbecause metastatic cells have acquired additional mutations,rendering them resistant to treatment or, possibly, because theBM and endosteal microenvironment provide additional pro-tection as observed with LSCs. Moreover, the CXCL12-CXCR4chemotactic axis plays a critical role in the metastasis ofprostate [65, 66] and breast cancer cells [67, 68] to the BMand bone, where they establish specific adhesive interactionswith the extracellular matrix of the BM environment [69, 70].There is, therefore, a rationale to test the use of drugs designedto disrupt interactions with the BM microenvironment toincrease sensitivity of metastatic cells to chemotherapies[71, 72] or to reduce the metastatic process by targeting pre-metastatic niches [73–75].
Tumor-associated macrophages are another potential tar-get of nichotherapies. Smoldering inflammation with persist-
J.-P. Levesque et al. Bio-engineering of stem/progenitor cells....
186 Bioessays 35: 183–190,� 2012 WILEY Periodicals, Inc.
Revie
wessays

ence of inflammatory macrophages favors carcinogenesis byreleasing factors that promote proliferation of neighboringcells, and create an oxidative environment that favors geneticlesions [76]. Once the tumor is established, macrophages arerecruited and infiltrate the tumor where they exert a numberof functions, such as supporting tumor cell invasion of theadjacent healthy tissue [77], neo-angiogenesis [78], epithelial–mesenchymal transition [79, 80], metastasis to distant sites[81], and suppression of anti-tumor immunity [82, 83]. Thesemacrophages are believed to be more the alternatively acti-vated M2 type rather than inflammatory macrophages [84].Therefore, specific populations of macrophages are critical toestablish and maintain a niche that supports cancer-initiatingcells in the tumor as well as invading cancer cells at theedge of the tumor. Importantly, in several mouse models ofsolid tumors, macrophage depletion or inactivation decreasedtumor growth and metastasis, prolonged mouse survival[85, 86], and reversed resistance to chemotherapy [87, 88].In these mouse models of tumors, macrophages were depletedwith liposomes loaded with the clodronate [77] or inactivatedwith function-blocking antibodies for the chemokine CCL2 orantagonists of the interaction between CSF-1 and its receptor[85, 86]. Finally, in many types of human tumors, includingHodgkin’s lymphoma [89, 90], high numbers of infiltratingmacrophages are associated with poor prognosis, poorresponse to treatment and reduced survival. The molecularmechanisms by which tumor-associated macrophages promotemetastasis and resistance to treatment are not well understood.To date, the only mechanism identified is the secretion of milkfat globule EGF-like 8 and interleukin-6 by tumor-infiltratingmacrophages. This triggers drug resistance of colon and lungcancer stem cells via a Stat3- and sonic hedgehog-dependentsignaling cascade [88]. Therefore, a more complete understand-ing of the mechanisms may facilitate targeting of tumor-infiltrating macrophages, or molecules that they secrete tocontrol potentially metastatic drug-resistant solid tumors.
Directing MSCs to bone surfaces to inducebone regeneration
Bone formation and homeostasis is controlled by the coupledaction of bone-forming osteoblasts, which are derived fromMSCs, and bone-degrading osteoclasts derived from HSCs.Many bone pathologies might benefit from the transplantationof MSCs to enhance regeneration of a pool of healthy osteo-blasts. Such pathologies include heritable bone disorders suchas osteogenesis imperfecta, mucopolysaccharidoses and mal-reunion following bone fractures. One of the main obstacles tothe use of MSCs to treat bone disorders is the poor homing ofinfused MSCs to the BM and bone surfaces in general. UnlikeHSCs, MSCs are not equipped with a repertoire of cell adhesionreceptors that enable rapid homing to the BM vasculatureand migration through the stroma to reach bone surfaces.In particular, MSCs do not tether and roll on BM endothelialcells due to their lack of expression of fucosyl transferases IVand VII necessary to form the fucosylated tetrasaccharide sialylLewis X [91] that mediates binding to endothelial selectinsE-selectin and P-selectin [92, 93]. As MSCs do not bind selectins,
their homing to the BM is very inefficient [91]. Although MSCscan be genetically modified to express these fucosyltransferases,widespread adoption would likely involve non-geneticallymodified cells. A first strategy to enhance MSC engraftmentinvolves the ex vivo incubation of human BM-derived MSCswith recombinant fucosyl transferase VI, resulting in theaddition of sialyl Lewis X motifs to CD44 and acquisition ofE-selectin binding [94]. Fucosyltransferase-treated human MSCsare able to home into the BM of immunodeficient mice but theircontribution to bone formation [94] remains to be defined.
