Embed Size (px)
Citation preview

Nutzen und Risiken der
zulassungsüberschreitenden Arzneimittelanwendung
in der ambulant-ärztlichen Versorgung
Dissertation zur Erlangung des akademischen Grades
„Doktor Public Health“ (Dr. P.H.)
vorgelegt der
Universität Bremen SOCIUM
Soziale Ungleichheit und Sozialpolitik Abteilung Gesundheit, Pflege und Alter
von Daniela Boeschen
Bremen, im Mai 2015

1. Gutachter: Prof. Dr. rer. nat. Gerd Glaeske 2. Gutachter: Prof. Dr. med. Michael Freitag, MPH Datum der Disputation: 02. Oktober 2015

Nele und meinen Eltern gewidmet

In dieser Welt gibt es nur zwei Tragödien.
Die eine ist, nicht zu bekommen, was man möchte,
und die andere ist, es zu bekommen.
(Oscar Wilde)

Danksagung Eine Arbeit wie diese ist nur durch die Unterstützung Vieler möglich. Deswegen geht an dieser Stelle
mein Dank an meine (ehemaligen) Kolleginnen und Kollegen der Arbeitsgruppe, an meine Familie
sowie an Freundinnen und Freunde.
Besonders danken möchte ich meinem Doktorvater, Herrn Prof. Dr. Gerd Glaeske: nicht nur für die
vielen hilfreichen Anmerkungen und seine Unterstützung während der Fertigstellung dieser Arbeit,
sondern insbesondere für die berufliche wie persönliche Freiheit, die er mir während der Zeit des
Forschungsvorhabens gewährt hat. Danken möchte ich außerdem Herrn Prof. Dr. Michael Freitag für
die spontane Zusage und die Erstellung des Zweitgutachtens.
Besonderer Dank gilt meinem ehemaligen Kollegen Prof. Dr. Falk Hoffmann, der mir mit vielen wert-
vollen Anmerkungen zu jeder erdenklichen Tages- und Nachtzeit unermüdlich zur Seite stand. Eben-
falls danken möchte ich Dr. Roland Windt und Dr. Judith Günther für die Jahre wertvoller Zusam-
menarbeit.
Ein ganz besonderer Dank für viele konstruktive Gespräche sowie die Entwirrung von Hirn- und
Wortgef(l)echten gilt ebenso meiner Schwester Karen, Dr. Michael Dörks, Angela Fritsch und vor
allem Frieda Höfel, die mich neben dem Korrigieren auch immer nach vorne schauen ließ. Lieben
Dank für gute Ideen, organisatorische Mithilfe und moralische Unterstützung in vielerlei freund-
schaftlicher Hinsicht schulde ich außerdem meinen Brüdern Jörg, Philipp und Tim, Rainer Schmidt,
Anja Höpfner, Susanne Pels, Kerstin Ubl, Bettina Schaper, Lore von Hollen, Renate Kelbling und nicht
zuletzt Antje Heinicke.
Und Martin vir ons jaar saam en die beste in my lewe: ons dogter Nele.
Der größte Dank jedoch geht an meine Eltern, Anne und Herbert Hetkamp. Sie haben mir nicht nur
die nötigen Freiräume zum Schreiben ermöglicht und mich stets motiviert, sondern standen mir
unzählige Male zu den möglichsten und unmöglichsten Momenten zur Seite. Auf ihren bedingungs-
losen Rückhalt kann ich jederzeit vertrauen. Ihnen sowie meiner Tochter Nele habe ich die vorlie-
gende Arbeit gewidmet.
Bremen, im Mai 2015


Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Inhaltsverzeichnis
Einleitung ..................................................................................................................... 7
1 Rechtliche Aspekte der Off-Label-Anwendung ........................................................ 9
1.1 Die Definition des Off-Label Use und seine Abgrenzung zum Unlicensed bzw. Compassionate Use ........................................................................................................................ 12
1.2 Die Vereinbarkeit des Off-Label Use mit den arzneimittelrechtlichen Zulassungsbestimmungen .................................................................................... 13
1.2.1 Die arzneimittelrechtliche Zulassung und ihre Grundlagen ...................... 13
2 Die Vereinbarkeit des Off-Label Use mit dem Krankenversicherungsrecht ............. 18
2.1 Der Off-Label Use und seine Ausnahmen in der Rechtsprechung ........................ 19
2.1.1 Das „Sandoglobulin®-Urteil“ des BSG vom 19.03.2002 ............................ 19
2.1.2 Die „Visudyne®-Entscheidung“ des BSG vom 10.10.2004 ......................... 21
2.1.3 Die „Nikolausentscheidung“ des BVerfG vom 06.12.2005 ........................ 21
2.2 Festlegung des medizinischen Standards durch Richtlinien in der GKV ............... 23
2.3 Der Off-Label Use aus ökonomischen Interessen: SGB V vs. AMG ...................... 25
3 Die Vereinbarkeit des Off-Label Use mit dem Arzthaftungsrecht ............................ 30
4 Die Vereinbarkeit des Off-Label Use mit der Haftung des pharmazeutischen Unternehmers .......................................................................................................................... 34
5 Nutzen und Risiko neuer Arzneimittel .................................................................... 36
5.1 Das Gesetz zur Neuordnung des Arzneimittelmarktes ......................................... 36
5.2 Wirksamkeit und patientenorientierter Nutzen neuer Arzneimittel.................... 37
5.3 Indikationserweiterungen im Bestandsmarkt ...................................................... 40
6 Zwischenfazit .......................................................................................................... 44
7 Analyse des Off-Label Use in der ambulant-ärztlichen Versorgung ......................... 46
7.1 Methodik zur Bewertung des Off-Label Use neuer und etablierter Arzneimittel 48
7.1.1 Methodisches Vorgehen zur Analyse der systematischen Übersichtsarbeiten 49
7.1.2 Leitlinienempfehlungen ............................................................................ 52
7.1.3 Fachinformationen .................................................................................... 52
7.1.4 Nutzenbewertung nach § 35a SGB V......................................................... 53
7.1.5 Methodik der Routinedaten-Analyse ........................................................ 53
8 Gründe für einen möglichen Off-Label Use: Analyse der Versorgungssituation ...... 55
8.1 Off-Label Use aufgrund eines möglichen Diagnose-Bias: COPD vs. Asthma bronchiale 56
8.1.1 COPD und Asthma bronchiale: Definition, Epidemiologie und sozialökonomische Aspekte ................................................................................................................... 57
8.1.1.1 Leitlinien und Therapieempfehlungen ................................................ 65
8.1.1.2 Diskussion der Ergebnisse ................................................................... 69
8.1.2 Neue Mittel bei obstruktiven Atemwegserkrankungen ............................ 75
8.1.2.1 Indacaterol ........................................................................................... 82
8.1.2.2 Roflumilast ........................................................................................... 90
1

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.1.2.3 Aclidiniumbromid ................................................................................ 96
8.1.2.4 Diskussion der Ergebnisse ................................................................... 100
8.2 Off-Label Use aufgrund fehlender Therapieoptionen am Beispiel der chronischen Hepatitis C .............................................................................................................................. 108
8.2.1 Hepatitis C: Definition, Epidemiologie und sozialökonomische Aspekte . 109
8.2.1.1 Leitlinien und Therapieempfehlungen vs. Zulassungsstatus nach Fachinformationen ............................................................................................... 116
8.2.1.2 Diskussion der Ergebnisse ................................................................... 131
8.2.2 Neue Mittel zur Therapie der chronischen Hepatitis C ............................. 138
8.2.2.1 Protease-Inhibitoren ............................................................................ 145
8.2.2.1.1 Boceprevir ................................................................................ 145
8.2.2.1.2 Telaprevir.................................................................................. 150
8.2.2.1.3 Simeprevir ................................................................................ 155
8.2.2.2 NS5A-Inhibitoren ................................................................................. 160
8.2.2.2.1 Daclatasvir ................................................................................ 160
8.2.2.2.2 Ledipasvir ................................................................................. 165
8.2.2.3 Nukleosidische Polymerase (NS5B)-Inhibitoren .................................. 170
8.2.2.3.1 Sofosbuvir ................................................................................. 170
8.2.2.4 Fixkombination aus NS5A-Inhibitor und Protease-Inhibitor + nicht-nukleosidischer Polymeraseinhibitor ............................................................................................. 176
8.2.2.4.1 Ombitasvir + Paritaprevir + Ritonavir (Fix) in Kombination mit Dasabuvir .................................................................................................. 176
8.2.2.5 Diskussion der Ergebnisse ................................................................... 181
9 Fazit und Ausblick ................................................................................................... 191
10 Literaturverzeichnis ................................................................................................ 195
11 Abkürzungen .......................................................................................................... 224
12 Abstract .................................................................................................................. 225
2

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Abbildungsverzeichnis
Abbildung 1: Aufbau des rechtlichen Teils der Arbeit ......................................................................................... 9
Abbildung 2: Verteilung der Arzneimittelzulassungen mit Orphan Drug-Status nach therapeutischem Gebiet (Zeitraum 2000-2014) ............................................................................................................. 42
Abbildung 3: Orphan Drug-Zulassungen pro Jahr in der Europäischen Union ................................................... 43
Abbildung 4: Ursachen-Analyse des Off-Label Use in der ambulant-ärztlichen Versorgung am Beispiel etablierter und neuer Arzneimittel ......................................................................................... 48
Abbildung 5: Aufbau des versorgungsanalytischen Teils der Arbeit .................................................................. 55
Abbildung 6: Häufigkeitsverteilung der Lebenszeitprävalenz von chronischer Bronchitis bei erwachsenen Frauen ..................................................................................................................................... 60
Abbildung 7: Häufigkeitsverteilung der Lebenszeitprävalenz von chronischer Bronchitis bei erwachsenen Männern ................................................................................................................................. 60
Abbildung 8: Häufigkeitsverteilung der Lebenszeitprävalenz von Asthma bei erwachsenen Frauen ................ 63
Abbildung 9: Häufigkeitsverteilung der Lebenszeitprävalenz von Asthma bei erwachsenen Männern............. 63
Abbildung 10: Stufenschema der medikamentösen Therapie im Erwachsenenalter: Asthma bronchiale vs. COPD ....................................................................................................................................... 67
Abbildung 11: Auswirkungen eines diagnoseinduzierten OLU auf die Therapieziele einer Behandlung ............ 71
Abbildung 12: Fehlerarten bzw. Verantwortungsbereiche bei bestätigten Behandlungsfehlern ........................ 73
Abbildung 13: Flowchart der Literaturrecherche zur Fragestellung des Off-Label Use aufgrund möglicher Fehldiagnosen am Beispiel COPD und Asthma bronchiale ..................................................... 76
Abbildung 14: Verteilung der Atemwegstherapeutika-Verordnungen (Anzahl Packungen) je Wirkstoffklasse bei COPD-Patienten (N = 210.250) nach Quartalen (2010-2011) ................................................. 86
Abbildung 15: Anzahl verordneter Packungen Indacaterol je Monat nach Packungsgrößen (2010-2011) ......... 86
Abbildung 16: Versicherte mit mindestens einer Verordnung Indacaterol nach Alter und Geschlecht (2011) ....... ................................................................................................................................................ 87
Abbildung 17: Prozentuale Verteilung der COPD-Patienten mit Indacaterol-Verordnungen nach Anzahl verordneter Packungen im Zeitraum 1 Jahr ab Erstverordnung ............................................. 88
Abbildung 18: Anzahl verordneter Packungen Roflumilast je Monat nach Packungsgrößen (2010-2011) .......... 93
Abbildung 19: Versicherte mit mindestens einer Verordnung Roflumilast nach Alter und Geschlecht (2011) ....... ................................................................................................................................................ 93
Abbildung 20: Prozentuale Verteilung der COPD-Patienten mit Roflumilast-Verordnungen nach Anzahl verordneter Packungen im Zeitraum 1 Jahr ab Erstverordnung ............................................. 94
Abbildung 21: Anzahl verordneter Packungen Aclidiniumbromid je Monat nach Packungsgrößen (2012-2013) .... ................................................................................................................................................ 98
3

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Abbildung 22: Versicherte mit mindestens einer Verordnung Aclidiniumbromid nach Alter und Geschlecht (2013) ...................................................................................................................................... 99
Abbildung 23: Prozentuale Verteilung der COPD-Patienten mit Aclidiniumbromid-Verordnungen nach Anzahl verordneter Packungen im Zeitraum 1 Jahr ab Erstverordnung ............................................. 99
Abbildung 24: Übertragungsweg der gemeldeten Fälle nach Referenzdefinition mit belastbaren Angaben zum wahrscheinlichsten Übertragungsweg .................................................................................. 110
Abbildung 25: Prävalenz in der erwachsenen Bevölkerung – Europäische Länder im Vergleich ....................... 111
Abbildung 26: Übermittelte Hepatitis C-Erstdiagnosen – Deutschland aus den Jahren 2001 bis 2013 ............. 112
Abbildung 27: Übermittelte Hepatitis C-Erstdiagnosen pro 100.000 Einwohner nach Bundesland für das Jahr 2013 ...................................................................................................................................... 113
Abbildung 28: Flowchart der Literaturrecherche zur Fragestellung des Off-Label Use aufgrund fehlender Therapieoptionen am Beispiel der chronischen Hepatitis C ................................................. 139
4

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabellenverzeichnis
Tabelle 1: Die Phasen der klinischen Prüfung ............................................................................................... 17
Tabelle 2: Vergleich der BSG- und BVerfG-Urteile hinsichtlich des Off-Label Use und seine Erstattungsvoraussetzung ....................................................................................................... 22
Tabelle 3: Beispiele für Wirkstoffe mit unterschiedlichen Indikationen ....................................................... 27
Tabelle 4: Risikofaktoren für die Entwicklung einer COPD ............................................................................ 58
Tabelle 5: Differentialdiagnose zur Unterscheidung des Asthma bronchiale von der COPD ......................... 61
Tabelle 6: Vergleich der medikamentösen Behandlung: COPD vs. Asthma bronchiale ................................. 66
Tabelle 7: Übersicht aller eingeschlossenen klinischen Studien .................................................................... 77
Tabelle 8: Übersicht der in der Datenbank des U.S. National Institutes of Health gelisteten Studien .......... 78
Tabelle 9: Versicherte mit Verordnungen des COPD-Medikaments Indacaterol in den Jahren 2010 und 2011 ................................................................................................................................................ 89
Tabelle 10: Versicherte mit Verordnungen des COPD-Medikaments Roflumilast in den Jahren 2010 und 2011 ................................................................................................................................................ 95
Tabelle 11: Umsatz und Absatz der neuen Arzneimittel zur Behandlung der cHC ........................................ 115
Tabelle 12: Übersicht verschiedener Therapieoptionen für die Erst- und Re-Therapie der chronischen Hepatitis C vom Genotyp 1-6 nach Empfehlungen der Fachinformationen ......................... 118
Tabelle 13: Übersicht über nationale und internationale Leitlinien zur Therapie der chronischen Hepatitis C .. .............................................................................................................................................. 119
Tabelle 14: Übersicht verschiedener Therapieregime für die Behandlung der cHC vom Genotyp 1 nach den Empfehlungen der aktuellen DGVS-Leitlinie im Vergleich zum Zulassungsstatus der entsprechenden Fachinformationen .................................................................................... 121
Tabelle 15: Übersicht verschiedener Therapieregime für die Behandlung der cHC vom Genotyp 2 nach den Empfehlungen der aktuellen DGVS-Leitlinie im Vergleich zum Zulassungsstatus der entsprechenden Fachinformationen .................................................................................... 123
Tabelle 16: Übersicht verschiedener Therapieregime für die Behandlung der cHC vom Genotyp 3 nach den Empfehlungen der aktuellen DGVS-Leitlinie im Vergleich zum Zulassungsstatus der entsprechenden Fachinformationen .................................................................................... 125
Tabelle 17: Übersicht verschiedener Therapieregime für die Behandlung der cHC vom Genotyp 4 nach den Empfehlungen der aktuellen DGVS-Leitlinie im Vergleich zum Zulassungsstatus der entsprechenden Fachinformationen .................................................................................... 127
Tabelle 18: Übersicht verschiedener Therapieregime für die Behandlung der cHC vom Genotyp 5/6 nach den Empfehlungen der aktuellen DGVS-Leitlinie im Vergleich zum Zulassungsstatus der entsprechenden Fachinformationen .................................................................................... 129
Tabelle 19: Übersicht verschiedener Therapieregime für die Behandlung der cHC bei dekompensierter Leberzirrhose sowie vor und nach Lebertransplantation vom Genotyp 1-6 nach den Empfehlungen der aktuellen DGVS-Leitlinie im Vergleich zum Zulassungsstatus der entsprechenden Fachinformationen .................................................................................... 130
5

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 20: Übersicht aller eingeschlossenen Studien der Phase I-III ............................................................ 140
Tabelle 21: Top 5 der Fachzeitschriften nach der Anzahl der Publikationen klinischer Studien Phase I-III zwischen 01.01.2009-01.04.2015 zur Therapie der chronischen Hepatitis C ....................... 144
Tabelle 22: Boceprevir: Main Study nach dem Assessment Report der EMA ................................................ 145
Tabelle 23: Boceprevir: Angaben nach Fachinformation ............................................................................... 148
Tabelle 24: Telaprevir: Main Study nach dem Assessment Report der EMA ................................................. 151
Tabelle 25: Telaprevir: Angaben nach Fachinformation ................................................................................ 153
Tabelle 26: Simeprevir: Main Study nach dem Assessment Report der EMA ................................................ 156
Tabelle 27: Simeprevir: Angaben nach Fachinformation ............................................................................... 158
Tabelle 28: Daclatasvir: Main Study nach dem Assessment Report der EMA ............................................... 160
Tabelle 29: Daclatasvir: Angaben nach Fachinformation .............................................................................. 163
Tabelle 30: Ledipasvir: Main Study nach dem Assessment Report der EMA ................................................. 165
Tabelle 31: Ledipasvir: Angaben nach Fachinformation ................................................................................ 168
Tabelle 32: Sofosbuvir: Main Study nach dem Assessment Report der EMA ................................................ 172
Tabelle 33: Sofosbuvir: Angaben nach Fachinformation ............................................................................... 174
Tabelle 34: Ombitasvir + Paritaprevir + Ritonavir plus Dasabuvir: Main Study nach dem Assessment Report der EMA ................................................................................................................................ 177
Tabelle 35: Ombitasvir + Paritaprevir + Ritonavir plus Dasabuvir: Angaben nach Fachinformation ............. 180
6

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Einleitung
Unter einem Off-Label Use (OLU) versteht man die nicht zulassungskonforme Anwendung eines
Arzneimittels, das auf dem Markt zugelassen ist, jedoch anders als in der Zulassung entschieden
eingesetzt wird. Off-Label-Anwendungen sind nicht nur gängige Praxis, sondern gehören oftmals zum
Standard bzw. zur Notwendigkeit einer patientengerechten und patientenindividuellen Therapie
(Hill, 2005; Janzen & Ludwig, 2012; Grell & Rieger, 2012; Poole & Dooley, 2004). Seit langem finden
sie aufgrund des rasanten Fortschritts medizinischer Erkenntnisse in nahezu allen ärztlichen Fachbe-
reichen statt. So stellt der Off-Label Use beispielsweise im Bereich der Onkologie aufgrund der hohen
Krebsinzidenz, der Vielfalt der Tumoren, Krankheitsstadien und Malignitätsgrade sowie Komorbiditä-
ten einen wesentlichen Therapieansatz dar (Müller, 2009; Joerger et al., 2014; Mellor et al., 2012;
Poole & Dooley, 2004). Grund dafür ist, dass sich die Zulassung häufig nur auf bestimmte Tumorenti-
täten und -stadien bezieht, auf die in klinischen Studien spezifisch eine Wirksamkeit nachgewiesen
werden konnte (Müller, 2009). Auch die gängige Praxis der Off-Label-Anwendung in der Pädiatrie hat
in den vergangenen Jahren immer wieder zu Diskussionen und schließlich auch zu Änderungen in der
Gesetzgebung geführt (Mühlbauer et al., 2009; Afentaki, 2014). Ethische, rechtliche und ökonomi-
sche Vorbehalte gegenüber der Forschung an nicht oder nur bedingt einwilligungsfähigen Patienten
erschweren allerdings nach wie vor die Einbeziehung von Kindern in klinische Prüfungen. Wissen-
schaftliche Erkenntnisse aus Untersuchungen an Erwachsenen können jedoch aufgrund entwick-
lungsspezifischer Unterschiede nicht uneingeschränkt auf den pädiatrischen Bereich übertragen
werden. So bleiben Kindern potentiell nützliche Wirkstoffe vorenthalten, wenn auf eine systemati-
sche Forschung verzichtet wird (Steinbrook, 2002).
Die vorliegende Arbeit beschäftigt sich mit dieser Thematik der zulassungsüberschreitenden Anwen-
dung aus Sicht der Versorgungsforschung. Die Frage danach ist von besonderem Interesse, weil sich
zivil- und strafrechtliche Anforderungen und Einschränkungen einerseits und das Leistungsrecht der
gesetzlichen Krankenversicherung andererseits gegenüberstehen und somit unmittelbare Auswir-
kungen auf den Arzt als Leistungserbringer und den Patienten als Leistungsempfänger haben. Wäh-
rend im ersten Teil der Dissertation (Kapitel 1-6) die rechtlichen Aspekte dargestellt werden, analy-
siert der zweite Teil (Kapitel 7-9) mögliche Rahmenbedingungen, die einen Off-Label Use begünsti-
gen. Die Ebene der Arzneimittelzulassung dient dabei als Ausgangspunkt einer vertiefenden Betrach-
tung. Der Fokus richtet sich in erster Linie auf mögliche strukturelle sowie administrative Lücken als
Ursache eines OLU.
Neben der Epidemiologie chronischer Erkrankungen (hier im Speziellen obstruktive Lungenerkran-
kungen sowie Hepatitis C), die eine hohe Relevanz im Public Health-Bereich aufweisen, wird vor
allem die Patientenversorgung in der ambulant-ärztlichen Praxis unter dem besonderen Aspekt der
7

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
zulassungsüberschreitenden Anwendung untersucht. Ziel dieser Arbeit ist es, weitere Gründe eines
OLU zu identifizieren, dadurch den Grenzbereich transparenter zu gestalten und so zur Verbesserung
einer qualitätsgesicherten Versorgung beizutragen.
8

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
1 Rechtliche Aspekte der Off-Label-Anwendung
Im Folgenden (Kapitel 1.1 bis 6) werden die rechtlichen Aspekte einer Arzneimittelanwendung au-
ßerhalb ihrer Zulassungsbestimmung dargestellt. Dabei werden sowohl die zivil- und strafrechtlichen
Anforderungen einerseits und das Leistungsrecht der gesetzlichen Krankenversicherung andererseits
gegenübergestellt (Abbildung 1), um das Spannungsfeld des OLU unter Berücksichtigung aller betei-
ligten Institutionen bzw. Personen aufzuzeigen.
Abbildung 1: Aufbau des rechtlichen Teils der Arbeit
Der Aufbau des rechtlichen Teils der Arbeit ergibt sich folgendermaßen: nach Klärung der Begrifflich-
keiten sowie der zulassungsrechtlichen Grundlagen (Kapitel 1.1), wird die Vereinbarkeit des OLU mit
dem Krankenversicherungsrecht aufgezeigt (Kapitel 2). Schließlich rückt die Frage nach Kriterien der
Erstattungsfähigkeit von Therapien im Allgemeinen und von Arzneimitteln im Speziellen unter der
Bedingung eines begrenzten Ausgabevolumens immer mehr in den Blickpunkt der gesetzlichen Kran-
kenversicherungen (GKV). So soll in diesem Abschnitt der Frage nachgegangen werden, welche Vo-
raussetzungen erfüllt sein müssen, damit ein Arzneimittel in den Bereich der GKV-Leistungspflicht
fällt und ob es möglicherweise Ausnahmen gibt oder der OLU geduldet wird. Jüngste Entwicklungen
zeigen, dass einige Krankenkassen bereit sind, einen Off-Label Use möglicherweise sogar zu fördern,
wenn dadurch wirtschaftliche Vorteile entstehen (Gaßner, 2013). Die gesetzlich verankerten Rabatt-
verträge zwischen den GKVen und den pharmazeutischen Unternehmen (pU) spielen für den Off-
9

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Label Use ebenfalls eine Rolle, die allerdings weit weniger im öffentlichen Fokus steht. Hinsichtlich
der Substituierung von Arzneimitteln aufgrund bestehender Rabattverträge geht es in erster Linie
um den gleichen Wirkstoff in der gleichen Dosierung und der gleichen oder einer vergleichbaren
galenischen Zubereitung. Aufgrund von nicht identischen Zulassungen in manchen Indikationen kann
es aber auch hier zu einem vertraglich induzierten Off-Label Use kommen (Sickmüller & Lietz, 2011).
Das 3. Kapitel setzt sich mit der Vereinbarkeit des OLU und dem Arzthaftungsrecht auseinander.
Schließlich ist es dem Arzt weder arzneimittel- noch strafrechtlich verwehrt, ein Medikament in ei-
nem zulassungsüberschreitenden Bereich einzusetzen. Eher kann der wissentliche Verzicht auf eine
Off-Label-Behandlung strafrechtliche Konsequenzen nach sich ziehen, wenn sich der Zustand eines
Patienten in der Folgezeit verschlechtert (OLG Köln, 1990). Für den medizinischen Alltag bedeutet
außerdem eine spezifische und detaillierte Festlegung des Anwendungsgebietes eine Einschränkung
des ärztlichen (Be-)Handlungsspielraums, der Arzt kann aber im Allgemeinen den aktuellen Kenntnis-
stand schneller berücksichtigen und umsetzen, als eine zunächst einmal mehrere Verfahren durch-
laufende Zulassungserweiterung, die unter Umständen viel Zeit in Anspruch nimmt. Haftungsrechtli-
che Konsequenzen wie die wirtschaftliche Haftung im Prüfverfahren, Haftung für eventuelle Kompli-
kationen durch die verordneten Medikamente, zivilrechtliche und strafrechtliche Haftung sowie
ethische Fragestellungen spielen für den Arzt dennoch eine entscheidende Rolle, wenn es um die
Frage einer Off-Label-Therapie geht (Kölch et al., 2009; Augustin, 2013; Wetterling, 2004).
Off-Label-Anwendungen haben auch für den pU haftungsrechtliche Konsequenzen, der aufgrund
seiner Produktbeobachtungspflicht in der Regel Kenntnis von der Praxis des zulassungsüberschrei-
tenden Einsatzes seines Produkts hat bzw. haben sollte. So befasst sich das 4. Kapitel mit dem OLU
aus Sicht der pharmazeutischen Industrie. Denn aus einem rein ökonomischen Aspekt ist es nach-
vollziehbar, dass im Rahmen von arzneimittelrechtlichen Erstzulassungen diese vorrangig in umsatz-
trächtigen und gewinnbringenden Märkten bzw. Indikationsbereichen angestrebt werden (Müller,
2009). Die hohen Kosten für klinische Entwicklungsprogramme von Zweitindikationen oder das ein-
geschränkte Marktpotential im Bereich des OLU führen oftmals zu einer ökonomisch betrachtet
wenig „rentablen“ Zulassung (Müller, 2009). Die Realität zeigt nämlich, dass die Umsätze der be-
troffenen Produkte durch ihren Einsatz außerhalb der eigentlichen Zulassung auch ohne ein zeit- und
kostenintensives Zulassungs- bzw. Änderungsverfahren ansteigen (Rückeshäuser, 2011).
Abschließend wird im 5. Kapitel kurz das Nutzen-Risiko-Potential von (neueren) Arzneimitteln aufge-
griffen. Nicht nur die Kosten-, sondern auch die Nutzenbewertungen sowohl der unterschiedlichen
Arzneimittel als auch der Therapieformen gewinnen im Gesundheitssektor als ein ökonomisch be-
deutsamer Wirtschaftsfaktor zunehmend an Bedeutung. Mit Wirkung zum 1. Januar 2011 ist das
Gesetz zur Neuordnung des Arzneimittelmarktes (AMNOG) nach § 35a SGB V in Kraft getreten. Die
Preise eines Arzneimittels sollen damit an seinen (Zusatz-)Nutzen für den Patienten gekoppelt wer-
10

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
den. Das Ergebnis der in diesem Gesetz geregelten (frühen) Nutzenbewertung durch den G-BA hat
möglicherweise einen Einfluss auf das Verordnungsverhalten von Ärzten. Des Weiteren soll analy-
siert werden, ob mit der Einführung einer solchen industrieunabhängigen Bewertung ein OLU ver-
hindert oder möglicherweise gefördert wird.
11

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
1.1 Die Definition des Off-Label Use und seine Abgrenzung zum Unlicensed bzw. Compassionate Use
Ausgehend vom Wortlaut kann der Off-Label Use als ein Gebrauch abseits des Etiketts übersetzt
werden (Becker, 2004). Gemeint ist eine von der arzneimittelrechtlichen Zulassung nicht umfasste
Anwendung eines zulassungspflichtigen Arzneimittels. Da sich die Zulassung nach § 22 Abs. 1 des
Arzneimittelgesetzes (AMG) nicht nur auf einzelne Indikationsgebiete bezieht, sondern auch auf
Dosierung, Häufigkeit und Dauer der Anwendung, Kontraindikationen, Alter etc., sind darüber hinaus
auch andere Abweichungen außerhalb des in der Zulassung definierten Bereichs unter den Begriff
Off-Label Use zusammenzufassen.
Nach § 35c Abs. 1 SGB V beruft das Bundesministerium für Gesundheit (BMG) Expertengruppen beim
Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) ein, die Bewertungen zum Stand der
wissenschaftlichen Erkenntnis über die Anwendung von zugelassenen Arzneimitteln für solche Indi-
kationsbereiche abgeben sollen, für die sie nach dem Arzneimittelgesetz nicht zugelassen sind. Nach
Auffassung von Plate et al. (2008) könnte durch die Bezeichnung dieser Gremien als Off-Label-
Expertengruppen die Definition des Off-Label Use auf die indikationsüberschreitende Anwendung
begrenzt werden. Der Begriff des Unlicensed Use ist vom Off-Label Use insofern abzugrenzen, als
dass hierbei ein Arzneimittel zur Therapie eingesetzt wird, für das keine Zulassung vorliegt, obwohl
es der Zulassungspflicht unterliegt (Bücheler et al., 2003). Ein Unlicensed Use liegt beispielsweise
dann vor, wenn die zulassungspflichtige Darreichungsform verändert werden würde (z.B. statt der
zugelassenen oralen Applikationsform eine parenterale). Ein bereits zugelassenes Arzneimittel ver-
liert damit seinen Status, da es somit von der ursprünglichen Zulassung nicht mehr erfasst wird
(Knöppel et al., 2000). Diese Vorgehensweise wird häufig in der Pädiatrie angewendet, da viele Me-
dikamente hinsichtlich ihrer Darreichungsform nicht für die Kinderheilkunde geeignet sind. Das
Schlucken von Tabletten oder der unangenehme Geschmack eines Arzneimittels können dazu füh-
ren, dass Kinder die Einnahme verweigern. Durch die Herstellung eines Safts oder Sirups aus einer
Tablette kann eine kindgerechte Darreichungsform hergestellt und die Adhärenz dieses Patienten-
kollektivs erhöht werden (Bajcetic & Gorodischer, 2005; Collier, 1999; Khdour et al., 2011). Von den
Begriffen des Off-Label Use und des Unlicensed Use ist ferner der Begriff des Compassionate Use
abzugrenzen. Darunter versteht man die Anwendung eines noch nicht zugelassenen Arzneimittels,
für das die klinischen Prüfungen so weit vorangeschritten sind, dass auf Seiten des pharmazeuti-
schen Unternehmens Wirksamkeit, Sicherheit und Qualität des Arzneimittels ausreichend belegt
werden konnten (Buhles, 2011). Bei einem Compassionate Use handelt es sich somit um eine schon
vor der Zulassung angesetzte Arzneimittelanwendung, um den Bedürfnissen der Patienten nach
einer raschen Pharmakotherapie gerecht zu werden.
12

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
1.2 Die Vereinbarkeit des Off-Label Use mit den arzneimittel-rechtlichen Zulassungsbestimmungen
Sowohl die Zulassung als solche als auch die Anwendungsbeobachtungen nach erfolgtem Marktein-
tritt sind für die rechtlichen Konsequenzen des OLU und der daraus folgenden Problematik bedeu-
tend. Die Grundzüge des arzneimittelrechtlichen Zulassungsverfahrens als unabdingbare Vorausset-
zung des OLU sollen im Weiteren erläutert werden.
1.2.1 Die arzneimittelrechtliche Zulassung und ihre Grundlagen
Der Arzneimittelbegriff nach § 2 AMG
Gemäß § 2 Abs. 1 des Arzneimittelgesetzes sind Arzneimittel Stoffe und Zubereitungen aus Stoffen,
die dazu bestimmt sind, durch Anwendung am oder im menschlichen oder tierischen Körper die in §
2 Abs. 1 AMG genannten Funktionen zu erfüllen. Hierzu zählen insbesondere Zubereitungen und
Stoffe, die dazu bestimmt sind, Krankheiten, Leiden, Körperschäden oder krankhafte Beschwerden
zu heilen, zu lindern, zu verhüten oder zu erkennen. Darüber hinaus gelten gemäß § 2 Abs. 2 AMG
Gegenstände als Arzneimittel, die ein Arzneimittel nach § 2 Abs. 1 enthalten oder auf denen ein
Arzneimittel nach § 2 Abs. 1 aufgebracht ist und die dazu bestimmt sind, dauernd oder vorüberge-
hend mit dem menschlichen oder tierischen Körper in Berührung gebracht zu werden. Dabei wird
der Arzneimittelbegriff objektiv-funktional statt subjektiv-funktional bestimmt (Müller, 2009). Das
heißt, dass ein Mittel, das nach seiner objektiven Zweckbestimmung ein Arzneimittel ist, diese Eigen-
schaft nicht durch die Angabe des Herstellers kein Arzneimittel verlieren kann (Schiedermair & Pohl,
2012). Wird im umgekehrten Fall die Zulassung vom BfArM mit der Begründung abgelehnt, dass es
sich bei dem Mittel nicht um ein Arzneimittel im arzneimittelrechtlichen Sinn handelt, dann hat auch
diese Negativentscheidung des BfArM eine allgemeinverbindliche Wirkung (Schiedermair & Pohl,
2012).
13

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Die Zulassungspflicht nach § 21 AMG und ihre Ausnahmen
Mit der Zulassung eines Arzneimittels wird von Seiten der zuständigen Behörden gewährleistet, dass
bei dem zu untersuchenden Wirkstoff bzw. bei der zu untersuchenden Wirkstoffkombination Quali-
tät, Wirksamkeit und Unbedenklichkeit in Übereinstimmung mit dem § 1 AMG nach dem derzeitigen
Erkenntnisstand gegeben sind und das Arzneimittel zur Verwendung in der breiten Bevölkerung
freigegeben werden kann. Nicht jedes Arzneimittel unterliegt jedoch dieser Zulassungspflicht. So
heißt es beispielsweise in § 21 Abs. 2 Nr. 1 AMG, dass es keiner Zulassung für Arzneimittel bedarf,
wenn sie im Rahmen der Defektur hergestellt werden. Darunter fallen Arzneimittel in der Eigenher-
stellung, die häufig von einem Arzt verordnet werden (beispielsweise Ohrentropfen oder Nasensal-
ben) oder Zubereitungen zur Weiterverarbeitung wie Salbengrundlagen. Ebenfalls von der Zulassung
befreit sind Rezepturarzneimittel, die für den jeweiligen Patienten individuell, entweder aufgrund
ärztlicher Anweisung oder eines Patientenwunsches, hergestellt werden. Auch enthält § 36 AMG
eine Ermächtigung des zuständigen Bundesministeriums für Standardzulassungen. Hierin wird fest-
gehalten, dass „bestimmte Arzneimittel oder Arzneimittelgruppen oder Arzneimittel in bestimmten
Abgabeformen von der Pflicht zur Zulassung freizustellen [sind], soweit eine unmittelbare oder mit-
telbare Gefährdung der Gesundheit von Mensch oder Tier nicht zu befürchten ist, weil die Anforde-
rungen an die erforderliche Qualität, Wirksamkeit und Unbedenklichkeit erwiesen sind“. In den so-
genannten Monographien findet man eine Zusammenfassung der Angaben zu den freizustellenden
Arzneimitteln, beispielsweise hinsichtlich der Zusammensetzung, Herstellung oder Haltbarkeit. Nach
wie vor ist eine Zulassung weder für homöopathische Arzneimittel (§§ 38, 39 AMG) noch für die
meisten traditionell angewandten pflanzlichen Arzneimittel (§§ 39a bis 39d AMG) notwendig, da hier
nur eine Registrierungspflicht gilt. Alle diese genannten Arzneimittel sind nicht vom OLU betroffen.
Die arzneimittelrechtlichen Voraussetzungen der Zulassung
Gemäß § 21 Abs. 3 AMG ist die Zulassung eines Arzneimittels vom pU bei der zuständigen Behörde
schriftlich zu beantragen. Die für die Zulassungserteilung verantwortliche Behörde ist gemäß § 77
Abs. 1 AMG grundsätzlich das BfArM. Für bestimmte Stoffe1 ist gemäß § 77 Abs. 2 AMG das Paul-
Ehrlich-Institut zuständig. Dem Zulassungsantrag sind alle für die Bewertung des Arzneimittels
zweckdienlichen Angaben und Unterlagen beizufügen, § 22 Abs. 2 Satz 3 AMG. Die wichtigsten Un-
terlagen sind in § 22 Abs. 1 und 2 AMG aufgeführt.
1 Darunter werden laut AMG Sera, Impfstoffe, Blutzubereitungen, Knochenmarkzubereitungen, Gewebezubereitungen, Ge-webe, Allergene, Arzneimittel für neuartige Therapien, xenogene Arzneimittel und gentechnisch hergestellte Blutbestandteile verstanden.
14

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Für die spätere Beurteilung eines möglichen Off-Label Use sind insbesondere folgende Unterlagen
und die daraus resultierenden Pflichtangaben von Bedeutung: die Darreichungsform, die Anwen-
dungsgebiete, die Kontraindikationen, die Dosierung, die Art der Anwendung und die Dauer der
Anwendung. Nach § 22 Abs. 2 Satz 1-3 AMG sind ebenfalls die Ergebnisse physikalischer, chemischer,
biologischer oder mikrobiologischer Versuche und die zu ihrer Ermittlung angewandten Methoden
(analytische Prüfung), die Ergebnisse der pharmakologischen und toxikologischen Versuche sowie
die Ergebnisse der klinischen Prüfungen oder sonstigen ärztlichen, zahnärztlichen oder tierärztlichen
Erprobung vorzulegen und durch Unterlagen zu belegen. Des Weiteren beinhaltet § 22 AMG Abs. 2
die verpflichtende Einreichung einer detaillierten Beschreibung des Pharmakovigilanz- und, soweit
zutreffend, desjenigen Risiko-Management-Systems, das der Antragsteller einführen wird. Auf die
Inhalte der Prüfungen soll im Folgenden näher eingegangen werden.
Die analytische Prüfung
Der Zulassungsantrag sieht die Ergebnisse physikalischer, chemischer, biologischer oder mikrobiolo-
gischer Versuche sowie die angewandten Methoden vor, um die Qualität eines Fertigarzneimittels
(Wirkstoff bzw. Wirkstoffe plus Hilfsstoffe) gewährleisten zu können.
Nach § 4 Abs. 15 AMG ist die Qualität eines Medikaments im arzneimittelrechtlichen Sinn lediglich
die Beschaffenheit eines Arzneimittels, welche nach Identität, Gehalt, Reinheit und weiteren chemi-
schen, physikalischen und biologischen Eigenschaften oder durch das Herstellungsverfahren be-
stimmt wird. Nicht nur die Wirksamkeit, sondern auch die Unbedenklichkeit eines Arzneimittels wird
dadurch maßgeblich beeinflusst. Eine Bewertung ist mit dem Begriff Qualität nicht verbunden.
Die pharmakologisch-toxikologische Prüfung
Die pharmakologisch-toxikologische Prüfung dient dem Nachweis der Unbedenklichkeit des Arznei-
mittels und stellt eine zwingende Voraussetzung für den Beginn der klinischen Arzneimittelprüfung
am Menschen dar (§ 40 Abs. 1 Nr. 6 AMG). Sollten in dieser Prüfungsphase erhebliche unerwünschte
Nebenwirkungen auftreten, so ist noch vor Beginn der klinischen Prüfung eine Zulassungserteilung
ausgeschlossen.
15

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Die klinische Prüfung
Klinische Studien am Menschen sind der letzte Schritt in einer langen Entwicklung zum fertigen Me-
dikament, sie werden nach dem Abschluss von Tierversuchen durchgeführt. Als wichtiger Bestandteil
der Forschung geht es hierbei primär um die Verträglichkeit und bzw. oder die Wirksamkeit des Arz-
neimittels und seiner Darreichungsform in festgelegter Dosierung am Individuum (Stapff, 2008).
Die klinischen Prüfungen, deren Ergebnisse für die Zulassungsentscheidung relevant sind, unterglie-
dern sich in drei Phasen. In der Phase I wird das Arzneimittel zunächst an gesunden Probanden er-
probt, danach in den Phasen II und III an Patienten, die an der Krankheit leiden und für die der pU
eine Indikation in der Zulassung beanspruchen will. Im Rahmen dieser Studien werden die Medika-
mente und ihre damit verbundenen Therapien bzw. Behandlungsformen überprüft (Stapff, 2008;
Röhrig et al., 2009). In Abhängigkeit von der Schwere der Erkrankung bzw. dem Nutzen-Risiko-
Verhältnis kann eine klinische Prüfung sowohl im stationären als auch im ambulanten Bereich durch-
geführt werden. Als zwingende Voraussetzung für die klinische Prüfung eines Arzneimittels bei Men-
schen bedarf es der Zustimmung der Ethik-Kommission einerseits und der zuständigen Bundesbe-
hörde andererseits (§§ 40-42 AMG). Des Weiteren müssen die vorhersehbaren Risiken und Nachteile
gegenüber dem Nutzen für die Person, bei der sie durchgeführt werden soll, und der voraussichtli-
chen Bedeutung des Arzneimittels für die Heilkunde ärztlich vertretbar sein (§ 40 Abs. 1 Satz 2 AMG).
Betrachtet man diese rechtlichen Rahmenbedingungen für eine Zulassung, so erklärt sich auch der
Off-Label Use in der Pädiatrie. Generell erschweren ethische Vorbehalte die Forschung an nicht oder
nur bedingt einwilligungsfähigen Patienten. Hinzu kommt, dass klinische Studien an Kindern auf-
grund ihrer entwicklungsabhängigen unterschiedlichen Physiologie und Stoffwechselfunktion ge-
trennte Untersuchungen nach Altersstufen erfordern (Bücheler et al., 2003) und dadurch sehr auf-
wendig und kostenintensiv sind. Liegen jedoch keine Studien zur Sicherheit oder zu den therapeu-
tisch sinnvollen Dosisbereichen vor, führt das Fehlen von systematisch durchgeführten klinischen
Prüfungen jedoch dazu, dass Kindern potentiell nützliche Wirkstoffe im Rahmen des In-Label Use
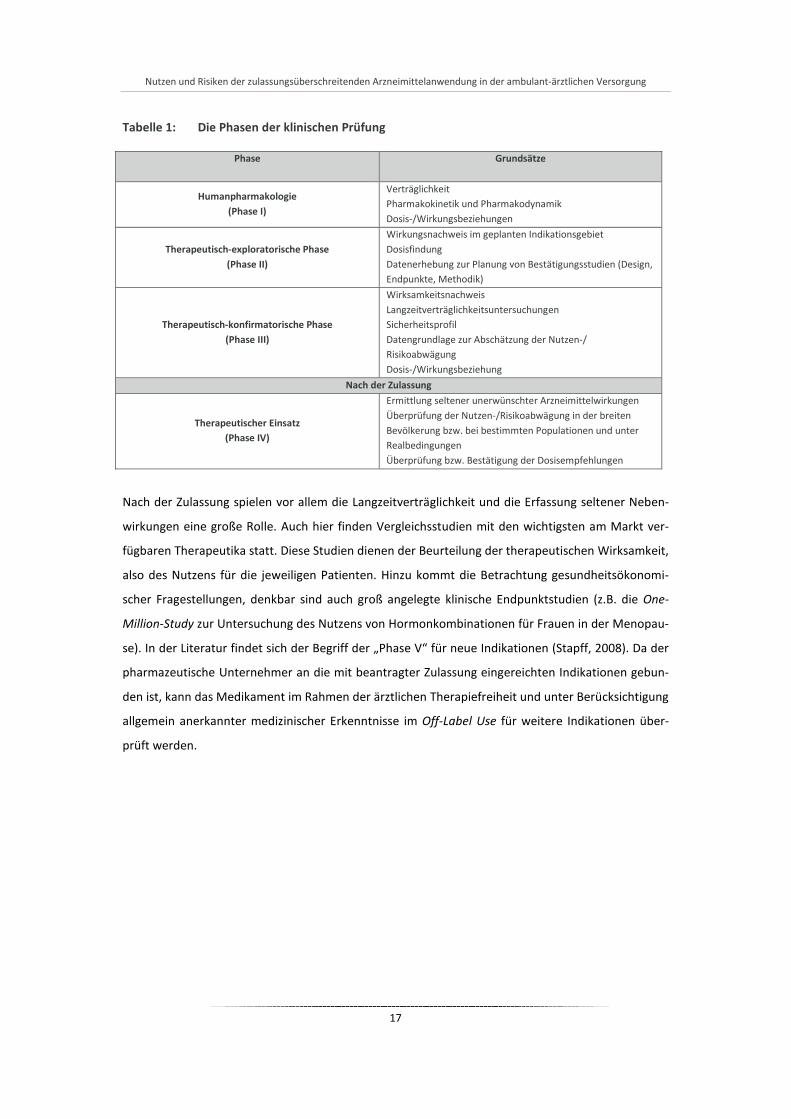
vorenthalten bleiben (Steinbrook, 2002). Tabelle 1 gibt einen Überblick über die Phasen der klini-
schen Prüfung und ihre Grundsätze.
16
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 1: Die Phasen der klinischen Prüfung
Phase
Grundsätze
Humanpharmakologie (Phase I)
Verträglichkeit Pharmakokinetik und Pharmakodynamik Dosis-/Wirkungsbeziehungen
Therapeutisch-exploratorische Phase (Phase II)
Wirkungsnachweis im geplanten Indikationsgebiet Dosisfindung Datenerhebung zur Planung von Bestätigungsstudien (Design, Endpunkte, Methodik)
Therapeutisch-konfirmatorische Phase (Phase III)
Wirksamkeitsnachweis Langzeitverträglichkeitsuntersuchungen Sicherheitsprofil Datengrundlage zur Abschätzung der Nutzen-/ Risikoabwägung Dosis-/Wirkungsbeziehung
Nach der Zulassung
Therapeutischer Einsatz (Phase IV)
Ermittlung seltener unerwünschter Arzneimittelwirkungen Überprüfung der Nutzen-/Risikoabwägung in der breiten Bevölkerung bzw. bei bestimmten Populationen und unter Realbedingungen Überprüfung bzw. Bestätigung der Dosisempfehlungen
Nach der Zulassung spielen vor allem die Langzeitverträglichkeit und die Erfassung seltener Neben-
wirkungen eine große Rolle. Auch hier finden Vergleichsstudien mit den wichtigsten am Markt ver-
fügbaren Therapeutika statt. Diese Studien dienen der Beurteilung der therapeutischen Wirksamkeit,
also des Nutzens für die jeweiligen Patienten. Hinzu kommt die Betrachtung gesundheitsökonomi-
scher Fragestellungen, denkbar sind auch groß angelegte klinische Endpunktstudien (z.B. die One-
Million-Study zur Untersuchung des Nutzens von Hormonkombinationen für Frauen in der Menopau-
se). In der Literatur findet sich der Begriff der „Phase V“ für neue Indikationen (Stapff, 2008). Da der
pharmazeutische Unternehmer an die mit beantragter Zulassung eingereichten Indikationen gebun-
den ist, kann das Medikament im Rahmen der ärztlichen Therapiefreiheit und unter Berücksichtigung
allgemein anerkannter medizinischer Erkenntnisse im Off-Label Use für weitere Indikationen über-
prüft werden.
17

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
2 Die Vereinbarkeit des Off-Label Use mit dem Kran-kenversicherungsrecht
Während das AMG wie im ersten Kapitel beschrieben die Sicherheit des Arzneimittelverkehrs ge-
währleisten soll und nicht die therapeutisch möglichst effiziente Arzneimittelversorgung gesetzlich
Krankenversicherter (Janda, 2010), liefert das 5. Sozialgesetzbuch (SGB V) die rechtlichen Vorausset-
zungen für die Leistungspflicht der GKV. Sie entsteht unter dem Vorbehalt der Wirtschaftlichkeit
bereits mit der arzneimittelrechtlichen Zulassung (Bruns & Herz, 2003). Unter Berücksichtigung eines
aus sozialrechtlichen Gesichtspunkten betrachteten Mindeststandards sowie eines vertretbaren
Finanzierungsbedarfs müssen Leistungen nach § 12 Abs. 1 SGB V ausreichend, zweckmäßig und wirt-
schaftlich sein und dürfen das Maß des Notwendigen nicht überschreiten2. Dabei versteht man unter
dem Begriff der Wirtschaftlichkeit die Erbringung der erforderlichen, ausreichenden und zweckmäßi-
gen Leistung mit dem geringstmöglichen Aufwand, der aufgrund einer Kosten-Nutzen-Bewertung
erfolgt (Huster & Bohmeier, 2012). Die Kosten-Nutzen-Bewertung bezieht sich dabei immer auf mög-
liche alternative Therapiemaßnahme. Das Behandlungsziel Heilung ist vorrangig gegenüber den Be-
handlungszielen Verhütung und Linderung. Eine alternative Therapiemaßnahme kann nur eine solche
sein, mit der mindestens das gleiche Behandlungsziel erreicht werden kann (Huster & Bohmeier,
2012). Im Umkehrschluss fehlt es bei einer Pharmakotherapie bereits an der Zweckmäßigkeit, falls
dem Arzneimittel beispielsweise in entsprechender Indikation keine Zulassung erteilt worden ist.
Schließlich entsprechen die Zulassungsvoraussetzungen den Mindestanforderungen, die an eine
wirtschaftliche Verordnungsweise gestellt werden (Ehlers & Wenke, 2011). Das Wirtschaftlichkeits-
gebot gilt ebenso für die Kostenübernahme durch die Krankenkassen. Speziell deren Geschäftsführer
treten in eine private Haftungsverpflichtung ein, sollte der gesetzlich vorgeschriebene Leistungsrah-
men überschritten worden sein (§ 12 Abs. 3 SGB V).
Generell ist die Frage nach der Verteilung der zur Verfügung stehenden Mittel für Leistungen im
Gesundheitswesen kein Deutschland-spezifisches Problem, sie stellt sich vielmehr in allen Europäi-
schen Staaten (Huster & Bohmeier, 2012). Die Erstattungsfähigkeit von Arzneimitteln im Off-Label
Use kann deshalb auch nach Jahren einer breiten Diskussion um die Frage der Voraussetzungen nicht
ohne Weiteres beantwortet werden. So bleibt die Verordnung eines Arzneimittels außerhalb seiner
Zulassungsbestimmung häufigster Anlass, die Expertise des Medizinischen Dienstes der Krankenkas-
sen (MDK) zur Rechtmäßigkeit der Kostenübernahme zu Rate zu ziehen (Grell & Rieger, 2012).
2 Nach Ehlers und Wenke stehen die Begriffselemente des Wirtschaftlichkeitsgebotes in einem untrennbaren inneren Zusam-menhang. So ist eine Maßnahme, die zum Erzielen eines Heilerfolges nicht notwendig oder zweckmäßig ist, begrifflich auch unwirtschaftlich. Eine Maßnahme, die zum Erzielen eines Heilerfolges nicht ausreicht, ist zugleich unzweckmäßig. Umgekehrt ist eine mehr als ausreichende Maßnahme zugleich nicht notwendig (Ehlers & Wenke, 2011).
18

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
2.1 Der Off-Label Use und seine Ausnahmen in der Rechtspre-chung
In Deutschland galt lange Zeit der Grundsatz, dass nur zugelassene Arzneimittel in ihren Indikationen
zur Leistungspflicht der GKV gehören. Im Jahr 2002 hat die Rechtsprechung jedoch erstmals Aus-
nahmen konkretisiert, die eine Kostenübernahme durch die GKV möglich machen und damit einen in
der Praxis oft unumgänglichen Bedarf eines Einsatzes von Arzneimitteln außerhalb der Zulassung
bestätigt. Im Folgenden werden die wichtigsten Urteile aufgeführt und die Voraussetzungen zur
Leistungspflicht im Fall eines OLU herausgearbeitet.
2.1.1 Das „Sandoglobulin®-Urteil“ des BSG vom 19.03.2002
In der Verhandlung des Bundessozialgerichtes (BSG), die zum „Sandoglobulin®-Urteil“ vom 19. März
2002 führte, ging es um den OLU eines Immunglobulins, das u.a. zur Substitutionstherapie bei pri-
mären Immunmangelkrankheiten zugelassen war. Durch die ausdrücklich als Heilversuch deklarierte
Therapie mit Sandoglobulin® erhofften sich die behandelnden Ärzte eine Besserung der durch Mul-
tiple Sklerose (MS) verursachten Ataxie. Die Therapie der MS, an der der Kläger seit 1987 litt, war im
Zulassungsantrag des Arzneimittels jedoch nicht mit aufgeführt. Im sogenannten „Sandoglobulin®-
Urteil“ hatte das BSG nun über den Anspruch des Klägers gegen die beklagte Versicherung bezüglich
der Erstattung der Kosten für eine bereits erfolgte Behandlung mit Sandoglobulin® sowie über die
Übernahme der in Zukunft entstehenden Kosten dieser Behandlung zu entscheiden (BSG, 2002).
Aufgrund der Tatsachen, dass es dem Arzt nicht verboten sei, ein Medikament außerhalb seiner
Zulassung einzusetzen, man dem Patienten unverzichtbare und wirksame Therapien nicht vorenthal-
ten dürfe und das Arzneimittel durch die Zulassung in anderen Indikationsgebieten bereits umfang-
reiche pharmakologisch-toxikologische Prüfungen an Tieren sowie klinische Prüfungen am Menschen
durchlaufen hatte, erkannte das BSG den „in bestimmten Versorgungbereichen der hausärztlichen
Praxis bzw. des klinischen Alltags bestehenden unverzichtbaren kontrollierten Einsatzes von Arznei-
mitteln im Off-Label an, ohne ihn allerdings pauschal zu bejahen“ (BSG, 2002). Das BSG legte mit der
Rechtsprechung vom 19.03.2002 (B1 KR 37/00R) somit klar definierte Kriterien für eine Verordnung
von Arzneimitteln außerhalb der zugelassenen Indikation zu Lasten der GKV fest. Eine Leistungs-
pflicht der GKV im Bereich des zulassungsüberschreitenden Einsatzes eines Arzneimittels besteht
demnach dann, wenn es
19

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
„(1) um die Behandlung einer schwerwiegenden (lebensbedrohlichen oder die Lebensqualität auf Dauer nachhaltig
beeinträchtigenden) Erkrankung geht, wenn
(2) keine andere Therapie verfügbar ist und wenn
(3) aufgrund der Datenlage die begründete Aussicht besteht, dass mit dem betreffenden Präparat ein Behandlungser-
folg (kurativ oder palliativ) erzielt werden kann.
Damit Letzteres angenommen werden kann, müssen Forschungsergebnisse vorliegen, die erwarten lassen, dass das
Arzneimittel für die betreffende Indikation zugelassen werden kann. Davon kann ausgegangen werden, wenn entwe-
der die Erweiterung der Zulassung bereits beantragt ist und die Ergebnisse einer kontrollierten klinischen Prüfung der
Phase III (gegenüber Standard oder Placebo) veröffentlicht sind und eine klinisch relevante Wirksamkeit respektive
einen klinisch relevanten Nutzen bei vertretbaren Risiken belegen oder außerhalb eines Zulassungsverfahrens gewon-
nene Erkenntnisse veröffentlicht sind, die über Qualität und Wirksamkeit des Arzneimittels in dem neuen Anwen-
dungsgebiet zuverlässige, wissenschaftlich nachprüfbare Aussagen zulassen und auf Grund derer in den einschlägigen
Fachkreisen Konsens über einen voraussichtlichen Nutzen in dem vorgenannten Sinne besteht“ (BSG, 2002).
Unter Berücksichtigung der Gesetzesvorgaben zur vertragsärztlichen Versorgung von Versicherten
müssen die Kriterien außerdem kumulativ erfüllt sein (Hafner, 2013). Die oben beschriebenen Vo-
raussetzungen waren im „Sandoglobulin®-Urteil“ nicht alle erfüllt – so fehlten gesicherte Erkenntnis-
se über die Wirksamkeit von Sandoglobulin® für die beim Kläger bestehende primär chronisch-
progrediente Verlaufsform dieser Erkrankung und es gab außerdem eine Behandlungsalternative mit
dem für die Therapie zugelassenen Betaferon®. Dennoch durfte hier nach Auffassung des BSG das
grundsätzliche Verbot des Off-Label Use nicht dazu führen, dass dem Patienten unverzichtbare und
erwiesenermaßen wirksame Therapien vorenthalten werden (BSG, 2002). Bei der Auslegung der vom
BSG aufgestellten Voraussetzungen für den Off-Label Use kam es in der Praxis immer wieder zu In-
terpretationsschwierigkeiten (Hafner, 2013; Müller, 2009; Dierks, 2003). Somit findet man beispiels-
weise keine allgemeingültige und einheitliche Definition der schwerwiegenden Erkrankung als Leis-
tungsvoraussetzung. Letztendlich sind immer Einzelfallentscheidungen notwendig, die Krankenkas-
sen oder Gerichte aufgrund einer Gesamtbewertung der Erkrankung unter Berücksichtigung der
damit verbundenen gesundheitlichen Beeinträchtigung sowie des Leidensdrucks des Patienten zu
fällen haben (Dierks, 2003).
20

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
2.1.2 Die „Visudyne®-Entscheidung“ des BSG vom 10.10.2004
Mit der sogenannten „Visudyne®-Entscheidung“ wurde der Therapie seltener, nicht systematisch
erforschter Krankheiten Rechnung getragen, indem das BSG Ausnahmen bzw. eine Herabsetzung des
Wirksamkeitsnachweises einräumte (BSG, 2004). In diesem konkreten Fall handelte es sich um die
Therapie eines zehnjährigen Jungen mit einer seltenen angeborenen Augenerkrankung. Bereits im
Jahr 2000 wurde der Junge mit dem Medikament Visudyne® (Vertreporfin) behandelt, welches zu
dem Zeitpunkt in Deutschland nicht zugelassen war. Den Antrag der Eltern auf Kostenerstattung in
Höhe von gut 2.100 Euro lehnte die Barmer Ersatzkasse (BEK) mit der Begründung ab, dass die Be-
handlungsmethode weder anerkannt noch hinreichend gesichert sei. Nach Auffassung des BSG dür-
fen aber lebensbedrohliche oder die Lebensqualität auf Dauer beeinträchtigende Erkrankungen, die
so selten auftreten, dass ihre systematische Erforschung praktisch nicht möglich ist und für deren
Behandlung keine anderen therapeutischen Möglichkeiten zur Verfügung stehen, nicht zwangsläufig
vom Leistungsumfang der GKV ausgeschlossen werden (BSG, 2004).
2.1.3 Die „Nikolausentscheidung“ des BVerfG vom 06.12.2005
Hinsichtlich der Behandlung lebensbedrohlicher Erkrankungen mit nicht zugelassenen Arzneimitteln
vertrat das Bundesverfassungsgericht (BVerfG), begründet auf Art. 2 Abs. 2 S. 1 des Grundgesetzes,
die gleiche Auffassung wie das BSG. Doch erst mit der „Nikolausentscheidung“ vom 6. Dezember
2005 übertrug das BVerfG diese Auffassung auch auf die Bereitstellung von Gesundheitsleistungen
als Leistungspflicht der gesetzlichen Krankenversicherung. Schließlich sei es nach Auffassung des
BVerfG nicht vertretbar, einen gesetzlich Krankenversicherten, für dessen lebensbedrohliche oder
regelmäßig tödlich verlaufende Erkrankung keine allgemein anerkannte, dem medizinischen Stand
entsprechende Behandlungsmethode zur Verfügung steht, von der Leistung einer von ihm gewähl-
ten, ärztlich angewandten Behandlungsmethode auszuschließen (hier: Bioresonanztherapie zur Be-
handlung der Muskeldystrophie des Typs Duchenne) (BVerfG, 2005). Durch diesen Beschluss erfolgte
eine entscheidende Ausweitung hinsichtlich des Off-Label Use und seiner Erstattungsvoraussetzun-
gen. In Tabelle 2 werden die Kernpunkte der beiden Urteile (BSG, 2002 und BVerfG, 2005) zum Off-
Label Use noch einmal zusammenfassend dargestellt.
21
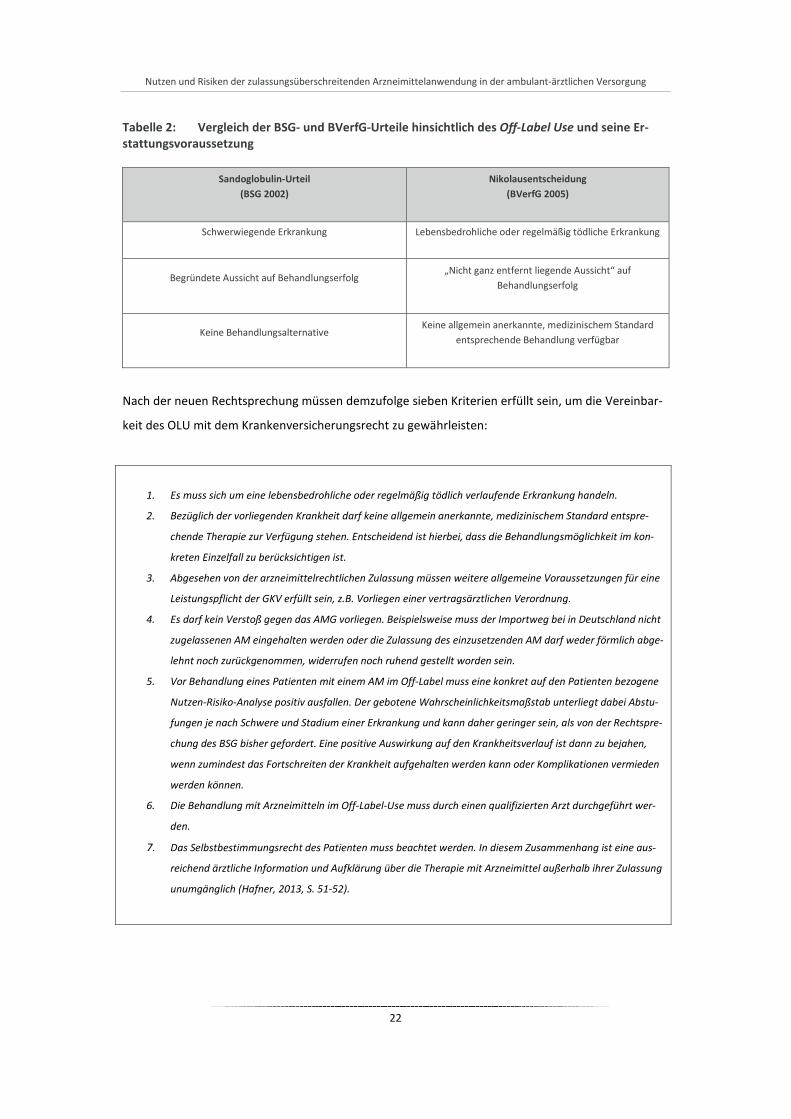
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 2: Vergleich der BSG- und BVerfG-Urteile hinsichtlich des Off-Label Use und seine Er-stattungsvoraussetzung
Sandoglobulin-Urteil (BSG 2002)
Nikolausentscheidung (BVerfG 2005)
Schwerwiegende Erkrankung
Lebensbedrohliche oder regelmäßig tödliche Erkrankung
Begründete Aussicht auf Behandlungserfolg
„Nicht ganz entfernt liegende Aussicht“ auf Behandlungserfolg
Keine Behandlungsalternative
Keine allgemein anerkannte, medizinischem Standard entsprechende Behandlung verfügbar
Nach der neuen Rechtsprechung müssen demzufolge sieben Kriterien erfüllt sein, um die Vereinbar-
keit des OLU mit dem Krankenversicherungsrecht zu gewährleisten:
1. Es muss sich um eine lebensbedrohliche oder regelmäßig tödlich verlaufende Erkrankung handeln.
2. Bezüglich der vorliegenden Krankheit darf keine allgemein anerkannte, medizinischem Standard entspre-
chende Therapie zur Verfügung stehen. Entscheidend ist hierbei, dass die Behandlungsmöglichkeit im kon-
kreten Einzelfall zu berücksichtigen ist.
3. Abgesehen von der arzneimittelrechtlichen Zulassung müssen weitere allgemeine Voraussetzungen für eine
Leistungspflicht der GKV erfüllt sein, z.B. Vorliegen einer vertragsärztlichen Verordnung.
4. Es darf kein Verstoß gegen das AMG vorliegen. Beispielsweise muss der Importweg bei in Deutschland nicht
zugelassenen AM eingehalten werden oder die Zulassung des einzusetzenden AM darf weder förmlich abge-
lehnt noch zurückgenommen, widerrufen noch ruhend gestellt worden sein.
5. Vor Behandlung eines Patienten mit einem AM im Off-Label muss eine konkret auf den Patienten bezogene
Nutzen-Risiko-Analyse positiv ausfallen. Der gebotene Wahrscheinlichkeitsmaßstab unterliegt dabei Abstu-
fungen je nach Schwere und Stadium einer Erkrankung und kann daher geringer sein, als von der Rechtspre-
chung des BSG bisher gefordert. Eine positive Auswirkung auf den Krankheitsverlauf ist dann zu bejahen,
wenn zumindest das Fortschreiten der Krankheit aufgehalten werden kann oder Komplikationen vermieden
werden können.
6. Die Behandlung mit Arzneimitteln im Off-Label-Use muss durch einen qualifizierten Arzt durchgeführt wer-
den.
7. Das Selbstbestimmungsrecht des Patienten muss beachtet werden. In diesem Zusammenhang ist eine aus-
reichend ärztliche Information und Aufklärung über die Therapie mit Arzneimittel außerhalb ihrer Zulassung
unumgänglich (Hafner, 2013, S. 51-52).
22

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
2.2 Festlegung des medizinischen Standards durch Richtlinien in der GKV
Der Anspruch einer ausreichenden, wirtschaftlichen und zweckmäßigen Versorgung aller Versicher-
ten sowie die Beachtung des medizinischen Standards stehen in einem engen Zusammenhang. Dabei
repräsentiert der Standard in der Medizin den jeweiligen Stand naturwissenschaftlicher Erkenntnis
und ärztlicher Erfahrung, der zur Erreichung des ärztlichen Behandlungszieles erforderlich ist und
sich in der Erprobung bewährt hat (Brunkhorst & Dittmayer, 2008). Erst durch die Festlegung dessel-
bigen kann die Frage nach der Verordnungsfähigkeit zu Lasten der GKV aufkommen und beantwortet
werden (Rückeshäuser, 2011). Inwieweit ein OLU zum medizinischen Standard gehört und daher
durch die GKV erstattungsfähig wird soll in diesem Abschnitt herausgearbeitet werden.
Der medizinische Standard dient der Qualitätssicherung ärztlicher Behandlung, indem er in umfang-
reichen Entscheidungssituationen über Nutzen und Risiken bestimmter Behandlungsmethoden in
entsprechenden Indikationsgebieten informiert und Therapieoptionen aufzeigt (Müller, 2009). So
wird der Begriff des medizinischen Standards nicht nur bei der vom Arzt zu verantwortenden Leis-
tung, sondern auch bei der Frage der Erstattungsfähigkeit der Therapie, der (Arzt-)Haftung und der
Therapieauswahl bzw. des Behandlungsortes gebraucht (Brunkhorst & Dittmayer, 2008; Laufs &
Uhlenbruck, 2002). Jedoch ändert sich der medizinische Standard laufend und bedarf aufgrund eines
raschen und stetigen Fortschritts einer kontinuierlichen Anpassung. Die Frage, inwieweit sozialrecht-
liche Entscheidungen Einfluss auf die Bestimmung des medizinischen Standards haben können und
dürfen, hat im Gesundheitssystem an Bedeutung gewonnen und zu dessen Reformierung beigetra-
gen (Brunkhorst & Dittmayer, 2008). So mussten mit dem Grundgesetzurteil des BSG im Jahr 2002
neue Rahmenbedingungen geschaffen werden, um unter Berücksichtigung des medizinischen Stan-
dards die Verordnungsmöglichkeiten und die Erstattungsfähigkeit des OL-Arzneimittels durch die
GKV zu regeln. Mit Erlass vom 17.09.2002 richtete das Bundesministerium für Gesundheit und Sozia-
le Sicherung (BMGS) erstmals eine Expertengruppe Off-Label ein (BfArM, 2013). Mit Inkrafttreten
des GKV-Modernisierungsgesetzes (GMG) am 01. Januar 2004 wurden schließlich die gesetzlichen
Grundlagen ihrer Arbeit geschaffen. Mitglieder dieser Expertengruppe sind neben Experten aus me-
dizinischen Fachgebieten u.a. auch zwei Vertreter des Medizinischen Dienstes der Krankenkassen.
Aufgabe der Expertengruppe Off-Label ist es festzustellen, in welchen Fällen für die Behandlung von
schweren Erkrankungen auch Arzneimittel eingesetzt werden können, die für diese Therapie noch
keine Zulassung nach dem AMG haben.
23

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Veröffentlichte Literatur und die Auffassung entsprechender Fachkreise bilden die Grundlage dieser
Entscheidungen, um unter Berücksichtigung der evidenzbasierten Medizin (EbM)3 ein fundiertes
Übereinkommen hinsichtlich Qualität und Wirksamkeit der zur Diskussion stehenden Therapie zu
erzielen. Auf der Basis dieser Bewertungen trifft der Gemeinsame Bundesausschuss eine Entschei-
dung, ob entsprechende Anwendungen in die Arzneimittelrichtlinien als erstattungsfähig oder nicht
erstattungsfähig aufgenommen werden. Eine Zustimmung des betroffenen pharmazeutischen Un-
ternehmers ist erforderlich, da er für den bestimmungsgemäßen Gebrauch haftet und deshalb die
Entscheidung des Gemeinsamen Bundesausschusses mittragen muss (Sickmüller & Lietz, 2011). So
werden in der Anlage VI zum Abschnitt K der Arzneimittel-Richtlinie Verordnungsfähigkeit von zuge-
lassenen Arzneimitteln in nicht zugelassenen Anwendungsgebieten (sog. Off-Label-Use) Arzneimittel
genannt, die unter Beachtung der dazu gegebenen Hinweise in nicht zugelassenen Anwendungsge-
bieten verordnungsfähig sind (AM-RL, Anlage VI). Dennoch gilt, dass eine Behandlungsmethode vor
ihrer Etablierung als medizinischer Standard letztendlich immer das Stadium des medizinischen Er-
probungshandelns (ein oder mehrere „individuelle Heilversuche“) zu durchlaufen hat (Müller, 2009).
So sind neben den wissenschaftlich nachweisbaren Belegen einer optionalen pharmakologischen
Therapie auch die Fachkompetenz des behandelnden Arztes sowie die patientenindividuellen Belan-
ge in der konkreten Behandlungssituation zu berücksichtigen (Gaßner & Strömer, 2012).
3 Bei der evidenzbasierten Medizin handelt es sich um ein wissenschaftstheoretisches Konzept, durch das der behandelnde Arzt die Möglichkeit erhält, für seine Entscheidungen bei der patientenindividuellen Therapie die derzeit wissenschaftlich am besten gesicherte Nachweisbarkeit (Evidenz) gewissenhaft, ausdrücklich und vernünftig anzuwenden (Gaßner & Strömer, 2012; Sackett et al., 1996). Allerdings darf die EbM nicht dahingehend missverstanden werden, dass nur Therapien, die mit einem hohen Evidenzgrad belegt sind (in der Regel mittels randomisierter klinischer Studien) zum medizinischen Standard gehören können (Kunz et. al., 2007). Denn nach Sackett kann mittels EbM auch unter mehreren Therapieoptionen mit kaum nachgewiesener Wirksamkeit „die am wenigsten schlechte Methode“ ermittelt werden (Sackett et al., 1996).
24

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
2.3 Der Off-Label Use aus ökonomischen Interessen: SGB V vs. AMG
Im Fokus der Frage nach der Vereinbarkeit des OLU mit dem Krankenversicherungsrecht stand bisher
die Darstellung der Ausnahmen, die die GKV aus ethischen Gründen zu einer Kostenübernahme
verpflichten. Dass ein Off-Label Use auch aus ökonomischen Interessen induziert sein kann, soll in
diesem Abschnitt dargestellt werden. Schließlich bilden neben stationären sowie ambulant-
ärztlichen Behandlungen die Ausgaben für Arzneimittel einen der größten Kostenfaktoren in der
gesetzlichen Krankenversicherung (GKV-Spitzenverband, 2015). Um diese Kosten zu reduzieren,
können die Krankenkassen und ihre Verbände gemäß § 130a Abs. 8 SGB V Rabattverträge mit phar-
mazeutischen Unternehmern abschließen4. Rabattverträge ermöglichen den GKVen seither, durch
einen direkten Vertragsabschluss mit dem pU die zu ihren Lasten zu verrechnenden Arzneimittel-
preise zu beeinflussen. Diese Rabattverträge sehen nämlich Rückvergütungen bei den jeweils be-
troffenen Arzneimitteln nach ihrer Verordnung vor, die in Höhe des ausgehandelten Rabatts auf den
Abgabepreis gewährt werden.
Im § 4 des Rahmenvertrags über die Arzneimittelversorgung nach § 129 Abs. 2 SGB V zwischen dem
GKV-Spitzenverband und dem Apothekerverband werden die Voraussetzungen für die Arzneimittel-
auswahl detailliert beschrieben, sollte der Arzt ein Arzneimittel nur unter seiner Wirkstoffbezeich-
nung verordnet oder die Ersetzung eines unter seinem Produktnamen verordneten Fertigarzneimit-
tels durch ein wirkstoffgleiches Arzneimittel nicht ausgeschlossen haben. Folgende Aspekte sind bei
der Auswahl zu berücksichtigen:
4 § 130a SGB V ist durch das Beitragssatzsicherungsgesetz (BSSichG) vom 23. Dezember 2002 mit Wirkung ab 1. Januar 2003 neu in das fünfte Sozialgesetzbuch aufgenommen worden. Mit dem GKV-Wirtschaftlichkeitsstärkungsgesetz (GKV-WSG) erhielten die Rabattverträge allerdings erst ihre eigentliche Wirksamkeit durch die Einführung der neuen Regelung zum Aut-idem-Austausch.
25

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
(a) gleicher Wirkstoff, dabei gelten die verschiedenen Salze, Ester, Ether, Isomere, Mischungen von
Isomeren, Komplexe und Derivate eines Wirkstoffes als ein und derselbe Wirkstoff, es sei denn ihre Eigen-
schaften unterscheiden sich nach wissenschaftlichen Erkenntnissen erheblich hinsichtlich der Unbedenklich-
keit und der Wirksamkeit,
(b) identische Wirkstärke,
(c) identische Packungsgröße,
(d) gleiche oder austauschbare Darreichungsform, dabei sind die Darreichungsformen mit identischer Bezeich-
nung in der Großen Deutschen Spezialitätentaxe (Lauer-Taxe) gleich, Darreichungsformen nach den Hinwei-
sen des Gemeinsamen Bundesausschusses nach § 129 Absatz 1a SGB V austauschbar,
(e) Zulassung für ein gleiches Anwendungsgebiet, die Übereinstimmung in einem von mehreren Anwendungs-
gebieten ist ausreichend,
(f) keine einer Ersetzung des verordneten Arzneimittels entgegenstehenden betäubungsmittelrechtlichen Vor-
schriften; insbesondere hat die abgegebene Menge der verordneten Menge zu entsprechen (GKV-
Spitzenverband, 2012).
Sofern rabattierte Arzneimittel die oben genannten Voraussetzungen erfüllen, ist die Apotheke zur
vorrangigen Abgabe verpflichtet. Im Rahmen der Rabattvertrags-bedingten Substitutionspflicht in
der Apotheke kann es dennoch in der Praxis vorkommen, dass Präparate, die sowohl den gleichen
Wirkstoff als auch die gleiche Wirkstoffmenge aufweisen, keine übereinstimmenden Indikationen
haben – begründet entweder in der Zulassung oder aber wegen bestehender gewerblicher Schutz-
rechte (Unterlagenschutz/Patente) (Sickmüller & Lietz, 2012). Der Patient erhält dann möglicher-
weise ein Präparat, das nicht für die Behandlung seiner Erkrankung zugelassen wurde. Durch von der
ärztlichen Verordnung abweichende Informationen (beispielsweise hinsichtlich der Dosierung) und
die nicht zutreffende Indikation in der Packungsbeilage kann der Patient zusätzlich verunsichert
werden, was sich schließlich negativ auf die Adhärenz auswirken kann. Tabelle 3 enthält Beispiele für
Wirkstoffe mit unterschiedlichen Indikationen.
26

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 3: Beispiele für Wirkstoffe mit unterschiedlichen Indikationen
Wirkstoffe
Handelspräparat (pU) Indikation
Handelspräparat (pU) Indikation
Doxazosin Doxazosin AbZ® 2 mg/4 mg Tabletten (AbZ Pharma) Essentielle Hypertonie
Uriduct® 2 mg (TAD) Benigne Prostatahyperplasie (BPH)
Methylphenidat Medikinet® adult (Medice) ADHS bei Erwachsenen
Ritalin® 10 mg (Novartis Pharma) ADHS bei Kindern und Jugendlichen
Acetylsalicylsäure
ASS-CT® 100 mg TAH Tabletten (AbZ Pharma) Zur Thrombozytenaggregations-hemmung Bei instabiler Angina pectoris Akute Myokardinfarkt Reinfarktprophylaxe Nach arteriellen gefäßchirurgischen oder interventionellen Eingriffen (z. B. nach ACVB, bei PTCA). Vorbeugung von transitorischen ischämischen Attacken (TIA) und Hirninfarkten, nachdem Vorläuferstadien aufgetreten sind
ASS 100 mg Heumann® (Heumann) Leichte bis mäßig starke Schmerzen und Fieber bei Kindern ab 6 Monate
Auch der aufgrund ökonomischer Interessen zulassungsüberschreitende Einsatz von Bevacizumab
zur Behandlung der altersabhängigen Makuladegeneration (AMD) wird kontrovers diskutiert (Lynch
& Cheng, 2007; Rosenfeld, 2011; Kirchhof et al., 2013; Gaßner, 2013). Bei dieser Erkrankung sind die
VEGF-Spiegel im Auge erhöht, was zu einer übermäßigen Gefäßneubildung der Choroidea führt.
Durch die erhöhte Permeabilität tritt aus den neu gebildeten Gefäßen Blut und Flüssigkeit aus, wes-
wegen sich die Netzhaut verdickt und Ödeme bilden. Ferner kommt es zu sub- oder intraretinalen
Blutungen, die die Sehschärfe beeinträchtigen. Letztendlich führt diese Erkrankung unbehandelt zur
Erblindung (Ferrara & Kerbel, 2005; Witmer et al., 2003; Penn et al., 2008). Derzeit stehen auf dem
deutschen Arzneimittelmarkt drei VEGF-Hemmstoffe für die Behandlung der AMD zur Verfügung:
Pegaptanib (Macugen®, Datum der Zulassungserteilung: Januar 2006), Ranibizumab (Lucentis®, Da-
tum der Zulassungserteilung: Januar 2007) und Aflibercept (Eylea®, Datum der Zulassungserteilung:
November 2012). Bis zum Frühjahr 2007 konzentrierte sich die medikamentöse Therapie allerdings
auf die intravitreale Gabe von Bevacizumab (Janzen & Ludwig, 2012) im sogenannten Off-Label Use.
Mit seiner Zulassung 2007 stand mit dem Wirkstoff Ranibizumab ein mit der Wirksamkeit von Beva-
cizumab vergleichbares zugelassenes Arzneimittel zur Verfügung, das allerdings auch um ein Vielfa-
ches teurer ist als Bevacizumab (Kirchhof et al., 2013). Im Hinblick auf die Arzneimitteltherapie ver-
langt das Wirtschaftlichkeitsgebot (§ 12 SGB V) unter anderem, dass bei gleicher Wirksamkeit und
Sicherheit die preiswertere Substanz verordnet wird. Die Kassenärztliche Vereinigung Nordrhein
informierte ihre Mitglieder im August 2008, dass der Einsatz von Bevacizumab zur Behandlung der
AMD im Rahmen eines Behandlungsvertrages möglich sei (KVNO, 2008a). Hiernach sehe der Vertrag
27

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
für den Patienten vor, dass neben den VEGF-Hemmern Ranibizumab und Pegaptanib auch Bevaci-
zumab zur intravitrealen Injektion eingesetzt werden könnte (KVNO, 2008a). Das Sozialgericht Düs-
seldorf hatte in seinem Urteil vom 02.07.2008 (S2 KA 181/07) zuvor den Antrag der Firma Novartis
(Hersteller von Ranibizumab) auf eine einstweilige Anordnung gegen diese Empfehlung zurückgewie-
sen. Nach seiner Auffassung sei der Vertrag rechtmäßig, da er die „volle Therapiefreiheit der behan-
delnden Ärzte und damit ihre Entscheidungsfreiheit“ für Ranibizumab, Bevacizumab oder ein ande-
res Produkt gewähre. Auch der „in dem streitbefangenen Vertrag vorrangig angestrebte Einsatz von
Avastin® [Bevacizumab]“ sei „ein legitimes Anliegen der Vertragspartner, das von der Rechtsordnung
gestützt“ werde (SG Düsseldorf, 2008). So ist nach Auffassung des Gerichts die „Erhaltung der finan-
ziellen Stabilität der gesetzlichen Krankenversicherung […] ein Gemeinwohlbelang von hohem Rang“
(SG Düsseldorf, 2008). Gut vier Jahre später entschied das BSG in seinem Urteil (B1 KR25/11 R) vom
3. Juli 2012, dass die zulassungsüberschreitende Anwendung von Bevacizumab in Anwendung der
Rechtsprechung zum OLU (BSG, 2002; BVerfG, 2005) nicht von der Leistungspflicht der GKV erfasst
sei (BSG, 2012)5.
BSG und BVerfG hatten in ihren Rechtsprechungen die medizinische Notwendigkeit betont, die einen
OLU aufgrund der Defizite im Arzneimittelrecht rechtfertigen. Das Wirtschaftlichkeitsgebot nach § 12
SGB V bildet hier hingegen keine Argumentationsgrundlage. Der „sozialversicherungsrechtliche off-
label use [dient] allein dem Interesse und dem Schutz der Versicherten und nicht dem Schutz seiner
Krankenversicherung vor finanzieller Überforderung oder der finanziellen Stabilität des Versiche-
rungssystems“ (Gaßner, 2013, S. A14756).
Ein Patient, der aufgrund einer AMD eine pharmakologische Therapie bedarf, hat demnach Anspruch
auf einen In-Label Use, in diesem Fall Lucentis®, auch wenn es die GKV finanziell stärker belastet. Das
ist gerade auch im Hinblick auf die arzneimittelrechtliche Gefährdungshaftung des pU (Kapitel 4) von
großer Bedeutung. Generell können weder Arzt noch Patient angehalten werden, sich in einem
rechtlichen Graubereich zu bewegen, um den ökonomischen Bedürfnissen der GKV entgegenzu-
5 In diesem Urteil ging es um die Klage einer Versicherten (* 1938), die bis August 2008 bei der BARMER GEK versichert war. Da sie an einer erheblichen Visuseinschränkung auf dem rechten Auge wegen rezidivierender Vaskulitis mit sekundärer epire-tinaler Membran und zystoidem Makulaödem litt – bei in diesem Zeitraum nahezu unbeeinträchtigtem Sehen auf dem linken Auge – beantragte sie im September 2006 bei ihrer GKV, dass diese die Kosten für die intravitreale Injektion von Avastin® übernehme. Die Barmer GEK lehnte den Antrag ab (Bescheid vom 5.10.2006). Im November 2006, im Januar 2007 und im März 2007 ließ sich die Klägerin dennoch ärztlich intravitreal Avastin® injizieren und wendete hierfür insgesamt 982,28 Euro auf. Ihr Überprüfungsantrag (12./15.10.2007) blieb ohne Erfolg (Bescheid vom 18.10.2007; Widerspruchsbescheid vom 10.1.2008). Das SG hatte ihre Klage auf Kostenerstattung ab- (Urteil vom 18.9.2008) und das LSG ihre Berufung zurückgewie-sen, mit der Begründung, es fehle für einen OLU an dem hierfür erforderlichen Wirksamkeitsnachweis. Des Weiteren beste-hen keine notstandsähnliche Situation und kein Seltenheitsfall. Das Makulaödem trete eher häufig auf und sei deshalb er-forschbar bzw. sogar erforscht. Das Krankheitsbild der Klägerin könne von entsprechenden Studien "mit umfasst sein" (BSG, 2012). 6 Von März 2010 bis Februar 2015 war Dr. Maximilian Gaßner Präsident des Bundesversicherungsamtes.
28

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
kommen. Schließlich dürfen sozialrechtliche Bestimmungen nicht „nach Bedarf“ ausgerichtet wer-
den, um je nach Auslegung dem Interesse der GKV zu nutzen.
29

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
3 Die Vereinbarkeit des Off-Label Use mit dem Arzthaf-tungsrecht
Die sozialrechtlichen Vorschriften des SGB V regeln in erster Linie die Fragen der Leistungspflicht der
GKV. Sind die Voraussetzungen einer Kostenübernahme (nach §§ 2 Abs. 1a, 31 und 35c SGB V bzw.
nach der Rechtsprechung von BSG und BVerfG) nicht erfüllt, hat der Versicherte keine Ansprüche auf
Sachleistungen und Kostenerstattung – nicht nur im OLU. Das SGB V hat jedoch zunächst einmal
keine unmittelbare Auswirkung auf das Arzthaftungsrecht. So ist es möglich, dass das Haftungsrecht
des Arztes über den sozialrechtlichen Standard hinausgeht. Die Vereinbarkeit des OLU mit dem Arzt-
haftungsrecht steht daher im Fokus des dritten Kapitels.
Die Therapiefreiheit des Arztes umfasst u.a. das Recht zur freien Entscheidung einer patientenindivi-
duell geeigneten Pharmakotherapie, dabei gilt es allerdings, den allgemeinen medizinischen Stan-
dard zu berücksichtigen7. Dieser muss nicht immer identisch mit einem in einer bestimmten Indika-
tion zugelassenen Arzneimittel sein (Walter, 2012). Nach dem Arzthaftungsrecht ist ein OLU somit
auch dann zulässig, wenn es nicht um die Behandlung einer schwerwiegenden Erkrankung geht
(Gaßner, 2013). Arzthaftungsrecht und SGB V kommen dann in Konflikt, wenn es um die Erstattungs-
frage und das Wirtschaftlichkeitsgebot geht. Beim Arzthaftungsrecht steht der vom Patienten durch
ein ärztliches Fehlverhalten erlittene Schaden im Vordergrund. Die Wirtschaftlichkeitsprüfung der
Krankenkasse knüpft an einen eventuell ihr entstandenen Schaden an, der durch eine unzureichende
Berücksichtigung der Verordnungsvorgaben des SGB V von Arzneimitteln verursacht wurde (Dierks,
2008). Dies kann eine Regressforderung an den behandelnden Arzt auslösen. Schließlich muss im
Sinne des § 12 SGB V das Medikament für seinen Einsatz ausreichend, zweckmäßig und wirtschaftlich
sein.
Aus arzneimittel- sowie strafrechtlicher Sicht ist es dem Arzt gestattet, ein Arzneimittel außerhalb
seiner dafür vorgesehenen Zulassung einzusetzen. Es gibt allerdings einen wesentlichen Unterschied
zwischen dem Arzneimittel- und dem Arzthaftungsrecht. Das Arzneimittelrecht soll die Arzneimittel-
sicherheit gewährleisten, das Arzthaftungsrecht die Anwendungssicherheit von Arzneimitteln.
Schließlich bietet eine Zulassung nicht automatisch die Gewähr für sorgfältige und standardgemäße
patientenindividuelle Therapieentscheidungen (Müller, 2009). Es ist also nicht nur der pharmazeuti-
sche Unternehmer, dem die Pflicht obliegt, die Sicherheit von Arzneimitteln zu gewährleisten. Der
Arzt hat ebenfalls eine weitgehende Produkt- und Marktbeobachtungspflicht (Hart, 1990) und hat
7 Ein Haftungsgrund im Schadensfall ist nicht etwa der unzureichende oder möglicherweise schlechte Ausgang einer (pharma-kologischen) Therapie, sondern das Abweichen vom medizinischen Standard (Gaßner & Strömer, 2012).
30

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
sich im Rahmen seiner Fortbildungspflicht über mögliche zulassungsüberschreitende Anwendungen
pharmakologischer Therapien zu informieren (Müller, 2009).
In vielen Facharztbereichen gehört der OLU zum Versorgungsalltag. Im Bereich der Arzthaftung
ergibt sich daraus die Frage, ob und wann ein Off-Label Use notwendigerweise stattfinden soll bzw.
muss. Die „Aciclovir-Entscheidung“ des Oberlandesgerichts (OLG) Köln (OLG, 1990) dient als oft zi-
tiertes Beispiel. Demnach gilt bei einer medizinischen Notwendigkeit ein Off-Label Use als verpflich-
tend, wenn ausreichende medizinische Kenntnisse sowohl über die Behandlungsrisiken als auch
Behandlungsvorteile bekannt sind. Im vorliegenden Fall besaß der Wirkstoff Aciclovir zwar keine
Zulassung für die Behandlung der Herpes-Enzephalitis, allerdings wurde diese Pharmakotherapie als
gewisser Standard im medizinischen Alltag angesehen. Der Arzt ist grundsätzlich an seine Pflicht zur
Behandlung nach dem aktuellen medizinischen Standard gebunden. So kann im Unterlassen eines
Off-Label Use ein Behandlungsfehler liegen. Für den daraus entstandenen Schaden haftet der Arzt
(Wegner, 2013), wenn eine begründete Aussicht auf eine erfolgreiche Behandlung im OLU bestanden
hätte und zugelassene wirksame und sichere Arzneimittel nicht zur Verfügung standen (Clemens,
2011; Schimmelpfennig-Schütte, 2004). Verzichtet der Arzt hingegen auf den zulassungsüberschrei-
tenden Einsatz eines Arzneimittels, welches sich zu diesem Zeitpunkt noch in der Erprobungsphase
befindet und noch nicht dem medizinischen Standard entspricht, kann dem Arzt bei einer ansonsten
vorliegenden Therapiealternative kein Behandlungsfehler unterstellt werden (Müller, 2009)8. Zivil-
rechtlich betrachtet kann der Off-Label Use somit in zwei Kategorien unterschieden werden: dem
„etablierten“ und dem „nicht-etablierten“ Off-Label Use (Walter, 2012).
Von einem „etablierten“ Off-Label Use wird dann gesprochen, wenn sich die Anwendung eines Arz-
neimittels außerhalb seines bestimmungsgemäßen Gebrauchs mittlerweile zum Facharzt-Standard
etabliert hat, wie es zum Beispiel in der Pädiatrie oder Onkologie der Fall sein kann. Auch kann es
vorkommen, dass Arzneimittel, die bereits den ärztlichen Standard in anderen Ländern erreicht ha-
ben, von deutschen wissenschaftlichen Fachgesellschaften in ihre Leitlinien übernommen werden
(Walter, 2012). Voraussetzung für die Anerkennung als Facharzt-Standard ist allerdings, dass das
betreffende Arzneimittel dem Stand der wissenschaftlichen Erkenntnis in Deutschland entspricht
(Walter, 2012).
8 Die evidenzbasierte Medizin dient dem Krankenversicherungsrecht, den Anspruch des Versicherten auf eine Behandlung nach dem allgemeinen Stand der medizinischen Erkenntnisse zu konkretisieren. Im Vergleich dazu werden ihre Grundsätze im Arzthaftungsrecht insbesondere zur Bestimmung des medizinischen Standards herangezogen (Gaßner & Strömer, 2012).
31

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Von einem „nicht-etablierten“ Off-Label Use spricht man hingegen, wenn der Facharzt-Standard für
diese Behandlung nicht belegbar ist. Im Gegensatz zum „etablierten“ Standard besteht somit auch
kein genereller Rechtsanspruch des Patienten auf die Behandlung mit einem Arzneimittel außerhalb
seiner Zulassung, was zur Konsequenz hat, dass grundsätzlich eine Einzelfallprüfung erforderlich ist
(Walter, 2012)9.
Die Pflicht zur umfassenden medizinischen Aufklärung
Ein OLU setzt grundsätzlich eine Patienteneinwilligung voraus. Diese muss vor Therapiebeginn ein-
geholt werden, da es sich hierbei immer um einen Eingriff in das Selbstbestimmungsrecht des Pati-
enten handelt. Die Therapie, ihre Risiken, Nutzen und mögliche Heilungschancen müssen dabei vom
Arzt umfassend erläutert werden, damit der Patient eine Entscheidung durch Abwägung von Vor-
und Nachteilen treffen kann (Wegner, 2013). Zudem übernimmt der Arzt im OLU auch die Rolle des
pU in der klinischen Prüfung mit Blick auf die Übernahme der Haftpflicht bei auftretenden uner-
wünschten Wirkungen oder Ereignissen. Grundsätzlich hat der Arzt auf eine fehlende arzneimittel-
rechtliche Zulassung hinzuweisen. Sofern es sich bei der Pharmakotherapie nicht um einen etablier-
ten Facharzt-Standard handelt, müssen Nutzen und Risiken eingehend evaluiert und gegenüber dem
Patienten vermittelt werden (Müller, 2009). Eine zulassungsübergreifende Pharmakotherapie bringt
nicht nur den erhofften Nutzen, sondern geht auch mit einem erhöhten Risiko einher, da in der je-
weiligen OLU-Indikation noch keine Ergebnisse aus systematisch durchgeführten klinischen Prüfun-
gen vorliegen (Bücheler et al., 2003; Janzen & Ludwig, 2012).
Patientenorientierte ökonomische Aspekte müssen beim OLU ebenfalls berücksichtigt werden. So ist
der Arzt dazu verpflichtet, auch auf Kosten hinzuweisen, die unter Umständen nicht von der GKV
übernommen werden und somit vom Patient selbst getragen werden müssen. Grundsätzlich hat
aber jeder Patient ein Anrecht auf eine solche, über den sozialrechtlichen Standard hinausgehende
Behandlung10.
9 Dort, wo der medizinische Standard verlassen wird, beginnt der so genannte „Heilversuch“. Er ist geprägt durch das Abwei-chen bzw. Fehlen eines medizinischen Standards im Rahmen einer therapeutischen Zielsetzung (Müller, 2009) und wird defi-niert als innovative Variante einer Standardbehandlung oder als erstmalige Anwendung einer neuen Behandlung bis zur systemisch-wissenschaftlichen Überprüfung ihrer Wirksamkeit (Hart, 1994). 10 Arzneimittelverordnungen liegen grundsätzlich in der Verantwortung des Vertragsarztes. Eine Genehmigung von Arzneimit-tel-Verordnungen durch die Krankenkasse ist demnach unzulässig (BMV-Ä, § 29).
32

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Die Vereinbarkeit des OLU sowohl mit dem Arzthaftungs- als auch mit dem Krankenversicherungs-
recht kann unter Berücksichtigung nachfolgender Anforderungen ermöglicht werden:
(1) Beachtung der Kriterien des BSG bzw. BVerfG.
(2) Anzeigen des OLU gegenüber der Krankenkasse mit der Bitte um Bestätigung, dass sie keine Feststellung ei-
nes sonstigen Schadens beantragen wird. Liegt diese Bestätigung nicht vor, sollte ein Privatrezept ausge-
stellt werden.
(3) Dokumentierte Aufklärung des Patienten über das übliche Maß hinaus und Übernahme der Haftpflicht bei
auftretenden unerwünschten Wirkungen und Ereignissen (KVNO, 2008).
33

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
4 Die Vereinbarkeit des Off-Label Use mit der Haftung des pharmazeutischen Unternehmers
Auf Basis der Überlegungen zur Vereinbarkeit eines OLU mit dem Sozialversicherungs- bzw. Arzthaf-
tungsrecht sollen im vierten Kapitel mögliche haftungsrechtliche Konsequenzen für den pharmazeu-
tischen Unternehmer aufgezeigt werden, wenn sein Arzneimittel außerhalb der dafür vorgesehenen
Zulassung angewendet wird. Schließlich kann die Verordnung eines Arzneimittels auch außerhalb
seiner eigentlichen Bestimmung durchaus gewinnbringend für den pU sein (Rückeshäuser, 2011).
Wie für den Arzt stellt sich auch für den pU die Frage, welche rechtlichen Haftungsrisiken mit dem
geduldeten OLU seines Arzneimittels verbunden sein können.
Die arzneimittelrechtliche Gefährdungshaftung und der bestimmungswidrige Ge-brauch eines Arzneimittels
Aufgrund der Erfahrungen aus dem Contergan-Skandal wurde durch das Gesetz zur Neuordnung des
Arzneimittelrechts 1976 der Gefährdungshaftungstatbestand11 in das AMG aufgenommen. Schließ-
lich können Arzneimittelrisiken nach erfolgter Marktzulassung nicht generell ausgeschlossen werden,
da die zuvor durchlaufenen klinischen Prüfungen nur an einem kleinen und sehr selektierten Patien-
tenkollektiv durchgeführt werden. Dem wirtschaftlichen Schutz zum Ausgleich für Personenschäden,
die trotz Sicherheitsvorkehrungen eingetreten sind, wird durch den Gefährdungshaftungstatbestand
Rechnung getragen (Müller, 2009). Allerdings haftet der pharmazeutische Unternehmer für seine
Arzneimittel nur im Rahmen des bestimmungsgemäßen Gebrauchs (Wegner, 2013).
Bei der Definition des bestimmungsgemäßen Gebrauchs eines Arzneimittels stellt sich vor allem die
Frage, ob dieser allein durch den pharmazeutischen Unternehmer erwirkt wird (subjektiver Ausle-
gungsmaßstab) oder ob ein Arzneimittel auch nach Auffassung der Fachkreise bestimmungsgemäß
eingesetzt werden kann (objektiver Auslegungsmaßstab) (Müller, 2009). Würde der bestimmungs-
gemäße Gebrauch allein durch den pU festgelegt werden, so würde es sich im Falle eines Off-Label
Use um einen bestimmungswidrigen Gebrauch handeln, der nicht vom Tatbestand des § 84 umfasst
wird (Müller, 2009). Ein nach Auffassung des pharmazeutischen Unternehmers bestimmungswidriger
11 Im § 84 Abs. 1 AMG heißt es: Wird infolge der Anwendung eines zum Gebrauch bei Menschen bestimmten Arzneimittels, das im Geltungsbereich dieses Gesetzes an den Verbraucher abgegeben wurde und der Pflicht zur Zulassung unterlegt oder durch Rechtsverordnung von der Zulassung befreit worden ist, ein Mensch getötet oder der Körper oder die Gesundheit eines Men-schen nicht unerheblich verletzt, so ist der pharmazeutische Unternehmer, der das Arzneimittel im Geltungsbereich dieses Gesetzes in den Verkehr gebracht hat, verpflichtet, dem Verletzten den daraus entstandenen Schaden zu ersetzen.
34

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Gebrauch kann aber auch dann bestimmungsgemäß sein, wenn dieser nicht explizit durch Kenn-
zeichnung, Packungsbeilage und Fachinformation ausgeschlossen wurde (Buchner & Jäkel, 2003;
Meyer & Grunert, 2005). Somit verbleibt dem pU zur Vermeidung eines „Fehlgebrauchs“ (Räpple,
1991) nur die Möglichkeit, entsprechende Warnhinweise anzugeben. Er kann den bestimmungswid-
rigen Arzneimittelgebrauch durch Kontraindikationen einschränken bzw. ausschließen, um seinen
Haftungsumfang einzugrenzen (Göben, 2009).
Der pU ist über den Stufenplanbeauftragten (§ 63a AMG) und den Pharmaberater (§ 76 AMG) ver-
pflichtet, Informationen zum Arzneimittelgebrauch zu sammeln, zu bewerten und zu dokumentieren.
Er wird vor allem dann Kenntnis von einem bestimmungswidrigen Gebrauch eines Arzneimittels
haben, wenn dieser im Sinne eines medizinischen Standards zwar indikationsfremd, wissenschaftlich
aber anerkannt ist. Schließt er die Anwendung nicht aus, erstreckt sich seine Haftung somit auch auf
den Off-Label Use (Wegner, 2013). Da auf die Möglichkeit des zulassungsüberschreitenden Einsatzes
von Arzneimitteln weder in der Kennzeichnung bzw. Packungsbeilage noch in der Fachinformation
hingewiesen werden darf, kommt eine Haftung des pharmazeutischen Unternehmers für eine nicht
den wissenschaftlichen Erkenntnissen entsprechende Information über den Off-Label Use nicht in
Betracht (Francke & Hart, 2003). Sofern Arzneimittel von der Expertengruppe als solche eingestuft
werden, die außerhalb ihrer Zulassungsbestimmung eingesetzt werden dürfen, werden auch diese
von der Gefährdungshaftung erfasst. Aus diesem Grund muss der pharmazeutische Unternehmer
seine Zustimmung zur Bewertung des Medikaments durch die Expertengruppe erteilen (§ 35b Abs. 3
S. 3 SGB V). Einer Zustimmung bedarf es grundsätzlich nicht, wenn in bewiesenen Ausnahmefällen
(beispielsweise bei lebensbedrohlichen Erkrankungen ohne Therapiemöglichkeit) ein Off-Label Use
einen Therapieerfolg verspricht (Buchner & Jäckel, 2003).
Gerade mangelnde Informationsmöglichkeiten durch die erwähnten Hinweis-Verbote sowohl der
jeweiligen Fachkreise über zusätzliche Indikationen als auch des Patienten über die korrekte Ein-
nahme seines Arzneimittels im OLU erschweren den Anspruch einer patientengerechten Versorgung.
Unerwünschte Arzneimittelwirkungen können bei einem geduldeten OLU auch nicht ausreichend
erfasst werden.
35

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
5 Nutzen und Risiko neuer Arzneimittel
Der Begriff der Nutzenbewertung findet sich unter anderem im AMG, im SGB V sowie der Verfah-
rensanordnung des G-BA (G-BA, 2008) und dem Methodenpapier des Institutes für Qualität und
Wirtschaftlichkeit im Gesundheitswesen (IQWiG, 2015a). Grundlage für die Bewertung eines Nutzens
ist die Festlegung eines Verfahrens zur Quantifizierung von Nutzen und Schaden. Dies ist umso rele-
vanter, will man eine einheitliche Verbindlichkeit als Grundlage für politische Entscheidungen schaf-
fen (Wegscheider et al., 2015).
Nach den Ausführungen der rechtlichen Rahmenbedingungen hinsichtlich einer zulassungsüber-
schreitenden Arzneimittelverordnung (Kapitel 1-4) soll mit dem fünften Kapitel der Aspekt der Nut-
zenbewertung neuer Arzneimittel aufgegriffen werden. Dabei soll der Frage nachgegangen werden,
inwiefern auch die Nutzenbewertung neuer Arzneimittel zu einem OLU beitragen kann.
5.1 Das Gesetz zur Neuordnung des Arzneimittelmarktes
Mit Wirkung zum 1. Januar 2011 ist das Gesetz zur Neuordnung des Arzneimittelmarktes nach § 35a
SGB V in Kraft getreten. Eines der Hauptziele des Gesetzes war hierbei, die in den letzten Jahren
angestiegenen Arzneimittelkosten vor allem im festbetragsfreien Marktsegment zu begrenzen und
die Kosten eines Arzneimittels an seinen therapeutischen sowie patientenrelevanten (Zusatz-)Nutzen
zu koppeln. Innerhalb eines Jahres soll auf Basis der Bewertungen zum Zusatznutzen der Erstat-
tungsbetrag des Arzneimittels zwischen pU und GKV-Spitzenverband ausgehandelt werden. Bis zum
endgültigen Beschluss durch den G-BA bestimmen zunächst die pharmazeutischen Unternehmen
den Preis ihres Arzneimittels. Beauftragt durch den G-BA bewertet das IQWiG den Zusatznutzen
eines neu in den Markt eingeführten Arzneimittels zunächst anhand der Herstellerunterlagen, um
festzustellen, ob und in welchem Ausmaß ein Zusatznutzen gegenüber der zweckmäßigen Ver-
gleichstherapie vorliegt. Diese Ergebnisse bilden die Grundlage für die Verhandlungen über den
Erstattungsbetrag. Das IQWiG folgt dabei den aus dem Gesetz und den zugehörigen Verordnungen
abgeleiteten Grundsätzen.
Als höchstes Organ der Selbstverwaltung hat der G-BA schließlich die Aufgabe, die entsprechenden
Beschlüsse zu den einzelnen Arzneimitteln zu fassen, die dann Eingang in die Arzneimittel-Richtlinien
finden. Konstatiert der G-BA dem Arzneimittel keinen Zusatznutzen, kann es in eine Festbetrags-
gruppe mit vergleichbaren Arzneimitteln und einer gemeinsamen Erstattungshöchstgrenze eingrup-
36

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
piert werden. Wenn eine Einordnung aufgrund einer fehlenden Festbetragsgruppe nicht möglich
sein sollte, wird ein Erstattungsbetrag der gesetzlichen Krankenkasse auf Grundlage der jährlichen
Kosten der Vergleichstherapie vereinbart. Wird hingegen ein Zusatznutzen festgestellt, vereinbart
der Spitzenverband Bund als Vertreter der GKVen mit den Arzneimittelherstellern auf Basis des Be-
schlusses der Nutzenbewertung Erstattungsbeträge, die für alle Kassen gültig sind. Der Preis darf
dabei höher liegen als der einer vergleichbaren Therapien (Windt et al., 2014). Mit Einführung des
AMNOG verpflichtet das Gesetz erstmals die pharmazeutischen Unternehmen, für Arzneimittel mit
neuen Wirkstoffen einen Zusatznutzen nachzuweisen, so dass sich der Erstattungsbetrags am thera-
peutischen Wert des Arzneimittels im Vergleich zur bisherigen Standardtherapie orientiert. Durch die
frühe Nutzenbewertung stehen den Ärzten sowie Patienten außerdem detaillierte und von der
pharmazeutischen Industrie unabhängig erstellte Informationen zur Verfügung. G-BA und IQWiG
verfolgen damit das Ziel, primär monetär orientierten Innovationen den Marktzugang zu erschwe-
ren, um so die Solidargemeinschaft finanziell zu entlasten.
5.2 Wirksamkeit und patientenorientierter Nutzen neuer Arz-neimittel
Die Wirksamkeit und der patientenorientierte Nutzen eines neuen Arzneimittels haben schon vor
Einführung des AMNOG im deutschen Gesundheitswesen zunehmend an Bedeutung gewonnen (BÄK
& KBV, 1996). Während die Bestimmung der Wirksamkeit eines Arzneimittels im Rahmen von expe-
rimentellen, präklinischen und klinischen Studien erfolgt, erweist sich der Nachweis eines zusätzli-
chen Nutzens gerade neuer Arzneimittel aufgrund der begrenzt vorliegenden Erfahrungswerte sowie
patientenindividueller Bedingungen als weitaus schwieriger. Aus diesem Grund kann man sich bei
der Nutzenbewertung in der Versorgungsforschung nicht ausschließlich auf randomisiert kontrollier-
te Vergleiche aus großen Studien beschränken. Patientenrelevante Endpunkte wie Mortalität, Mor-
bidität und Lebensqualität, die in klinischen Studien primäre Endpunkte darstellen können, werden
zwar bei der Beurteilung des (Zusatz-)Nutzens berücksichtigt, ein weitaus wichtigerer Faktor in der
Nutzenbeurteilung ist allerdings der Verlauf der Zeit. Krankheiten durchlaufen unterschiedliche Sta-
dien, verändern sich, bedingen unterschiedliche Prioritäten mit weitreichenden Auswirkungen auf
die Lebensumstände des Patienten. Dadurch verschieben sich auch seine individuellen Bedürfnisse.
Was ihm heute nutzt, kann morgen schon nutzlos sein. Auch aus diesem Grund müssen Nutzenstu-
dien in der realen Patientenversorgung an anderen Kriterien ausgerichtet werden als Wirksamkeits-
studien im Zulassungsprozess an ausgesuchten und vorher definierten Patientenpopulationen.
37

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Neben der Nutzen- spielt auch die Kosten-Bewertung eine wesentliche Rolle. Kosten und Nutzen
stehen dabei in synergistischer Beziehung zueinander, um Angemessenheit und Zumutbarkeit einer
finanziellen Übernahme durch die Versichertengemeinschaft entsprechend beurteilen zu können
(Windt et al., 2014). Problematisch ist auch hier, dass sich zum Zeitpunkt der Zulassung eines Arz-
neimittels dieser patientenorientierter Zusatznutzen nach § 35b SGB V (Reduktion der Mortalität,
der Morbiditätsbelastung und der unerwünschten Wirkungen sowie Erhöhung der Lebensqualität)
im vollen Umfang nicht ohne Weiteres finanziell bewerten lässt.
Kriterien für die Beurteilung von neu auf den Markt gekommenen Arzneimitteln
Die Deutsche Pharmazeutische Gesellschaft (DPhG) stellt in ihrem Positionspapier Kriterien für die
Beurteilung von neu auf den Markt gekommenen Arzneimitteln vor, um den möglicherweise innova-
tiven Charakter – und somit auch den damit verbundenen (Patienten-)Nutzen – eines neuen Wirk-
stoffes oder einer neuen Arzneiform besser beurteilen und einschätzen zu können (DPhG, 2005).
Neue Arzneistoffe können demnach in die Kategorien Sprunginnovation, Schrittinnovation und
Scheininnovation eingruppiert werden. So werden Arzneistoffe oder auch Arzneiformen grundsätz-
lich dann als Sprunginnovation bezeichnet, wenn sie als erste Vertreter einer neuen bisher nicht
bestehenden Wirkstoffklasse einen wertvollen therapeutischen Fortschritt darstellen. Schrittinnova-
tionen sind hingegen durch eine schrittweise Verbesserung bereits bekannter Wirkstoffe gekenn-
zeichnet und weisen im Vergleich zu den Scheininnovationen eine therapeutische Verbesserung auf.
Kriterien, die die Einordnung in die drei Kategorien ermöglichen, sind neben den Stoff- (z.B. chemi-
sche Struktur) und pharmakodynamischen Kriterien (z.B. Selektivität für Zielstrukturen im Körper)
auch pharmakokinetische (z.B. Bioverfügbarkeit im Körper), pharmazeutisch-technologische (z.B. Art
der Anwendung, Stabilität) oder Interaktions-Kriterien (z.B. Wechselwirkungen mit anderen Arznei-
mitteln) (Windt et al., 2014). Mit der Kategorie der Schrittinnovation berücksichtigt die DPhG auch
den Umstand, dass ein therapeutischer Mehrwert zum Zeitpunkt der Markteinführung noch nicht
ausreichend abschätzbar ist, sich der Zusatznutzen eines Arzneimittels möglicherweise erst nach
Jahren der Vermarktung zeigt.
38

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Im Vergleich zur DPhG geht man bei einer Arzneimittel-Bewertung nach Fricke und Klaus von phar-
makologischen Aspekten aus und bewertet für den potentiellen (Zusatz-)Nutzen in erster Linie die
Neuartigkeit der Wirkmechanismen der Arzneistoffe (Fricke & Beck, 2013). Die neuen Arzneistoffe
werden dabei – unter Berücksichtigung der aktuellen Marktsituation im jeweiligen Indikationsgebiet
sowie basierend auf den publizierten Ergebnissen der pharmakologischen und klinischen (Zulas-
sungs-)Studien – nach folgendem Klassifikationsschema eingeteilt:
A für innovative Struktur oder neuartiges Wirkprinzip mit therapeutischer Relevanz,
B für eine Verbesserung pharmakodynamischer oder pharmakokinetischer Eigenschaften be-
reits bekannter Wirkprinzipien,
C für Analogpräparat mit keinen oder marginalen Unterschieden zu bereits eingeführten Prä-
paraten und
D bei einem nicht ausreichend gesicherten Wirkprinzip oder unklarem therapeutischer Stel-
lenwert.
Auch die International Society of Drug Bulletins (ISDB) verbindet mit dem Begriff der Arzneimittelin-
novation in ihrer Declaration on therapeutic advance in the use of medicines (ISDB, 2001) drei Kon-
zepte. Mit dem kommerziellen Konzept wird zunächst einmal jede Markteinführung eines neuen
Wirkstoffes, einer neuen Indikation eines (bereits etablierten) Wirkstoffes, einer neuen Darrei-
chungsform oder einer neuen Behandlungsmethode als innovativ bezeichnet. Bei dem technologi-
schen Konzept steht die technische Innovation im Vordergrund. So gelten beispielsweise neue bio-
technologische Herstellungen bekannter Wirkstoffe als innovativ. Das Konzept des therapeutischen
Fortschritts hingegen bewertet ein Arzneimittel dann als innovativ, wenn ein Zusatznutzen bzw. eine
bessere therapeutische Wirksamkeit gegenüber bestehenden Therapien vorliegt.
Schon allein die unterschiedlichen Bewertungsverfahren machen deutlich, dass es zum Zeitpunkt der
Zulassung nicht ohne Weiteres beurteilbar ist, ob ein (patientenindividueller) Zusatznutzen vorliegt.
39

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
5.3 Indikationserweiterungen im Bestandsmarkt
Zulassungsstudien sind aufgrund der begrenzten Patientenzahl, der Aufnahme bzw. des Ausschlusses
bestimmter Patientenkollektive sowie des oftmals kurzen Beobachtungszeitraums nicht ausreichend,
um das Potential des neuen Wirkstoffes aufzudecken. Dieser Aspekt wird u.a. auch durch die Bewer-
tung eines Arzneimittels als Schrittinnovation berücksichtigt. Der therapeutische Zusatznutzen of-
fenbart sich oftmals erst nach Jahren der Vermarktung, auch durch Erkenntnisse, die außerhalb des
Zulassungsbereichs im OLU gesammelt werden konnten. Unabhängig von der Dauer ihres Marktzu-
ganges erfahren Arzneimittel so nicht nur Anwendungseinschränkungen, sondern auch Indikations-
erweiterungen – entweder auf dem gleichen therapeutischen Gebiet oder sie führen zu Zulassungen
neuer Fertigarzneimittel in anderen Anwendungsbereichen. Während Anwendungseinschränkungen
aufgrund neuer Risikoerkenntnisse nach erfolgtem Marktzugang nicht unmittelbar vorhersehbar
sind, stellt sich die Frage, inwiefern Indikationserweiterungen nicht auch zur Marktstrategie eines pU
gehören. Im Jahr 1994 wurde beispielsweise Gabapentin zur Therapie der Epilepsie zugelassen. Seit-
dem erfährt der Wirkstoff immer weitere Therapiemöglichkeiten (Steinman et al., 2006) und wird
mittlerweile u.a. auch bei peripheren neuropathischen Schmerzen wie schmerzhafter diabetischer
Neuropathie und postherpetischer Neuralgie bei Erwachsenen eingesetzt. In der Zwischenzeit befin-
det sich das Antiepileptikum auf der Anlage VI der Arzneimittel-Richtlinie Verordnungsfähigkeit von
zugelassenen Arzneimitteln in nicht zugelassenen Anwendungsgebieten zur Behandlung der Spastik
im Rahmen der Multiplen Sklerose (Stand: 08.10.2014).
Vergleichbar erfolgreich in ökonomischer Hinsicht sind Arzneimittel zur Therapie seltener Erkrankun-
gen (sogenannte Orphan Drugs). In Deutschland gilt eine Krankheit dann als selten, wenn weniger als
40.000 Menschen (< 5/10.000 Einwohner) an ihr leiden. Dementsprechend ist das Interesse der
pharmazeutischen Unternehmen, Zeit und finanziellen Aufwand in die Entwicklung solcher Arznei-
mittel zu investieren, gering, da Marktzulassungen in Indikationsgebieten, die eine höhere Morbidi-
tätsrate haben oder umsatzstärker sind, ökonomisch gesehen erfolgsversprechender sind. Aus die-
sem Grund wurden von Seiten der Behörden auf diesem Forschungsgebiet Anreize geschaffen, um
für die pharmazeutischen Unternehmen die Forschung und Entwicklung zu erleichtern und zu för-
dern.
Im Januar 2000 trat die Verordnung des Europäischen Parlaments und des Europäischen Rates über
Arzneimittel für seltene Leiden (Orphan-Arzneimittel) in Kraft (EU-Richtlinie EG 141/2000). Ein Arz-
neimittel wird im Sinne dieser Verordnung dann als Orphan Drug eingestuft, wenn es für die Diagno-
se, Prävention oder Therapie eines Leidens bestimmt ist, das lebensbedrohlich ist oder eine chroni-
sche Invalidität nach sich zieht und von dem nicht mehr als fünf von 10.000 Menschen betroffen sind
40

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
(Europäisches Parlament & Europäischer Rat, 1999)12. Weitere Anforderungen sind neben den defizi-
tären Behandlungsmöglichkeiten auch ein erheblicher patientenindividueller Nutzen (Schwabe &
Paffrath, 2011). Finanzielle Anreize für den pU wurden in dieser Verordnung ebenfalls festgehalten,
um die Entwicklung von Arzneimitteln für seltene Krankheiten zu unterstützen (Forschungsunter-
stützung, teilweise oder vollständige Befreiung von den Zulassungsgebühren oder exklusive Ver-
marktungsrechte für das entsprechende Indikationsgebiet (vfa, 2013; Kesselheim et al., 2011).
Abbildung 2 zeigt die Verteilung der Arzneimittelzulassungen mit einem Orphan Drug-Status nach
therapeutischem Gebiet im Zeitraum 2000 bis 2014. Gerade im onkologischen Bereich sind viele
seltene Erkrankungen zu erkennen. Das bedeutet allerdings nicht, dass diese Arzneimittel nach ihrem
Marktzugang nicht auch Indikationserweiterungen erfahren. Als Beispiel dafür wurde Imatinib
(Glivec®) im Jahr 2001 zunächst zur Therapie der Chronischen Myeloischen Leukämie zugelassen.
Weitere Indikationserweiterungen mit Orphan Drug-Status folgten (Akute Lymphatische Leukämie,
Gastrointestinaler Stromatumor, Dermatofibrosarcoma protuberans) (Glaeske et al., 2010).
Zu bedenken gilt, dass in klinischen Studien für Orphan Drugs häufiger kleine Patientenzahlen unter-
sucht werden. Auch wird statt einer randomisierten, placebokontrollierten und doppelblind angeleg-
ten Studie häufig ein nichtrandomisiertes, unverblindetes Design gewählt. Surrogatparameter wer-
den zur Bewertung der Wirksamkeit herangezogen (Kesselheim et al., 2011; Kesselheim et al.,
2011a). Unerwünschte Ereignisse treten in solchen Studien signifikant häufiger auf (Kesselheim et al.,
2011; Kesselheim et al., 2011a).
12 Die FDA-Richtlinien für Orphan Drugs sehen dagegen vor, dass nicht mehr als 200.000 Menschen der gesamten US-amerikanischen Bevölkerung von der Krankheit betroffen sein dürfen, damit ein Medikament den Orphan Drug-Status erhält. Zudem fehlt die Einschränkung, dass es noch keine zufriedenstellende Therapie gibt und es sich um Arzneimittel oder Dia-gnostika handeln muss (vfa bio, 2015).
41
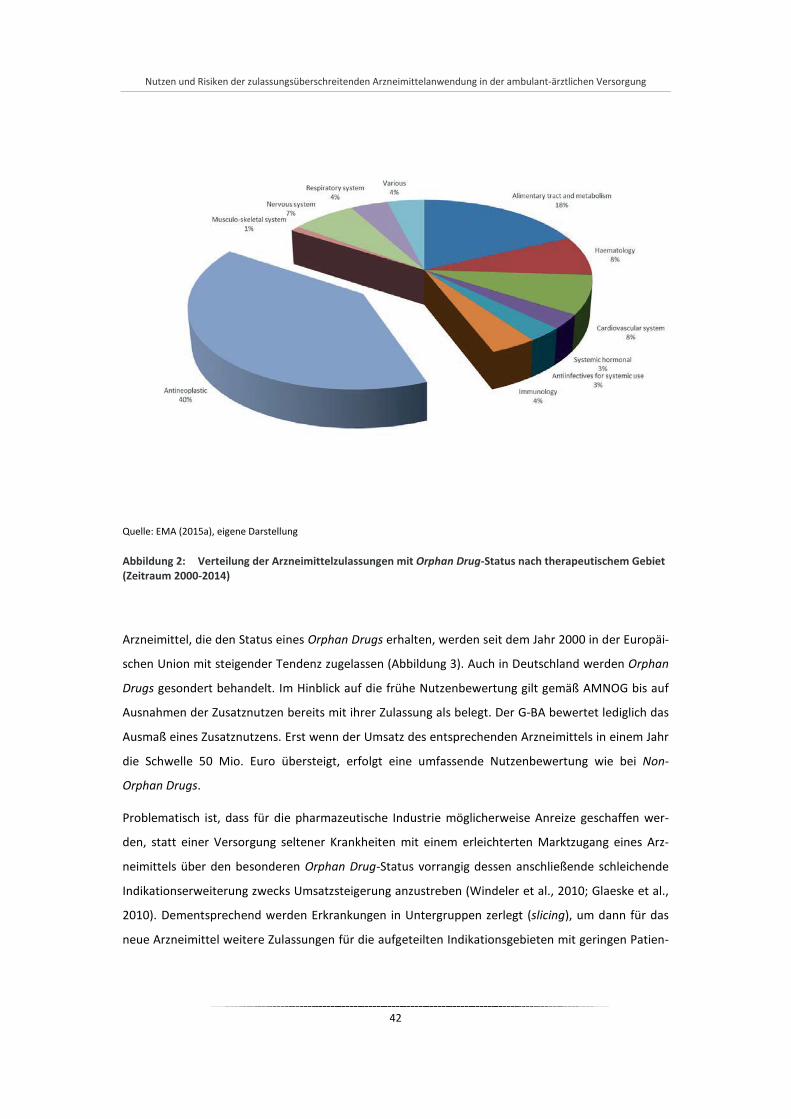
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Quelle: EMA (2015a), eigene Darstellung
Abbildung 2: Verteilung der Arzneimittelzulassungen mit Orphan Drug-Status nach therapeutischem Gebiet (Zeitraum 2000-2014)
Arzneimittel, die den Status eines Orphan Drugs erhalten, werden seit dem Jahr 2000 in der Europäi-
schen Union mit steigender Tendenz zugelassen (Abbildung 3). Auch in Deutschland werden Orphan
Drugs gesondert behandelt. Im Hinblick auf die frühe Nutzenbewertung gilt gemäß AMNOG bis auf
Ausnahmen der Zusatznutzen bereits mit ihrer Zulassung als belegt. Der G-BA bewertet lediglich das
Ausmaß eines Zusatznutzens. Erst wenn der Umsatz des entsprechenden Arzneimittels in einem Jahr
die Schwelle 50 Mio. Euro übersteigt, erfolgt eine umfassende Nutzenbewertung wie bei Non-
Orphan Drugs.
Problematisch ist, dass für die pharmazeutische Industrie möglicherweise Anreize geschaffen wer-
den, statt einer Versorgung seltener Krankheiten mit einem erleichterten Marktzugang eines Arz-
neimittels über den besonderen Orphan Drug-Status vorrangig dessen anschließende schleichende
Indikationserweiterung zwecks Umsatzsteigerung anzustreben (Windeler et al., 2010; Glaeske et al.,
2010). Dementsprechend werden Erkrankungen in Untergruppen zerlegt (slicing), um dann für das
neue Arzneimittel weitere Zulassungen für die aufgeteilten Indikationsgebieten mit geringen Patien-
42
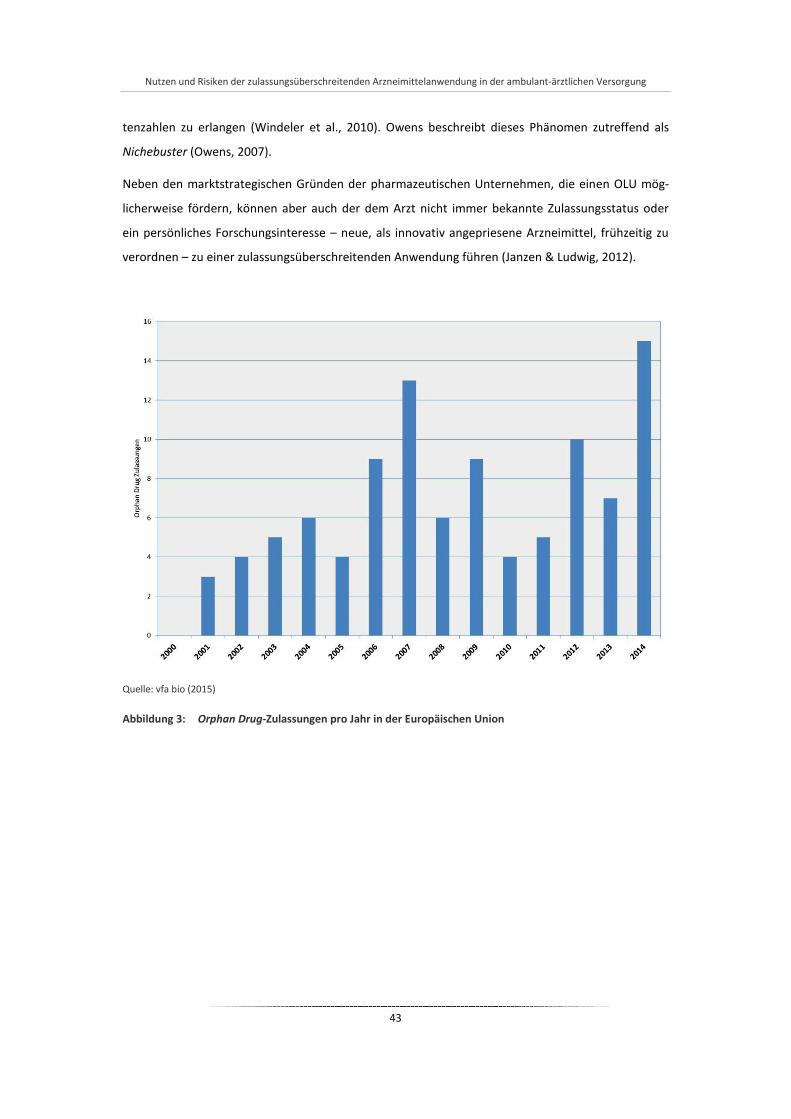
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
tenzahlen zu erlangen (Windeler et al., 2010). Owens beschreibt dieses Phänomen zutreffend als
Nichebuster (Owens, 2007).
Neben den marktstrategischen Gründen der pharmazeutischen Unternehmen, die einen OLU mög-
licherweise fördern, können aber auch der dem Arzt nicht immer bekannte Zulassungsstatus oder
ein persönliches Forschungsinteresse – neue, als innovativ angepriesene Arzneimittel, frühzeitig zu
verordnen – zu einer zulassungsüberschreitenden Anwendung führen (Janzen & Ludwig, 2012).
Quelle: vfa bio (2015)
Abbildung 3: Orphan Drug-Zulassungen pro Jahr in der Europäischen Union
43

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
6 Zwischenfazit
Die bestmögliche Therapie einer Erkrankung ist das grundsätzlich unterstellte Anliegen aller Akteure
im Gesundheitswesen, für den Patienten hat die wirksamste Therapie oberste Priorität. Neben den
rechtlichen Unsicherheiten, die sich bei einer zulassungsüberschreitenden Anwendung ergeben, ist
gerade die potentielle Gefährdung durch ein für die Indikation des Patienten ungeprüftes bzw. nicht
hinreichend geprüftes Arzneimittel nicht auszublenden.
Gerade wenn es sich um einen „nicht-etablierten“ Off-Label Use handelt, fehlen den behandelnden
Ärzten entscheidende Informationen hinsichtlich Dosierung, Applikationsweg und bzw. oder Kome-
dikation. Unerwünschte Arzneimittelwirkungen beim zulassungsüberschreitenden Einsatz sind daher
häufig (Jonville-Béra et al., 2005; Bennett et al., 2005) und stellen ein ernstzunehmendes Problem
dar. Neben dem Ausbleiben der erhofften Behandlungserfolge kann unter Umständen auch die wei-
tere Therapie des Patienten gefährdet werden, wenn unerwünschte Arzneimittelwirkungen den
Patienten zusätzlich physisch und psychisch belasten. Dennoch gilt die Anwendung von Arzneimitteln
außerhalb ihrer Zulassung seit Jahrzehnten gerade in den Bereichen der Onkologie, Neurologie und
Pädiatrie als fester und somit auch unverzichtbarer Bestandteil der Therapien. Schließlich ist der Off-
Label Use unter engen Voruassetzungen auch rechtmäßig. Das kann zur Konsequenz haben, dass
Forschungsanstrengungen der Arzneimittelindustrie immer mehr in den Hintergrund treten, oder
dass es durch Entwicklung sogenannter Nichebuster zu einer weiteren Zunahme des Off-Label Use
kommt (Janzen & Ludwig, 2012).
Auf der anderen Seite zeigt sich am Beispiel Avastin® (Bevacizumab) und Lucentis® (Ranibizumab) die
„Lücke“ in diesem komplexen System. Beide Wirkstoffe wurden von Genentech entwickelt, einem
Tochterunternehmen des Pharmakonzerns Roche (Avastin®), während das pharmazeutische Unter-
nehmen Novartis – das knapp ein Drittel des stimmberechtigten Kapitals von Roche hält – der Zulas-
sungsinhaber von Lucentis® ist. Beide Wirkstoffe werden im (geduldeten) OLU (Avastin®) und im ILU
(Lucentis®) zur Therapie der AMD eingesetzt. Letztgenannter Wirkstoff ist allerdings im Vergleich zu
Avastin® um ein Vielfaches teurer.
Eine Lösung des Problems kann also letztendlich nur aufgrund eines engen Zusammenspiels ver-
schiedener Regularien greifen. Gefordert sind vor allem pharmazeutische Unternehmen, bei Er-
kenntnissen zur Sicherheit und Wirksamkeit ihres Arzneimittels in einer zulassungsüberschreitenden
Anwendung eine zeitnahe Zulassungserweiterung zu beantragen. Nur dann ist auch eine komplikati-
onsfreie Versorgung der Patienten auf der Basis von Daten aus klinischen Studien möglich. Das Ge-
sundheitswesen hat als Wirtschafts- und Wachstumsfaktor einen hohen Stellenwert in der Volkswirt-
schaft: durch den Schutz des Einzelnen und seiner Gesundheit wird auch der Arbeitsmarkt erheblich
beeinflusst. Umsätze und Gewinne im gesamtwirtschaftlichen Sinn können somit auch als Erfolg des
44

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Gesundheitswesens verbucht werden. Ökonomische Interessen dürfen aber nicht so weit reichen,
dass der Schutz und das Wohl des Einzelnen eine untergeordnete Rolle spielen. Diese haben in unse-
rem Sozialstaat nach wie vor oberste Priorität.
45

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
7 Analyse des Off-Label Use in der ambulant-ärztlichen Versorgung
Hintergrund und Ziele
Der Off-Label Use wird nach wie vor als Notwendigkeit einer patientengerechten und patientenindi-
viduellen Therapie betrachtet. Arzneimittel, die erst kürzlich den Marktzugang erhalten haben und
für deren Anwendung weniger Erfahrung besteht, besitzen ein höheres Risikopotential. Die Anwen-
dung dieser Arzneimittel außerhalb ihrer eigentlichen Zulassungsbestimmungen sollte besonders
kritisch betrachtet werden. Aber auch bei Wirkstoffen, die schon seit Jahrzehnten auf dem deut-
schen Arzneimittelmarkt etabliert sind, findet die Anwendung nicht immer zulassungskonform statt.
So hat der Wirkstoff Bevacizumab für die Therapie der altersbedingten Makuladegeneration (AMD)
trotz therapeutischer Alternativen durch Einführung zugelassener monoklonaler Antikörper seinen
Off-Label-Status bei ophthalmologischen Verordnungen über die Jahre nie ganz verloren, nicht zu-
letzt aufgrund der Therapieempfehlungen unterschiedlicher Fachgesellschaften und ökonomisch
bedingter Interessen. Bei dem Wirkstoff Metamizol, der während seiner fast 100-jährigen Vermark-
tungsgeschichte aufgrund schwerer UAW eine immer stärkere Indikationseinschränkung erfahren
musste, findet dies in der alltäglichen Praxis allerdings offenbar ebenso wenig Berücksichtigung und
steht einem breiten Einsatz außerhalb seiner Zulassungsbestimmung nicht entgegen (Hoffmann et
al., 2015).
Vorrangiges Ziel dieser Arbeit ist es, mögliche Gründe für den Off-Label Use in der ambulant-
ärztlichen Versorgung zu ermitteln. Nachdem zu Beginn der Arbeit die rechtlichen Aspekte aufge-
zeigt und die Bedeutung einer zulassungsüberschreitenden Anwendung für die beteiligten Institutio-
nen (GKV, pU) und bzw. oder (verantwortlichen) Personen (Arzt, Patient) dargelegt wurden, soll im
zweiten Teil der Arbeit der Ursachen-Frage nachgegangen werden.
Zwei Gründe können zu einem OLU führen:
1. ein möglicher Diagnose-Bias (aufgezeigt am Beispiel der chronisch obstruktiven Lun-
genkrankheiten COPD und Asthma) und
2. fehlende Therapieoptionen (am Beispiel der chronischen Hepatitis C).
Diese Aspekte sind Gegenstand der Untersuchung.
46

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Die Frage nach einem möglichen OLU aufgrund fehlerhafter Diagnosestellungen soll anhand der
chronischen Lungenkrankheiten Asthma bronchiale und COPD analysiert werden. Auch wenn sich die
beiden Erkrankungen in ihrer Symptomatik ähneln, weisen sie Unterschiede in der Pathologie auf.
Abweichungen in den Therapiestrategien und -zielen erfordern eine präzise Differenzierung der
beiden Erkrankungen im medizinischen Alltag. Untersuchungsgegenstand sind dabei die drei Arznei-
-2-Sympathomimetikum, Roflumilast aus der Gruppe der
Phosphodiesterase-4-Hemmer sowie Aclidiniumbromid als lang wirkender Muscarin-Rezeptor-
antagonist. Alle drei Wirkstoffe wurden ausschließlich zur Therapie der COPD zugelassen, so dass
eine eindeutige Zuweisung eines OLU erfolgen kann, wenn einem Asthmatiker dieses Arzneimittel
verordnet wird.
Die Frage nach einem möglichen OLU aufgrund fehlender Therapieoptionen soll anhand der chroni-
schen Hepatitis C analysiert werden. Schließlich änderten sich mit der Markteinführung von neuen,
direkt antiviral wirkenden Substanzen (DAA) in den Jahren 2014 und 2015 auch die Therapieempfeh-
lungen. Der Erfolg einer Therapie ist u.a. vom identifizierten Genotyp abhängig, so dass nicht alle
Patienten von der Zulassung der neuen DAA profitieren. Hier stellt sich die Frage, ob Fachgesellschaf-
ten auch bei Wirkstoffen, die erst kürzlich den Marktzugang erhalten haben, eine Empfehlung für
ihre zulassungsüberschreitende Anwendung aussprechen und inwiefern Forschungsinhalte und ihre
Ergebnisse aus Zulassungsstudien mit der späteren Zulassung übereinstimmen.
Verschiedene eigene bereits ausgewiesene (Windt et al., 2013; Boeschen et al., 2015a) bzw. noch zur
Veröffentlichung ausstehende (Boeschen et al., 2015b) Publikationen fließen in die Erörterung ver-
schiedener thematischer Aspekte dieser Dissertation. Umfangreiche zusätzliche und vertiefende
Auswertungen ergänzen die betreffenden Themen.
47

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
7.1 Methodik zur Bewertung des Off-Label Use neuer und eta-blierter Arzneimittel
Ziel dieser Arbeit ist es, die Ursachen eines OLU in der ambulant-ärztlichen Versorgung zu ermitteln,
Abbildung 4 umreißt kurz die methodische Vorgehensweise zur Bewertung der entsprechenden
Fragestellungen.
Abbildung 4: Ursachen-Analyse des Off-Label Use in der ambulant-ärztlichen Versorgung am Beispiel etab-lierter und neuer Arzneimittel
Um der Frage eines möglichen OLU aufgrund fehlerhafter Diagnosen nachzugehen, wurden zunächst
die relevanten Leitlinien gesichtet, um Therapieunterschiede der Erkrankungen Asthma bronchiale
und COPD herauszuarbeiten. Die Ergebnisse der Update-Recherchen, die im Auftrag des G-BA vom
IQWiG durchgeführt wurden, fanden ebenfalls Berücksichtigung, um mögliche Änderungen von The-
rapieempfehlungen in Deutschland zu erfassen. Mit den Update-Recherchen wurde das Ziel verfolgt,
aus aktuellen sowie evidenzbasierten Leitlinien Kernempfehlungen zu synthetisieren, die für eine
Überarbeitung der entsprechenden Disease-Management-Programme (DMP) von Bedeutung sein
könnten. Im Anschluss daran wurden Studien gesichtet, in denen die Wirkstoffe Indacaterol, Roflu-
48

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
milast und Aclidiniumbromid – in Deutschland zugelassen zur Therapie der COPD – auch in der
Asthma-Therapie unterschiedlicher Genese eingesetzt wurden. Für diese Fragestellung wurde nicht
nur in der Meta-Datenbank PubMed, sondern zusätzlich in der Datenbank des U.S. National Institu-
tes of Health (ClinicalTrials.gov)13 recherchiert, um auch nicht veröffentlichte Studien zu erfassen.
Die Ergebnisse von Routinedaten-Auswertungen wurden am Ende der entsprechenden Wirkstoffka-
pitel aufgeführt, um das Verordnungsverhalten in der ambulant-ärztlichen Praxis aufzuzeigen.
Auch für die Fragestellung eines möglichen OLU aufgrund fehlender Behandlungsoptionen wurden
Leitlinienempfehlungen herausgearbeitet und mit dem Zulassungsstatus (anhand der entsprechen-
den Fachinformationen) der neuen Wirkstoffe zur Therapie der cHC verglichen. Untersucht werden
sollte, ob Fachgesellschaften auch bei Wirkstoffen, die erst kürzlich den Marktzugang erhalten ha-
ben, eine Empfehlung für ihre zulassungsüberschreitende Anwendung aussprechen. Daran anschlie-
ßend wurden die Zulassungs-relevanten Studien der neuen antiviralen Wirkstoffe zur Therapie der
cHC (Boceprevir, Telaprevir, Simeprevir, Daclatasvir, Ledipasvir, Sofosbuvir, Dasabuvir sowie die
Fixkombination zweier Wirkstoffklassen bestehend aus Ombitasvir und dem Ritonavir-geboosterten
Paritaprevir) gesichtet und mit dem Zulassungsstatus verglichen. Hier dienten die Beurteilungsbögen
der EMA als Literaturquelle zur Ausarbeitung möglicher Divergenzen.
7.1.1 Methodisches Vorgehen zur Analyse der systematischen Über-sichtsarbeiten
Bewertung der Studienlage
Für das Review wurde einerseits Medline (Medical Literature Analysis and Retrieval System Online)
als öffentlich zugängliche Datenbank des US-amerikanischen National Center for Biotechnology In-
formation (NCBI) für medizinische Fachliteratur und andererseits die von der United States National
Library of Medicine aufgebaute Datenbank PubMed Central (PMC) genutzt. Recherchen in beiden
Datenbanken wurden über die Meta-Datenbank PubMed zwischen Januar 2013 und April 2015
durchgeführt. Die Bewertung der Studienlage basierte auf den Ergebnissen der relevanten Zulas-
sungsstudien bzw. auf Studien, die nach der Markteinführung veröffentlicht wurden, sowie auf den
Beurteilungsberichten der verantwortlichen Zulassungsbehörden.
13 ClinicalTrials.gov listet gegenwärtig 189.718 Studien mit Standorten in allen 50 Staaten und in 190 Ländern (Stand: Mai 2015).
49

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Um keine relevanten Publikationen zu übersehen, wurde zum einen eine breite Suchstrategie ge-
wählt, zum anderen wurden alle identifizierten Abstracts oder Volltexte zweimal im Abstand von
mindestens 14 Tagen gesichtet. Der Publikationszeitraum der Studien wurde in Abhängigkeit von der
jeweiligen Fragestellung eingegrenzt. Es musste sich dabei um originäre Studien (Primärstudien)
handeln.
Um die eingangs aufgeführte Frage zu beantworten, wurden die Abstracts gesichtet und, falls dies
für die Ermittlung der notwendigen Informationen nicht ausreichend war, auch die Volltexte. Da es
das primäre Ziel des Reviews war, eine Übersicht der gelisteten Artikel zu erstellen, wurden Doppel-
publikationen nicht ausgeschlossen.
Im Folgenden sollen die Suchstrategien sowie die Ein- und Ausschlusskriterien der gefundenen Publi-
kationen aufgeführt werden.
(1) Off-Label Use aufgrund fehlerhafter Diagnosestellungen
Für das Review zur Fragestellung eines OLU aufgrund fehlerhafter Diagnosestellungen wurde folgen-
der Suchstring eingegeben
asthma AND (Indacaterol OR Roflumilast OR Aclidinium bromide) AND (therapy OR treat-
ment OR prophylaxis OR prevention OR “clinical trial”) NOT COPD
Die Literraturrecherchen erfolgten letztmalig am 01.04.2015. Der Publikationszeitraum wurde für
diese Fragestellung nicht eingeschränkt. Mit dieser ersten Suche konnten insgesamt 38 Publikatio-
nen identifiziert werden. Die Ergebnisse der Recherche werden im Kapitel 8.1 aufgeführt.
Im Folgenden wurden Publikationen ausgeschlossen, wenn
1. kein Abstract oder kein Volltext vorlag,
2. der Abstract oder der Volltext nicht auf Englisch oder Deutsch verfasst wurde,
3. das Thema der Publikation die Erkrankung Asthma nicht schwerpunktmäßig mit einbezog,
4. der Fokus nicht auf Indacaterol, Roflumilast oder Aclidiniumbromid gelegt wurde, entweder
als Monotherapie oder in Kombination mit anderen Wirkstoffen,
5. es sich nicht um eine klinische Studie handelte.
50

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
(2) Off-Label Use aufgrund fehlender Therapieoptionen
Für das Review zur Fragestellung eines OLU aufgrund fehlender Therapieoptionen wurde folgender
Suchstring eingegeben
“hepatitis c” AND (Boceprevir OR Telaprevir OR Simeprevir OR TMC435 OR Daclatasvir OR
BMS790052 OR Ledipasvir OR GS-5885 OR Sofosbuvir OR GS-7977 OR PSI-7977 OR Om-
bitasvir OR ABT-267 OR Paritaprevir OR ABT-450 OR Ritonavir OR Dasabuvir OR ABT-333)
AND (therapy OR treatment OR prophylaxis OR prevention) AND “clinical trial”
Die Literraturrecherchen erfolgten letztmalig am 03.01.2015. Der Publikationszeitraum wurde für
diese Fragestellung auf den 01.01.2009 bis 01.04.2015 beschränkt. Dieser Zeitraum garantierte den
Einschluss zulassungsrelevanter Studien. Mit der ersten Suche konnten insgesamt 402 Publikationen
identifiziert werden. Die Ergebnisse der Recherche werden im Kapitel 8.2 aufgeführt.
Im Folgenden wurden Publikationen ausgeschlossen, wenn
kein Abstract oder kein Volltext vorlag,
der Abstract oder der Volltext nicht auf Englisch oder Deutsch verfasst wurde,
es sich nicht um eine klinische Studie der Phasen I-III handelte,
das Thema der Studie die Erkrankung chronische Hepatitis C nicht schwerpunktmäßig ein-
bezog,
der Fokus nicht auf die Protease-Inhibitoren Boceprevir, Telaprevir, Simeprevir, die NS5A-
Inhibitoren Daclatasvir und Ledipasvir, den NS5B-Inhibitor Sofosbuvir, die Fixkombination
bestehend aus Ombitasvir, Paritaprevir und Ritonavir sowie den nicht-nukleosidischen Po-
lymerase-Inhibitor Dasabuvir gelegt wurde,
Wirkstoffkombinationen mit in Deutschland (noch nicht) zugelassenen Substanzen zur The-
rapie der chronischen Hepatitis C im Fokus der Studie standen.
51

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Für die Literaturrecherche wurden außerdem Leitlinienempfehlungen (für COPD und Asthma bron-
chiale die Nationalen Versorgungsleitlinien, für die cHC das Addendum der DGVS), Fachinformatio-
nen sowie – wenn vorhanden – die Nutzenbewertung des G-BA hinzugezogen. Die vom IQWiG vorge-
legten Ergebnisse der Update-Recherchen zum Thema Asthma bronchiale und COPD wurden eben-
falls berücksichtigt, um mögliche Änderungen von Therapieempfehlungen in Deutschland zu erfas-
sen. Beurteilungsbögen der EMA dienten der Erfassung von Unterschieden zwischen den Zulassungs-
studien und dem Zulassungsstatus.
7.1.2 Leitlinienempfehlungen
Nach Definition des Institute of Medicine handelt es sich bei Leitlinien um systematisch entwickelte
Entscheidungshilfen für Leistungserbringer und Patienten, die bei speziellen Gesundheitsproblemen
Therapieoptionen aufzeigen (Field & Lohr, 1995). Sie sollen als Orientierungshilfen verstanden wer-
den, von denen in begründeten Fällen ein Abweichen erlaubt oder sogar verlangt wird (BÄK & KBV,
1997).
Für die Empfehlungen zu den einzelnen Arzneimitteln und ihren jeweiligen Indikationsgebieten wur-
den Leitlinien der entsprechenden Fachgesellschaften genutzt, die im Register der Arbeitsgemein-
schaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF) eingetragen sind. Soll-
te zu den bestimmten Indikationsgebieten keine AWMF-Leitlinie zu finden gewesen sein, wurde nach
anderen nationalen Handlungsempfehlungen, beispielsweise Empfehlungen der Arzneimittelkom-
mission der deutschen Ärzteschaft (AkdÄ), gesucht und die Ergebnisse gegebenenfalls berücksichtigt.
7.1.3 Fachinformationen
Im AMG ist die Pflicht zur Erstellung einer Fachinformation in § 11a gesetzlich verankert. Danach
muss der pharmazeutische Unternehmer für in Deutschland zugelassene Arzneimittel den Fachkrei-
sen auf Anforderung Fachinformationen bzw. für in Europa zugelassene Arzneimittel die entspre-
chenden Summary of Product Characteristics (SPC) zur Verfügung stellen (Fachinfo-Service®).
Aktuelle Fachinformationen der in dieser Arbeit untersuchten Wirkstoffe aus den Jahren 2011 bis
2015 wurden gesichtet, um den derzeitigen Zulassungsstatus zu ermitteln. Des Weiteren wurden alle
Fachinformationen aus den Jahren 2011-2015 untersucht, um mögliche Unterschiede aufgrund von
Änderungsanzeigen (Indikation, Dosierung, Kontraindikationen etc.) zu erfassen.
52

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
7.1.4 Nutzenbewertung nach § 35a SGB V
Seit dem 1. Januar 2011 hat der Gemeinsame Bundesausschuss im Rahmen des AMNOG die Aufga-
be, eine (Zusatz-)Nutzenbewertung für alle neu zugelassenen Arzneimittel mit neuen Wirkstoffen
durchzuführen (§ 35a SGB V). Die Bewertung des G-BA dient als Grundlage für die Entscheidung, wie
hoch der Erstattungsbetrag ausfällt, den die gesetzliche Krankenversicherung für ein neues Arznei-
mittel mit einem neuen Wirkstoff zu zahlen hat.
Die Ergebnisse dieser Nutzenbewertungen haben möglicherweise Einfluss auf das Verordnungsver-
halten der Ärzte – gerade auch im Hinblick auf einen möglichen OLU. Arzneimittel, die nach der Nut-
zenbewertung besonders gut abschneiden, wecken eventuell eher das Forschungsinteresse, diese
Wirkstoffe auch in anderen Indikationsgebieten einzusetzen, als Arzneimittel, die weniger positiv
beurteilt werden.
7.1.5 Methodik der Routinedaten-Analyse
Um das Verordnungsverhalten in der ambulant-ärztlichen Praxis aufzuzeigen, wurden Routinedaten
der GKV-Erstattung hinzugezogen. Allein die drei Wirkstoffe Indacaterol, Roflumilast und Aclidinium-
bromid lassen Rückschlüsse auf einen möglichen OLU zu, da eine Verordnung außerhalb der Indika-
tion COPD nicht zulässig ist. Routinedaten der Wirkstoffe zur antiviralen Therapie der cHC lassen
diese Rückschlüsse nicht zu, da die Identifizierung des Genotyps nicht durch die ICD-Codierung er-
fasst wird.
Für die Sekundärdaten-Analyse zur Versorgung mit neuen Arzneimitteln wurden ambulant-ärztliche
Abrechnungsdaten von Versicherten der Techniker Krankenkasse (TK) aus den Jahren 2010-2013
genutzt (bereits veröffentlicht im Innovationsreport 2014 (Windt et al., 2014) bzw. noch zur Veröf-
fentlichung ausstehend (Innovationsreport 2015, Boeschen et al., 2015)). Neben den Daten zu Arz-
neimittelverordnungen wurden u.a. auch die erfassten Diagnosedaten niedergelassener Ärzte her-
angezogen. Die Auswertung der Daten erfolgte mit dem Statistikprogrammpaket SAS.
Die zur Erstellung der Arbeit benötigten Daten wurden bereits seitens der TK selektiert und anony-
misiert an die Universität Bremen weitergegeben. Dabei erfolgte die Anonymisierung sämtlicher
Daten unter Verwendung einer Fallnummer, die es ermöglichte, anonymisierte Datensätze mitein-
ander zu verknüpfen, ohne dass die Identität der Versicherten festgestellt werden konnte. Für die
allgemeinen Auswertungen wurden ausschließlich Daten berücksichtigt, bei denen auch Angaben zu
Alter und Geschlecht der Versicherten vorlagen.
53

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Die Bezugspopulation bestand aus Versicherten der TK, die im Untersuchungszeitraum versichert
waren. Analog zum Arzneiverordnungsreport (u.a. Schwabe & Paffrath, 2011) entsprach eine ärztli-
che Verordnung genau einer Arzneimittelpackung, d.h. zwei Arzneimittelverordnungen auf einem
Rezeptblatt zählten als zwei Arzneimittelpackungen.
Ambulante und Arzneimittel(verordnungs)daten
Die Grundlage der gesetzlich vorgeschriebenen Diagnoseverschlüsselung im ambulanten und statio-
nären Sektor stellt die von der WHO entwickelte Internationale Klassifikation der Krankheiten (Inter-
national Classification of Diseases: ICD) dar. Die Diagnosen werden in Deutschland als Behandlungs-
anlass nach der Internationalen Klassifikation von Krankheiten in der deutschen Fassung (ICD-GM;
german modification) verschlüsselt und übermittelt. Diagnosen werden pro Behandlungsfall und
quartalsweise abgerechnet. Des Weiteren enthalten die ambulanten Daten der TK u.a. Behandlungs-
zeitraum, Abrechnungstag, Abrechnungsziffer und die Arztnummer des jeweiligen Leistungserbrin-
gers. Diagnostische Maßnahmen sind hingegen nur begrenzt abbildbar, da diese als fakultative Leis-
tung über eine Pauschale abgerechnet werden. Bei den Verordnungsanalysen wurden ausschließlich
zugelassene Fertigarzneimittel berücksichtigt, da bei der Frage einer möglichen Anwendung eines
Arzneimittels im Off-Label Use eine behördliche Zulassung vorausgegangen sein muss. Dabei richtete
sich die Fragestellung nach den Zulassungsbestimmungen für den deutschen Arzneimittelmarkt.
Der Arzneimittelverordnungsdatensatz enthält die Daten aller zu Lasten der TK übermittelten Rezep-
te, die über Apotheken abgerechnet worden sind. Für die vorliegende Arbeit wurden nur die Verord-
nungen der zu untersuchenden Arzneimittel erhoben.
Zur Verarbeitung und Aggregierung der Daten wurde die Codierung des Anatomisch-Therapeutisch-
Chemischen (ATC)-Arzneimittelklassifikationssystems genutzt, welches Arzneimittel in hierarchischer
Form nach therapeutischen und chemischen Kriterien gliedert. Dabei unterteilen sich die Arzneistof-
fe nach diesem System in 14 Hauptgruppen (Ebene 1) mit zwei therapeutisch-pharmakologischen
Untergruppen (Ebene 2 und 3). Eine anatomisch-therapeutisch-chemische Untergruppe bildet die 4.
Ebene, während die 5. Ebene den chemischen Wirkstoff benennt. Einige Wirkstoffe besitzen z.B. bei
unterschiedlichen Indikationsgebieten auch unterschiedliche ATC-Codierungen.
54

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8 Gründe für einen möglichen Off-Label Use: Analyse der Versorgungssituation
Nach Klärung der rechtlichen Aspekte eines OLU werden in den folgenden Kapiteln dieser Arbeit
Gründe analysiert, die zu einem Off-Label Use in der ambulant-ärztlichen Versorgung führen können.
Der Aufbau der Kapitel ist jeweils identisch: Das Kapitel beginnt mit der Charakterisierung des Krank-
heitsbildes, seinen epidemiologischen und sozialökonomischen Aspekten und der sich daraus erge-
benden Relevanz für den Bereich Public Health, der Darstellung der Leitlinien und Therapieempfeh-
lungen. Im Anschluss daran werden neuere Arzneimittel auf einen möglichen OLU untersucht. Im
Hinblick auf die methodische Vorgehensweise und der dadurch angestrebten Ziele unterscheiden
sich die einzelnen Kapitel jedoch aufgrund ihrer unterschiedlichen Forschungsfragen.
Abbildung 5: Aufbau des versorgungsanalytischen Teils der Arbeit
55

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.1 Off-Label Use aufgrund eines möglichen Diagnose-Bias: COPD vs. Asthma bronchiale
Wie Asthma bronchiale zählt die COPD (chronic obstructive pulmonary disease) zu den chronischen
Krankheiten der unteren Atemwege (ICD-10-GM J40-J44). Trotz unterschiedlicher Pathologie ähneln
sich die beiden Erkrankungen in ihrer Symptomatik. Da sich Therapiestrategien und -ziele von COPD
und Asthma aber teilweise erheblich unterscheiden, ist eine exakte Differenzierung im medizinischen
Alltag zwar notwendig, jedoch auf Basis des klinischen Bildes nicht immer eindeutig möglich. So kann
auch neben der COPD ein Asthma bronchiale bestehen. Betroffene Patienten weisen in solchen Fäl-
len neben einer bronchialen Hyperreagibilität eine größere Variabilität bzw. Reversibilität der Atem-
obstruktion auf (Windt, 2010). Im umgekehrten Fall kann nicht ausgeschlossen werden, dass langan-
haltendes Asthma durch Remodelling-Vorgänge14 in eine COPD mit irreversibler Obstruktion und
veränderter Hyperreagibilität übergeht (Windt, 2010). Ein durch Fehldiagnosen induzierter OLU führt
im schlimmsten Fall dazu, dass ein erwarteter Therapieerfolg ausbleibt und sich somit negativ auf
Lebenszeit und Lebensqualität des Patienten auswirkt. Zulassungsüberschreitende Anwendungen
aufgrund einer fehlerhaften Diagnosestellung bzw. bestehender Mischformen eines Krankheitsbildes
und eines möglicherweise daraus resultierenden Bias in der ICD-Codierung sind daher naheliegend
und somit Forschungsgegenstand dieses Kapitels.
Nach Klärung der Begrifflichkeiten COPD und Asthma bronchiale sowie ihrem sozialökonomischen
Stellenwert in der Bevölkerung im ersten Teil, werden nachfolgend die therapeutischen Unterschie-
de zur Behandlung der beiden Erkrankungen herausgearbeitet und diskutiert. Auf dem Verständnis
dieser Grundlagen -2-
Sympathomimetikum, Roflumilast aus der Gruppe der Phosphodiesterase-4-Hemmer sowie Aclidini-
umbromid als lang wirkender Muscarin-Rezeptorantagonist. Alle drei Wirkstoffe wurden allein zur
Therapie der COPD zugelassen. Nach Darstellung der Studienlage im Rahmen ihrer zulassungskon-
formen Anwendung, werden für die entsprechenden Wirkstoffe die anhand der Literaturrecherche
ermittelten klinischen Studien aufgezeigt, bei denen es um die Therapie des Asthma bronchiale un-
terschiedlicher Genese geht. Zum Schluss wird das Verordnungsverhalten in der ambulant-ärztlichen
Praxis durch Auswertungen von Routinedaten aufgezeigt und diskutiert.
14 Remodelling: Vermehrte Einlagerung von Kollagen in die Bronchialwand durch wiederholte Schädigung und nachfolgende Reparaturvorgänge.
56

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.1.1 COPD und Asthma bronchiale: Definition, Epidemiologie und sozialökonomische Aspekte
Die chronische Bronchitis zählt zu den häufigen Erkrankungen und wird „als Husten und Auswurf
über wenigstens drei Monate in mindestens zwei aufeinanderfolgenden Jahren“ definiert (Vogel-
meier et al., 2007; Kim & Criner, 2013; Konietzko & Fabel, 2005). Geht sie mit einer dauerhaften
Verengung der Atemwege einher, so stellt sie ein klinisch bedeutsames Krankheitsbild, die chronisch
obstruktive Bronchitis, dar. Sie wird in der Literatur auch als COPD (chronic obstructive pulmonary
disease) bezeichnet (Vogelmeier et al., 2007; Forey et al., 2011). Hauptsymptome einer COPD sind
neben dem Auswurf von zähem Schleim und chronischem Husten auch die Atemnot, die zu Beginn
nur unter Belastung auftritt. Mittels Spirometrie wird die Einschränkung der Lungenfunktion gemes-
sen und die COPD kann so in verschiedene Schweregrade aufgeteilt werden, wonach sich letztend-
lich auch ihre Therapie richtet (Vogelmeier et al., 2007). Im Gegensatz zum Asthma bronchiale ist die
chronische Obstruktion auch nach Gabe von Bronchodilatatoren und bzw. oder Corticosteroiden
nicht mehr voll reversibel (van Schayck et al., 1991; Burge et al., 2000; Vestbo et al., 1999).
Charakteristisch für die COPD ist eine Folge pathophysiologischer Veränderungen in der Lunge, die
insgesamt zu einer andauernden Reduzierung des Atemstroms und somit zu einer schnellen Abnah-
me der Lungenfunktionsparameter führen. Sowohl die chronische Bronchitis als auch das Lungen-
emphysem stellen Verlaufsformen der COPD dar. Somit ist die COPD als Sammelbegriff zu verstehen,
mit dem chronische Lungenerkrankungen beschrieben werden, bei denen die Atmung infolge einer
Schädigung und chronischer Entzündungsprozesse der Lunge hochgradig beeinträchtigt ist (Mutsch-
ler et al., 2008). Im Allgemeinen nehmen die entzündlichen und strukturellen Veränderungen mit
dem Schweregrad der Erkrankung zu. Dabei beruht die Entstehung der chronisch obstruktiven Bron-
chitis möglicherweise auf Wechselwirkungen zwischen genetischen und erworbenen Faktoren (Hall-
berg et al., 2008; Svartengren et al., 2009). Zwar gehört der Tabakkonsum zu den wichtigsten modifi-
zierbaren Risikofaktoren der COPD (Forey et al., 2011; Kim & Criner, 2013) und das Erkrankungsrisiko
steigt mit der Zahl gerauchter Zigaretten (sogenannte pack years) kumulativ an (Forey et al., 2011;
Meteran et al., 2012), trotzdem führt das Einstellen des Rauchens nicht zu einem Stillstand der Er-
krankung (Hogg et al., 2004).
57

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 4 fasst die genuinen und erworbenen Risikofaktoren für die Entwicklung einer COPD zusam-
men. Danach ist das Zigarette-Rauchen zwar nicht als der einzige, jedoch zweifellos der weltweit
bedeutendste Risikofaktor (GOLD, 2015) für die Entwicklung einer COPD, der die aufgrund des Alters
bedingte Abnahme der Lungenfunktion stark beschleunigt (Mutschler et al., 2008).
Tabelle 4: Risikofaktoren für die Entwicklung einer COPD
Genuine Faktoren
Erworbene Faktoren
genetische Prädisposition -1-Protease-Inhibitor-Mangel)
inhalativer Tabakkonsum
bronchiale Überempfindlichkeit
berufsbedingte Stäube
Störungen des Lungenwachstums
häufige Atemwegsinfektionen in der Kindheit
Quelle: NVL (2006)
Eine Reihe von kardiopulmonalen Krankheiten wie beispielsweise Bronchiektasen, Bronchialkarzi-
nom, Lungentuberkulose, Asthma bronchiale, aber auch die Linksherzinsuffizienz können ähnliche
Symptome hervorrufen, weswegen die Diagnose COPD stets eines Ausschlusses bedarf (Mutschler et
al., 2008). Andererseits handelt es sich hierbei um eine komplexe Systemerkrankung, die nicht nur
die Atemwege und die Lunge betrifft (Lorenz et al., 2013). Betroffene Patienten weisen deutlich
erhöhte Raten an Komorbiditäten auf, die mit der sytemischen Komponente der COPD-spezifischen
Entzündung in Zusammenhang gebracht werden (Fabbri & Rabe, 2007).
Die COPD ist nicht nur eine der häufigsten Erkrankungen weltweit, sie gehört mittlerweile auch zu
den führenden krankheitsbedingten Todesursachen. Lag sie 1990 noch an sechster Stelle, so wird sie
bis zum Jahr 2020 voraussichtlich auf den dritten Platz vorrücken (NVL, 2006). Pritzkuleit et al. (2010)
schätzen in ihrer aktuellen Studie, dass die COPD-Erkrankungszahl bis 2050 um 1,2 Millionen Fälle
ansteigen wird (Vergleich: 2010 = 6,8 Millionen vs. 2050 = 8,0 Millionen). Da das Erkrankungs-Risiko
nicht nur mit dem Raucherstatus, sondern auch mit zunehmendem Alter steigt, muss aufgrund der
immer älter werdenden Bevölkerung in Zukunft auch mit einer höheren Erkrankungshäufigkeit ge-
rechnet werden muss (Aumann & Prenzler, 2013).
58

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Nach Aussagen des Robert Koch-Instituts ist die Prävalenz der COPD in Deutschland nicht exakt bezif-
ferbar, da sich die Definition der Erkrankung aus der Feststellung einer chronischen Bronchitis in
Kombination mit einer dauerhaften Verengung der unteren Atemwege herleitet (RKI, 2014b). Das
erklärt auch, warum in Studien die Prävalenz der COPD zwischen 1,3-13,2 Prozent schwankt, in Ab-
hängigkeit von den verwendeten Krankheitsdefinitionen, der Erhebungsmethode und der einbezo-
genen Population (Aumann & Prenzler, 2013; Gingter et al., 2009; Geldmacher et al., 2008). Die vom
RKI initiierte Studie „Gesundheit in Deutschland aktuell“ (RKI, 2014c) erfasste u.a. die Lebenszeit-
prävalenz der chronischen Bronchitis in der erwachsenen Bevölkerung. Deren Ermittlung erfolgte in
zwei Befragungs-Stufen. Wurde die erste Frage („Wurde bei Ihnen jemals von einem Arzt eine chro-
nische Bronchitis festgestellt?“, gegebenenfalls mit dem Hinweis „unter chronischer Bronchitis ver-
stehen wir Husten und Auswurf mindestens drei Monate lang pro Jahr“) bejaht, erfolgte die Frage,
ob die chronische Bronchitis auch in den letzten zwölf Monaten bestand. Die Ergebnisse der Umfrage
werden in den Abbildungen 6 und 7 dargestellt. Durch die Einschränkung, dass die Schätzung der
Lebenszeitprävalenz nicht auf bevölkerungsrepräsentativen Daten beruhte15, lag sie mit ihren ermit-
telten knapp neun Prozent niedriger als die Schätzung des Weißbuchs Lunge für Deutschland (RKI,
2014b). Die vorliegenden Daten der GEDA 2012 zeigen einen Anstieg der Prävalenz mit zunehmen-
dem Lebensalter sowie eine höhere Prävalenz bei Frauen als bei Männern (9,9 vs. 7,3 Prozent). In-
nerhalb der Gruppen nimmt diese Prävalenz mit zunehmendem Bildungsgrad ab. Gründe für die
hohen Raten in bestimmten Bevölkerungssubgruppen sehen die Autoren der Untersuchung in der
höheren Wahrscheinlichkeit von Lungenschäden (Tabakexposition, andere Luftschadstoffe) mit stei-
gendem Alter. Dass gerade Frauen eine höhere Prävalenz aufweisen als Männer, hängt vermutlich
mit einer stärkeren Vulnerabilität der weiblichen Lunge gegenüber Umweltschadstoffen und Tabak-
rauch zusammen (van Haren-Willems & Heijdra, 2010). Der Gesundheitssurvey bestätigte allgemein
den engen Zusammenhang zwischen Bildung und Gesundheit (RKI, 2005). So findet man in bildungs-
ferneren Gesellschaftsschichten oft auch einen höheren Anteil an Rauchern bzw. Arbeitnehmern, die
berufsbedingt lungenschädigenden Faktoren wie beispielsweise Steinstaub oder Chemikalien ausge-
setzt sind. Grundsätzlich gilt jedoch, dass sich nicht aus jeder chronischen Bronchitis auch zwangsläu-
fig eine COPD entwickeln muss.
15Das RKI berücksichtigt dabei den Hinweis der Deutschen Gesellschaft für Pneumologie, dass durch die bestehende Begriffs-vielfalt für COPD im Hinblick auf die uneinheitliche Definition und der daraus resultierenden uneinheitlichen Zuordnung zu den in den vergangenen Jahren wiederholt neu definierten ICD-Ziffern (Deutsche Gesellschaft für Pneumologie, 2000, S. 10) die Datenerhebung limitiert sei.
59
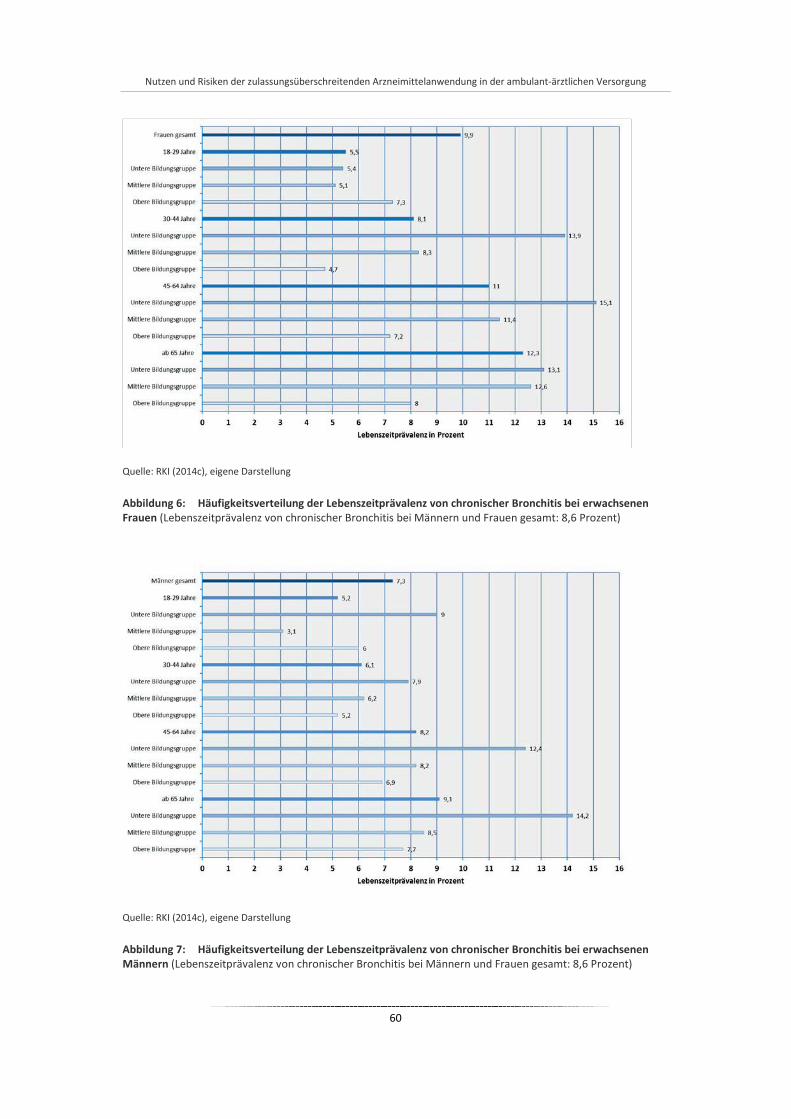
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Quelle: RKI (2014c), eigene Darstellung
Abbildung 6: Häufigkeitsverteilung der Lebenszeitprävalenz von chronischer Bronchitis bei erwachsenen Frauen (Lebenszeitprävalenz von chronischer Bronchitis bei Männern und Frauen gesamt: 8,6 Prozent)
Quelle: RKI (2014c), eigene Darstellung
Abbildung 7: Häufigkeitsverteilung der Lebenszeitprävalenz von chronischer Bronchitis bei erwachsenen Männern (Lebenszeitprävalenz von chronischer Bronchitis bei Männern und Frauen gesamt: 8,6 Prozent)
60
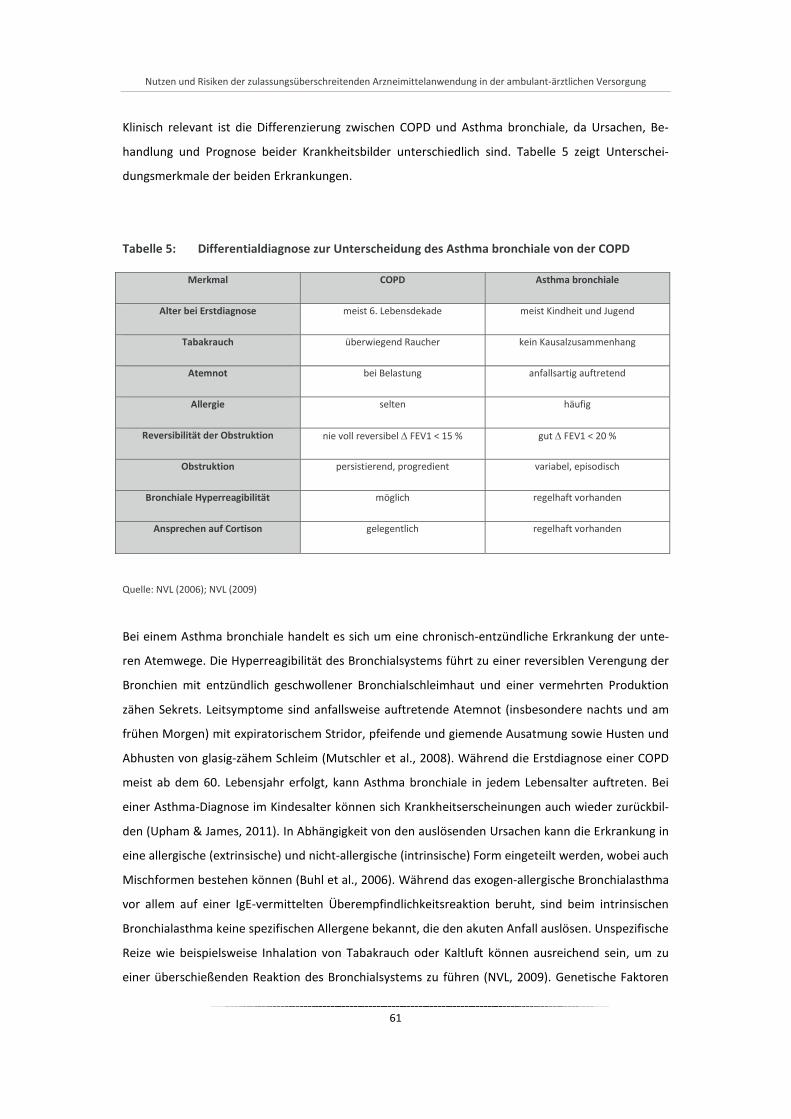
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Klinisch relevant ist die Differenzierung zwischen COPD und Asthma bronchiale, da Ursachen, Be-
handlung und Prognose beider Krankheitsbilder unterschiedlich sind. Tabelle 5 zeigt Unterschei-
dungsmerkmale der beiden Erkrankungen.
Tabelle 5: Differentialdiagnose zur Unterscheidung des Asthma bronchiale von der COPD
Merkmal COPD
Asthma bronchiale
Alter bei Erstdiagnose meist 6. Lebensdekade meist Kindheit und Jugend
Tabakrauch überwiegend Raucher kein Kausalzusammenhang
Atemnot bei Belastung anfallsartig auftretend
Allergie selten häufig
Reversibilität der Obstruktion nie voll reversibel FEV1 < 15 %
gut FEV1 < 20 %
Obstruktion
persistierend, progredient variabel, episodisch
Bronchiale Hyperreagibilität
möglich regelhaft vorhanden
Ansprechen auf Cortison
gelegentlich regelhaft vorhanden
Quelle: NVL (2006); NVL (2009)
Bei einem Asthma bronchiale handelt es sich um eine chronisch-entzündliche Erkrankung der unte-
ren Atemwege. Die Hyperreagibilität des Bronchialsystems führt zu einer reversiblen Verengung der
Bronchien mit entzündlich geschwollener Bronchialschleimhaut und einer vermehrten Produktion
zähen Sekrets. Leitsymptome sind anfallsweise auftretende Atemnot (insbesondere nachts und am
frühen Morgen) mit expiratorischem Stridor, pfeifende und giemende Ausatmung sowie Husten und
Abhusten von glasig-zähem Schleim (Mutschler et al., 2008). Während die Erstdiagnose einer COPD
meist ab dem 60. Lebensjahr erfolgt, kann Asthma bronchiale in jedem Lebensalter auftreten. Bei
einer Asthma-Diagnose im Kindesalter können sich Krankheitserscheinungen auch wieder zurückbil-
den (Upham & James, 2011). In Abhängigkeit von den auslösenden Ursachen kann die Erkrankung in
eine allergische (extrinsische) und nicht-allergische (intrinsische) Form eingeteilt werden, wobei auch
Mischformen bestehen können (Buhl et al., 2006). Während das exogen-allergische Bronchialasthma
vor allem auf einer IgE-vermittelten Überempfindlichkeitsreaktion beruht, sind beim intrinsischen
Bronchialasthma keine spezifischen Allergene bekannt, die den akuten Anfall auslösen. Unspezifische
Reize wie beispielsweise Inhalation von Tabakrauch oder Kaltluft können ausreichend sein, um zu
einer überschießenden Reaktion des Bronchialsystems zu führen (NVL, 2009). Genetische Faktoren
61