Despite their inability to bind selectin, human MSCs expresshigh levels of integrin a4 (CD49d) [91] that mediates adhesion toVCAM-1, a cell adhesion molecule expressed by BM endothelialcells [95], and to osteopontin [96], an abundant component ofthe bone matrix. A peptidomimetic ligand of integrin a4 wasdesigned and conjugated to alendronate, a bisphosphonatethat strongly binds to the mineralized hydroxyapatite matrixof the bone. Indeed, the oxygen atoms of the two phosphonategroups from the bisphosphonate molecules (such as alendro-nate) engage in a strong coordination bond with the Ca2þ
cations in hydroxyapatite crystals [97] (Fig. 2C). Ex vivo pre-incubation of human MSCs with alendronate-conjugatedpeptidomimetic (LLP2A-Ale), promoted engraftment of intra-venously injected human MSCs at the periosteum andendosteum of bones in mucopolysaccharidosis type VII immu-nodeficient mice with a consequent increase in the rate of boneformation [98]. This effect is probably caused by the enhancedability of these MSCs to adhere to the bone matrix by bridgingMSCs with the bone matrix via integrin a4. More surprising wasthe effect of LLP2A-Ale in naıve or ovariectomized immuno-competent mice. Two intravenous injections of LLP2A-Ale weresufficient to boost osteoblast numbers on the endosteal surfaceand bone formation in healthy young (8-week-old) mice, and toreduce trabecular bone loss due to aging following ovariectomyin mice [98]. Although not proven, a possibility is that LLP2A-Ale promotes the migration of endogenous MSCs in the BM toendosteal bone surfaces with enhanced osteogenic differen-tiation in non-transplanted mice (Fig. 2C). If this hypothesisis confirmed, a new era of nichotherapy designed to guideendogenous stem cells to a specific site may be born.
The above form of reprogramming via nichotherapy couldbe used to promote commitment and differentiation of stemcells to a specific lineage. Indeed, when human cancer cells areinjected into mouse blastocysts prior to implantation, they canproduce apparently normal chimeric tissues. This surprisingresult indicates that niche-related epigenetic and pro-apoptoticforces can override oncogenic signals [99, 100]. Thus, futurenichotherapies might be harnessed to silence cancer.
Conclusions
The future for nichotherapies designed to mobilize normalstem cells, facilitate chemotherapy, or induce bone regener-ation looks very promising. A large number of clinical trialsarising from a firm foundation in understanding of the HSCniche have been initiated. An increasing awareness of themultiple components of the HSC niche has drawn attentionto the highly integrated homeostatic mechanisms that exist.The HSCs can no longer reasonably be viewed in isolation and
....Bio-engineering of stem/progenitor cells J.-P. Levesque et al.
Bioessays 35: 183–190,� 2012 WILEY Periodicals, Inc. 187
Revie
wessays

must be subjected to a contextual lens. Nichologists: welcometo the new era of nichotherapy!
AcknowledgmentsThe work of J.P.L. and I.G.W. is supported by funds from theNational Health and Medical Research Council (Project Grants# 604303) and from the Cancer Council of Queensland. I.G.W.is the recipient of a Career Development Fellowship from theNational Health and Medical Research Council. The work ofJ.E.J.R. is supported by the Cancer Council of NSW, Rebecca LCooper Medical Research Foundation, NHMRC (Project Grant #1027560) and Cure the Future.
References
1. Schofield R. 1978. The relationship between the spleen colony-formingcell and the haemopoietic stem cell. Blood Cells 4: 7–25.
2. Lymperi S, Ferraro F, Scadden DT. 2010. The HSC niche concepthas turned 31. Has our knowledge matured? Ann N Y Acad Sci 1192:12–8.
3. To LB, Levesque J-P, Herbert KE. 2011. How I treat patients whomobilize hematopoietic stem cells poorly. Blood 118: 4530–40.
4. Magee JA, Piskounova E, Morrison SJ. 2012. Cancer stem cells:impact, heterogeneity, and uncertainty. Cancer Cell 21: 283–96.