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
zur Entstehung und Entwicklung von Asthma sollten ebenfalls berücksichtigt werden (Bergmann et
al., 2002). Die weltweite Zunahme der Asthma-Prävalenz insbesondere in den Industrieländern un-
terstreicht jedoch die große Bedeutung exogener Faktoren. Polosa et al. (2008) konnten in ihrer
Studie den Konsum von Tabak als unabhängigen Risikofaktor für eine Asthma-Genese identifizieren.
Auch besteht zwischen einer vorbestehenden allergischen Rhinitis und der Entwicklung von Asthma
möglicherweise ein Kausalzusammenhang (Rzehak et al., 2008; Shaaban et al., 2008). Welche Rolle
die Allergenbelastung jedoch tatsächlich für die Inzidenz des Asthma bronchiale spielt, ist nach wie
vor Gegenstand der wissenschaftlichen Diskussion (Pearce et al, 2000; Lau et al., 2000; Torrent et al.,
2006; Salo et al., 2008).
Nach Angaben der WHO sind derzeit 235 Millionen Menschen weltweit von der Erkrankung Asthma
bronchiale betroffen (WHO, 2013). In der Literatur beläuft sich die Prävalenz in Deutschland auf ca.
fünf Prozent bei Erwachsenen und zehn Prozent bei Kindern (Konietzko & Fabel, 2000, Fabel & Ko-
nietzko, 2005; Buhl et al., 2006) und zählt damit zu den häufigsten chronischen Erkrankungen im
Kindesalter (Bacharier et al., 2008). Allerdings nehmen die Asthma-Prävalenz und besonders die
Prävalenz des allergischen Asthmas mit zunehmendem Alter ab. Ergebnisse früherer Studien weisen
auf einen signifikanten regionalen und genderspezifischen Prävalenz-Unterschied in Deutschland hin
(Hermann-Kunz, 1999; Hermann-Kunz, 2000). Wie bei der Erhebung der COPD-Prävalenz gilt auch für
Asthma, dass die Feststellung je nach angewandter Methodik der Datenerhebung variieren kann. So
erfolgen seit dem Bundes-Gesundheitssurvey 1998 regelmäßig Erhebungen zum ärztlich-
diagnostizierten Asthma bronchiale. Durch die Querschnittsanalyse „Gesundheit in Deutschland
aktuell“ konnten erneut repräsentative Daten zur Asthma-Prävalenz in der erwachsenen Bevölke-
rung in Deutschland erhoben werden (RKI, 2014d). In Analogie zur Datenerhebung der COPD-
Prävalenz wurde eine telefonische Befragung durchgeführt, in der die Personen sowohl nach dem
Vorliegen einer Asthma-Diagnose in den vergangenen zwölf Monaten als auch nach einer irgend-
wann in ihrem Leben gestellten Asthma-Diagnose (Lebenszeitprävalenz) befragt wurden. Die Abbil-
dungen 8 und 9 fassen die Ergebnisse der Untersuchung zusammen und bestätigen damit die Resul-
tate anderer Studien (Langen et al., 2013; Stock et al., 2005; Schäfer et al., 2005; Demoly et al.,
2009). So sind Frauen eher von Asthma bronchiale betroffen als Männer (Lebenszeitprävalenz: 11,5
vs. 8,3 Prozent). Dabei liegt der Höchststand der Lebenszeitprävalenz bei Frauen ab dem 65. Lebens-
jahr, bei Männern hingegen in der Altersgruppe der 18- bis 29-Jährigen. Differenziert man die Ergeb-
nisse nach dem Bildungshintergrund so zeigt sich, dass eine höhere Bildung mit einer geringeren
Asthma-Lebenszeitprävalenz einhergeht. Allerdings zeigt sich dieser Zusammenhang nur bei den
Frauen ab dem dritten Lebensjahrzehnt.
62
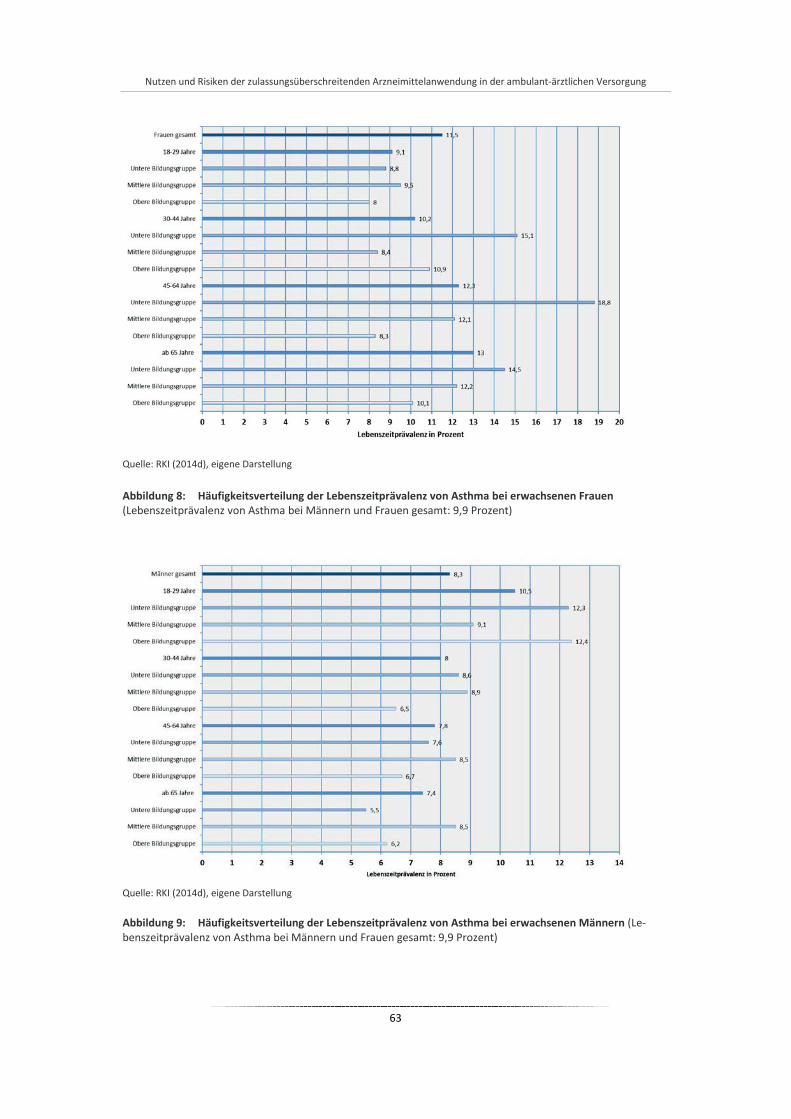
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Quelle: RKI (2014d), eigene Darstellung
Abbildung 8: Häufigkeitsverteilung der Lebenszeitprävalenz von Asthma bei erwachsenen Frauen (Lebenszeitprävalenz von Asthma bei Männern und Frauen gesamt: 9,9 Prozent)
Quelle: RKI (2014d), eigene Darstellung
Abbildung 9: Häufigkeitsverteilung der Lebenszeitprävalenz von Asthma bei erwachsenen Männern (Le-benszeitprävalenz von Asthma bei Männern und Frauen gesamt: 9,9 Prozent)
63

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Vergleicht man die Daten aus den letzten zehn bis fünfzehn Jahren, so ist die Asthma-Mortalität in
Deutschland um etwa ein Drittel zurückgegangen (Fabel & Konietzko, 2005). Allerdings sind Mortali-
tätsdaten zum Asthma häufig nur von begrenzter Aussagekraft, da die Todesursachenstatistik mono-
kausal aufgebaut ist und viele Personen mit diagnostiziertem Asthma letztendlich nicht aufgrund
dieser Erkrankung sterben (Windt, 2010). Neben Neubildungen sowie Erkrankungen des Herzens und
des Stoffwechsels zählen Atemwegserkrankungen nach wie vor zu den großen Volkskrankheiten in
Deutschland (Kirsch et al., 2013). Durch die zunehmende Umweltbelastung, den chronischen Verlauf
der Erkrankung sowie die steigenden Ausgaben im Gesundheitswesen besitzen COPD und Asthma
eine große gesundheitsökonomische Bedeutung. Im Bereich der direkten Kosten sind Arzneimittel
sowie die stationäre Versorgung die größten Kostenverursacher, während die Arbeitsunfähigkeit
neben der Frühberentung den größten Anteil an indirekten Kosten hat (Kirsch et al., 2013; Aumamn
& Prenzler, 2014). Im Jahr 2008 verursachten allein Erkrankungen der Atemwege (ICD-10-Code J00
bis J99) Kosten von 13.189 Mio. Euro (IQWiG, 2013). Im Jahr 2010 wurden 231.764 Frauen und Män-
ner stationär wegen einer chronischen Erkrankung der unteren Atemwege (ICD-10-Code J40 bis J47)
behandelt. Patienten mit Asthma bronchiale machten gut zehn Prozent der Behandelten aus, Patien-
ten mit COPD waren nicht explizit ausgewiesen (IQWiG, 2013). Für Asthma konnten die indirekten
Kosten mittlerweile gesenkt werden, wobei dies möglicherweise in einer konsequenteren ambulan-
ten Therapie begründet liegt (Windt, 2010).
64

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.1.1.1 Leitlinien und Therapieempfehlungen
Um den medikamentösen Standard in der Therapie des Asthma bronchiale und der COPD zu ermit-
teln, werden die wichtigsten Behandlungsempfehlungen vorgestellt. Im Bereich der Therapieemp-
fehlungen sind im Wesentlichen die interdisziplinär angelegten Nationalen Versorgungsleitlinien
Asthma (NVL, 2009) und COPD (NVL, 2006) zu nennen, die sich derzeit beide in Überarbeitung befin-
den. Um mögliche Änderungen von Therapieempfehlungen in Deutschland zu erfassen, wurden die
Recherche-Ergebnisse des IQWiG ebenso berücksichtigt (IQWiG, 2013a; IQWiG, 2013b). Beauftragt
durch den G-BA verfolgte das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen mit
den beiden Update-Recherchen das Ziel, aus aktuellen sowie evidenzbasierten Leitlinien Kernemp-
fehlungen zu synthetisieren, die für eine Überarbeitung der entsprechenden Disease-Management-
Programme16 (DMP) von Bedeutung sein könnten. Da nicht nur nationale, sondern auch internatio-
nale Leitlinien in die Recherche des IQWiG miteinflossen, wurden Therapiebesonderheiten anderer
Länder berücksichtigt und für die Verwendung in Deutschland adaptiert (OLU, Lifestyle-Arzneimittel).
Für den Versorgungsaspekt Medikamentöse Maßnahmen bei der Asthma-Therapie sah das IQWiG
keinen Aktualisierungs- bzw. Ergänzungsbedarf, da die Empfehlungen im Wesentlichen mit der DMP-
Richtlinie Asthma übereinstimmen (IQWiG, 2013a). Für die medikamentöse Therapie der COPD
stimmen die Empfehlungen ebenfalls im Wesentlichen mit der DMP-Richtlinie überein. Mögliche
Aktualisierungs- bzw. Ergänzungsbedarfe sieht das IQWiG vor allem in einer Ergänzung bzw. Spezifi-
zierung des DMP im Hinblick auf die medikamentöse Therapie mit Bronchodilatatoren, Glucocorti-
coidsteroiden und Phosphodiesterase-Hemmern (IQWiG, 2013a). Ziele der medikamentösen COPD-
Therapie sind u.a. neben der Verminderung der Progression der Erkrankung und der Steigerung der
körperlichen Belastbarkeit auch eine allgemeine Verbesserung des Gesundheitsstatus sowie die
Vorbeugung und Behandlung von Exazerbationen (NVL, 2006). Ziele der medikamentösen Asthma-
Therapie sind vor allem neben der Vermeidung akuter und chronischer Krankheitserscheinungen
auch die Normalisierung bzw. das Anstreben der bestmöglichen Lungenfunktion sowie eine Redukti-
on der bronchialen Hyperreagibilität (NVL, 2009).
Auf Grundlage der Behandlungsempfehlungen von IQWiG und der NVL werden im Folgenden die
wesentlichen Pharmakotherapien zur Behandlung der COPD und des Asthma bronchiale herausgear-
beitet. Mögliche Schnittstellen und Abweichungen sollen dabei identifiziert, für eine schnellere Ori-
entierung tabellarisch festgehalten und im Anschluss diskutiert werden. Tabelle 6 gibt einen Über-
blick der vergleichbaren Bedarfs- und Dauertherapie sowohl zur Behandlung der COPD als auch des
16 Bei den Disease-Management-Programmen handelt es sich um strukturierte Behandlungsprogramme für chronisch Kranke, die auf den Erkenntnissen der evidenzbasierten Medizin beruhen. Vorrangig eingesetzte Behandlungsmethoden entsprechen dem Stand der Wissenschaft.
65
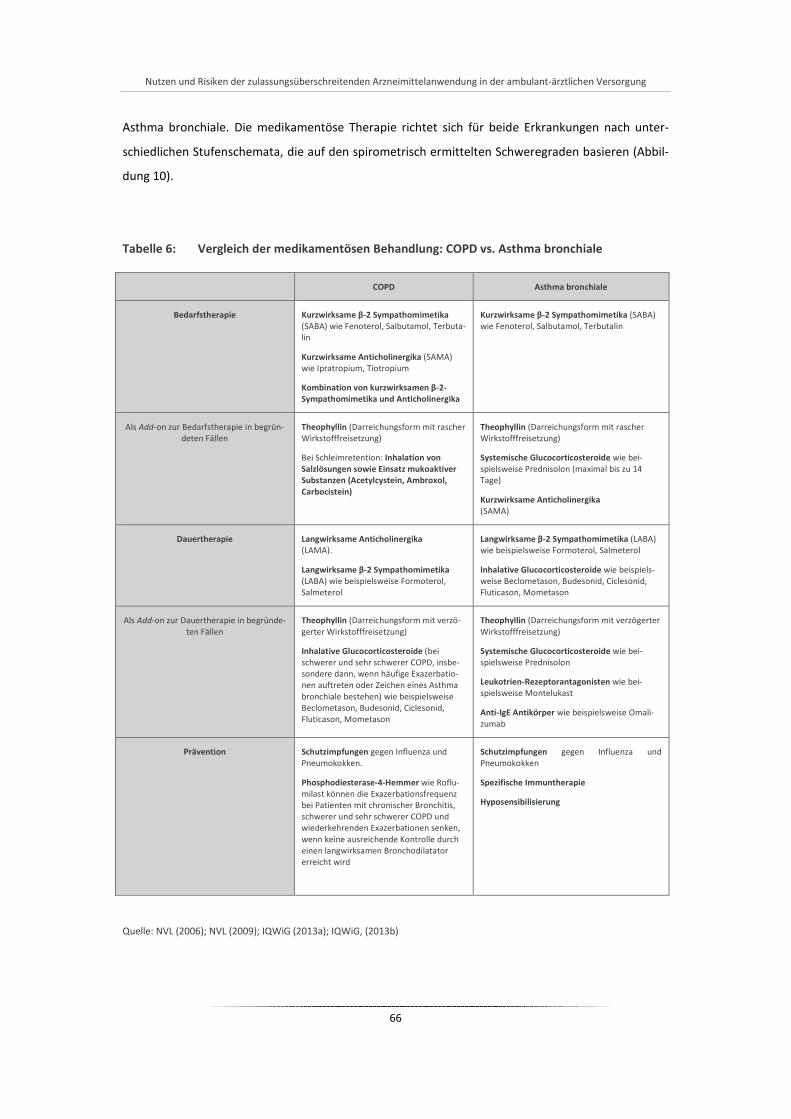
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Asthma bronchiale. Die medikamentöse Therapie richtet sich für beide Erkrankungen nach unter-
schiedlichen Stufenschemata, die auf den spirometrisch ermittelten Schweregraden basieren (Abbil-
dung 10).
Tabelle 6: Vergleich der medikamentösen Behandlung: COPD vs. Asthma bronchiale
COPD Asthma bronchiale
Bedarfstherapie Kurzwirksam -2 Sympathomimetika (SABA) wie Fenoterol, Salbutamol, Terbuta-lin
Kurzwirksame Anticholinergika (SAMA) wie Ipratropium, Tiotropium
Kombination von kurzwirksamen -2-Sympathomimetika und Anticholinergika
Kurzwirksam -2 Sympathomimetika (SABA) wie Fenoterol, Salbutamol, Terbutalin
Als Add-on zur Bedarfstherapie in begrün-deten Fällen
Theophyllin (Darreichungsform mit rascher Wirkstofffreisetzung)
Bei Schleimretention: Inhalation von Salzlösungen sowie Einsatz mukoaktiver Substanzen (Acetylcystein, Ambroxol, Carbocistein)
Theophyllin (Darreichungsform mit rascher Wirkstofffreisetzung)
Systemische Glucocorticosteroide wie bei-spielsweise Prednisolon (maximal bis zu 14 Tage)
Kurzwirksame Anticholinergika (SAMA)
Dauertherapie Langwirksame Anticholinergika (LAMA).
Langwirksame -2 Sympathomimetika (LABA) wie beispielsweise Formoterol, Salmeterol
Langwirksame -2 Sympathomimetika (LABA) wie beispielsweise Formoterol, Salmeterol
Inhalative Glucocorticosteroide wie beispiels-weise Beclometason, Budesonid, Ciclesonid, Fluticason, Mometason
Als Add-on zur Dauertherapie in begründe-ten Fällen
Theophyllin (Darreichungsform mit verzö-gerter Wirkstofffreisetzung)
Inhalative Glucocorticosteroide (bei schwerer und sehr schwerer COPD, insbe-sondere dann, wenn häufige Exazerbatio-nen auftreten oder Zeichen eines Asthma bronchiale bestehen) wie beispielsweise Beclometason, Budesonid, Ciclesonid, Fluticason, Mometason
Theophyllin (Darreichungsform mit verzögerter Wirkstofffreisetzung)
Systemische Glucocorticosteroide wie bei-spielsweise Prednisolon
Leukotrien-Rezeptorantagonisten wie bei-spielsweise Montelukast
Anti-IgE Antikörper wie beispielsweise Omali-zumab
Prävention Schutzimpfungen gegen Influenza und Pneumokokken.
Phosphodiesterase-4-Hemmer wie Roflu-milast können die Exazerbationsfrequenz bei Patienten mit chronischer Bronchitis, schwerer und sehr schwerer COPD und wiederkehrenden Exazerbationen senken, wenn keine ausreichende Kontrolle durch einen langwirksamen Bronchodilatator erreicht wird
Schutzimpfungen gegen Influenza und Pneumokokken
Spezifische Immuntherapie
Hyposensibilisierung
Quelle: NVL (2006); NVL (2009); IQWiG (2013a); IQWiG, (2013b)
66

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
In der Asthma- -2-Sympathomimetika zusätzlich als Bedarfsmedikation ab Stufe II gegeben werden.
Quelle: NVL (2006); NVL (2009)
Abbildung 10: Stufenschemata der medikamentösen Therapie im Erwachsenenalter: Asthma bronchiale vs. COPD
Wie aus Tabelle 6 ersichtlich, stellen bronchialerweiternde Medikamente – in der Regel als Dosierae-
rosol angewendet – die Basismedikation sowohl für die COPD als auch für Asthma dar. Kurzwirksame
Bronchodilatatoren werden bei Bedarf, Arzneimittel mit langer Wirkdauer hingegen in der Dauerthe-
rapie eingesetzt. So werden in Abhängigkeit des Schweregrades (siehe Abbildung 10) zu den rasch
wirksamen -2-Sympathomimetika (beispielsweise Salbutamol, Terbutalin) bei Bedarf auch langwirk-
same -2-Sympathomimetika wie Formoterol und Salmeterol zur Dauertherapie genutzt. Sie dienen
neben der Erleichterung von bronchialobstruktiven Beschwerden bei Patienten mit COPD auch der
symptomatischen Langzeitbehandlung des chronisch mäßigen bis schweren Asthma bronchiale.
Gleichzeitig soll bei letztgenannter Indikation eine regelmäßige Therapie mit entzündungshemmen-
den Arzneimitteln (inhalative und bzw. oder orale Glucocorticoidsteroide) sichergestellt werden. Der
-2-Sympathomimetika beruht auf einer selektiven -
2-Rezeptoren an den Zellen der glatten Bronchialmuskulatur (Spring et al., 1992; Barnes, 2002).
Dadurch wird die bronchiale Muskelspannung reduziert und es kommt zu einer Erweiterung der
Atemwege. In den vergangenen Jahren sind -2-Sympathomimetika zur Asthma-
67

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Therapie immer wieder in die Diskussion geraten. So wird ihre Anwendung mit einem erhöhten Risi-
ko für schwerste Exazerbationen von Asthmasymptomen assoziiert, die zu Hospitalisationen von
Kindern und Erwachsenen mit zum Teil letalem Ausgang führten (Cates & Cates, 2008; Castle et al.,
1993; Kuehn, 2009). Aus diesem Grund sollte bei einem Asthmatiker eine Langzeitmonotherapie
dieser Wirkstoffgruppe – ohne gleichzeitige Anwendung eines antientzündlich wirkenden Antiasth-
matikums – vermieden werden. Andererseits ist unklar, ob nicht auch eine duale Kombinationsthe-
rapie, -2-Sympathomimetika und Glucocorticoidsteroiden, mit ähnlichen Risiken
einhergeht (Chowdhury & Dal Pan, 2010).
Während für die Dauertherapie des Asthma bronchiale die zusätzliche Gabe entzündungshemmen-
der Wirkstoffe angezeigt ist, kommen bei der COPD langwirksame Anticholinergika zum Einsatz. Ihre
Wirkung beruht dabei auf einer Blockade von Muscarin-Rezeptoren, die sich an der glatten Muskula-
tur der Atemwege befinden. Während die inhalativen Anticholinergika Tiotropiumbromid, Aclidini-
umbromid und Glycopyrroniumbromid in Deutschland ausschließlich zur Therapie der COPD zugelas-
-2-Agonisten auch zur Behandlung einer Asth-
ma-induzierten reversiblen Atemwegsobstruktion geeignet. Die Wahl zwischen -2-Sympa-
thomimetika und Anticholinergika hängt dabei vom individuellen Ansprechen des Patienten und den
unerwünschten Wirkungen ab (NVL, 2006; NVL, 2009). Gleiches gilt für die Auswahl der inhalativen
Glucocorticosteroide, die sich aufgrund ihrer Rezeptoraffinität in der Wirkstärke unterscheiden, nicht
aber in ihren pharmakodynamischen Eigenschaften. Ein weiterer wenn auch schwächerer Bronchodi-
latator ist Theophyllin, der zur Therapie chronischer Lungenerkrankungen nur noch in begründeten
Einzelfällen als Wirkstoff der dritten Wahl eingesetzt werden sollte. Neben zahlreichen Interaktionen
besitzt er eine relativ geringe, vom Alter abhängige (Scholz et al., 2005) therapeutische Breite. So
kann es bei zu hoher Dosierung zu Krampfanfällen kommen, weswegen die Einnahme des Wirkstof-
fes eine regelmäßige Kontrolle des Blutspiegels erfordert.
Der monoklonale Anti-IgE-Antikörper Omalizumab ist bei schwerem persistierendem allergischem
Asthma indiziert. Seine Wirkung beruht auf der Bindung von freiem IgE, welches dann für die Auslö-
sung einer IgE-vermittelten allergischen Reaktion nicht mehr zur Verfügung steht (Easthope & Jarvis,
2001). Der Leukotrien-Rezeptorantagonist Montelukast ist ebenfalls nur zur Asthma-Therapie ange-
zeigt. Entweder indiziert als Zusatzbehandlung für Patienten, bei denen ein leichter bis mittelgradi-
ger persistierender Asthma bronchiale diagnostiziert wurde und der nicht mit anderen Medikamen-
ten ausreichend behandelt werden kann, zur Vorbeugung eines Belastungsasthmas oder um bei
Asthmatikern eine Linderung der saisonalen allergischen Rhinitis zu bewirken.
COPD-Exazerbationen sind durch eine akute und anhaltende Verschlimmerung des Krankheitszu-
standes gekennzeichnet. Sie gehen über die für den Patienten normale Variation seiner Erkrankung
hinaus und erfordern eine Intensivierung der Therapie (NVL, 2006). Phosphodiesterase-4-Hemmer
68

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
wie Roflumilast sollten dann bei Patienten mit schwerer COPD und wiederkehrenden Exazerbationen
eingesetzt werden, wenn keine ausreichende Kontrolle durch einen langwirksamen Bronchodilatator
erreicht wird (NVL, 2006).
8.1.1.2 Diskussion der Ergebnisse
Asthma bronchiale und COPD zählen zu den chronischen Krankheiten der unteren Atemwege. Wie
aufgezeigt werden konnte, ähneln sich die beiden Erkrankungen trotz unterschiedlicher Pathologie.
Zu beachten gilt außerdem, dass neben Asthma bronchiale auch eine Reihe von kardiopulmonalen
Krankheiten wie beispielsweise Bronchiektasen, Bronchialkarzinom, Lungentuberkulose sowie die
Linksherzinsuffizienz ähnliche Symptome hervorrufen können. Begründet in ihrem chronischen Cha-
rakter und ihrer Mortalität schränken beide Erkrankungen nicht nur die Lebensqualität der betroffe-
nen Personen ein, sondern besitzen auch einen hohen gesundheitsökonomischen und volkswirt-
schaftlichen Stellenwert. Während einerseits die Asthma-Mortalität in den vergangenen Jahren stark
zurückgegangen ist, zählt die COPD nicht nur zu den häufigsten Erkrankungen weltweit, sondern
mittlerweile auch zu den führenden Todesursachen. Da das Erkrankungsrisiko der COPD nicht nur
mit dem Raucherstatus, sondern auch mit zunehmendem Alter steigt, muss in Zukunft aufgrund der
demographischen Entwicklung der Bevölkerung mit einer höheren Erkrankungshäufigkeit gerechnet
werden. Im Bereich der direkten Kosten zählen Arzneimittel sowie die stationäre Versorgung zu den
größten Kostenverursachern, während die Arbeitsunfähigkeit neben der Frühberentung den größten
Anteil an indirekten Kosten aufweist.
Wie die Analyse der Leitlinienempfehlungen für Asthma bronchiale und COPD gezeigt hat, unter-
scheiden sich die Therapiestrategien aufgrund unterschiedlicher Pathomechanismen teilweise er-
heblich voneinander. So erfordern gerade Krankheitsbilder mit ähnlicher Symptomatik eine exakte
Differenzierung in der Diagnosestellung.
Bis auf wenige identifizierte Wirkstoffgruppen (Leukotrien-Antagonisten, Anti-IgE-Antikörper, Phos-
phodiesterase-4-Hemmer, langwirksame Anticholinergika) sind alle anderen Arzneimittel bzw.
pharmakotherapeutischen Gruppen sowohl für die Asthma- als auch für die COPD-Therapie als Dau-
er- bzw. Bedarfsmedikation zugelassen. Bei den zugelassenen Wirkstoffen bzw. Wirkstoffgruppen
lassen sich aber Unterschiede im Stellenwert der Arzneimittel in der Therapie der entsprechenden
Erkrankung aufzeigen. Basismedikation sowohl für die COPD als auch für Asthma stellen bronchial-
erweiternde Medikamente dar. Kurzwirksame Bronchodilatatoren werden bei Bedarf, Arzneimittel
mit langer Wirkdauer hingegen in der Dauertherapie eingesetzt. In Abhängigkeit vom Schweregrad
der Erkrankung werden zu den rasch wirksamen -2-Sympathomimetika auch langwirksame -2-
69

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Sympathomimetika zur Dauertherapie genutzt. Gerade langwirksam -2-Sympathomimetika sind
jedoch in den vergangenen Jahren immer wieder in die Diskussion geraten, wenn es um die Therapie
des Asthma bronchiale ging (Cates & Cates, 2008; Castle et al., 1993; Kuehn, 2009). Aus diesem
Grund sollte bei einem Asthmatiker eine Langzeitmonotherapie dieser Wirkstoffgruppe ohne gleich-
zeitige Anwendung eines antientzündlich wirkenden Antiasthmatikums vermieden werden. Auch die
Wirkstoffgruppe der Anticholinergika sollte im Rahmen der Asthma-Bedarfstherapie lediglich als
Mittel zweiter Wahl eingesetzt werden, während sie bei der COPD Arzneimittel der ersten Wahl in
der Dauer- und Bedarfstherapie sind. Glucocorticoide unterdrücken chronische Entzündungen, die
beim Asthma bronchiale maßgeblich an der Entstehung und Chronifizierung der Atemwegsobstrukti-
on beteiligt sind. Aus diesem Grund bildet diese Wirkstoffgruppe die therapeutische Stütze in der
Dauertherapie des Asthmas. Neben der Tatsache, dass eine bedarfs- und therapiegerechte Basisver-
sorgung auch die Nezessität einer Add-on-Therapie begrenzt, sind vermeidbare Exazerbationen von
hoher Tragweite in der Therapie. So verursachen häufig vorkommende Exazerbationen auch in stabi-
len Krankheitsphasen bei COPD-Patienten eine gesteigerte Entzündungsaktivität in den Atemwegen
(Bhowmik et al., 2000). Zudem verschlechtern sie nicht nur den Allgemeinzustand, sondern be-
schleunigen auch die Erkrankungsprogression durch zunehmende Einschränkung der Lungenfunktion
und gleichzeitige Erhöhung des Mortalitätsrisikos (Wedzicha & Seemungal, 2007). Niedrig dosiertes
Glucocorticoid verbessert hingegen schon bei einem leichten Asthma der Therapiestufe II nicht nur
die Symptomatik und die Lungenfunktion, sondern reduziert auch die Exazerbations- und Mortali-
tätsraten (Adams et al., 2001; Adams et al., 2005). Da die genannten Wirkstoffe für beide Erkrankun-
gen zugelassen sind, kann schlussfolgernd nicht von einem OLU gesprochen werden, sondern von
einem Abweichen in einer leitliniengerechten Therapie. Leitlinien bzw. Therapieempfehlungen sollen
als Orientierungshilfen verstanden werden, daher kann der Arzt in begründeten Fällen auch von
ihnen abweichen.
Ein OLU entsteht erst dann, wenn Medikamente außerhalb ihrer Zulassung verordnet werden. Der
monoklonale Anti-IgE-Antikörper Omalizumab ist nur bei schwerem persistierendem allergischem
Asthma indiziert. Der Leukotrien-Rezeptorantagonist Montelukast ist ebenfalls nur zur Asthma-
Therapie angezeigt: Entweder indiziert als Zusatzbehandlung für Patienten, bei denen ein leichter bis
mittelgradiger persistierender Asthma bronchiale diagnostiziert wurde und durch andere Medika-
mente nicht ausreichend behandelt werden kann, zur Vorbeugung eines Belastungsasthmas oder um
bei Asthmatikern eine Linderung der saisonalen allergischen Rhinitis zu bewirken. COPD-
Exazerbationen sind durch eine akute und anhaltende Verschlimmerung des Krankheitszustandes
gekennzeichnet und erfordern eine Intensivierung der Therapie. In diesem Fall sollten Phosphodies-
terase-4-Hemmer wie Roflumilast bei Patienten mit schwerer COPD und wiederkehrenden Exazerba-
70

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
tionen eingesetzt werden, wenn keine ausreichende Kontrolle durch einen langwirksamen Broncho-
dilatator erreicht wird.
Betrachtet man das Stufenschema der medikamentösen Therapie im Erwachsenenalter für Asthma
bronchiale und COPD (Abbildung 10) der Nationalen Versorgungsleitlinien, so kann ein OLU schon in
der zweiten Therapiestufe zum Tragen kommen. Je schwerwiegender die Erkrankung, desto weitrei-
chender sind die Konsequenzen für den betroffenen Patienten. Vor allem dann, wenn die Fehldia-
gnose nicht erkannt wird, der erwartete Therapierfolg ausbleibt, sich die Erkrankung weiter ver-
schlimmert und letztendlich eine Kaskade von Fehlentscheidungen nach sich zieht, die unmittelbare
Konsequenzen auf Lebenszeit und Lebensqualität des Patienten haben können (siehe Abbildung 11).
Quelle: eigene Darstellung
Abbildung 11: Auswirkungen eines diagnoseinduzierten OLU auf die Therapieziele einer Behandlung
Zusammenfassend lässt sich sagen, dass ein durch Fehldiagnosen induzierter OLU nicht grundsätzlich
auf jeder Therapiestufe zu finden ist und auch keine unmittelbare Auswirkung auf die Morbidität wie
Mortalität des Patienten hat. Festzuhalten ist allerdings, dass ein Diagnose-Bias eine leitliniengerech-
te Therapie ausschließen kann, was im weiteren Verlauf zu nicht zulassungskonformen Pharmako-
71

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
therapien führt. Der Einsatz eines Arzneimittels – ob im ILU oder OLU – hat zum Ziel, Krankheiten zu
heilen, ihre Symptome zu lindern, das Fortschreiten der Erkrankung zu stoppen oder zumindest zu
verlangsamen und die Lebensqualität zu steigern. Im Idealfall werden mehrere Therapieziele er-
reicht. Ein durch Fehldiagnosen induzierter OLU (zulassungskonforme Anwendung eines Arzneimit-
tels bei falsch-positiver Diagnose) kann andererseits mit Unter-, Über- und Fehlversorgungen und
den daraus resultierenden Folgen für den Patienten einhergehen, wie Verschlechterung der Erkran-
kung und Einbußen in der Lebensqualität. Da die Therapieentscheidungen nicht bedarfsgemäß ge-
troffen werden, stellen sie außerdem eine Grenzverletzung der Wirtschaftlichkeit dar (Glaeske,
2007).
Bereits vor 15 Jahren thematisierte der Sachverständigenrat für die Konzentrierte Aktion im Gesund-
heitswesen (SVR) in seinem Gutachten Unter-, Über- und Fehlversorgungen in der Pharmakotherapie
von Asthma- und COPD-Patienten (SVR, 2001). Nach Auffassung des SVR erfolgten Diagnostik und
Therapie häufig zu spät und bzw. oder nicht leitliniengerecht. Ursache für das oben beschriebene
Nebeneinander sah der Sachverständigenrat in erster Linie in einer mangelhaften Diagnostik auf-
grund von Qualifikationsdefiziten in der Weiter- und Fortbildung bei den behandelnden Ärzten, und
dort vor allem bei Nicht-Pneumologen. Rabe et al. (2000) zeigten in ihrer Arbeit, dass im Rahmen der
Untersuchung bei 50 Prozent der Befragten die Lungenfunktion nicht geprüft worden war. Eine wei-
tere Analyse zeigte, dass bei nur gut einem Drittel der diagnostizierten Asthma-Patienten und knapp
60 Prozent der Patienten mit diagnostizierter COPD eine spirometrische Untersuchung durchgeführt
wurde (Schneider et al., 2005). Spirometrische Lungenfunktionsmessungen sind jedoch das unent-
behrliche Verfahren für die (Erst-)Diagnose (NVL, 2006; NVL, 2009), nach ihnen richtet sich der diag-
nostische Algorithmus der Leitlinien und Therapieempfehlungen. In einer weiteren Untersuchung
musste bei 18 Prozent der untersuchten Patienten eine falsch-positive Asthmadiagnose festgestellt
werden (Hellmann, 2007). Ähnliche Hinweise lieferte auch eine schwedische Studie: tatsächlich Er-
krankte wurden nicht als Asthmatiker identifiziert (Montnémery et al., 2002).
Lungenfachärzte stehen jedoch nicht überall in ausreichender Zahl zur Verfügung, so dass hier auch
strukturelle Defizite für eine mangelhafte Versorgung verantwortlich gemacht werden können. Da
die Pneumologie an Universitäten unterrepräsentiert ist und sich daraus zwangsläufig Defizite in
Forschung, Lehre und Versorgung ableiten lassen (Fabel & Konietzko, 2005), werden weniger Diag-
nosen durch Fachärzte gestellt und potentiell mehr Raum für mangelhafte Diagnosen geboten.
Wird von einem OLU gesprochen, geht man in der Regel von einer richtigen Diagnose aus, die auf-
grund fehlender Therapieoptionen eine nicht zulassungskonforme Anwendung eines Arzneimittels
nach sich zieht (therapieinduzierter OLU). Wenn der Rat des MDK eingeholt wird, ist die Verordnung
eines Arzneimittels außerhalb seiner Zulassungsbestimmung der häufigste Anlass (Grell & Rieger,
2012). Untersuchungen zeigen jedoch, dass Fehldiagnosen in der ärztlichen Praxis nicht selten sind
72
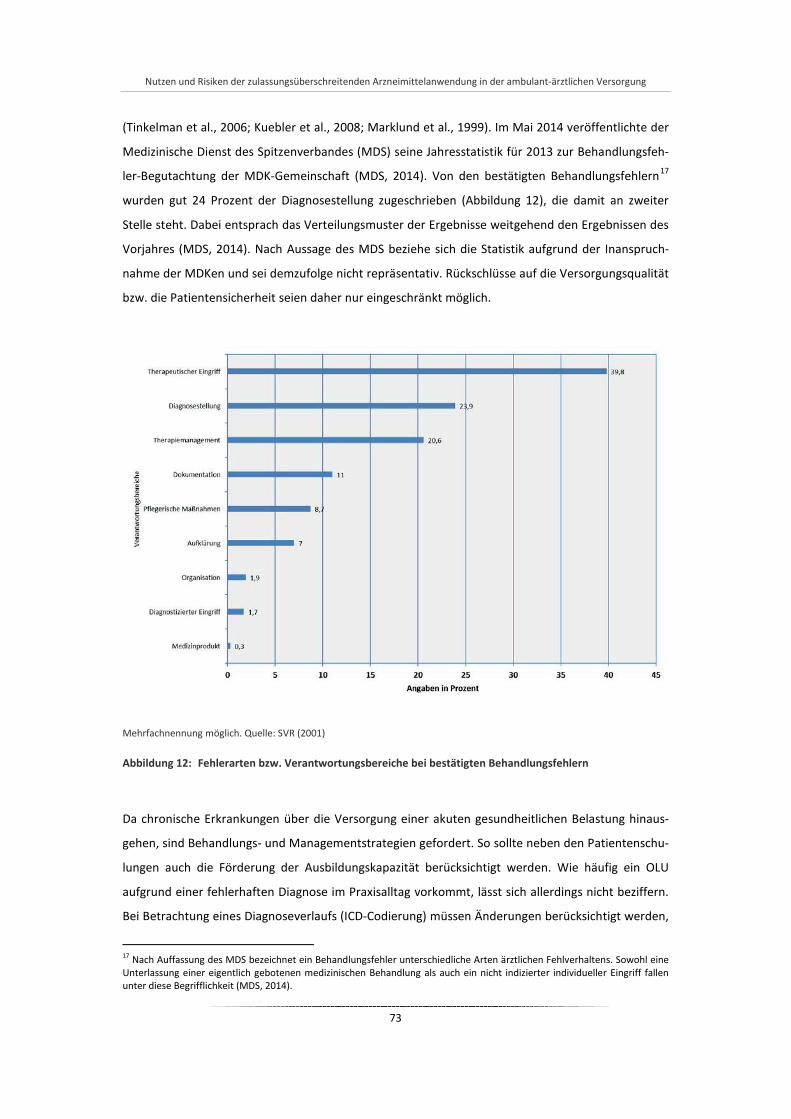
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
(Tinkelman et al., 2006; Kuebler et al., 2008; Marklund et al., 1999). Im Mai 2014 veröffentlichte der
Medizinische Dienst des Spitzenverbandes (MDS) seine Jahresstatistik für 2013 zur Behandlungsfeh-
ler-Begutachtung der MDK-Gemeinschaft (MDS, 2014). Von den bestätigten Behandlungsfehlern17
wurden gut 24 Prozent der Diagnosestellung zugeschrieben (Abbildung 12), die damit an zweiter
Stelle steht. Dabei entsprach das Verteilungsmuster der Ergebnisse weitgehend den Ergebnissen des
Vorjahres (MDS, 2014). Nach Aussage des MDS beziehe sich die Statistik aufgrund der Inanspruch-
nahme der MDKen und sei demzufolge nicht repräsentativ. Rückschlüsse auf die Versorgungsqualität
bzw. die Patientensicherheit seien daher nur eingeschränkt möglich.
Mehrfachnennung möglich. Quelle: SVR (2001)
Abbildung 12: Fehlerarten bzw. Verantwortungsbereiche bei bestätigten Behandlungsfehlern
Da chronische Erkrankungen über die Versorgung einer akuten gesundheitlichen Belastung hinaus-
gehen, sind Behandlungs- und Managementstrategien gefordert. So sollte neben den Patientenschu-
lungen auch die Förderung der Ausbildungskapazität berücksichtigt werden. Wie häufig ein OLU
aufgrund einer fehlerhaften Diagnose im Praxisalltag vorkommt, lässt sich allerdings nicht beziffern.
Bei Betrachtung eines Diagnoseverlaufs (ICD-Codierung) müssen Änderungen berücksichtigt werden,
17 Nach Auffassung des MDS bezeichnet ein Behandlungsfehler unterschiedliche Arten ärztlichen Fehlverhaltens. Sowohl eine Unterlassung einer eigentlich gebotenen medizinischen Behandlung als auch ein nicht indizierter individueller Eingriff fallen unter diese Begrifflichkeit (MDS, 2014).
73

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
die dann letztendlich auch in eine andere pharmakologische Therapie (ATC-Codierung) münden.
Mängel in der Codierung müssten zu 100 Prozent ausgeschlossen werden. Dazu bedarf es gezielter
Studien, um über das Auftreten von Behandlungsfehlern sowie der Vermeidbarkeit ihrer daraus
folgenden Ereignisse Erkenntnisse zu erlangen.
74

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.1.2 Neue Mittel bei obstruktiven Atemwegserkrankungen
Der überwiegende Teil der zur Therapie der obstruktiven Atemwegserkrankungen eingesetzten
Wirkstoffe findet seine therapeutische Verwendung sowohl in der Asthma- als auch in der COPD-
Behandlung. Die bei der COPD zu beobachtende Entzündung weist jedoch Unterschiede von derjeni-
gen bei Asthma auf und liefert möglicherweise eine Erklärung für das unterschiedliche Ansprechen
beider Erkrankungen auf die medikamentöse Therapie (Barnes, 2000; Barnes, 2004).
Im Fokus des dritten Abschnittes steht die Versorgungsanalyse der neuen Mittel zur Therapie der
COPD (Indacaterol, Roflumilast und Aclidiniumbromid). Zu Beginn werden kurz die Ergebnisse der
Literaturrecherche umrissen, auf die dann in den entsprechenden Wirkstoff-Abschnitten detaillierter
eingegangen werden soll. Zusätzlich erfolgte eine Recherche nach möglicherweise noch nicht veröf-
fentlichten Studien in der Datenbank des U.S. National Institutes of Health (ClinicalTrials.gov), deren
Ergebnisse ebenfalls tabellarisch dargestellt werden.
Nach anfänglicher Wirkstoffcharakterisierung sowie Darstellung der Evidenzlage im In-Label Use und
– für das ärztliche Verordnungsverhalten möglicherweise mitunter entscheidend – der zusammen-
fassenden Nutzenbewertung durch den G-BA, sollen die Ergebnisse der Studien kurz umrissen wer-
den, bei denen diese Wirkstoffe auch bei Patienten mit diagnostiziertem Asthma eingesetzt wurden.
Ein mögliches Forschungsinteresse auch im OLU soll so aufgezeigt werden. Werden solche Daten
publiziert? Und hängt die Publikation dieser Daten von der Art der Finanzierung ab? Dadurch sollen
Gründe identifiziert werden, warum ein pharmazeutischer Unternehmer die eine Indikation zulässt
und die andere ausschließt.
Im Anschluss daran wird das Verordnungsverhalten in der ambulant-ärztlichen Praxis mit Hilfe von
Routinedaten dargestellt. Für die Versorgungsanalyse von Indacaterol und Roflumilast wurden am-
bulant-ärztliche Abrechnungsdaten von Versicherten der Techniker Krankenkasse (TK) aus den Jah-
ren 2010 und 2011 genutzt, die zu dem Zeitpunkt mit ca. 7,4 Mio. (2010) bzw. 7,8 Mio. (2011) Versi-
cherten die zweitgrößte gesetzliche Krankenversicherung Deutschland darstellte. Für die Versor-
gungsanalyse von Aclidiniumbromid standen ambulant-ärztliche Abrechnungsdaten von Versicher-
ten der TK aus den Jahren 2012 und 2013 zur Verfügung. Die TK ist mit 8,1 Mio. (2012) bzw. 8,5 Mio.
(2013) Versicherten mittlerweile die größte GKV Deutschlands. Abschließend werden die Ergebnisse
dieser Untersuchungen diskutiert.
75
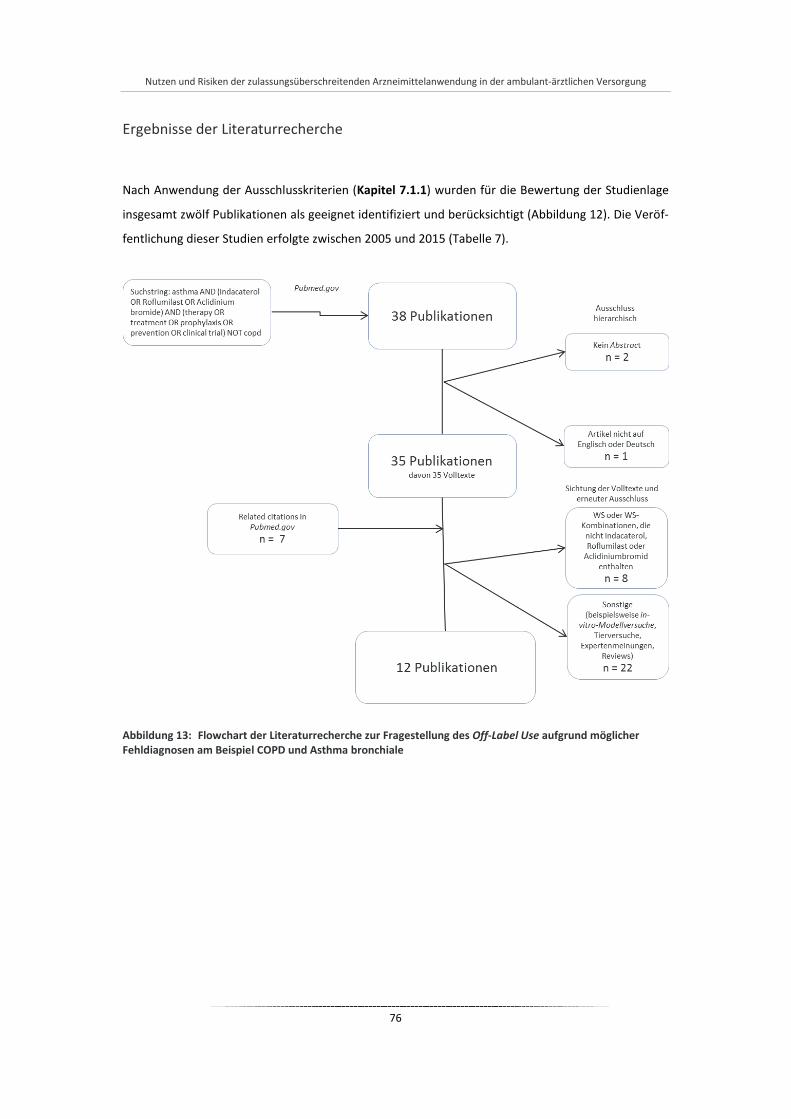
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Ergebnisse der Literaturrecherche
Nach Anwendung der Ausschlusskriterien (Kapitel 7.1.1) wurden für die Bewertung der Studienlage
insgesamt zwölf Publikationen als geeignet identifiziert und berücksichtigt (Abbildung 12). Die Veröf-
fentlichung dieser Studien erfolgte zwischen 2005 und 2015 (Tabelle 7).
Abbildung 13: Flowchart der Literaturrecherche zur Fragestellung des Off-Label Use aufgrund möglicher Fehldiagnosen am Beispiel COPD und Asthma bronchiale
76
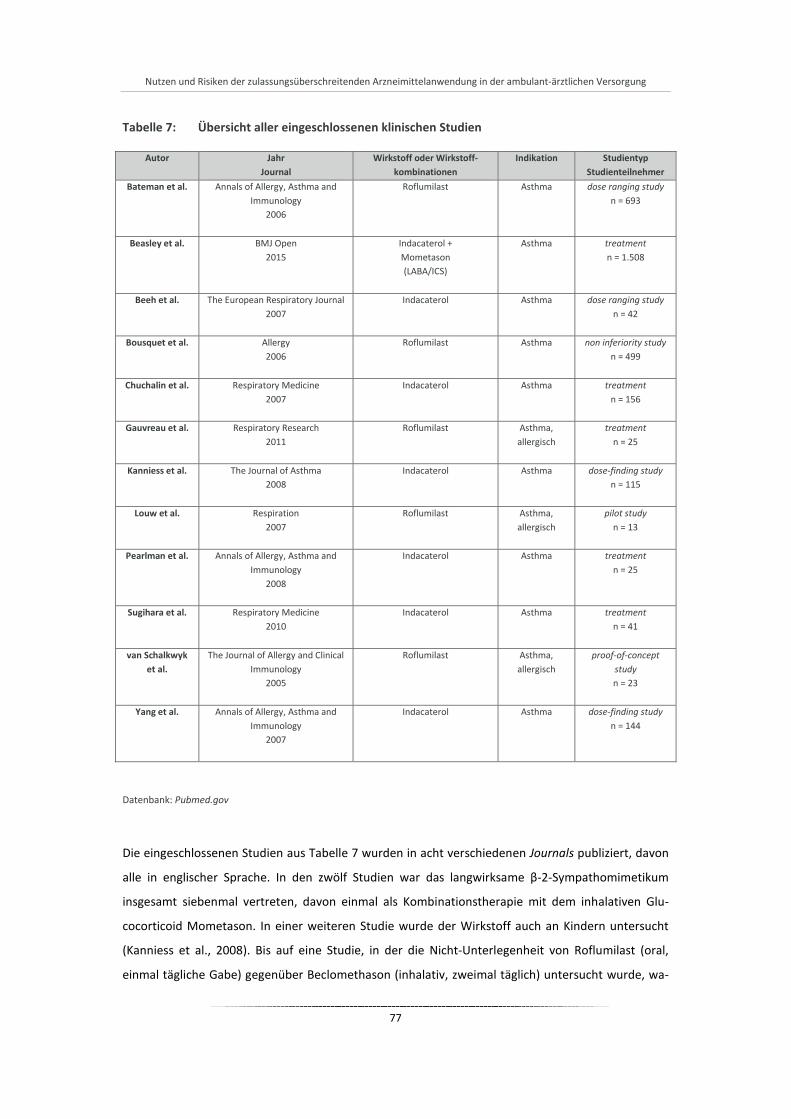
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 7: Übersicht aller eingeschlossenen klinischen Studien
Autor Jahr Journal
Wirkstoff oder Wirkstoff-kombinationen
Indikation Studientyp Studienteilnehmer
Bateman et al. Annals of Allergy, Asthma and Immunology
2006
Roflumilast Asthma dose ranging study n = 693
Beasley et al. BMJ Open 2015
Indacaterol + Mometason (LABA/ICS)
Asthma treatment n = 1.508
Beeh et al. The European Respiratory Journal 2007
Indacaterol Asthma dose ranging study n = 42
Bousquet et al. Allergy 2006
Roflumilast Asthma non inferiority study n = 499
Chuchalin et al. Respiratory Medicine 2007
Indacaterol Asthma treatment n = 156
Gauvreau et al. Respiratory Research 2011
Roflumilast Asthma, allergisch
treatment n = 25
Kanniess et al. The Journal of Asthma 2008
Indacaterol Asthma
dose-finding study n = 115
Louw et al. Respiration 2007
Roflumilast Asthma, allergisch
pilot study n = 13
Pearlman et al. Annals of Allergy, Asthma and Immunology
2008
Indacaterol Asthma treatment n = 25
Sugihara et al. Respiratory Medicine 2010
Indacaterol Asthma treatment n = 41
van Schalkwyk et al.
The Journal of Allergy and Clinical Immunology
2005
Roflumilast Asthma, allergisch
proof-of-concept study n = 23
Yang et al. Annals of Allergy, Asthma and Immunology
2007
Indacaterol Asthma dose-finding study n = 144
Datenbank: Pubmed.gov
Die eingeschlossenen Studien aus Tabelle 7 wurden in acht verschiedenen Journals publiziert, davon
alle in englischer Sprache. In den zwölf Studien war -2-Sympathomimetikum
insgesamt siebenmal vertreten, davon einmal als Kombinationstherapie mit dem inhalativen Glu-
cocorticoid Mometason. In einer weiteren Studie wurde der Wirkstoff auch an Kindern untersucht
(Kanniess et al., 2008). Bis auf eine Studie, in der die Nicht-Unterlegenheit von Roflumilast (oral,
einmal tägliche Gabe) gegenüber Beclomethason (inhalativ, zweimal täglich) untersucht wurde, wa-
77
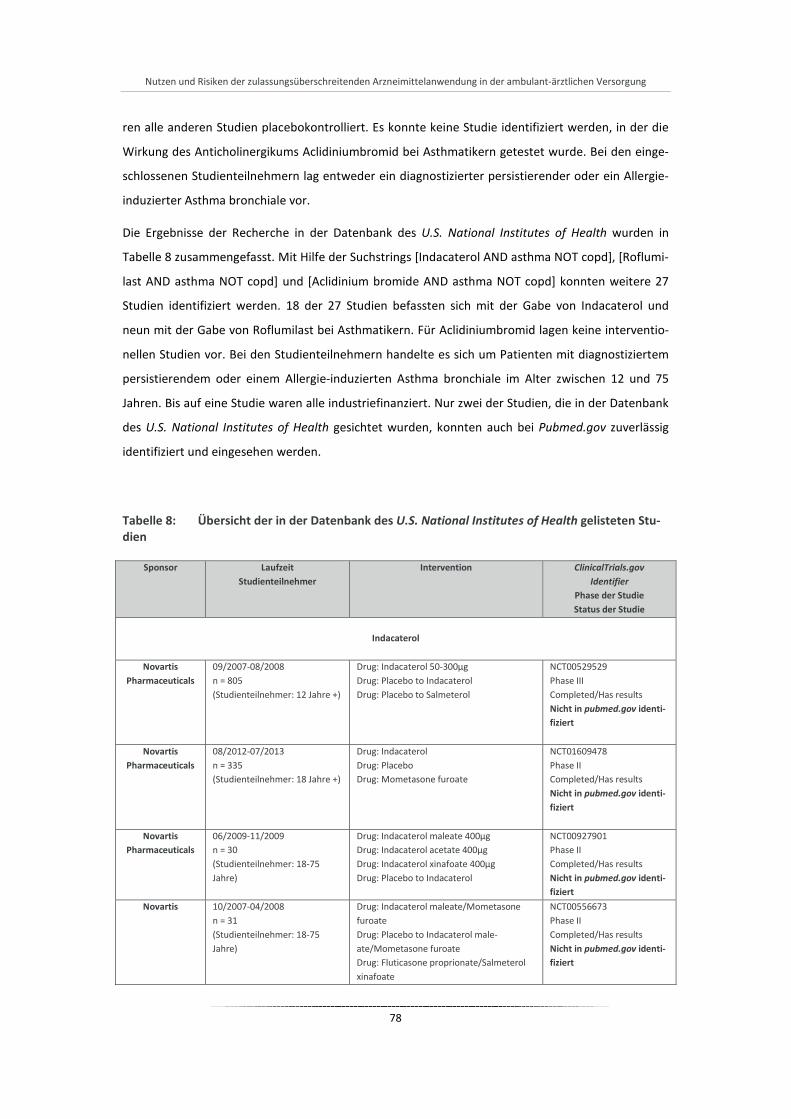
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
ren alle anderen Studien placebokontrolliert. Es konnte keine Studie identifiziert werden, in der die
Wirkung des Anticholinergikums Aclidiniumbromid bei Asthmatikern getestet wurde. Bei den einge-
schlossenen Studienteilnehmern lag entweder ein diagnostizierter persistierender oder ein Allergie-
induzierter Asthma bronchiale vor.
Die Ergebnisse der Recherche in der Datenbank des U.S. National Institutes of Health wurden in
Tabelle 8 zusammengefasst. Mit Hilfe der Suchstrings [Indacaterol AND asthma NOT copd], [Roflumi-
last AND asthma NOT copd] und [Aclidinium bromide AND asthma NOT copd] konnten weitere 27
Studien identifiziert werden. 18 der 27 Studien befassten sich mit der Gabe von Indacaterol und
neun mit der Gabe von Roflumilast bei Asthmatikern. Für Aclidiniumbromid lagen keine interventio-
nellen Studien vor. Bei den Studienteilnehmern handelte es sich um Patienten mit diagnostiziertem
persistierendem oder einem Allergie-induzierten Asthma bronchiale im Alter zwischen 12 und 75
Jahren. Bis auf eine Studie waren alle industriefinanziert. Nur zwei der Studien, die in der Datenbank
des U.S. National Institutes of Health gesichtet wurden, konnten auch bei Pubmed.gov zuverlässig
identifiziert und eingesehen werden.
Tabelle 8: Übersicht der in der Datenbank des U.S. National Institutes of Health gelisteten Stu-dien
Sponsor Laufzeit Studienteilnehmer
Intervention
ClinicalTrials.gov Identifier
Phase der Studie Status der Studie
Indacaterol
Novartis
Pharmaceuticals 09/2007-08/2008 n = 805 (Studienteilnehmer: 12 Jahre +)
Drug: Indacaterol 50-300 Drug: Placebo to Indacaterol Drug: Placebo to Salmeterol
NCT00529529 Phase III Completed/Has results Nicht in pubmed.gov identi-fiziert
Novartis Pharmaceuticals
08/2012-07/2013 n = 335 (Studienteilnehmer: 18 Jahre +)
Drug: Indacaterol Drug: Placebo Drug: Mometasone furoate
NCT01609478 Phase II Completed/Has results Nicht in pubmed.gov identi-fiziert
Novartis Pharmaceuticals
06/2009-11/2009 n = 30 (Studienteilnehmer: 18-75 Jahre)
Drug: Indacaterol maleate 400 g Drug: Indacaterol acetate 400 Drug: Indacaterol xinafoate 400 Drug: Placebo to Indacaterol
NCT00927901 Phase II Completed/Has results Nicht in pubmed.gov identi-fiziert
Novartis 10/2007-04/2008 n = 31 (Studienteilnehmer: 18-75 Jahre)
Drug: Indacaterol maleate/Mometasone furoate Drug: Placebo to Indacaterol male-ate/Mometasone furoate Drug: Fluticasone proprionate/Salmeterol xinafoate
NCT00556673 Phase II Completed/Has results Nicht in pubmed.gov identi-fiziert
78
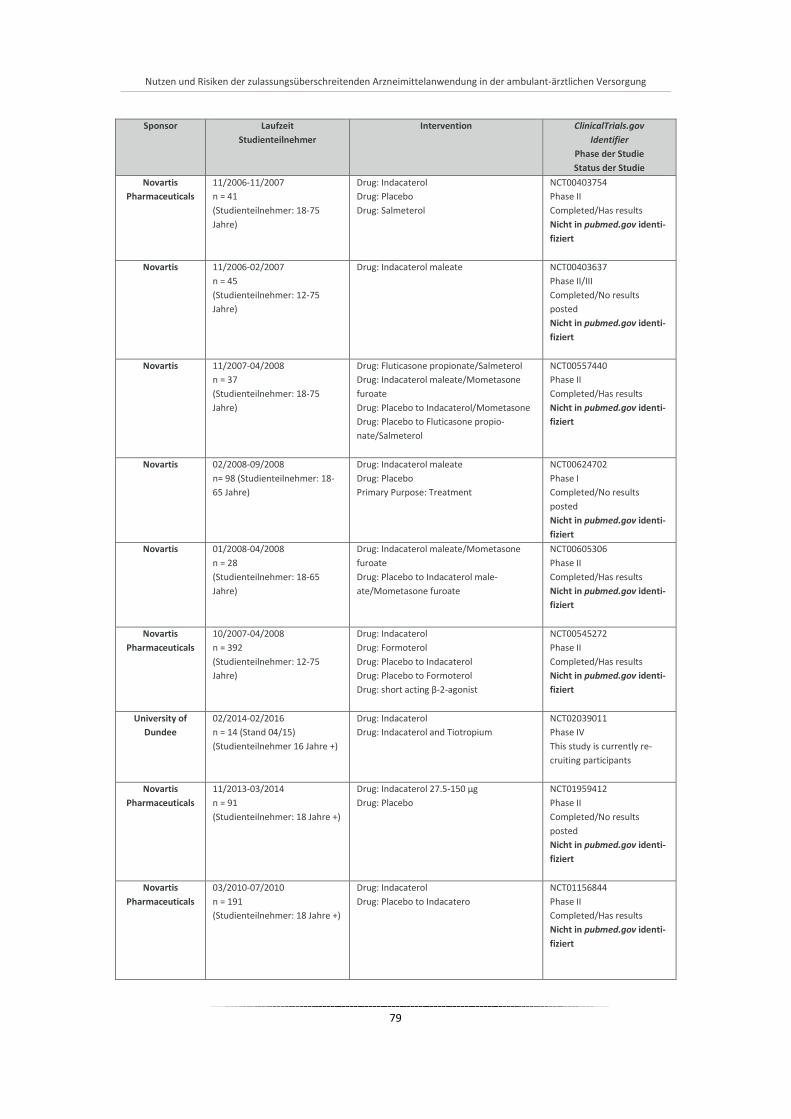
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Sponsor Laufzeit Studienteilnehmer
Intervention
ClinicalTrials.gov Identifier
Phase der Studie Status der Studie
Novartis Pharmaceuticals
11/2006-11/2007 n = 41 (Studienteilnehmer: 18-75 Jahre)
Drug: Indacaterol Drug: Placebo Drug: Salmeterol
NCT00403754 Phase II Completed/Has results Nicht in pubmed.gov identi-fiziert
Novartis 11/2006-02/2007 n = 45 (Studienteilnehmer: 12-75 Jahre)
Drug: Indacaterol maleate
NCT00403637 Phase II/III Completed/No results posted Nicht in pubmed.gov identi-fiziert
Novartis 11/2007-04/2008 n = 37 (Studienteilnehmer: 18-75 Jahre)
Drug: Fluticasone propionate/Salmeterol Drug: Indacaterol maleate/Mometasone furoate Drug: Placebo to Indacaterol/Mometasone Drug: Placebo to Fluticasone propio-nate/Salmeterol
NCT00557440 Phase II Completed/Has results Nicht in pubmed.gov identi-fiziert
Novartis 02/2008-09/2008 n= 98 (Studienteilnehmer: 18-65 Jahre)
Drug: Indacaterol maleate Drug: Placebo Primary Purpose: Treatment
NCT00624702 Phase I Completed/No results posted Nicht in pubmed.gov identi-fiziert
Novartis 01/2008-04/2008 n = 28 (Studienteilnehmer: 18-65 Jahre)
Drug: Indacaterol maleate/Mometasone furoate Drug: Placebo to Indacaterol male-ate/Mometasone furoate
NCT00605306 Phase II Completed/Has results Nicht in pubmed.gov identi-fiziert
Novartis Pharmaceuticals
10/2007-04/2008 n = 392 (Studienteilnehmer: 12-75 Jahre)
Drug: Indacaterol Drug: Formoterol Drug: Placebo to Indacaterol Drug: Placebo to Formoterol
-2-agonist
NCT00545272 Phase II Completed/Has results Nicht in pubmed.gov identi-fiziert
University of Dundee
02/2014-02/2016 n = 14 (Stand 04/15) (Studienteilnehmer 16 Jahre +)
Drug: Indacaterol Drug: Indacaterol and Tiotropium
NCT02039011 Phase IV This study is currently re-cruiting participants
Novartis Pharmaceuticals
11/2013-03/2014 n = 91 (Studienteilnehmer: 18 Jahre +)
Drug: Indacaterol 27.5-150 μg Drug: Placebo
NCT01959412 Phase II Completed/No results posted Nicht in pubmed.gov identi-fiziert
Novartis Pharmaceuticals
03/2010-07/2010 n = 191 (Studienteilnehmer: 18 Jahre +)
Drug: Indacaterol Drug: Placebo to Indacatero
NCT01156844 Phase II Completed/Has results Nicht in pubmed.gov identi-fiziert
79
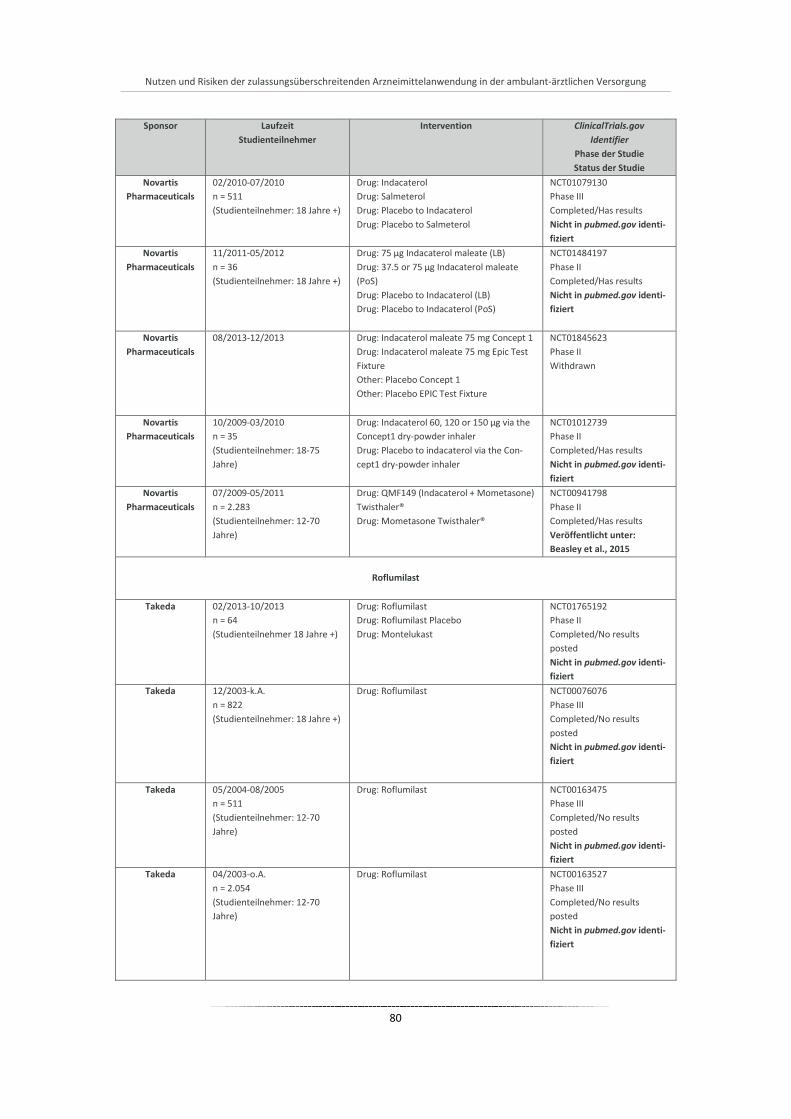
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Sponsor Laufzeit Studienteilnehmer
Intervention
ClinicalTrials.gov Identifier
Phase der Studie Status der Studie
Novartis Pharmaceuticals
02/2010-07/2010 n = 511 (Studienteilnehmer: 18 Jahre +)
Drug: Indacaterol Drug: Salmeterol Drug: Placebo to Indacaterol Drug: Placebo to Salmeterol
NCT01079130 Phase III Completed/Has results Nicht in pubmed.gov identi-fiziert
Novartis Pharmaceuticals
11/2011-05/2012 n = 36 (Studienteilnehmer: 18 Jahre +)
Drug: 75 μg Indacaterol maleate (LB) Drug: 37.5 or 75 μg Indacaterol maleate (PoS) Drug: Placebo to Indacaterol (LB) Drug: Placebo to Indacaterol (PoS)
NCT01484197 Phase II Completed/Has results Nicht in pubmed.gov identi-fiziert
Novartis Pharmaceuticals
08/2013-12/2013 Drug: Indacaterol maleate 75 mg Concept 1 Drug: Indacaterol maleate 75 mg Epic Test Fixture Other: Placebo Concept 1 Other: Placebo EPIC Test Fixture
NCT01845623 Phase II Withdrawn
Novartis Pharmaceuticals
10/2009-03/2010 n = 35 (Studienteilnehmer: 18-75 Jahre)
Drug: Concept1 dry-powder inhaler Drug: Placebo to indacaterol via the Con-cept1 dry-powder inhaler
NCT01012739 Phase II Completed/Has results Nicht in pubmed.gov identi-fiziert
Novartis Pharmaceuticals
07/2009-05/2011 n = 2.283 (Studienteilnehmer: 12-70 Jahre)
Drug: QMF149 (Indacaterol + Mometasone) Twisthaler® Drug: Mometasone Twisthaler®
NCT00941798 Phase II Completed/Has results Veröffentlicht unter: Beasley et al., 2015
Roflumilast
Takeda 02/2013-10/2013
n = 64 (Studienteilnehmer 18 Jahre +)
Drug: Roflumilast Drug: Roflumilast Placebo Drug: Montelukast
NCT01765192 Phase II Completed/No results posted Nicht in pubmed.gov identi-fiziert
Takeda 12/2003-k.A. n = 822 (Studienteilnehmer: 18 Jahre +)
Drug: Roflumilast
NCT00076076 Phase III Completed/No results posted Nicht in pubmed.gov identi-fiziert
Takeda 05/2004-08/2005 n = 511 (Studienteilnehmer: 12-70 Jahre)
Drug: Roflumilast
NCT00163475 Phase III Completed/No results posted Nicht in pubmed.gov identi-fiziert
Takeda 04/2003-o.A. n = 2.054 (Studienteilnehmer: 12-70 Jahre)
Drug: Roflumilast
NCT00163527 Phase III Completed/No results posted Nicht in pubmed.gov identi-fiziert
80
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Sponsor Laufzeit Studienteilnehmer
Intervention
ClinicalTrials.gov Identifier
Phase der Studie Status der Studie
Nycomed 12/2004-07/2005 n = 48 (Studienteilnehmer: 18-55 Jahre)
Drug: Roflumilast Drug: Placebo
NCT01365533 Phase II Completed/No results posted Veröffentlicht unter: Gauvreau et al.2011
Takeda 12/2004-10/2007 n = 150 (Studienteilnehmer: 20-71 Jahre)
Drug: Roflumilast
NCT00246922 Phase III Completed/No results posted Nicht in pubmed.gov identi-fiziert
Takeda 05/2004-06/2007 n = 450 (Studienteilnehmer: 20-70 Jahre)
Drug: Roflumilast
NCT00242307 Phase II/III Completed/No results posted Nicht in pubmed.gov identi-fiziert
Takeda 11/2003-o.A: n = 819 (Studienteilnehmer: 18-70 Jahre)
Drug: Roflumilast
NCT00073177 Phase II Completed/No results posted Nicht in pubmed.gov identi-fiziert
Nycomed 04/2004-12/2004 n = 27 (Studienteilnehmer: 18-45 Jahre, Männer, gesund)
Drug: Roflumilast
NCT00940329 Phase I Completed/No results posted Veröffentlicht unter: de Mey et al., 2011
Datenbank: ClinicalTrials.gov
Die Ergebnisdarstellung beider Suchstrategien erfolgt in den entsprechenden Abschnitten der drei
Wirkstoffe (siehe Kapitel 8.1.2.1-8.1.2.3).
81

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.1.2.1 Indacaterol
Wirkstoff und Pharmakologie
Indacaterol zählt zu den langwirksamen -2-Sympathomimetika. Als partieller Agonist am
-2-Rezeptor wirkt er bronchodilatierend. Der Eintritt der Wirkung erfolgt nach etwa
fünf Minuten und ist damit vergleichbar (mit Salbutamol, Formoterol) oder schneller als andere
Vertreter aus dieser Substanzklasse (Sturton et al., 2008). Gleichzeitig ist die Wirkdauer mit etwa 24
Stunden besonders lang (im Vergleich Formoterol: ca. zwölf Stunden), vermutlich aufgrund des
lipophilen Wirkstoffcharakters. Eine tägliche Gabe ist somit ausreichend (Battram et al., 2006).
Indacaterol ist für die bronchialerweiternde Erhaltungstherapie Erwachsener mit chronisch-
obstruktiver Lungenerkrankung (COPD) angezeigt. Die empfohlene Dosis beträgt dabei 150 μg
einmal täglich. Die Inhalation von 300 μg einmal täglich kann einen zusätzlichen Nutzen hinsichtlich
der Kurzatmigkeit vor allem bei Patienten mit schwerer COPD bieten (Novartis Pharma, 2014). Laut
Hersteller fehlen Langzeitdaten, die den klinischen Einsatz von Indacaterol bei Asthma bronchiale
rechtfertigen. Aus diesem Grund ist – im Gegensatz zu den anderen marktverfügbaren
-2-Agonisten Formoterol und Salmeterol – das Arzneimittel nicht für die Asthma-
Therapie zugelassen, genauso wenig für die Notfallbehandlung bei COPD (Novartis Pharma, 2014).
Seit September 2013 steht Indacaterol jetzt auch als fixe Kombination mit Glycopyrronium, einem
Wirkstoff aus der Gruppe der Parasympatholytika, zur Verfügung – ebenfalls ausschließlich
zugelassen zur Therapie der COPD (Novartis Pharma, 2015).
Studienlage im In-Label Use
Nach den Daten der INLIGHT-1-Studie zeigte sich nach zwölf Wochen für Indacaterol (1x täglich
150 μg) eine statistisch signifikante Verbesserung der Lungenfunktion gegenüber Placebo. So lag das
forcierte exspiratorische Volumen in der ersten Sekunde (FEV1) um 130 ml höher als unter Placebo.
Eine Zunahme um 120 ml gilt bereits als klinisch relevant (Feldman et al., 2010). Die INVOLVE-Studie
zeigte für Indacaterol (1x täglich 300 oder 600 μg) eine Verbesserung der Lungenfunktion nach zwölf
Wochen, die in beiden Dosierungen stärker war als Formoterol (2x täglich 12 μg). Auch nach einem
Jahr ließ sich dieser Vorteil noch nachweisen. Der stärker patientenrelevante Endpunkt Reduzierung
von Exazerbationen verbesserte sich hingegen nur im Placebo-, nicht jedoch im Formoterol-Vergleich
82

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
(Dahl et al., 2010). Auch in der INHANCE-Studie wurde als primärer Endpunkt der FEV1-Wert nach
zwölf Wochen erhoben. Im Hinblick auf die Lungenfunktion und den Gesundheitsstatus konnte eine
Nicht-Unterlegenheit von Indacaterol gegenüber Tiotropium (nicht verblindet) demonstriert werden.
Zum Endpunkt Exazerbationen lieferte die Studie keine belastbaren Ergebnisse (Donohue et al.,
2010). Kornmann et al. (2011) zeigten in der Therapie von Patienten mit mittelschwerer bis schwerer
COPD für Indacaterol (1x täglich 150 μg) eine Erhöhung des FEV1-Wertes um 170 ml gegenüber Pla-
cebo und 60 ml gegenüber dem aktiven Komparator Salmeterol (2x täglich 50 μg) auf. Ebenso lag der
FEV1-Wert in der INSIST-Studie nach zwölf Wochen um 57 ml höher als bei Salmeterol, was ein sta-
tistisch signifikantes, klinisch aber nicht relevantes Ergebnis darstellt (Korn et al., 2011). Nach den
Daten zweier placebokontrollierter Studien (INTRUST-1 und INTRUST-2) mit identischem Studiende-
sign und einer Laufzeit von zwölf Wochen verbesserte die duale Kombinationstherapie Indacaterol
(1x täglich 150 μg) plus Tiotropium (1x täglich 18 μg) die Lungenfunktion (FEV1) von Patienten mit
mittelschwerer bis schwerer COPD stärker als die Monotherapie mit dem langwirksamen Anticholi-
nergikum Tiotropium (18 μg). Dieser Effekt ging allerdings mit einer höheren Nebenwirkungsrate
(Husten) einher (zehn bzw. neun Prozent vs. vier Prozent) (Mahler et al., 2012). Andererseits konnte
die Untersuchung von Buhl et al. (2011) für die duale Kombinationstherapie Hinweise auf eine ver-
besserte Symptomatik (Atemnot) und Lebensqualität geben. Hinsichtlich unerwünschter Arzneimit-
telwirkungen zeigen sich im Vergleich zu anderen Bronchodilatatoren (Decramer et al., 2013) keine
Unterschi -2-Agonisten wie Hyperglykämie und Hypokaliämie fielen
relativ gering aus (Rodrigo & Neffen, 2012). Die Verbesserung der Lungenfunktion unter der Therapie
von Indacaterol in fixer Kombination mit Glycopyrronium gegenüber Glycopyrronium als Monothe-
rapie bzw. Salmeterol und Fluticason wurde als signifikant höher eingestuft (Vogelmeier et al., 2013;
Wedzicha et al., 2013). Generell erweist sich jedoch die Vergleichbarkeit der vorliegenden Studiener-
gebnisse aufgrund heterogener Patientenkollektive, unterschiedlicher primärer Endpunkte sowie
Abweichungen in der Therapiedauer als schwierig. Zwar kamen die Autoren in einer systematischen
Übersichtsarbeit zum Schluss, dass Indacaterol insbesondere im Vergleich zu langwirksamen Bron-
chodilatatoren wie Formoterol Vorteile nicht nur im Hinblick auf die Lungenfunktion, sondern auch
auf Symptomatik und Lebensqualität biete (Rodrigo & Neffen, 2012), um jedoch belastbare Aussagen
zu patientenrelevanten Endpunkten wie der Exazerbations- und COPD-bedingten Hospitalisierungs-
raten treffen zu können, sind weitere Studien nötig.
83

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Studienlage im Off-Label Use
Durch die Suche in einschlägigen Datenbanken konnten mehrere Studien gesichtet werden, in denen
die Gabe des COPD-Wirkstoffes auch an Patienten mit diagnostiziertem Asthma bronchiale unter-
schiedlicher Genese untersucht wurde. So erwies sich Indacaterol dem Placebo signifikant überlegen
(Sugihara et al., 2010; Kannies et al., 2008) und zeigte bei einer einmaligen täglichen Gabe einen
bronchodilatatorischen Effekt über 24 Stunden (Kanniess et al., 2008, Beeh et al., 2007; Pearlman et
al., 2008) bei einem sicheren Wirkprofil, unabhängig von der eingesetzten Dosis des Wirkstoffes
(Beeh et al., 2007; Yang et al., 2007; Sugihara et al., 2010). Sicherheitsbedenken hinsichtlich kardialer
oder metabolischer Effekte konnten nicht geteilt werden (Chuchalin et al., 2007). Unerwünschte
Wirkungen wie Nasopharyngitis, Kopfschmerzen oder Husten wurden unter der Gabe von In-
dacaterol häufiger dokumentiert als unter Placebo (Chuchalin et al., 2007), insgesamt waren jedoch
die aufgetretenen Nebenwirkungen vergleichbar mit denen in der zulassungskonformen Anwen-
dung, die sich auch nach höherer Wirkstoff-Gabe von Indacaterol nicht änderten (Yang et al., 2007;
Chuchalin et al., Beeh et al., 2007). In allen publizierten Studien wurde der Bronchodilatator als wirk-
sam, sicher und gut toleriert bezeichnet – unabhängig von der untersuchten Altersgruppe, in der
Indacaterol zum Einsatz kam. Zu bedenken gilt jedoch, dass es sich dabei primär um placebokontrol-
lierte Studien mit niedrigen Fallzahlen handelt. Allein die industriefinanzierte Studie von Beasley et
al. (2015) schloss eine größere Patientenzahl ein. Die Fixkombination, bestehend aus Indacaterol und
dem inhalativen Glucocorticoid Mometason, reduzierte im Vergleich zur Glucocorticoid-Mono-
therapie signifikant die jährliche Rate schwerer Exazerbationen, die in der Regel die Gabe eines sys-
-2-
Sympathomimetikums hatte hingegen keinen signifikanten Einfluss auf den Zeitpunkt bis zur ersten
schwerwiegenden Exazerbation (Hospitalisierung, Intubation, Tod), der hier den primären Stu-
dienendpunkt darstellt.
Veröffentlichte Studien aus der Datenbank des U.S. National Institutes of Health (NIH) kamen zu
vergleichbaren Ergebnissen was das Nebenwirkungsprofil angeht, unabhängig der eingesetzten
Wirkstoffstärke (bis zu 600 μg täglich). Statistische Auswertungen zu patientenrelevanten Endpunk-
ten wurden nicht zur Verfügung gestellt, so dass belastbare Aussagen derzeit nicht getroffen werden
können. In diesen Studien wurde der Wirkstoff an teilweise sehr geringen Fallzahlen (n < 30) unter-
sucht und die Patienten waren in den meisten Fällen zwischen 40 und 50 Jahre alt.
84

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Nutzenbewertung nach § 35a SGB V
Eine Nutzenbewertung nach § 35a SGB V liegt nur für die duale Kombinationstherapie, bestehend
aus Indacaterol und Glycopyrronium, vor. Für diese Wirkstoffkombination konstatierte der G-BA in
seinem Beschluss vom 8. Mai 2014 für Patienten mit COPD Stufe II einen Anhaltspunkt für einen
geringen Zusatznutzen gegenüber der zweckmäßigen Vergleichstherapie (für diese sowie für die
anderen Patientenpopulationen) -2-Sympathomimetika (Formoterol
oder Salmeterol) oder langwirksamen Anticholinergika (Tiotropium) oder der Kombination beider
Wirkstoffklassen. Der G-BA hält die Herabstufung der Bewertung von einem Hinweis auf einen An-
haltspunkt durch eine möglicherweise geringere Effektstärke für Patienten mit COPD Stufe II für
gerechtfertigt. Für Patienten mit COPD Stufe III, mit höchstens einer Exazerbation pro Jahr, sieht der
G-BA keine Anzeichen einer geringeren Effektstärke und stellt deshalb für diese Population einen
Hinweis für einen geringen Zusatznutzen fest. Für die anderen beiden Untersuchungsfragen „Pati-
enten mit COPD Stufe IV, mit höchstens einer Exazerbation pro Jahr“ sowie „Patienten mit Stufe III
und IV, mit zwei und mehr Exazerbationen pro Jahr“ konnte nach Angaben des G-BA kein Zusatznut-
zen belegt werden, da in der vom pU vorgelegten Studie für den Nachweis eines Zusatznutzens die
Anzahl der eingeschlossenen Patienten zu gering war (G-BA, 2014b).
Versorgungsanalyse
Im Untersuchungszeitraum codierten die Ärzte im ambulanten Bereich für 290.790 TK-Versicherte
mindestens einmal eine COPD-Diagnose mit dem Diagnosekennzeichen „G“ für gesichert. Dies ent-
sprach einer Prävalenz von 3,7 Prozent in der betrachteten Versichertenpopulation. Liegen für Versi-
cherte in mindestens zwei Abrechnungsquartalen entsprechende Diagnosen vor, gilt dies als Hinweis
auf eine chronische Erkrankung. Im Jahr 2010 traf dies für 167.671 und im Jahr 2011 für insgesamt
177.784 TK-Versicherte zu. Von den 290.790 Versicherten mit mindestens einer COPD-Diagnose
erhielten 72,3 Prozent (n = 210.250) mindestens einmal ein Arzneimittel zur Behandlung dieser Er-
krankung. Nach wie vor besitze -2-Sympathomimetika Cortisonsprays bzw. fixe Kombi-
nationen aus beiden Wirkstoffgruppen den größten Verordnungsanteil (Abbildung 14). Der Verord-
nungsanteil der neuen COPD-Medikamente (Indacaterol und Roflumilast) fiel vergleichsweise gering
aus und erzielte im dritten Quartal des Jahres 2011 mit 4.539 Verordnungen das Maximum. Von den
4.539 Verordnungen fielen alleine gut 84 Prozent auf Indacaterol. Mit rund 1.400 Packungen wurden
im Juli 2011 für den gesamten Untersuchungszeitraum die meisten Indacaterol-Verordnungen aus-
gestellt (insgesamt: 20.148 Packungen) (Abbildung 15). Dass die Verordnungszahlen Ende 2011 ein-
85
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
brachen, kann als Folge der Eingruppierung in eine Festbetragsgruppe mit den daraus resultierenden
Mehrkosten für die Versicherten gesehen werden.
Abbildung 14: Verteilung der Atemwegstherapeutika-Verordnungen (Anzahl Packungen) je Wirkstoffklasse bei COPD-Patienten (N = 210.250) nach Quartalen (2010-2011)
Abbildung 15: Anzahl verordneter Packungen Indacaterol je Monat nach Packungsgrößen (2010-2011)
86
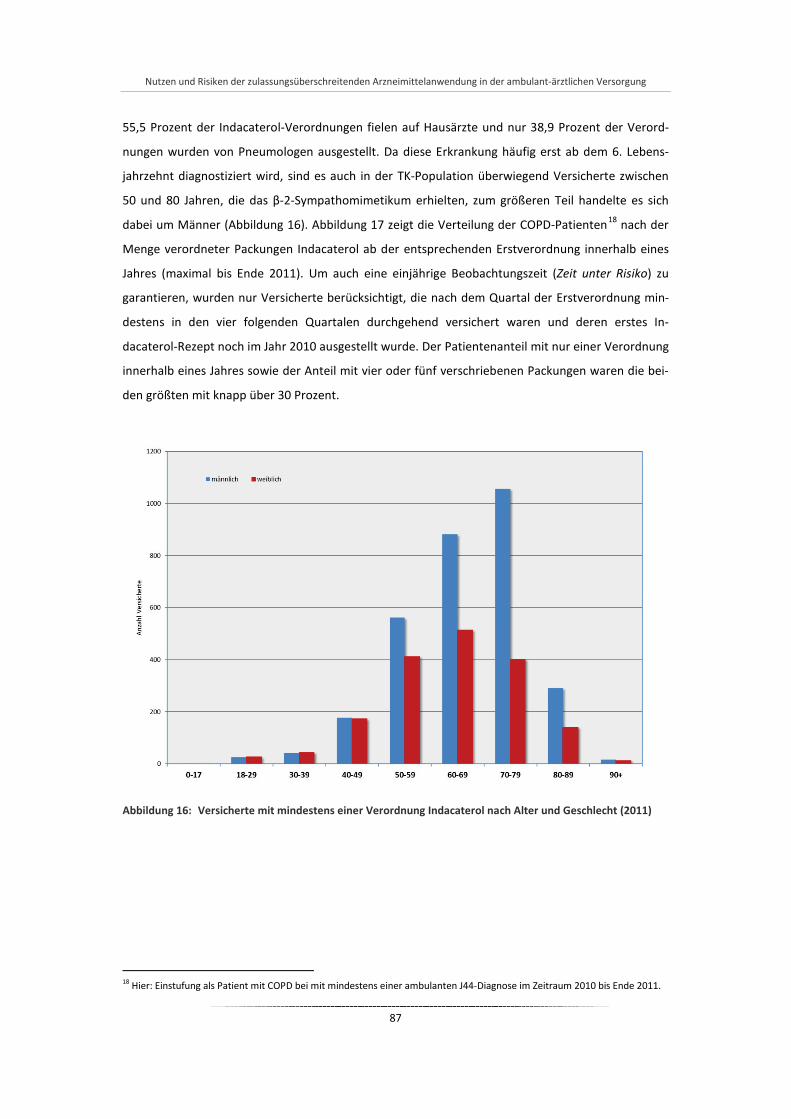
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
55,5 Prozent der Indacaterol-Verordnungen fielen auf Hausärzte und nur 38,9 Prozent der Verord-
nungen wurden von Pneumologen ausgestellt. Da diese Erkrankung häufig erst ab dem 6. Lebens-
jahrzehnt diagnostiziert wird, sind es auch in der TK-Population überwiegend Versicherte zwischen
50 und 80 Jahren, die das -2-Sympathomimetikum erhielten, zum größeren Teil handelte es sich
dabei um Männer (Abbildung 16). Abbildung 17 zeigt die Verteilung der COPD-Patienten18 nach der
Menge verordneter Packungen Indacaterol ab der entsprechenden Erstverordnung innerhalb eines
Jahres (maximal bis Ende 2011). Um auch eine einjährige Beobachtungszeit (Zeit unter Risiko) zu
garantieren, wurden nur Versicherte berücksichtigt, die nach dem Quartal der Erstverordnung min-
destens in den vier folgenden Quartalen durchgehend versichert waren und deren erstes In-
dacaterol-Rezept noch im Jahr 2010 ausgestellt wurde. Der Patientenanteil mit nur einer Verordnung
innerhalb eines Jahres sowie der Anteil mit vier oder fünf verschriebenen Packungen waren die bei-
den größten mit knapp über 30 Prozent.
Abbildung 16: Versicherte mit mindestens einer Verordnung Indacaterol nach Alter und Geschlecht (2011)
18 Hier: Einstufung als Patient mit COPD bei mit mindestens einer ambulanten J44-Diagnose im Zeitraum 2010 bis Ende 2011.
87
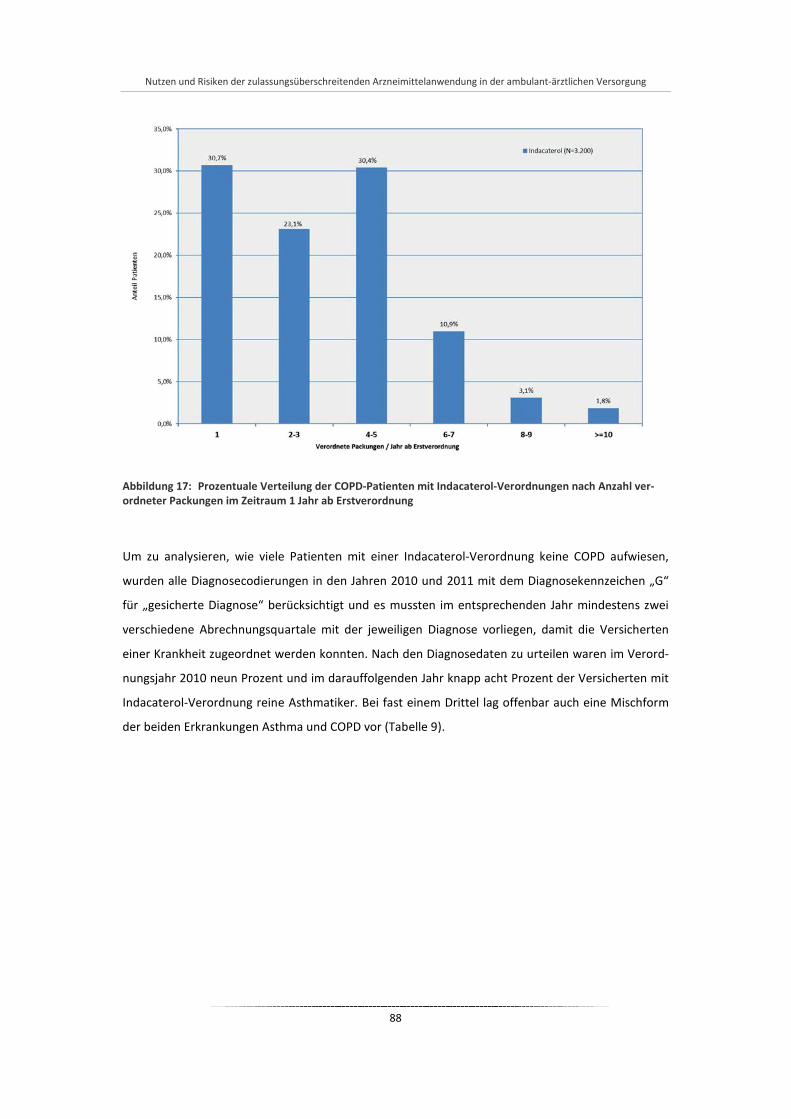
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Abbildung 17: Prozentuale Verteilung der COPD-Patienten mit Indacaterol-Verordnungen nach Anzahl ver-ordneter Packungen im Zeitraum 1 Jahr ab Erstverordnung
Um zu analysieren, wie viele Patienten mit einer Indacaterol-Verordnung keine COPD aufwiesen,
wurden alle Diagnosecodierungen in den Jahren 2010 und 2011 mit dem Diagnosekennzeichen „G“
für „gesicherte Diagnose“ berücksichtigt und es mussten im entsprechenden Jahr mindestens zwei
verschiedene Abrechnungsquartale mit der jeweiligen Diagnose vorliegen, damit die Versicherten
einer Krankheit zugeordnet werden konnten. Nach den Diagnosedaten zu urteilen waren im Verord-
nungsjahr 2010 neun Prozent und im darauffolgenden Jahr knapp acht Prozent der Versicherten mit
Indacaterol-Verordnung reine Asthmatiker. Bei fast einem Drittel lag offenbar auch eine Mischform
der beiden Erkrankungen Asthma und COPD vor (Tabelle 9).
88
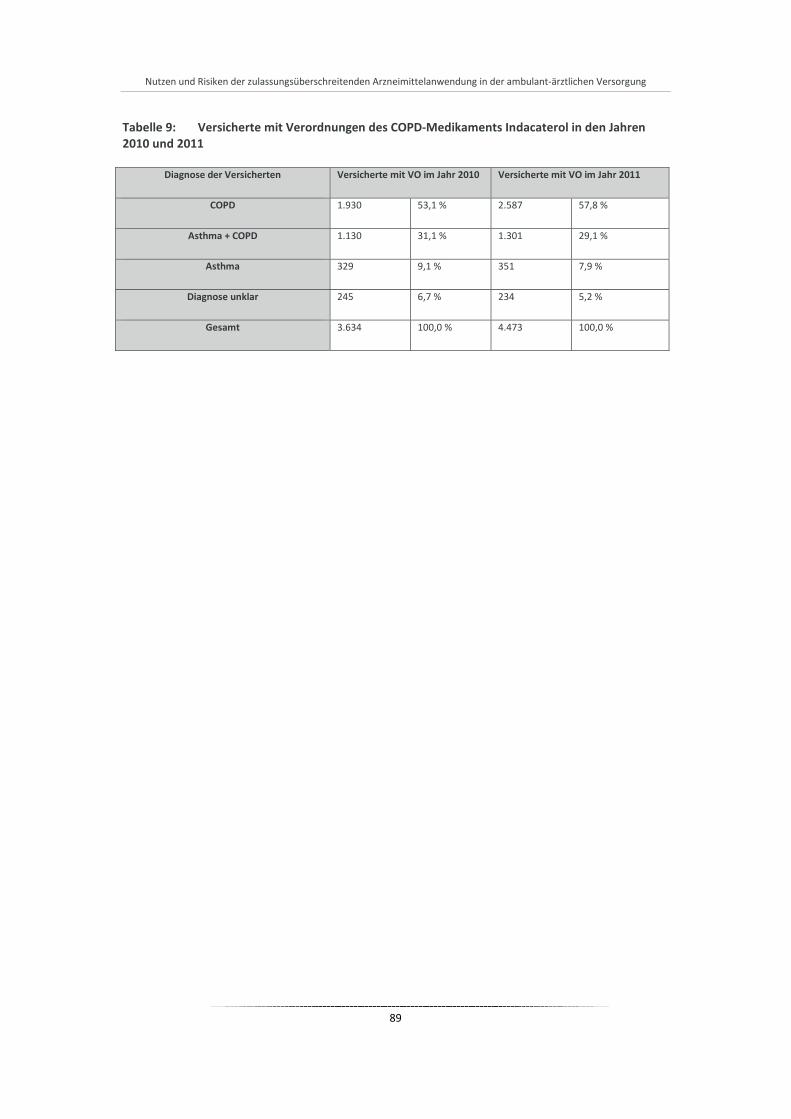
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 9: Versicherte mit Verordnungen des COPD-Medikaments Indacaterol in den Jahren 2010 und 2011
Diagnose der Versicherten
Versicherte mit VO im Jahr 2010 Versicherte mit VO im Jahr 2011
COPD
1.930 53,1 % 2.587 57,8 %
Asthma + COPD
1.130 31,1 % 1.301 29,1 %
Asthma
329 9,1 % 351 7,9 %
Diagnose unklar
245 6,7 % 234 5,2 %
Gesamt
3.634 100,0 % 4.473 100,0 %
89

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.1.2.2 Roflumilast
Wirkstoff und Pharmakologie
Im Gegensatz zum nicht-selektiven Phosphodiesterase (PDE)-4-Inhibitor Theophyllin, hemmt
Roflumilast das Enzym selektiv. PDE 4 stellt in strukturellen und inflammatorischen Zellen eines der
wichtigsten Enzyme dar, welches für den Abbau von cAMP verantwortlich ist. Durch Blockade des
Enzyms wird ein Anstieg der cAMP-Konzentration in Lungenzellen erzielt, wodurch Entzündungs-
prozesse zurückgedrängt und die Lungenfunktion verbessert wird (Lippworth, 2005). Roflumilast
wurde 2010 zugelassen und ist indiziert zur Behandlung Erwachsener mit schwerer COPD, definiert
als FEV1 nach Anwendung eines Bronchodilatators weniger als 50 Prozent vom Soll, die gleichzeitig
eine chronische Bronchitis und häufigen Exazerbationen in der Vergangenheit aufweisen. Die
Behandlung sollte als Add-on zu einer bronchodilatatorischen Therapie eingesetzt werden (Takeda,
2013).
Studienlage im In-Label Use
Die Ergebnisse (gepoolte Analyse) der beiden zulassungsrelevanten placebokontrollierten klinischen
Studien (M2-124 und M2-125) zeigten eine Verbesserung der Lungenfunktion in der
Interventionsgruppe, die ein Jahr lang einmal täglich Roflumilast erhielten. Trotz statistischer
Signifikanz bedeutet es jedoch eine klinisch nicht relevante Verbesserung der Lungenfunktion. Die
mittlere Rate an Exazerbationen konnte durch Roflumilast ebenfalls nur geringfügig gesenkt werden
(Calverley et al., 2009). Da die Auswertung der Exazerbationrate nur bei denjenigen durchgeführt
wurde, die nach Studienbeginn eine entsprechende Verschlimmerung aufwiesen, ist eine
Überschätzung dieses Effekts nicht auszuschließen. In zwei weiteren placebokontrollierten Studien
(M2-127 und M2-128) wurde die Wirksamkeit von Roflumilast als Add-on zu einer bestehenden
bronchienerweiternden Salmeterol- (M2-127) oder Tiotropium-Therapie (M2-128) untersucht. Auch
-2-Sympathomimetikum bzw. mit einem Anticholinergikum
konnte kein klinisch relevantes Ergebnis erzielen oder die Exazerbationsrate signifikant senken
(Fabbri et al., 2009). In die erst kürzlich veröffentlichte REACT-Studie wurden Patienten
eingeschlossen, bei denen eine schwere COPD diagnostiziert wurde und die trotz Gabe von
Corticosteroiden und bzw. oder langwirksam -2-Sympathimimetika ein hohes Exazerbations-
90

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Risiko hatten. Unter der Gabe von Roflumilast konnte nicht nur die Exazerbations-, sondern auch die
damit verbundenen Hospitalisierungsraten gesenkt werden (Martinez et al., 2015). Inwiefern sich
Roflumilast nicht nur positiv auf kardiovaskuläre Ereignisse auswirkt (White et al., 2013), sondern
auch Einfluss auf eine COPD-bedingte Dyspnoe hat, muss erst noch in weiteren Studien untersucht
werden (Pan et al., 2013). Nach wie vor mangelt es jedoch an adäquaten Untersuchungen, die einen
Zusatznutzen gegenüber der Standardbehandlung bei schwerer COPD (bronchienerweiternde
Medikamente + inhalatives Cortison) belegen könnten. Vergleichsstudien mit der pharmakologisch
verwandten Substanz Theophyllin fehlen ebenfalls.
Eine neuere Übersichtsarbeit, die insgesamt 8.698 Patienten aus acht Studien berücksichtigte, kam
ebenfalls zu dem Ergebnis, dass Roflumilast im Vergleich zu anderen für die Behandlung der COPD
eingesetzten Wirkstoffe keine Verbesserung aufweist (Oba & Lone, 2013). In dieser Arbeit wurde
außerdem auf das höhere Nebenwirkungsrisiko (Vorhofflimmern und Suizidalität) hingewiesen.
Studienlage im Off-Label Use
Auch bei dem PDE-4-Inhibitor Roflumilast liegen publizierte sowie nicht publizierte Studien vor, die
die Wirksamkeit und Sicherheit des Wirkstoffes auch bei Asthmatikern untersuchten. So erwies sich
die einmal tägliche, orale (500 μg) Gabe von Roflumilast nicht unterlegen im Vergleich zur zweimal
täglichen Inhalation mit dem Glucocorticoid Beclomethason (400 μg/Tag) (Bousquet et al., 2006). Die
Wirkung der beiden Substanzen war bezüglich der Verbesserung der Lungenfunktion sowie der
Reduzierung der Asthma-Symptome vergleichbar. Auch konnte unter der Einnahme von Roflumilast
die Gabe von Notfallmedikamenten reduziert werden (Bousquet et al., 2006). Eine weitere Studie (n
= 25) zeigte eine schützende Wirkung von Roflumilast hinsichtlich Allergen-induzierter
Atemwegsentzündungen (Gauvreau et al., 2011). Van Schalkwyk et al. (2005) untersuchten schon
Jahre zuvor eine mögliche positive Wirkung von Roflumilast bei Asthmatikern. Hier zeigte die einmal
tägliche Gabe eine moderate Linderung asthmatischer Reaktionen und in einem größeren Ausmaß
eine Verbesserung Allergen-induzierter asthmatischer Spätreaktionen bei Patienten mit mildem
allergischen Asthma. Diese Ergebnisse wurden in einer späteren Studie bestätigt (Louw et al., 2007).
Wie schon Indacaterol erwies sich auch Roflumilast als insgesamt sicher und wirksam.
Von den insgesamt zehn Studien, die in der NIH-Datenbank veröffentlicht wurden, fand sich keine
mit publizierten Daten.
91

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Nutzenbewertung nach § 35a SGB V
Aufgrund des Termins seiner Markteinführung im August 2010 und somit vor dem Umsetzen der
frühen Nutzenbewertung durch den G-BA liegt kein entsprechender Beschluss für Roflumilast vor.
Versorgungsanalyse
Analog zu Indacaterol wurde die Versorgungsanalyse auch für den selektiven PDE-4-Hemmer durch-
geführt. Erst Mitte des Jahres 2010 in den deutschen Arzneimittelmarkt eingeführt, lag die Anzahl an
Verordnungen von Roflumilast mit insgesamt 4.857 Packungen deutlich niedriger als bei Indacaterol
(Abbildung 18). Der November war mit 415 Packungen der verordnungsstärkste Monat für diesen
Wirkstoff. Möglicherweise hatte hier die Einordnung von Indacaterol in eine Festbetragsgruppe
Auswirkungen auf das Verordnungsverhalten niedergelassener Ärzte. 46,5 Prozent der Roflumilast-
Verordnungen fielen auf Hausärzte und 47,0 Prozent der Verordnungen wurden von Pneumologen
ausgestellt. Wie bei Indacaterol gab es die meisten Verordnungen bei männlichen Versicherten zwi-
schen 50 und 80 Jahren (Abbildung 19). Abbildung 20 zeigt die Verteilung der COPD-Patienten nach
der Menge verordneter Packungen Roflumilast analog zu Indacaterol ab der entsprechenden Erst-
verordnung innerhalb eines Jahres (maximal bis Ende 2011). Auch hier war der Patientenanteil mit
nur einer Verordnung innerhalb eines Jahres ebenso wie der Anteil mit vier oder fünf verschriebenen
Packungen jeweils am größten (knapp über 30 Prozent).
92
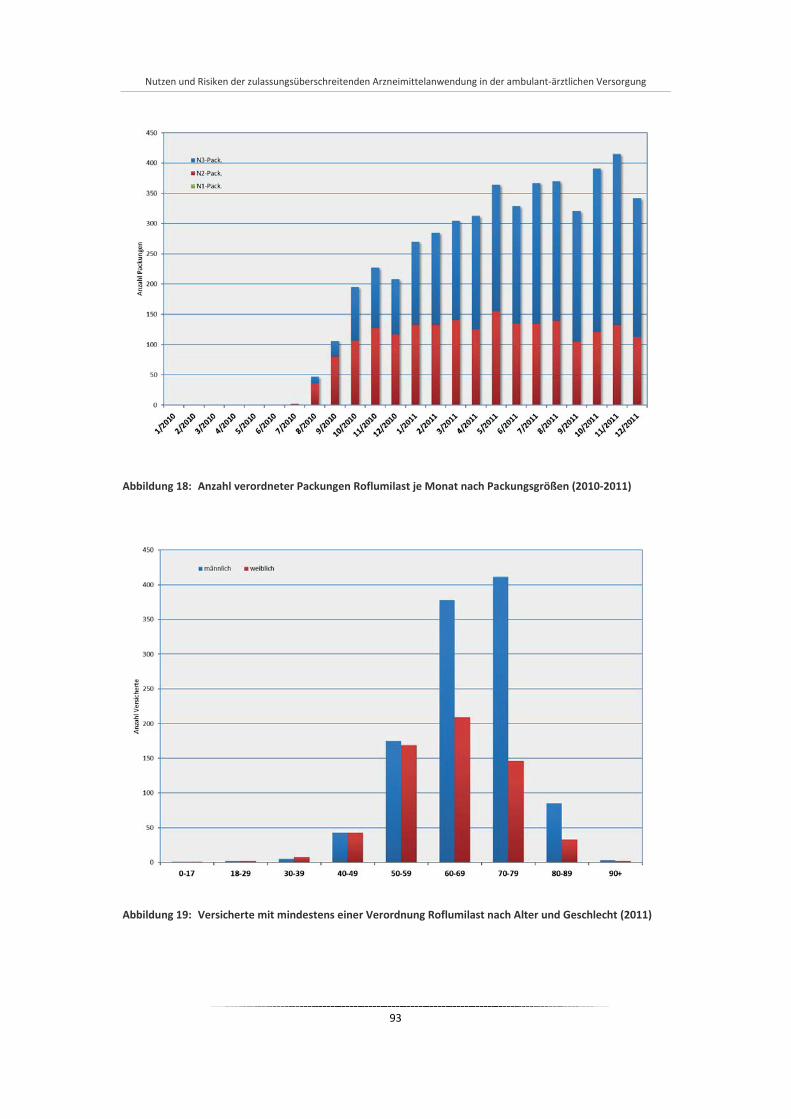
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Abbildung 18: Anzahl verordneter Packungen Roflumilast je Monat nach Packungsgrößen (2010-2011)
Abbildung 19: Versicherte mit mindestens einer Verordnung Roflumilast nach Alter und Geschlecht (2011)
93
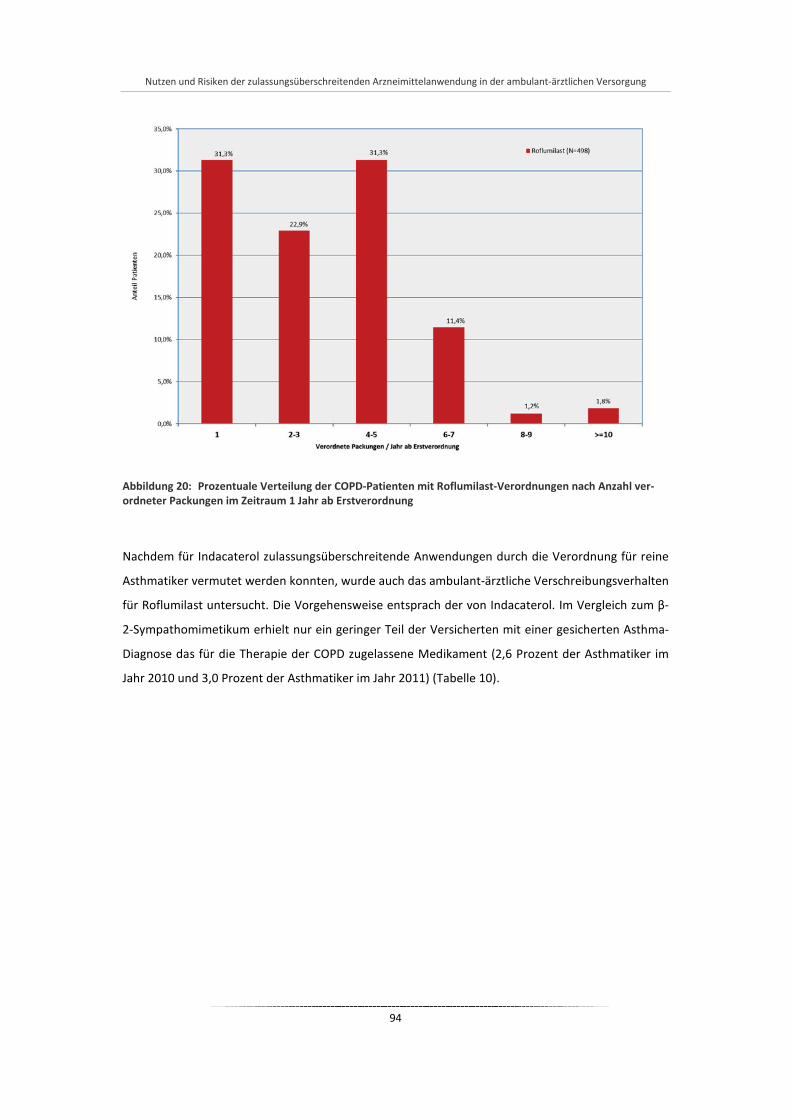
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Abbildung 20: Prozentuale Verteilung der COPD-Patienten mit Roflumilast-Verordnungen nach Anzahl ver-ordneter Packungen im Zeitraum 1 Jahr ab Erstverordnung
Nachdem für Indacaterol zulassungsüberschreitende Anwendungen durch die Verordnung für reine
Asthmatiker vermutet werden konnten, wurde auch das ambulant-ärztliche Verschreibungsverhalten
für Roflumilast untersucht. Die Vorgehensweise entsprach der von Indacaterol. -
2-Sympathomimetikum erhielt nur ein geringer Teil der Versicherten mit einer gesicherten Asthma-
Diagnose das für die Therapie der COPD zugelassene Medikament (2,6 Prozent der Asthmatiker im
Jahr 2010 und 3,0 Prozent der Asthmatiker im Jahr 2011) (Tabelle 10).
94
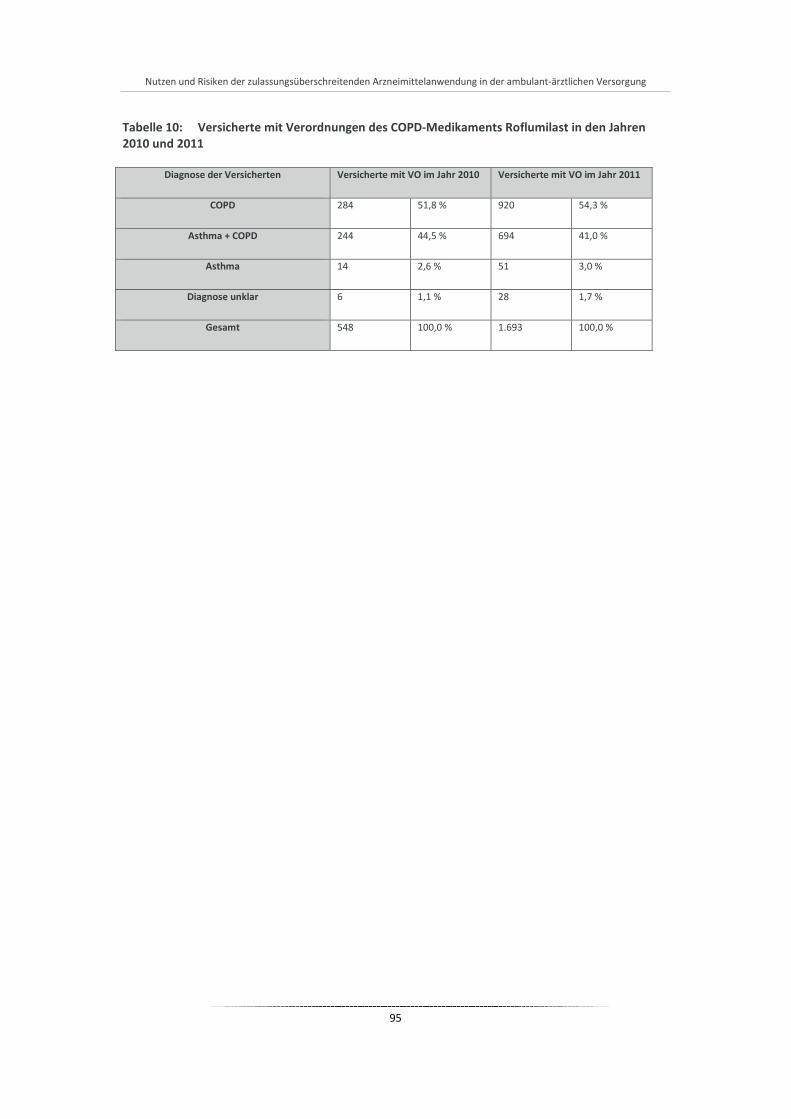
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 10: Versicherte mit Verordnungen des COPD-Medikaments Roflumilast in den Jahren 2010 und 2011
Diagnose der Versicherten
Versicherte mit VO im Jahr 2010 Versicherte mit VO im Jahr 2011
COPD
284 51,8 % 920 54,3 %
Asthma + COPD
244 44,5 % 694 41,0 %
Asthma
14 2,6 % 51 3,0 %
Diagnose unklar
6 1,1 % 28 1,7 %
Gesamt
548 100,0 % 1.693 100,0 %
95

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.1.2.3 Aclidiniumbromid
Wirkstoff und Pharmakologie
Neben Tiotropiumbromid und Glycopyrroniumbromid ist Aclidiniumbromid ein weiterer Vertreter
der lang wirkenden Muscarin-Rezeptorantagonisten zur symptomatischen bronchodilatatorischen
Dauertherapie der chronisch obstruktiven Lungenkrankheit. Der kompetitive, selektive Antagonist
bewirkt durch Bindung an die M3-Rezeptoren der glatten Bronchialmuskulatur eine Broncho-
dilatation und verbessert so die Lungenfunktion. Nach rascher pulmonaler Resorption erreichen
COPD-Patienten in der Regel innerhalb von 15 Minuten die maximale Plasmakonzentration. Da
Aclidiniumbromid im Plasma rasch zu einem inaktiven Alkohol und einem Carbonsäuremetaboliten
hydrolysiert wird, ist die Rate an systemischen anticholinergen Nebenwirkungen gering (EMA,
2014f). Ursprünglich als einmal tägliche Dosierung entwickelt, muss der Wirkstoff aufgrund klinisch
relevanter Wirkungen zweimal täglich inhaliert werden (Almirall, 2014). In der Europäischen Union
erfolgte inzwischen eine Zulassung für die Fixkombination mit Formoterol. Eine Anwendung des
Wirkstoffes zur Therapie des Asthma bronchiale wird grundsätzlich ausgeschlossen.
Studienlage im In-Label Use
Da die einmal tägliche Inhalation von Aclidiniumbomid (1x täglich 200 μg) nicht den Zulas-
sungsanforderungen einer FEV1-Verbesserung gegenüber dem Ausgangswert erfüllte (Jones et al.,
2012; Cazzola et al., 2008; Donohue, 2005), wurde eine weitere placebokontrollierte Studie
durchgeführt. In der ATTAIN-Studie (Jones et al., 2012), erhielten die Studienteilnehmer nach einer
zweiwöchigen Run-in-Phase entweder eine zweimal tägliche Gabe von 200 bzw. 400 μg
Aclidiniumbromid oder Placebo. Die Beibehaltung der Komedikation (inhalative und bzw. oder
systemische Glucocorticoide (entsprechend 10 mg Prednison), retardiertes Theophyllin, Sauerstoff (<
15 Stunden täglich) sowie Salbutamol bei Bedarf) war erlaubt. Primärer Endpunkt dieser Studie war
die Änderung der morgens vor der Anwendung gemessenen FEV1 nach 24 Wochen gegenüber dem
Ausgangswert. Die Differenz von 128 ml (2 x täglich 400 μg Aclidiniumbromid vs. Placebo) wurde als
signifikant eingestuft. Dieses Ergebnis konnte von einer weiteren Studie (ACCORD, Kerwin et al.,
2012) bestätigt werden. Hier verringerte Aclidiniumbromid außerdem frühmorgendliche Symptome
der Kurzatmigkeit in der ersten Stunde nach dem Aufstehen. Auch waren nächtliche Symptome wie
96

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Dyspnoe, Husten, Auswurf oder Keuchen signifikant seltener. In einem Review konnte eine ähnliche
Verbesserung der Lungenfunktion sowie gesundheitsbezogener Lebensqualität zwischen
Aclidiniumbromid, Tiotropium und Glycopyrronium aufgezeigt werden (Karabis et al., 2013). In einer
sechswöchigen randomisierten Studie war eine Verbesserung COPD-bedingter Symptome unter Acli-
diniumbromid im Vergleich zu Tiotropium nur numerisch höher (Beier et al., 2013).
Nutzenbewertung nach § 35a SGB V
Unter Berücksichtigung des Stufenschemas der Nationalen Versorgungsleitlinie COPD ergab die
frühe Nutzenbewertung für den Wirkstoff Aclidiniumbromid durch den G-BA sowohl für die
Patienten ab Therapiestufe II als auch für die Patienten ab Therapiestufe III/IV mit mehr als zwei
Exazerbationen, dass Ausmaß und Wahrscheinlichkeit des Zusatznutzens nicht belegt seien. Die
-2-Sympathomimetika
(Formoterol, Salmeterol) und bzw. oder langwirksamen Anticholinergika (Tiotropiumbromid) sowie
zusätzlich für die Patienten ab Therapiestufe III/IV aus inhalativen Corticosteroiden. Der G-BA
begründet seine Entscheidung u.a. darin, dass die Studiendauer (zwei und sechs Wochen) der
vorgelegten (unveröffentlichten) Studien nicht ausreichend lang waren, um einen Zusatznutzen für
patientenrelevante Endpunkte zu belegen. Schließlich handele es sich bei Aclidiniumbromid um ein
dauerhaft einzusetzendes Arzneimittel (G-BA, 2013). Für die Verbesserung der Lungenobstruktion
schlagen die derzeitigen Empfehlungen sowohl der EMA als auch der FDA eine Behandlungsdauer
von mindestens drei Monaten vor und hinsichtlich der Symptomverbesserung eine
Behandlungsdauer von mindestens sechs Monaten. Bei kurzfristigen Effekten in Studien (Dauer zwölf
bis 24 Wochen) ist eine nachhaltige Wirksamkeit von mindestens zwölf Monaten nachzuweisen,
ebenfalls belegt durch Studien (G-BA, 2013). Zusätzlich zu den beiden Studien reichte der
pharmazeutische Unternehmer einen indirekten Vergleich von Aclidiniumbromid und
Tiotropiumbromid ein, der jedoch eine valide Bewertung des Zusatznutzens aufgrund mangelhafter
Qualität (Diskrepanzen zwischen den aus den Originaldokumenten und den im Modul 4 des Dossiers
aufbereiteten Daten) nicht zuließ (G-BA, 2013).
97
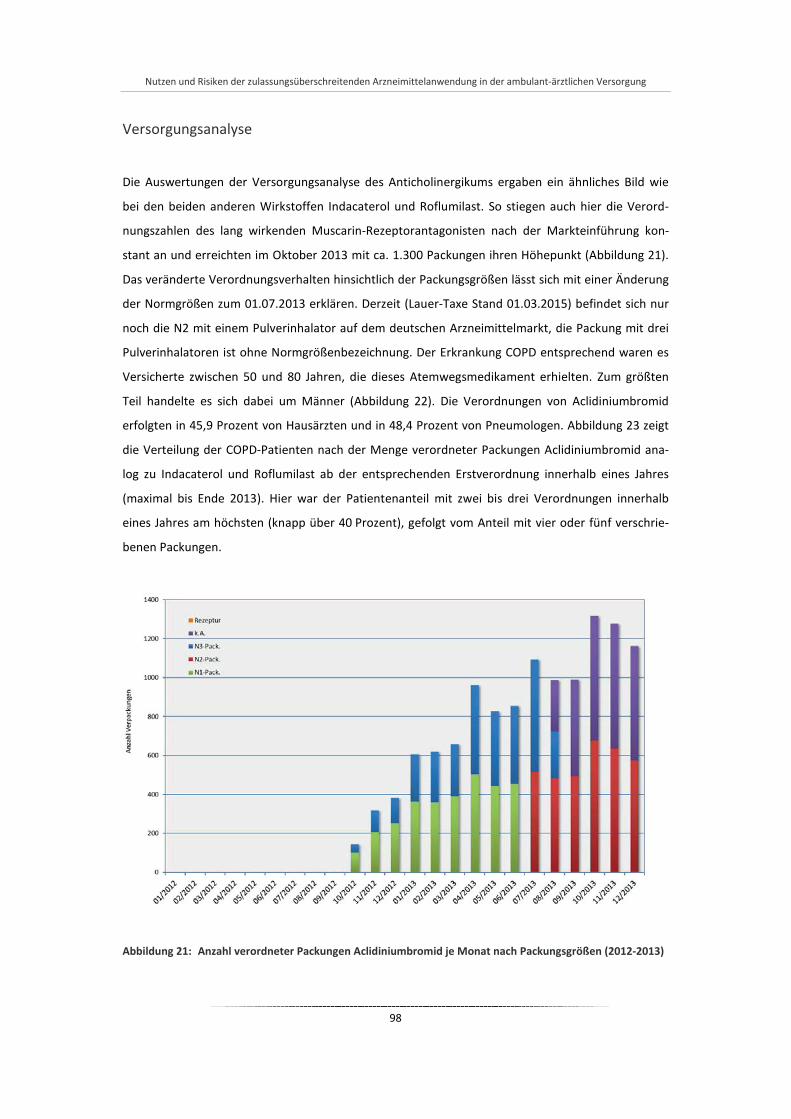
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Versorgungsanalyse
Die Auswertungen der Versorgungsanalyse des Anticholinergikums ergaben ein ähnliches Bild wie
bei den beiden anderen Wirkstoffen Indacaterol und Roflumilast. So stiegen auch hier die Verord-
nungszahlen des lang wirkenden Muscarin-Rezeptorantagonisten nach der Markteinführung kon-
stant an und erreichten im Oktober 2013 mit ca. 1.300 Packungen ihren Höhepunkt (Abbildung 21).
Das veränderte Verordnungsverhalten hinsichtlich der Packungsgrößen lässt sich mit einer Änderung
der Normgrößen zum 01.07.2013 erklären. Derzeit (Lauer-Taxe Stand 01.03.2015) befindet sich nur
noch die N2 mit einem Pulverinhalator auf dem deutschen Arzneimittelmarkt, die Packung mit drei
Pulverinhalatoren ist ohne Normgrößenbezeichnung. Der Erkrankung COPD entsprechend waren es
Versicherte zwischen 50 und 80 Jahren, die dieses Atemwegsmedikament erhielten. Zum größten
Teil handelte es sich dabei um Männer (Abbildung 22). Die Verordnungen von Aclidiniumbromid
erfolgten in 45,9 Prozent von Hausärzten und in 48,4 Prozent von Pneumologen. Abbildung 23 zeigt
die Verteilung der COPD-Patienten nach der Menge verordneter Packungen Aclidiniumbromid ana-
log zu Indacaterol und Roflumilast ab der entsprechenden Erstverordnung innerhalb eines Jahres
(maximal bis Ende 2013). Hier war der Patientenanteil mit zwei bis drei Verordnungen innerhalb
eines Jahres am höchsten (knapp über 40 Prozent), gefolgt vom Anteil mit vier oder fünf verschrie-
benen Packungen.
Abbildung 21: Anzahl verordneter Packungen Aclidiniumbromid je Monat nach Packungsgrößen (2012-2013)
98
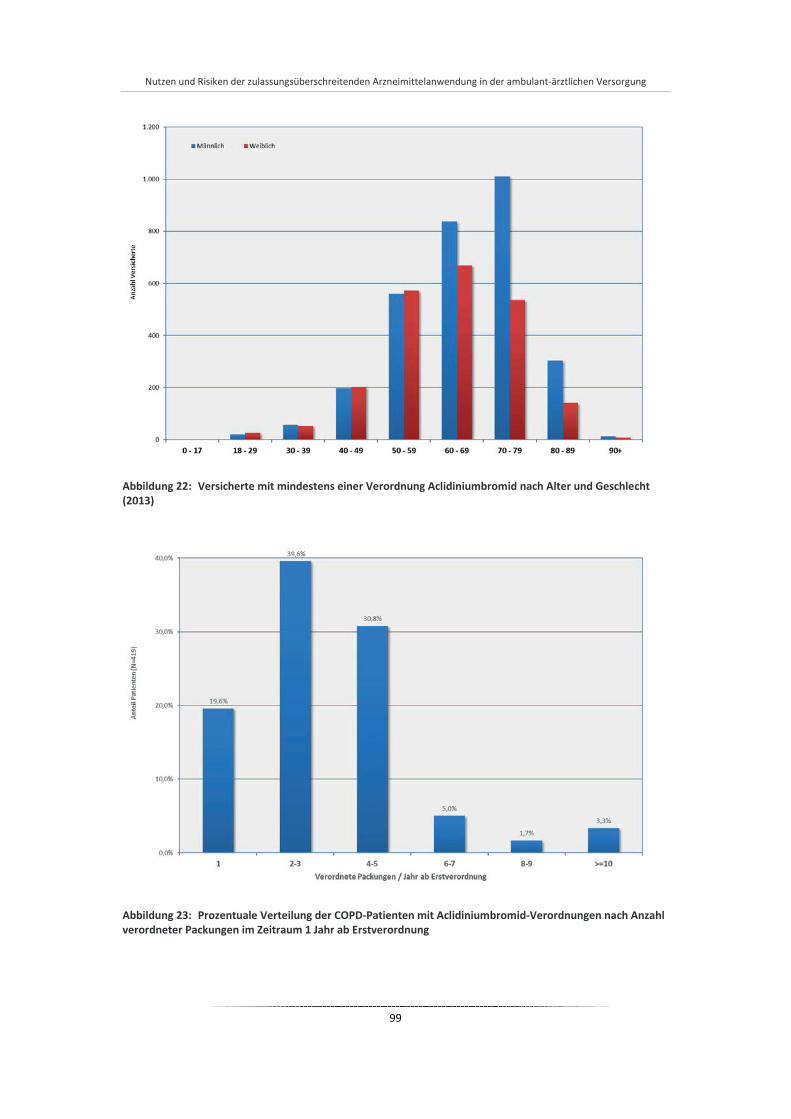
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Abbildung 22: Versicherte mit mindestens einer Verordnung Aclidiniumbromid nach Alter und Geschlecht (2013)
Abbildung 23: Prozentuale Verteilung der COPD-Patienten mit Aclidiniumbromid-Verordnungen nach Anzahl verordneter Packungen im Zeitraum 1 Jahr ab Erstverordnung
99

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.1.2.4 Diskussion der Ergebnisse
-2-Sympathomimetika sind nach wie vor die effektivsten Bronchodilatatoren – sowohl
zur Prävention als auch in der Behandlung eines akuten Anfalls einer chronisch obstruktiven Lun-
generkrankung (Ram & Sestini, 2003). Langwirksam -2-Sympathomimetika wie Salmeterol und
Formoterol mit einer Wirkdauer von bis zu zwölf Stunden werden in der Asthma-Therapie nur in
Kombination mit inhalativen Corticoiden empfohlen. Die Unsicherheit in der Datenlage führt aller-
dings nach wie vor zu kontroversen Diskussionen über den Nutzen (Cates & Cates, 2008; Castle et al.,
1993; Kuehn, 2009; Chowdhury & Dal Pan, 2010; Cates et al., 2014; Chowdhury et al., 2011). Kurz-
wirksame Anticholinergika wie beispielsweise Ipratropium wurden auch über Jahre in der Asthma-
Therapie eingesetzt (Restrepo, 2007; Meier & Jick, 1997). Ihre Anwendung wurde allerdings nicht
forciert, da sie zur Therapie einer akuten Bronchodilatation als weniger effektiv angesehen wurden
als kurzwirksam -2- -2-Sympathomimetika und inhalative Glucocorti-
coide sind bis dato die gängigsten Mittel, wenn es um die Therapie chronisch obstruktiver Lungener-
krankungen geht. Resistenzentwicklungen gegen Glucocorticoide gerade beim schweren Asthma
bronchiale stellen allerdings ein zunehmendes Problem dar, so dass die Unterscheidung in Subtypen
in der Symptomatik in Zukunft einen höheren Stellenwert einnehmen wird (Schwabe & Paffrath,
2014; Barnes, 2012).
Für die Untersuchung eines möglichen Off-Label Use wurden Arzneimittel gewählt, die ausschließlich
für die Therapie der COPD zugelassen sind. Die Begründung für diese Entscheidung liegt in der be-
schränkten Spezifität der Medikamente, die bei akuten und chronischen Erkrankungen der Atemwe-
ge eingesetzt werden. So lässt eine Verordnung aus dem ATC-Code-Bereich R03 keine zuverlässige
Zuordnung auf eine Indikation zu. Indacaterol (mit dem ATC-Code R03AC18), Roflumilast (mit dem
ATC-Code R03DX07) sowie Aclidiniumbromid (mit dem ATC-Code R03BB05) hingegen ermöglichen
aufgrund ihrer Zulassungseinschränkung eine spezifischere Zuweisung, die es möglich macht, eine
nicht zulassungskonforme Anwendung bei der Therapie eines diagnostizierten Asthma bronchiale zu
identifizieren.
Mit Indacaterol wurde die Wirkstoffgruppe der lan -2-Sympathomimetika zur Therapie
der COPD erweitert. In klinischen ILU-Studien zeigte sich eine statistisch signifikante Verbesserung
der Lungenfunktion bei COPD-Patienten gegenüber Placebo, die sich auch noch nach einem Jahr
nachweisen ließ. Der stärker patientenrelevante Endpunkt Reduzierung von Exazerbationen verbes-
serte sich hingegen nur im Placebo-, nicht jedoch im Formoterol-Vergleich. Gegenüber Tiotropium
konnte – bezogen auf Lungenfunktion und Gesundheitsstatus – eine Nicht-Unterlegenheit demons-
triert werden. In ihrer systematischen Übersichtsarbeit kamen die Autoren zum Schluss, dass In-
100

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
dacaterol insbesondere im Vergleich zu langwirksamen Bronchodilatatoren wie Formoterol Vorteile
nicht nur im Hinblick auf die Lungenfunktion, sondern auch auf Symptomatik und Lebensqualität
biete (Rodrigo & Neffen, 2012).
Die Suche in einschlägigen Datenbanken ergab mehrere Treffer, in denen der COPD-Wirkstoff auch
an Patienten mit diagnostiziertem Asthma bronchiale unterschiedlicher Genese untersucht wurde.
Auch in diesen Studien erwies sich Indacaterol im Vergleich zu Placebo als signifikant überlegen und
zeigte bei einer einmaligen täglichen Gabe einen bronchodilatatorischen Effekt über 24 Stunden. In
den primär placebokontrollierten Studien mit kleiner Fallzahl wurde der Bronchodilatator als wirk-
sam, sicher und gut toleriert bezeichnet. Veröffentlichte Studien aus der Datenbank des U.S. Natio-
nal Institutes of Health kamen zu vergleichbaren Ergebnissen bei der Bewertung des Nutzen-Risiko-
Profils. Statistische Auswertungen zu patientenrelevanten Endpunkten wurden jedoch nicht zur
Verfügung gestellt, so dass dazu keine gesicherten Aussagen getroffen werden können.
-2-Sympathomimetika zunächst für Patienten mit
Asthma bronchiale entwickelt und später – in gleicher Dosierung – auch für die Therapie der COPD
zugelassen (Chowdhury et al., 2011a). Novartis, Zulassungsinhaber von Onbrez® Breezhaler® (In-
dacaterol) fokussierte hingegen die Zulassung des Wirkstoffes ausschließlich auf die Therapie der
COPD und beantragte zwei unterschiedliche Dosierungen (150 μg und 300 μg), die durch die EMA
zugelassen, hingegen von der FDA aufgrund von Sicherheitsbedenken abgelehnt wurden (hier erfolg-
te die Zulassung schließlich für 75 μg Indacaterol) (Chowdhury et al., 2011a). -2-
Sympathomimetika stehen unter Verdacht, das Risiko von Asthma-bezogenen schwerwiegenden
unerwünschten Ereignissen, einschließlich Asthma-Todesfällen, zu erhöhen, wenn sie für die Be-
handlung dieser Erkrankung verwendet werden. Aus diesem Grund hat die amerikanische Zulas-
-2-Sympathomimetika als Monothe-
rapie bei Asthma von Kindern und Erwachsenen untersagt (FDA, 2010). Aufgrund der insgesamt
schlechten Datenlage dieser Wirkstoffgruppe spielen für den pU auch ökonomische Gründe eine
Rolle dafür, die Zulassung in einem bestimmten Indikationsgebiet erst gar nicht zu beantragen. Wie
aus der Datenbank des NIH ersichtlich, wurden Studien mit Indacaterol an Asthmatikern (allerdings
mit kleiner Fallzahl) ungefähr zur gleichen Zeit durchgeführt wie an Patienten mit diagnostizierter
COPD. Wenn bereits im Vorfeld Sicherheitsbedenken für ein Arzneimittel oder eine Wirkstoffgruppe
für die Anwendung innerhalb eines bestimmten Patientenkollektivs geäußert bzw. publiziert werden,
müssen die Fallzahlen groß genug sein, um einen zu erwartenden unerwünschten Effekt, der syste-
matisch vorhanden ist, sichtbar zu machen. Die Größe der Stichprobe korreliert dementsprechend
auch immer mit Geld und Aufwand, die der pU aufbringen muss, und erklärt, warum Indikationen
möglicherweise nicht im Fokus einer möglichen Zulassung stehen. Problematisch ist jedoch, dass in
der Praxis neben einer reinen COPD auch Mischformen (COPD + Asthma) bestehen können. So muss
101

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
grundsätzlich von einem Restrisiko ausgegangen werden, wenn dieser Wirkstoff bzw. allgemein die-
se Wirkstoffgruppe als Monotherapie verordnet wird.
Der Wirkstoff Roflumilast, zur pharmakotherapeutischen Gruppe der Phosphodiesterase-4-Hemmer
gehörend, zeigte in den Studien, die die entscheidenden Daten zur Zulassung für die Therapie der
COPD lieferten, keine klinisch relevante Verbesserung der Lungenfunktion. Ebenso konnte die mittle-
re Rate an Exazerbationen nur geringfügig gesenkt werden. Auch fünf Jahre nach seiner Marktein-
führung mangelt es noch an adäquaten Untersuchungen, die einen Zusatznutzen gegenüber der
Standardbehandlung bei schwerer COPD (bronchienerweiternde Medikamente + inhalatives Corti-
son) belegen könnten. Es liegen ebenfalls keine Vergleichsstudien mit dem Wirkstoff Theophyllin –
pharmakologisch verwandt mit Roflumilast – vor. Nach Sichtung der Studien im OLU zeigten sich
positive Effekte in der Asthma-Therapie. So konnte nicht nur die Gabe von Notfallmedikamenten
reduziert werden, Roflumilast bewirkte auch eine moderate Linderung asthmatischer Reaktionen
und in einem größeren Ausmaß eine Verbesserung Allergen-induzierter asthmatischer Spätreaktio-
nen bei Patienten mit mildem allergischen Asthma. Die OLU-Ergebnisse der neueren Studien bestä-
tigten damit die Ergebnisse aus der Übersichtsarbeit von Lipworth (2005). Als Kritikpunkt führte der
Autor an, dass es sich um keine echten randomisierten placebokontrollierten Studien handele. Statt-
dessen beruhten die Vergleiche auf einer ein- bis dreiwöchigen nicht-randomisierten, auf Placebo
basierenden Run-in-Periode – begründet mit ethischen und praktischen (immer weniger Asthmatiker
sind Corticoid-naiv) Aspekten. Dennoch zeigte sich Roflumilast so effektiv wie niedrig dosiertes inha-
latives Corticoid. Die Suche nach Studien zu Roflumilast in der Therapie des Asthma unterschiedli-
cher Genese lieferte zwar insgesamt neun Treffer in der NIH-Datenbank (alle Studien waren indus-
triefinanziert), allerdings wurden durch keine Studie Aussagen zu einem möglichen Wirksamkeitspro-
fil oder dem Risiko-Nutzen-Verhältnis publiziert. Theophyllin galt lange Zeit als effektiv in der Thera-
pie der chronisch obstruktiven Lungenerkrankungen (Asthma und COPD) und allein deshalb ist eine
Therapie mit einem PDE-4-Hemmer sowohl bei COPD als auch bei Asthma bronchiale wahrscheinlich
(Page, 2014). Dieser Wirkstoff wird allerdings nur noch in begründeten Einzelfällen als Wirkstoff der
dritten Wahl eingesetzt, da er neben zahlreichen Interaktionen eine relativ geringe, vom Alter der
Patienten abhängige therapeutische Breite besitzt. Inhalative und vom pharmakologischen Wirkprin-
zip her besser verträgliche Substanzen haben mittlerweile Vorrang in der Therapie der obstruktiven
Lungenerkrankungen.
Die Therapie des Asthma bronchiale wird vom pU in seiner Fachinformation nicht explizit ausge-
schlossen. So finden sich Hinweise dazu weder unter Punkt 4.3 (Gegenanzeigen) noch unter Punkt
4.4 (Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung) (Takeda, 2013). Allein
der Hinweis, dass es sich bei Roflumilast um einen Wirkstoff handelt, der nicht zur Therapie des
akuten Bronchospasmus indiziert ist, wird vom pU aufgegriffen. Das lässt die Frage aufkommen,
warum der Hersteller die Anwendung des Phosphodiesterase-Hemmers in der Therapie des Asthma
102

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
bronchiale nicht explizit ausschließt. Ein bestimmungswidriger Gebrauch wird auf diese Weise mög-
licherweise von den Ärzten als bestimmungsgemäß gesehen, mögliche Haftungsfragen können dann
von Seiten des pU nicht ohne Weiteres ausgeschlossen werden. Gerade aber bei Erkrankungen, die
sich von der Symptomatik her ähneln, ist ein Fehlgebrauch nicht auszuschließen. Eine Unkenntnis
über mögliche Anwendungen in der Asthma-Therapie seitens des pU kann – unabhängig vom Phar-
makovigilanz-System – allein deshalb ausgeschlossen werden, weil vom Hersteller finanzierte Stu-
dien existieren, die auf diese Thematik fokussieren. Das legt den Schluss nahe, dass der pU den OLU
und das Haftungsrisiko wissentlich in Kauf nimmt.
Aclidiniumbromid ist ein weiterer Vertreter der langwirkenden Muscarin-Rezeptorantagonisten.
Mittlerweile liegen klinische Studien vor, die die Wirkung von Aclidiniumbromid auf die Lungenfunk-
tion im Vergleich zu Placebo und Tiotropium geprüft haben. Im Direktvergleich mit Tiotropium waren
die bronchodilatatorischen Effekte der beiden Wirkstoffe ähnlich gut. Die nächtliche Lungenfunktion
konnte unter Aclidiniumbromid allerdings nur numerisch verbessert werden. Es werden allerdings
Studien mit einer größeren Patientenzahl sowie längerer Dauer benötigt, um den Nachweis dieses
Effekts zu bestärken.
Wie bei Indacaterol wird die Anwendung dieses Wirkstoffes zur Asthma-Therapie nicht vom pU emp-
fohlen. Im Gegensatz -2-Sympathomimetikum gab es für Aclidiniumbromid in den einschlägi-
gen Datenbanken zu dieser Fragestellung dementsprechend auch keine Publikation. Bedingt durch
den Zeitpunkt der Zulassung (Markteinführung nach Verpflichtung zur AMNOG-Nutzenbewertung)
ist Aclidiniumbromid von den drei untersuchten Arzneimitteln zur Behandlung der COPD der einzige
Wirkstoff, der in der Monotherapie bzw. als Einzelsubstanz einer frühen Nutzenbewertung unterzo-
gen wurde. Im Vergleich zu anderen inhalativen Bronchodilatatoren konstatierte der Gemeinsame
Bundesausschuss in seinem Beschluss über die Nutzenbewertung von Arzneimitteln mit neuen Wirk-
stoffen nach § 35a SGB V Aclidiniumbromid keinen Zusatznutzen. In seinem Dossier legte der Her-
steller keine belastbaren Ergebnisse vor. So fehlten für den direkten Vergleich Studien mit einer
ausreichenden Studiendauer, der indirekte Vergleich ließ hingegen eine valide Bewertung des Zu-
satznutzens aufgrund mangelhafter Qualität nicht zu.
Grundsätzlich gilt bei der Recherche nach Publikationen, dass es aufgrund der Wahl der Literaturda-
tenbank, der Suchstrategie und auch der Ausschlusskriterien möglich ist, Veröffentlichungen nicht zu
erfassen. Auch eine zusätzliche Suche in der Datenbank des U.S. National Institutes of Health kann
keinen Anspruch auf Vollständigkeit garantieren. Dennoch lassen sich in der Literatur – überwiegend
industriefinanzierte – Studien auffinden, in denen die beiden Wirkstoffe Indacaterol und Roflumilast
auch in der Asthma-Therapie eingesetzt wurden.
Zu bedenken gilt, dass nicht alle klinischen Studien der Öffentlichkeit zugänglich gemacht werden,
unabhängig von ihren Ergebnissen. So zeigen Untersuchungen, dass teilweise mehr als die Hälfte der
103

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
industriefinanzierten Studien, die für die Zulassung eines Arzneimittels durchgeführt werden, unver-
öffentlicht bleiben (Schott et al., 2010). Außerdem konnten diese Untersuchungen Mehrfach-
Publikationen derselben Ergebnisse, selektives Publizieren von ausgewählten Daten einer Studie
sowie das Zurückhalten von Daten zu UAW identifizieren (Schott et al., 2010).
Das Veröffentlichen von Studienergebnissen in Abhängigkeit vom „gewünschten“ Ergebnis stellt
hinsichtlich der Patientenversorgung tatsächlich ein relevantes Problem dar. Das Zurückhalten von
Erkenntnissen aus klinischen Studien ebenso wie das selektive Publizieren „guter Studienergebnisse“
kann zu einer Fehleinschätzung des Nutzen-Risiko-Verhältnisses führen (Turner et al., 2008; Hart et
al., 2012) und so auch indirekt einen OLU fördern. Auch wird durch die Verzerrung der Ergebnisse
das Planen und Durchführen weiterer klinischer Studien erschwert, da die auf Publikationen basie-
rende, angenommene (Wirksamkeit, Sicherheit und Unbedenklichkeit einer Substanz) möglicher-
weise nicht der eigentlichen Ausgangssituation entspricht. Studien mit ein und derselben Fragestel-
lung werden so möglicherweise aufgrund mangelhafter oder fehlender Information mehrfach durch-
geführt, da Ergebnisse aus vorherigen Untersuchungen nicht veröffentlicht wurden. Gerade dieser
Punkt stellt auch ein nicht zu unterschätzendes ethisches Problem dar, weil nicht ausgeschlossen
werden kann, dass Patienten mit schweren und schwerwiegenden Nebenwirkungen belastet wer-
den, die schon in anderen Untersuchungen zu Studienabbrüchen geführt hatten. Bei der Ergebnisin-
terpretation der zum OLU aufgefunden Studien zur Anwendung der COPD-Therapeutika müssen
diese Punkte berücksichtigt werden.
Um den sogenannten Publication-Bias zu verhindern oder zumindest zu minimieren, wurde im Jahr
1999 das ClinicalTrials.gov-Register eingerichtet, in dem klinische Studien der Phase I-IV erfasst wer-
den sollen. Seit 2007 besteht für die USA eine gesetzliche Registrierungspflicht von klinischen Stu-
dien, verbunden mit der Aufforderung, die Ergebnisse innerhalb eines Jahres nach Abschluss der
Studie zu publizieren (FDA, 2014). Trotz der gesetzlichen Vorgabe wird diese Aufforderung nur unzu-
reichend befolgt. So zeigte sich, dass bei etwa 75 Prozent der Studien die Ergebnisse nicht veröffent-
licht wurden (Gill, 2012) oder die Ein-Jahres-Frist nicht eingehalten wurde (78 Prozent) (Prayle et al.,
2012). Auffällig ist auch, dass die Ergebnisse aus frühen Phase-I/II-Studien seltener publiziert werden
als aus Phase-III-Studien (Anderson et al., 2015). Mit EudraCT gibt es ein vergleichbares Register auf
europäischer Ebene. In diesem Register werden alle Studien erfasst, die seit Mai 2004 in der europä-
ischen Union durchgeführt werden. Die Tatsache, dass nur Zulassungsbehörden und Studiensponso-
ren Zugriff auf die Daten erhielten, wurde kritisiert und führte letztendlich dazu, dass Anfang des
Jahres 2011 das EU Clinical Trials Register auch für die Allgemeinheit freigeschaltet wurde (Wieseler,
2010).
Selbst wenn also die Registrierung klinischer Studien mittlerweile akzeptiert worden ist, wird die
Diskussion um die Veröffentlichung von Studienergebnissen vorerst andauern. Demnach ist noch
nicht wirklich geklärt, ob ein pU verpflichtet werden kann, seine Ergebnisse zu publizieren und wenn
104

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
ja, in welchem Umfang dies erfolgen muss (Wieseler, 2010). Die Wahrung des geistigen Eigentums
kann schlussendlich dazu führen, dass der Nutzen des neuen Arzneimittels über-, potentielle Risiken
hingegen unterschätzt werden. Problematisch an diesen Registern ist außerdem, dass nicht über-
prüft werden kann, ob die Studienregistrierung eines pharmazeutischen Unternehmens wirklich in
vollem Umfang erfolgte. Unabhängig davon sagt eine Registrierung nichts über Quantität und Quali-
tät der Daten aus.
Weitreichende Folgen hat ein Publication-Bias auch deshalb, weil nicht ausgeschlossen werden kann,
dass die veröffentlichten Ergebnisse – als Grundlage für die Erstellung von Reviews und Meta-
Analysen – auch in Therapieempfehlungen der Fachgesellschaften einfließen. Bekanntes Beispiel ist
hier das Antiepileptikum Gabapentin: durch Veränderungen des primären Endpunktes bzw. Nicht-
Veröffentlichung ungünstiger Daten erweckte der pU den Eindruck u.a. einer Wirksamkeit des Arz-
neimittels bei neuropathischen Schmerzen (Vedula et al., 2009). Das verzerrte Ergebnis der darge-
stellten Gabapentin-Wirksamkeit spiegelte sich schlussendlich in unterschiedlichen Beurteilungen in
Übersichtsarbeiten und Leitlinien wider (Schott et al., 2010).
Die Forderung, Studienergebnisse nur zu veröffentlichen, wenn der Zugang zu allen Daten gewähr-
leistet wird (Barbui & Cipriani, 2007), muss insofern kritisch hinterfragt werden, weil dadurch die
Motivation, zu publizieren, möglicherweise noch weiter absinkt.
Auch die Forderung, klinische Studien vermehrt durch öffentliche Gelder zu finanzieren – insbeson-
dere dann, wenn es um die Erfassung von Arzneimittelrisiken geht (Avorn, 2006) – kann kontrovers
diskutiert werden. Für eine industrieunabhängige Darstellung der Studiendaten spricht die Notwen-
digkeit, eine möglichst objektive Darstellung der Ergebnisse publizieren zu können, da sie nicht durch
ökonomische Interessen beeinflusst wird. Auf der anderen Seite könnte dies aber auch als Signal an
den pU missverstanden werden, von seiner Verantwortung entlastet zu werden, Ergebnis-treu statt
Ergebnis-geleitet zu publizieren.
Festzuhalten ist, dass publizierte Daten ebenso wie unpublizierte Daten einen OLU fördern können,
entweder offensichtlich durch (falsch-)positive Ergebnisdarstellungen motiviert oder indirekt durch
das Zurückhalten von wichtigen Informationen. So werden Studien, die Forschungsinteressen auch
im OLU berücksichtigen, zwar publiziert, eine Bewertung dieser Studien erweist sich jedoch vielfach
als schwierig.
Hinweise, dass anscheinend COPD-Wirkstoffe auch im OLU bei Asthmatikern eingesetzt werden,
ergaben sich nicht nur durch die Sichtung von Publikationen, sondern zeigten sich auch bei der Aus-
wertung von Routinedaten, mit denen das Verordnungsverhalten in der ambulant-ärztlichen Praxis
dargestellt werden konnte. Bei der Interpretation der ambulanten Diagnosedaten muss allerdings
berücksichtigt werden, dass sie in erster Linie zum Zweck der ambulanten bzw. stationären Abrech-
nung generiert werden. Bei Routinedaten handelt es sich somit immer um Informationssammlungen,
105

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
die im Rahmen der Leistungserbringung bzw. Kostenerstattung anfallen (Windt, 2010). Die Daten
wurden nicht speziell für die benötigten Auswertungen erhoben, was in der Ergebnisinterpretation
von Routinedatenanalysen berücksichtigt werden muss (Hoffmann, 2008). Andererseits stellen Kran-
kenkassenroutinedaten eine wichtige Informationsquelle dar, wenn es um patienten- oder arztbezo-
gene Arzneimittelverbrauchs- bzw. Versorgungsstudien geht (Windt, 2010).
Für die COPD-Jahresprävalenz in der TK-Population konnte im Rahmen der vorliegenden Arbeit auf
der Basis von ambulanten Diagnosedaten über alle Altersgruppen hinweg Werte von 3,7 Prozent
(Untersuchungszeitraum 2010 und 2011) ermittelt werden. Dieser Wert liegt damit deutlich unter
dem der GEDA 2012 (8,6 Prozent), die die Lebenszeitprävalenz einer chronischen Bronchitis in der
erwachsenen Bevölkerung erfasste. Zwar bestärken die Daten aus der TK-Population damit die Aus-
sage, dass sich nicht aus jeder chronischen Bronchitis zwangsläufig auch eine COPD entwickeln muss.
Andererseits ist eine Jahresprävalenz immer niedriger als eine Lebenszeitprävalenz, die aus Primär-
daten erhoben wird. Für diese Analyse19 wurden alle Diagnosecodierungen in den Jahren 2010 und
2011 (Indacaterol und Roflumilast) mit dem Diagnosekennzeichen „G“ für „gesicherte Diagnose“
berücksichtigt und es mussten im entsprechenden Jahr mindestens zwei verschiedene Abrechnungs-
quartale mit der jeweiligen Diagnose vorliegen, damit die Versicherten einer Krankheit zugeordnet
werden konnten. Die meisten Verordnungen von Indacaterol, Roflumilast und Aclidiniumbromid
erfolgten in der Altersgruppe der 60- bis 80-Jährigen. Die Verschreibung auch in jüngeren Alters-
gruppen lässt jedoch nicht grundsätzlich den Schluss zu, dass sie fehldiagnostiziert bzw. fehlcodiert
worden sind. Neuere Untersuchungen zeigen, dass die spätere Entwicklung einer COPD möglicher-
weise schon ihren Ursprung in der Kindheit erfährt (Chan et al., 2015; Postma et al., 2015), was mög-
licherweise auch Auswirkungen auf einen früheren Eintritt in das Erkrankungsalter hat.
Der prozentual höhere Verordnungsanteil von Indacaterol bei Asthmatikern durch Hausärzte kann
eventuell mit den in Abschnitt 8.1.2 bereits beschriebenen Defiziten zusammenhängen. Ursachen
für eine Indikations-überschreitende Verordnung können so neben einer mangelhaften Diagnostik
aufgrund von Qualifikationsdefiziten in der Weiter- und Fortbildung bei den behandelnden Ärzten
auch auf strukturelle Defizite hinweisen, da Lungenfachärzte nicht in ausreichender Zahl flächende-
ckend zur Verfügung stehen und der betroffene Patient somit eher seinen Hausarzt anstelle eines
Facharztes aufsucht. Generell legen die vorliegenden Ergebnisse aber trotz der genannten Ein-
schränkungen der Routinedaten-Analyse einen OLU der drei Wirkstoffe Indacaterol, Roflumilast und
Aclidiniumbromid nahe. Dies gilt nicht nur für codierte Asthmatiker, sondern auch für unklare Dia-
gnosestellungen. Möglich ist aber auch, dass bei der Codierung Fehler unterlaufen sind. So kann
einerseits neben der COPD auch ein Asthma bronchiale bestehen, andererseits kann nicht ausge-
schlossen werden, dass langanhaltendes Asthma durch Remodelling-Vorgänge in eine COPD über-
19 Für den Wirkstoff Aclidiniumbromid wurden seitens der TK keine Diagnosedaten bereitgestellt.
106

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
geht. Frühere Untersuchungen zeigen, dass Codierungen in Routinedaten oft fehlerbehaftet sind
(Gerste & Gutschmidt, 2006). Bei der Interpretation dieser Daten muss dementsprechend auch eine
fehlerbehaftete Codierung in Betracht gezogen werden. Möglicherweise sind also die Patienten mit
einer Asthma-Codierung keine „reinen“ Asthmatiker.
Ein Diagnose-Bias aufgrund falscher ICD-Codierungen kann aber durch die alleinige Betrachtung der
Routinedaten nicht ermittelt werden, da klinische Daten in den Abrechnungsdaten fehlen. Auch
lassen die Auswertungen der Routinedaten keine Rückschlüsse zu, ob die richtige Diagnose gestellt
und das falsche Medikament verordnet oder – im umgekehrten Fall – möglicherweise eine falsche
Diagnose gestellt und mit dem richtigen Arzneimittel therapiert wurde. So wie sich Ärzte also nicht
grundsätzlich darauf verlassen können, dass ihnen die medizinische Literatur valide und verlässliche
Informationen bietet, kann sich auch die GKV nicht nur auf die Analyse von Routinedaten beziehen,
wenn es um mögliche Regressansprüche im Fall einer zulassungsüberschreitenden Anwendung geht.
Die Ergänzung um zusätzliche Patientendaten aus der ärztlichen Praxis wäre in diesem Fall unver-
zichtbar.
107

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.2 Off-Label Use aufgrund fehlender Therapieoptionen am
Beispiel der chronischen Hepatitis C
Während im vorausgegangenen Kapitel (Kapitel 8.1) der OLU aufgrund struktureller Defizite und
fehlerhafter Diagnosen am Beispiel von Asthma bronchiale und COPD diskutiert wurde, befasst sich
das folgende Kapitel (Kapitel 8.2) mit den Ursachen eines OLU aufgrund fehlender Therapieoptionen
und möglicher administrativer Defizite am Beispiel der chronischen Hepatitis C.
Bei der Hepatitis C handelt es sich um eine durch das Hepatitis C-Virus (HCV) verursachte Entzün-
dung des Leberparenchyms. Die ausgeprägte genetische Variabilität ist auf eine hohe Replikationsra-
te des HCV zurückzuführen. Mittlerweile sind sechs Genotypen (GT1-6) mit einer größeren unter-
schiedlichen Zahl von Subtypen bekannt (Simmonds et al., 2005). Während GT4 vorwiegend in Nord-
afrika sowie im mittleren Osten und GT5 und GT6 vor allem in Südafrika und Ostasien identifiziert
werden, finden sich in Deutschland insbesondere die Genotypen 1 bis 3 (Wedemeyer et al., 2012;
Cornberg et al., 2014).
Mit der Markteinführung von neuen, direkt antiviral wirkenden Substanzen (DAA) in den Jahren 2014
und 2015 haben sich auch die Therapieempfehlungen geändert. So ergibt sich mit dem erweiterten
Behandlungsspektrum der HCV-Infektionen erstmals auch eine Interferon-freie Therapie. Allerdings
ist der Erfolg einer Therapie u.a. vom identifizierten Genotyp abhängig, so dass nicht alle Patienten
von der Zulassung der neuen DAA profitieren.
Nach den Ausführungen zur epidemiologischen sowie sozialökonomischen Bedeutung der chroni-
schen Hepatitis C (cHC), werden nachfolgend Leitlinienempfehlungen herausgearbeitet und mit dem
Zulassungsstatus der neuen Wirkstoffe zur Therapie der cHC verglichen. Dabei wird der Frage nach-
gegangen, ob Fachgesellschaften auch bei Wirkstoffen, die erst kürzlich den Marktzugang erhalten
haben, eine Empfehlung für ihre zulassungsüberschreitende Anwendung aussprechen. Mögliche
Ursachen dafür sollen identifiziert und anschließend diskutiert werden. Darauf aufbauend widmet
sich der dritte Teil den aus den Jahren 2011 bis 2015 zugelassenen Wirkstoffen zur Therapie der cHC.
Zu diesen zählen die Protease-Inhibitoren Boceprevir, Telaprevir und Simeprevir, die NS5A-
Inhibitoren Daclatasvir und Ledipasvir, der nukleosidische Polymerase (NS5B)-Inhibitor Sofosbuvir,
der nicht-nukleosidische Polymeraseinhibitor Dasabuvir sowie die Fixkombination zweier Wirkstoff-
klassen, bestehend aus Ombitasvir und dem Ritonavir-geboosterten Paritaprevir. In diesem Teil der
Arbeit soll der Frage nachgegangen werden, ob Forschungsfragen und ihre Ergebnisse aus Zulas-
108

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
sungsstudien mit der späteren Zulassung übereinstimmen. Mögliche Abweichungen sollen aufgezeigt
und im Anschluss diskutiert werden. Diese Untersuchung verfolgt das Ziel, die Kriterien der Zulas-
sungsbehörden transparenter zu gestalten. Ihre Entscheidungen haben unmittelbare Auswirkungen
auf die Versorgungsstruktur der Bevölkerung und ihren rechtlichen Konsequenzen.
8.2.1 Hepatitis C: Definition, Epidemiologie und sozialökonomische Aspekte
Nach Infizierung mit dem Virus kann der Krankheitsverlauf der Hepatitis C in zwei Phasen eingeteilt
werden. Die akute Hepatitis C verläuft nach einer Inkubationszeit (in der Regel 2-26 Wochen) meist
asymptomatisch, so dass diese Phase häufig nicht diagnostiziert wird (Thimme et al., 2014). Lediglich
ein Fünftel der infizierten Personen entwickeln grippeartige Symptome, rechtsseitige Beschwerden
im Oberbauch, Appetitlosigkeit sowie Müdigkeit und Abgeschlagenheit. Mit einem Ikterus oder einer
Transaminaseerhöhung auf > 400 IU/L reagieren 25 Prozent. Symptomatisch verlaufende akute He-
patitiden zeigen eine Immunreaktion der Betroffenen an, was die Spontanheilungsrate (ca. 50 Pro-
zent) erhöht, während es bei den asymptomatischen Verläufen lediglich ca. 30 Prozent sind. Auf-
grund einer hohen Chronifizierungsrate (50-85 Prozent) entwickelt sich die akute Hepatitis C in den
meisten Fällen zu einer dauerhaften Erkrankung, die langfristig zu einer Leberfibrose führen kann.
Etwa 2-35 Prozent der betroffenen Personen mit diagnostizierter chronischer Hepatitis C-Erkrankung
entwickeln innerhalb von 20 bis 30 Jahren eine Leberzirrhose, aus der bei ca. zwei bis vier Prozent
der Patienten ein Leberzellkarzinom entstehen kann (RKI, 2014a). Auch wenn das Fortschreiten der
Leberschädigung individuell sehr variabel ist, gibt es dennoch Faktoren, die den Verlauf ungünstig
beeinflussen und die Zirrhoseentstehung fördern. Zu diesen Faktoren zählen unter anderem ein
höheres Lebensalter der Patienten zum Infektionszeitpunkt, männliches Geschlecht, HIV-Koinfektion
sowie ein übermäßiger Konsum von Alkohol. Genetische Grundlagen, die das Fortschreiten der Le-
berschädigung beeinflussen, werden zunehmend erforscht (Iwata et al., 2011).
Vor der Einführung der Untersuchung von Blutkonserven auf Hepatitis C-Antikörper waren Bluttrans-
fusionen eine bedeutende Infektionsquelle. Einen wichtigen Schritt zur Verhinderung von Virusüber-
tragungen durch Blutkomponenten leitete das Paul-Ehrlich-Institut (PEI) ein: so dürfen seit dem 1.
April 1999 Konzentrate von Blutplättchen (Thrombozyten) nur noch aus Blutspenden hergestellt
werden, die mittels Nukleinsäure-Amplifikations-Technik (NAT) einen negativen Nachweis des HCV
erbringen (PEI, 1998). Bis zur Einführung der NAT bestand für zelluläre Präparate (Erythrozyten und
Thrombozyten) immer noch ein Restrisiko eines falsch-negativen oder nicht identifizierten Virus-
Nachweises (PEI, 1998). Seit dem Einsatz gentechnologisch hergestellter Blutbestandteile gilt das
109
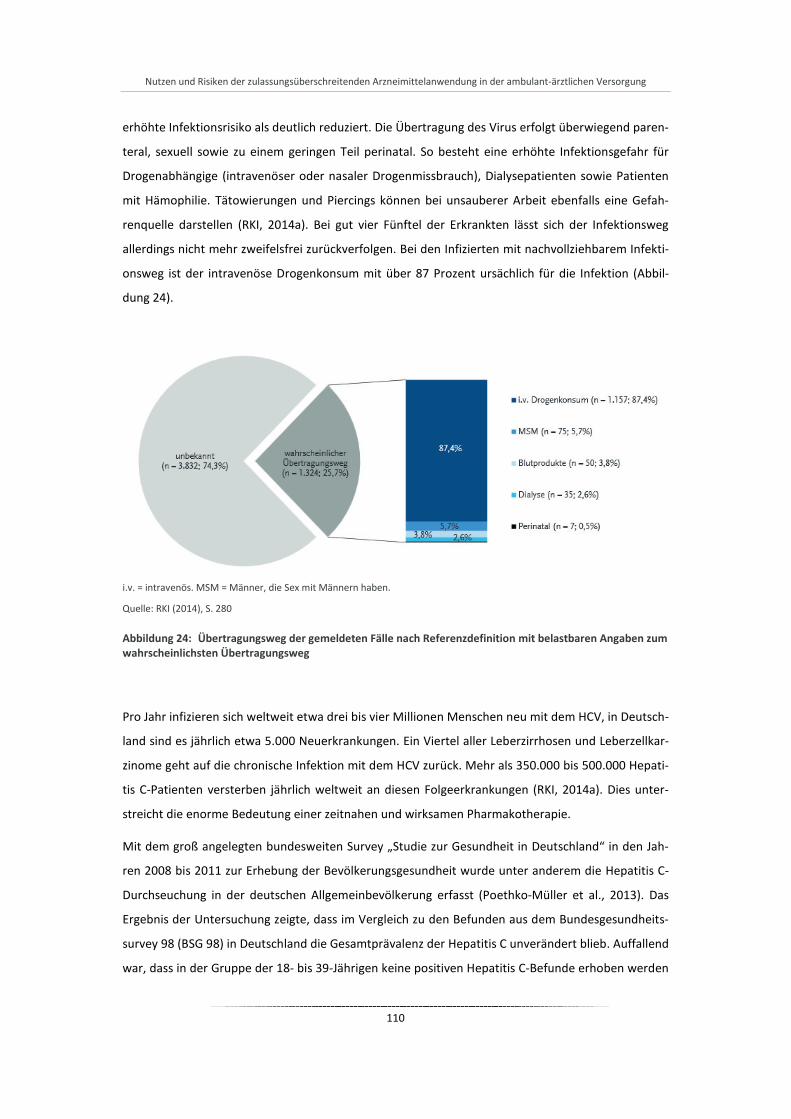
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
erhöhte Infektionsrisiko als deutlich reduziert. Die Übertragung des Virus erfolgt überwiegend paren-
teral, sexuell sowie zu einem geringen Teil perinatal. So besteht eine erhöhte Infektionsgefahr für
Drogenabhängige (intravenöser oder nasaler Drogenmissbrauch), Dialysepatienten sowie Patienten
mit Hämophilie. Tätowierungen und Piercings können bei unsauberer Arbeit ebenfalls eine Gefah-
renquelle darstellen (RKI, 2014a). Bei gut vier Fünftel der Erkrankten lässt sich der Infektionsweg
allerdings nicht mehr zweifelsfrei zurückverfolgen. Bei den Infizierten mit nachvollziehbarem Infekti-
onsweg ist der intravenöse Drogenkonsum mit über 87 Prozent ursächlich für die Infektion (Abbil-
dung 24).
i.v. = intravenös. MSM = Männer, die Sex mit Männern haben.
Quelle: RKI (2014), S. 280
Abbildung 24: Übertragungsweg der gemeldeten Fälle nach Referenzdefinition mit belastbaren Angaben zum wahrscheinlichsten Übertragungsweg
Pro Jahr infizieren sich weltweit etwa drei bis vier Millionen Menschen neu mit dem HCV, in Deutsch-
land sind es jährlich etwa 5.000 Neuerkrankungen. Ein Viertel aller Leberzirrhosen und Leberzellkar-
zinome geht auf die chronische Infektion mit dem HCV zurück. Mehr als 350.000 bis 500.000 Hepati-
tis C-Patienten versterben jährlich weltweit an diesen Folgeerkrankungen (RKI, 2014a). Dies unter-
streicht die enorme Bedeutung einer zeitnahen und wirksamen Pharmakotherapie.
Mit dem groß angelegten bundesweiten Survey „Studie zur Gesundheit in Deutschland“ in den Jah-
ren 2008 bis 2011 zur Erhebung der Bevölkerungsgesundheit wurde unter anderem die Hepatitis C-
Durchseuchung in der deutschen Allgemeinbevölkerung erfasst (Poethko-Müller et al., 2013). Das
Ergebnis der Untersuchung zeigte, dass im Vergleich zu den Befunden aus dem Bundesgesundheits-
survey 98 (BSG 98) in Deutschland die Gesamtprävalenz der Hepatitis C unverändert blieb. Auffallend
war, dass in der Gruppe der 18- bis 39-Jährigen keine positiven Hepatitis C-Befunde erhoben werden
110
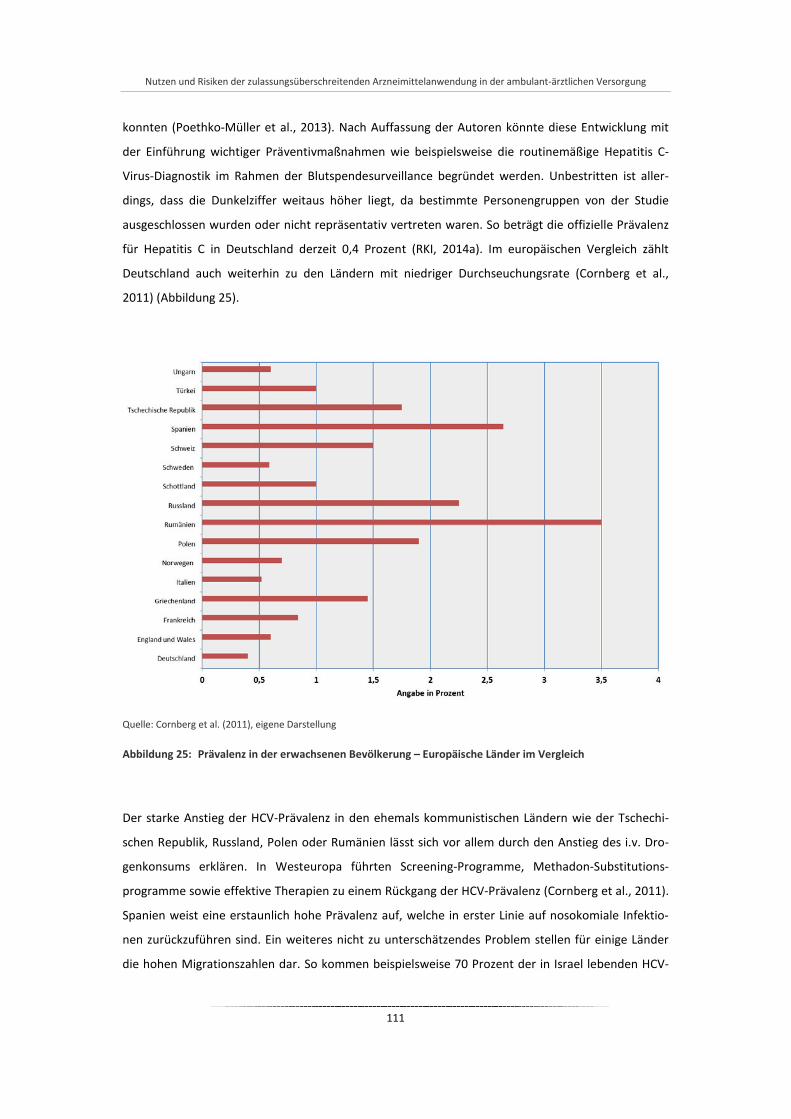
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
konnten (Poethko-Müller et al., 2013). Nach Auffassung der Autoren könnte diese Entwicklung mit
der Einführung wichtiger Präventivmaßnahmen wie beispielsweise die routinemäßige Hepatitis C-
Virus-Diagnostik im Rahmen der Blutspendesurveillance begründet werden. Unbestritten ist aller-
dings, dass die Dunkelziffer weitaus höher liegt, da bestimmte Personengruppen von der Studie
ausgeschlossen wurden oder nicht repräsentativ vertreten waren. So beträgt die offizielle Prävalenz
für Hepatitis C in Deutschland derzeit 0,4 Prozent (RKI, 2014a). Im europäischen Vergleich zählt
Deutschland auch weiterhin zu den Ländern mit niedriger Durchseuchungsrate (Cornberg et al.,
2011) (Abbildung 25).
Quelle: Cornberg et al. (2011), eigene Darstellung
Abbildung 25: Prävalenz in der erwachsenen Bevölkerung – Europäische Länder im Vergleich
Der starke Anstieg der HCV-Prävalenz in den ehemals kommunistischen Ländern wie der Tschechi-
schen Republik, Russland, Polen oder Rumänien lässt sich vor allem durch den Anstieg des i.v. Dro-
genkonsums erklären. In Westeuropa führten Screening-Programme, Methadon-Substitutions-
programme sowie effektive Therapien zu einem Rückgang der HCV-Prävalenz (Cornberg et al., 2011).
Spanien weist eine erstaunlich hohe Prävalenz auf, welche in erster Linie auf nosokomiale Infektio-
nen zurückzuführen sind. Ein weiteres nicht zu unterschätzendes Problem stellen für einige Länder
die hohen Migrationszahlen dar. So kommen beispielsweise 70 Prozent der in Israel lebenden HCV-
111
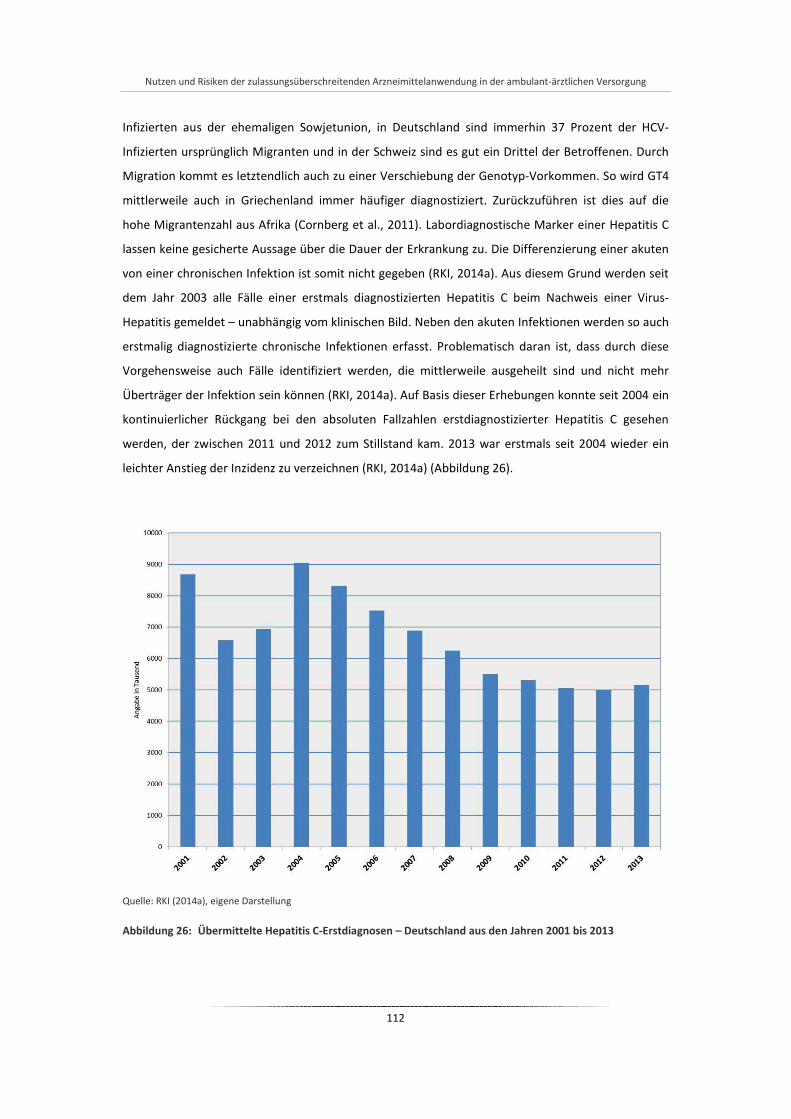
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Infizierten aus der ehemaligen Sowjetunion, in Deutschland sind immerhin 37 Prozent der HCV-
Infizierten ursprünglich Migranten und in der Schweiz sind es gut ein Drittel der Betroffenen. Durch
Migration kommt es letztendlich auch zu einer Verschiebung der Genotyp-Vorkommen. So wird GT4
mittlerweile auch in Griechenland immer häufiger diagnostiziert. Zurückzuführen ist dies auf die
hohe Migrantenzahl aus Afrika (Cornberg et al., 2011). Labordiagnostische Marker einer Hepatitis C
lassen keine gesicherte Aussage über die Dauer der Erkrankung zu. Die Differenzierung einer akuten
von einer chronischen Infektion ist somit nicht gegeben (RKI, 2014a). Aus diesem Grund werden seit
dem Jahr 2003 alle Fälle einer erstmals diagnostizierten Hepatitis C beim Nachweis einer Virus-
Hepatitis gemeldet – unabhängig vom klinischen Bild. Neben den akuten Infektionen werden so auch
erstmalig diagnostizierte chronische Infektionen erfasst. Problematisch daran ist, dass durch diese
Vorgehensweise auch Fälle identifiziert werden, die mittlerweile ausgeheilt sind und nicht mehr
Überträger der Infektion sein können (RKI, 2014a). Auf Basis dieser Erhebungen konnte seit 2004 ein
kontinuierlicher Rückgang bei den absoluten Fallzahlen erstdiagnostizierter Hepatitis C gesehen
werden, der zwischen 2011 und 2012 zum Stillstand kam. 2013 war erstmals seit 2004 wieder ein
leichter Anstieg der Inzidenz zu verzeichnen (RKI, 2014a) (Abbildung 26).
Quelle: RKI (2014a), eigene Darstellung
Abbildung 26: Übermittelte Hepatitis C-Erstdiagnosen – Deutschland aus den Jahren 2001 bis 2013
112
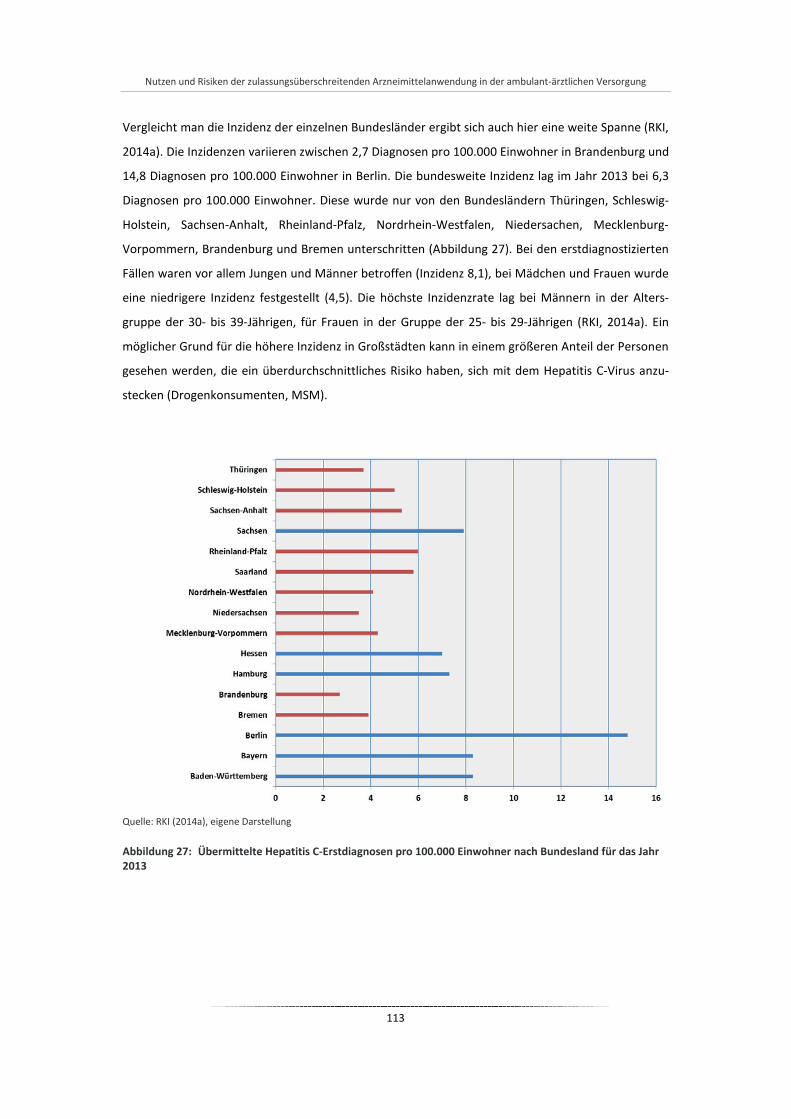
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Vergleicht man die Inzidenz der einzelnen Bundesländer ergibt sich auch hier eine weite Spanne (RKI,
2014a). Die Inzidenzen variieren zwischen 2,7 Diagnosen pro 100.000 Einwohner in Brandenburg und
14,8 Diagnosen pro 100.000 Einwohner in Berlin. Die bundesweite Inzidenz lag im Jahr 2013 bei 6,3
Diagnosen pro 100.000 Einwohner. Diese wurde nur von den Bundesländern Thüringen, Schleswig-
Holstein, Sachsen-Anhalt, Rheinland-Pfalz, Nordrhein-Westfalen, Niedersachen, Mecklenburg-
Vorpommern, Brandenburg und Bremen unterschritten (Abbildung 27). Bei den erstdiagnostizierten
Fällen waren vor allem Jungen und Männer betroffen (Inzidenz 8,1), bei Mädchen und Frauen wurde
eine niedrigere Inzidenz festgestellt (4,5). Die höchste Inzidenzrate lag bei Männern in der Alters-
gruppe der 30- bis 39-Jährigen, für Frauen in der Gruppe der 25- bis 29-Jährigen (RKI, 2014a). Ein
möglicher Grund für die höhere Inzidenz in Großstädten kann in einem größeren Anteil der Personen
gesehen werden, die ein überdurchschnittliches Risiko haben, sich mit dem Hepatitis C-Virus anzu-
stecken (Drogenkonsumenten, MSM).
Quelle: RKI (2014a), eigene Darstellung
Abbildung 27: Übermittelte Hepatitis C-Erstdiagnosen pro 100.000 Einwohner nach Bundesland für das Jahr 2013
113