5. Joyce JA, Pollard JW. 2009. Microenvironmental regulation of meta-stasis. Nat Rev Cancer 9: 239–52.
6. Bissell MJ, Hines WC. 2011. Why don’t we get more cancer? A pro-posed role of the microenvironment in restraining cancer progression.Nat Med 17: 320–9.
7. Raaijmakers MHGP. 2011. Niche contributions to oncogenesis:emerging concepts and implications for the hematopoietic system.Haematologica 96: 1041–8.
8. Cabarcas SM, Mathews LA, Farrar WL. 2011. The cancer stem cellniche – there goes the neighborhood? Int J Cancer 129: 2315–27.
9. Ishikawa F, Yoshida S, Saito Y, Hijikata A, et al. 2007. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol 25: 1315–21.
10. Iwamoto S, Mihara K, Downing JR, Pui C-H, et al. 2007. Mesenchymalcells regulate the response of acute lymphoblastic leukemia cells toasparaginase. J Clin Invest 117: 1049–57.
11. Lane SW, Scadden DT, Gilliland DG. 2009. The leukemic stem cell niche:current concepts and therapeutic opportunities. Blood 114: 1150–7.
12. Butler JM, Gars EJ, James DJ, Nolan DJ, et al. 2012. Development of avascular niche platform for expansion of repopulating human cord bloodstem and progenitor cells. Blood 120: 1344–7.
13. Sacchetti B, Funari A, Michienzi S, Di Cesare S, et al. 2007. Self-renewing osteoprogenitors in bone marrow sinusoids can organize ahematopoietic microenvironment. Cell 131: 324–36.
14. Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, et al. 2010.Mesenchymal and haematopoietic stem cells form a unique bone mar-row niche. Nature 466: 829–34.
15. Ding L, Saunders TL, Enikolopov G, Morrison SJ. 2012. Endothelialand perivascular cells maintain haematopoietic stem cells. Nature 481:457–62.
16. Calvi LM, Adams GB, Weibrecht KW, Weber JM, et al. 2003.Osteoblastic cells regulate the haematopoietic stem cell niche. Nature425: 841–6.
17. Raaijmakers MHGP, Mukherjee S, Guo S, Zhang S, et al. 2010. Boneprogenitor dysfunction induces myelodysplasia and secondary leukae-mia. Nature 464: 852–7.
18. Visnjic D, Kalajzic Z, Rowe DW, Katavic V, et al. 2004. Hematopoiesisis severely altered in mice with an induced osteoblast deficiency. Blood103: 3258–64.
19. Butler JM, Nolan DJ, Vertes EL, Varnum-Finney B, et al. 2010.Endothelial cells are essential for the self-renewal and repopulationof Notch-dependent hematopoietic stem cells. Cell Stem Cell 6:251–64.
20. Yamazaki S, Ema H, Karlsson G, Yamaguchi T, et al. 2011.Nonmyelinating Schwann cells maintain hematopoietic stem cell hiber-nation in the bone narrow niche. Cell 147: 1146–58.
21. Winkler IG, Sims NA, Pettit AR, Barbier V, et al. 2010. Bone marrowmacrophages maintain hematopoietic stem cell (HSC) niches and theirdepletion mobilizes HSCs. Blood 116: 4815–28.
22. Ludin A, Itkin T, Gur-Cohen S, Mildner A, et al. 2012. Monocytes-macrophages that express alpha-smooth muscle actin preserve primi-tive hematopoietic cells in the bone marrow. Nat Immunol 13: 1072–82.
23. Kiel MJ, Morrison SJ. 2008. Uncertainty in the niches that maintainhaematopoietic stem cells. Nat Rev Immunol 8: 290–301.
24. Trumpp A, Essers M, Wilson A. 2010. Awakening dormant haemato-poietic stem cells. Nat Rev Immunol 10: 201–9.
25. Wilson A, Laurenti E, Oser G, van der Wath RC, et al. 2008.Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135: 1118–29.
26. Winkler IG, Barbier V, Nowlan B, Jacobsen RN, et al. 2012. Vascularniche E-selectin regulate hematopoietic stem cell dormancy, self-renewaland chemoresistance. Nat Med, in press, DOI: 10.1038/nm.2969.
27. Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, et al. 2005. SLAM familyreceptors distinguish hematopoietic stem and progenitor cells andreveal endothelial niches for stem cells. Cell 121: 1109–21.
28. Lo Celso C, Fleming HE, Wu JW, Zhao CX, et al. 2009. Live-animaltracking of individual haematopoietic stem/progenitor cells in their niche.Nature 457: 92–7.