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Aufgrund der hohen Chronifizierungswahrscheinlichkeit, der schwerwiegenden Folgeerkrankungen
und der hohen Kosten der antiviralen Therapie besitzt die Hepatitis C auch eine enorme gesund-
heitsökonomische Relevanz. Dabei umfasst das Spektrum der direkten Krankheitskosten die Kosten
für Präventivmaßnahmen sowie Kosten für ambulante und stationäre Behandlungen, Pflege und
Rehabilitation. Häufig können diese jedoch nicht beziffert werden, da differenzierte Daten für die
Hepatitis C fehlen. Belastbare Daten zu Morbidität, Mortalität und Krankheitslast von Hepatitis C-
Infektionen und ihren Folgeerkrankungen in der Allgemeinbevölkerung sowie in besonders betroffe-
nen Gruppen werden benötigt (RKI, 2014a). Das RKI plant daher eine Bestandsaufnahme der aktuel-
len Datenlage in Deutschland. Dabei soll zwischen der Allgemeinbevölkerung und besonders be-
troffenen Gruppen differenziert werden. Ein etwaiger Forschungsbedarf soll identifiziert und eine
Empfehlung für die nächsten Schritte gegeben werden.
Der dominierende Kostenfaktor einer HCV-Therapie ist allerdings nach wie vor die pharmakologische
Behandlung (Stahmeyer et al., 2013). Durch die Zulassung von Sofosbuvir als ersten nukleosidischen
Polymerase (NS5B)-Inhibitor im Januar 2014 sowie weiteren neuen antiviralen Mitteln zur Therapie
der cHC hat sich die Debatte um die Kosten verschärft. Die hohen Preise dieser neuen Arzneimittel
werfen die Frage auf, inwiefern die Behandlung einer einzigen Krankheit die GKV vor neue finanzielle
Probleme stellen darf. Während beim bisherigen, konventionellen Therapieansatz mit der dualen
Standard-Therapie, bestehend aus PEG- bavirin, die Kosten der Behandlung je
nach Dauer der antiviralen Therapie 7.800 bis 35.000 Euro betrugen (Stahmeyer et al., 2013), wer-
den durch die neuen Arzneimittel die Kosten erheblich steigen. Tabelle 11 stellt zusammenfassend
den Umsatz und Absatz des deutschen Apothekenmarktes der bis zu dem Zeitpunkt der Datenerfas-
sung (Stand: Dezember 2014) verfügbaren neuen antiviralen Mittel zur Therapie der cHC dar. Auffal-
lend sind dabei die enormen Umsatz- und Absatzeinbußen für Boceprevir und Telaprevir nach
Markteinführung der neuen DAA.
Allerdings profitieren nicht alle Patienten gleichermaßen von den neuen Wirkstoffen. Vor diesem
Hintergrund ist eine Betrachtung der Behandlungskosten nur in Bezug auf die hierfür in Frage kom-
menden Patientengruppen sinnvoll.
114
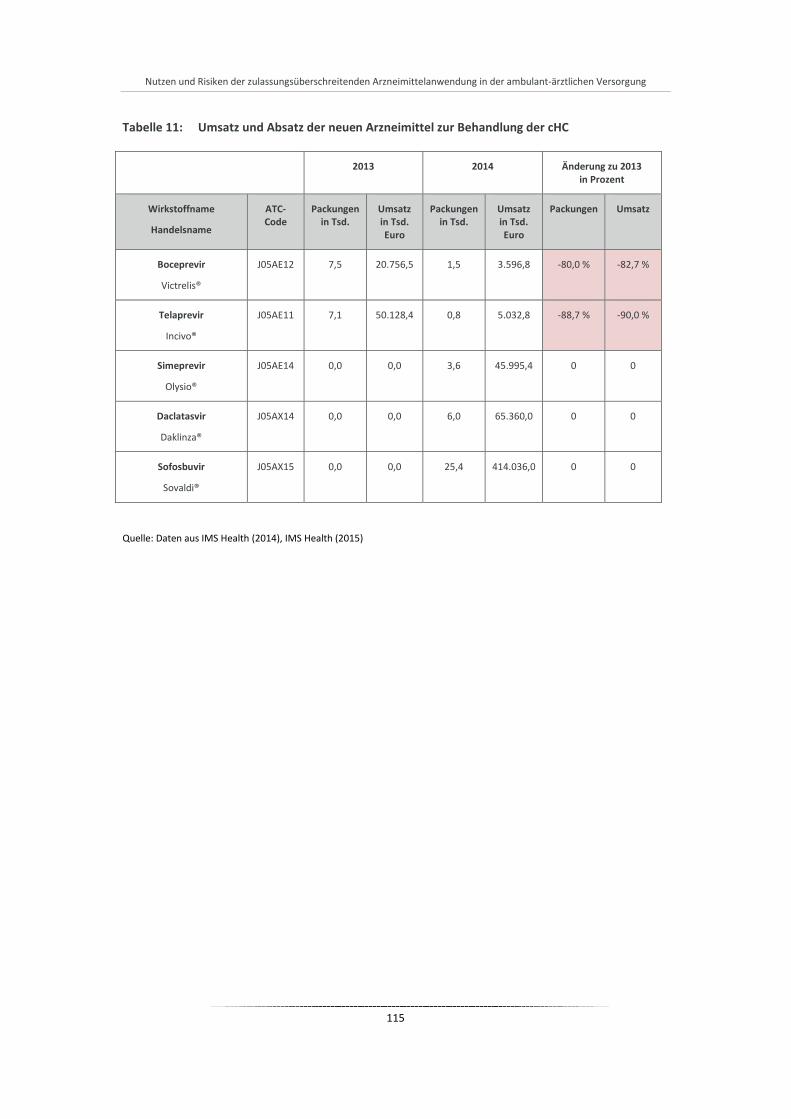
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 11: Umsatz und Absatz der neuen Arzneimittel zur Behandlung der cHC
2013 2014 Änderung zu 2013 in Prozent
Wirkstoffname
Handelsname
ATC-Code
Packungen in Tsd.
Umsatz in Tsd. Euro
Packungen in Tsd.
Umsatz in Tsd. Euro
Packungen Umsatz
Boceprevir
Victrelis®
J05AE12 7,5 20.756,5 1,5 3.596,8 -80,0 % -82,7 %
Telaprevir
Incivo®
J05AE11 7,1 50.128,4 0,8 5.032,8 -88,7 % -90,0 %
Simeprevir
Olysio®
J05AE14 0,0 0,0 3,6 45.995,4 0 0
Daclatasvir
Daklinza®
J05AX14 0,0 0,0 6,0 65.360,0 0 0
Sofosbuvir
Sovaldi®
J05AX15 0,0 0,0 25,4 414.036,0 0 0
Quelle: Daten aus IMS Health (2014), IMS Health (2015)
115

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.2.1.1 Leitlinien und Therapieempfehlungen vs. Zulassungsstatus nach Fachinformationen
In diesem Abschnitt soll der Frage nachgegangen werden, inwiefern Fachgesellschaften auch bei
Wirkstoffen, die erst kürzlich den Marktzugang erhalten haben, eine Empfehlung für ihre zulas-
sungsüberschreitende Anwendung aussprechen. Mögliche Ursachen wie beispielsweise fehlende
Therapieoptionen sollen identifiziert und anschließend diskutiert werden. Nachfolgend werden die
Leitlinienempfehlungen herausgearbeitet und mit dem Zulassungsstatus der neuen Wirkstoffe zur
Therapie der cHC verglichen.
Aufgrund des fortschreitenden Charakters der chronischen Hepatitis C mit ihren schweren Folgeer-
krankungen sollte in Abhängigkeit des Risikoprofils der betroffenen Patienten immer eine antivirale
Therapie in Betracht gezogen werden. Allerdings können sich wegen der langsam fortschreitenden
Erkrankung Zeitpunkt und Auswahl der Behandlung gemäß den Empfehlungen der europäischen und
amerikanischen Fachgesellschaften durchaus an den individuellen Gegebenheiten der Betroffenen
orientieren. So wird beispielsweise eine rechtzeitige Behandlung bei Patienten mit fortgeschrittener
Fibrose (METAVIR Score 3 und 4), Lebertransplantaten oder extrahepatischen Manifestationen emp-
fohlen, während bei Patienten mit weniger schwerwiegendem Krankheitsstadium, mit minimaler
oder keiner Fibrose der Therapiebeginn kritisch diskutiert werden kann (EASL, 2014; AALSD, 2014).
Im Gegensatz dazu legen die Autoren der deutschen Leitlinie eine frühzeitige Behandlung aller Pati-
enten nahe (DGVS, 2015)20. Da sich sowohl das Therapieschema als auch die Therapiedauer danach
richten, ist eine Bestimmung des HCV-Genotyps unerlässlich. Auch sind das Stadium der Leberer-
krankung, der Grad der Transaminasenerhöhung, die Frage, ob es sich um einen therapienaiven oder
therapieerfahrenen Patienten handelt (Relapser, partieller Responder, Null-Responder)21 sowie das
Vorhandensein von HCV-Resistenzvarianten entscheidende Faktoren für die differentialtherapeuti-
schen Vorgehensweisen (awmf, 2009).
20 Für die Autoren der deutschen Leitlinie stellen die cHC sowie extrahepatische Manifestationen eine Indikation zur antivira-len Therapie dar. Patienten mit einer fortgeschrittenen Lebererkrankung sowie Patienten mit einer Leberzirrhose haben hingegen eine dringliche Therapieindikation (DGVS, 2015). 21 Patientenkollektive, aufgeteilt in Therapienaive Patienten: Patienten, die noch nie eine Behandlung erhalten haben und therapieerfahrene Patienten: Patienten, die bereits mit direkt antiviral wirkenden Mitteln oder mit (PEG-) Interferon be-handelt wurden und entweder keine oder eine nicht ausreichende Therapieantwort zeigten bzw. nach Abschluss der Behand-lung einen Rückfall aufwiesen, diese werden unterschieden in: Relapser: Patienten, die am Ende der Behandlung keine virale Belastung mehr aufweisen, können diese nach Abschluss der Behandlung nicht aufrechterhalten. Partielle (Non)-Responder: Patienten, deren Viruslast unter der Behandlung bis Woche 12 zwar deutlich absinkt, aber bis zum Therapieende nicht unter die Nachweisgrenze sinkt. Null-Responder: Patienten, deren Viruslast trotz Behandlung nicht absinkt. (Partielle (Non)-Responder und Null-Responder werden in manchen Studien auch als Non-Responder zusammengefasst). Patienten mit Relapse (im Vergleich zum Non-Response), mit niedriger Virusbelastung und ohne Zirrhose haben bessere Heilungsaussichten.
116

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Primäres Ziel einer Hepatitis C-Therapie ist ein anhaltendes virologisches Ansprechen (sustained
virological response, SVR22). Darunter versteht man das Fehlen eines HCV-RNA-Nachweises im Serum
24 Wochen nach Therapieende. Mit einer derartigen SVR ist eine Heilung der Lebererkrankung asso-
ziiert. Die Chancen auf eine dauerhafte Viruseradikation nach einer adäquaten Therapie haben mitt-
lerweile mehr als 95 Prozent der Patienten. Diese Patienten entwickeln in der Regel seltener klini-
sche Endpunkte als diejenigen, bei denen eine dauerhafte Viruseradikation nicht möglich ist (Veldt et
al., 2007; Morgan et al., 2013).
it Ausheilungsraten von 15-20 Prozent
Erst durch die duale Therapie in Kombination mit Ribavirin, einem Wirkstoff, der Ende der 90er Jahre
auch in Deutschland seine Zulassung erhielt, wurden schließlich höhere Ausheilungsraten erzielt, so
dass 40-50 Prozent der Patienten mit Genotyp 1- sowie 70-80 Prozent der Patienten mit Genotyp 2-
oder 3-Infektion geheilt werden konnten (Thimme et al., 2014). Kontraindikationen gegen eine Inter-
feron -basierte Therapie, hohe Interaktionspotentiale mit anderen Substanzen sowie eine lange
Behandlungsdauer verbunden mit hohen Nebenwirkungsraten bei vergleichsweise unzureichenden
Heilungschancen, gerade für Patienten mit HCV GT1-Infektion, erschwerten jedoch die Therapie
(Maasoumy et al., 2013a; Maasoumy et al., 2013b).
Mit der Zulassung der beiden Protease-Inhibitoren Telaprevir und Boceprevir für die erste Tripel-
Therapie in Kombination mit PEG- im Jahr 2011 verbesserten sich die Hei-
lungschancen für HCV-Infizierte. Für therapienaive Patienten mit HCV GT1-Infektion konnten nun
SVR-Raten von bis zu 70 Prozent erreicht und gleichzeitig die Therapiedauer verkürzt werden
(Neumann-Haefelin et al., 2012). Bei therapieerfahrenen Patienten ist das Ansprechen auf diese
Tripel-Therapie allerdings stark von der Therapieantwort auf vorangehende Medikamente abhängig.
So liegen die SVR-Raten für Relapser bei ca. 80 Prozent, für sogenannte partielle Responder bei ca. 50
Prozent und für sogenannte Null-Responder bei ca. 30 Prozent (Bacon et al., 2011). Ausgeprägte
Medikamenteninteraktionen bei speziellen Patientengruppen wie beispielsweise bei HIV-
Koinfizierten sowie Lebertransplantat-Empfängern gestalten die Tripel-Therapie mit einem der bei-
den Protease-Inhibitoren als schwierig. Im Vergleich zu anderen Substanzen weisen Boceprevir und
Telaprevir eine geringere Resistenzbarriere auf, weswegen strenge Stopp-Regeln im Therapieverlauf
einzuhalten sind. Im Rahmen der Zulassungsstudien sind daher auch in Abhängigkeit der jeweiligen
Substanz und dem Ansprechen der Patienten individuelle Therapiealgorithmen etabliert worden, die
im klinischen Alltag unbedingt beachtet werden müssen (Geier, 2014).
22 Das IQWiG wertet die SVR als validen Surrogatparameter.
117
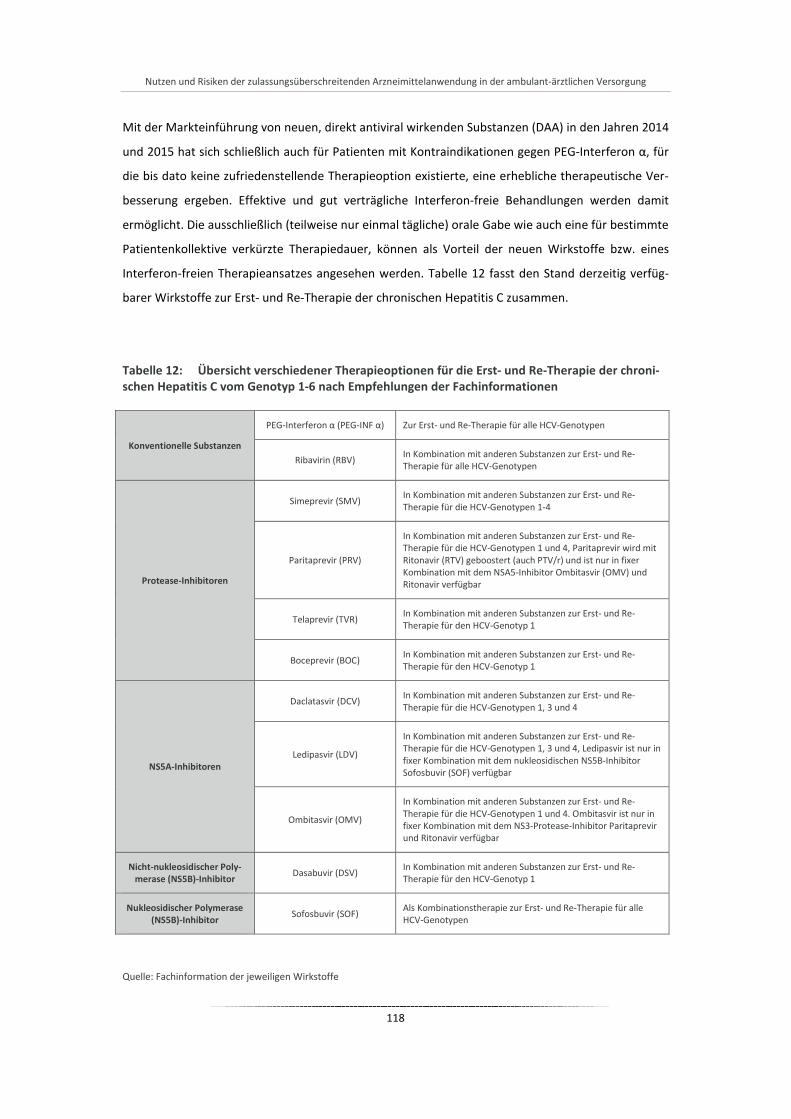
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Mit der Markteinführung von neuen, direkt antiviral wirkenden Substanzen (DAA) in den Jahren 2014
und 2015 hat sich schließlich auch für Patienten mit Kontraindikationen gegen PEG- , für
die bis dato keine zufriedenstellende Therapieoption existierte, eine erhebliche therapeutische Ver-
besserung ergeben. Effektive und gut verträgliche Interferon-freie Behandlungen werden damit
ermöglicht. Die ausschließlich (teilweise nur einmal tägliche) orale Gabe wie auch eine für bestimmte
Patientenkollektive verkürzte Therapiedauer, können als Vorteil der neuen Wirkstoffe bzw. eines
Interferon-freien Therapieansatzes angesehen werden. Tabelle 12 fasst den Stand derzeitig verfüg-
barer Wirkstoffe zur Erst- und Re-Therapie der chronischen Hepatitis C zusammen.
Tabelle 12: Übersicht verschiedener Therapieoptionen für die Erst- und Re-Therapie der chroni-schen Hepatitis C vom Genotyp 1-6 nach Empfehlungen der Fachinformationen
Konventionelle Substanzen
PEG- - ) Zur Erst- und Re-Therapie für alle HCV-Genotypen
Ribavirin (RBV) In Kombination mit anderen Substanzen zur Erst- und Re-Therapie für alle HCV-Genotypen
Protease-Inhibitoren
Simeprevir (SMV) In Kombination mit anderen Substanzen zur Erst- und Re-Therapie für die HCV-Genotypen 1-4
Paritaprevir (PRV)
In Kombination mit anderen Substanzen zur Erst- und Re-Therapie für die HCV-Genotypen 1 und 4, Paritaprevir wird mit Ritonavir (RTV) geboostert (auch PTV/r) und ist nur in fixer Kombination mit dem NSA5-Inhibitor Ombitasvir (OMV) und Ritonavir verfügbar
Telaprevir (TVR) In Kombination mit anderen Substanzen zur Erst- und Re-Therapie für den HCV-Genotyp 1
Boceprevir (BOC) In Kombination mit anderen Substanzen zur Erst- und Re-Therapie für den HCV-Genotyp 1
NS5A-Inhibitoren
Daclatasvir (DCV) In Kombination mit anderen Substanzen zur Erst- und Re-Therapie für die HCV-Genotypen 1, 3 und 4
Ledipasvir (LDV)
In Kombination mit anderen Substanzen zur Erst- und Re-Therapie für die HCV-Genotypen 1, 3 und 4, Ledipasvir ist nur in fixer Kombination mit dem nukleosidischen NS5B-Inhibitor Sofosbuvir (SOF) verfügbar
Ombitasvir (OMV)
In Kombination mit anderen Substanzen zur Erst- und Re-Therapie für die HCV-Genotypen 1 und 4. Ombitasvir ist nur in fixer Kombination mit dem NS3-Protease-Inhibitor Paritaprevir und Ritonavir verfügbar
Nicht-nukleosidischer Poly-merase (NS5B)-Inhibitor Dasabuvir (DSV) In Kombination mit anderen Substanzen zur Erst- und Re-
Therapie für den HCV-Genotyp 1
Nukleosidischer Polymerase (NS5B)-Inhibitor Sofosbuvir (SOF) Als Kombinationstherapie zur Erst- und Re-Therapie für alle
HCV-Genotypen
Quelle: Fachinformation der jeweiligen Wirkstoffe
118
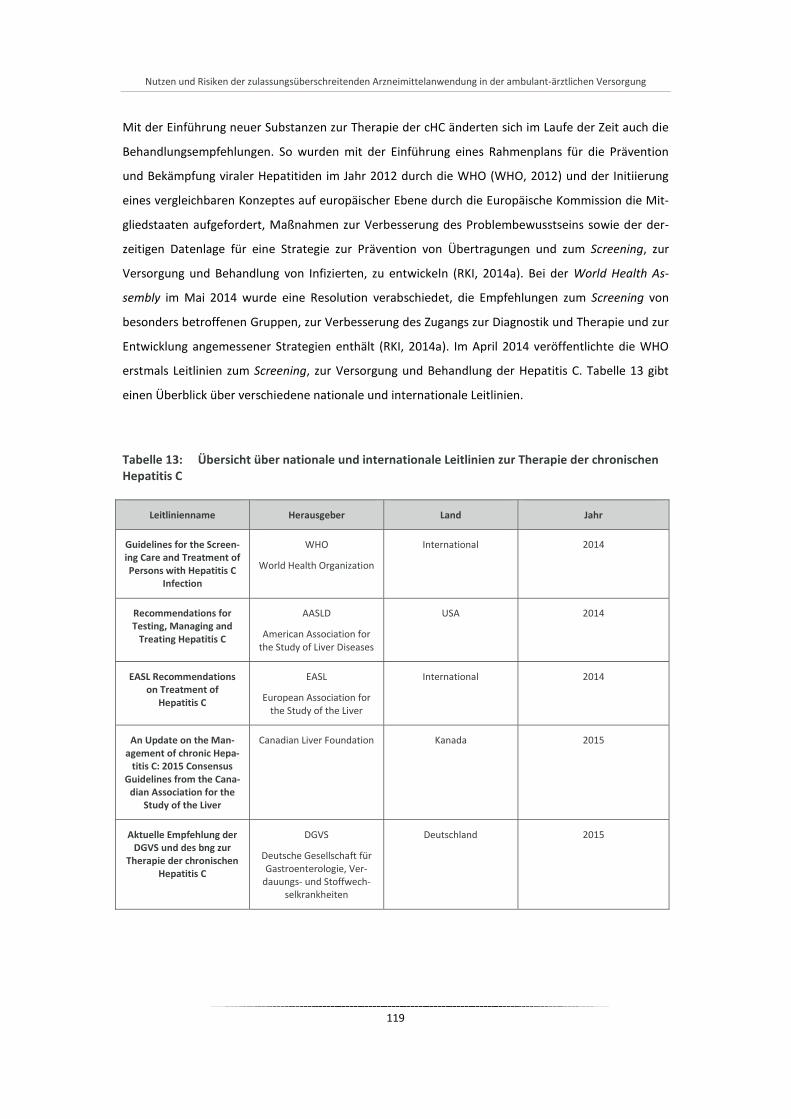
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Mit der Einführung neuer Substanzen zur Therapie der cHC änderten sich im Laufe der Zeit auch die
Behandlungsempfehlungen. So wurden mit der Einführung eines Rahmenplans für die Prävention
und Bekämpfung viraler Hepatitiden im Jahr 2012 durch die WHO (WHO, 2012) und der Initiierung
eines vergleichbaren Konzeptes auf europäischer Ebene durch die Europäische Kommission die Mit-
gliedstaaten aufgefordert, Maßnahmen zur Verbesserung des Problembewusstseins sowie der der-
zeitigen Datenlage für eine Strategie zur Prävention von Übertragungen und zum Screening, zur
Versorgung und Behandlung von Infizierten, zu entwickeln (RKI, 2014a). Bei der World Health As-
sembly im Mai 2014 wurde eine Resolution verabschiedet, die Empfehlungen zum Screening von
besonders betroffenen Gruppen, zur Verbesserung des Zugangs zur Diagnostik und Therapie und zur
Entwicklung angemessener Strategien enthält (RKI, 2014a). Im April 2014 veröffentlichte die WHO
erstmals Leitlinien zum Screening, zur Versorgung und Behandlung der Hepatitis C. Tabelle 13 gibt
einen Überblick über verschiedene nationale und internationale Leitlinien.
Tabelle 13: Übersicht über nationale und internationale Leitlinien zur Therapie der chronischen Hepatitis C
Leitlinienname Herausgeber Land Jahr
Guidelines for the Screen-ing Care and Treatment of Persons with Hepatitis C
Infection
WHO
World Health Organization
International 2014
Recommendations for Testing, Managing and
Treating Hepatitis C
AASLD
American Association for the Study of Liver Diseases
USA 2014
EASL Recommendations on Treatment of
Hepatitis C
EASL
European Association for the Study of the Liver
International 2014
An Update on the Man-agement of chronic Hepa-
titis C: 2015 Consensus Guidelines from the Cana-
dian Association for the Study of the Liver
Canadian Liver Foundation Kanada 2015
Aktuelle Empfehlung der DGVS und des bng zur
Therapie der chronischen Hepatitis C
DGVS
Deutsche Gesellschaft für Gastroenterologie, Ver-
dauungs- und Stoffwech-selkrankheiten
Deutschland 2015
119

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Die Off-Label-Frage muss grundsätzlich auf nationaler Ebene betrachtet werden. Allein der (hier
deutsche) Zulassungsstatus besitzt bindenden Charakter bei juristischen Fragestellungen im deut-
schen Gesundheitswesen (Haftung des pU sowie des Arztes, Leistungspflicht der GKV). Aus diesem
Grund beschränkt sich die nachfolgend aufgeführte Analyse auf die Empfehlungen der DGVS, die mit
Erstellung ihres Addendums aktuelle Therapieempfehlungen ausgearbeitet hat (DGVS, 2015).
Mit Fokussierung auf neue Wirkstoffe und basierend auf den aktuellen Empfehlungen der Fachge-
sellschaften soll im Folgenden der Forschungsfrage nachgegangen werden, inwiefern ein OLU durch
fehlende Therapieoptionen begünstigt wird. In welchen Aspekten der Behandlung der chronischen
Hepatitis C stimmen Empfehlungen der Fachgesellschaften mit dem Zulassungsstatus der diskutier-
ten Wirkstoffe überein und wo weichen diese von einer zulassungskonformen Anwendung (Thera-
pieregime und bzw. oder Behandlungsdauer) ab?
Die Ergebnisse wurden entsprechend des jeweiligen Genotyps 1-6 in den nachstehenden Tabellen
14-19 dargestellt. Die Betrachtung fand auf Ebene der Patientenkollektive statt, die ihrerseits in
Therapienaive und Therapieerfahrene mit und ohne Zirrhose unterteilt wurden. Behandlungen kön-
nen sowohl mit als auch ohne Ribavirin empfohlen bzw. zugelassen sein. Die zusätzliche Gabe der
konventionellen Substanz verlängert in der Regel die Therapie. Die Therapiedauer wird im Folgenden
in Behandlungswochen angegeben. Erfolgte für ein bestimmtes Kollektiv kein Eintrag, so war dazu
keine Angabe in der Leitlinie bzw. in der Fachinformation zu finden.
Für eine visuelle und somit schnellere Erfassung der Resultate wurden empfohlene Standardthera-
pien (DGVS) grün, nur in Einzelfällen empfohlene Therapien (DGVS) dagegen rot gekennzeichnet.
Wurde aufgrund vorhandener Therapieoptionen eine Behandlung seitens der DGVS gar nicht mehr
empfohlen, wurde dies durch die Farbe grau kenntlich gemacht.
120
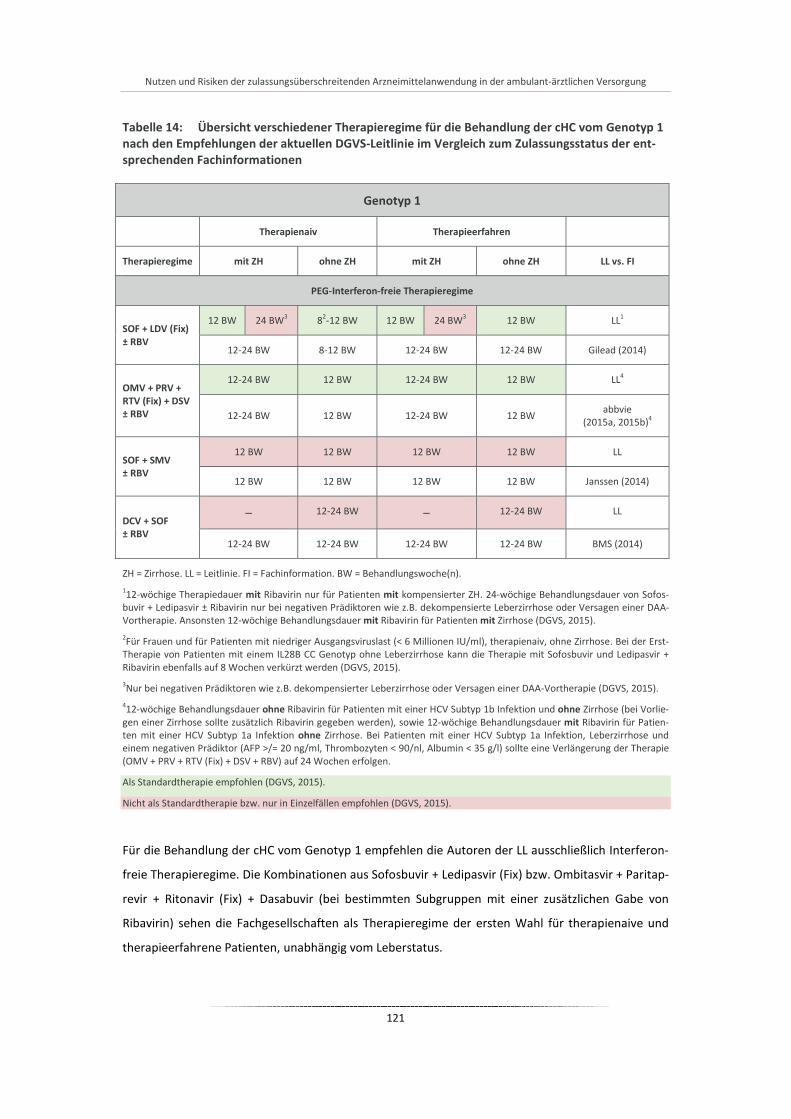
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 14: Übersicht verschiedener Therapieregime für die Behandlung der cHC vom Genotyp 1 nach den Empfehlungen der aktuellen DGVS-Leitlinie im Vergleich zum Zulassungsstatus der ent-sprechenden Fachinformationen
Genotyp 1
Therapienaiv Therapieerfahren
Therapieregime mit ZH ohne ZH mit ZH ohne ZH LL vs. FI
PEG-Interferon-freie Therapieregime
SOF + LDV (Fix) ± RBV
12 BW 24 BW3 82-12 BW 12 BW 24 BW3 12 BW LL1
12-24 BW 8-12 BW 12-24 BW 12-24 BW Gilead (2014)
OMV + PRV + RTV (Fix) + DSV ± RBV
12-24 BW 12 BW 12-24 BW 12 BW LL4
12-24 BW 12 BW 12-24 BW 12 BW abbvie (2015a, 2015b)4
SOF + SMV ± RBV
12 BW 12 BW 12 BW 12 BW LL
12 BW 12 BW 12 BW 12 BW Janssen (2014)
DCV + SOF ± RBV
– 12-24 BW – 12-24 BW LL
12-24 BW 12-24 BW 12-24 BW 12-24 BW BMS (2014)
ZH = Zirrhose. LL = Leitlinie. FI = Fachinformation. BW = Behandlungswoche(n). 112-wöchige Therapiedauer mit Ribavirin nur für Patienten mit kompensierter ZH. 24-wöchige Behandlungsdauer von Sofos-buvir + Ledipasvir ± Ribavirin nur bei negativen Prädiktoren wie z.B. dekompensierte Leberzirrhose oder Versagen einer DAA-Vortherapie. Ansonsten 12-wöchige Behandlungsdauer mit Ribavirin für Patienten mit Zirrhose (DGVS, 2015). 2Für Frauen und für Patienten mit niedriger Ausgangsviruslast (< 6 Millionen IU/ml), therapienaiv, ohne Zirrhose. Bei der Erst-Therapie von Patienten mit einem IL28B CC Genotyp ohne Leberzirrhose kann die Therapie mit Sofosbuvir und Ledipasvir + Ribavirin ebenfalls auf 8 Wochen verkürzt werden (DGVS, 2015). 3Nur bei negativen Prädiktoren wie z.B. dekompensierter Leberzirrhose oder Versagen einer DAA-Vortherapie (DGVS, 2015). 412-wöchige Behandlungsdauer ohne Ribavirin für Patienten mit einer HCV Subtyp 1b Infektion und ohne Zirrhose (bei Vorlie-gen einer Zirrhose sollte zusätzlich Ribavirin gegeben werden), sowie 12-wöchige Behandlungsdauer mit Ribavirin für Patien-ten mit einer HCV Subtyp 1a Infektion ohne Zirrhose. Bei Patienten mit einer HCV Subtyp 1a Infektion, Leberzirrhose und einem negativen Prädiktor (AFP >/= 20 ng/ml, Thrombozyten < 90/nl, Albumin < 35 g/l) sollte eine Verlängerung der Therapie (OMV + PRV + RTV (Fix) + DSV + RBV) auf 24 Wochen erfolgen.
Als Standardtherapie empfohlen (DGVS, 2015).
Nicht als Standardtherapie bzw. nur in Einzelfällen empfohlen (DGVS, 2015).
Für die Behandlung der cHC vom Genotyp 1 empfehlen die Autoren der LL ausschließlich Interferon-
freie Therapieregime. Die Kombinationen aus Sofosbuvir + Ledipasvir (Fix) bzw. Ombitasvir + Paritap-
revir + Ritonavir (Fix) + Dasabuvir (bei bestimmten Subgruppen mit einer zusätzlichen Gabe von
Ribavirin) sehen die Fachgesellschaften als Therapieregime der ersten Wahl für therapienaive und
therapieerfahrene Patienten, unabhängig vom Leberstatus.
121

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Eine 24-wöchige Behandlungsdauer des erstgenannten Therapieregimes sollte nach Auffassung der
LL-Autoren nur bei negativen Prädiktoren wie z.B. bei Diagnose einer dekompensierten Leberzirrho-
se oder nach Versagen einer DAA-Vortherapie erfolgen. Auch bei Patienten mit kompensierter Zir-
rhose sollte ihrer Meinung nach eine Verlängerung auf 24 Wochen nur für den Ausnahmefall vorge-
sehen sein. Die Empfehlung der Fachinformation lautet hingegen eine standardmäßige 24-wöchige
Behandlung ohne Ribavirin für Patienten mit kompensierter Zirrhose (Gilead, 2014). Die Verkürzung
der Behandlungszeit (acht statt zwölf Wochen) bei der Gabe von Sofosbuvir + Ledipasvir (Fix) wird
sowohl von den Fachgesellschaften als auch von der Fachinformation unterstützt (DGVS, 2015;
Gilead, 2014). Die Autoren der LL geben allerdings eine detailliertere Beschreibung für bestimmte
Subgruppen. Demnach sollte eine Behandlungsdauer von acht Wochen als Ersttherapie von Patien-
ten ohne Zirrhose mit bestätigter Ausgangsviruslast < 6 Millionen IU/ml und bei der Ersttherapie von
Frauen ohne Leberzirrhose erfolgen. Bei der Ersttherapie von Patienten mit einem IL28B CC Genotyp
ohne Leberzirrhose kann die Therapie mit Sofosbuvir und Ledipasvir plus Ribavirin ebenfalls auf acht
Wochen verkürzt werden. Diese Unterscheidung der Subgruppen findet sich nicht in der Fachinfor-
mation (Gilead, 2014). Eine achtwöchige Behandlungsdauer der fixen Kombination aus Sofosbuvir
und Ledipasvir ohne Ribavirin kann nach ihrer Angabe generell für therapienaive Patienten ohne
Zirrhose in Betracht gezogen werden (Gilead, 2014).
Die duale bzw. Tripel-Therapie, bestehend aus Sofosbuvir + Simeprevir ± Ribavirin bzw. Daclatasvir in
Kombination mit Sofosbuvir ± Ribavirin, sollten nach Auffassung der LL-Autoren nur in Einzelfällen
und letztgenannte nur bei Patienten ohne Leberzirrhose (als Erst- und Re-Therapie) gegeben werden.
Die duale bzw. Tripel-Therapie bestehend aus Sofosbuvir + Simeprevir ± Ribavirin ist auch gemäß
ihrer Zulassung nur auf den Einsatz bei Patienten mit Kontraindikationen oder Unverträglichkeit von
(PEG- r Therapieindikation beschränkt (Janssen, 2014).
Die Zulassung von Daclatasvir zur Therapie des HCV GT1 ist mit limitierten Daten verbunden. So
liegen für das 12-wöchige Behandlungsregime Daclatasvir + Sofosbuvir nur Daten für therapienaive
Patienten mit Genotyp-1-Infektion vor (BMS, 2014). Für Daclatasvir + Sofosbuvir mit oder ohne
Ribavirin irrhose vor.
Für das Regime von Daclatasvir + PEG- + Ribavirin gibt es Daten für behandlungsnaive
Patienten (BMS, 2014).
122
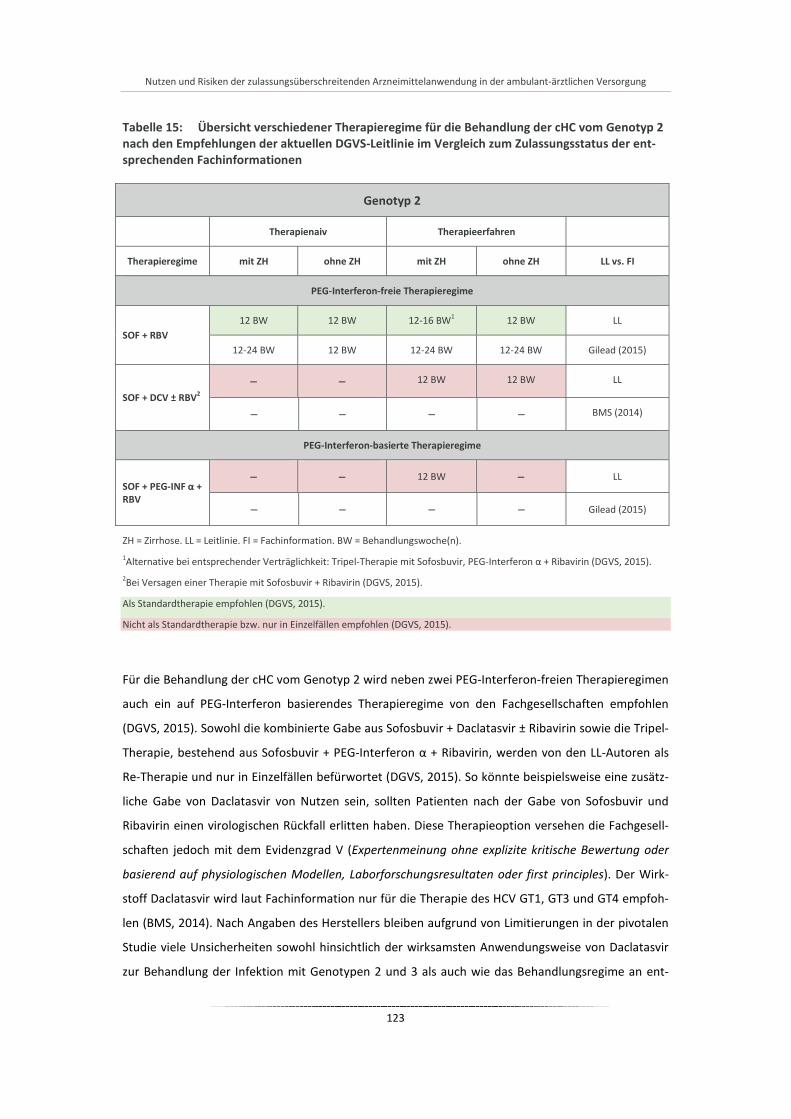
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 15: Übersicht verschiedener Therapieregime für die Behandlung der cHC vom Genotyp 2 nach den Empfehlungen der aktuellen DGVS-Leitlinie im Vergleich zum Zulassungsstatus der ent-sprechenden Fachinformationen
Genotyp 2
Therapienaiv Therapieerfahren
Therapieregime mit ZH ohne ZH mit ZH ohne ZH LL vs. FI
PEG-Interferon-freie Therapieregime
SOF + RBV 12 BW 12 BW 12-16 BW1 12 BW LL
12-24 BW 12 BW 12-24 BW 12-24 BW Gilead (2015)
SOF + DCV ± RBV2 – – 12 BW 12 BW LL
– – – – BMS (2014)
PEG-Interferon-basierte Therapieregime
SOF + PEG-RBV
– – 12 BW – LL
– – – – Gilead (2015)
ZH = Zirrhose. LL = Leitlinie. FI = Fachinformation. BW = Behandlungswoche(n). 1Alternative bei entsprechender Verträglichkeit: Tripel-Therapie mit Sofosbuvir, PEG- (DGVS, 2015). 2Bei Versagen einer Therapie mit Sofosbuvir + Ribavirin (DGVS, 2015).
Als Standardtherapie empfohlen (DGVS, 2015).
Nicht als Standardtherapie bzw. nur in Einzelfällen empfohlen (DGVS, 2015).
Für die Behandlung der cHC vom Genotyp 2 wird neben zwei PEG-Interferon-freien Therapieregimen
auch ein auf PEG-Interferon basierendes Therapieregime von den Fachgesellschaften empfohlen
(DGVS, 2015). Sowohl die kombinierte Gabe aus Sofosbuvir + Daclatasvir ± Ribavirin sowie die Tripel-
Therapie, bestehend aus Sofosbuvir + PEG-Interferon , werden von den LL-Autoren als
Re-Therapie und nur in Einzelfällen befürwortet (DGVS, 2015). So könnte beispielsweise eine zusätz-
liche Gabe von Daclatasvir von Nutzen sein, sollten Patienten nach der Gabe von Sofosbuvir und
Ribavirin einen virologischen Rückfall erlitten haben. Diese Therapieoption versehen die Fachgesell-
schaften jedoch mit dem Evidenzgrad V (Expertenmeinung ohne explizite kritische Bewertung oder
basierend auf physiologischen Modellen, Laborforschungsresultaten oder first principles). Der Wirk-
stoff Daclatasvir wird laut Fachinformation nur für die Therapie des HCV GT1, GT3 und GT4 empfoh-
len (BMS, 2014). Nach Angaben des Herstellers bleiben aufgrund von Limitierungen in der pivotalen
Studie viele Unsicherheiten sowohl hinsichtlich der wirksamsten Anwendungsweise von Daclatasvir
zur Behandlung der Infektion mit Genotypen 2 und 3 als auch wie das Behandlungsregime an ent-
123

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
sprechend wichtige Faktoren, die das virologische Ansprechen beeinflussen können, demzufolge
anzupassen sei (BMS, 2014).
Die duale Kombinationstherapie aus Sofosbuvir und Ribavirin kann laut Fachinformation über zwölf
Wochen erfolgen. Allerdings ist zu erwägen, die Dauer der Therapie möglicherweise über zwölf Wo-
chen hinaus auf bis zu 24 Wochen zu verlängern; dies gilt insbesondere für Subgruppen mit einem
oder mehreren der negativen prädiktiven Faktoren, die in der Vergangenheit mit niedrigeren An-
sprechraten auf Interferon-haltige Therapien (z. B. fortgeschrittene Fibrose/Zirrhose, hohe Aus-
gangsviruslast, schwarze Hautfarbe, IL28B-Non-CC-Genotyp, früheres Nichtansprechen auf PEG-
und Ribavirin) assoziiert waren (Gilead, 2015). Die DGVS empfiehlt keine Therapiever-
längerung bei negativen Prädiktoren, da ihrer Ansicht nach keine Studiendaten vorliegen, die dieses
Vorgehen rechtfertigen (DGVS, 2015). Die Empfehlung einer Therapieverlängerung auf 16 Wochen
für Patienten mit Re-Therapie und Leberzirrhose bewerten die Autoren der LL ebenfalls nur mit dem
Evidenzgrad V (DGVS, 2015).
124
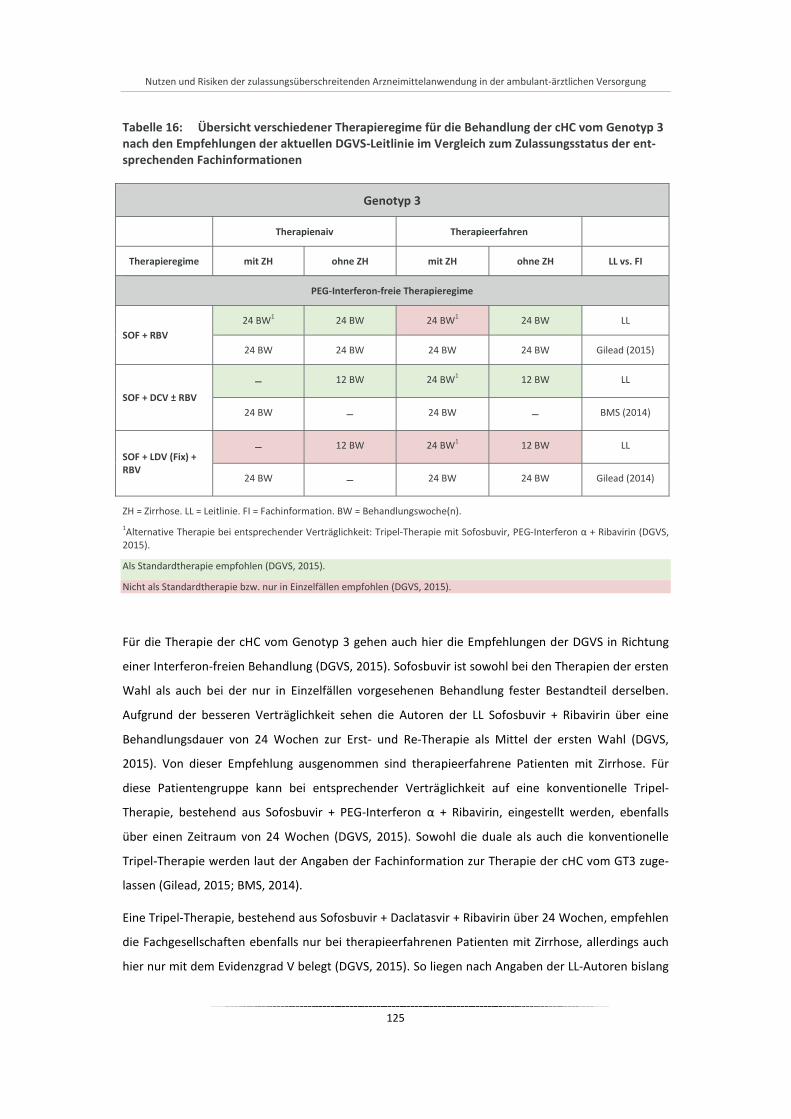
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 16: Übersicht verschiedener Therapieregime für die Behandlung der cHC vom Genotyp 3 nach den Empfehlungen der aktuellen DGVS-Leitlinie im Vergleich zum Zulassungsstatus der ent-sprechenden Fachinformationen
Genotyp 3
Therapienaiv Therapieerfahren
Therapieregime mit ZH ohne ZH mit ZH ohne ZH LL vs. FI
PEG-Interferon-freie Therapieregime
SOF + RBV 24 BW1 24 BW 24 BW1 24 BW LL
24 BW 24 BW 24 BW 24 BW Gilead (2015)
SOF + DCV ± RBV – 12 BW 24 BW1 12 BW LL
24 BW – 24 BW – BMS (2014)
SOF + LDV (Fix) + RBV
– 12 BW 24 BW1 12 BW LL
24 BW – 24 BW 24 BW Gilead (2014)
ZH = Zirrhose. LL = Leitlinie. FI = Fachinformation. BW = Behandlungswoche(n). 1Alternative Therapie bei entsprechender Verträglichkeit: Tripel-Therapie mit Sofosbuvir, PEG- (DGVS, 2015).
Als Standardtherapie empfohlen (DGVS, 2015).
Nicht als Standardtherapie bzw. nur in Einzelfällen empfohlen (DGVS, 2015).
Für die Therapie der cHC vom Genotyp 3 gehen auch hier die Empfehlungen der DGVS in Richtung
einer Interferon-freien Behandlung (DGVS, 2015). Sofosbuvir ist sowohl bei den Therapien der ersten
Wahl als auch bei der nur in Einzelfällen vorgesehenen Behandlung fester Bestandteil derselben.
Aufgrund der besseren Verträglichkeit sehen die Autoren der LL Sofosbuvir + Ribavirin über eine
Behandlungsdauer von 24 Wochen zur Erst- und Re-Therapie als Mittel der ersten Wahl (DGVS,
2015). Von dieser Empfehlung ausgenommen sind therapieerfahrene Patienten mit Zirrhose. Für
diese Patientengruppe kann bei entsprechender Verträglichkeit auf eine konventionelle Tripel-
Therapie, bestehend aus Sofosbuvir + PEG-Interferon + Ribavirin, eingestellt werden, ebenfalls
über einen Zeitraum von 24 Wochen (DGVS, 2015). Sowohl die duale als auch die konventionelle
Tripel-Therapie werden laut der Angaben der Fachinformation zur Therapie der cHC vom GT3 zuge-
lassen (Gilead, 2015; BMS, 2014).
Eine Tripel-Therapie, bestehend aus Sofosbuvir + Daclatasvir + Ribavirin über 24 Wochen, empfehlen
die Fachgesellschaften ebenfalls nur bei therapieerfahrenen Patienten mit Zirrhose, allerdings auch
hier nur mit dem Evidenzgrad V belegt (DGVS, 2015). So liegen nach Angaben der LL-Autoren bislang
125

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
noch keine Daten zur Effektivität der Kombinationstherapie bei dieser Patientensubgruppe vor, auf-
grund der zusätzlichen antiviralen Aktivität von Daclatasvir sei eine Steigerung der Effektivität jedoch
zu vermuten (DGVS, 2015). Die Empfehlung einer Gabe von Sofosbuvir und Daclatasvir über zwölf
Wochen für Patienten ohne Zirrhose zur Erst- und Re-Therapie stellt auch hier die Behandlung der
ersten Wahl dar (DGVS, 2015). Mit dieser Empfehlung weicht die DGVS von der Fachinformation ab.
Diese sieht für die Therapie der cHC vom GT3 grundsätzlich die Tripel-Kombination bestehend aus
Daclatasvir + Sofosbuvir + Ribavirin über eine Behandlungsdauer von 24 Wochen vor (BMS, 2014).
Wie schon für die Behandlung der Infektion mit GT2 bestehen auch für die Therapie der cHC mit GT3
viele Unsicherheiten hinsichtlich der wirksamsten Anwendungsweise von Daclatasvir (BMS, 2014).
Ein ähnliches Bild zeigt sich bei der Tripel-Therapie mit Sofosbuvir + Ledipasvir + Ribavirin. Nach An-
gaben der Fachinformation liegen nur begrenzt klinische Daten vor, die eine Anwendung der fixen
Kombination + Ribavirin bei Patienten mit einer HCV-Infektion vom GT3 unterstützen (Gilead, 2014).
So wurde die relative Wirksamkeit eines zwölfwöchigen Behandlungsregimes nicht untersucht. Aus
diesem Grund empfiehlt sich die konservative Behandlungsdauer über 24 Wochen bei allen vorbe-
handelten Genotyp-3-Patienten sowie bei therapienaiven Genotyp-3-Patienten mit Zirrhose (Gilead,
2014). Aus Sicht der LL-Autoren stellt – mit Einschränkung – die Gabe von Sofosbuvir, Ledipasvir und
Ribavirin bei Patienten ohne Zirrhose über zwölf Wochen eine Therapiealternative zur 24-wöchigen
Behandlung mit Sofosbuvir + Ribavirin bzw. zur zwölfwöchigen Therapie mit Sofosbuvir und Dacla-
tasvir dar (DGVS, 2015).
126
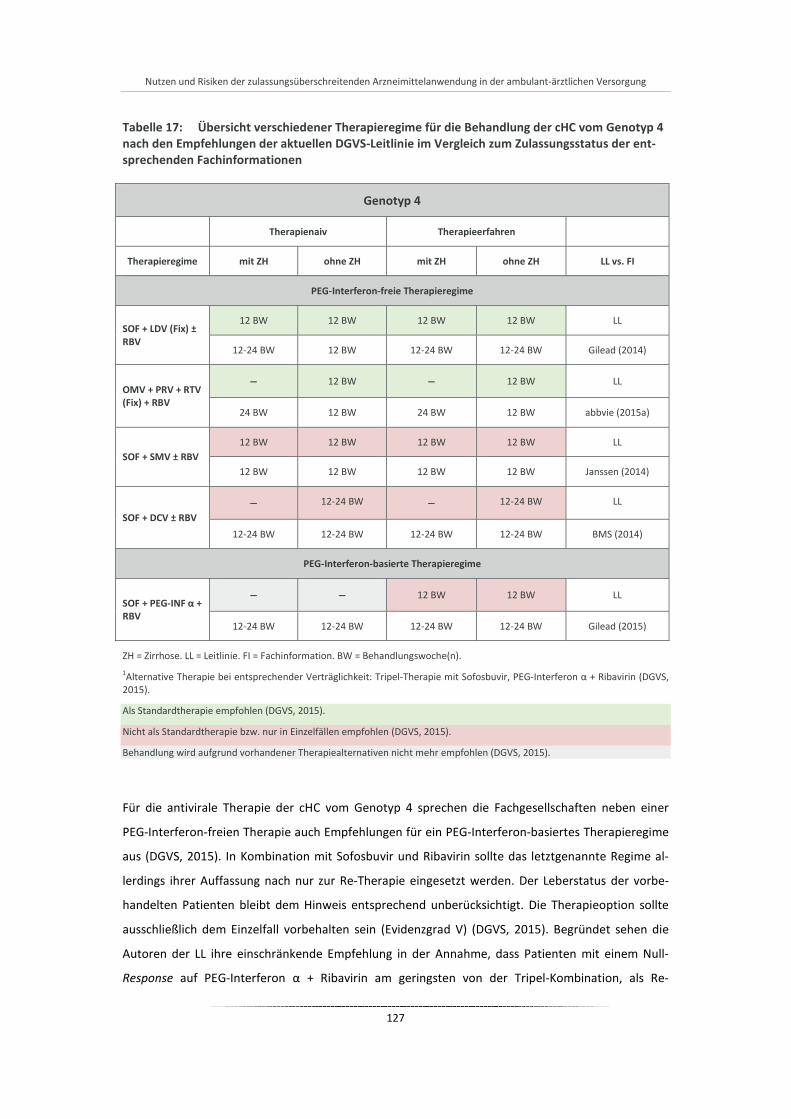
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 17: Übersicht verschiedener Therapieregime für die Behandlung der cHC vom Genotyp 4 nach den Empfehlungen der aktuellen DGVS-Leitlinie im Vergleich zum Zulassungsstatus der ent-sprechenden Fachinformationen
Genotyp 4
Therapienaiv Therapieerfahren
Therapieregime mit ZH ohne ZH mit ZH ohne ZH LL vs. FI
PEG-Interferon-freie Therapieregime
SOF + LDV (Fix) ± RBV
12 BW 12 BW 12 BW 12 BW LL
12-24 BW 12 BW 12-24 BW 12-24 BW Gilead (2014)
OMV + PRV + RTV (Fix) + RBV
– 12 BW – 12 BW LL
24 BW 12 BW 24 BW 12 BW abbvie (2015a)
SOF + SMV ± RBV 12 BW 12 BW 12 BW 12 BW LL
12 BW 12 BW 12 BW 12 BW Janssen (2014)
SOF + DCV ± RBV – 12-24 BW – 12-24 BW LL
12-24 BW 12-24 BW 12-24 BW 12-24 BW BMS (2014)
PEG-Interferon-basierte Therapieregime
SOF + PEG-RBV
– – 12 BW 12 BW LL
12-24 BW 12-24 BW 12-24 BW 12-24 BW Gilead (2015)
ZH = Zirrhose. LL = Leitlinie. FI = Fachinformation. BW = Behandlungswoche(n). 1Alternative Therapie bei entsprechender Verträglichkeit: Tripel-Therapie mit Sofosbuvir, PEG- Ribavirin (DGVS, 2015).
Als Standardtherapie empfohlen (DGVS, 2015).
Nicht als Standardtherapie bzw. nur in Einzelfällen empfohlen (DGVS, 2015).
Behandlung wird aufgrund vorhandener Therapiealternativen nicht mehr empfohlen (DGVS, 2015).
Für die antivirale Therapie der cHC vom Genotyp 4 sprechen die Fachgesellschaften neben einer
PEG-Interferon-freien Therapie auch Empfehlungen für ein PEG-Interferon-basiertes Therapieregime
aus (DGVS, 2015). In Kombination mit Sofosbuvir und Ribavirin sollte das letztgenannte Regime al-
lerdings ihrer Auffassung nach nur zur Re-Therapie eingesetzt werden. Der Leberstatus der vorbe-
handelten Patienten bleibt dem Hinweis entsprechend unberücksichtigt. Die Therapieoption sollte
ausschließlich dem Einzelfall vorbehalten sein (Evidenzgrad V) (DGVS, 2015). Begründet sehen die
Autoren der LL ihre einschränkende Empfehlung in der Annahme, dass Patienten mit einem Null-
Response auf PEG-Interferon + Ribavirin am geringsten von der Tripel-Kombination, als Re-
127

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Therapie über eine Behandlungszeit von zwölf Wochen profitieren, da hier die additiven Effekte der
Kombination aus direkt antiviraler und konventioneller Therapie am geringsten ausgeprägt seien
(DGVS, 2015). Indes empfiehlt die Fachinformation für alle Patienten mit cHC vom GT4 die Therapie-
dauer möglicherweise über zwölf Wochen hinaus auf bis zu 24 Wochen zu verlängern, insbesondere
für Subgruppen mit einem oder mehreren der negativen prädiktiven Faktoren (Gilead, 2015). Über
diese Empfehlung hinaus enthält die Fachinformation die Angabe, dass keine Phase-III-Studien vor-
liegen, in denen therapieerfahrene Patienten zur Studienpopulation gehörten. Eine optimale Be-
handlungsdauer wurde für diese Patientengruppen nicht bestimmt (Gilead, 2015).
Auch für die PEG-Interferon-freien Therapieregime gehen die Empfehlungen der DGVS im Vergleich
zum vorliegenden Zulassungsstatus in Richtung einer kürzeren Therapiedauer. Sowohl die Fachin-
formation als auch die Fachgesellschaften sehen eine zusätzliche Gabe von Ribavirin zur fixen Kom-
bination von Sofosbuvir und Ledipasvir nur bei Patienten mit Zirrhose indiziert (DGVS, 2015; Gilead,
2014). Der Leberstatus als Ausgangssituation für diese Tripel-Therapie ist aber jeweils ein anderer.
Während die Autoren der LL die Indikation durch den Oberbegriff der Leberzirrhose als gegeben
sehen (DGVS, 2015), unterscheidet die Fachinformation genauer: eine kompensierte Zirrhose wird
hier nur mit der dualen Fixkombination therapiert, bei Vorliegen einer dekompensierten Zirrhose
bzw. vor oder nach einer Lebertransplantation sollte laut Zulassungsstatus die zusätzliche Gabe von
Ribavirin erfolgen (Gilead, 2014).
Aufgrund fehlender Daten befürworten die Autoren der LL die Anwendung der fixen Kombination
bestehend aus Ombitasvir und Ritonavir-geboostertem Paritaprevir + Ribavirin nur bei nicht-
zirrhotischen Patienten (DGVS, 2015). Demgegenüber empfiehlt die Fachinformation eine 24-
wöchige Behandlungsdauer auch bei Patienten mit Zirrhose (abbvie, 2015a). Zwar sei aufgrund feh-
lender Daten die optimale Therapiedauer bei Vorliegen einer kompensierten Zirrhose nicht erwie-
sen, basierend auf In-vitro-Daten zur antiviralen Aktivität und den verfügbaren klinischen Daten zum
HCV-Genotyp 1 werde aber für Patienten mit einer HCV-Infektion vom Genotyp 4 und kompensierter
Zirrhose eine konservative Therapiedauer von 24 Wochen empfohlen (abbvie, 2015a).
Die dualen Kombinationen bestehend aus Sofosbuvir + Simeprevir bzw. Sofosbuvir + Daclatasvir und
möglicherweise einer zusätzlichen Gabe von Ribavirin werden von den Fachgesellschaften nur noch
in Einzelfällen befürwortet (DGVS, 2015). Das letztgenannte Regime soll dabei ausschließlich zur Erst-
und Re-Therapie bei nicht-zirrhotischen Patienten indiziert sein (DGVS, 2015).
128
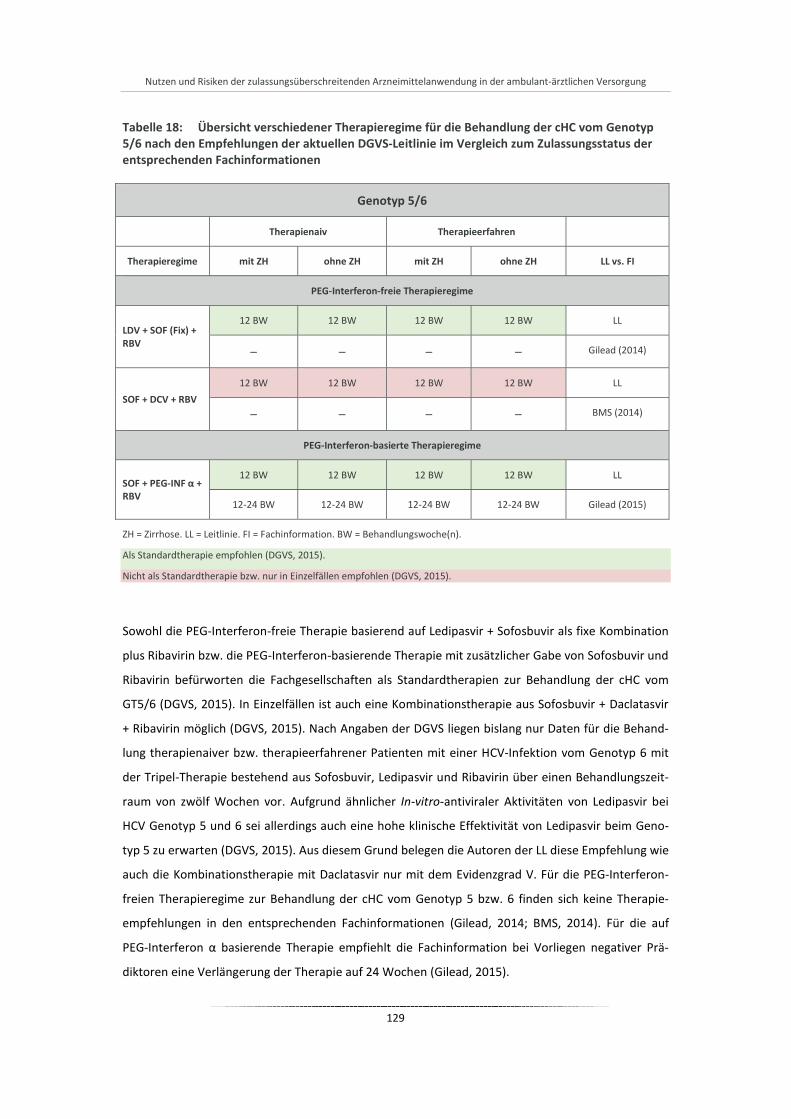
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 18: Übersicht verschiedener Therapieregime für die Behandlung der cHC vom Genotyp 5/6 nach den Empfehlungen der aktuellen DGVS-Leitlinie im Vergleich zum Zulassungsstatus der entsprechenden Fachinformationen
Genotyp 5/6
Therapienaiv Therapieerfahren
Therapieregime mit ZH ohne ZH mit ZH ohne ZH LL vs. FI
PEG-Interferon-freie Therapieregime
LDV + SOF (Fix) + RBV
12 BW 12 BW 12 BW 12 BW LL
– – – – Gilead (2014)
SOF + DCV + RBV 12 BW 12 BW 12 BW 12 BW LL
– – – – BMS (2014)
PEG-Interferon-basierte Therapieregime
SOF + PEG-RBV
12 BW 12 BW 12 BW 12 BW LL
12-24 BW 12-24 BW 12-24 BW 12-24 BW Gilead (2015)
ZH = Zirrhose. LL = Leitlinie. FI = Fachinformation. BW = Behandlungswoche(n).
Als Standardtherapie empfohlen (DGVS, 2015).
Nicht als Standardtherapie bzw. nur in Einzelfällen empfohlen (DGVS, 2015).
Sowohl die PEG-Interferon-freie Therapie basierend auf Ledipasvir + Sofosbuvir als fixe Kombination
plus Ribavirin bzw. die PEG-Interferon-basierende Therapie mit zusätzlicher Gabe von Sofosbuvir und
Ribavirin befürworten die Fachgesellschaften als Standardtherapien zur Behandlung der cHC vom
GT5/6 (DGVS, 2015). In Einzelfällen ist auch eine Kombinationstherapie aus Sofosbuvir + Daclatasvir
+ Ribavirin möglich (DGVS, 2015). Nach Angaben der DGVS liegen bislang nur Daten für die Behand-
lung therapienaiver bzw. therapieerfahrener Patienten mit einer HCV-Infektion vom Genotyp 6 mit
der Tripel-Therapie bestehend aus Sofosbuvir, Ledipasvir und Ribavirin über einen Behandlungszeit-
raum von zwölf Wochen vor. Aufgrund ähnlicher In-vitro-antiviraler Aktivitäten von Ledipasvir bei
HCV Genotyp 5 und 6 sei allerdings auch eine hohe klinische Effektivität von Ledipasvir beim Geno-
typ 5 zu erwarten (DGVS, 2015). Aus diesem Grund belegen die Autoren der LL diese Empfehlung wie
auch die Kombinationstherapie mit Daclatasvir nur mit dem Evidenzgrad V. Für die PEG-Interferon-
freien Therapieregime zur Behandlung der cHC vom Genotyp 5 bzw. 6 finden sich keine Therapie-
empfehlungen in den entsprechenden Fachinformationen (Gilead, 2014; BMS, 2014). Für die auf
PEG- basierende Therapie empfiehlt die Fachinformation bei Vorliegen negativer Prä-
diktoren eine Verlängerung der Therapie auf 24 Wochen (Gilead, 2015).
129
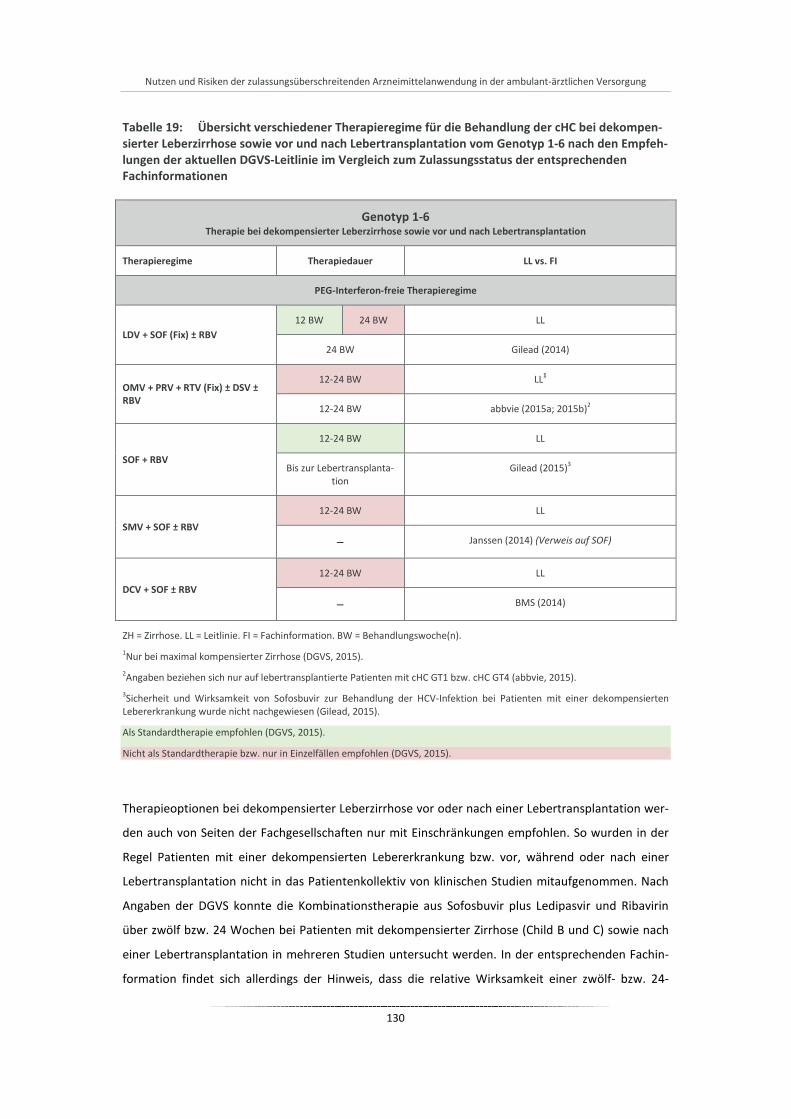
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 19: Übersicht verschiedener Therapieregime für die Behandlung der cHC bei dekompen-sierter Leberzirrhose sowie vor und nach Lebertransplantation vom Genotyp 1-6 nach den Empfeh-lungen der aktuellen DGVS-Leitlinie im Vergleich zum Zulassungsstatus der entsprechenden Fachinformationen
Genotyp 1-6 Therapie bei dekompensierter Leberzirrhose sowie vor und nach Lebertransplantation
Therapieregime Therapiedauer LL vs. FI
PEG-Interferon-freie Therapieregime
LDV + SOF (Fix) ± RBV 12 BW 24 BW LL
24 BW Gilead (2014)
OMV + PRV + RTV (Fix) ± DSV ± RBV
12-24 BW LL1
12-24 BW abbvie (2015a; 2015b)2
SOF + RBV
12-24 BW LL
Bis zur Lebertransplanta-tion
Gilead (2015)3
SMV + SOF ± RBV 12-24 BW LL
– Janssen (2014) (Verweis auf SOF)
DCV + SOF ± RBV 12-24 BW LL
– BMS (2014)
ZH = Zirrhose. LL = Leitlinie. FI = Fachinformation. BW = Behandlungswoche(n). 1Nur bei maximal kompensierter Zirrhose (DGVS, 2015). 2Angaben beziehen sich nur auf lebertransplantierte Patienten mit cHC GT1 bzw. cHC GT4 (abbvie, 2015). 3Sicherheit und Wirksamkeit von Sofosbuvir zur Behandlung der HCV-Infektion bei Patienten mit einer dekompensierten Lebererkrankung wurde nicht nachgewiesen (Gilead, 2015).
Als Standardtherapie empfohlen (DGVS, 2015).
Nicht als Standardtherapie bzw. nur in Einzelfällen empfohlen (DGVS, 2015).
Therapieoptionen bei dekompensierter Leberzirrhose vor oder nach einer Lebertransplantation wer-
den auch von Seiten der Fachgesellschaften nur mit Einschränkungen empfohlen. So wurden in der
Regel Patienten mit einer dekompensierten Lebererkrankung bzw. vor, während oder nach einer
Lebertransplantation nicht in das Patientenkollektiv von klinischen Studien mitaufgenommen. Nach
Angaben der DGVS konnte die Kombinationstherapie aus Sofosbuvir plus Ledipasvir und Ribavirin
über zwölf bzw. 24 Wochen bei Patienten mit dekompensierter Zirrhose (Child B und C) sowie nach
einer Lebertransplantation in mehreren Studien untersucht werden. In der entsprechenden Fachin-
formation findet sich allerdings der Hinweis, dass die relative Wirksamkeit einer zwölf- bzw. 24-
130

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
wöchigen Therapie in diesem speziellen Patientenkollektiv nicht untersucht wurde. Aus dem Grund
wird eine generelle Behandlung über 24 Wochen empfohlen (Gilead, 2014).
8.2.1.2 Diskussion der Ergebnisse
Vor dem Beginn einer Behandlung mit PEG-Interferon a-
ren Diagnose der chronischen Infektion mit Hepatitis C-Viren, einschließlich der Bestimmung des
verursachenden Genotyps und des Leberstatus. Die Dringlichkeit einer medikamentösen Behand-
lung, beeinflusst durch Alter, Geschlecht und Risikoprofil des Patienten, durch Leberwerte und
Krankheitsstadium zum Diagnose-Zeitpunkt und bestehende Komorbiditäten, ist zunächst grundsätz-
lich zu diskutieren. Sicher sollte bei der Entscheidung auch bedacht werden, dass die Erfolgsaussich-
ten einer antiviralen Behandlung mit dem Alter, dem Leberstatus und steigender Viruslast sinken
(Foster et al., 2007). Aufgrund der langsam fortschreitenden Erkrankung können sich Zeitpunkt und
Auswahl der Behandlung gemäß den Empfehlungen der europäischen und amerikanischen Fachge-
sellschaften durchaus an den individuellen Gegebenheiten der Betroffenen orientieren. Eine soforti-
ge Behandlung wird bei Patienten mit fortgeschrittener Fibrose (METAVIR Score 3 und 4), Leber-
transplantaten oder extrahepatischen Manifestationen empfohlen, während bei Patienten mit weni-
ger schwerwiegendem Krankheitsstadium, mit minimaler oder keiner Fibrose der Therapiebeginn
kritisch diskutiert werden kann. Die Autoren der deutschen Leitlinie raten indes zu einer frühzeitigen
Behandlung aller Patienten.
Für die Therapie der HCV-Infektion sind neben den bereits etablierten Wirkstoffen PEG-
und Ribavirin mit ihren langen Vermarktungszyklen nun auch mehrere direkt antiviral wirksame
Substanzen zugelassen, deren Wirksamkeit auf der Interaktion mit unterschiedlichen drug targets
beruht. Bis auf wenige Ausnahmen werden von den Autoren der LL PEG-Interferon-freie Therapiere-
gime empfohlen. Gründe dafür sind neben den höheren SVR-Raten auch die bessere Verträglichkeit
sowie die verkürzte Therapiedauer und verlängerten Dosierungsintervalle. Folglich fehlen die Wirk-
stoffe Boceprevir und Telaprevir, deren Markteinführung gerade vier Jahre zurückliegt und die ge-
mäß Zulassung nur als Tripel-Therapie in Kombination mit PEG-
werden dürfen, in der Leitlinie.
Nach Gegenüberstellung der unterschiedlichen Sachverhalte konnten bei allen Genotypen Abwei-
chungen der Leitlinienempfehlungen im Vergleich zu den Hinweisen der entsprechenden Fachinfor-
mationen identifiziert werden. Unterschiede fanden sich entweder in der Behandlungsdauer (wobei
sich die Autoren der LL in der Regel für kürzere Therapiezeiten aussprachen) oder in der Anwendung
eines antiviralen Wirkstoffes bzw. einer Wirkstoffkombination für bestimmte HCV-Genotypen. Zu
131

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
gleichen Ergebnissen führte die Analyse im Hinblick auf die Behandlung der Patienten mit dekom-
pensierter Zirrhose sowie für Leber-transplantierte Patienten oder solche, für die ein Spenderorgan
langfristig die einzige Therapieoption darstellt. Für die Beurteilung eines OLU kann die Abweichung
von der Dauer einer Anwendung gehören, die mit zu den Pflichtangaben der Zulassungsunterlagen
zählt (siehe Kapitel 1 Abschnitt 1.2.1). Auch hier weichen die Empfehlungen der DGVS teils erheblich
von denen der entsprechenden Fachinformation ab. Die Einschätzungen der DGVS gründen sich
gerade bei Patientenkollektiven mit unsicherer Datenlage auf Studien mit kleineren Fallzahlen, post
hoc-Subgruppenanalysen für bestimmte Patientenuntergruppen sowie auf Annahmen und Rück-
schlüsse von bereits etablierten Therapieoptionen anderer Genotypen.
Zu bedenken ist in diesem Zusammenhang aber, dass die neuen Therapieverfahren zwar außerge-
wöhnlich hohe Heilungsraten versprechen, aber dass lange nicht so viele klinische Erfahrungen zur
Wirksamkeit, Nachhaltigkeit der Behandlung und Verträglichkeit vorliegen wie für die bislang emp-
fohlenen – und in offiziellen Bewertungsverfahren des G-BA und IQWiG immer noch als Vergleichs-
therapien verwendeten – Behandlungsoptionen. Vor allem für die in 2014 zugelassenen direkt antivi-
ral wirkenden Mittel wie Ledipasvir, Dasabuvir, Ombitasvir und Paritaprevir sind lediglich für kleine
Patientenkollektive und über kurze Behandlungszeiträume Daten vorhanden. Eine statistische Absi-
cherung der gefundenen Therapieeffekte PEG-Interferon-freier Behandlungen gegenüber der bishe-
rigen Standardtherapie steht somit aus. Langzeitstudien fehlen aufgrund des erst seit kurzem erfolg-
ten Marktzugangs ebenfalls. Fragen zu möglichen Resistenzentwicklungen können genauso wenig
zufriedenstellend beantwortet werden. Trotz ihrer Hinweise auf die verbesserungsbedürftige Daten-
lage und obwohl direkte Vergleichsaussagen zur dualen oder Tripel-Standardtherapie für die neuen
Therapieoptionen aufgrund fehlender Studien nicht möglich sind, forcieren die Autoren der LL die
neuen Therapien und das in der Regel unter der Angabe kürzerer Behandlungszeiten bzw. Missach-
tung der zugelassenen Genotypen. Auffällig dabei ist, dass die Leitlinien-Autoren zum Teil Erst- und
bzw. oder Co-Autoren der überwiegend industriefinanzierten pivotalen Studien waren. Die Unpartei-
lichkeit von Leitlinienautoren mit angegebenen oder unterstellten Interessenkonflikt sollte somit
grundsätzlich kritisch hinterfragt werden, wenn es um die Bewertung der ausgesprochenen Empfeh-
lungen geht. Solche Hinweise geben Anlass, hinter den LL-Bewertungen auch industriegeleitete Inte-
ressen zu vermuten. Einzig die LL-Empfehlungen, Therapien möglichst zu verkürzen, sprechen dage-
gen. Schließlich ist eine längere Behandlungszeit auch mit höheren Absätzen der entsprechenden
Wirkstoffe verbunden und käme einem ökonomisch geleiteten Interesse entgegen. Andererseits
wird in der LL auch immer wieder darauf hingewiesen, dass kürzere Therapiedauern für bestimmte
Patientensubgruppen mit höheren Relapse-Raten verbunden sind. Die Empfehlungen sind auch
schlecht mit einem Forschungsinteresse zu begründen, da die Fallzahlen, die von einer kürzeren
132

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Behandlungsdauer profitieren würden, zu gering sind, um sie letztendlich auf eine größere Populati-
on zu beziehen.
Im vierten Kapitel dieser Arbeit wurde beschrieben, dass der pU einen nach seiner Auffassung be-
stimmungswidrigen Gebrauch explizit durch Kennzeichnung, Packungsbeilage und Fachinformation
ausschließen muss (Buchner & Jäkel, 2003; Meyer & Grunert, 2005). Er kann den bestimmungswidri-
gen Arzneimittelgebrauch durch die Nennung von Kontraindikationen einschränken bzw. ausschlie-
ßen, um seinen Haftungsumfang einzugrenzen (Göben, 2009). Bei den hier untersuchten Wirkstoffen
wurde die Therapie eines anderen als der empfohlenen Genotypen zwar nicht als Kontraindikation
deklariert, es fanden sich allerdings Hinweise dazu unter dem Abschnitt 4.4 der Fachinformation
(Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung). Allein bei der fixen Kombi-
nation Viekirax® (Ombitasvir + Paritaprevir + Ritonavir) ist die Anwendung dieser Wirkstoffkombina-
tion bei Patienten mit schwerer Leberfunktionsstörung kontraindiziert (Child-Pugh C) (abbvie,
2015a). Eine Kontraindikation wird also nur im schädlichsten Fall vom pU ausgesprochen, ein in Fra-
ge kommender OLU möglicherweise dadurch erleichtert.
In der Gesamtbetrachtung der Ergebnisse kann somit von einer breiten Befürwortung eines OLU von
Seiten der Fachgesellschaften gesprochen werden – und zwar ohne dass sich entsprechende Hinwei-
se im Addendum finden. Zwar belegen die Autoren der LL ihre Empfehlungen mit einem entspre-
chenden Evidenzgrad (V) und weisen zu Beginn ihrer Ausführungen darauf hin, dass es sich bei dem
schlechtesten Evidenzgrad V um Expertenmeinungen ohne explizite kritische Bewertung oder basie-
rend auf physiologischen Modellen, Laborforschungsresultaten oder first principles handelt, das allein
lässt allerdings keine automatischen Rückschlüsse auf einen OLU zu. Wenn Befürwortungen ausge-
sprochen werden, die eine zulassungsüberschreitende Anwendung nach sich ziehen, sollten diese a
priori auch als solche kenntlich gemacht werden. Bemerkenswert auch, dass eine breit aufgestellte
Fürsprache für einen OLU in sämtlichen Bereichen (Genotyp, Therapieregime, Behandlungsdauer) in
den meisten Fällen nicht etwa aufgrund mangelnder Therapieoptionen erfolgt – wie zu Beginn ver-
mutet – da Therapiealternativen vorhanden sind. Das bestärkt ebenfalls den Verdacht, dass in Emp-
fehlungen, die im Rahmen von Leitlinien ausgesprochen werden, nicht nur Forschungsinteressen von
Seiten der Ärzteschaft, sondern auch Interessen pharmazeutischer Unternehmer miteinfließen. Der
OLU sollte ausschließlich dem Interesse und Wohl des Patienten dienen und nicht aufgrund falscher
Anreize ins Leben gerufen werden. Wenn sich eine zulassungsüberschreitende Anwendung in For-
schungsinteresse begründet, so bietet § 35 c SGB V in Verbindung mit § 31 AM-RL genügend Spiel-
raum, diese im Rahmen einer klinischen Prüfung durchzuführen23.
23 So gilt mit Einführung des § 35 c SGB V durch das GKV-Wettbewerbsstärkungsgesetz vom 1. April 2007 ein OLU im Rahmen einer klinischen Prüfung als zulässig und von der GKV zu finanzieren.
133

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Entscheiden sich Ärzte, im Fall der Therapie der chronischen Hepatitis C dennoch „leitlinienkonform“
nach der DGVS zu behandeln, sollten sie sich auch über die Bedeutung medizinischer Leitlinien und
der daraus folgenden rechtlichen Konsequenz (Haftungsanspruch des Patienten im Schadensfall,
mögliche Regress-Ansprüche seitens der GKV) im Klaren sein. Bei Leitlinien bzw. Therapieempfeh-
lungen handelt es sich um systematisch entwickelte Entscheidungshilfen für Leistungserbringer und
Patienten. Sie sollen als Orientierungshilfen verstanden werden, von denen in begründeten Fällen
ein Abweichen erlaubt oder sogar verlangt wird (BÄK & KBV, 1997). Leitlinien stellen im Gegensatz zu
Richtlinien24 Orientierungshilfen dar. Sie sind somit nicht juristisch, sondern nur normativ verbindlich
(Ollenschläger et al., 2000). Medizinische Handlungsempfehlungen werden mittlerweile von den
verschiedenen Ärzteorganisationen, Fachgesellschaften sowie von regionalen (Ärztekammern, Fach-
organisation der Länder) und lokalen Institutionen (Krankenhausabteilungen, Praxisnetzen) aufge-
stellt und herausgegeben (Müller, 2009). Durch formalisierte Konsensusverfahren (z.B. Delphi-
Methode oder Konsensuskonferenzen), in denen innerhalb einer ausgewählten Expertengruppe
Mehrheitsmeinungen gebildet werden, kommt medizinischen Leitlinien insbesondere mit Blick auf
den Grad der professionellen Akzeptanz einer Behandlungsmethode eine erhebliche Bedeutung zu
(Müller, 2009). Das Deutsche Instrument zur methodischen Leitlinien-Bewertung (DELBI) soll die Be-
wertung der methodischen Qualität medizinischer Leitlinien ermöglichen und dient als Hilfe für Ent-
wickler und Anwender medizinischer Leitlinien zur Beurteilung deren methodologischer Qualität
(awmf & äzq, 2008). Von einer endgültigen Harmonisierung des Leitlinienverfahrens kann dennoch
nicht ausgegangen werden (Müller, 2009). Auch muss berücksichtigt werden, dass sich der medizini-
sche Standard laufend ändert, ohne dass dieser gleichzeitig Eingang in Leitlinien findet. Im umge-
kehrten Fall gilt, dass sich allein durch die Aufnahme einer (pharmakologischen) Behandlungsoption
in eine Leitlinie noch lange kein medizinischer Standard ergibt. So heißt es in einem aktuellen Urteil
des Bundesgerichtshofes (BGH, 2014; Urteil vom 15.04.2014. Az: VI ZR 382/12):
Handlungsanweisungen in Leitlinien ärztlicher Fachgremien oder Verbände dürfen nicht unbesehen mit dem medizinischen
Standard gleichgesetzt werden. (…) Leitlinien ersetzen kein Sachverständigengutachten. Zwar können sie im Einzelfall den
medizinischen Standard für den Zeitpunkt ihres Erlasses zutreffend beschreiben; sie können aber auch Standards ärztlicher
Behandlung fortentwickeln oder ihrerseits veralten (BGH, 2014).
24 Während Leitlinien eine gewisse Verbindlichkeit zukommen soll, können Sanktionen bei vorliegenden Verstößen gegen Richtlinien verhängt werden (Brunkhorst & Dittmayer, 2008).
134

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
In betreffendem BGH-Urteil ging es um den Beweiswert von Leitlinien ärztlicher Fachgremien oder
Verbände für die Bestimmung des medizinischen Standards bei der Versorgung einer Frau mit Hoch-
risikoschwangerschaft (Wiegard, 2014). Der Kerngedanke dieses Urteils ist, dass Leitlinien, die erst
nach der zu beurteilenden medizinischen Behandlung veröffentlicht worden sind, nicht für die Beur-
teilung von Sachverhalten herangezogen werden dürfen, die vor dem Veröffentlichungsdatum dieser
Leitlinien liegen (Wiegard, 2014). Für die Beurteilung der Angemessenheit einer Behandlung ist somit
entscheidend, was Kenntnisstand der medizinischen Wissenschaft zum Zeitpunkt der Behandlung ist.
Nach wie vor gilt also, dass ein Haftungsgrund im Schadensfall nicht etwa der unzureichende oder
möglicherweise schlechte Ausgang einer (pharmakologischen) Therapie ist, sondern das Abweichen
vom medizinischen Standard (Gaßner & Stömer, 2012). Vor Gericht ist somit allein der medizinische
Standard zum Zeitpunkt der Behandlung relevant. Leitlinien können zwar helfen, den medizinischen
Standard zu ermitteln, sie bestimmen ihn aber nicht konstitutiv (Nölling, 2014). Wenn Arzneimittel
erst kurz zuvor auf den Markt gekommen sind, kann und darf nicht von einem medizinischen Stan-
dard ausgegangen werden. So hat eine Behandlungsmethode vor ihrer Etablierung als medizinischer
Standard das Stadium des medizinischen Erprobungshandelns (individueller Heilversuch) zu durchlau-
fen. Ein OLU im sogenannten individuellen Heilversuch kann dann erfolgen, wenn zugelassene Alter-
nativen fehlen oder sich der Erfolg einer Behandlung nicht eingestellt hat. Der Therapieansatz dieses
Heilversuches muss schlüssig sein und darf nur unter strengen Nutzen-Risiko-Abwägungen erfolgen.
Dem Arzt unterliegt eine umfassende Informationspflicht, um das Selbstbestimmungsrecht des ein-
zelnen Patienten hinreichend zu berücksichtigen (siehe Kapitel 3).
Ein Abweichen von den Anweisungen aus der Fachinformation ist somit nur dann berechtigt, wenn
es dem derzeitigen Stand der medizinischen Wissenschaft entspricht. Um eine sichere Arzneimittel-
therapie gewährleisten zu können, übermittelt die Fachinformation notwendige wissenschaftliche
Informationen. In der Anwendung eines Arzneimittels entgegen ihrer Beschreibungen, Warnhinwei-
se und Kontraindikationen kann ein Behandlungsfehler gesehen werden. Zumindest dann, wenn das
Abweichen von der Fachinformation mit einer Missachtung des ärztlichen Therapiestandards einher-
geht (Gaßner & Strömer, 2012). Wird der Therapiestandard verlassen, kann dies nicht nur zivilrecht-
liche Folgen wie Schadensersatz und Schmerzensgeld, sondern auch strafrechtliche Konsequenzen
(Verurteilung wegen vorsätzlicher Körperverletzung) nach sich ziehen25.
Aber auch in den Fachinformationen lassen Empfehlungen häufig Diskrepanzen erkennen. Gerade
dann, wenn einerseits – wie beim Beispiel der chronischen Hepatitis C dargestellt – die antivirale
Therapie eines spezifischen Genotyps empfohlen wird (inklusive der Angabe der Behandlungsdauer),
25 Im Juli 2007 verurteilte das Landgericht Ellwangen (Urteil vom 18.07.2007, Az.: 2KLs 11 Js 21209/02) einen Arzt zu einer zweijährigen Freiheitsstrafe auf Bewährung wegen vorsätzlicher Körperverletzung. Entgegen den Regeln der ärztlichen Kunst und entgegen den Angaben auf der Packungsbeilage hatte er das Narkotikum Propofol mehrmalig und längerfristig im Ge-brauch, obwohl die Fachinformation ausdrücklich darauf hinwies, dass die Flasche nur zur einmaligen Anwendung bestimmt und nach Ende der Verabreichung an einen Patienten zu vernichten sei (LG Ellwangen, 2007).
135

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
sich jedoch in dem Abschnitt 4.4 Hinweise ergeben, dass der zu therapierende Patient nicht Untersu-
chungsgegenstand in klinischen Studien war. Interessant auch deshalb, weil der Umgang mit solchen
„Studienlücken“ unterschiedlich gehandhabt wird. So lassen sich auch Fachinformationen auffinden,
die Therapieeinschränkungen unter dem Abschnitt 4.4 aufführen und konsequenterweise keine
Therapieempfehlungen unter dem Abschnitt 4.2 (Dosierung und Art der Anwendung) angeben.
Eine Zulassung für die Anwendung bei erwachsenen Patienten mit dem HCV vom Genotyp 1 konnte
für alle neuen Substanzen erreicht werden. Bis auf Telaprevir, Boceprevir und Dasabuvir gilt dies
auch für erwachsene Patienten mit dem HCV vom Genotyp 4. Für die anderen Genotypen (GT 2/3,
5/6) sind die empfohlenen Therapieoptionen hingegen weitaus eingeschränkter: Der NS5A-Inhibitor
Daclatasvir und der nukleosidische Polymerase-Inhibitor Sofosbuvir sind die einzigen neuen Wirk-
stoffe, die in Kombination mit anderen Substanzen zur Erst- und Re-Therapie für die HCV-Genotypen
1 bis 6 befürwortet wurden. Es ist unbestritten, dass durch eine verbesserte Zulassungssituation
folgerichtig auch Empfehlungen von Seiten der Fachgesellschaften herausgegeben werden können,
die mit den Auffassungen der Zulassungsbehörden und pharmazeutischen Unternehmen überein-
stimmen.
Ein Grund der mangelhaften Datenlage für die Genotypen 2/3 bzw. 5/6 liegt möglicherweise darin,
dass der HCV vom Genotyp 1 mit seinen beiden Subtypen 1a und 1b weltweit die höchste Prävalenz
hat, wobei der Subtyp 1a in erster Linie in den USA und der Subtyp 1b in Europa zu finden ist (We-
demeyer et al., 2012). Für die westlichen Industrieländer sind in erster Linie die ubiquitär vorkom-
menden Genotypen 1 bis 3 von Bedeutung, während die verbleibenden Genotypen 4 bis 6 mit einer
Infektionsrate von drei Prozent eine untergeordnete Rolle spielen (Zipprich & Dollinger, 2010).
Weltweit sind aber die letztgenannten Genotypen für 20 Prozent der Hepatitis C-Infektionen ver-
antwortlich (Antaki et al., 2010). Es ist somit auch nicht überraschend, dass sich gerade in westlichen
Industrieländern der überwiegende Teil der klinischen Studien, daraus resultierende Publikationen
sowie Therapieempfehlungen mit den in diesen Regionen häufigsten Genotypen beschäftigen. Zu
bedenken gilt, dass eine zunehmende Migration auch zu einer Verschiebung der Genotyp-
Vorkommen führt. Eigentlich eher selten vorkommende Genotypen der entsprechenden Region
finden auf einmal Einzug in den Therapiealltag und konfrontieren Ärzte mit fehlenden Behand-
lungsoptionen. Gerade die oft fehlenden zuverlässigen Aussagen zu Sicherheit und Wirksamkeit der
aufgeführten Therapieregime zur Behandlung von Patienten, die auch in der Praxis an ihre Behand-
lungsgrenzen stoßen, sind nicht unerheblich. Dies meint nicht nur „exotische“ Genotypen, sondern
beispielsweise auch Patienten mit dekompensierter Leberzirrhose oder solche nach einer Leber-
transplantation. In den Fachinformationen der betreffenden Arzneimittel sind für diese Patientenkol-
lektive oftmals nur wenige Angaben zu finden, in erster Linie wegen des Ausschlusses bestimmter
Patientenkollektive aus klinischen Studien. Ein Rückfall sowie der Verlust des Transplantats stellen
136

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
aber ein ernstzunehmendes Problem dar. Wechselwirkungen mit Immunsuppressiva schränken hier
mögliche Therapieoptionen weiter ein. Auch aus diesem Grund können Fachgesellschaften unter
Nutzen-Risiko-Aspekten Empfehlungen abgeben, die sich unter Umständen nur auf kleine Fallzahlen
berufen. Dennoch: durch den Mangel an randomisierten Studien, der oftmals geringen Zahl der Pati-
enten und der Heterogenität der klinischen Untersuchungen müssen diese Ergebnisse kritisch hinter-
fragt werden.
Unbestritten ist auch die Tatsache, dass Zulassungsstatus und ärztliche Behandlung nicht immer
denselben Konsens aufweisen müssen oder sogar sollen. Dies allein bedingt schon die rechtliche
Auflage für die Bewilligung einer Zulassung einerseits (siehe Kapitel 1) und die Therapiefreiheit des
Arztes andererseits (siehe Kapitel 3). Schließlich bedeutet eine spezifische und detaillierte Festle-
gung des Anwendungsgebietes eine Einschränkung des ärztlichen (Be)Handlungsspielraums, der im
Allgemeinen den aktuellen Kenntnisstand schneller berücksichtigen und umsetzen kann als eine
nicht nur kostenintensive (und damit für den pU möglicherweise uninteressante), sondern auch eine
zeitlich verzögernde Zulassungserweiterung. Eine behördliche Zulassungserteilung eines Arzneimit-
tels in einer bestimmten Indikation schließt nicht automatisch Wirksamkeit und Unbedenklichkeit in
anderen Anwendungsgebieten aus. Festzuhalten bleibt schlussendlich, dass selbst dann, wenn sich
durch die Zulassung neuer Wirkstoffe gerade für schwer therapierbare Patienten neue pharmakolo-
gische Ansatzpunkte aufzeigen, die Sicherheit einer evidenzbasierten Bewertung oft nicht Schritt
hält. Wird zusätzlich der zulassungsüberschreitende Einsatz von Arzneimitteln aufgrund der Empfeh-
lung unterschiedlicher Fachgesellschaften befürwortet oder sogar forciert, muss kritisch angemerkt
werden, dass auch ein kontrollierter OLU außerhalb klinischer Prüfungen keine wissenschaftlichen
Erkenntnisse liefert, die über den Einzelfall hinausgehen. Eine systematische Erfassung unerwünsch-
ter Arzneimittelwirkungen, die in der breiten Anwendung und unter realen Bedingungen auftreten
und die den Kern eines jeden Pharmakovigilanz-Systems darstellt, wird außerdem durch einen ge-
duldeten OLU ad absurdum geführt.
137

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.2.2 Neue Mittel zur Therapie der chronischen Hepatitis C
Weichen Empfehlungen der Fachgesellschaften von dem Zulassungsstatus eines Arzneimittels ab,
wird dies häufig mit dem Fortschritt medizinischer Erkenntnisse begründet, der durch Leitlinien
schneller erfasst und umgesetzt werden kann, als eine Zulassungserweiterung. Klinische Forschung
stellt die Grundlage des medizinischen Fortschritts dar und ist zugleich die Voraussetzung einer evi-
denzbasierten Medizin (Kabisch et al., 2011). Goldstandard dieser angewandten Wissenschaft sind
randomisierte und klinische Studien (randomised controlled trials, RCTs). Zum Vergleich dienen be-
reits etablierte Therapien oder Placebo, um die Wirksamkeit und Sicherheit eines neuen Wirkstoffes
nachweisen zu können (Kabisch et al., 2011). Während RCTs in der klinischen Forschung primär das
Ziel haben, patientenrelevante Forschungsfragen zu untersuchen, dienen sie der Arzneimittelent-
wicklung als Grundlage der behördlichen Zulassungsentscheidung (Kabisch et al., 2011). Die Genera-
lisierbarkeit der Ergebnisse solcher Studien auf die breite Bevölkerung und unter realen Bedingungen
muss allerdings kritisch hinterfragt werden. Begleiterkrankungen und Medikationen im klinischen
Alltag werden und können oft nicht Berücksichtigung im therapeutischen Entwicklungsprogramm
finden. Auch das homogene Patientenkollektiv reflektiert nicht die Versorgungsrealität.
Während zuvor aufgezeigt werden konnte, dass die Empfehlungen der Fachgesellschaften trotz vor-
handener Behandlungsoptionen – und deshalb mitunter nicht immer nachvollziehbar – von der
Fachinformation und somit auch vom Zulassungsstatus abweichen, soll in diesem Abschnitt der Fra-
ge nachgegangen werden, wie sich die Ergebnisse von Zulassungsstudien im späteren Zulassungssta-
tus wiederfinden. Erfolgt die Zulassung „Ergebnis-orientiert“ oder ergeben sich Hinweise für eine
theory of the second best. Aus sozial- wie auch aus haftungsrechtlicher Sicht nimmt die Zulassung
eine entscheidende Rolle ein und beeinflusst damit unmittelbar eine angemessene patientenindivi-
duelle Versorgung.
Im Fokus des dritten Teils steht die Studienanalyse der für die Zulassung entscheidenden Wirkstoffe
zur Therapie der chronischen Hepatitis C. Analysiert werden sollen vor allem Unterschiede bezüglich
des Patientenkollektivs, der Studienmedikation, der Behandlungsdauer sowie die SVR als primären
Endpunkt. Im Fokus steht dabei nicht die Qualität der pivotalen Studien. Zu Beginn werden kurz die
Ergebnisse der Literaturrecherche umrissen, auf die dann in den entsprechenden Wirkstoff-
Abschnitten detaillierter eingegangen werden soll. Neben den Zulassungsstudien dienen auch hier
die Hinweise aus den Fachinformationen bzw. der EMA Assessment Reports als Grundlage für die
spätere Diskussion. Für die individuelle Versorgung des betroffenen Patienten können außerdem die
Ergebnisse der G-BA-Nutzenbewertung einen nicht zu unterschätzenden Einfluss haben. Diese kön-
138

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
nen die Therapieentscheidung der Ärzte maßgeblich beeinflussen, wenn es um persönliche For-
schungsinteressen geht, die als innovativ bewerteten Wirkstoffe auch in der Praxis anzuwenden.
Ergebnisse der Literaturrecherche
Nach Anwendung der Ausschlusskriterien (siehe Kapitel 7.1.1) wurden für die Bewertung der Stu-
dienlage insgesamt 64 Publikationen (davon 59 mit Volltext) berücksichtigt (Abbildung 28). Tabelle
20 enthält die Basischarakteristika der zusammengefassten Studien.
Abbildung 28: Flowchart der Literaturrecherche zur Fragestellung des Off-Label Use aufgrund fehlender Therapieoptionen am Beispiel der chronischen Hepatitis C
139
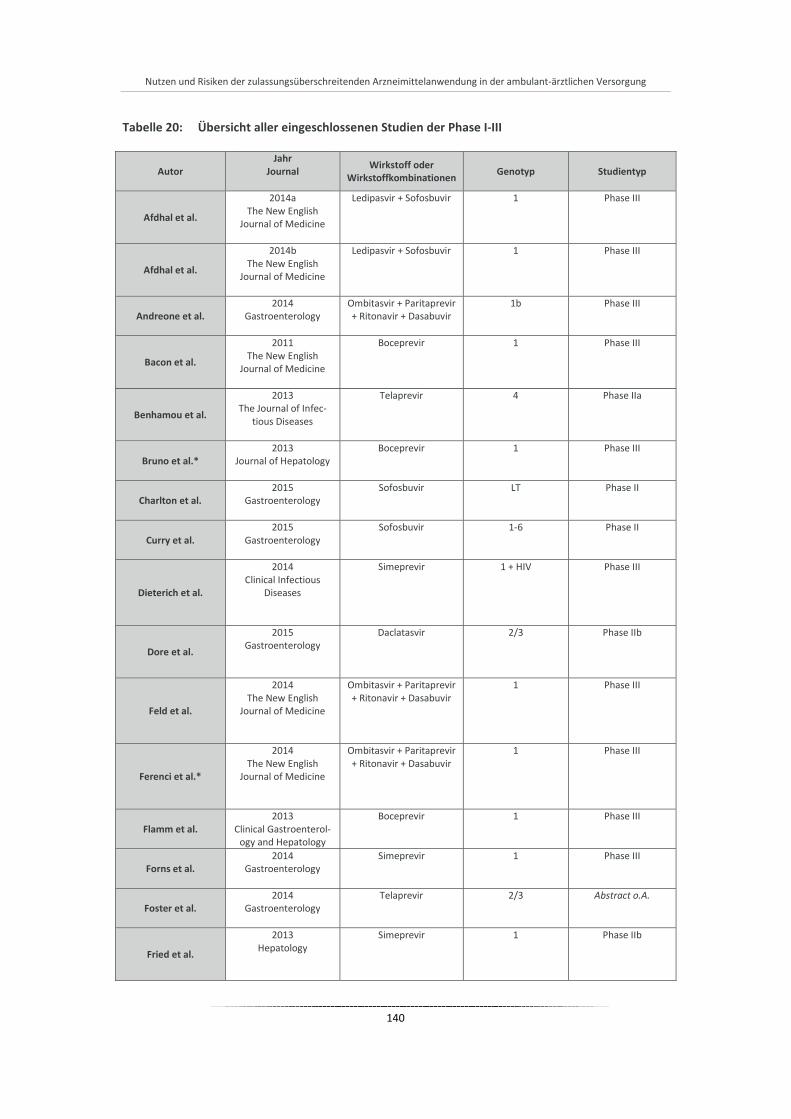
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 20: Übersicht aller eingeschlossenen Studien der Phase I-III
Autor Jahr
Journal
Wirkstoff oder Wirkstoffkombinationen Genotyp Studientyp
Afdhal et al.
2014a The New English
Journal of Medicine
Ledipasvir + Sofosbuvir 1 Phase III
Afdhal et al.
2014b The New English
Journal of Medicine
Ledipasvir + Sofosbuvir 1 Phase III
Andreone et al. 2014
Gastroenterology
Ombitasvir + Paritaprevir + Ritonavir + Dasabuvir
1b Phase III
Bacon et al.
2011 The New English
Journal of Medicine
Boceprevir 1 Phase III
Benhamou et al.
2013 The Journal of Infec-
tious Diseases
Telaprevir 4 Phase IIa
Bruno et al.* 2013
Journal of Hepatology
Boceprevir 1 Phase III
Charlton et al. 2015
Gastroenterology
Sofosbuvir LT Phase II
Curry et al. 2015
Gastroenterology
Sofosbuvir 1-6 Phase II
Dieterich et al.
2014 Clinical Infectious
Diseases
Simeprevir 1 + HIV Phase III
Dore et al.
2015 Gastroenterology
Daclatasvir 2/3 Phase IIb
Feld et al.
2014 The New English
Journal of Medicine
Ombitasvir + Paritaprevir + Ritonavir + Dasabuvir
1 Phase III
Ferenci et al.*
2014 The New English
Journal of Medicine
Ombitasvir + Paritaprevir + Ritonavir + Dasabuvir
1 Phase III
Flamm et al. 2013
Clinical Gastroenterol-ogy and Hepatology
Boceprevir 1 Phase III
Forns et al. 2014
Gastroenterology
Simeprevir 1 Phase III
Foster et al. 2014
Gastroenterology
Telaprevir 2/3 Abstract o.A.
Fried et al.
2013 Hepatology
Simeprevir 1 Phase IIb
140
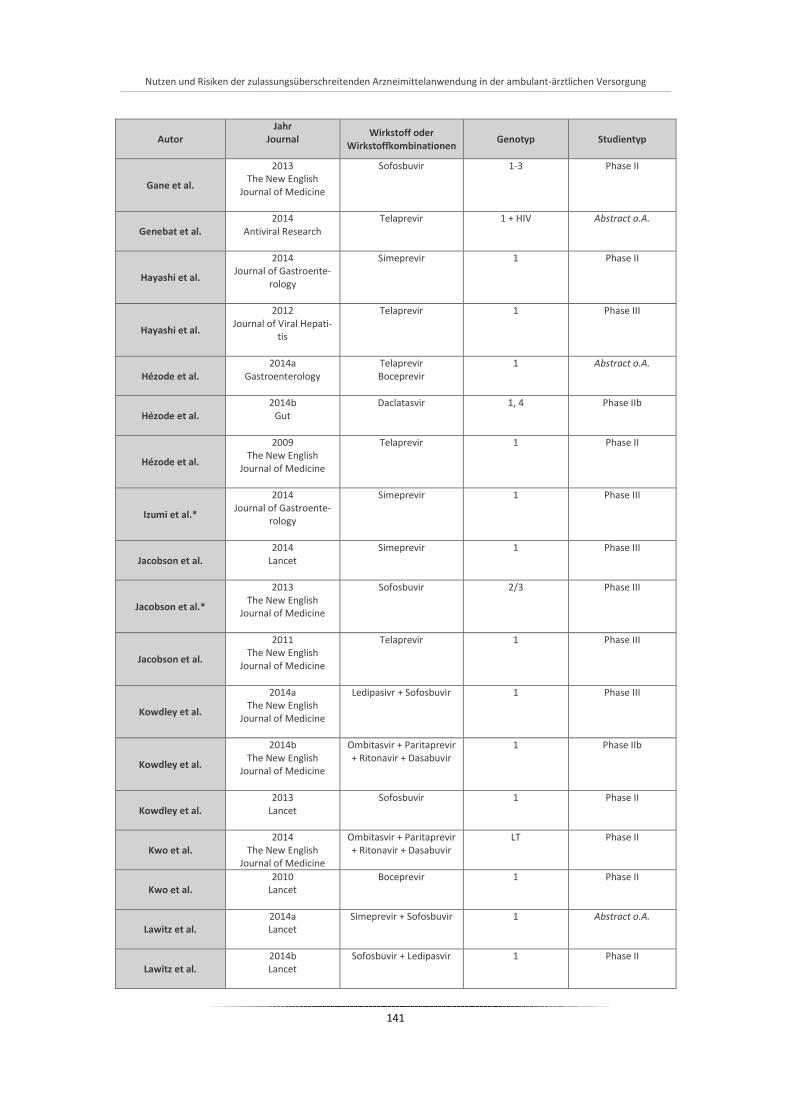
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Autor Jahr
Journal
Wirkstoff oder Wirkstoffkombinationen Genotyp Studientyp
Gane et al.
2013 The New English
Journal of Medicine
Sofosbuvir 1-3 Phase II
Genebat et al. 2014
Antiviral Research
Telaprevir 1 + HIV Abstract o.A.
Hayashi et al.
2014 Journal of Gastroente-
rology
Simeprevir 1 Phase II
Hayashi et al.
2012 Journal of Viral Hepati-
tis
Telaprevir 1 Phase III
Hézode et al. 2014a
Gastroenterology
Telaprevir Boceprevir
1 Abstract o.A.
Hézode et al. 2014b
Gut
Daclatasvir 1, 4 Phase IIb
Hézode et al.
2009 The New English
Journal of Medicine
Telaprevir 1 Phase II
Izumi et al.*
2014 Journal of Gastroente-
rology
Simeprevir 1 Phase III
Jacobson et al. 2014
Lancet
Simeprevir 1 Phase III
Jacobson et al.*
2013 The New English
Journal of Medicine
Sofosbuvir 2/3 Phase III
Jacobson et al.
2011 The New English
Journal of Medicine
Telaprevir 1 Phase III
Kowdley et al.
2014a The New English
Journal of Medicine
Ledipasivr + Sofosbuvir 1 Phase III
Kowdley et al.
2014b The New English
Journal of Medicine
Ombitasvir + Paritaprevir + Ritonavir + Dasabuvir
1 Phase IIb
Kowdley et al. 2013
Lancet
Sofosbuvir 1 Phase II
Kwo et al. 2014
The New English Journal of Medicine
Ombitasvir + Paritaprevir + Ritonavir + Dasabuvir
LT Phase II
Kwo et al. 2010
Lancet
Boceprevir 1 Phase II
Lawitz et al. 2014a Lancet
Simeprevir + Sofosbuvir 1 Abstract o.A.
Lawitz et al. 2014b Lancet
Sofosbuvir + Ledipasvir 1 Phase II
141
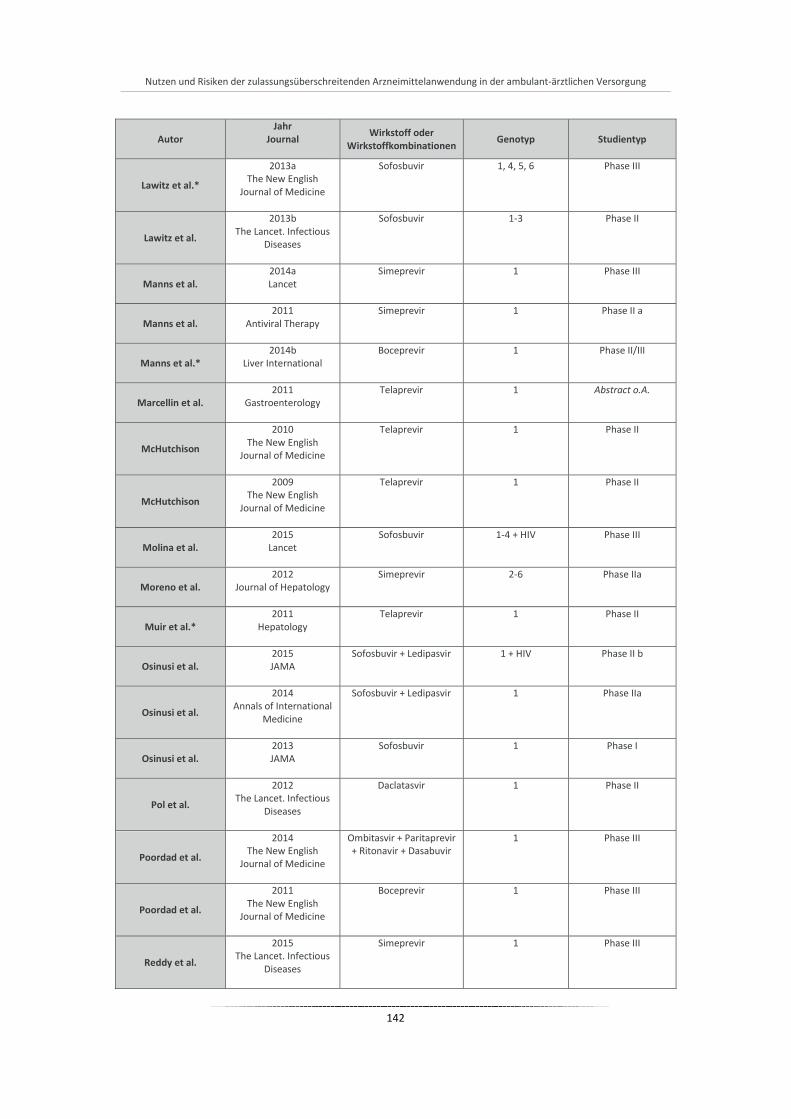
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Autor Jahr
Journal
Wirkstoff oder Wirkstoffkombinationen Genotyp Studientyp
Lawitz et al.*
2013a The New English
Journal of Medicine
Sofosbuvir 1, 4, 5, 6 Phase III
Lawitz et al.
2013b The Lancet. Infectious
Diseases
Sofosbuvir 1-3 Phase II
Manns et al. 2014a Lancet
Simeprevir 1 Phase III
Manns et al. 2011
Antiviral Therapy
Simeprevir 1 Phase II a
Manns et al.* 2014b
Liver International
Boceprevir 1 Phase II/III
Marcellin et al. 2011
Gastroenterology
Telaprevir 1 Abstract o.A.
McHutchison
2010 The New English
Journal of Medicine
Telaprevir 1 Phase II
McHutchison
2009 The New English
Journal of Medicine
Telaprevir 1 Phase II
Molina et al. 2015
Lancet
Sofosbuvir 1-4 + HIV Phase III
Moreno et al. 2012
Journal of Hepatology
Simeprevir 2-6 Phase IIa
Muir et al.* 2011
Hepatology
Telaprevir 1 Phase II
Osinusi et al. 2015 JAMA
Sofosbuvir + Ledipasvir 1 + HIV Phase II b
Osinusi et al.
2014 Annals of International
Medicine
Sofosbuvir + Ledipasvir 1 Phase IIa
Osinusi et al. 2013 JAMA
Sofosbuvir 1 Phase I
Pol et al.
2012 The Lancet. Infectious
Diseases
Daclatasvir 1 Phase II
Poordad et al.
2014 The New English
Journal of Medicine
Ombitasvir + Paritaprevir + Ritonavir + Dasabuvir
1 Phase III
Poordad et al.
2011 The New English
Journal of Medicine
Boceprevir 1 Phase III
Reddy et al.
2015 The Lancet. Infectious
Diseases
Simeprevir 1 Phase III
142
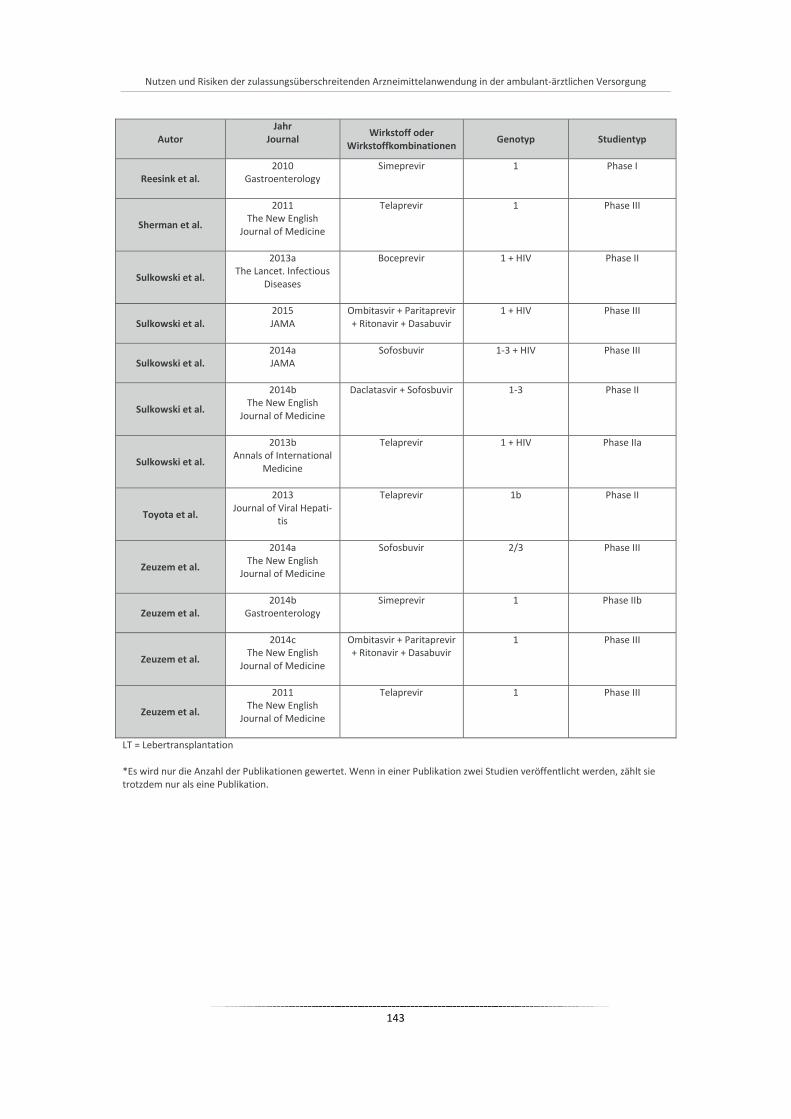
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Autor Jahr
Journal
Wirkstoff oder Wirkstoffkombinationen Genotyp Studientyp
Reesink et al. 2010
Gastroenterology
Simeprevir 1 Phase I
Sherman et al.
2011 The New English
Journal of Medicine
Telaprevir 1 Phase III
Sulkowski et al.
2013a The Lancet. Infectious
Diseases
Boceprevir 1 + HIV Phase II
Sulkowski et al. 2015 JAMA
Ombitasvir + Paritaprevir + Ritonavir + Dasabuvir
1 + HIV Phase III
Sulkowski et al. 2014a JAMA
Sofosbuvir 1-3 + HIV Phase III
Sulkowski et al.
2014b The New English
Journal of Medicine
Daclatasvir + Sofosbuvir 1-3 Phase II
Sulkowski et al.
2013b Annals of International
Medicine
Telaprevir 1 + HIV Phase IIa
Toyota et al.
2013 Journal of Viral Hepati-
tis
Telaprevir 1b Phase II
Zeuzem et al.
2014a The New English
Journal of Medicine
Sofosbuvir 2/3 Phase III
Zeuzem et al. 2014b
Gastroenterology
Simeprevir 1 Phase IIb
Zeuzem et al.
2014c The New English
Journal of Medicine
Ombitasvir + Paritaprevir + Ritonavir + Dasabuvir
1 Phase III
Zeuzem et al.
2011 The New English
Journal of Medicine
Telaprevir 1 Phase III
LT = Lebertransplantation
*Es wird nur die Anzahl der Publikationen gewertet. Wenn in einer Publikation zwei Studien veröffentlicht werden, zählt sie trotzdem nur als eine Publikation.
143
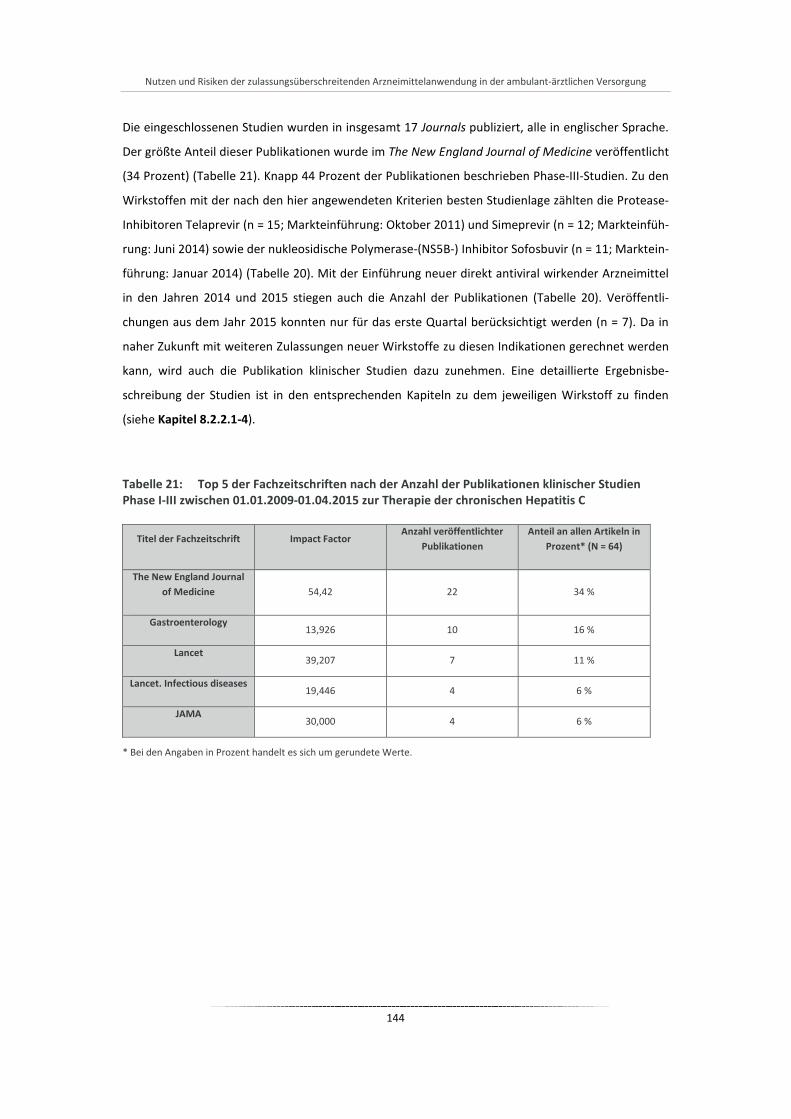
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Die eingeschlossenen Studien wurden in insgesamt 17 Journals publiziert, alle in englischer Sprache.
Der größte Anteil dieser Publikationen wurde im The New England Journal of Medicine veröffentlicht
(34 Prozent) (Tabelle 21). Knapp 44 Prozent der Publikationen beschrieben Phase-III-Studien. Zu den
Wirkstoffen mit der nach den hier angewendeten Kriterien besten Studienlage zählten die Protease-
Inhibitoren Telaprevir (n = 15; Markteinführung: Oktober 2011) und Simeprevir (n = 12; Markteinfüh-
rung: Juni 2014) sowie der nukleosidische Polymerase-(NS5B-) Inhibitor Sofosbuvir (n = 11; Marktein-
führung: Januar 2014) (Tabelle 20). Mit der Einführung neuer direkt antiviral wirkender Arzneimittel
in den Jahren 2014 und 2015 stiegen auch die Anzahl der Publikationen (Tabelle 20). Veröffentli-
chungen aus dem Jahr 2015 konnten nur für das erste Quartal berücksichtigt werden (n = 7). Da in
naher Zukunft mit weiteren Zulassungen neuer Wirkstoffe zu diesen Indikationen gerechnet werden
kann, wird auch die Publikation klinischer Studien dazu zunehmen. Eine detaillierte Ergebnisbe-
schreibung der Studien ist in den entsprechenden Kapiteln zu dem jeweiligen Wirkstoff zu finden
(siehe Kapitel 8.2.2.1-4).
Tabelle 21: Top 5 der Fachzeitschriften nach der Anzahl der Publikationen klinischer Studien Phase I-III zwischen 01.01.2009-01.04.2015 zur Therapie der chronischen Hepatitis C
Titel der Fachzeitschrift
Impact Factor
Anzahl veröffentlichter Publikationen
Anteil an allen Artikeln in Prozent* (N = 64)
The New England Journal
of Medicine
54,42 22 34 %
Gastroenterology
13,926 10 16 %
Lancet
39,207 7 11 %
Lancet. Infectious diseases
19,446 4 6 %
JAMA
30,000 4 6 %
* Bei den Angaben in Prozent handelt es sich um gerundete Werte.
144
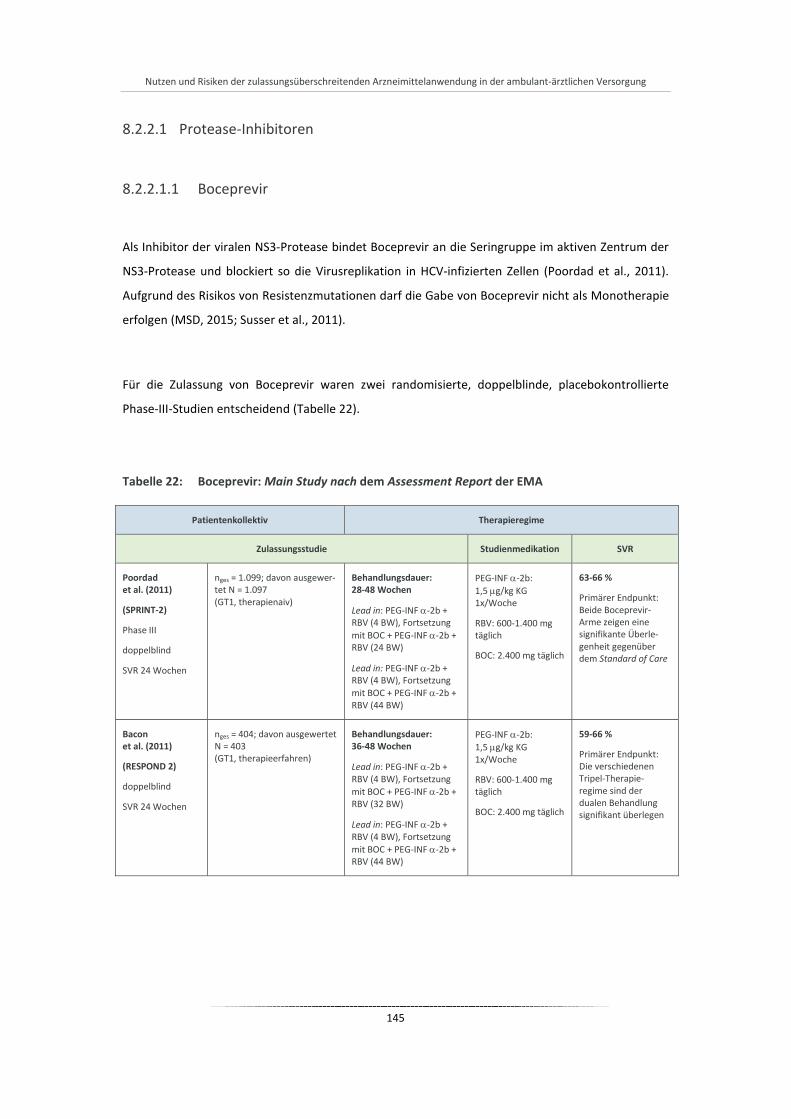
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.2.2.1 Protease-Inhibitoren
8.2.2.1.1 Boceprevir
Als Inhibitor der viralen NS3-Protease bindet Boceprevir an die Seringruppe im aktiven Zentrum der
NS3-Protease und blockiert so die Virusreplikation in HCV-infizierten Zellen (Poordad et al., 2011).
Aufgrund des Risikos von Resistenzmutationen darf die Gabe von Boceprevir nicht als Monotherapie
erfolgen (MSD, 2015; Susser et al., 2011).
Für die Zulassung von Boceprevir waren zwei randomisierte, doppelblinde, placebokontrollierte
Phase-III-Studien entscheidend (Tabelle 22).
Tabelle 22: Boceprevir: Main Study nach dem Assessment Report der EMA
Patientenkollektiv Therapieregime
Zulassungsstudie Studienmedikation SVR
Poordad et al. (2011)
(SPRINT-2)
Phase III
doppelblind
SVR 24 Wochen
nges = 1.099; davon ausgewer-tet N = 1.097 (GT1, therapienaiv)
Behandlungsdauer: 28-48 Wochen
Lead in: PEG-INF -2b + RBV (4 BW), Fortsetzung mit BOC + PEG-INF -2b + RBV (24 BW)
Lead in: PEG-INF -2b + RBV (4 BW), Fortsetzung mit BOC + PEG-INF -2b + RBV (44 BW)
PEG-INF -2b: 1,5 g/kg KG 1x/Woche
RBV: 600-1.400 mg täglich
BOC: 2.400 mg täglich
63-66 %
Primärer Endpunkt: Beide Boceprevir-Arme zeigen eine signifikante Überle-genheit gegenüber dem Standard of Care
Bacon et al. (2011)
(RESPOND 2)
doppelblind
SVR 24 Wochen
nges = 404; davon ausgewertet N = 403 (GT1, therapieerfahren)
Behandlungsdauer: 36-48 Wochen
Lead in: PEG-INF -2b + RBV (4 BW), Fortsetzung mit BOC + PEG-INF -2b + RBV (32 BW)
Lead in: PEG-INF -2b + RBV (4 BW), Fortsetzung mit BOC + PEG-INF -2b + RBV (44 BW)
PEG-INF -2b: 1,5 g/kg KG 1x/Woche
RBV: 600-1.400 mg täglich
BOC: 2.400 mg täglich
59-66 %
Primärer Endpunkt: Die verschiedenen Tripel-Therapie-regime sind der dualen Behandlung signifikant überlegen
145

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Poordad et al. (2011; SPRINT-2) untersuchten die Wirksamkeit von Boceprevir als Therapie für eine
chronische HCV-Infektion vom Genotyp 1 bei therapienaiven Erwachsenen. Nicht mit in das Studien-
kollektiv aufgenommen wurden u.a. Patienten mit HIV-Koinfektion oder dekompensierter Zirrhose.
Die Patienten wurden in drei Gruppen eingeteilt. Neben der placebokontrollierten Gruppe (erste
Gruppe) erhielten die Patienten entweder Boceprevir + PEG-Interfer -2b + Ribavirin über 24
Wochen (zweite Gruppe) oder über eine Dauer von 44 Wochen (dritte Gruppe). Wenn in der zweiten
Gruppe Patienten eine nachweisbare HCV-RNA in Behandlungswoche 8 und 24 aufwiesen, erhielten
sie für weitere 20 Wochen PEG-Interferon -2b + Ribavirin. In allen drei Gruppen wurde die Behand-
lung über vier Wochen mit PEG- -2b + Ribavirin eingeleitet (Lead-in). Die Studienmedika-
tion wurde unterbrochen, wenn in der 24. Behandlungswoche noch der Nachweis einer HCV-RNA
erbracht werden konnte. Der primäre Endpunkt war ein anhaltendes virologisches Ansprechen, defi-
niert als nicht nachweisbare HCV-RNA im Serum 24 Wochen nach Beendigung der Therapie. Beide
Boceprevir-Arme zeigten eine signifikante Überlegenheit gegenüber dem Standard of Care (PEG-
). Das Kollektiv in der Studie von Bacon et al. (2011; RESPOND-2) umfasste
hingegen erwachsene Patienten (cHC GT1), die nicht auf eine vorangegangene Therapie angespro-
chen oder einen Rückfall erlitten hatten. Aus diesem Kollektiv ausgeschlossen wurden ebenfalls u.a.
Patienten mit HIV-Koinfektion oder dekompensierter Zirrhose. Null-Responder (historisch definiert
als Abnahme der HCV-RNA-Viruslast < 2 log10 in Woche 12 gegenüber Behandlungsbeginn) wurden
ebenfalls ausgeschlossen. Primärer Endpunkt war jeweils das anhaltende virologische Ansprechen
(SVR), definiert als fehlende Nachweisbarkeit der HCV-RNA im Blut mindestens 24 Wochen nach dem
Ende der Therapie. Auch in dieser Studie wurde das Kollektiv in drei Gruppen eingeteilt. Die Studi-
enmedikation entsprach der von SPRINT-2, jedoch mit dem Unterschied, dass in der zweiten Gruppe
die Behandlungsdauer der Studienmedikation Boceprevir + PEG-Interferon -2b + Ribavirin über 32
Wochen verabreicht wurde. Wiesen die Patienten in dieser Gruppe einen HCV-RNA in Behandlungs-
woche 8 auf, erhielten sie für weitere zwölf Wochen PEG-Interferon -2b + Ribavirin. Auch hier zeig-
ten sich die Tripel-Therapieregime gegenüber der dualen Behandlung als signifikant überlegen.
Im Juli 2011 wurde Boceprevir als erster Protease-Inhibitor von der EMA in einem beschleunigten
Verfahren zugelassen. Der Wirkstoff ist indiziert zur Behandlung der cHC vom Genotyp 1 in Kombina-
tion mit PEG- ompensierter Lebererkran-
kung, die nicht vorbehandelt sind oder die nicht auf eine vorangegangene Therapie angesprochen
bzw. einen Rückfall erlitten haben (MSD, 2015). Die empfohlene Dosis von Boceprevir beträgt drei-
mal täglich 800 mg (maximale Tagesdosis 2.400 mg).
146

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Zulassungsrelevante Studien vs. Zulassungsstatus
Wie bereits in den Studien ermittelt, müssen auch laut Fachinformation (Tabelle 23) Lead-in sowie
Stopp-Regeln beachtet werden. Die theoretische Begründung der vorgeschalteten einleitenden Pha-
se beruht auf der Annahme, dass durch eine vierwöchige Gabe der Standardtherapie ein steady state
erreicht wird. Die sich daraus ergebende niedrigere Viruslast zu Beginn der Therapie mit Boceprevir
soll das Risiko der Ausbildung von Resistenzvarianten und daraus resultierenden Virusdurchbrüchen
minimieren (EMA, 2011). Da in den beiden relevanten Zulassungsstudien unterschiedliche Stopp-
Regeln angewandt wurden, vereinheitlichte der pU im Verlauf des Zulassungsverfahrens die Stopp-
Regeln, die sowohl in der Erst- als auch in der Re-Therapie dringend eingehalten werden müssen
(Beendigung der Tripel-Therapie, wenn in Behandlungswoche 12 die HCV-RNA bei gleich oder größer
100 IU/ml liegt und Beendigung der Tripel-Therapie bei einer nachweisbaren HCV-RNA in Behand-
lungswoche 24). Die Daten aus den beiden Schlüsselstudien wurden mit Hilfe retrospektiver Analy-
sen miteinander verknüpft, weswegen die empfohlene Dosierung geändert wurde – im Vergleich zu
den Dosierungen, die in einigen Patientensubgruppen geprüft wurden (MSD, 2015).
Obwohl Null-Responder zunächst von der Studienpopulation ausgeschlossen wurden (RESPOND-2),
beantragte der pU im Nachhinein die Erweiterung der Indikation auch für diese Subgruppe mit der
Begründung, dass Patienten mit einer Abnahme der HCV-RNA-Viruslast < 1 log10 am Ende der vier-
wöchigen Einleitungsphase mit vorherigen Null-Respondern gleichzusetzen seien (EMA, 2011). Des
Weiteren wird in der Zulassung für die Re-Therapie mit Boceprevir grundsätzlich eine Therapie über
48 Wochen ohne die Möglichkeit einer Therapieverkürzung empfohlen, da im Boceprevir-Arm über
die längere Behandlungsdauer im Vergleich zur individualisierten Therapie höhere SVR-Raten er-
reicht wurden. Da der pU Daten zu der vergleichbaren Wirksamkeit mit der Gabe von PEG-Interferon
-2a nachreichen konnte, ist auch dieses pegylierte Interferon in Kombination mit Ribavirin und
-2a und -2b unterscheiden sich auf struktureller
Ebene nur durch eine Aminosäure. Diese beeinträchtigt weder erwünschte noch unerwünschte Arz-
neimittelwirkungen (EMA, 2011). Ein vergleichbarer Einsatz ist somit möglich. Abweichungen in den
Zulassungen sind mit Unterschieden in den vom Hersteller eingereichten Unterlagen aus den klini-
schen Studien zu erklären. Dagegen hat die Bindung des Interferon-Moleküls mit Polyethylenglykol
(PEG) Auswirkungen auf die Elimination des Interferons. Durch die strukturelle Veränderung ist die
Eliminationshalbwertszeit auf bis zu 70 Stunden verlängert, was eine einmalige Injektion pro Woche
ermöglicht (Mutschler et al., 2008). Zu beachten gilt, dass sich die lange Wirkdauer bei Unverträg-
lichkeitsreaktionen auch nachteilig auswirken kann.
147
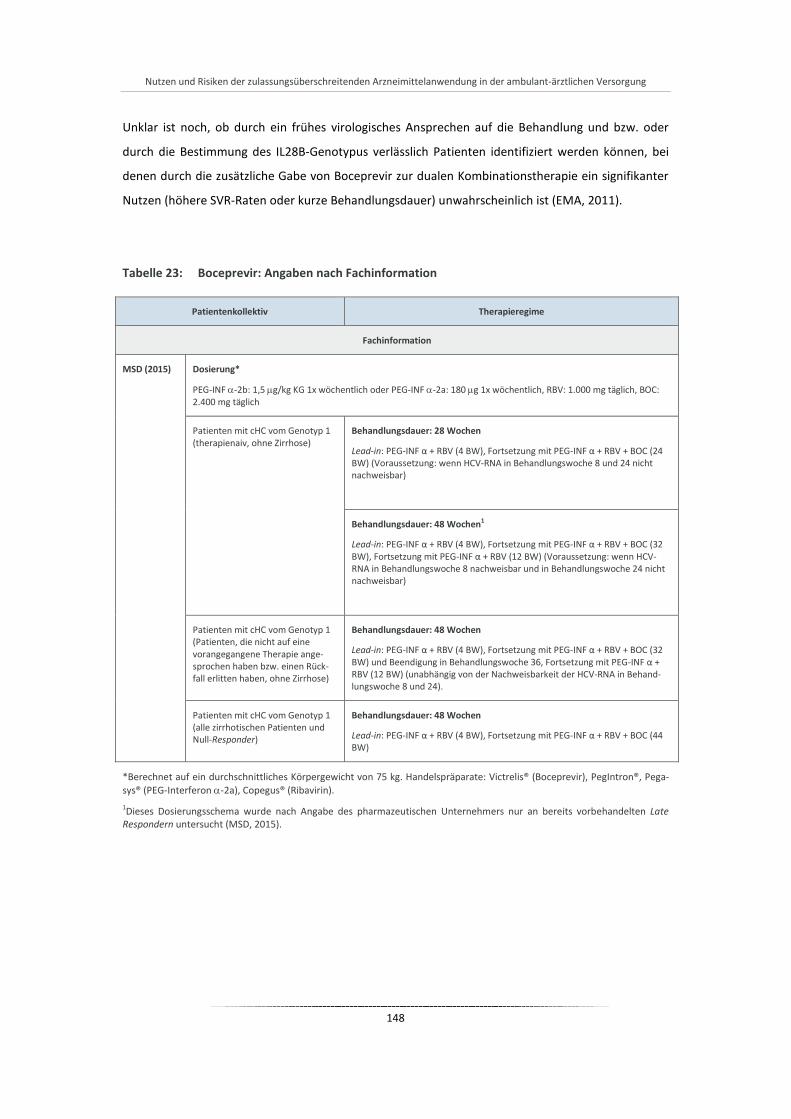
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Unklar ist noch, ob durch ein frühes virologisches Ansprechen auf die Behandlung und bzw. oder
durch die Bestimmung des IL28B-Genotypus verlässlich Patienten identifiziert werden können, bei
denen durch die zusätzliche Gabe von Boceprevir zur dualen Kombinationstherapie ein signifikanter
Nutzen (höhere SVR-Raten oder kurze Behandlungsdauer) unwahrscheinlich ist (EMA, 2011).
Tabelle 23: Boceprevir: Angaben nach Fachinformation
Patientenkollektiv Therapieregime
Fachinformation
MSD (2015) Dosierung*
PEG-INF -2b: 1,5 g/kg KG 1x wöchentlich oder PEG-INF -2a: 180 g 1x wöchentlich, RBV: 1.000 mg täglich, BOC: 2.400 mg täglich
Patienten mit cHC vom Genotyp 1 (therapienaiv, ohne Zirrhose)
Behandlungsdauer: 28 Wochen
Lead-in: PEG- -BW) (Voraussetzung: wenn HCV-RNA in Behandlungswoche 8 und 24 nicht nachweisbar)
Behandlungsdauer: 48 Wochen1
Lead-in: PEG- - BW), Fortsetzung mit PEG- -RNA in Behandlungswoche 8 nachweisbar und in Behandlungswoche 24 nicht nachweisbar)
Patienten mit cHC vom Genotyp 1 (Patienten, die nicht auf eine vorangegangene Therapie ange-sprochen haben bzw. einen Rück-fall erlitten haben, ohne Zirrhose)
Behandlungsdauer: 48 Wochen
Lead-in: PEG- -BW) und Beendigung in Behandlungswoche 36, Fortsetzung mit PEG-RBV (12 BW) (unabhängig von der Nachweisbarkeit der HCV-RNA in Behand-lungswoche 8 und 24).
Patienten mit cHC vom Genotyp 1 (alle zirrhotischen Patienten und Null-Responder)
Behandlungsdauer: 48 Wochen
Lead-in: PEG- - BOC (44 BW)
*Berechnet auf ein durchschnittliches Körpergewicht von 75 kg. Handelspräparate: Victrelis® (Boceprevir), PegIntron®, Pega-sys® (PEG-Interferon -2a), Copegus® (Ribavirin). 1Dieses Dosierungsschema wurde nach Angabe des pharmazeutischen Unternehmers nur an bereits vorbehandelten Late Respondern untersucht (MSD, 2015).
148

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Nutzenbewertung nach § 35a SGB V
Der G-BA bewertete den Zusatznutzen von Boceprevir im Verhältnis zur zweckmäßigen Vergleichs-
therapie bestehend aus PEG- seinem Beschluss vom 1. März 2012 kon-
statierte der G-BA sowohl bei therapienaiven als auch bei therapieerfahrenen Patienten mit chroni-
scher HCV-Infektion (Genotyp 1) einen Hinweis auf einen Zusatznutzen von Boceprevir, allerdings
mit nicht quantifizierbarem Ausmaß (G-BA, 2012a). Begründet sieht der G-BA seinen Beschluss
darin, dass in den Patientengruppen der therapienaiven und therapieerfahrenen Patienten, für die
jeweils ein Zusatznutzen festgestellt wurde, auch die Gruppe der Patienten mit Zirrhose sowie Pati-
enten mit Koinfektionen enthalten waren. Auch wurden in der Gruppe der Therapieerfahrenen die
Gruppe der Null-Responder mit erfasst. Da für diese Patientengruppen keine ausreichenden Daten
zur Bewertung des Zusatznutzens vorliegen, können dementsprechend keine gesicherten Aussagen
im Hinblick auf den primären Endpunkt SVR gemacht werden (G-BA, 2012b). Das signifikant häufige-
re Auftreten von Anämien unter der Behandlung von Boceprevir führte in der Gesamtschau letztend-
lich ebenfalls zu dieser G-BA-Bewertung.
149

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.2.2.1.2 Telaprevir
Wirkstoff und Pharmakologie
Telaprevir ist ein Inhibitor der HCV-NS3/4A-Serinprotease, die für die virale Replikation essentiell ist.
Da der HC-Virus eine hohe genetische Variabilität besitzt, können sich beim Einsatz direkt antiviral
wirkender Mittel häufig Resistenzen entwickeln. Aus diesem Grund darf auch dieser Wirkstoff nur in
Kombination mit der bisherigen Standardtherapie bestehend aus PEG-
angewendet werden (MSD, 2014).
Zulassungsrelevant waren drei Phase-III-Studien, die Telaprevir in Kombination mit der Standardthe-
rapie bestehend aus PEG-Interferon und Ribavirin an therapienaiven (Jacobson et al., 2011; Sher-
man et al., 2011) sowie an therapieerfahrenen Patienten (mit Relapse, partiellen Respondern und
Null-Respondern) (Zeuzem et al., 2011) untersuchten (Tabelle 24).
Die erste Schlüsselstudie (ADVANCE) war eine randomisierte, doppelblinde und placebokontrollierte
Studie der Phase III, in der zwei Behandlungsregime von Telaprevir (acht Behandlungswochen im
Vergleich zu zwölf Behandlungswochen) in Kombination mit der konventionellen Therapie, beste-
hend aus PEG- -2a und Ribavirin, getestet worden sind (Jacobson et al., 2011). Einge-
schlossen waren therapienaive Patienten mit cHC vom Genotyp 1, Ausschlusskriterien waren wie
zuvor schon bei Boceprevir u.a. eine diagnostizierte dekompensierte Leber und bzw. oder Koinfekti-
onen mit HIV oder Hepatitis B. Diese Studie war die erste, in der die Wirksamkeit des Protease-
Inhibitors durch ein kürzeres Behandlungsregime nachgewiesen werden sollte. Bei Nachweis einer
HCV-RNA nach vier Wochen wurde die Gabe von Telaprevir beendet. Wurde der Nachweis nach
zwölf Behandlungswochen erbracht, wurde der Therapie ein virologisches Versagen konstatiert. Der
primäre Endpunkt war ein anhaltendes virologisches Ansprechen definiert als nicht nachweisbare
HCV-RNA im Serum 24 Wochen nach Beendigung der Therapie. Die geprüften Tripel-Regime über
acht bzw. zwölf Wochen waren der Kontrollbehandlung über 48 Wochen signifikant überlegen. Al-
lerdings wurde häufiger ein virologisches Versagen im achtwöchigen Telaprevir-Arm dokumentiert
(Jacobson et al., 2011).
150
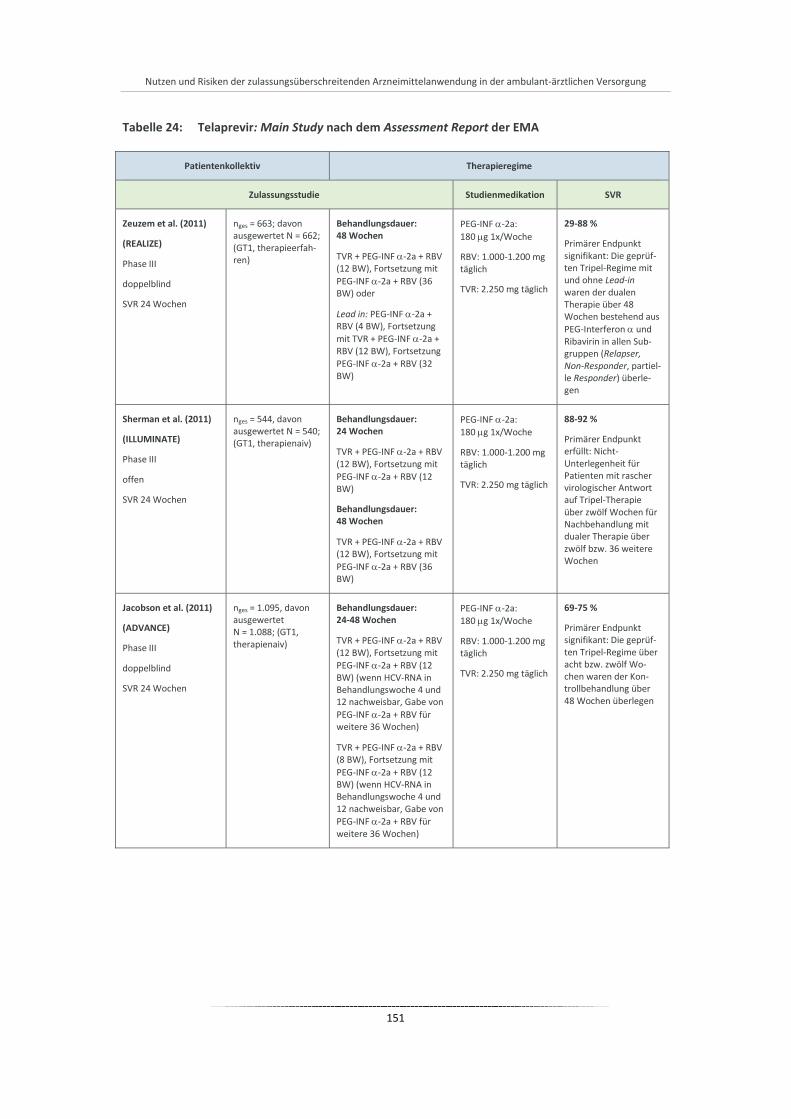
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 24: Telaprevir: Main Study nach dem Assessment Report der EMA
Patientenkollektiv Therapieregime
Zulassungsstudie Studienmedikation SVR
Zeuzem et al. (2011)
(REALIZE)
Phase III
doppelblind
SVR 24 Wochen
nges = 663; davon ausgewertet N = 662; (GT1, therapieerfah-ren)
Behandlungsdauer: 48 Wochen
TVR + PEG-INF -2a + RBV (12 BW), Fortsetzung mit PEG-INF -2a + RBV (36 BW) oder
Lead in: PEG-INF -2a + RBV (4 BW), Fortsetzung mit TVR + PEG-INF -2a + RBV (12 BW), Fortsetzung PEG-INF -2a + RBV (32 BW)
PEG-INF -2a: 180 g 1x/Woche
RBV: 1.000-1.200 mg täglich
TVR: 2.250 mg täglich
29-88 %
Primärer Endpunkt signifikant: Die geprüf-ten Tripel-Regime mit und ohne Lead-in waren der dualen Therapie über 48 Wochen bestehend aus PEG-Interferon und Ribavirin in allen Sub-gruppen (Relapser, Non-Responder, partiel-le Responder) überle-gen
Sherman et al. (2011)
(ILLUMINATE)
Phase III
offen
SVR 24 Wochen
nges = 544, davon ausgewertet N = 540; (GT1, therapienaiv)
Behandlungsdauer: 24 Wochen
TVR + PEG-INF -2a + RBV (12 BW), Fortsetzung mit PEG-INF -2a + RBV (12 BW)
Behandlungsdauer: 48 Wochen
TVR + PEG-INF -2a + RBV (12 BW), Fortsetzung mit PEG-INF -2a + RBV (36 BW)
PEG-INF -2a: 180 g 1x/Woche
RBV: 1.000-1.200 mg täglich
TVR: 2.250 mg täglich
88-92 %
Primärer Endpunkt erfüllt: Nicht-Unterlegenheit für Patienten mit rascher virologischer Antwort auf Tripel-Therapie über zwölf Wochen für Nachbehandlung mit dualer Therapie über zwölf bzw. 36 weitere Wochen
Jacobson et al. (2011)
(ADVANCE)
Phase III
doppelblind
SVR 24 Wochen
nges = 1.095, davon ausgewertet N = 1.088; (GT1, therapienaiv)
Behandlungsdauer: 24-48 Wochen
TVR + PEG-INF -2a + RBV (12 BW), Fortsetzung mit PEG-INF -2a + RBV (12 BW) (wenn HCV-RNA in Behandlungswoche 4 und 12 nachweisbar, Gabe von PEG-INF -2a + RBV für weitere 36 Wochen)
TVR + PEG-INF -2a + RBV (8 BW), Fortsetzung mit PEG-INF -2a + RBV (12 BW) (wenn HCV-RNA in Behandlungswoche 4 und 12 nachweisbar, Gabe von PEG-INF -2a + RBV für weitere 36 Wochen)
PEG-INF -2a: 180 g 1x/Woche
RBV: 1.000-1.200 mg täglich
TVR: 2.250 mg täglich
69-75 %
Primärer Endpunkt signifikant: Die geprüf-ten Tripel-Regime über acht bzw. zwölf Wo-chen waren der Kon-trollbehandlung über 48 Wochen überlegen
151

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Sherman et al. (2011) untersuchten in ihrer Studie die SVR-Raten von Patienten, die einen extended
rapid viral response (eRVR) – definiert als nicht detektierbare HCV RNA in der vierten und zwölften
Behandlungswoche (Wedemeyer et al., 2012a) – unter der Tripel-Therapie Telaprevir + PEG-
– erzielten. Ein- und Ausschlusskriterien entsprachen der ADVANCE-Studie.
Patienten mit einer eRVR beendeten die Studie entweder nach 24 Wochen Behandlung oder erhiel-
ten wie die Patienten ohne eRVR die duale Standardtherapie bis zur Behandlungswoche 48. Der
primäre Endpunkt war ein anhaltendes virologisches Ansprechen, definiert als nicht nachweisbare
HCV-RNA im Serum 24 Wochen nach Beendigung der Therapie. Das Ergebnis der Studie zeigte eine
Nicht-Unterlegenheit für Patienten mit rascher virologischer Antwort auf die Tripel-Therapie über
zwölf Wochen im Vergleich zur Nachbehandlung mit der dualen Standardtherapie über zwölf bzw. 36
weitere Wochen (response guided algorithm). Zirrhotische Patienten mit eRVR erzielten hingegen
höhere SVR-Raten nach 48 Wochen Behandlungsdauer im Vergleich zu der verkürzten Therapie über
24 Wochen. Die Studie unterstützt damit eine Antwort-geführte Therapie in der Erstbehandlung.
In ihrer REALIZE-Studie untersuchten Zeuzem et al. die Nicht-Unterlegenheit eines Behandlungsre-
gimes ohne gegenüber eines Behandlungsregimes mit Lead-in (Zeuzem et al., 2011). Das Studienkol-
lektiv bestand aus therapieerfahrenen Patienten mit cHC vom Genotyp 1. Der primäre Endpunkt war
ein anhaltendes virologisches Ansprechen definiert als nicht nachweisbare HCV-RNA im Serum 24
Wochen nach Beendigung der Therapie. Die geprüften Tripel-Regime mit und ohne Lead-in waren
der dualen Therapie über 48 Wochen bestehend aus PEG-Interferon -2a und Ribavirin in allen Sub-
gruppen (Relapser, Non-Responder, partielle Responder) überlegen. Die Wahrscheinlichkeit eines
erneuten virologischen Versagens hing von einem vorherigen Ansprechen ab und zeigt die Bedeu-
tung von PEG- in der Therapie mit einem DAA (Zeuzem et al., 2011).
Nach Boceprevir erfolgte im September 2011 die Zulassung des zweiten Protease-Hemmstoffes
Telaprevir zur Behandlung der cHC vom Genotyp 1 bei erwachsenen Patienten mit kompensierter
Lebererkrankung (einschließlich Zirrhose). Das Mittel ist indiziert zur Behandlung der cHC-Infektion
vom Genotyp 1 in Kombination mit der dualen Standardtherapie bei erwachsenen Patienten mit
kompensierter Lebererkrankung, die nicht vorbehandelt sind oder die nicht auf eine vorangegangene
Therapie angesprochen bzw. einen Rückfall erlitten haben (MSD, 2014). Die empfohlene Gesamt-
tagesdosis von Telaprevir beträgt 2.250 mg. Die Gesamtdauer der Behandlung ist abhängig vom
Krankheitsbild und -verlauf und kann insgesamt bis zu 48 Wochen betragen.
152

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Zulassungsrelevante Studien vs. Zulassungsstatus
Aufgrund der Ergebnisse aus der REALIZE-Studie und dem daraus folgenden Verzicht eines Lead-in ist
die Tripel-Therapie mit Telaprevir im Vergleich zur Therapie mit Boceprevir um zwölf Wochen kürzer.
Anders als in der Studie von Zeuzem et al. (2011) beschrieben, wird in der Fachinformation (MSD,
2014) die verkürzte Therapie von einer Gesamtdauer über 24 Wochen auch für Relapser angezeigt.
Voraussetzung dafür ist eine nicht nachweisbare HCV-RNA in Behandlungswoche 4 und 12 (Tabelle
25).
Tabelle 25: Telaprevir: Angaben nach Fachinformation
Patientenkollektiv Therapieregime
Fachinformation
MSD (2014)
Dosierung*
PEG-INF -2b: 1,5 g/kg KG 1x wöchentlich oder PEG-INF -2a: 180 g 1x wöchentlich, RBV: 1.000 mg täglich, TVR: 2.250 mg täglich
Patienten mit cHC vom Genotyp 1 (therapienaive Erwachsene und vorbehandelte Patienten mit Relap-se)1
Behandlungsdauer: 24 Wochen
TVR + PEG- -(Voraussetzung: wenn HCV-RNA in Behandlungswoche 4 und 12 nicht nach-weisbar)
Behandlungsdauer: 48 Wochen
TVR + PEG- -(Voraussetzung: wenn HCV-RNA in Behandlungswoche 4 und 12 nachweisbar)
Patienten mit cHC vom Genotyp 1 (vorbehandelte Erwachsene mit vorherigem partiel-lem Ansprechen oder Null-Responder)
Behandlungsdauer: 48 Wochen
TVR + PEG- -weitere 36 Wochen (Gesamtbehandlung 48 Wochen)
**Berechnet auf ein durchschnittliches Körpergewicht von 75 kg. Handelspräparate: Incivo® (Telaprevir), PegIntron®, Pega-sys® (PEG-Interferon -2a), Copegus® (Ribavirin).
Nach Auffassung des CHMP (EMA, 2011a) waren die Rezidiv-Raten vorheriger Relapser mit einer
nicht nachweisbaren HCV-RNA in Behandlungswoche vier und zwölf niedrig oder vergleichbar mit
den Rezidiv-Raten zuvor unbehandelter Patienten. Des Weiteren wurde vom pU eine pharmakomet-
rische Datenanalyse nachgereicht, aus der hervorging, dass „mögliche Relapser“ allein durch die
Gabe der konventionellen Therapie (PEG- e-
rücksichtigung dieser Daten betrachtete das CHMP die Gründe für eine verkürzte Behandlungszeit
153

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
als hinreichend überzeugend. Die bei der Arzneimittel-Entwicklung eingesetzten Stopp-Regeln variie-
ren zwischen den klinischen Studien. Optimale Stopp-Regeln können nicht innerhalb einer Studie in
vollem Umfang erfasst werden. Mit seinem Zulassungsantrag reichte der pU Stopp-Regeln ein, die in
keiner der Pivotalstudien angewendet wurden. Diese stellen eine Simplifikation der verschiedenen
Stopp-Regeln dar, die in den Phase-III-Studien angewendet wurden (EMA, 2011a).
Nutzenbewertung nach § 35a SGB V
Mit dem Hinweis, dass das Ausmaß des Zusatznutzens aufgrund fehlender Daten (virologisches
Ansprechen gegenüber zweckmäßiger Vergleichstherapie verbessert, Vermeidung von Folgekompli-
kationen nicht belegt) nicht quantifizierbar sei, konstatierte der G-BA in seinem Beschluss vom
29.03.2012 für die Kombination von Telaprevir mit PEG- -
n-
ten mit chronischer Hepatitis C Genotyp 1 einen Hinweis auf einen Zusatznutzen (G-BA, 2012c).
Zuvor attestierte das IQWiG in seinem Abschlussbericht einen Zusatznutzen nur bei bestimmten
Patientengruppen: So fehlte gegenüber der zweckmäßigen Vergleichstherapie ein Zusatznutzen bei
therapienaiven Patienten mit niedriger Viruslast bei Therapiebeginn, wohingegen bei hoher Viruslast
ein Vorteil anerkannt wurde (IQWiG, 2012). Auch erreichten Relapser durch die Tripel-Therapie un-
ter Anwendung von Telaprevir zusätzlich zur Standardtherapie häufiger eine virologische Freiheit. Bei
Null-Respondern sieht das IQWiG vor allem einen Vorteil für Patienten, die noch keine Zirrhose ent-
wickelt haben (IQWiG, 2012).
154

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.2.2.1.3 Simeprevir
Wie bei anderen HCV-Proteasehemmern beruht die Wirkung auf einer nicht-kovalente Hemmung
der viralen NS3/4A-Protease, welche für die Virusvermehrung von Bedeutung ist.
Zulassungsrelevant waren drei randomisierte Studien der Phase III (Forns et al., 2014; Jacobson et
al., 2014; Manns et al., 2014a). Primärer Endpunkt war jeweils das anhaltende virologische Anspre-
chen, definiert als fehlende Nachweisbarkeit der HCV-RNA im Blut mindestens zwölf Wochen nach
dem Ende der Therapie (Tabelle 26). Der ursprünglich geplante primäre Endpunkt (SVR 24) wurde –
basierend auf der engen Korrelation zwischen SVR 12 und SVR 24 aus zwei Phase-IIb-Studien – ver-
lassen und in SVR 12 geändert (EMA, 2014).
Jacobson et al. (2014; QUEST-1) untersuchten in ihrer doppelblinden Phase-III-Studie die Wirksam-
keit und Sicherheit an therapienaiven Erwachsenen, die mit dem HCV vom Genotyp 1 infiziert waren.
Nicht mit in diese Studie aufgenommen wurden u.a. Patienten mit dekompensierter Zirrhose oder
einer HCV-Infektion eines anderen Genotypus. Die Dauer der Therapie richtete sich nach dem virolo-
gischen Ansprechen auf die Behandlung. Demnach erhielten die Studienteilnehmer einmal täglich
150 mg Simeprevir über zwölf Wochen in Kombination mit der konventionellen Therapie (PEG-
-2a + Ribavirin) über insgesamt 24 oder 48 Wochen. Gut 85 Prozent der Studienpopula-
tion aus der Verum-Gruppe erfüllten die im Studienprotokoll vorgesehenen Kriterien26 einer Ant-
wort-gesteuerten Therapie und verkürzten die Studienmedikation auf eine gesamte Behandlungs-
dauer von 24 Wochen. 91 Prozent dieser Subgruppe erzielten den primären Endpunkt (SVR, definiert
als fehlende Nachweisbarkeit der HCV-RNA im Blut mindestens zwölf Wochen nach dem Ende der
Therapie). Der primäre Endpunkt in der Gesamtpopulation war signifikant gegenüber Placebo (Ja-
cobson et al., 2014).
In der Pivotalstudie QUEST-2 (Manns et al., 2014), die ein vergleichbares Design aufwies wie QUEST-
1, wurde neben PEG- -2a auch PEG- -2b eingesetzt, limitiert auf eine ausge-
wählte Anzahl von Ländern und mit einer vorgeschalteten 1:1-Randomisierung des Studienkollektivs.
In dieser Studie erfüllten 91 Prozent des Verum-Kollektivs die im Studienprotokoll vorgesehenen
Kriterien einer Antwort-gesteuerten Therapie und konnten so die Behandlungsdauer auf 24 Wochen
verkürzen. Von ihnen erzielten 86 Prozent den primären Endpunkt. Auch in dieser Studie war dieser
signifikant gegenüber Placebo (Manns et al., 2014).
26 i.e. HCV RNA < 25 IU/ml in der vierten Behandlungswoche und keine nachweisbare HCV RNA in Behandlungswoche 12
155
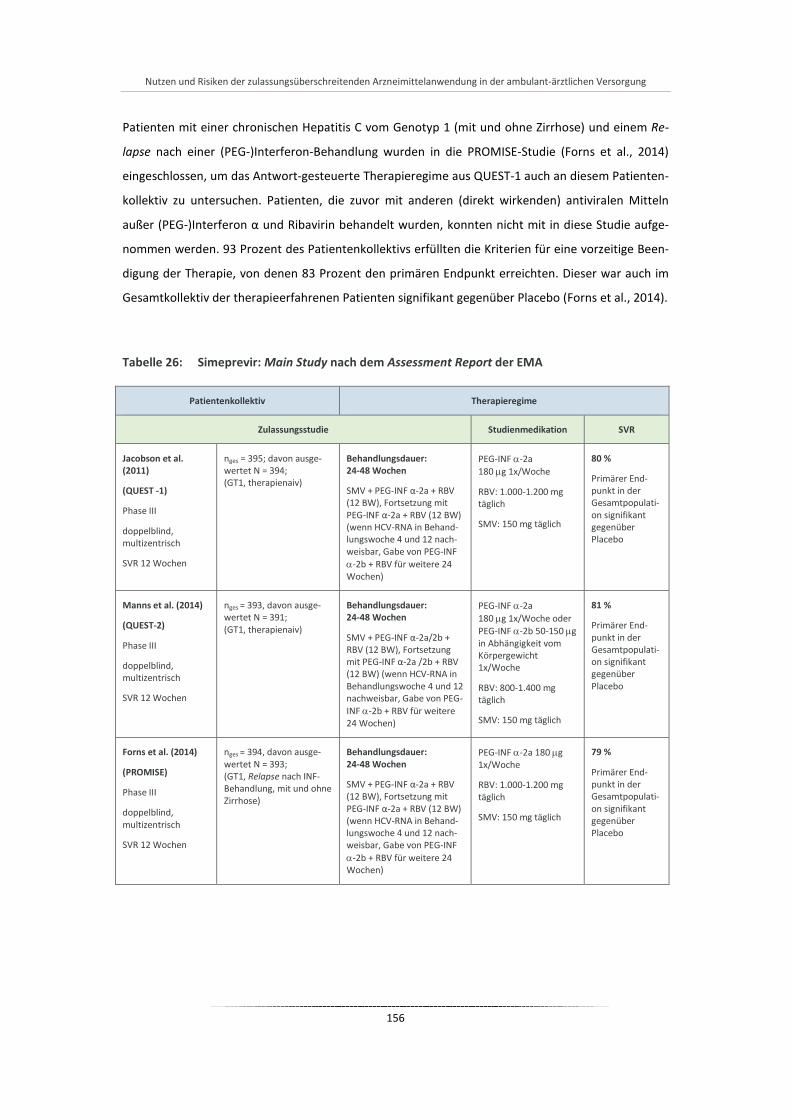
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Patienten mit einer chronischen Hepatitis C vom Genotyp 1 (mit und ohne Zirrhose) und einem Re-
lapse nach einer (PEG-)Interferon-Behandlung wurden in die PROMISE-Studie (Forns et al., 2014)
eingeschlossen, um das Antwort-gesteuerte Therapieregime aus QUEST-1 auch an diesem Patienten-
kollektiv zu untersuchen. Patienten, die zuvor mit anderen (direkt wirkenden) antiviralen Mitteln
außer (PEG-)Interferon und Ribavirin behandelt wurden, konnten nicht mit in diese Studie aufge-
nommen werden. 93 Prozent des Patientenkollektivs erfüllten die Kriterien für eine vorzeitige Been-
digung der Therapie, von denen 83 Prozent den primären Endpunkt erreichten. Dieser war auch im
Gesamtkollektiv der therapieerfahrenen Patienten signifikant gegenüber Placebo (Forns et al., 2014).
Tabelle 26: Simeprevir: Main Study nach dem Assessment Report der EMA
Patientenkollektiv Therapieregime
Zulassungsstudie Studienmedikation SVR
Jacobson et al. (2011)
(QUEST -1)
Phase III
doppelblind, multizentrisch
SVR 12 Wochen
nges = 395; davon ausge-wertet N = 394; (GT1, therapienaiv)
Behandlungsdauer: 24-48 Wochen
SMV + PEG-INF -2a + RBV (12 BW), Fortsetzung mit PEG-INF -2a + RBV (12 BW) (wenn HCV-RNA in Behand-lungswoche 4 und 12 nach-weisbar, Gabe von PEG-INF
-2b + RBV für weitere 24 Wochen)
PEG-INF -2a 180 g 1x/Woche
RBV: 1.000-1.200 mg täglich
SMV: 150 mg täglich
80 %
Primärer End-punkt in der Gesamtpopulati-on signifikant gegenüber Placebo
Manns et al. (2014)
(QUEST-2)
Phase III
doppelblind, multizentrisch
SVR 12 Wochen
nges = 393, davon ausge-wertet N = 391; (GT1, therapienaiv)
Behandlungsdauer: 24-48 Wochen
SMV + PEG-INF -2a/2b + RBV (12 BW), Fortsetzung mit PEG-INF -2a /2b + RBV (12 BW) (wenn HCV-RNA in Behandlungswoche 4 und 12 nachweisbar, Gabe von PEG-INF -2b + RBV für weitere 24 Wochen)
PEG-INF -2a 180 g 1x/Woche oder PEG-INF -2b 50-150 g in Abhängigkeit vom Körpergewicht 1x/Woche
RBV: 800-1.400 mg täglich
SMV: 150 mg täglich
81 %
Primärer End-punkt in der Gesamtpopulati-on signifikant gegenüber Placebo
Forns et al. (2014)
(PROMISE)
Phase III
doppelblind, multizentrisch
SVR 12 Wochen
nges = 394, davon ausge-wertet N = 393; (GT1, Relapse nach INF-Behandlung, mit und ohne Zirrhose)
Behandlungsdauer: 24-48 Wochen
SMV + PEG-INF -2a + RBV (12 BW), Fortsetzung mit PEG-INF -2a + RBV (12 BW) (wenn HCV-RNA in Behand-lungswoche 4 und 12 nach-weisbar, Gabe von PEG-INF
-2b + RBV für weitere 24 Wochen)
PEG-INF -2a 180 g 1x/Woche
RBV: 1.000-1.200 mg täglich
SMV: 150 mg täglich
79 %
Primärer End-punkt in der Gesamtpopulati-on signifikant gegenüber Placebo
156