29. Xie Y, Yin T, Wiegraebe W, He XC, et al. 2009. Detection of functionalhaematopoietic stem cell niche using real-time imaging. Nature 457: 97–101.
30. Sugiyama T, Kohara H, Noda M, Nagasawa T. 2006. Maintenance ofthe hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signal-ing in bone marrow stromal cell niches. Immunity 25: 977–88.
31. Winkler IG, Barbier V, Wadley R, Zannettino ACW, et al. 2010.Positioning of bone marrow hematopoietic and stromal cells relativeto blood flow in vivo: serially reconstituting hematopoietic stem cellsreside in distinct nonperfused niches. Blood 116: 375–85.
32. Branemark P-I. 1961. Experimental investigation of microcirculation inbone marrow. Angiology 12: 293–305.
33. Levesque J-P, Winkler IG, Hendy J, Williams B, et al. 2007.Hematopoietic progenitor cell mobilization results in hypoxia withincreased hypoxia-inducible transcription factor-1a and vascular endo-thelial growth factor A in bone marrow. Stem Cells 25: 1954–65.
34. Parmar K, Mauch P, Vergilio J-A, Sackstein R, et al. 2007. Distributionof hematopoietic stem cells in the bone marrow according to regionalhypoxia. Proc Natl Acad Sci USA 104: 5431–6.
35. Takubo K, Goda N, Yamada W, Iriuchishima H, et al. 2010. Regulationof the HIF-1alpha level is essential for hematopoietic stem cells. CellStem Cell 7: 391–402.
36. Christopher MJ, Rao M, Liu F, Woloszynek JR, et al. 2011. Expressionof the G-CSF receptor in monocytic cells is sufficient to mediate hem-atopoietic progenitor mobilization by G-CSF in mice. J Exp Med 208:251–60.
37. Chow A, Lucas D, Hidalgo A, Mendez-Ferrer S, et al. 2011. Bonemarrow CD169þ macrophages promote the retention of hematopoieticstem and progenitor cells in the mesenchymal stem cell niche. J ExpMed 208: 261–71.
38. Levesque JP, Hendy J, Takamatsu Y, Simmons PJ, et al. 2003.Disruption of the CXCR4/CXCL12 chemotactic interaction during hem-atopoietic stem cell mobilization induced by GCSF or cyclophospha-mide. J Clin Invest 111: 187–96.
39. Levesque JP, Hendy J, Winkler IG, Takamatsu Y, et al. 2003.Granulocyte colony-stimulating factor induces the release in the bonemarrow of proteases that cleave c-KIT receptor (CD117) from the sur-face of hematopoietic progenitor cells. Exp Hematol 31: 109–17.
40. Levesque JP, Takamatsu Y, Nilsson SK, Haylock DN, et al. 2001.Vascular cell adhesion molecule-1 (CD106) is cleaved by neutrophilproteases in the bone marrow following hematopoietic progenitorcell mobilization by granulocyte colony-stimulating factor. Blood 98:1289–97.
41. Winkler IG, Levesque JP. 2006. Mechanisms of hematopoietic stemcell mobilization: when innate immunity assails the cells that make bloodand bone. Exp Hematol 34: 996–1009.
42. Katayama Y, Battista M, Kao WM, Hidalgo A, et al. 2006. Signals fromthe sympathetic nervous system regulate hematopoietic stem cellegress from bone marrow. Cell 124: 407–21.
43. Levesque JP, Helwani FM, Winkler IG. 2010. The endosteal ‘‘osteo-blastic’’ niche and its role in hematopoietic stem cell homing and mobil-ization. Leukemia 24: 1979–92.
44. Ratajczak MZ, Lee H, Wysoczynski M, Wan W, et al. 2010. Novelinsight into stem cell mobilization-plasma sphingosine-1-phosphate is amajor chemoattractant that directs the egress of hematopoietic stem
J.-P. Levesque et al. Bio-engineering of stem/progenitor cells....
188 Bioessays 35: 183–190,� 2012 WILEY Periodicals, Inc.
Revie
wessays

progenitor cells from the bone marrow and its level in peripheral bloodincreases during mobilization due to activation of complement cascade/membrane attack complex. Leukemia 24: 976–85.