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Mit der Zulassung von Simeprevir steht seit Mai 2014 der dritte spezifische Hemmstoff der NS3/4A-
Serinprotease von Hepatitis C-Viren zur Verfügung. Der Wirkstoff ist bei erwachsenen Patienten in
Kombination mit anderen Arzneimitteln (PEG-Interferon + Ribavirin oder Sofosbuvir ± Ribavirin) zur
Behandlung der chronischen Hepatitis C vom GT1 oder GT4 indiziert (Janssen, 2014). Im Unterschied
zu den bis dato verfügbaren Wirkstoffen aus der Substanzgruppe muss Simeprevir nur einmal täglich
in einer Dosis von 150 mg mit einer Mahlzeit eingenommen werden (Janssen, 2014).
Zulassungsrelevante Studien vs. Zulassungsstatus
Studiendaten zufolge lässt ein viraler NS3-Q80K-Polymorphismus die Ansprechraten auf Simeprevir
absinken (Forns et al., 2014; Jacobson et al., 2014; Manns et al., 2014), weswegen vor Therapiebe-
ginn einer Tripel-Therapie mit PEG-Interferon und Ribavirin bei HCV Genotyp-1a-Patienten ein Test
auf Viren mit NS3-Q80K-Polymorphismus durchzuführen ist, da diese Erreger gegen Simeprevir pri-
mär resistent sind (Janssen, 2014).
Partielle und Null-Responder waren nicht im Patientenkollektiv der konfirmatorischen Phase-III-
Studien enthalten. Der pU beantragte dennoch die Erweiterung der Indikation auch für diese Patien-
tengruppe und berief sich dabei auf Wirksamkeitsbelege aus einer Phase-IIb-Studie (C206; EMA,
2014). Aufgrund des hohen Anteils an Patienten, die die Kriterien für eine kurze Behandlungszeit
erfüllten, wurde von Seiten des pU die Therapiedauer von insgesamt 24 Wochen beantragt. Partielle
und Null-Responder wurden davon ausgenommen (Tabelle 27). Daten, die die Wirksamkeit bei cHC-
Patienten mit GT4 belegen sollten und auf deren (Zwischen-)Ergebnisse die Zulassung beruht, wur-
den aus einer zum Zeitpunkt des Zulassungsantrags noch laufenden Studie entnommen (HPC 3011)
(EMA, 2014). Laut Assessment Report der EMA liegen mittlerweile auch Daten der COSMOS-Studie
vor (Lawitz et al., 2014a), die die Wirksamkeit und Sicherheit von Simeprevir in Kombination mit
Sofosbuvir und eventuell zusätzlicher Gabe von Ribavirin bei Patienten mit cHC vom GT1 untersuch-
ten. Die Wirkstoffkombination wurde über zwölf und 24 Wochen getestet. Aufgrund vergleichbarer
hoher SVR-Raten wurde eine antivirale Therapie über zwölf Wochen als Standard gewählt. Eine 24-
wöchige Behandlung sollte nur im Einzelfall in Erwägung gezogen werden (Tabelle 27). Die zusätzli-
che Gabe von Ribavirin hatte keinen Einfluss auf eine bessere SVR-Rate (Lawitz et al., 2014a). Auf-
grund der limitierten Daten sollte sie dennoch in Abhängigkeit der patientenindividuellen Situation
erfolgen. Daten für die Therapie der HCV vom GT4 lagen zum Zeitpunkt der Zulassung nicht vor
(EMA, 2014).
157

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 27: Simeprevir: Angaben nach Fachinformation
Patientenkollektiv Therapieregime
Fachinformation
Janssen (2014) Dosierung*
PEG-INF -2b: 1,5 g/kg KG 1x wöchentlich oder PEG-INF -2a: 180 g 1x wöchentlich, RBV: 1.000 mg täglich, SOF: 400 mg täglich, SIM 150 mg täglich über 12 Wochen
Patienten mit cHC vom Genotyp 1 oder 4 (therapienaive und therapieerfahrene Patienten, mit/ohne Zirrhose, mit/ohne HIV-Koinfektion)
Behandlungsdauer: 24 Wochen1
SIM + PEG- -INF + RBV (12 BW)
Patienten mit cHC vom Genotyp 1 oder 4 (vorherige Non-Responder, einschließlich partieller und Null-Responder, mit/ohne Zirrhose, mit/ohne HIV-Koinfektion)
Behandlungsdauer: 48 Wochen
SIM + PEG- -INF + RBV (36 BW)
Patienten mit cHC vom Genotyp 1 oder 4 (unabhängig von vorherigen Behandlungen, mit/ohne Zirrhose)
Behandlungsdauer: 12 Wochen
SIM + SOF (± RBV)
*Berechnet auf ein durchschnittliches Körpergewicht von 75 kg. Handelspräparate: Olysio (Simeprevir), Sovaldi (Sofos-buvir), PegIntron , Pegasys (PEG-Interferon -2a), Copegus (Ribavirin). 1Therapienaive Patienten und vorherige Relapser mit Zirrhose und HIV-Koinfektion sollten 48 Wochen behandelt werden. Die Therapie mit Simeprevir muss in Kombination mit PEG- e-leitet werden, gefolgt von einer weiteren 36-wöchigen Behandlung mit PEG-
Nutzenbewertung nach § 35a SGB V
Auf Basis der vorliegenden Daten kam das IQWiG in seiner Nutzenbewertung (IQWiG, 2014) zu dem
Ergebnis, dass der Zusatznutzen für Simeprevir für Patienten mit Genotyp 4 und für Patienten mit
Koinfektionen aus HCV und HIV wegen unzureichender Datenlage gegenüber der zweckmäßigen
Vergleichstherapie nicht bestimmbar sei. Bei Patienten mit HCV-Infektionen Genotyp 1 ohne Vorbe-
handlung oder mit einem Relapse nach Vorbehandlung sah das IQWiG hingegen einen Hinweis auf
einen Zusatznutzen gegenüber PEG-Int n, allerdings ohne quantifizierbaren Aus-
maß. So konnte ein deutlicher Effekt beim virologischen Ansprechen aufgezeigt werden, allerdings
ließe sich wegen fehlender Studiendaten keine Aussage zu den Auswirkungen auf relevante Folgeer-
krankungen treffen. Einen Hinweis auf einen erheblichen Zusatznutzen sah das IQWiG bei Non-
Respondern mit HCV Genotyp 1 im Vergleich zu einer Tripel-Therapie aus PEG-
Ribavirin und einem weiteren Protease-Inhibitor (Telaprevir).
158

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Der Gemeinsame Bundesausschuss wich in seiner Beschlussfassung zum Zusatznutzen von Simepre-
vir von den Bewertungen des IQWiG ab. Er sah für therapienaive Patienten (mit und ohne Zirrhose)
mit Genotyp 1 sowie für therapieerfahrene Patienten (Relapser) mit Genotyp 1 gegenüber der
zweckmäßigen Vergleichstherapie, bestehend aus Ribavirin + PEG- Hinweis auf
einen beträchtlichen Zusatznutzen (G-BA, 2014). Diesen konstatierte der G-BA auch für die Tripel-
Therapie bestehend aus Simeprevir + PEG-
(Non-Responder) im Vergleich zu einer Behandlung mit Telaprevir oder Boceprevir + PEG-Interferon
-basierte Tripel-Therapie gegenüber PEG-
+ Ribavirin ein Anhaltspunkt auf einen geringen Zusatznutzen bei therapienaiven und therapieer-
fahrenen Patienten mit Genotyp 4 gesehen (G-BA, 2014).
159

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.2.2.2 NS5A-Inhibitoren
8.2.2.2.1 Daclatasvir
Das virale Nichtstrukturprotein NS5A (Non-Structural Protein 5A) fungiert als multifunktionelles Pro-
tein als wesentlicher Bestandteil des HCV-Replikationskomplexes. Daclatasvir hemmt als NS5A-
Inhibitor selektiv die virale RNA-Replikation wie auch die Viruszusammensetzung (virus assembly).
Erkenntnisse zu klinischen Erfahrungen des NS5A-Inhibitors lieferten bislang in erster Linie Phase-II-
Studien und eine bis dato unveröffentlichte Phase-III-Studie (EMA, 2014a) mit teilweise geringen
Patientenzahlen.
Tabelle 28: Daclatasvir: Main Study nach dem Assessment Report der EMA
Patientenkollektiv Therapieregime
Zulassungsstudie Studienmedikation SVR
Sulkowski et al. (2014b)
Phase IIb
offen
SVR 12 Wochen
nges = 211, davon ausge-wertet N = 211, (davon GT1 n = 126 therapienaiv, n = 41 therapieerfahren; GT2/3 n = 44 therapiena-iv)
Behandlungsdauer: 12-24 Wochen
DCV + SOF ± RBV
RBV: 1.000-1.200 mg täglich
DCV: 60 mg täglich
SOF: 400 mg täglich
86-100 %
Keine spezifischen statisti-schen Angaben für die ver-schiedenen Genotypen.
PE = Primärer Endpunkt. GK = Gesamtkollektiv.
Die Pivotalstudie von Sulkowski et al. (2014b) (Tabelle 28) evaluierte die Sicherheit, Pharmakokinetik
und Pharmakodynamik von Sofosbuvir in Kombination mit Daclatasvir sowie mit bzw. ohne Ribavirin
in therapienaiven Patienten mit cHC vom Genotyp 1-3. Patienten mit cHC vom Genotyp 1 und vorhe-
rigem Versagen auf eine Telaprevir- oder Boceprevir-Tripel-Therapie wurden später mit eingeschlos-
sen. Allerdings machten die therapieerfahrenen Patienten nur knapp ein Fünftel des Patientenkollek-
tivs aus. Primärer Endpunkt war auch hier die SVR, definiert als fehlende Nachweisbarkeit der HCV-
RNA im Blut mindestens zwölf Wochen nach dem Ende der Therapie. Die Studie bestand aus insge-
samt zehn Behandlungsgruppen. Zwei Gruppen erhielten ein siebentägiges Lead-in mit Sofosbuvir.
Ziel war, durch Erreichen eines steady state einer späteren Resistenzvariante vorzubeugen. Da diese
Strategie keine Auswirkung zeigte, wurde sie kurze Zeit später verworfen und die Patienten aus den
160

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
beiden Behandlungsarmen in die nachfolgenden Gruppen aufgeteilt. Verglichen wurde zum einen
die Behandlungsdauer (12 vs. 24 Wochen) sowie unterschiedliche Therapieregime (Daclatasvir +
Sofosbuvir als duale Therapie bzw. als Tripel-Therapie mit Ribavirin). Es wurde auf einen Behand-
lungsarm mit der derzeitigen Standardtherapie bestehend aus PEG- + Ribavirin und ge-
gebenenfalls einem weiteren, direkt antiviral wirkenden Protease-Inhibitor wie Boceprevir oder
Telaprevir verzichtet. Patienten mit cHC vom Genotyp 2 und 3 und vorherigem Therapieversagen auf
eine konventionelle Therapie mit bzw. ohne Gabe eines zusätzlichen Protease-Inhibitors (Boceprevir
oder Telaprevir) sowie zirrhotische Patienten waren nicht im Studienkollektiv enthalten (Sulkowski et
al., 2014b). Die Ansprechraten ließen sich durch den Ribavirin-Zusatz gegenüber der dualen Therapie
nicht steigern. Auf Basis vergleichsweise geringer Patientenzahlen ergeben sich unter Daclatasvir
hohe Heilungsraten nach Behandlungsende bei Patienten mit chronischer Hepatitis C vom Genotyp
1, mit und ohne Vorbehandlung. Aussagen zur Wirksamkeit bei Patienten mit fortgeschrittener Le-
bererkrankung lassen sich aus dieser Studie nicht ziehen. Für eine 24-wöchige Daclatasvir-
Behandlung bei nicht vorbehandelten Patienten findet sich bei Patienten mit HCV vom Genotyp 4
gegenüber der Standardtherapie aus PEG-Int
Überlegenheit in Bezug auf die Virusfreiheit zwölf bzw. 24 Wochen nach Abschluss der Behandlung
(Sulkowski et al., 2014b).
Mit Daclatasvir wurde im August 2014 der erste NS5A-Inhibitor zur Behandlung der chronischen
Hepatitis C zugelassen. In Abhängigkeit vom klinischen Bild der Erkrankung kann der Wirkstoff als
Erst- und Eskalationstherapie bei Infektion mit HCV Genotyp 1, 3 und 4 eingesetzt werden. Auch bei
Daclatasvir besteht die Gefahr sich leicht ausbildender Resistenzen. So besitzt der Wirkstoff, ange-
wendet als einzige antivirale Substanz, eine niedrige Resistenzbarriere aufgrund sind schnell ausbil-
dender Punktmutationen. Dies gilt vor allem in Bezug auf Genotyp 1a (McPhee et al., 2014; Wong et
al., 2013; Murakami et al., 2014). Für den Genotyp 3 steht mit Daclatasvir nun auch eine Interferon-
freie Tripel-Therapie (bestehend aus Daclatasvir, Sofosbuvir und Ribavirin) zur Verfügung (BMS,
2014). Die zugelassene Dosis liegt bei einmal täglich 60 mg.
Zulassungsrelevante Studien vs. Zulassungsstatus
Die Interferon-freie Anwendung bei Genotyp-4-Infektionen beruht bislang nur auf eine Extrapolation
der Genotyp-1-Daten (EMA, 2014a). Einer zusätzlichen Gabe von Ribavirin zur Optimierung der The-
rapie bedarf bei HCV-Infektionen mit den Genotypen 1 und 4 nur in Einzelfällen (Tabelle 29). Zum
Zulassungszeitpunkt können nur begrenzte Behandlungsempfehlungen für die Genotypen 2 und 3
161

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
ausgesprochen werden. Zugelassen wurde Daclatasvir letztendlich nur zur Therapie des HVC vom
Genotyp 3. Das CHMP begründet seine Empfehlung mit dem zugelassen Interferon-freien Therapie-
Regime bestehend aus Sofosbuvir und Ribavirin über eine Behandlungszeit von 24 Wochen für diese
Subpopulation (EMA, 2014a). Für Patienten mit negativen prognostischen Faktoren stellt eine zusätz-
liche Gabe von Daclatasvir eine weitere Behandlungsoption dar, sollte eine Gabe von PEG-Interferon
Einer Therapie-Verkürzung auf zwölf Behandlungswochen wurde nicht stattge-
geben. Auch wurde aufgrund von Limitierungen in der pivotalen Studie von Seiten des CHMP eine
Behandlung von Patienten mit cHC vom Genotyp 2 nicht empfohlen (EMA, 2014a).
Bei nicht zirrhotischen Patienten mit cHC vom Genotyp 1 oder 4 wird eine zwölfwöchige Behand-
lungsdauer empfohlen, die nur in Einzelfällen auf insgesamt 24 Wochen verlängert werden sollte
(Tabelle 29). Da die Pivotalstudie keinen Nachweis eines Benefits durch zusätzliche Gabe von
Ribavirin erbringen konnte, sollte auch hier die Tripel-Therapie nur in Ausnahmefällen eingesetzt
werden (EMA, 2014a).
Da die verfügbaren Daten zu Wirksamkeit und Sicherheit der dualen Kombination (Daclatasvir +
Sofosbuvir) zur Behandlung zirrhotischer Patienten nur begrenzt vorliegen und die optimale Behand-
lungsdauer sowie das Nutzenpotential einer zusätzlichen Ribavirin-Gabe durch die Pivotalstudie bis
zum Zeitpunkt der Zulassung noch nicht in vollem Umfang abgeklärt werden konnte, wurden von
Seiten des CHMP Daten aus anderen Studien mit vergleichbaren Wirkstoffkombinationen hinzugezo-
gen. Diese belegen zwar eine Wirksamkeit von Daclatasvir bei diagnostizierter Leberzirrhose, geben
allerdings keinen Aufschluss bezüglich der Behandlungsdauer bzw. dem Nutzen einer Ribavirin-Gabe.
Die Datenlage zur Behandlung zirrhotischer Patienten lässt keine gesicherten Aussagen zu. Allerdings
konnten auch keine Nebenwirkungen dokumentiert werden, die sich bei dieser Patientenpopulation
als schwerwiegender herausstellen würden. Aus diesem Grund wurde der Behandlung von Patienten
mit kompensierter Zirrhose stattgegeben (EMA, 2014a).
Daten für die Behandlung therapieerfahrener Patienten liegen nur für Genotyp-1-Infektionen vor.
162

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 29: Daclatasvir: Angaben nach Fachinformation
Patientenkollektiv Therapieregime
Fachinformation
BMS (2014) Dosierung*
PEG-INF -2b: 1,5 g/kg KG 1x wöchentlich oder PEG-INF -2a: 180 g 1x wöchentlich, RBV: 1.000 mg täglich, DCV: 60 mg täglich
Duale Therapie/Tripel-Therapie
Patienten mit cHC vom Genotyp 1 oder 4 (ohne Zirrhose)
Behandlungsdauer: 12 Wochen1:
DCV + SOF (BW 12)1
Patienten mit cHC vom Genotyp 1 oder 4 (mit kompensierter Zirrhose)
Behandlungsdauer: 24 Wochen2:
DCV + SOF (24 BW)
Patienten mit cHC vom Genotyp 3 (mit kompen-sierter Zirrhose und/oder therapieerfahren)
Behandlungsdauer: 24 Wochen:
DCV + SOF + RBV (24 BW)
Patienten mit cHC vom Genotyp 4
Behandlungsdauer: 24-48 Wochen:
DCV + PEG- (Voraussetzung: wenn HCV-RNA in Behandlungswoche 4 und 12 nicht nachweisbar)
DCV + PEG- ung mit PEG- + RBV (24 BW) (Voraussetzung: wenn HCV-RNA in Behandlungswoche 4 und 12 nachweisbar, sollte Daclatasvir nach 24 Wochen abgesetzt und die duale Standard-Therapie für eine Ge-samtdauer von 48 Wochen weitergeführt werden)
*Berechnet auf ein durchschnittliches Körpergewicht von 75 kg. Handelspräparate: Daklinza® (Daclatasvir), PegIntron®, Pe-gasys® (PEG-Interferon -2a), Copegus® (Ribavirin). 1Bei vorbehandelten Patienten, deren Therapie auch einen NS3/4A-Proteaseinhibitor beinhaltet, ist zu erwägen, die Behand-lung auf 24 Wochen zu verlängern. (BMS, 2014). 2Bei vorher unbehandelten Patienten mit Zirrhose und positiven Prognosefaktoren, wie IL28B-CC-Genotyp und/oder niedrige Ausgangsvirenlast, kann erwogen werden, die Behandlung auf 12 Wochen zu verkürzen. Bei Patienten mit weit fortgeschritte-ner Lebererkrankung oder anderen negativen Prognosefaktoren, wie z.B. Vorbehandlung, kann die zusätzliche Anwendung von Ribavirin erwogen werden (BMS, 2014).
163

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Nutzenbewertung nach § 35a SGBV
Im Februar 2015 veröffentlichte der G-BA in seiner Beschlussfassung die frühe Nutzenbewertung von
Daclatasvir. Während der Ausschuss für therapienaive Patienten vom Genotyp 4 gegenüber der
Tripel-Therapie (Daclatasvir in Kombination mit PEG-Interferon plus Ribavirin) einen Anhaltspunkt
für einen beträchtlichen Zusatznutzen im Verhältnis zur zweckmäßigen Vergleichstherapie (hier:
PEG-Interferon plus Ribavirin) aussprach, sah er für therapienaive Patienten vom Genotyp 1 und
ohne Zirrhose nur einen Anhaltspunkt für einen geringen Zusatznutzen (zweckmäßige Vergleichs-
therapie bestehend aus PEG-Interferon + Ribavirin + Boceprevir oder Telaprevir) (G-BA, 2015).
Für alle anderen Subgruppen (therapienaive Patienten mit kompensierter Zirrhose vom Genotyp 1,
therapieerfahrene Patienten vom Genotyp 1, therapienaive Patienten mit kompensierter Zirrhose
und therapieerfahrene Patienten vom Genotyp 3, therapienaive und therapieerfahrene Patienten
vom Genotyp 4) die Gegenstand der Fragestellung waren, sah der G-BA den Zusatznutzen von Dacla-
tasvir mit Sofosbuvir und gegebenenfalls einer zusätzlichen Gabe von Ribavirin im Verhältnis zur
zweckmäßigen Vergleichstherapie (PEG-Interferon + Ribavirin (+ Boceprevir/Telaprevir) als nicht
belegt an (G-BA, 2015). Mit seinem Beschluss geht der G-BA in zwei Entscheidungen nicht konform
mit der Auffassung des IQWiG. Das Institut konnte aufgrund mangelnder Daten und methodischer
Mängel (fehlende Daten für bestimmte Patientenkollektive, methodische Anforderungen des IQWiG
an indirekte Vergleiche nicht erfüllt, Studiendaten wiesen teilweise ein hohes Verzerrungspotential
aufgrund schwerwiegender Mängel bei der Studiendurchführung auf) Daclatasvir in keinem der zu-
gelassenen Indikationen einen Zusatznutzen attestieren (IQWiG, 2014a).
164
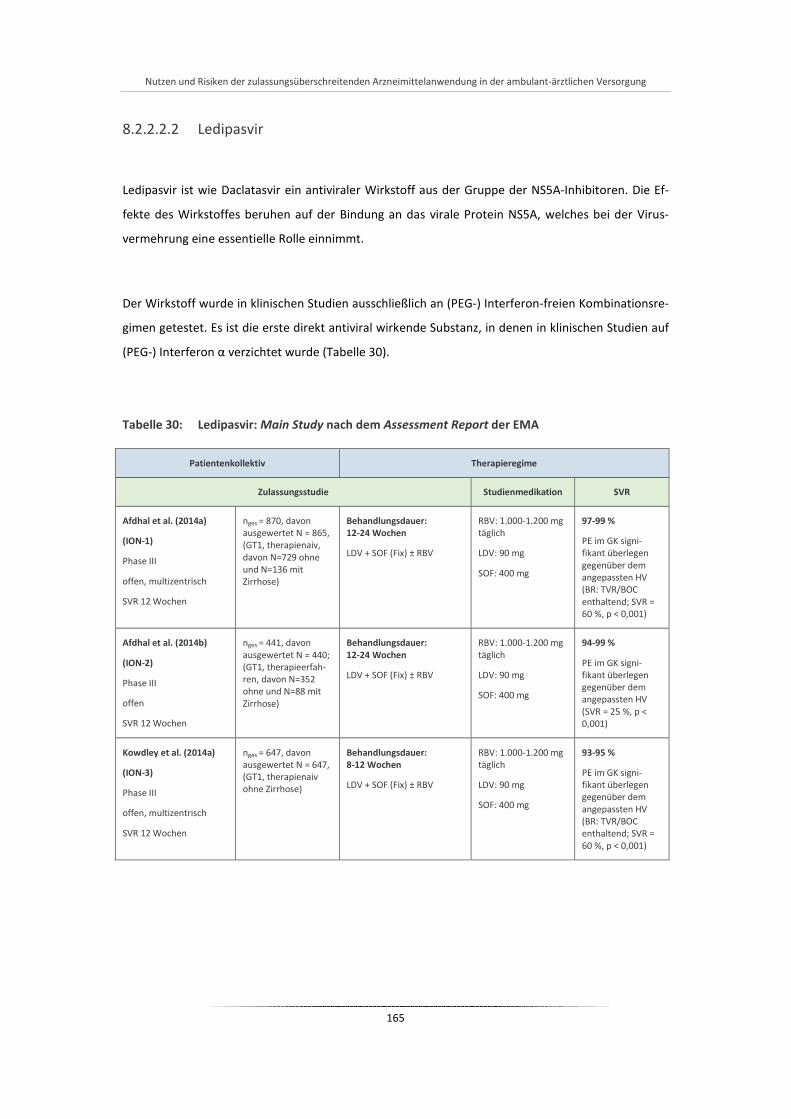
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.2.2.2.2 Ledipasvir
Ledipasvir ist wie Daclatasvir ein antiviraler Wirkstoff aus der Gruppe der NS5A-Inhibitoren. Die Ef-
fekte des Wirkstoffes beruhen auf der Bindung an das virale Protein NS5A, welches bei der Virus-
vermehrung eine essentielle Rolle einnimmt.
Der Wirkstoff wurde in klinischen Studien ausschließlich an (PEG-) Interferon-freien Kombinationsre-
gimen getestet. Es ist die erste direkt antiviral wirkende Substanz, in denen in klinischen Studien auf
(PEG-) Interferon verzichtet wurde (Tabelle 30).
Tabelle 30: Ledipasvir: Main Study nach dem Assessment Report der EMA
Patientenkollektiv Therapieregime
Zulassungsstudie Studienmedikation SVR
Afdhal et al. (2014a)
(ION-1)
Phase III
offen, multizentrisch
SVR 12 Wochen
nges = 870, davon ausgewertet N = 865, (GT1, therapienaiv, davon N=729 ohne und N=136 mit Zirrhose)
Behandlungsdauer: 12-24 Wochen
LDV + SOF (Fix) ± RBV
RBV: 1.000-1.200 mg täglich
LDV: 90 mg
SOF: 400 mg
97-99 %
PE im GK signi-fikant überlegen gegenüber dem angepassten HV (BR: TVR/BOC enthaltend; SVR = 60 %, p < 0,001)
Afdhal et al. (2014b)
(ION-2)
Phase III
offen
SVR 12 Wochen
nges = 441, davon ausgewertet N = 440; (GT1, therapieerfah-ren, davon N=352 ohne und N=88 mit Zirrhose)
Behandlungsdauer: 12-24 Wochen
LDV + SOF (Fix) ± RBV
RBV: 1.000-1.200 mg täglich
LDV: 90 mg
SOF: 400 mg
94-99 %
PE im GK signi-fikant überlegen gegenüber dem angepassten HV (SVR = 25 %, p < 0,001)
Kowdley et al. (2014a)
(ION-3)
Phase III
offen, multizentrisch
SVR 12 Wochen
nges = 647, davon ausgewertet N = 647, (GT1, therapienaiv ohne Zirrhose)
Behandlungsdauer: 8-12 Wochen
LDV + SOF (Fix) ± RBV
RBV: 1.000-1.200 mg täglich
LDV: 90 mg
SOF: 400 mg
93-95 %
PE im GK signi-fikant überlegen gegenüber dem angepassten HV (BR: TVR/BOC enthaltend; SVR = 60 %, p < 0,001)
165

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Die klinischen Untersuchungen (Afdhal et al., 2014a; Afdhal et al., 2014b; Kowdley et al., 2014a), die
letztendlich die zulassungsrelevanten Daten lieferten, waren allesamt unverblindet. Die Fixkombina-
tion aus Sofosbuvir und Ledipasvir mit und ohne Ribavirin wurde in den o.g. Studien an therapienai-
ven wie auch an vorbehandelten Patienten mit cHC Genotyp 1 getestet (Tabelle 30).
In den Studien ION-1 (Afdhal et al., 2014a) und ION-3 (Kowdley et al., 2014a) wurden therapienaive
Patienten mit cHC vom Genotyp 1 eingeschlossen, um Wirksamkeit und Sicherheit von Ledipasvir in
fixer Kombination mit Sofosbuvir zu untersuchen. 20 Prozent der ION-1-Studienpopulation wiesen
eine Zirrhose auf. Hohe Response-Raten konnten sowohl in ION-1 (Behandlungsdauer: zwölf bis 24
Wochen) als auch in ION-3 (Behandlungsdauer: acht bis zwölf Wochen) erzielt werden. Die Anzahl
der Patienten, die eine Zirrhose aufwiesen und zwölf Wochen therapiert wurden, war allerdings zu
gering, um definitive Rückschlüsse hinsichtlich einer geeigneten Therapiedauer (zwölf oder 24 Wo-
chen) ziehen zu können. Sehr gute Ergebnisse erzielte hingegen die zwölfwöchige Behandlung in
ION-3 (Kowdley et al., 2014b), die Relapse-Rate nach achtwöchiger Behandlung war allerdings höher
als die nach zwölf Wochen (5,1 bzw. 4,2 Prozent vs. 0,9 Prozent), ungeachtet einer zusätzlichen Gabe
von Ribavirin. Diese Studie gab allerdings Hinweise darauf, dass bei therapienaiven Patienten mög-
licherweise eine Verkürzung der Behandlungsdauer auf acht Wochen denkbar ist, ohne dass die
Erfolgsraten sinken. In der ION-2-Studie (Afdhal et al., 2014b) wurde die Wirksamkeit der dualen
Kombination an Patienten untersucht, die ein Therapieversagen auf eine vorangegangene Therapie
(konventionelle Therapie, eventuell in Kombination mit Boceprevir oder Telaprevir) aufwiesen. In
allen drei ION-Studien wurden hohe SVR-Raten von bis zu 99 Prozent erzielt. Der primäre Endpunkt
im Gesamtkollektiv war signifikant überlegen gegenüber dem angepassten historischen Vergleich. An
einem kleinen Patientenkollektiv mit dekompensierter Zirrhose (n = 20) wurde die duale Fixkombina-
tion ohne einer zusätzlichen Gabe von Ribavirin getestet. Mit 35 Prozent war der Anteil der Relapser
überdurchschnittlich hoch (ELECTRON-2, EMA, 2014b). Bei der größer angelegten SOLAR-1-Studie
(EMA, 2014b) entschied man sich stattdessen für die Fixkombination plus Ribavirin. Eingeschlossen
waren Patienten mit cHC vom Genotyp 1 oder 4, dekompensierter Zirrhose oder Lebertransplantati-
on. Die Behandlung über zwölf bzw. 24 Wochen erwies sich auch in dieser Patientengruppe als effek-
tiv. Zu anderen HCV-Genotypen lagen zur Zeit der Zulassung nur begrenzte Daten vor. Bei Versagen
einer Behandlung mit Ledipasvir und Sofosbuvir konnte ferner eine Selektion von HCV mit Mutatio-
nen im Nicht-Strukturproteins NS5A beobachtet werden, die zu einer deutlichen Herabsetzung der
Empfindlichkeit gegenüber Ledipasvir führt und sich auch in der langfristigen Nachbeobachtung nicht
zurückbildeten (EMA, 2014b).
Ledipasvir besitzt seit dem 17. November 2014 in Deutschland als fixe Kombination zusammen mit
Sofosbuvir eine Zulassung (Gilead, 2014). Das Mittel ist zur Erst- und Re-Therapie von Patienten mit
166

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
chronischer Hepatitis C Genotyp 1 und 4 mit und ohne Zirrhose zugelassen. Liegt eine schwere Le-
bererkrankung oder eine Lebertransplantation vor, kann die kombinierte Anwendung mit Ribavirin
(Tripel-Therapie) eingesetzt werden. Dies gilt auch für Patienten mit chronischer Hepatitis C Genotyp
3, bei denen ein Therapieversuch bereits versagt hat (Tabelle 31). Die fixe Kombination aus 90 mg
Ledipasvir und 400 mg Sofosbuvir wird einmal täglich verabreicht (Gilead, 2014). Bereits seit Februar
2014 lag eine positive Stellungnahme der Europäischen Zulassungsbehörde zur kombinierten An-
wendung von Ledipasvir zusammen mit dem NS5B-Polymeraseinhibitor Sofosbuvir vor. Die Gutach-
ter befürworteten damals die Fixkombination für den Einsatz bei Patienten mit sehr weit fortge-
schrittener Lebererkrankung aufgrund einer chronischen Hepatitis C vom Genotyp 1.
Zulassungsrelevante Studien vs. Zulassungsstatus
In den drei zulassungsrelevanten Studien zeigte sich, dass die Zweierkombination unabhängig von
der Ausprägung der Lebererkrankung (mit/ohne Zirrhose), den Polymorphismen beim Patientenkol-
lektiv (IL28b-Genotyp) sowie dem vorangehenden Therapieversagen zu hohen Erfolgsraten führte.
Obwohl eine Kombination mit Ribavirin die Erfolgsraten der Zweierkombination aus Sofosbuvir und
Ledipasvir nicht relevant steigern konnte, dies sogar vermehrt zu unerwünschten Wirkungen wie
Anämie, Hyperbilirubinämie und Rash führte (EMA, 2014b), wird die Tripel-Therapie als vertretbar
angesehen, wenn eine weit fortgeschrittene Lebererkrankung vorliegt, bei der kein Therapieversa-
gen in Kauf genommen werden kann (EMA, 2014b) (Tabelle 31). Eine Kreuzresistenz zwischen
NS3/4A-Protease-Inhibitoren und der fixen Kombination kann ausgeschlossen werden (EMA, 2014b).
Gerade für Relapser nach vorangegangener Therapie mit einem Protease-Inhibitor kann so die
Fixkombination eine weitere Behandlungsoption darstellen. Die pivotalen Studien haben gezeigt,
dass eine kurze Therapiedauer auch mit einer höheren Relapse-Rate einhergehen kann. So sollten
nach Angaben der Fachinformation beispielsweise Patienten mit kompensierter Zirrhose grundsätz-
lich über 24 Wochen behandelt werden (Gilead, 2014). Eine kürzere Behandlungsdauer sollte nach
Auffassung des CHMP nur unter Nutzen-Risiko-Betrachtung für bestimmte Subgruppen (Tabelle 31)
vorbehalten sein (EMA, 2014b). Die Zulassung für die Behandlung (schwerer) cHC-Infektionen mit
den Genotypen 3 oder 4 beruht auf einer sehr begrenzten Anzahl von Patienten aus Phase-II-Studien
bzw. aus noch laufenden Untersuchungen. So wurde die klinische Wirksamkeit bei einer kleinen
Patientenzahl (n = 51) mit cHC vom Genotyp 3 in einer offenen Phase-II-Studie mit oder ohne
Ribavirin bei therapienaiven Patienten mit oder ohne Zirrhose untersucht. Noch geringer ist die in
den klinischen Studien untersuchten Anzahl an Patienten mit cHC vom Genotyp 4: in einer Phase-II-
Studie zur Beurteilung von Ledipasvir/Sofosbuvir über zwölf Wochen wurden insgesamt nur 21 Pati-
enten behandelt (EMA, 2014b).
167

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Virale klinische Daten, die eine direkte Wirkung von Ledipasvir auf den Genotyp 3 belegen, sind nicht
vorhanden. Auch gibt es keine eindeutigen Beweise für einen Selektionsdruck. Rückschlüsse auf eine
Wirksamkeit der Fixkombination zieht das Komitee aus Querschnittsstudien (EMA, 2014b). So erziel-
ten in Phase-III-Studien 60 Prozent der therapienaiven Patienten mit cHC vom Genotyp 3 nach allei-
niger Gabe von Sofosbuvir + Ribavirin über eine Therapiedauer von zwölf Wochen eine SVR (EMA,
2014b). Das erlaubt nach Auffassung des CHMP den Rückschluss auf eine klinisch relevante Aktivität
von Ledipasvir in der antiviralen Therapie des GT3. Eine Zulassung zur Therapie der HCV-Infektion
vom Genotyp 4 wird ebenfalls mit in-vitro-Daten und Ergebnissen aus klinischen Studien mit ver-
gleichbaren Therapieregimen begründet (EMA, 2014b).
Tabelle 31: Ledipasvir: Angaben nach Fachinformation
Patientenkollektiv Therapieregime
Fachinformation
Gilead (2014)
Dosierung*
RBV: 1.000-1.200 mg täglich, LDV: 90 mg, SOF:400 mg
Duale Therapie / Tripel-Therapie
Patienten mit cHC vom Genotyp 1,4, ohne Zirrhose
Behandlungsdauer: 8-24-Wochen
Ledipasvir + Sofosbuvir
Patienten mit cHC vom Genotyp 1,4, mit kompensierter Zirrhose
Behandlungsdauer: 12-24 Wochen
Ledipasvir + Sofosbuvir
Patienten mit cHC vom Genotyp 1,4, mit dekompensierter Zirrhose bzw. vor oder nach Leber-transplantation
Behandlungsdauer: 24 Wochen
Ledipasvir + Sofosbuvir + Ribavirin
Patienten mit cHC vom Genotyp 3, mit Zirrhose und/oder Versagen einer vorherigen Behand-lung
Behandlungsdauer: 24-48 Wochen
Ledipasvir + Sofosbuvir + Ribavirin
PE = Primärer Endpunkt. HV = Historischer Vergleich. BR = Behandlungsregime. TVR = Telaprevir. BOC = Boceprevir. GK = Gesamtkollektiv.
*Berechnet auf ein durchschnittliches Körpergewicht von 75 kg. Handelspräparate:Harvoni® (Ledipasvir + Sofosbuvir), Cope-gus® (Ribavirin).
168

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Nutzenbewertung nach § 35a SGB V
Derzeit wird die Beschlussfassung des G-BA für das Nutzenbewertungsverfahren der fixen Wirkstoff-
kombination, bestehend aus Ledipasvir und Sofosbuvir, vorbereitet. Sie wird Ende Mai 2015 erwar-
tet.
Die Stellungnahme des IQWiG, welches zuvor mit der Nutzenbewertung beauftragt worden war,
liegt mittlerweile vor. Das Institut bewertete den Zusatznutzen für mehrere Subgruppen. Darunter
fielen Patienten mit cHC und Genotyp 1 (therapienaiv mit bzw. ohne Zirrhose, therapieerfahren,
therapienaiv und therapieerfahren mit HIV-Koinfektion), Patienten mit dekompensierter Zirrhose
(Genotyp 1/4), therapienaive Patienten vom Genotyp 3 mit Zirrhose oder therapieerfahrene Patien-
ten vom Genotyp 3 sowie therapienaive und therapieerfahrene Patienten mit chronischer Hepatitis
C vom Genotyp 4 (IQWiG, 2015). Der pharmazeutische Unternehmer legte jedoch nur zu einem Teil
der Fragestellungen Daten vor.
Für therapienaive Patienten (cHC GT1) ohne Zirrhose sowie für therapieerfahrene Patienten mit
chronischer Hepatitis C vom Genotyp 1 sah das IQWiG in der Fixkombination Sofosbuvir/Ledipasvir
einen Anhaltspunkt für einen nicht quantifizierbaren Zusatznutzen in der Kategorie „schwerwiegen-
de Folgekomplikationen“ im Verhältnis zur zweckmäßigen Vergleichstherapie (hier duale Therapie
bestehend aus PEG-Interferon + Ribavirin oder Tripel-Therapie bestehend aus PEG-Interferon +
Ribavirin + Boceprevir/Telaprevir). Für alle anderen Subgruppen und Kategorien sah das IQWiG auf-
grund mangelnder Daten den Zusatznutzen als nicht belegt an (IQWiG, 2015).
169

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.2.2.3 Nukleosidische Polymerase (NS5B)-Inhibitoren
8.2.2.3.1 Sofosbuvir
Bei Sofosbuvir handelt es sich um ein Nukleotid-Prodrug, das nach intrazellulärer Metabolisierung in
das pharmakologisch wirksame Uridin-Analogon-Triphosphat mittels der NS5B-Polymerase in die
HCV-RNA eingebaut wird und im Rahmen der Replikation zum Kettenabbruch führt (Gilead, 2015).
Mittlerweile liegen eine Vielzahl von klinischen Phase-II- und Phase-III-Studien vor, die die Wirksam-
keit und Sicherheit sowohl an therapienaiven als auch an therapieerfahrenen Patienten untersuch-
ten und zulassungsrelevante Daten lieferten (siehe Tabelle 32).
In der NEUTRINO-Studie (Lawitz et al., 2013a) wurde die Wirksamkeit und Sicherheit von Sofosbuvir
in Kombination mit der konventionellen Therapie über eine Behandlungsdauer von zwölf Wochen
getestet. Eingeschlossen waren therapienaive Patienten mit chronischer cHC vom Genotyp 1, 4, 5
oder 6. Primärer Endpunkt dieser Studie war die SVR 12, die höher liegen sollte als 60 Prozent (histo-
rische SVR-Rate, abgeleitet von Daten aus der ADVANCE- (Telaprevir) bzw. SPRINT-2- (Boceprevir)
Studie). Bei dieser Untersuchung handelte es sich um eine offene, einarmige Studie ohne Randomi-
sierung der Patienten. Knapp 90 Prozent der Studienteilnehmer hatten eine diagnostizierte cHC vom
Genotyp 1, 8,6 Prozent wiesen eine Infektion mit dem Genotyp 4, und nur gut 2 Prozent vom Geno-
typ 5 oder 6 auf. Eine kompensierte Zirrhose konnte bei knapp 17 Prozent aller Studienteilnehmer
nachgewiesen werden. Der primäre Endpunkt war im Gesamtkollektiv signifikant überlegen gegen-
über dem historischen Vergleich (Lawitz et al., 2013a). Basierend auf historischen Präzendenzfällen
und Daten aus Phase-II-Studien lassen auf eine vergleichbare Wirksamkeit der Genotypen 2 und 3
auch in Interferon-freien Therapieregimen schließen (EMA, 2013).
So untersuchten Lawitz et al. (2013a) Sicherheit und Wirksamkeit der o.g. Tripel-Therapie bei thera-
pienaiven Patienten mit Infektionen vom Genotyp 2 und 3 (FISSION) über zwölf Wochen im Vergleich
zur Standardtherapie über eine Behandlungsdauer von 24 Wochen. Das Studienkollektiv wies sowohl
kompensierte als auch keine Zirrhose auf. Auch in dieser Studie zeigte der primäre Endpunkt im
Gesamtkollektiv gemäß den Vorannahmen der Autoren eine Nicht-Unterlegenheit zwischen den
Behandlungsgruppen. Jacobson et al. (2013) betrachteten in ihrer doppelblinden und placebokon-
trollierten Studie das Interferon-freie Therapieregime (Sofosbuvir + Ribavirin) ebenfalls ausschließ-
lich an Patienten mit cHC vom Genotyp 2 oder 3 (POSITRON). Dieses Regime stellte entweder eine
Erst- oder Re-Therapie für Personen dar, die eine auf (PEG-)Interferon-basierende Behandlung aus
170

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
unterschiedlichen Gründen nicht erhalten durften. Das Originalprotokoll wurde vor Beginn der Stu-
die noch einmal geändert, was jedoch nicht die Integrität der Untersuchung beeinflusste (EMA,
2013). In dieser Studie lag der primäre Endpunkt signifikant höher als im Vergleich zu Placebo. In
einer weiteren Studie der Autoren (FUSION) wurden ausschließlich therapieerfahrene Patienten mit
cHC vom Genotyp 2 oder 3 involviert. Beschrieben werden sollten Wirksamkeit und Sicherheit der
Interferon-freien dualen Therapie (Sofosbuvir + Ribavirin) bei einer Behandlungsdauer über zwölf
Wochen im Vergleich zu einer 16-wöchigen Therapie. Auch hier wurden Änderungen am Studienpro-
tokoll vorgenommen, bevor die ersten Studienteilnehmer gescreent wurden. In der Gesamtpopula-
tion lag die SVR-Rate nach 16 Wochen höher (73 Prozent) als nach zwölf Wochen (50 Prozent) und
im Gesamtkollektiv signifikant höher im Vergleich zur historischen Kontrolle.
Sofosbuvir zeigte sowohl in Kombination mit PEG-Interferon + Ribavirin wie auch in der alleinigen
Kombination mit Ribavirin bei HCV-Infektionen vom Genotyp 1-6 hohe Heilungsraten von meist über
90 Prozent zwölf bzw. 24 Wochen nach Behandlungsende. Lediglich Infektionen mit GT3 bzw. Zirrho-
se schienen mit SVR-Raten von ca. 80 Prozent etwas schlechter auf die hier beschriebenen Therapie-
optionen mit Sofosbuvir anzusprechen.
Inzwischen gibt es eine Reihe von Studien, in denen Sofosbuvir in Kombination mit aktuell zugelasse-
nen direkt antiviral wirkenden Mitteln eingesetzt wird. So wird der pangenotypische Wirkstoff zu-
sammen mit Simeprevir (± Ribavirin), mit Daclatasvir (± Ribavirin) oder in einer Fixkombination mit
Ledipasvir bei Patienten mit cHC vom Genotyp 1 zur Therapie eingesetzt.
171
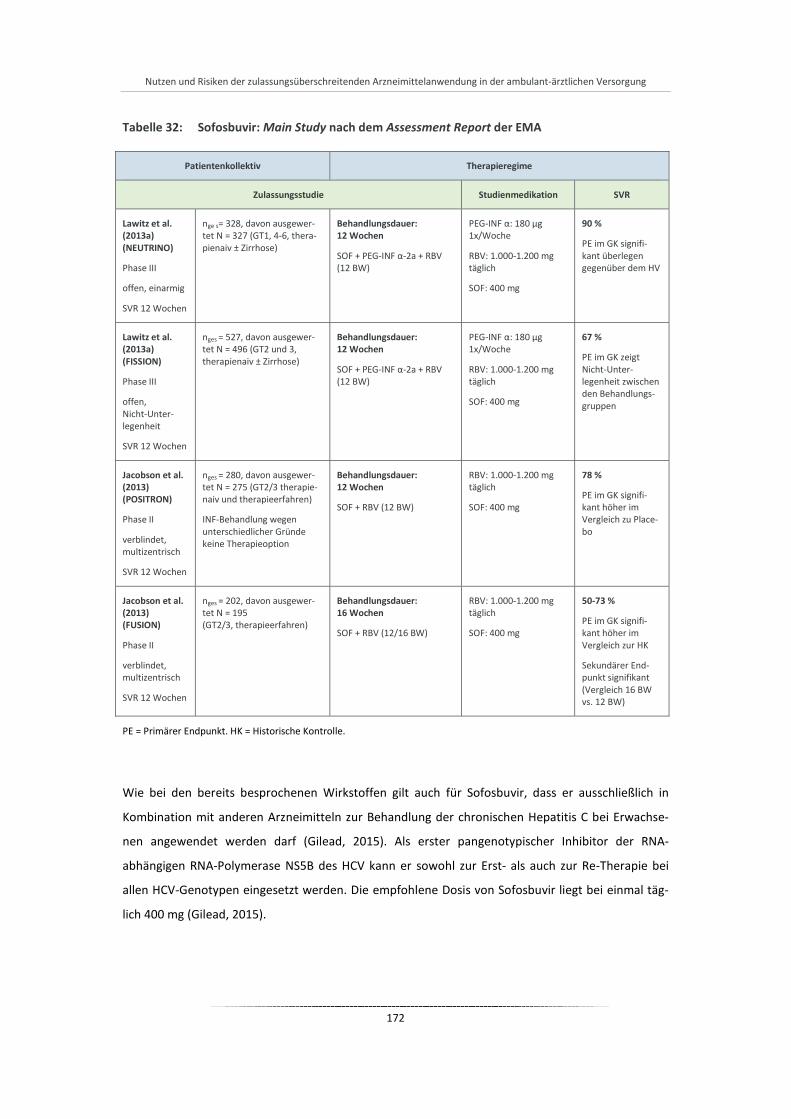
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 32: Sofosbuvir: Main Study nach dem Assessment Report der EMA
Patientenkollektiv Therapieregime
Zulassungsstudie Studienmedikation SVR
Lawitz et al. (2013a) (NEUTRINO)
Phase III
offen, einarmig
SVR 12 Wochen
nge s= 328, davon ausgewer-tet N = 327 (GT1, 4-6, thera-pienaiv ± Zirrhose)
Behandlungsdauer: 12 Wochen
SOF + PEG-INF -2a + RBV (12 BW)
PEG-1x/Woche
RBV: 1.000-1.200 mg täglich
SOF: 400 mg
90 %
PE im GK signifi-kant überlegen gegenüber dem HV
Lawitz et al. (2013a) (FISSION)
Phase III
offen, Nicht-Unter- legenheit
SVR 12 Wochen
nges = 527, davon ausgewer-tet N = 496 (GT2 und 3, therapienaiv ± Zirrhose)
Behandlungsdauer: 12 Wochen
SOF + PEG-INF -2a + RBV (12 BW)
PEG-1x/Woche
RBV: 1.000-1.200 mg täglich
SOF: 400 mg
67 %
PE im GK zeigt Nicht-Unter-legenheit zwischen den Behandlungs-gruppen
Jacobson et al. (2013) (POSITRON)
Phase II
verblindet, multizentrisch
SVR 12 Wochen
nges = 280, davon ausgewer-tet N = 275 (GT2/3 therapie-naiv und therapieerfahren)
INF-Behandlung wegen unterschiedlicher Gründe keine Therapieoption
Behandlungsdauer: 12 Wochen
SOF + RBV (12 BW)
RBV: 1.000-1.200 mg täglich
SOF: 400 mg
78 %
PE im GK signifi-kant höher im Vergleich zu Place-bo
Jacobson et al. (2013) (FUSION)
Phase II
verblindet, multizentrisch
SVR 12 Wochen
nges = 202, davon ausgewer-tet N = 195 (GT2/3, therapieerfahren)
Behandlungsdauer: 16 Wochen
SOF + RBV (12/16 BW)
RBV: 1.000-1.200 mg täglich
SOF: 400 mg
50-73 %
PE im GK signifi-kant höher im Vergleich zur HK
Sekundärer End-punkt signifikant (Vergleich 16 BW vs. 12 BW)
PE = Primärer Endpunkt. HK = Historische Kontrolle.
Wie bei den bereits besprochenen Wirkstoffen gilt auch für Sofosbuvir, dass er ausschließlich in
Kombination mit anderen Arzneimitteln zur Behandlung der chronischen Hepatitis C bei Erwachse-
nen angewendet werden darf (Gilead, 2015). Als erster pangenotypischer Inhibitor der RNA-
abhängigen RNA-Polymerase NS5B des HCV kann er sowohl zur Erst- als auch zur Re-Therapie bei
allen HCV-Genotypen eingesetzt werden. Die empfohlene Dosis von Sofosbuvir liegt bei einmal täg-
lich 400 mg (Gilead, 2015).
172

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Zulassungsrelevante Studien vs. Zulassungsstatus
In der Gesamtbetrachtung erweist sich Sofosbuvir als insgesamt sicher und wirksam. Das gilt auch für
die Therapie von Patienten mit cHC und gleichzeitiger HIV-Infektion (PHOTON-1) sowie für Patienten,
für die eine Lebertransplantation langfristig die einzige Therapieoption bleibt (EMA, 2013).
Unklarheiten bestehen allerdings im Hinblick auf die Therapiedauer von zirrhotischen Patienten. So
wird u.a. die Gabe der Tripel-Kombination über zwölf Wochen für therapienaive Patienten mit cHC
vom Gentoyp 1 als wirkungsvoll erachtet, sollte zusätzlich die Standardtherapie verabreicht werden
(Tabelle 33). Nach Auffassung des CHMP gilt dies nicht automatisch auch für zirrhotische Patienten,
so dass für diese Patientenpopulation eine Behandlung über 24 Wochen in Betracht gezogen werden
sollte (EMA, 2013). Die Begründung liegt in der Annahme, dass mit einer längeren Therapie auch
höhere SVR-Raten erreicht werden könnten. Gleiches gilt für vorherige Null-Responder, die in der
Studienpopulation der klinischen Prüfung nur in geringer Zahl vertreten waren (EMA, 2013). Auch ist
der Nutzen für Patienten, die Sofosbuvir vor ihrer Lebertransplantation erhalten, noch unklar, da die
Studienpopulation der Interim-Analyse, die diese Indikation erfüllte, zu gering war. Eine 24-wöchige
Therapie war außerdem mit hohen Relapse-Raten verbunden. Ob eine Verlängerung der Therapie
auf 48 Wochen bessere Ergebnisse erzielen würde, ist derzeit Untersuchungsgegenstand.
Die Zulassung weist für alle Genotypen eine Interferon-freie Therapieoption aus (Tabelle 33). Ange-
merkt werden muss allerdings, dass Interferon-freie Behandlungsregime in Phase-III-Studien nur an
Patienten mit cHC vom Genotyp 2 und 3 getestet wurden. Die optimale Behandlungsdauer wurde
nicht bestimmt, gleiches gilt für das Therapieregime im Allgemeinen (EMA, 2013). Aus diesem Grund
wird die Zulassung mit dem Hinweis versehen, dass Interferon-freie Regime nur Patienten vorbehal-
ten bleiben sollten, für die eine zusätzliche Gabe von (PEG-)Interferon aus unterschiedlichen Grün-
den keine Behandlungsalternative darstellt (Gilead, 2015).
173
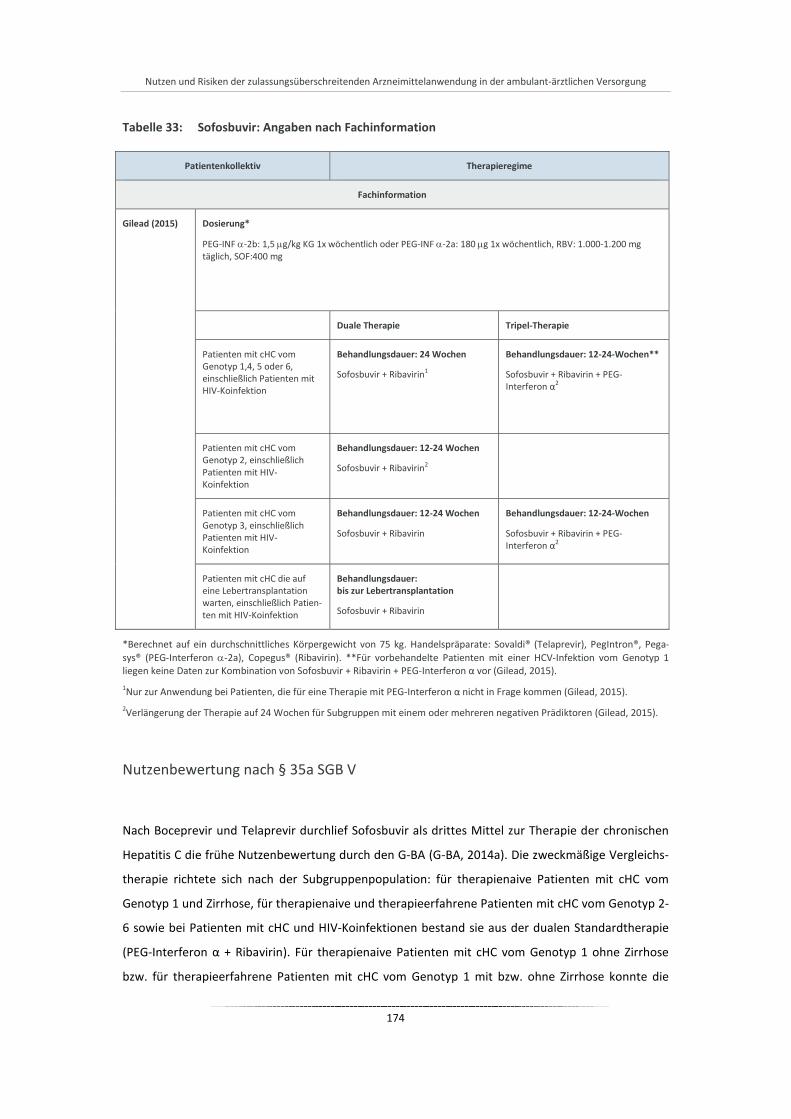
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 33: Sofosbuvir: Angaben nach Fachinformation
Patientenkollektiv Therapieregime
Fachinformation
Gilead (2015) Dosierung*
PEG-INF -2b: 1,5 g/kg KG 1x wöchentlich oder PEG-INF -2a: 180 g 1x wöchentlich, RBV: 1.000-1.200 mg täglich, SOF:400 mg
Duale Therapie Tripel-Therapie
Patienten mit cHC vom Genotyp 1,4, 5 oder 6, einschließlich Patienten mit HIV-Koinfektion
Behandlungsdauer: 24 Wochen
Sofosbuvir + Ribavirin1
Behandlungsdauer: 12-24-Wochen**
Sofosbuvir + Ribavirin + PEG-Interferon 2
Patienten mit cHC vom Genotyp 2, einschließlich Patienten mit HIV-Koinfektion
Behandlungsdauer: 12-24 Wochen
Sofosbuvir + Ribavirin2
Patienten mit cHC vom Genotyp 3, einschließlich Patienten mit HIV-Koinfektion
Behandlungsdauer: 12-24 Wochen
Sofosbuvir + Ribavirin
Behandlungsdauer: 12-24-Wochen
Sofosbuvir + Ribavirin + PEG-Interferon 2
Patienten mit cHC die auf eine Lebertransplantation warten, einschließlich Patien-ten mit HIV-Koinfektion
Behandlungsdauer: bis zur Lebertransplantation
Sofosbuvir + Ribavirin
*Berechnet auf ein durchschnittliches Körpergewicht von 75 kg. Handelspräparate: Sovaldi® (Telaprevir), PegIntron®, Pega-sys® (PEG-Interferon -2a), Copegus® (Ribavirin). **Für vorbehandelte Patienten mit einer HCV-Infektion vom Genotyp 1 liegen keine Daten zur Kombination von Sofosbuvir + Ribavirin + PEG-Interferon vor (Gilead, 2015). 1Nur zur Anwendung bei Patienten, die für eine Therapie mit PEG- rage kommen (Gilead, 2015). 2Verlängerung der Therapie auf 24 Wochen für Subgruppen mit einem oder mehreren negativen Prädiktoren (Gilead, 2015).
Nutzenbewertung nach § 35a SGB V
Nach Boceprevir und Telaprevir durchlief Sofosbuvir als drittes Mittel zur Therapie der chronischen
Hepatitis C die frühe Nutzenbewertung durch den G-BA (G-BA, 2014a). Die zweckmäßige Vergleichs-
therapie richtete sich nach der Subgruppenpopulation: für therapienaive Patienten mit cHC vom
Genotyp 1 und Zirrhose, für therapienaive und therapieerfahrene Patienten mit cHC vom Genotyp 2-
6 sowie bei Patienten mit cHC und HIV-Koinfektionen bestand sie aus der dualen Standardtherapie
(PEG- + Ribavirin). Für therapienaive Patienten mit cHC vom Genotyp 1 ohne Zirrhose
bzw. für therapieerfahrene Patienten mit cHC vom Genotyp 1 mit bzw. ohne Zirrhose konnte die
174

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
zweckmäßige Vergleichstherapie auch aus einer Tripel-Therapie, bestehend aus PEG-
Ribavirin und einem Protease-Hemmstoff (Telaprevir oder Boceprevir), akzeptiert werden. Während
das IQWiG aufgrund „inadäquater Vorgehensweisen“ in der Dossiererstellung (u.a. hohes Verzer-
rungspotential auf Studienebene aufgrund inadäquater Umsetzung des ITT-Prinzips27) nur für thera-
pienaive Patienten mit cHC vom Genotyp 2 einen nicht quantifizierbaren Zusatznutzen (Daten lagen
nur zum Surrogatendpunkt SVR 24 und nicht zum patientenrelevanten Endpunkt hepatozelluläres
Karzinom vor) sah (IQWiG, 2014b), ging der G-BA teilweise über die Empfehlungen des IQWiG hin-
weg.
So konstatierte der G-BA für therapienaive Patienten mit cHC vom Genotyp 1 mit bzw. ohne Zirrhose
einen Anhaltspunkt für einen geringen Zusatznutzen im Hinblick auf eine Sofosbuvir-basierte Tripel-
Therapie, im Vergleich zur Boceprevir- oder Telaprevir-basierten Tripel-Therapie bzw. zur dualen
Therapie. Für therapienaive Patienten mit cHC vom Genotyp 2 sah der G-BA sogar einen Hinweis auf
einen beträchtlichen Zusatznutzen für die Kombination aus Sofosbuvir + Ribavirin gegenüber der
angemessenen Vergleichstherapie. Für therapieerfahrene Patienten mit cHC vom Genotyp 2 ergab
sich hingegen nur ein Anhaltspunkt auf einen geringen Zusatznutzen, zu einem gleichen Ergebnis
führte die Bewertung der Subgruppenpopulationen bestehend aus Patienten mit cHC vom Genotyp 3
bzw. für HIV-koinfizierten Patienten (G-BA, 2014a).
27 Es handelt sich dabei um ein Analyse-Prinzip von Daten aus einer randomisierten klinischen Studie. Das sogenannte Intenti-on-to-Treat-Kollektiv (ITT) besteht aus allen randomisierten Patienten (Verum- und Placebo-Gruppe), für die eine Behandlung vorgesehen war, und dient der Auswertung. So werden für die Auswertung auch Daten von Patienten berücksichtigt, die beispielsweise die Studienbehandlung nicht erhielten, unterbrachen oder vorzeitig beendeten. Das hat zur Konsequenz, dass der therapeutische Effekt tendenziell unterschätzt wird (Kabisch et al., 2011).
175

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.2.2.4 Fixkombination aus NS5A-Inhibitor und Protease-Inhibitor + nicht-nukleosidischer Polymeraseinhibitor
8.2.2.4.1 Ombitasvir + Paritaprevir + Ritonavir (Fix) in Kombination mit Dasabuvir
Wie Daclatasvir und Ledipasvir wirkt Ombitasvir als Hemmstoff des viralen Nicht-Strukturproteins 5A
(NS5A). Das Phosphoprotein NS5A spielt eine wichtige Rolle bei der Virus-Replikation und bei der
intrazellulären Virusassemblierung. Paritaprevir (ABT-450) ist ein HCV-Proteaseinhibitor. Das Mittel
bindet an den NS3/4A-Proteasekomplex und hemmt so die NS3-Serinprotease, die eine wichtige
Rolle bei der Virusvermehrung spielt. Um die Verfügbarkeit von Paritaprevir zu erhöhen, wird der
Wirkstoff mit Ritonavir, einem starken CYP3A4-Hemmstoff, kombiniert. Durch die gemeinsame Gabe
wird eine einmal tägliche Verabreichung ermöglicht (Ferenci et al., 2014). Dasabuvir ist der erste
nicht-nukleosidische Polymerase-Inhibitor, welcher durch die Hemmung des viralen Enzyms RNA-
abhängige RNA-Polymerase NS5B die Virusvermehrung unterbindet (EMA, 2014c).
Inzwischen liegen eine Reihe randomisiert kontrollierter Studien vor, die die Fixkombination beste-
hend aus Ombitasvir in Kombination mit Ritonavir-geboostertem Paritaprevir als antivirale Thera-
piestrategie zur Behandlung der chronischen Hepatitis C untersuchten (Tabelle 34). Die allesamt
Interferon-freie Studienmedikation wurde zusätzlich mit dem direkt antiviral wirkenden Dasabuvir
kombiniert. Untersucht wurden therapienaive wie auch (standard-) therapieerfahrene Patienten mit
cHC vom Genotyp 1, mit und ohne Zirrhose (Tabelle 34). SAPHIRE-I (Feld et al., 2014) und SAPHIRE-II
(Zeuzem et al., 2014c) untersuchten die Wirksamkeit und Sicherheit der Kombination bestehend aus
den drei direkt antiviral wirkenden Substanzen plus Ribavirin über eine Behandlungsdauer von zwölf
Wochen gegenüber Placebo. Drei weitere Studien (PEARL-II, PEARL-III und PEARL-IV) untersuchten
die o.g. drei DAA mit bzw. ohne Ribavirin über zwölf Wochen. Die sechste Studie (TURQUOISE-II)
verglich unterschiedliche Therapiedauern (12 vs. 24 Wochen) der Studienmedikation aus den SAPHI-
RE-Studien (Tabelle 34). Diese Studie ist die erste Zulassungsstudie im Bereich der HCV-Therapie, die
ausschließlich Patienten mit (kompensierter) Zirrhose gewidmet wurde. In den anderen Pivotal-
Studien stellte eine diagnostizierte Zirrhose ein Ausschlusskriterium dar. Nach logistischer Regression
scheinen weder Alter, Geschlecht, Ethnie, IL28B-Genotyp, Fibrosestatus noch Viruslast zu Behand-
lungsbeginn einen Einfluss auf die virale Therapieantwort zwölf Wochen nach Behandlungsbeginn zu
haben (Ferenci et al., 2004; Feld et al., 2014). Eine Infektion mit Genotyp 1a oder ein Non-Response
auf PEG- -Vorbehandlung in der Vorgeschichte senken möglicherweise die
Heilungsraten. In allen Studien war der primäre Endpunkt im Gesamtkollektiv gemäß den Festlegun-
176

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
gen der Studienautoren nicht unterlegen und überlegen gegenüber dem historischen Vergleich.
Poordad et al. (2014; TURQUOISE-II) ermittelten keinen statistisch signifikanten Unterschied zwi-
schen den Behandlungsgruppen (p = 0,09).
Für Patienten mit zusätzlicher HIV-Infektion bzw. mit Lebertransplantation liegen derzeit nur unkon-
trollierte Beobachtungen vor (Kwo et al., 2014; Sulkowski et al., 2015), die jedoch beachtliche Er-
folgsraten vermuten lassen.
Tabelle 34: Ombitasvir + Paritaprevir + Ritonavir plus Dasabuvir: Main Study nach dem Assess-ment Report der EMA
Patientenkollektiv Therapieregime
Zulassungsstudie Studienmedikation SVR
Feld et al. (2014)
(SAPHIRE-I)
Phase III
doppelblind, multizentrisch
SVR 12 Wochen
nges = 636, davon ausge-wertet N = 631, (GT1a und GT1b, therapienaiv, ohne Zirrhose)
Behandlungs-dauer: 12 Wochen
OMV + PRV + RTV (Fix) + DSV + RBV
(OMV:25 mg , PRV:150 mg, RTV:100 mg) als fixe Kombination 1x täglich
DSV:500 mg täglich
RBV: 1.000/1.200 mg täglich
96 %
PE im GK nicht unterlegen und überlegen gegenüber dem HV (Patientenkollektiv des HV: TN mit GT1 ohne Zirrhose; Behandlungsre-gime: TVR + PEG-RBV)
Zeuzem et al. (2014)
(SAPHIRE-II)
Phase III
doppelblind, multizentrisch
SVR 12 Wochen
nges = 395, davon ausge-wertet N = 394 (GT1a und GT1b, therapieerfahrene Patien-ten mit Relapse, partiel-lem Ansprechen oder Non-Response, ohne Zirrhose)
Behandlungs-dauer: 12-24 Wochen
OMV + PRV + RTV (Fix) + DSV + RBV
(OMV:25 mg , PRV:150 mg, RTV:100 mg) als fixe Kombination 1x täglich
DSV:500 mg täglich
RBV: 1.000/1.200 mg täglich
96 %
PE im GK nicht unterlegen und überlegen gegenüber dem HV (Patientenkollektiv des HV: TE mit GT1 ohne Zirrhose; Behandlungsre-gime: TVR + PEG-INF + RBV)
Poordad et al. (2014)
(TURQUOISE-II)
Phase III
offen
SVR 12 Wochen
nges = 381, davon ausge-wertet N = 380 (GT1a und GT1b, therapienaive und therapieerfahrene Patien-ten, mit Zirrhose (Metavir-Score 3,4))
Behandlungs-dauer:12-24 Wochen
OMV + PRV + RTV (Fix) + DSV + RBV
(OMV:25 mg , PRV:150 mg, RTV:100 mg) als fixe Kombination 1x täglich
DSV:500 mg täglich
RBV: 1.000/1.200 mg täglich
92-96 %
PE im GK nicht unterlegen und überlegen gegenüber dem HV (Patientenkollektiv des HV: TE mit GT 1 mit Zirrhose; Behandlungsre-gime: TVR + PEG-INF + RBV) Kein statistisch signifi-kanter Unterschied zwischen den Behandlungsgruppen (p = 0,09)
177
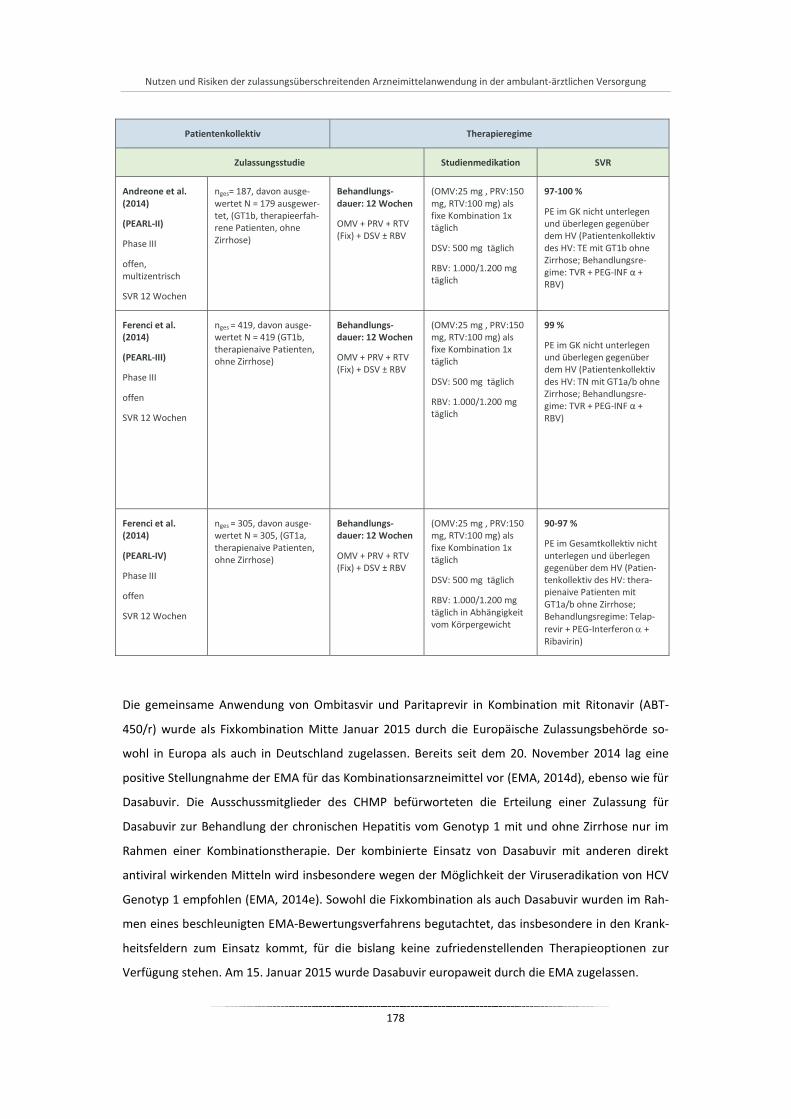
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Patientenkollektiv Therapieregime
Zulassungsstudie Studienmedikation SVR
Andreone et al. (2014)
(PEARL-II)
Phase III
offen, multizentrisch
SVR 12 Wochen
nges= 187, davon ausge-wertet N = 179 ausgewer-tet, (GT1b, therapieerfah-rene Patienten, ohne Zirrhose)
Behandlungs-dauer: 12 Wochen
OMV + PRV + RTV (Fix) + DSV ± RBV
(OMV:25 mg , PRV:150 mg, RTV:100 mg) als fixe Kombination 1x täglich
DSV: 500 mg täglich
RBV: 1.000/1.200 mg täglich
97-100 %
PE im GK nicht unterlegen und überlegen gegenüber dem HV (Patientenkollektiv des HV: TE mit GT1b ohne Zirrhose; Behandlungsre-gime: TVR + PEG-INF + RBV)
Ferenci et al. (2014)
(PEARL-III)
Phase III
offen
SVR 12 Wochen
nges = 419, davon ausge-wertet N = 419 (GT1b, therapienaive Patienten, ohne Zirrhose)
Behandlungs-dauer: 12 Wochen
OMV + PRV + RTV (Fix) + DSV ± RBV
(OMV:25 mg , PRV:150 mg, RTV:100 mg) als fixe Kombination 1x täglich
DSV: 500 mg täglich
RBV: 1.000/1.200 mg täglich
99 %
PE im GK nicht unterlegen und überlegen gegenüber dem HV (Patientenkollektiv des HV: TN mit GT1a/b ohne Zirrhose; Behandlungsre-gime: TVR + PEG-INF + RBV)
Ferenci et al. (2014)
(PEARL-IV)
Phase III
offen
SVR 12 Wochen
nges = 305, davon ausge-wertet N = 305, (GT1a, therapienaive Patienten, ohne Zirrhose)
Behandlungs-dauer: 12 Wochen
OMV + PRV + RTV (Fix) + DSV ± RBV
(OMV:25 mg , PRV:150 mg, RTV:100 mg) als fixe Kombination 1x täglich
DSV: 500 mg täglich
RBV: 1.000/1.200 mg täglich in Abhängigkeit vom Körpergewicht
90-97 %
PE im Gesamtkollektiv nicht unterlegen und überlegen gegenüber dem HV (Patien-tenkollektiv des HV: thera-pienaive Patienten mit GT1a/b ohne Zirrhose; Behandlungsregime: Telap-revir + PEG-Interferon + Ribavirin)
Die gemeinsame Anwendung von Ombitasvir und Paritaprevir in Kombination mit Ritonavir (ABT-
450/r) wurde als Fixkombination Mitte Januar 2015 durch die Europäische Zulassungsbehörde so-
wohl in Europa als auch in Deutschland zugelassen. Bereits seit dem 20. November 2014 lag eine
positive Stellungnahme der EMA für das Kombinationsarzneimittel vor (EMA, 2014d), ebenso wie für
Dasabuvir. Die Ausschussmitglieder des CHMP befürworteten die Erteilung einer Zulassung für
Dasabuvir zur Behandlung der chronischen Hepatitis vom Genotyp 1 mit und ohne Zirrhose nur im
Rahmen einer Kombinationstherapie. Der kombinierte Einsatz von Dasabuvir mit anderen direkt
antiviral wirkenden Mitteln wird insbesondere wegen der Möglichkeit der Viruseradikation von HCV
Genotyp 1 empfohlen (EMA, 2014e). Sowohl die Fixkombination als auch Dasabuvir wurden im Rah-
men eines beschleunigten EMA-Bewertungsverfahrens begutachtet, das insbesondere in den Krank-
heitsfeldern zum Einsatz kommt, für die bislang keine zufriedenstellenden Therapieoptionen zur
Verfügung stehen. Am 15. Januar 2015 wurde Dasabuvir europaweit durch die EMA zugelassen.
178

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Zulassungsrelevante Studien vs. Zulassungsstatus
Insgesamt erweist sich die Tripel-Therapie aus den drei DAA ohne Ribavirin über zwölf Wochen bei
zirrhotischen und nicht-zirrhotischen Patienten mit cHC vom Genotyp 1b als effektiv, gleiches gilt für
nicht-zirrhotische Patienten mit cHC vom Genotyp 1a (EMA, 2014c). In der letztgenannten Subgrup-
pe sollte allerdings eine zwölfwöchige Therapie nur mit zusätzlicher Gabe von Ribavirin verbunden
sein. Patienten mit cHC vom Genotyp 1a besitzen auch ohne fortgeschrittene Lebererkrankung ein
höheres Risiko eines viralen Durchbruchs bzw. eines Rückfalls nach Beendigung der Therapie, sollte
auf die zusätzliche Gabe von Ribavirin verzichtet werden (EMA, 2014c). Unklar ist, ob dies auch für
Patienten mit cHC vom Genotyp 1b gilt.
Patienten mit einer HCV-Infektion vom Genotyp 1 und nach einer Lebertransplantation wird eine
Behandlung mit der Fixkombination bestehend aus Ombitasvir + Ritonavir-geboostertem Paritaprevir
plus Dasabuvir in Kombination mit Ribavirin über 24 Wochen hinweg empfohlen (Tabelle 35). Bei
einer Infektion vom Genotyp 4 wird die Fixkombination mit einer zusätzlichen Gabe von Ribavirin
befürwortet, ebenfalls über 24 Wochen. Daten zur Anwendung der Fixkombination + Ribavirin bei
Patienten mit einer HCV-Infektion vom Genotyp 4 und kompensierter Zirrhose liegen nicht vor. Eine
optimale Therapiedauer ist somit nicht erwiesen. Basierend auf In-vitro-Daten zur antiviralen Aktivi-
tät und den verfügbaren klinischen Daten zum HCV-Genotyp 1 wird für Patienten mit einer HCV-
Infektion vom Genotyp 4 und kompensierter Zirrhose eine konservative Therapiedauer von 24 Wo-
chen empfohlen (abbvie, 2015a; abbvie, 2015b).
179
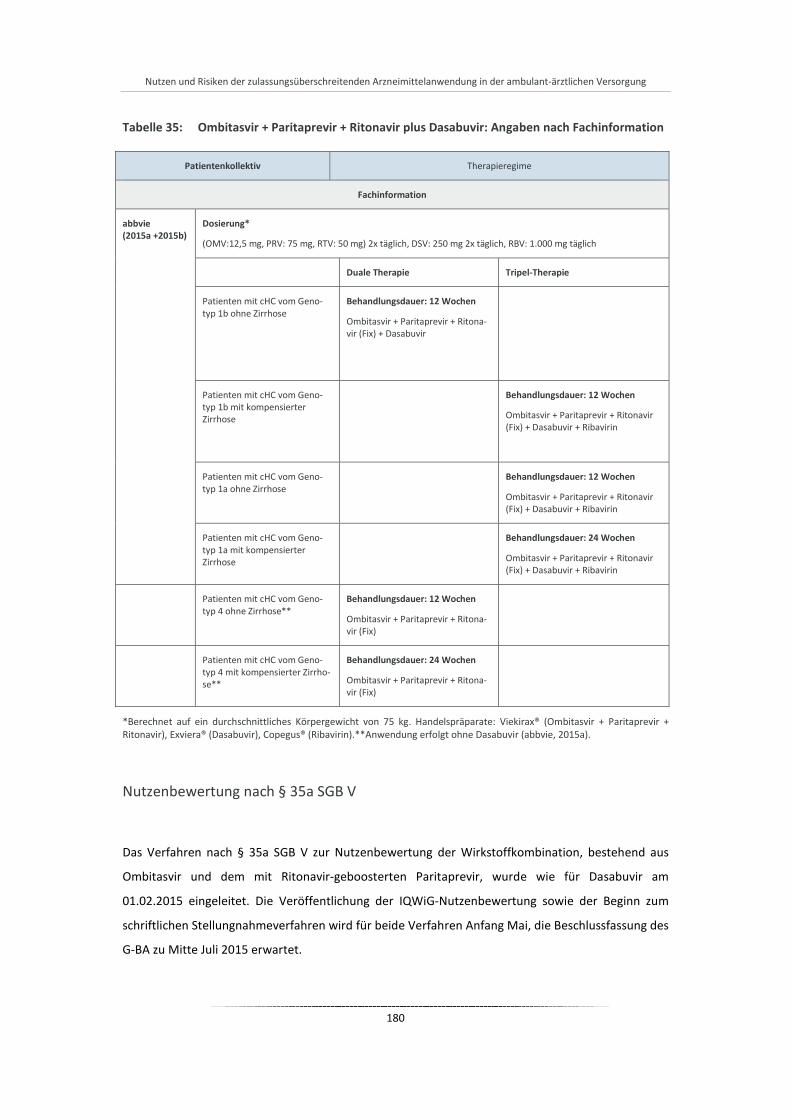
Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Tabelle 35: Ombitasvir + Paritaprevir + Ritonavir plus Dasabuvir: Angaben nach Fachinformation
Patientenkollektiv Therapieregime
Fachinformation
abbvie (2015a +2015b)
Dosierung*
(OMV:12,5 mg, PRV: 75 mg, RTV: 50 mg) 2x täglich, DSV: 250 mg 2x täglich, RBV: 1.000 mg täglich
Duale Therapie Tripel-Therapie
Patienten mit cHC vom Geno-typ 1b ohne Zirrhose
Behandlungsdauer: 12 Wochen
Ombitasvir + Paritaprevir + Ritona-vir (Fix) + Dasabuvir
Patienten mit cHC vom Geno-typ 1b mit kompensierter Zirrhose
Behandlungsdauer: 12 Wochen
Ombitasvir + Paritaprevir + Ritonavir (Fix) + Dasabuvir + Ribavirin
Patienten mit cHC vom Geno-typ 1a ohne Zirrhose
Behandlungsdauer: 12 Wochen
Ombitasvir + Paritaprevir + Ritonavir (Fix) + Dasabuvir + Ribavirin
Patienten mit cHC vom Geno-typ 1a mit kompensierter Zirrhose
Behandlungsdauer: 24 Wochen
Ombitasvir + Paritaprevir + Ritonavir (Fix) + Dasabuvir + Ribavirin
Patienten mit cHC vom Geno-typ 4 ohne Zirrhose**
Behandlungsdauer: 12 Wochen
Ombitasvir + Paritaprevir + Ritona-vir (Fix)
Patienten mit cHC vom Geno-typ 4 mit kompensierter Zirrho-se**
Behandlungsdauer: 24 Wochen
Ombitasvir + Paritaprevir + Ritona-vir (Fix)
*Berechnet auf ein durchschnittliches Körpergewicht von 75 kg. Handelspräparate: Viekirax® (Ombitasvir + Paritaprevir + Ritonavir), Exviera® (Dasabuvir), Copegus® (Ribavirin).**Anwendung erfolgt ohne Dasabuvir (abbvie, 2015a).
Nutzenbewertung nach § 35a SGB V
Das Verfahren nach § 35a SGB V zur Nutzenbewertung der Wirkstoffkombination, bestehend aus
Ombitasvir und dem mit Ritonavir-geboosterten Paritaprevir, wurde wie für Dasabuvir am
01.02.2015 eingeleitet. Die Veröffentlichung der IQWiG-Nutzenbewertung sowie der Beginn zum
schriftlichen Stellungnahmeverfahren wird für beide Verfahren Anfang Mai, die Beschlussfassung des
G-BA zu Mitte Juli 2015 erwartet.
180

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
8.2.2.5 Diskussion der Ergebnisse
Klinische Forschung stellt die Grundlage des medizinischen Fortschritts dar. Neben dem primären
Ziel, patientenrelevante Forschungsfragen zu untersuchen, dienen klinische Studien der Arzneimit-
telentwicklung als Grundlage der behördlichen Zulassungsentscheidung (Kabisch et al., 2011).
Diskrepanzen zwischen Zulassungsstudien und Zulassungsstatus lassen sich – wie zuvor schon bei
den Empfehlungen der Fachgesellschaften – auch nach Analyse der relevanten Publikationen (Zulas-
sungsstudien, EMA Assessment Reports und Fachinformationen) aufzeigen.
So wurden unterschiedliche Stopp-Regeln, die während der klinischen Studien des Wirkstoffes Boce-
previr aufgestellt wurden, im Laufe des Zulassungsverfahrens vereinheitlicht, neue Dosierungsemp-
fehlungen auf Basis retrospektiver Analysen erteilt, und obwohl Null-Responder zunächst von der
Boceprevir-Studienpopulation ausgeschlossen wurden, wurde dem Antrag des pU auf eine Erwei-
terung der Indikation auch für diese Subgruppe stattgegeben. Auch für Telaprevir stellen die ange-
gebenen Stopp-Regeln in der Fachinformation eine Simplifikation dar, die in den Phase-III-Studien
angewendet wurden. Die Telaprevir-Therapie von Relapsern lässt – entgegen der Behandlungszeit in
der pivotalen Studie – nach Auffassung des CHMP auch ein verkürztes Behandlungsregime zu, be-
gründet auch hier u.a. durch das Nachreichen pharmakometrischer Datenanalysen28 durch den pU
während des laufenden Verfahrens. Bei Simeprevir wurde der für die Schlüsselstudie ursprünglich
geplante primäre Endpunkt (SVR 24) verlassen und in SVR 12 geändert – basierend auf der engen
Korrelation zwischen den beiden SVR. Partielle und Null-Responder waren auch hier nicht im Patien-
tenkollektiv der konfirmatorischen Phase-III-Studien für Simeprevir enthalten. Der pU beantragte
dennoch die Erweiterung der Indikation auch für diese Patienten und berief sich dabei auf Belege aus
einer Phase-IIb-Studie. Daten, die die Wirksamkeit von Simeprevir bei cHC-Patienten mit Genotyp 4
aufzeigen sollen und auf deren (Zwischen-)Ergebnissen die Zulassung beruht, wurden aus einer zum
Zeitpunkt des Zulassungsantrags noch laufenden Studie entnommen. Somit lagen Daten für die The-
rapie der HCV-Infektion mit Genotyp 4 zum Zeitpunkt der Zulassung noch nicht vor. Belastbare Daten
aus randomisiert-kontrollierten und verblindeten Studien sind bis dato nur für die Infektion mit dem
HCV vom Genotyp 1 verfügbar. Ebenso liegen – trotz Zulassung durch die EMA – nur geringe Erfah-
rungen zum Einsatz von Simeprevir im Rahmen Interferon-freier Behandlungsregime vor. Auch für
den Wirkstoff Daclatasvir beruht die Interferon-freie Anwendung bei Genotyp-4-Infektionen bislang
nur auf einer Extrapolation der Genotyp-1-Daten. Zum Zulassungszeitpunkt können nur begrenzte
Behandlungsempfehlungen für den Genotyp 3 ausgesprochen werden. Trotzdem wird der Wirkstoff
auch für diese Patientengruppe zugelassen. Das CHMP begründet seine Empfehlung mit dem zuge-
28 Unter einer pharmakometrischen Datenanalyse versteht man das Beschreiben und Vorhersagen möglicher Auswirkungen eines Arzneimittels auf einen Patienten mit Hilfe mathematischer Modelle.
181