45. Golan K, Vagima Y, Ludin A, Itkin T, et al. 2012. S1P promotes murineprogenitor cell egress and mobilization via S1P1-mediated ROS signal-ing and SDF-1 release. Blood 119: 2478–88.
46. Larsen SR, Chng K, Battah F, Martiniello-Wilks R, et al. 2008.Improved granulocyte colony-stimulating factor mobilization of hemo-poietic progenitors using cytokine combinations in primates. Stem Cells26: 2974–80.
47. Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, et al. 2005.Rapid mobilization of murine and human hematopoietic stem andprogenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med 201:1307–18.
48. DiPersio JF, Micallef IN, Stiff PJ, Bolwell BJ, et al. 2009. Phase IIIprospective randomized double-blind placebo-controlled trial of plerix-afor plus granulocyte colony-stimulating factor compared with placeboplus granulocyte colony-stimulating factor for autologous stem-cellmobilization and transplantation for patients with non-Hodgkin’s lym-phoma. J Clin Oncol 27: 4767–73.
49. DiPersio JF, Stadtmauer EA, Nademanee A, Micallef IN, et al. 2009.Plerixafor and G-CSF versus placebo and G-CSF to mobilize hemato-poietic stem cells for autologous stem cell transplantation in patientswith multiple myeloma. Blood 113: 5720–6.
50. Adams GB, Martin RP, Alley IR, Chabner KT, et al. 2007. Therapeutictargeting of a stem cell niche. Nat Biotechnol 25: 238–43.
51. Takamatsu Y, Simmons PJ, Moore RJ, Morris HA, et al. 1998.Osteoclast-mediated bone resorption is stimulated during short-termadministration of granulocyte colony-stimulating factor but is notresponsible for hematopoietic progenitor cell mobilization. Blood 92:3465–73.
52. Ballen KK, Shpall EJ, Avigan D, Yeap BY, et al. 2007. Phase I trial ofparathyroid hormone to facilitate stem cell mobilization. Biol BloodMarrow Transplant 13: 838–43.
53. Holst J, Watson S, Lord MS, Eamegdool SS, et al. 2010. Substrateelasticity provides mechanical signals for the expansion of hemopoieticstem and progenitor cells. Nat Biotechnol 28: 1123–8.
54. Bonnet D, Dick JE. 1997. Human acute myeloid leukemia is organizedas a hierarchy that originates from a primitive hematopoietic cell. NatMed 3: 730–7.
55. Kennedy JA, Barabe F, Poeppl AG, Wang JC, et al. 2007. Comment on‘‘tumor growth need not be driven by rare cancer stem cells.’’ Science318: 1722; author reply 1722.
56. Tang M, Gonen M, Quintas-Cardama A, Cortes J, et al. 2011.Dynamics of chronic myeloid leukemia response to long-term targetedtherapy reveal treatment effects on leukemic stem cells. Blood 118:1622–31.
57. Marcucci G, Haferlach T, Dohner H. 2011. Molecular genetics of adultacute myeloid leukemia: prognostic and therapeutic implications. J ClinOncol 29: 475–86.
58. Yates J, Glidewell O, Wiernik P, Cooper MR, et al. 1982. Cytosinearabinoside with daunorubicin or adriamycin for therapy of acute mye-locytic leukemia: a CALGB study. Blood 60: 454–62.
59. Lowenberg B, Pabst T, Vellenga E, van Putten W, et al. 2011.Cytarabine dose for acute myeloid leukemia. N Engl J Med 364:1027–36.
60. Burnett A, Wetzler M, Lowenberg B. 2011. Therapeutic advances inacute myeloid leukemia. J Clin Oncol 29: 487–94.
61. Ninomiya M, Abe A, Katsumi A, Xu J, et al. 2006. Homing, proliferationand survival sites of human leukemia cells in vivo in immunodeficientmice. Leukemia 21: 136–42.
62. Nervi B, Ramirez P, Rettig MP, Uy GL, et al. 2009. Chemosensitizationof acute myeloid leukemia (AML) following mobilization by the CXCR4antagonist AMD3100. Blood 113: 6206–14.
63. Zeng Z, Shi YX, Samudio IJ, Wang R-Y, et al. 2009. Targeting theleukemia microenvironment by CXCR4 inhibition overcomes resistanceto kinase inhibitors and chemotherapy in AML. Blood 113: 6215–24.
64. Schmidt T, Kharabi Masouleh B, Loges S, Cauwenberghs S, et al.2011. Loss or Inhibition of stromal-derived PlGF prolongs survivalof mice with imatinib-resistant Bcr-Abl1þ leukemia. Cancer Cell 19:740–53.
65. Shiozawa Y, Pedersen EA, Havens AM, Jung Y, et al. 2011. Humanprostate cancer metastases target the hematopoietic stem cellniche to establish footholds in mouse bone marrow. J Clin Invest 121:1298–312.
66. Sun YX, Schneider A, Jung Y, Wang J, et al. 2005. Skeletal localizationand neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate
cancer metastasis and growth in osseous sites in vivo. J Bone Miner Res20: 318–29.
67. Huang EH, Singh B, Cristofanilli M, Gelovani J, et al. 2009. A CXCR4antagonist CTCE-9908 inhibits primary tumor growth and metastasis ofbreast cancer. J Surg Res 155: 231–6.
68. Muller A, Homey B, Soto H, Ge N, et al. 2001. Involvement of chemo-kine receptors in breast cancer metastasis. Nature 410: 50–6.
69. Sloan EK, Pouliot N, Stanley KL, Chia J, et al. 2006. Tumor-specificexpression of alphavbeta3 integrin promotes spontaneous metastasis ofbreast cancer to bone. Breast Cancer Res 8: R20.
70. Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, et al. 2012.Interactions between cancer stem cells and their niche govern meta-static colonization. Nature 481: 85–9.
71. Schuettpelz LG, Link DC. 2011. Niche competition and cancer meta-stasis to bone. J Clin Invest 121: 1253–5.
72. Sun Y, Campisi J, Higano C, Beer TM, et al. 2012. Treatment-induceddamage to the tumor microenvironment promotes prostate cancertherapy resistance through WNT16B. Nat Med, in press, DOI: 10.1038/nm.2890.
73. Erler JT, Bennewith KL, Cox TR, Lang G, et al. 2009. Hypoxia-inducedlysyl oxidase is a critical mediator of bone marrow cell recruitment toform the premetastatic niche. Cancer Cell 15: 35–44.
74. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, et al. 2005.VEGFR1-positive haematopoietic bone marrow progenitors initiate thepre-metastatic niche. Nature 438: 820–7.
75. Eckhardt BL, Francis PA, Parker BS, Anderson RL. 2012. Strategiesfor the discovery and development of therapies for metastatic breastcancer. Nat Rev Drug Discov 11: 479–97.
76. Ben-Neriah Y, Karin M. 2011. Inflammation meets cancer, withNF-kappaB as the matchmaker. Nat Immunol 12: 715–23.
77. Qian B, Deng Y, Im JH, Muschel RJ, et al. 2009. A distinct macrophagepopulation mediates metastatic breast cancer cell extravasation, estab-lishment and growth. PLoS ONE 4: e6562.
78. Pucci F, Venneri MA, Biziato D, Nonis A, et al. 2009. A distinguishinggene signature shared by tumor-infiltrating Tie2-expressing monocytes,blood ‘‘resident’’ monocytes, and embryonic macrophages suggestscommon functions and developmental relationships. Blood 114: 901–14.
79. Bonde AK, Tischler V, Kumar S, Soltermann A, et al. 2012.Intratumoral macrophages contribute to epithelial-mesenchymal tran-sition in solid tumors. BMC Cancer 12: 35.
80. Kawata M, Koinuma D, Ogami T, Umezawa K, et al. 2012. TGF-beta-induced epithelial-mesenchymal transition of A549 lung adenocarci-noma cells is enhanced by pro-inflammatory cytokines derived fromRAW 264.7 macrophage cells. J Biochem 151: 205–16.
81. Sangaletti S, Di Carlo E, Gariboldi S, Miotti S, et al. 2008.Macrophage-derived SPARC bridges tumor cell-extracellular matrixinteractions toward metastasis. Cancer Res 68: 9050–9.
82. Cheng P, Corzo CA, Luetteke N, Yu B, et al. 2008. Inhibition ofdendritic cell differentiation and accumulation of myeloid-derived sup-pressor cells in cancer is regulated by S100A9 protein. J Exp Med 205:2235–49.
83. Marigo I, Bosio E, Solito S, Mesa C, et al. 2010. Tumor-inducedtolerance and immune suppression depend on the C/EBPbeta tran-scription factor. Immunity 32: 790–802.
84. Qian BZ, Pollard JW. 2010. Macrophage diversity enhances tumorprogression and metastasis. Cell 141: 39–51.
85. Abraham D, Zins K, Sioud M, Lucas T, et al. 2010. Stromal cell-derivedCSF-1 blockade prolongs xenograft survival of CSF-1-negative neuro-blastoma. Int J Cancer 126: 1339–52.
86. Lucas T, Abraham D, Untergasser G, Zins K, et al. 2009. Adenoviral-mediated endothelial precursor cell delivery of soluble CD115 sup-presses human prostate cancer xenograft growth in mice. Stem Cells27: 2342–52.
87. Paulus P, Stanley ER, Schafer R, Abraham D, et al. 2006. Colony-stimulating factor-1 antibody reverses chemoresistance in humanMCF-7 breast cancer xenografts. Cancer Res 66: 4349–56.
88. Jinushi M, Chiba S, Yoshiyama H, Masutomi K, et al. 2011. Tumor-associated macrophages regulate tumorigenicity and anticancer drugresponses of cancer stem/initiating cells. Proc Natl Acad Sci USA 108:12425–30.
89. Kamper P, Bendix K, Hamilton-Dutoit S, Honore B, et al. 2011. Tumor-infiltrating macrophages correlate with adverse prognosis and Epstein–Barr virus status in classical Hodgkin’s lymphoma. Haematologica 96:269–76.
90. Steidl C, Lee T, Shah SP, Farinha P, et al. 2010. Tumor-associatedmacrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med362: 875–85.
....Bio-engineering of stem/progenitor cells J.-P. Levesque et al.
Bioessays 35: 183–190,� 2012 WILEY Periodicals, Inc. 189
Revie
wessays

91. Brooke G, Tong H, Levesque JP, Atkinson K. 2008. Moleculartrafficking mechanisms of multipotent mesenchymal stem cellsderived from human bone marrow and placenta. Stem Cells Dev 17:929–40.
92. Homeister JW, Thall AD, Petryniak B, Maly P, et al. 2001. Thealpha(1,3)fucosyltransferases FucT-IV and FucT-VII exert collaborativecontrol over selectin-dependent leukocyte recruitment and lymphocytehoming. Immunity 15: 115–26.
93. Frenette PS, Subbarao S, Mazo IB, von Andrian UH, et al. 1998.Endothelial selectins and vascular cell adhesion molecule-1 promotehematopoietic progenitor homing to bone marrow. Proc Natl Acad Sci USA95: 14423–8.
94. Sackstein R, Merzaban JS, Cain DW, Dagia NM, et al. 2008. Ex vivoglycan engineering of CD44 programs human multipotent mesenchymalstromal cell trafficking to bone. Nat Med 14: 181–7.
95. Jacobsen K, Kravitz J, Kincade PW, Osmond DG. 1996. Adhesionreceptors on bone marrow stromal cells: in vivo expression of vascular
cell adhesion molecule-1 by reticular cells and sinusoidal endothelium innormal and gamma-irradiated mice. Blood 87: 73–82.
96. Grassinger J, Haylock DN, Storan MJ, Haines GO, et al. 2009.Thrombin-cleaved osteopontin regulates hemopoietic stem and progen-itor cell functions through interactions with {alpha}9{beta}1 and{alpha}4{beta}1 integrins. Blood 114: 49–59.
97. Rogers MJ. 2003. New insights into the molecular mechanisms of actionof bisphosphonates. Curr Pharm Des 9: 2643–58.
98. Guan M, Yao W, Liu R, Lam KS, et al. 2012. Directing mesenchymalstem cells to bone to augment bone formation and increase bone mass.Nat Med 18: 456–62.
99. Postovit LM, Margaryan NV, Seftor EA, Kirschmann DA, et al. 2008.Human embryonic stem cell microenvironment suppresses the tumorigenicphenotype of aggressive cancer cells. Proc Natl Acad Sci USA 105: 4329–34.
100. Ruiz-Vela A, Aguilar-Gallardo C, Simon C. 2009. Building a frameworkfor embryonic microenvironments and cancer stem cells. Stem Cell Rev5: 319–27.
J.-P. Levesque et al. Bio-engineering of stem/progenitor cells....
190 Bioessays 35: 183–190,� 2012 WILEY Periodicals, Inc.
Revie
wessays





