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
lassenen Interferon-freien Therapie-Regime, bestehend aus Sofosbuvir und Ribavirin über eine Be-
handlungszeit von 24 Wochen für diese Subpopulation. Nur gut ein Fünftel des Patientenkollektivs
des Interferon-freien Regimes zur Therapie der cHC Genotyp 1 waren mit anderen Protease-
Inhibitoren vorbehandelt worden. Daten zu Wirksamkeit und Sicherheit der dualen Kombination
(Daclatasvir + Sofosbuvir) zur Behandlung zirrhotischer Patienten liegen zum Zeitpunkt der Zulassung
nur begrenzt vor, so dass die optimale Behandlungsdauer sowie das Nutzenpotential einer zusätzli-
chen Ribavirin-Gabe bis zum Zeitpunkt der Zulassung noch nicht in vollem Umfang abgeklärt werden
konnte. Auch hier wurden von Seiten des CHMP Daten aus anderen Studien mit vergleichbaren
Wirkstoffkombinationen hinzugezogen, die zwar die Wirksamkeit von Daclatasvir bei diagnostizierter
Leberzirrhose belegen, allerdings keinen Aufschluss bezüglich der Behandlungsdauer bzw. dem Nut-
zen einer Ribavirin-Gabe geben. Ledipasvir in Kombination mit Sofosbuvir und Ribavirin ist u.a. zuge-
lassen zur Behandlung von Patienten mit cHC vom Genotyp 3, mit Zirrhose und bzw. oder Versagen
einer vorherigen Behandlung, obwohl valide klinische Daten, die eine direkte Wirkung von Ledipasvir
auf den Genotyp 3 belegen, nicht vorhanden sind, ebenso wenig wie eindeutige Belege für einen
Selektionsdruck. Rückschlüsse auf eine Wirksamkeit der Fixkombination zieht das Komitee aus Quer-
schnittsstudien, die eine klinisch relevante Aktivität von Ledipasvir in der antiviralen Therapie des
Genotypus 3 belegen. Eine Zulassung zur Therapie der HCV-Infektion vom Genotyp 4 wird ebenfalls
mit In-vitro-Daten und Ergebnissen aus klinischen Studien mit vergleichbarem Therapieregime be-
gründet. Als erster pangenotypischer Inhibitor der RNA-abhängigen RNA-Polymerase NS5B des HCV
kann Sofosbuvir sowohl zur Erst- als auch zur Re-Therapie bei allen HCV-Genotypen eingesetzt wer-
den. Die Zulassung weist für alle Genotypen eine Interferon-freie Therapieoption aus und das, ob-
wohl das Interferon-freie Behandlungsregime in Phase-III-Studien nur an Patienten mit cHC vom
Genotyp 2 und 3 getestet wurden. Weder die bestmögliche Behandlungsdauer noch das optimale
Therapieregime wurden bestimmt.
Letztendlich zeigt sich am Beispiel dieser Wirkstoffe zur Behandlung der cHC, dass die Zulassung für
bestimmte Genotypen oder auch das Behandlungsregime nicht immer auf evidenzbasierten und
validen Daten beruht. Berücksichtigt werden muss außerdem, dass es sich bei den klinischen Unter-
suchungen hauptsächlich um placebokontrollierte Studien handelt. Direkte Vergleichsstudien zum
bisherigen Therapiestandard fehlen hier, ebenso direkte Vergleichsstudien zu therapeutisch ähnli-
chen Behandlungsoptionen. Daher sind belastbare Aussagen zu diesen Alternativen nicht möglich.
Das zu beurteilen ist aber auch nicht Aufgabe einer Zulassungsbehörde. Mit der Zulassung eines
Arzneimittels soll von Seiten der zuständigen Behörden gewährleistet werden, dass bei dem zu un-
tersuchenden Wirkstoff, bzw. der zu untersuchenden Wirkstoffkombination, Qualität, Wirksamkeit
und Unbedenklichkeit nach dem derzeitigen Erkenntnisstand gegeben sind und das Arzneimittel zur
Verwendung in der breiten Bevölkerung freigegeben werden kann. Sind diese Bedingungen nicht
erfüllt, muss die Zulassung verweigert werden. Die Orientierung am derzeitigen Erkenntnisstand
182

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
suggeriert objektive Fakten, die mit der Erteilung einer Zulassung attestiert werden. Am Ende der
Zulassungserteilung steht jedoch eine gewisse Unsicherheit, da Ergebnisse solcher Studien nicht
generell auf die breite Bevölkerung übertragen werden können. Begleiterkrankungen und Medikati-
onen im klinischen Alltag finden selten Berücksichtigung im therapeutischen Entwicklungsprogramm.
Auch das homogene Patientenkollektiv reflektiert nicht die Versorgungsrealität. Die Zulassung eines
Arzneimittels ist also auch immer mit einem Abwägen des Nutzen-Risiko-Verhältnisses verbunden.
Das wird nicht nur allein durch abweichende Entscheidungen nationaler wie internationaler Zulas-
sungsbehörden deutlich, sondern auch mit dem Auftreten schwerer UAW, die sich erst nach Ertei-
lung des Marktzugangs offenbaren.
So informierte der pU im Februar 2012 – also kein Jahr nach Erteilung der Zulassung – in einem Rote-
Hand-Brief über Arzneimittelinteraktionen zwischen Boceprevir und Ritonavir-geboosteren HIV-
Protease-Inhibitoren (MSD, 2012). Da zum Zeitpunkt der Marktzulassung von Boceprevir nur Interak-
tionsdaten für Ritonavir vorlagen, die keine nennenswerten Wechselwirkungen zeigten, waren nach
Aussage des pU keine Interaktionen mit Ritonavir-geboosterten HIV-Protease-Inhibitoren zu erwar-
ten. Eine pharmakokinetische Studie bei 39 gesunden Freiwilligen zeigte allerdings, dass es bei einer
gleichzeitigen Anwendung von Boceprevir mit Ritonavir in Kombination mit Atazanavir, Darunavir
oder mit Lopinavir zu einer deutlichen Abnahme der Plasmakonzentrationen sowohl der HIV-
Proteaseinhibitoren als auch von Boceprevir kam. Welchen schätzbaren Wert die Erforschung aller
naheliegenden Interaktionen einnimmt, wird gerade durch dieses Beispiel deutlich. Ungefähr ein
Drittel der Patienten, die in den USA und Europa mit dem Humanen Immundefizienz Virus (HIV)
infiziert sind, haben auch eine HCV-Infektion. Im Vergleich zu Patienten, bei denen nur eine HCV-
Infektion diagnostiziert worden ist, zeigen Patienten mit HIV und HCV u.a. eine raschere Progression
der HCV-Erkrankung mit einem erhöhten Risiko von Leberzirrhose und Leberkarzinom.
Auch wurden schon während der klinischen Studien zu Telaprevir unerwünschte Arzneimittelwirkun-
gen registriert, insbesondere Anämien, die eine Dosisanpassung oder Bluttransfusion erforderten,
sowie schwere Hautausschläge (Rash), Stevens-Johnson-Syndrom oder DRESS (Arzneimittelexan-
them mit Eosinophilie und systemischen Symptomen) (EMA, 2015). Nach der Markteinführung tra-
ten in Japan zwei Fälle schwerster Hautreaktionen auf (einer davon tödlich), welche als Toxische
Epidermale Nekrolyse (TEN) klassifiziert wurden. Während im Rahmen des klinischen Entwicklungs-
programms von Telaprevir die o.g. UAW aufgetreten sind, war eine TEN im Zusammenhang mit dem
neuen Wirkstoff bislang unbekannt (Janssen, 2013). Entsprechend verwies der pharmazeutische
Hersteller auf die Notwendigkeit einer sofortigen Beendigung der Dreifach-Therapie bei schwerwie-
gender Hautreaktion mit systemischen Symptomen, progredientem schwerem Hautausschlag bzw.
Verdacht auf oder Diagnose eines generalisierten bullösen Exanthems.
Schließlich informierten sowohl die deutsche Zulassungsbehörde BfArM wie auch die amerikanische
Zulassungsbehörde FDA im März 2015 über schwerwiegende Herzrhythmusstörungen im Zusam-
183

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
menhang mit der Einnahme von Sofosbuvir-haltigen Mitteln. Bei Einnahme von Sovaldi® bzw. von
Harvoni® kam es in Einzelfällen zu schwerwiegenden Bradykardien, die bei einigen Betroffenen zum
Einsatz von Herzschrittmachern führte und in einem Fall sogar tödlich endete (BfArM, 2015; FDA,
2015). Alle Patienten erhielten neben anderen antiviralen Wirkstoffen zur Therapie der cHC (Sime-
previr, Daclatasvir) mit Amiodaron und Betablocker noch weitere Arzneimittel, die selbst Einfluss auf
den Herzrhythmus haben können.
Wenn die Zulassung eines Arzneimittels von Anfang an mit nicht vorhersehbaren Ereignissen belegt
ist, drängt sich umso mehr die Frage auf, warum dennoch Daten zur Beurteilung einer Zulassung
hinzugezogen werden, die nicht aus klinischen Studien bzw. nicht von dem Patientenkollektiv der
klinischen Studie stammen. Zumal – wie an diesen Wirkstoffbeispielen aufgezeigt – bei der Bewer-
tung der Wirksamkeit der jeweiligen Substanzen, getestet an teilweise sehr kleinen Patientenzahlen,
als primärer Endpunkt der klinischen Studien ein Surrogatparameter anstelle „harter klinischer End-
punkte“ hinzugezogen wurde.
Morbidität und Mortalität zählen zu den „harten klinischen Endpunkten“, die den Ansprüchen der
evidenzbasierten Medizin gerecht werden. Studien, die auf derartige Endpunkte ausgelegt sind, sind
jedoch mit hohen Kosten verbunden und benötigen eine entsprechende Laufzeit, da Aussagen zu
Morbidität und Mortalität in der Regel erst nach Jahren der Nachbeobachtungszeit getroffen werden
können. Ausnahmen können klinische Studien im onkologischen Bereich darstellen. So ist aufgrund
der Schwere der Erkrankungen bei zahlreichen Krebserkrankungen (beispielsweise Tumore der Le-
ber, der Lunge oder des Pankreas) mit einer sehr hohen Sterblichkeitsrate zu rechnen, die auch im
Rahmen klinischer Studien erfasst werden kann. Es steht außer Zweifel, dass die Arzneimittelthera-
pie und ihre Wirksamkeit nicht ausschließlich auf Morbidität und Mortalität ausgelegt werden kön-
nen und dürfen, wenn man bedenkt, dass zum Zeitpunkt der Zulassung eines Medikaments in der
Regel sechs bis acht Jahre der klinischen Arzneimittelentwicklung vergangen sind und diese Zeit nicht
ausreicht, um gesicherte Aussagen zur Mortalität treffen zu können (Böger, 2002). Primäre Endpunk-
te, die – wie es bei Surrogatparametern der Fall ist – Veränderungen kurzfristig und schnell aufzeigen
können, dürfen und müssen somit auch in die Bewertung der Wirksamkeit durch die Zulassungsbe-
hörde miteinfließen. Voraussetzung der Verwendung von Surrogatparametern als Endpunkt in klini-
schen Interventionsstudien ist natürlich, dass ein definierter Zusammenhang zwischen Surrogatpa-
rametern und der zu untersuchenden Erkrankung besteht und dass Änderungen der Ausprägung des
Surrogatparameters den Effekt der Therapie auf den eigentlich beabsichtigten Effekt und den klini-
schen Verlauf der Krankheit widergeben (Böger, 2002). Ein weiterer Grund für die Akzeptanz solcher
Surrogatparameter als klinischen Endpunkt kann aber auch in dem begründet Zeitdruck der Zulas-
sungsbehörden gesehen werden, neuen Arzneimitteln gerade für schwer therapierbare Erkrankun-
gen einen schnelleren Marktzugang zu gewähren (Glaeske, 2011). Mit dem gleichen Argument wer-
den – wie im Fall der Fixkombination bestehend aus Ombitasvir + Paritaprevir + Ritonavir sowie
184

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Dasabuvir – auch beschleunigte EMA-Bewertungsverfahren begründet, die insbesondere in den
Krankheitsfeldern zum Einsatz kommen, für die bislang keine zufriedenstellenden Therapieoptionen
zur Verfügung stehen.
Eine Verkürzung der Prüfverfahren reduziert die Arzneimittelsicherheit. Eine zuverlässige Analyse der
verfügbaren Daten benötigt grundsätzlich auch Zeit, schwere UAW können sonst möglicherweise
während des Zulassungsverfahrens nicht identifiziert werden, mit schwerwiegenden Folgen nach
erfolgter Marktzulassung. Gerade für Arzneimittel, die durch ein beschleunigtes EMA-
Bewertungsverfahren ihren frühen Marktzugang erhalten haben – und das vor dem Hintergrund,
dem Patienten mit schwerer oder seltener Erkrankung eine rechtzeitige Behandlung zu ermöglichen
– ist zwingend ein umfassenderer Risikomanagementplan zu fordern. Diese Arzneimittel sollten bei-
spielsweise nur in bestimmten Zentren oder innerhalb eines Registers eingesetzt werden, um engere
Nutzen-Risiko-Profile erstellen und bei Auftreten von (schweren und schwerwiegenden) UAW ent-
sprechend zeitnah angemessen handeln zu können.
Primäres Ziel einer Hepatitis C-Therapie ist ein anhaltendes virologisches Ansprechen, ein Surrogat-
parameter, der mit einer Heilung der Lebererkrankung assoziiert wird. Darunter versteht man das
Fehlen eines HCV-RNA-Nachweises im Serum 24 Wochen nach Therapieende. Mit einer derartigen
SVR ist eine Heilung der Lebererkrankung assoziiert, da diese Patienten in der Regel seltener klini-
sche Endpunkte entwickeln als diejenigen, bei denen eine dauerhafte Viruseradikation nicht möglich
ist (Veldt et al., 2007; Morgan et al., 2013). Kritisch hinterfragt werden sollte hier aber, dass in man-
chen Studien die SVR nach 24 Wochen, in anderen Studien allerdings schon nach zwölf Wochen er-
hoben wurde. So findet im fachlichen Umfeld noch eine Diskussion darüber statt, ob die Definition
einer SVR 12 Wochen nach Behandlungsende zielführend ist. Eine Verkürzung der Zeit bis zur End-
punkterhebung wird grundsätzlich positiv bewertet. Studienergebnisse können so schneller publi-
ziert und möglicherweise effektive Therapie-Regime schneller beim Patienten zum Einsatz kommen.
Andererseits wird darauf hingewiesen, dass die Untersuchungen, mit denen eine große Überein-
stimmung zwischen den Erfolgsraten in Woche 12 nach Behandlungsende und denen nach Woche 24
gezeigt werden konnte, mit anderen Therapie-Regimen (plus PEG-
es daher noch unklar ist, ob diese Ergebnisse auch auf Interferon-freie Behandlungsregime übertrag-
bar sind (Hellard et al., 2014). Auch zeigen klinisch virologische Untersuchungen, dass die gemessene
HCV-RNA-Konzentration zwischen unterschiedlichen HCV-RNA-Assays trotz der Standardisierung auf
internationale Einheiten erheblich abweichen kann. Daten für Grenzwerte der HCV-RNA-
Konzentration, die auf eine mögliche Therapieverkürzung hindeuten, liegen von den verschiedenen
in der klinischen Routine eingesetzten Assays bisher nicht vor. Auch konnte die Bedeutung einer
residualen Virämie für einen späteren virologischen Rückfall noch nicht hinreichend untersucht wer-
den.
185

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
In der Studie von Zeuzem et al. (2014a, VALENCE) wurde das Originalprotokoll aufgrund neuer wis-
senschaftlicher Erkenntnisse während der Studiendurchführung geändert und der ursprünglich defi-
nierte primäre Endpunkt verändert. Ursprünglich war die Gabe einer duale Therapie bestehend aus
Sofosbuvir + Placebo für alle Studienteilnehmer aus dem Verum-Arm geplant. Nach Initiierung der
Studie lagen Ergebnisse aus einer weiteren Phase-III-Studie (FUSION) vor, die eine Verlängerung der
Behandlung der Patienten mit HCV Genotyp 3 von zwölf auf 24 Wochen notwendig erscheinen ließ.
Die Studiengruppe wurde daraufhin entblindet, Patienten mit HCV vom Genotyp 2 erhielten die
geplanten zwölf Behandlungswochen, während Patienten mit HCV vom Genotyp 3 die Studienmedi-
kation über 24 Wochen erhielten. Ein Protokoll während einer laufenden Studie zu ändern sollte
grundsätzlich kritisch hinterfragt werden. Letztendlich resultiert die medizinisch-wissenschaftliche
Begründung für eine Studie aus dem primären Ziel und sollte und darf allein deswegen nicht ohne
Weiteres im Verlauf geändert werden. Aus statistischer Sicht ist auch die Unterscheidung zwischen
Haupt- und Nebenfragestellung schon unerlässlich, weil sich die Bestimmung der Fallzahl ausschließ-
lich am primären Zielkriterium orientiert (ICH E9, 1998). Andererseits kann eine geänderte Datenlage
dazu führen, dass die ursprünglich geplante Studie aus ethischen Gründen nicht mehr vertretbar ist.
Würde man an diesem Punkt die Studie abbrechen, um eine neue Studie auf Basis aktueller Erkennt-
nisse durchzuführen, wäre es nicht nur mit hohen Kosten verbunden, sondern auch mit einem nicht
zu unterschätzenden Zeitverlust, der gerade bei der Zulassung von Orphan Drugs oder Arzneimitteln
zur Therapie schwerer Erkrankungen eine große Bedeutung hat.
Bei der Gegenüberstellung der Ergebnisse und der offensichtlichen Unterschiede zwischen den Da-
ten aus pivotalen Studien und der dann daraus folgenden Zulassung stellt sich auch die Frage, inwie-
fern die pharmazeutische Industrie Einfluss auf entsprechende Zulassungsbehörden hat oder schon
im Vorfeld auf einen positiven Ausgang des Zulassungsantrags hinwirken kann. Der Einfluss der
pharmazeutischen Industrie auf Zulassungsbehörden stand in der Vergangenheit immer wieder zur
Diskussion. Dass eine Zulassung auch von einem hohen ökonomischen Interesse des pU getragen
wird, ist Fakt. Schließlich steigt und fällt der Börsenwert eines Unternehmens mit einem zugelasse-
nen Arzneimittel. So wurden wesentliche Kompetenzen des nationalen Arzneimittelzulassungsver-
fahrens mittlerweile auf die europäische Ebene verlagert, um die Wettbewerbsfähigkeit der europäi-
schen Pharmaindustrie gegenüber ihren internationalen Konkurrenten zu verbessern (Schmucker,
2005).
Welcher Beurteilungsmaßstab seitens der Behörden abschließend zur Zulassung auch in unzu-
reichend geprüften Patientenkollektiven führt, ist faktisch nicht alleine durch eine kritische Evaluati-
on der Zulassungsstudien zu ermitteln. Wurde im Vorfeld der auf breiter Ebene empfohlene OLU
seitens der Fachgesellschaften kritisch hinterfragt, so kann dies auch die behördlichen Entscheidun-
gen betreffen. Festzuhalten ist, dass viele Faktoren, die zwischen den Zeilen stehen, zu einem positi-
186

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
ven Zulassungsbescheid beitragen. So erfolgt die Zulassung eben nicht nur „Ergebnis-orientiert“
sondern auch als eine theory of the second best. Festzuhalten gilt aber auch, dass Studien keinem
Selbstzweck dienen, den Anforderungen des AMG Genüge zu tun. Das Contergan®-Beispiel aus den
1960er-Jahren hat gezeigt, dass Tierversuche und In-vitro-Daten keine ausreichende Basis dafür sein
können, um schwerwiegende unerwünschte Wirkungen zu erkennen.
Sicher ist, dass die Bewertung der Zulassungsbehörde keinen OLU einschließt. Tatsache ist aber auch,
dass ein OLU nur mit zugelassenen Arzneimitteln möglich ist. Insofern ist die In-Label-Zulassung eine
Voraussetzung dafür, dass Arzneimittel auch off-label angewendet werden können. In der Versor-
gung sollte auf diesen Zusammenhang vor allem deshalb geachtet werden, weil offensichtlich pU bei
der Zulassung eines Arzneimittels bereits die Anwendung im Off-Label Use ökonomisch miteinkalku-
lieren (Glaeske, 2015). Dass dadurch aber die Arzneimittelsicherheit zu Lasten der Patienten ge-
schwächt wird, sollte bei einem OLU immer bedacht werden. Ein OLU ist also nur dort akzeptabel,
wo die engen Auflagen der bisherigen Gerichtsurteile eingehalten werden.
Für die individuelle Versorgung des betroffenen Patienten spielen aber auch die Beschlüsse des Ge-
meinsamen Bundesausschusses – auf Basis der IQWiG-Nutzenbewertung – eine nicht zu unterschät-
zende Rolle. Während der Zulassungsbehörde die Aufgabe obliegt, Qualität, Sicherheit und Wirk-
samkeit eines Arzneimittels zu bewerten, liegt der Fokus von G-BA und IQWiG auf der Ermittlung des
patientenindividuellen (Zusatz-)Nutzens. Ihre Entscheidungen können die Therapie der Ärzte maß-
geblich beeinflussen, wenn es darum geht, zugunsten der Patienten die als innovativ bewerteten
Wirkstoffe auch möglichst zeitnah in der Praxis anzuwenden. IQWiG und G-BA benennen bei allen
HCV Genotypen von 1 bis 6 (therapienaive wie therapieerfahrene Patienten mit und ohne Zirrhose
und unabhängig von Komorbidität mit HIV) als zweckmäßige Vergleichstherapie nach wie vor die
Kombination von PEG- . Lediglich bei therapienaiven HCV-Genotyp 1-
Patienten ohne Zirrhose bzw. bei therapieerfahrenen HCV-Genotyp 1-Patienten mit und ohne Zirrho-
se geben IQWiG und G-BA seit Anfang 2014 die mit einem Zusatznutzen belegte Tripel-Therapie,
bestehend aus der Standardtherapie plus einem Protease-Inhibitor (Boceprevir oder Telaprevir), als
therapeutische Alternative an. In diesem Zusammenhang muss auch darauf hingewiesen werden,
dass mit dem Indikator anhaltendes virologisches Ansprechen erstmals ein Surrogatparameter vom
IQWiG bezüglich des patientenrelevanten Endpunkts „Entwicklung eines Leberkrebses“ als „ausrei-
chend valide“ akzeptiert wurde. In seinen Dossiers vermerkt das IQWiG allerdings, dass die SVR als
Surrogat nicht formal validiert ist und dass die Einschätzung der „ausreichenden Validität“ aus-
schließlich auf Daten aus Beobachtungsstudien beruht.
187

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
In seinem Methodenpapier weist das IQWiG darauf hin, dass „Surrogatendpunkte im Rahmen der
Nutzenbewertung des Instituts in der Regel nur dann in Betracht gezogen [werden], wenn sie zuvor
anhand geeigneter statistischer Methoden innerhalb einer hinreichend eingegrenzten Patientenpo-
pulation und innerhalb von vergleichbaren Interventionen (z. B. Arzneimittel mit vergleichbarem
Wirkmechanismus) validiert wurden.“ (IQWiG, 2015a, S. 40). Weiter heißt es, dass „Surrogatend-
punkte, die nicht valide sind oder für die kein adäquates Validierungsverfahren durchgeführt wurde,
[…] dennoch in den Berichten des Instituts dargestellt werden [können]. Derartige Endpunkte sind
aber unabhängig von den beobachtbaren Effekten nicht als Belege für einen Nachweis des zusätzli-
chen Nutzens einer Intervention geeignet.“ (IQWiG, 2015a, S. 42).
Arzneimittel, deren Zulassung auf Surrogatendpunkten beruht, können sich in der ärztlichen Praxis
auch als schädigend für den Patienten erweisen (Svensson et al., 2013, Glaeske, 2011). Zu diesen
Arzneimitteln zählen beispielsweise das Antidiabetikum Rosiglitazon (Surrogatparameter: Abnahme
des HbA1c-Wertes, in der Praxis: akute Myopathie, Frakturen der Füße, Hände und Oberarme, unkla-
res kardiovaskuläres Sicherheitsprofil), dessen EU-weite Marktrücknahme im September 2010 er-
folgte, sowie der Wirkstoff Erythropoietin zur Behandlung von renalen Anämien (Surrogatparameter:
Erhöhung des Hb-Wertes), der nach subkutaner Injektion bei Patienten mit chronischer Niereninsuf-
fizienz bzw. terminalem Nierenversagen zu einer Antikörper-vermittelten Erythroblastopenie (PRCA)
führte (Ortho Biotech, 2002). Kvitkina et al. (2014) untersuchten in ihrer Arbeit die Durchführbarkeit
der frühen Nutzenbewertung anhand patientenrelevanter Endpunkte, indem die in den Hersteller-
Dossiers verfügbaren Endpunkte systematisch charakterisiert sowie die Einschätzungen der Herstel-
ler bzw. des IQWiG bezüglich der Patientenrelevanz bzw. Surrogatvalidität verglichen wurden. Die
Autoren kamen zu dem Schluss, dass von insgesamt 214 durch das IQWiG bearbeiteten Endpunkten
nur 13 Prozent als Surrogate eingestuft werden konnten. Dabei handelte es sich in den meisten Fäl-
len um laborparameter-definierte Ansprechraten oder Auswertungen, die auf bildgebenden Verfah-
ren beruhen (wie beispielsweise die Tumor-Ansprechrate im Cabazitaxel-Dossier) (Kvitkina et al.,
2014). Nur zwei der insgesamt 27 Surrogatendpunkte wurden letztendlich vom IQWiG als ausrei-
chend valide angesehen. Dazu zählte neben der SVR auch das virologische Ansprechen bei HIV (Kvit-
kina et al., 2014).
Insgesamt konstatieren IQWiG und G-BA bei der Mehrzahl der bewerteten Arzneimittel im Rahmen
der frühen Nutzenbewertung einen Zusatznutzen als nicht belegt. Dabei wurde die Nutzenbewer-
tung nach einzelnen Subgruppen differenziert. Allein bei Sofosbuvir wurden neun Teilpopulationen
bei der Nutzenbewertung erfasst. Betrachtet man die bis dato abgeschlossenen Verfahren in ihrer
Gesamtschau, muss festgehalten werden, dass eine solche Aufteilung der Anwendungsgebiete durch
den G-BA in etwa jedem zweiten Fall stattfand (Rasch & Dintsios, 2015). Sowohl das SGB V als auch
die Arzneimittel-Nutzenverordnung (AM-NutzenV) enthalten die rechtlichen Rahmenbedingungen
188

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
für die Bewertung der einzelnen Patientengruppen. So müssen die Unterlagen des Herstellers u.a.
Angaben über Anzahl der Patienten und Patientengruppen, für die ein therapeutisch bedeutsamer
Zusatznutzen besteht, enthalten (§ 35a Abs. 1 Satz 3 Nr. 4 SGB V). Zudem muss geprüft werden, wel-
cher Zusatznutzen für welche Patientengruppen in welchem Ausmaß belegt ist (§ 7 Abs. 2 AM-
NutzenV). Des Weiteren soll das Gutachten des IQWiG unter Berücksichtigung alters-, geschlechts-
und lebenslagenspezifischer Besonderheiten durchgeführt werden (§ 139a SGB V).
Aktuell veröffentlichten Rasch und Dintsios (2015) die Ergebnisse ihrer jüngsten Untersuchung und
zeigten dabei wesentliche Problemfelder rund um Subgruppenanalysen, Subpopulationen sowie
Inkongruenzen zwischen den Vorgaben der frühen Nutzenbewertung und der Zulassung auf. Dabei
werden als Subpopulation die Patientengruppen bezeichnet, die sich aus dem zugelassenen Indikati-
onsgebiet ergeben, während unter dem Begriff Subgruppen Teilpopulationen erfasst werden, die
sich aufgrund der Unterschiede im Ausmaß eines Effekts aufzeigen (Rasch & Dintsios, 2015). Nach
Auffassung der Autoren führt die Festlegung einer zweckmäßigen Vergleichstherapie ebenfalls zu
einer differenzierten Nutzenbewertung. Dabei handele es sich nicht um Teilmengen einer Population,
sondern um alternative Einschätzungen zum Zusatznutzen je nach festgelegtem Komparator. In sei-
nem Methodenpapier führt das IQWiG selbst die Limitationen dieses Bewertungsverfahrens aus
(IQWiG, 2015a). Neben dem fehlenden Beweischarakter von Subgruppenanalysen und dem Problem
der geringen Power, wenn eine Subgruppe nicht eine adäquate Fallzahl aufweist, bezeichnet das
IQWiG die Ausführung multipler Tests von mehreren Subgruppenmerkmalen ebenfalls als einen
weiteren Limitationspunkt. So führen multiple Tests möglicherweise zu einer statistischen Signifi-
kanz, die auf einem Zufallsergebnis basiert (IQWiG, 2015). Auch wenn Subgruppenanalysen das Stu-
dienergebnis der primären Analyse nicht dominieren sollten, können sie supportiv zur Abstufung der
Wahrscheinlichkeit und des Ausmaßes des Zusatznutzens führen (Rasch & Dintsios, 2015). Erweist
sich also das Zerlegen von Erkrankungen in Untergruppen (slicing) zur sukzessiven Erlangung weite-
rer Zulassungen in Indikationsgebieten mit Seltenheitswert schon für die solidarische Erstattung in
der GKV als problematisch (Kapitel 5.3), so muss diese Vorgehensweise bei der Interpretation der
Nutzenbewertung durch IQWiG und G-BA ebenfalls kritisch hinterfragt werden.
Sowohl von den forschenden Pharmaunternehmen als auch von Seiten der Fachgesellschaften wer-
den die Entscheidungen der Behörden auch deshalb kontrovers diskutiert, weil Wissens- und Er-
kenntnisdefizite eine ausreichend sichere Beurteilung nicht immer möglich machen. So können wie
im Fall der chronischen Hepatitis C die Fragen nach klinisch relevanten Unterschieden zwischen
denkbaren Therapiealternativen, einer dauerhaften Virusfreiheit nach der Behandlung mit einem
Interferon-freien Therapie-Regime, dem richtigen Zeitpunkt einer Therapie, um eine höhere und
nachhaltigere Heilungsrate zu erzielen, oder aber mögliche virale Resistenzentwicklungen aufgrund
eines breiten Einsatzes von Kombinationsregimen derzeit nicht beantwortet werden.
189

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Inwiefern sich nun die wenig euphorischen Bewertungen durch G-BA und IQWiG im konkreten Fall
der chronischen Hepatitis C auch auf den Off-Label Use in der ärztlichen Praxis auswirken, kann zu-
mindest näherungsweise nur durch Routinedaten-Analysen ermittelt werden. Näherungsweise des-
halb, weil Routinedaten keine klinischen Daten zum Genotyp oder anderen anamnestischen Hinwei-
sen enthalten.
Dies ist allerdings nicht immer im Sinne der GKV, da die Analyse solcher Daten und der so identifi-
zierte OLU auch rechtliche Konsequenzen für die Krankenkassen hat, solange keine entsprechenden
Maßnahmen folgen. Das Bundesversicherungsamt wird in solchen Fällen die jeweiligen Kassen dazu
ermahnen, tätig zu werden – und dies trifft nicht immer auf eine willige Bereitschaft.
190

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
9 Fazit und Ausblick
So lange es die Zulassungspflicht für Arzneimittel gibt, die die Anwendung der Wirkstoffe aufgrund
der Entscheidung der Hersteller für die jeweils beanspruchte Indikation eingrenzt, so lange wird es
auch den Off-Label Use geben. Allein der Contergan®-Skandal aus den 1960er Jahren macht deutlich,
wie unumgänglich im Sinne des Patientenschutzes eine solche Zulassung ist, die insbesondere auch
die erkennbaren Risiken bei einer Arzneimittelanwendung gegenüber der nachgewiesenen Wirksam-
keit prüft. Der medizinische Fortschritt wird allerdings auch in Zukunft der evidenzbasierten Medizin
immer ein Stück voraus sein. Die gesetzlichen Rahmenbedingungen ermöglichen schon aus diesen
Gründen den Off-Label Use und stellen ihn unter engen Voraussetzungen auch als rechtmäßig dar.
Unter dem Vorbehalt der Wirtschaftlichkeit entsteht die Leistungspflicht der gesetzlichen Kranken-
versicherung bereits mit der arzneimittelrechtlichen Zulassung. Durch die Rechtsprechung wurden in
den vergangenen Jahren Ausnahmen konkretisiert, die eine Kostenübernahme bei einem OLU durch
die GKV möglich machen. Damit wird ein in der Praxis oft unumgänglicher bedarfsgerechter Einsatz
von Arzneimitteln außerhalb der Zulassung bestätigt. Doch gerade die Auslegung der Forderungen
nach einer begründeten Aussicht auf Behandlungserfolg sowie die Nicht-Verfügbarkeit einer alterna-
tiven Behandlungsoption erweist sich auch noch nach Jahren als schwierig. Schließlich können bei
einem Behandlungsbedarf Arzneimittel zur Verfügung stehen, die sich im konkreten Behandlungsfall
– beispielsweise aufgrund bestehender Unverträglichkeiten – als nicht zumutbar herausstellen.
Die Therapiefreiheit des Arztes umfasst u.a. das Recht zur freien Entscheidung einer patientenindivi-
duell geeigneten Pharmakotherapie unter Berücksichtigung des allgemeinen medizinischen Stan-
dards. Da dieser nicht immer identisch mit einem in einer bestimmten Indikation zugelassenen Arz-
neimittel sein muss, ist nach dem Arzthaftungsrecht ein OLU auch dann zulässig, wenn es nicht um
die Behandlung einer schwerwiegenden Erkrankung geht. Grundsätzlich muss der Arzt dabei die
Möglichkeiten einer zulassungsüberschreitenden Anwendung und die Notwendigkeit haftungsrecht-
licher Verpflichtungen berücksichtigen.
Wie in dieser Arbeit aufgezeigt werden konnte, liegt der OLU aber nicht ausschließlich in der Be-
gründung stetig neuer Erkenntnisse. Vielmehr können auch (gesetzliche) Rahmenbedingungen, die
den In-Label Use stärken sollen, den Off-Label Use fördern. Diese reichen von der Verpflichtung zur
Registrierung klinischer Studien und der Veröffentlichung ihrer Ergebnisse, die nicht unbedingt den
Zulassungsanspruch widerspiegeln, bis hin zu den Leitlinien- und Therapieempfehlungen, die Einzug
in die ärztliche Praxis finden, in denen hin und wieder Empfehlungen ausgesprochen werden, die
nicht die zugelassenen Indikationsansprüche berücksichtigen.
191

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
Eine positive Nutzenbewertung durch den G-BA kann ebenfalls dazu führen, das Forschungsinteresse
behandelnder Ärzte auch außerhalb der zugelassenen Indikation zu wecken und die als innovativ
angepriesenen Arzneimittel möglichst zeitnah in ihrer Praxis einzusetzen.
Letztendlich trägt der behandelnde Arzt in dieser Frage die alleinige Verantwortung für eine ange-
messene patientenindividuelle Therapie. Gerade in den Fällen, in denen der erhoffte Behandlungser-
folg jedoch ausbleibt oder – wie vor allem in der Anwendung von Arzneimitteln im OLU häufig do-
kumentiert – schwere unerwünschte Arzneimittelwirkungen eine weitere Behandlung erschweren
oder sogar ausschließen, „klopft“ das Haftungsrecht nicht an die Tür von Zulassungsbehörde, GKV,
pU oder LL-Autorenschaft, sondern vielmehr an die Tür zur ärztlichen Praxis. Schließlich stellt der
medizinische Standard den Kern einer Therapie dar, mit ihm steht und fällt das ärztliche Haftungsri-
siko. Er ist außerdem der Streitpunkt bei juristischen Auseinandersetzungen. Wie diese Arbeit jedoch
zeigt, kann sich der Arzt nicht grundsätzlich darauf verlassen, aus der medizinischen Literatur valide
und verlässliche Informationen entnehmen zu können. Kenntnisse über wissenschaftliche Studien
werden beim behandelnden Arzt vorausgesetzt und intra legem verlangt, wenn es um die zivilrecht-
liche Haftung geht. In der Praxis kann der Arzt dieser Anforderung in den meisten Fällen allein schon
deshalb nicht gerecht werden, weil die Veröffentlichung von Studienergebnissen inhomogen ge-
handhabt wird und die Menge der veröffentlichten Studien eine sorgfältige Kenntnisnahme neben
der Alltagspraxis kaum möglich macht. Auch der Verweis auf Leitlinienempfehlungen kann im Fall
einer gerichtlichen Auseinandersetzung nicht unbedingt standhalten. Hier sollte für Therapieempfeh-
lungen grundsätzlich die Verpflichtung bestehen, die Empfehlung eines OLU auch als solchen kennt-
lich zu machen und auf die Bedeutung solcher Befürwortungen nach dem Haftungsrecht hinzuwei-
sen.
Bei der Auswahl der Autorenschaft für die Erstellung solcher Leitlinien sollte außerdem darauf ge-
achtet werden, dass bei keinem der Autoren ein Interessenkonflikt besteht, um Industrie-
unabhängigen Empfehlungen für Therapien gerecht zu werden. Bei der Bewertung von Studiener-
gebnissen sollte generell auf die Autorenschaft geachtet werden. Gerade bei Publikationen dersel-
ben Ergebnisse ist auffällig, dass sich bei der Autorenschaft oft nur die Reihenfolge geändert hat und
der Erst-Autor der Studie X plötzlich zum Co-Autor der Studie Y wird und umgekehrt. Dies führt zu
Mehrfach-Publikationen derselben Ergebnisse und möglicherweise zu einer Überbewertung eines
positiven Effektes (Publication-Bias).
Ein OLU setzt grundsätzlich eine Patienteneinwilligung voraus. Diese muss vor Therapiebeginn ein-
geholt werden, da es sich bei einem OLU immer um einen Eingriff in das Selbstbestimmungsrecht
des Patienten handelt. Die Dokumentation darüber kann nicht nur im zivilen Haft-, sondern auch im
Strafrecht von unschätzbarem Wert sein, rechtfertigt allerdings noch lange nicht die rechtlichen
Lücken im Verantwortungsbereich. Um den Haftungsrisiken eines OLU auszuweichen, besteht eben-
so die Möglichkeit der Verordnung eines Arzneimittels im individuellen Heilversuch, verbunden mit
192

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
einer gesteigerten Dokumentationspflicht sowie einer erhöhten Aufklärungs- und Dokumentations-
pflicht sowie einer ausreichend hohen Haftpflichtversicherung, die der in klinischen Studien ent-
spricht ( 1 Mio. Euro pro Patient) (Glaeske, 2015).
In erster Linie sind nach wie vor die pharmazeutischen Unternehmen gefordert, bei Erkenntnissen
zur Sicherheit und Wirksamkeit ihres Arzneimittels in einer zulassungsüberschreitenden Anwendung
eine zeitnahe Zulassungserweiterung zu beantragen, anstatt ökonomische Interessen im Rahmen
einer Ausweitung des Absatzes in einer nicht zugelassenen Indikation vor das Patientenwohl zu stel-
len. Anreize wie ein vereinfachtes Verfahren sowie Patent- und Unterlagenschutz sollten auch bei
einer Zulassungserweiterung in Betracht gezogen werden. Eine positive Bewertung durch die Exper-
tengruppe Off-Label Use und auf diese Weise eines legitimierten OLU dank der Aufnahme eines
Wirkstoffes in die Arzneimittelrichtlinie Anlage VI mag zwar im Einzelfall angezeigt sein, löst aber
nicht das grundsätzliche Problem, dass Erkenntnisse über die richtige Anwendung, Wirksamkeit und
Sicherheit nicht durch „Einzelfall-Therapien“ hinreichend erfasst werden können. Die Beachtung der
Pharmakovigilanz gerade im Bereich der zulassungsüberschreitenden Anwendung muss daher eine
höhere Gewichtung erhalten, da Zwangszulassungen aufgrund verfassungsrechtlicher Grenzen keine
Option sind. So sollten auch Ärzte an ihre Aufgabe erinnert werden, unerwünschte Ereignisse und
Wirkungen nicht nur als solche zu erkennen, sondern auch konsequent zu melden – unabhängig von
ihrer Schwere. Schließlich lässt sich nicht bestreiten, dass der breite OLU in der Regel auf ungesicher-
ten wissenschaftlichen Erkenntnissen basiert – wissentlich geduldet vom pU, der so den gesetzlichen
Bestimmungen eines Zulassungsverfahrens ausweicht.
Vorstellbar wäre grundsätzlich, Informationen speziell zu den zulassungsüberschreitenden Anwen-
dungen zu sammeln und diese – ähnlich wie die Informationsseite des Pharmakovigilanz- und Bera-
tungszentrums für Embryonaltoxikologie (www.embryotox.de) – aufzubereiten und Fachgesellschaf-
ten zur Verfügung zu stellen. Diese Aufgabe sollte ein öffentlich gefördertes, autonomes Institut
übernehmen, welches unabhängige Informationen zum komplexen Thema OLU anbietet und den
Ärzten einen schnellen Zugriff auf wichtige Informationen liefert. So schafft man es auch, Arzneimit-
tel aus ihrer Grauzone des OLU herauszuholen und verpflichtet gleichzeitig den pU durch die Veröf-
fentlichung der Daten, Stellung zu beziehen.
Leider wird der wichtige Beitrag einer methodisch abgesicherten Versorgungsforschung auch bei
vielen GKVen noch immer nicht ausreichend erkannt. Dieser kann aber geleistet werden, indem die
Kassen Patientendaten „nicht nur verwalten“, sondern sie unter Beachtung des Datenschutzgesetzes
auch für Forschungszwecke zur Verfügung stellen – unabhängig von einer pressewirksamen Public
Relation. Schließlich sollten die bereits heute existierenden Möglichkeiten besser zur systematischen
Gewinnung von medizinischen Erkenntnissen genutzt werden, um patientenorientierte Lösungen zu
erhalten. Dazu zählt auch die Strategie, nach der Zulassung eines Arzneimittels wissenschaftlich gesi-
cherte Erkenntnisse zur Stärkung einer unabhängigen Versorgungsforschung zu erhalten, um Wirk-
193

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
samkeit und Sicherheit des zulassungsüberschreitenden Einsatzes von Arzneimitteln systematisch
erfassen und gewinnen zu können.
Die Verordnung eines Arzneimittels außerhalb der Zulassung ist der häufigste Anlass für die GKVen,
beim MDK eine medizinische Beurteilung für diesen OLU einzuholen. Aus diesem Grunde sollten
diese Beurteilungen – unter Berücksichtigung, dass es sich dabei immer um Einzelfallentscheidungen
handelt – auch für eine unabhängige Versorgungsforschung zur Verfügung gestellt werden. Ökono-
mische Interessen (§ 12 SGB V) dürfen grundsätzlich auch für GKVen nicht die Argumentationsbasis
für das Zurückhalten wichtiger Daten zur Erkenntnisgewinnung über einen OLU sein. Letztendlich
sollten auch nicht Regressansprüche seitens der GKVen der Motor für das Aufdecken eines OLU sein,
sondern der Anspruch auf eine wirksame und angemessene Arzneimitteltherapie, der allein durch
die Zulassung eines Arzneimittels nicht gegeben ist. Schlussendlich darf nicht verkannt werden, dass
durch eine zulassungsüberschreitende Anwendung neue Behandlungsperspektiven aufgezeigt wer-
den können. Aber auch diese durchaus sinnvolle Strategie kann nur innerhalb klarer Rahmenbedin-
gungen toleriert werden, die insbesondere das Patientenwohl und den Patientenschutz in den Vor-
dergrund stellen. Ein OLU zur Erweiterung der Indikationsansprüche ohne entsprechende Zulas-
sungsstudien, die nur dem pU nützt, darf nicht durch die behandelnden Ärzte leichtfertig unterstützt
werden.
Die Lösung des Problems kann also letztendlich nur aufgrund eines engen Zusammenspiels verschie-
dener Institutionen und Regularien greifen, wobei auf jeden Fall das Wohl des Patienten über den
ökonomischen Interessen der Anbieter steht. Neben Änderungen in der Gesetzgebung ist daher der
gute Wille aller Beteiligten notwendig, die das Ziel verfolgen, den Patienten die bestmögliche Ver-
sorgung zur Verfügung zu stellen.
194

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
10 Literaturverzeichnis
1. AALSD (2014). American Association For The Study Of Liver Disease/Infectious Diseases Society of America. Recommendations for testing, managing and treating Hepatitis C. http://www.hcvguidelines.org. Letzter Zugriff: 28.01.2015.
2. abbvie (2015a). Fachinformation – Zusammenfassung der Merkmale des Arzneimittels Viekirax® 12,5 mg/75 mg/50 mg. Stand der Information: Januar 2015.
3. abbvie (2015b). Fachinformation – Zusammenfassung der Merkmale des Arzneimittels Exviera® 250 mg Filmtabletten. Stand der Information: Januar 2015.
4. Adams NP, Bestall JB, Jones PW (2001). Budesonide for chronic asthma in children and adults. Cochrane Database Syst Rev (4):CD003274.
5. Adams NP, Bestall JB, Malouf R, Lasserson TJ, Jones PW (2005). Inhaled beclomethasone versus placebo for chronic asthma. Cochrane Database Syst Rev(1):CD002738.
6. Afdhal N, Reddy KR, Nelson DR, Lawitz E, Gordon SC, Schiff E, Nahass R, Ghalib R, Gitlin N, Herring R, Lalezari J, Younes ZH, Pockros PJ, Di Bisceglie AM, Arora S, Subramanian GM, Zhu Y, Dvory-Sobol H, Yang JC, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Sulkowski M, Kwo P; ION-2 Investigators (2014b). Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med. 370 (16): 1483-93.
7. Afdhal N, Zeuzem S, Kwo P, Chojkier M, Gitlin N, Puoti M, Romero-Gomez M, Zarski JP, Agarwal K, Buggisch P, Foster GR, Bräu N, Buti M, Jacobson IM, Subramanian GM, Ding X, Mo H, Yang JC, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Mangia A, Marcellin P; ION-1 Investigators (2014a). Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med. 370 (20): 1889-98.
8. Afentaki A (2014). Medicines for children and "off-label use" 5 years after implementation of the Paediatric Regulation (EC) No 1901/2006. An interim analyis. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 57 (9): 1111-9.
9. Almirall (2014). Fachinformation – Zusammenfassung der Merkmale des Arzneimittels Eklira® Genuair® 322 Mikrogramm Pulver zur Inhalation. Stand der Information: Mai 2014.
10. AMG (2005). Gesetz über den Verkehr mit Arzneimitteln (AMG – Arzneimittelgesetz) Arzneimittelgesetz in der Fassung der Bekanntmachung vom 12. Dezember 2005 (BGBl. I S. 3394), zuletzt geändert durch Artikel 3 des Gesetzes vom 17. Dezember 2014 (BGBl. I S. 2222).
11. Anderson ML, Chiswell K, Peterson ED, Tasneem A, Topping J, Califf RM (2015). Compliance with results reporting at ClinicalTrials.gov. N Engl J Med. 372 (11): 1031-9. doi: 10.1056/NEJMsa1409364.
12. Andreone P, Colombo MG, Enejosa JV, Koksal I, Ferenci P, Maieron A, Müllhaupt B, Horsmans Y, Weiland O, Reesink HW, Rodrigues L Jr, Hu YB, Podsadecki T, Bernstein B (2014). ABT-450, ritonavir, ombitasvir, and dasabuvir achieves 97% and 100% sustained virologic response with or without ribavirin in treatment-experienced patients with HCV genotype 1b infection. Gastroenterology. 147 (2): 359-365.e1.
195

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
13. Antaki N, Craxi A, Kamal S, Moucari R, Van der Merwe S, Haffar S, Gadano A, Zein N, Lai CL, Pawlotsky JM, Heathcote EJ, Dusheiko G, Marcellin P (2010). The neglected hepatitis C virus genotypes 4, 5 and 6: an international consensus report. Liver Int. 30 (3): 342-55.
14. Augustin M (2013). [Appropriate off-label prescription in practice]. Hautarzt. 64 (10): 728-35.
15. Aumann I & Prenzler A (2013). Epidemiologie und Kosten der COPD in Deutschland – Eine Literaturrecherche zu Prävalenz, Inzidenz und Krankheitskosten. Klinikarzt. 42 (4): 168-172.
16. Aumann I, Prenzler A, Welte T, Gillissen A (2014). [Epidemiology and costs of asthma in Germany - a systematic literature review]. Pneumologie. 68 (8): 557-67.
17. Avorn J (2006). Dangerous deception - hiding the evidence of adverse drug effects. N Engl J Med. 355 (21): 2169-71.
18. awmf & äzq (2008). Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften und Ärztliches Zentrum für Qualität in der Medizin. Deutsches Instrument zur methodischen Leitlinien-Bewertung (DELBI) in der Fassung 2005/2006 + Domäne 8 (2008). http://www.leitlinien.de/mdb/edocs/pdf/literatur/delbi-fassung-2005-2006-domaene-8-2008.pdf. Letzter Zugriff: 10.05.2015.
19. awmf (2009). Arbeitsgemeinschaft der wissenschaftlichen, medizinischen Fachgesellschaften: S3-Leitlinie Gastroenterologie: Prophylaxe, Diagnostik und Therapie der Hepatitis C-Virus (HCV)-Infektion. http://www.awmf.org/uploads/tx_szleitlinien/021-012l_S3_Hepatitis-C-Virus_HCV-Infektion_abgelaufen-mit-a.pdf. Gültigkeit bis 31.12.2012, derzeit in Überarbeitung. Letzter Zugriff: 21.01.2015.
20. Bacharier LB, Boner A, Carlsen KH, Eigenmann PA, Frischer T, Gotz M, Helms PJ, Hunt J, Liu A, Papadopoulos N, Platts-Mills T, Pohunek P, Simons FE, Valovirta E, Wahn U, Wildhaber J (2008). Diagnosis and treatment of asthma in childhood: a PRACTALL consensus report. Allergy. 63 (1): 5-34.
21. Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S, Poordad F, Goodman ZD, Sings HL, Boparai N, Burroughs M, Brass CA, Albrecht JK, Esteban R, for the HCV RESPOND-2-Investigators (2011). Boceprevir for Previously Treated Chronic HCV Genotype 1 Infection. N Engl J Med. 364: 1207-17.
22. Bajcetic M & Gorodischer R (2005). Off label and unlicensed drugs use in paediatric cardiology. European Journal of Clinical Pharmacology. 61, 775-779.
23. BÄK & KBV (1996). Qualitätssicherung und kontinuierliche Qualitätsverbesserung: Grundlagen einer bedarfsgerechten Gesundheitsversorgung. Gemeinsame Bestandsaufnahme der Bundesärztekammer und der Kassenärztlichen Bundesvereinigung über die Aktivitäten der Spitzenorganisationen der ärztlichen Selbstverwaltung auf dem Gebiet der Qualitätssicherung in der Medizin 1955 bis 1995. http://www.aezq.de/mdb/edocs/pdf/stellungnahmen/sn-qualitaetssicherung-1996.pdf. Letzter Zugriff: 11.01.2015.
24. BÄK & KBV (1997). Bundesärztekammer und Kassenärztliche Bundesvereinigung. Beurteilungskriterien für Leitlinien in der medizinischen Versorgung. Dtsch Ärztebl,94:A2154-2155,B-1622-1623,C-1754-1755.
25. Barbui C & Cipriani A (2007). Publication bias in systematic reviews. Arch Gen Psychiatry. 6 4(7): 868.
196

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
26. Barnes PJ (2000). Mechanisms in COPD. Chest. 117:10S-14S.
27. Barnes PJ (2002). Scientific rationale for inhaled combination therapy with long-acting beta2-agonists and corticosteroids. Eur Respir J. 19 (1): 182-91.
28. Barnes PJ (2004). Corticosteroid resistance in airway disease. Proc Am Thorac Soc. 1 (3): 264-8.
29. Barnes PJ (2012). Severe asthma: advances in current management and future therapy. J Allergy Clin Immunol. 129 (1): 48-59.
30. Bateman ED, Izquierdo JL, Harnest U, Hofbauer P, Magyar P, Schmid-Wirlitsch C, Leichtl S, Bredenbröker D (2006). Efficacy and safety of roflumilast in the treatment of asthma. Ann Allergy Asthma Immunol. 96 (5): 679-86.
31. Battram C, Charlton SJ, Cuenoud B, Dowling MR, Fairhurst RA, Farr D, Fozard JR, Leighton-Davies JR, Lewis CA, McEvoy L, Turner RJ, Trifilieff A (2006). In vitro and in vivo pharmacological characterization of 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-1-hydroxy-ethyl]-8-hydroxy-1H-quinolin-2-one (indacaterol), a novel inhaled beta(2) adrenoceptor agonist with a 24-h duration of action. J Pharmacol Exp Ther. 317 (2): 762-70.
32. Beasley RW, Donohue JF, Mehta R, Nelson HS, Clay M, Moton A, Kim HJ, Hederer BM (2015). Effect of once-daily indacaterol maleate/mometasone furoate on exacerbation risk in adolescent and adult asthma: a double-blind randomised controlled trial. BMJ Open. 5(2): e006131.
33. Becker J (2004). Off-label-Use: Arzneimittelversorgung in der gesetzlichen Krankenversicherung nur bei Todesgefahr? SGb. 594-599.
34. Beeh KM, Derom E, Kanniess F, Cameron R, Higgins M, van As A (2007). Indacaterol, a novel inhaled beta2-agonist, provides sustained 24-h bronchodilation in asthma. Eur Respir J. 29 (5): 871-8.
35. Beier J, Kirsten AM, Mróz R, Segarra R, Chuecos F, Caracta C, Gil EG (2013). Efficacy and safety of aclidinium bromide compared with placebo and tiotropium in patients with moderate-to-severe chronic obstructive pulmonary disease: results from a 6-week, randomized, controlled Phase IIIb study. COPD. 10 (4): 511-22.
36. Benhamou Y, Moussalli J, Ratziu V, Lebray P, De Backer K, De Meyer S, Ghys A, Luo D, Picchio GR, Beumont M (2013). Telaprevir activity in treatment-naive patients infected hepatitis C virus genotype 4: a randomized trial. J Infect Dis. 208 (6): 1000-7.
37. Bennett CL, Nebeker JR, Lyons EA, Samore MH, Feldman MD, McKoy JM, Carson KR, Belknap SM, Trifilio SM, Schumock GT, Yarnold PR, Davidson CJ, Evens AM, Kuzel TM, Parada JP, Cournoyer D, West DP, Sartor O, Tallman MS, Raisch DW (2005). The Research on Adverse Drug Events and Reports (RADAR) project. JAMA. 293 (17): 2131-40.
38. Bergmann R, Forster J, Bauer CP, Illi S, von Mutius E, Wahn V, Bergmann K, Wahn U (2002). Zehn Jahre Multizentrische Allergie Studie MAS 90. Pädiatrische Allergologie. 3:6-11.
39. BfArM (2013). Bundesinstitut für Arzneimittel und Medizinprodukte – Historie der Expertengruppe Off-Label von 2002-2013. http://www.bfarm.de/DE/Arzneimittel/zul/BereitsZugelAM/offLabel/Historie/historie-startseite.html. Letzter Zugriff: 03.01.2015.
197

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
40. BfArM (2015). Bundesinstitut für Arzneimittel und Medizinprodukte - Sofosbuvir: Herzrhythmusstörungen als mögliche Wechselwirkung mit anderen direkt wirksamen antiviralen Arzneimitteln (DAA) in der Behandlung der Hepatitis C in Kombination mit Arzneimitteln, die eine Bradykardie verursachen können (z.B. Amiodaron). Stand der Information: 27.03.2015.
41. BGH (2014). Bundesgerichtshof. Urteil vom 15.04.2014. Az: VI ZR 382/12.
42. Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA (2000). Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax. 55 (2): 114-20.
43. BMS (2014). Bristol-Myers Squibb. Fachinformation - Zusammenfassung der Merkmale des Arzneimittels Daklinza® Filmtabletten. Stand der Information: September 2014.
44. BMV-Ä (2015). Bundesmantelvertrag Ärzte. Stand Januar 2015.
45. Böger RH (2002). Endpunkte klinischer Studien: Surrogatparameter oder “harte klinische Endpunkte”?. Internist 43: 493-497.
46. Boeschen D, Günther J, Glaeske G (2015a). Chronische Hepatitis C – Ein Statusbericht zur antiviralen Therapie. Projektbericht der Universität Bremen. Zentrum für Sozialpolitik; Techniker Krankenkasse, Hamburg; Eigenverlag.
47. Boeschen D, Fuchs D, Günther J, Glaeske G (2015b). Innovationsreport 2015. Auswertungsergebnisse von Routinedaten der Techniker Krankenkasse aus den Jahren 2012 und 2013 (Langfassung), SOCIUM; Techniker Krankenkasse, Hamburg; Eigenverlag (bislang unveröffentlicht).
48. Bousquet J, Aubier M, Sastre J, Izquierdo JL, Adler LM, Hofbauer P, Rost KD, Harnest U, Kroemer B, Albrecht A, Bredenbröker D (2006). Comparison of roflumilast, an oral anti-inflammatory, with beclomethasone dipropionate in the treatment of persistent asthma. Allergy. 61 (1): 72-8.
49. Brunkhorst H & Dittmayer M (2008). Der „medizinische Standard“: Wie bestimmt sich eigentlich der „richtige“ Standard? Seminararbeit im „Gesundheitsrechtlichen Seminar“ von Prof. Dr. Benedikt Buchner. Universität Bremen.
50. Bruno S, Vierling JM, Esteban R, Nyberg LM, Tanno H, Goodman Z, Poordad F, Bacon B, Gottesdiener K, Pedicone LD, Albrecht JK, Brass CA, Thompson S, Burroughs MH (2013). Efficacy and safety of boceprevir plus peginterferon-ribavirin in patients with HCV G1 infection and advanced fibrosis/cirrhosis. J Hepatol. 58 (3): 479-87.
51. Bruns J & Herz E (2003). Off label use: from the viewpoint of the social health insurance funds. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 46: 477-482.
52. BSG (2002). Bundessozialgericht - Urteil vom 19.03.2002. Az. B1 KR 37/00 R. http://lexetius.com/2002,1157. Letzter Zugriff: 03.05.2014.
53. BSG (2004). Bundessozialgericht - Urteil vom 19.10.2004, Az. B 1 KR 27/02 R. http://lexetius.com/2004,3714. Letzter Zugriff: 03.05.2014.
54. BSG (2012). Bundessozialgericht – Urteil vom 03.07.2012. Az.: B 1 KR 25/11 R. https://sozialgerichtsbarkeit.de/sgb/esgb/show.php?modul=esgb&id=154929. Letzter Zugriff: 01.04.2015.
198

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
55. Bücheler R, Schwoerer P, Gleiter CH (2003). Off-Label-Verordnungen in der Pädiatrie. Bundesgesundheitsblatt – Gesundheitsforschung – Gesundheitsschutz. 46: 467-476.
56. Buchner R & Jäckel Chr (2003). Off-Label-Use von Arzneimitteln in der gesetzlichen Krankenversicherung – neue Regelungen durch das GKV-Modernisierungsgesetz. PharmR. 433-438.
57. Buhl R, Berdel D, Criee C, Gillissen A, Kardos P, Kroegel C, Leupold W, Lindemann H, Magnussen H, Nowak D, Pfeiffer-Kascha D, Rabe K, Rolke M, Schultze-Werninghaus G, Sitter H, Ukena D, Vogelmeier C, Welte T, Wettengel R, Worth H (2006). Leitlinie zur Diagnostik und Therapie von Patienten mit Asthma. Herausgegeben von der Deutschen Atemwegsliga und der Deutschen Gesellschaft für Pneumologie und Beatmungsmedizin e. V. Pneumologie. 60 (3): 139-77.
58. Buhl R, Dunn LJ, Disdier C, Lassen C, Amos C, Henley M, Kramer B; INTENSITY study investigators (2011). Blinded 12-week comparison of once-daily indacaterol and tiotropium in COPD. Eur Respir J. 38 (4): 797-803.
59. Buhles WC (2011). Compassionate use: a story of ethics and science in the development of a new drug. Perspect Biol Med. 54 (3): 304-15.
60. Burge PS, Calverley PM, Jones PW, Spencer S, Anderson JA, Maslen TK (2000). Randomised, double blind, placebo controlled study of fluticasone propionate in patients with moderate to severe chronic obstructive pulmonary disease: the ISOLDE trial. BMJ. 320 (7245): 1297-303.
61. BVerfG (2005). Bundesverfassungsgericht – Urteil vom 06.12.2005, Az. 1 BvR 347/98. http://lexetius.com/2005,2827. Letzter Zugriff: 03.05.2014.
62. Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ, M2-124 and M2-125 study groups (2009): Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet. 374 (9691): 685-94.
63. Castle W, Fuller R, Hall J, Palmer J (1993). Serevent nationwide surveillance study: comparison of salmeterol with salbutamol in asthmatic patients who require regular bronchodilator treatment. BMJ. 306 (6884): 1034-7.
64. Cates CJ & Cates MJ (2008). Regular treatment with salmeterol for chronic asthma: serious adverse events. Cochrane Database Syst Rev. (3): CD006363. doi: 10.1002/14651858.CD006363.pub2.
65. Cates CJ, Wieland LS, Oleszczuk M, Kew KM (2014). Safety of regular formoterol or salmeterol in adults with asthma: an overview of Cochrane reviews. Cochrane Database Syst Rev. 2: CD010314.
66. Cazzola M, MacNee W, Martinez FJ, Rabe KF, Franciosi LG, Barnes PJ, Brusasco V, Burge PS, Calverley PM, Celli BR, Jones PW, Mahler DA, Make B, Miravitlles M, Page CP, Palange P, Parr D, Pistolesi M, Rennard SI, Rutten-van Mölken MP, Stockley R, Sullivan SD, Wedzicha JA, Wouters EF; American Thoracic Society; European Respiratory Society Task Force on outcomes of COPD (2008). Outcomes for COPD pharmacological trials: from lung function to biomarkers. Eur Respir J. 31 (2): 416-69.
67. Chan JY, Stern DA, Guerra S, Wright AL, Morgan WJ, Martinez FD (2015). Pneumonia in childhood and impaired lung function in adults: a longitudinal study. Pediatrics. 135 (4): 607-16.
199

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
68. Charlton M, Gane E, Manns MP, Brown RS Jr, Curry MP, Kwo PY, Fontana RJ, Gilroy R, Teperman L, Muir AJ, McHutchison JG, Symonds WT, Brainard D, Kirby B, Dvory-Sobol H, Denning J, Arterburn S, Samuel D, Forns X, Terrault NA (2015). Sofosbuvir and ribavirin for treatment of compensated recurrent hepatitis C virus infection after liver transplantation. Gastroenterology. 148 (1): 108-17.
69. Chowdhury BA & Dal Pan G (2010). The FDA and safe use of long-acting beta-agonists in the treatment of asthma. N Engl J Med. 362 (13): 1169-71.
70. Chowdhury BA, Seymour SM, Levenson MS (2011). Assessing the safety of adding LABAs to inhaled corticosteroids for treating asthma. N Engl J Med. 364 (26): 2473-5.
71. Chowdhury BA, Seymour SM, Michele TM, Durmowicz AG, Liu D, Rosebraugh CJ (2011a). The risks and benefits of indacaterol--the FDA's review. N Engl J Med. 365 (24): 2247-9.
72. Chuchalin AG, Tsoi AN, Richter K, Krug N, Dahl R, Luursema PB, Cameron R, Bao W, Higgins M, Woessner R, van As A (2007). Safety and tolerability of indacaterol in asthma: a randomized, placebo-controlled 28-day study. Respir Med. 101 (10): 2065-75.
73. Clemens T (2011). Zulässigkeit von Arzneiverordnungen und Kostenregresse durch Ärzte – Off-Label-Use und Unlicensed Use. GesR. 397-409.
74. Collier J (1999). Paediatric prescribing: using unlicensed drugs and medicinesoutside their licensed indications. Br J Clin Pharmacol. 48(1): 5-8.
75. Cornberg M, Höner zu Siederdissen C, Maasoumy B, Manns MP (2014). New direct-acting antiviral agents for the treatment of chronic hepatitis C in 2014. Internist (Berl). 55 (4): 390-400.
76. Cornberg M, Razavi HA, Alberti A, Bernasconi E, Buti M, Cooper C, Dalgard O, Dillion JF, Flisiak R, Forns X, Frankova S, Goldis A, Goulis I, Halota W, Hunyady B, Lagging M, Largen A, Makara M, Manolakopoulos S, Marcellin P, Marinho RT, Pol S, Poynard T, Puoti M, Sagalova O, Sibbel S, Simon K, Wallace C, Young K, Yurdaydin C, Zuckerman E, Negro F, Zeuzem S (2011). A systematic review of hepatitis C virus epidemiology in Europe, Canada and Israel. Liver Int. 31 Suppl 2:30-60.
77. Curry MP, Forns X, Chung RT, Terrault NA, Brown R Jr, Fenkel JM, Gordon F, O'Leary J, Kuo A, Schiano T, Everson G, Schiff E, Befeler A, Gane E, Saab S, McHutchison JG, Subramanian GM, Symonds WT, Denning J, McNair L, Arterburn S, Svarovskaia E, Moonka D, Afdhal N (2015). Sofosbuvir and ribavirin prevent recurrence of HCV infection after liver transplantation: an open-label study. Gastroenterology. 148(1): 100-107.e1.
78. Dahl R, Chung KF, Buhl R, Magnussen H, Nonikov V, Jack D, Bleasdale P, Owen R, Higgins M, Kramer B; INVOLVE (INdacaterol: Value in COPD: Longer Term Validation of Efficacy and Safety) Study Investigators (2010). Efficacy of a new once-daily long-acting inhaled beta2-agonist indacaterol vs. twice-daily formoterol in COPD. Thorax. 65 (6): 473-9.
79. Decramer ML, Chapman KR, Dahl R, Frith P, Devouassoux G, Fritscher C, Cameron R, Shoaib M, Lawrence D, Young D, McBryan D; INVIGORATE investigators (2013). Once-daily indacaterol versus tiotropium for patients with severe chronic obstructive pulmonary disease (INVIGORATE), a randomised, blinded, parallel-group study. Lancet Respir Med. 1 (7): 524-33.
80. de Mey C, Nassr N, Lahu G (2011). No relevant cardiac, pharmacokinetic or safety interactions between roflumilast and inhaled formoterol in healthy subjects: an open-label, randomised, actively controlled study. BMC Clin Pharmacol. 11:7.
200

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
81. Demoly P, Paggiaro P, Plaza V, Bolge SC, Kannan H, Sohier B, Adamek L (2009). Prevalence of asthma control among adults in France, Germany, Italy, Spain and the UK. Eur Respir Rev. 18 (112): 105-12.
82. Deutsche Gesellschaft für Pneumologie (2000). Stellungnahme der Deutschen Gesellschaft für Pneumologie an den Sachverständigenrat für die Konzentrierte Aktion im Gesundheitswesen Über-, Unter- und Fehlversorgung in der Pneumologie. Stand der Information: 29.08.2000.
83. DGVS (2015). Deutsche Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselerkrankungen: Aktuelle Empfehlung zur Therapie der chronischen Hepatitis C. Addendum zur Hepatitis C Leitlinie. http://www.dgvs.de/fileadmin/user_upload/Leitlinien/Hepatitis_C/Addendum_Leitlinie_Therapie_Hepatitis_C_18_02_2015.pdf. Letzter Zugriff: 15.03.2015.
84. Dierks Chr (2003). Rechtliche Aspekte der Off-Label-Verordnung in der Praxis. BGesundhBl. 46/6, 458-460.
85. Dierks Chr (2008). Rechtliche Konsequenzen suboptimaler Arzneimitteltherapue. Z Evid Fortbild Qual Gesundh.wesen. 102: 591-593.
86. Dieterich D, Rockstroh JK, Orkin C, Gutiérrez F, Klein MB, Reynes J, Shukla U, Jenkins A, Lenz O, Ouwerkerk-Mahadevan S, Peeters M, De La Rosa G, Tambuyzer L, Jessner W (2014). Simeprevir (TMC435) With Pegylated Interferon/Ribavirin in Patients Coinfected With HCV Genotype 1 and HIV-1: A Phase 3 Study. Clin Infect Dis. 9 (11): 1579-87.
87. Donohue JF (2005). Minimal clinically important differences in COPD lung function. COPD. 2 (1): 111-24.
88. Donohue JF, Fogarty C, Lötvall J, Mahler DA, Worth H, Yorgancioglu A, Iqbal A, Swales J, Owen R, Higgins M, Kramer B; INHANCE Study Investigators (2010). Once-daily bronchodilators for chronic obstructive pulmonary disease: indacaterol vs. tiotropium. Am J Respir Crit Care Med. 182(2): 155-62.
89. Dore GJ, Lawitz E, Hézode C, Shafran SD, Ramji A, Tatum HA, Taliani G, Tran A, Brunetto MR, Zaltron S, Strasser SI, Weis N, Ghesquiere W, Lee SS, Larrey D, Pol S, Harley H, George J, Fung SK, de Lédinghen V, Hagens P, McPhee F, Hernandez D, Cohen D, Cooney E, Noviello S, Hughes EA (2015). Daclatasvir plus peginterferon and ribavirin is noninferior to peginterferon and ribavirin alone, and reduces the duration of treatment for HCV genotype 2 or 3 infection. Gastroenterology. 148 (2): 355-366.e1.
90. DPhG (2005). „Kriterien für die Beurteilung von Arzneimittelinnovationen“ - Positionspapier der Deutschen Pharmazeutischen Gesellschaft unter Mitarbeit der Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik http://dphg.de/includes/upload/DPhG-Positionspapier.pdf. Letzter Zugriff: 14.01.2014.
91. EASL (2014). European Association for Study of Liver. EASL Clinical Practice Guidelines: Management of hepatitis C virus infection. J Hepatol. 60 (2): 392-420.
92. Easthope S & Jarvis B (2001). Omalizumab. Drugs. 61 (2): 253-60; discussion 261.
93. Ehlers AP & Wenke A (2011). [Reimbursement of new medical therapy]. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 54 (7): 849-55.
94. EMA (2011). European Medicines Agency – Assessment Report Victrelis (Boceprevir). EMA/CHMP/314280/2011. Stand der Information: 26.05.2011.
201

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
95. EMA (2011a). European Medicines Agency – Assessment Report Incivo (Telaprevir). EMA/CHMP/475470/2011. Stand der Information: 21.07.2011.
96. EMA (2013). European Medicines Agency – Assessment Report Sovaldi (Sofosbuvir). EMA/CHMP/688774/2013. Stand der Information: 21.11.2013.
97. EMA (2014). European Medicines Agency – Assessment Report Olysio (Simeprevir). EMA/CHMP/285947/2014. Stand der Information: 20.03.2014.
98. EMA (2014a). European Medicines Agency – Assessment Report Daklinza (Daclatasvir). EMA/419836/2014. Stand der Information: 26.06.2014.
99. EMA (2014b). European Medicines Agency – Assessment Report Harvoni (Ledipasvir/Sofosbuvir). EMA/702742/2014. Stand der Information: 25.09.2014.
100. EMA (2014c). European Medicines Agency – Assessment Report Exviera (Dasabuvir). EMA/768319/2014. Stand der Information: 20.11.2014.
101. EMA (2014d). European Medicines Agency – Committee for Medicinal Products for Human Use (CHMP) Summary of opinion1 (initial authorisation) Viekirax: ombitasvir / paritaprevir / ritonavir. 20 November 2014. EMA/CHMP/688255/2014. Letzter Zugriff: 21.01.2015.
102. EMA (2014e). European Medicines Agency – Summary of opinion Exviera. http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_-_Initial_authorisation/human/003837/WC500177625.pdf. Letzter Zugriff: 01.12.2014.
103. EMA (2014f) European Medicines Agency – Zusammenfassung der Merkmale des Arzneimittels Bretaris Genuair. http://www.ema.europa.eu/docs/de_DE/document_library/EPAR_-_Product_Information/human/002706/WC500132732.pdf. Stand: 18.12.2014. Letzter Zugriff: 29.12.2014
104. EMA (2015). European Medicines Agency – EPAR Product Information Incivo http://www.ema.europa.eu/docs/de_DE/document_library/EPAR_-_Product_Information/human/002313/WC500115529.pdf. Letzter Zugriff: 08.04.2015.
105. EMA (2015a). European Medicines Agency – Orphan Medicines Figures 2000/2014. Update 01.01.2015. http://www.ema.europa.eu/docs/en_GB/document_library/Other/2015/04/WC500185766.pdf. Letzter Zugriff: 05.05.2015.
106. Europäisches Parlament & Europäischer Rat (1999). Verordnung (EG) Nr. 141/2000 vom 16. Dezember 1999 über Arzneimittel für seltene Leiden geändert durch Verordnung (EG) Nr. 596/2009 vom 18. Juni 2009.
107. Fabbri LM & Rabe KF (2007). From COPD to chronic systemic inflammatory syndrome? Lancet.; 370 (9589): 797-9.
108. Fabbri LM, Calverley PM, Izquierdo-Alonso JL, Bundschuh DS, Brose M, Martinez FJ, Rabe KF; M2-127 and M2-128 study groups (2009). Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet. 374 (9691): 695-703.
202

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
109. FDA (2010). U.S. Food and Drug Administration. FDA Drug Safety Communication: New safety requirements for long-acting inhaled asthma medications called Long-Acting Beta-Agonists (LABAs). http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm200776.htm. Letzter Zugriff: 03.02.2015.
110. FDA (2014). U.S. Food and Drug Administration. Certifications To Accompany Drug, Biological Product, and Device Applications/Submissions: Compliance with Section 402(j) of The Public Health Service Act, Added By Title VIII of The Food and Drug Administration Amendments Act of 2007. Guidance for Sponsors, Industry, Researchers, Investigators, and Food and Drug Administration Staff. http://www.fda.gov/RegulatoryInformation/Guidances/ucm125335.htm. Stand: Oktober 2014. Letzter Zugriff: 27.04.2015.
111. FDA (2015). U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA warns of serious slowing of the heart rate when antiarrhythmic drug amiodarone is used with hepatitis C treatments containing sofosbuvir (Harvoni) or Sovaldi in combination with another Direct Acting Antiviral drug. Stand der Information: 24. März 2015.
112. Feld JJ, Kowdley KV, Coakley E, Sigal S, Nelson DR, Crawford D, Weiland O, Aguilar H, Xiong J, Pilot-Matias T, DaSilva-Tillmann B, Larsen L, Podsadecki T, Bernstein B (2014). Treatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med. 370 (17): 1594-603.
113. Feldman G, Siler T, Prasad N, Jack D, Piggott S, Owen R, Higgins M, Kramer B; INLIGHT 1 study group (2010). Efficacy and safety of indacaterol 150 microg once-daily in COPD: a double-blind, randomised, 12-week study. BMC Pulm Med. 10: 11.
114. Ferenci P, Bernstein D, Lalezari J, Cohen D, Luo Y, Cooper C, Tam E, Marinho RT, Tsai N, Nyberg A, Box TD, Younes Z, Enayati P, Green S, Baruch Y, Bhandari BR, Caruntu FA, Sepe T, Chulanov V, Janczewska E, Rizzardini G, Gervain J, Planas R, Moreno C, Hassanein T, Xie W, King M, Podsadecki T, Reddy KR; PEARL-III Study; PEARL-IV Study (2014). ABT-450/r-ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med. 370 (21): 1983-92.
115. Ferrara N & Kerbel RS (2005). Angiogenesis as a therapeutic target. Nature. 438 (7070): 967-74.
116. Field MJ & Lohr KN (1995). Health services research: an expanding field of inquiry. J Eval Clin Pract. 1 (1): 61-5.
117. Flamm SL, Lawitz E, Jacobson I, Bourlière M, Hezode C, Vierling JM, Bacon BR, Niederau C, Sherman M, Goteti V, Sings HL, Barnard RO, Howe JA, Pedicone LD, Burroughs MH, Brass CA, Albrecht JK, Poordad F (2013). Boceprevir with peginterferon alfa-2a-ribavirin is effective for previously treated chronic hepatitis C genotype 1 infection. Clin Gastroenterol Hepatol. 11 (1): 81-87.e4; quiz e5.
118. Forey BA, Thornton AJ, Lee PN (2011). Systematic review with meta analysis of the epidemiological evidence relating smoking to COPD, chronic bronchitis and emphysema. BMC Pulm Med. 11: 36.
119. Forns X, Lawitz E, Zeuzem S, Gane E, Bronowicki JP, Andreone P, Horban A, Brown A, Peeters M, Lenz O, Ouwerkerk-Mahadevan S, Scott J, De La Rosa G, Kalmeijer R, Sinha R, Beumont-Mauviel M (2014). Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a Phase III trial. Gastroenterology. 146: 1669-79.
120. Foster GR, Fried MW, Hadziyannis SJ, et al. (2007). Prediction of sustained virological response in chronic hepatitis C patients treated with peginterferon alfa-2a (40KD) and ribavirin. Scand J Gastroenterol. 42: 247-255.
203

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
121. Francke R & Hart D (2003). Off-Label-Use – Arzneimittelrechtliche, haftungsrechtliche, berufsrechtliche und sozialrechtliche Fragen. SGb 653 ff.
122. Fricke U & Beck T (2013). Neue Arzneimittel. Band 20: Fakten und Bewertungen. Stuttgart: Wissenschaftliche Verlagsgesellschaft.
123. Fried MW, Buti M, Dore GJ, Flisiak R, Ferenci P, Jacobson I, Marcellin P, Manns M, Nikitin I, Poordad F, Sherman M, Zeuzem S, Scott J, Gilles L, Lenz O, Peeters M, Sekar V, De Smedt G, Beumont-Mauviel M (2013). Once-daily simeprevir (TMC435) with pegylated interferon and ribavirin in treatment-naïve genotype 1 hepatitis C: the randomized PILLAR study. Hepatology. 58 (6): 1918-29.
124. Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT, Hindes RG, Berrey MM (2013). Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med. 368(1): 34-44.
125. Gaßner M & Strömer JM (2012). Die Arzthaftung bei der Behandlung gesetzlich krankenversicherter Patienten. MedR. 30: 159-169.
126. Gaßner M (2013). Off-label-Gebrauch: Instrumentalisierung für wirtschaftliche Zwecke. Dtsch Arztebl. 110 (31–32): A 1474–6.
127. Gauvreau GM, Boulet LP, Schmid-Wirlitsch C, Côté J, Duong M, Killian KJ, Milot J, Deschesnes F, Strinich T, Watson RM, Bredenbröker D, O'Byrne PM (2011). Roflumilast attenuates allergen-induced inflammation in mild asthmatic subjects. Respir Res. 12: 140.
128. G-BA (2008). Gemeinsamer Bundesausschuss – Verfahrensanordnung, in der Fassung vom 18. Dezember 2008, zuletzt geändert am 20. März 2014, in Kraft getreten am 8. Mai 2014. Richtlinien-Version ist nicht mehr in Kraft.
129. G-BA (2012a). Gemeinsamer Bundesausschuss – Beschlüsse über die Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen nach § 35a SGB V: Boceprevir. Stand der Information: 01.03.2012.
130. G-BA (2012b). Gemeinsamer Bundesausschuss – Tragende Gründe zum Beschluss des Gemeinsamen Bundesausschusses über die Änderung der Arzneimittel-Richtlinie (AM-RL): Anlage XII – Beschlüsse über die Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen nach § 35a SGB V: Boceprevir. Stand der Information: 01.03.2012.
131. G-BA (2012c). Gemeinsamer Bundesausschuss – Beschlüsse über die Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen nach § 35a SGB V: Telaprevir. Stand der Information: 29.03.2012.
132. G-BA (2013): Gemeinsamer Bundesausschuss – Beschlüsse über die Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen nach § 35a SGB V: Aclidiniumbromid. Stand der Information: 21.03.2013.
133. G-BA (2014). Gemeinsamer Bundesausschuss – Beschlüsse über die Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen nach § 35a SGB V: Simeprevir. Stand der Information: 20.11.2014.
134. G-BA (2014a). Gemeinsamer Bundesausschuss – Beschlüsse über die Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen nach § 35a SGB V: Sofosbuvir. Stand der Information: 17.07.2014.
204

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
135. G-BA (2014b). Gemeinsamer Bundesausschuss – Beschlüsse über die Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen nach § 35a SGB V: Indacaterol/Glycopyrronium. Stand der Information: 08.05.2014.
136. G-BA (2015). Gemeinsamer Bundesausschuss – Beschlüsse über die Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen nach § 35a SGB V: Daclatasvir. Stand der Information: 19.02.2015.
137. Geier A (2014). Update Hepatitis C: Was hat sich geändert? Was kommt demnächst? MMW - Fortschritte der Medizin. 19: 59-63.
138. Geldmacher H, Biller H, Herbst A, Urbanski K, Allison M, Buist AS, Hohlfeld JM, Welte T (2008). [The prevalence of chronic obstructive pulmonary disease (COPD) in Germany. Results of the BOLD study]. Dtsch Med Wochenschr. 133(50): 2609-14.
139. Genebat M, Vera F, Hernández-Quero J, Domingo P, Guardiola JM, Martínez-Madrid O, Martínez L, de la Llana FG, Sánchez-Villegas J, Alvarez H, Mariño A, Lluch JF, Martínez-Pérez MA, Marín J, Ruiz-Mateos E, Leal M (2014). Efficacy and tolerability after 24 weeks of treatment with telaprevir, pegylated interferon and ribavirin in cirrhotic HIV-HCV coinfected subjects. Antiviral Res. 104: 59-61.
140. Gerste B & Gutschmidt S (2006). Datenqualität von Diagnosen aus dem ambulanten Bereich. Gesundheits- und Sozialpolitik. (34): 29-43.
141. Gilead (2014). Fachinformation – Zusammenfassung der Merkmale des Arzneimittels Harvoni® 90 mg/ 400 mg Filmtabletten (Sofosbuvir/Ledipasvir). Stand der Information: November 2014.
142. Gilead (2015). Fachinformation – Zusammenfassung der Merkmale des Arzneimittels Sovaldi® 400 mg Filmtabletten (Sofosbuvir). Stand der Information: Januar 2015.
143. Gill CJ (2012). How often do US-based human subjects research studies register on time, and how often do they post their results? A statistical analysis of the Clinicaltrials.gov database. BMJ Open. 2 (4). pii: e001186. doi: 10.1136/bmjopen-2012-001186.
144. Gingter C, Wilm S, Abholz HH (2009). Is COPD a rare disease? Prevalence and identification rates in smokers aged 40 years and over within general practice in Germany. Fam Pract. 26(1): 3-9.
145. GKV-Spitzenverband (2012). Rahmenvertrag über die Arzneimittelversorgung nach § 129 Absatz 2 SGB V in der Fassung vom 15. Juni 2012 zwischen dem Spitzenverband Bund der Krankenkassen und dem Deutschen Apothekerverband e.V. http://www.gkv-spitzenverband.de/media/dokumente/krankenversicherung_1/arzneimittel/rahmenvertraege/apotheken/AM_20120615_S_RVtg_129_Abs2.pdf. Letzter Zugriff: 15.04.2015.
146. GKV-Spitzenverband (2015). Kennzahlen der gesetzlichen Krankenversicherung. Zuletzt aktualisiert: März 2015. http://www.gkv-spitzenverband.de/media/grafiken/gkv_kennzahlen/kennzahlen_gkv_2014_q4/GKV_Kennzahlen_Booklet_Q4-2014_300dpi_2015-03-26.pdf. Letzter Zugriff: 15.04.2015.
147. Glaeske G (2007). [The limits of economic efficiency--medication provision and "border violations" through structures and interests]. Z Arztl Fortbild Qualitatssich. 101 (5): 347-55.
205

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
148. Glaeske G, Höffken K, Ludwig WD, Schrappe M, Weißbach L, Wille E (2010). Sicherstellung einer effizienten Arzneimittelversorgung in der Onkologie. http://www.bmg.bund.de/fileadmin/redaktion/pdf_allgemein/ Gutachten_Sicherstellung_einer_effizienten_Arzneimittelversorgung_in_der_Onkologie.pdf. Letzter Zugriff: 09.01.2015.
149. Glaeske G (2011). Methodik zwischen Regeln und Willkür. Surrogatendpunkte – irgendwo zwischen unverzichtbar und indiskutabel. IQWiG-Herbst-Symposium 25.11. und 26.11.2011. https://www.iqwig.de/download/11-11-25_Glaeske_Surrogatendpunkte.pdf. Letzter Zugriff: 05.05.2015.
150. Glaeske G (2015). eMail-Auskunft zum Thema „Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung.“ vom 25.05.2015.
151. Göben J (2009). Der „Off-Label-Use” von Fertigarzneimitteln: Offene Fragen an der Schnittstelle von Standard, Humanität und Wirtschaftlichkeitsgebot, in: Ahrens HJ, von Bar Chr, Fischer G, Spickhoff A, Taupitz J (Hrsg.): Medizin und Haftung, Festschrift für Erwin Deutsch zum 80. Geburtstag, Berlin/Heidelberg, 2009.
152. GOLD (2015). Global Initiative For Chronic Obstructive Lung Disease. Pocket Guide To COPD Diagnosis, Management and Prevention. Updated 2015. http://www.goldcopd.org/uploads/users/files/GOLD_Pocket_2015_Feb18.pdf. Letzter Zugriff: 25.04.2015.
153. Grell L & Rieger M (2012). [Off-label therapy from the perspective of the medical insurance service]. Z Rheumatol. 71 (2): 101-4, 106-7.
154. Hafner K (2013). Off-Label-Use von Arzneimitteln in der Palliativmedizin. Saarbrücken: Südwestdeutscher Verlag für Hochschulschriften.
155. Hallberg J, Dominicus A, Eriksson UK, Gerhardsson de Verdier M, Pedersen NL, Dahlbäck M, Nihlén U, Higenbottam T, Svartengren M (2008). Interaction between smoking and genetic factors in the development of chronic bronchitis. Am J Respir Crit Care Med. 177(5): 486-90.
156. Hart B, Lundh A, Bero L (2012). Effect of reporting bias on meta-analyses of drug trials: reanalysis of meta-analyses. BMJ. 344: d7202. doi: 10.1136/bmj.d7202.
157. Hart D (1990). Arzneimitteltherapie und ärztliche Verantwortung. Thieme. Stuttgart.
158. Hart D (1994). Heilversuch, Entwicklung therapeutischer Strategien, klinische Prüfung und Humanexperiment – Grundsätze ihrer arzneimittel-, arzthaftungs- und berufsrechtlichen Beurteilung. MedR. 94-105.
159. Hayashi N, Okanoue T, Tsubouchi H, Toyota J, Chayama K, Kumada H (2012). Efficacy and safety of telaprevir, a new protease inhibitor, for difficult-to-treat patients with genotype 1 chronic hepatitis C. J Viral Hepat. 19 (2) :e134-42.
160. Hayashi N, Seto C, Kato M, Komada Y, Goto S (2014). Once-daily simeprevir (TMC435) with peginterferon/ribavirin for treatment-naïve hepatitis C genotype 1-infected patients in Japan: the DRAGON study. J Gastroenterol. 49 (1): 138-47.
206

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
161. Hellard ME, Doyle JS (2014). Interferon-free hepatitis C treatment: one pill to fit all? Lancet. 383 (9916): 491-2.
162. Hellmann A (2007). Fachärztliche ambulante und stationäre Versorgung von Asthma bronchiale. In: Lingner H, Schultz K, Schwartz F (Hrsg.). Volkskrankheit Asthma: Bestandsaufnahme und Perspektiven. 355-370. Heidelberg: Springer.
163. Hermann-Kunz E (1999) Häufigkeit allergischer Krankheiten in Ost- und Westdeutschland. Das Gesundheitswesen. 61 (Sonderheft 2): 100-105.
164. Hermann-Kunz E (2000) Allergische Krankheiten in Deutschland. Ergebnisse einer repräsentativen Studie. Bundesgesundheitsblatt - Gesundheitsforschung – Gesundheitsschutz. 43: 400-406.
165. Hézode C, Fontaine H, Dorival C, Zoulim F, Larrey D, Canva V, De Ledinghen V, Poynard T, Samuel D, Bourliere M, Alric L, Raabe JJ, Zarski JP, Marcellin P, Riachi G, Bernard PH, Loustaud-Ratti V, Chazouilleres O, Abergel A, Guyader D, Metivier S, Tran A, Di Martino V, Causse X, Dao T, Lucidarme D, Portal I, Cacoub P, Gournay J, Grando-Lemaire V, Hillon P, Attali P, Fontanges T, Rosa I, Petrov-Sanchez V, Barthe Y, Pawlotsky JM, Pol S, Carrat F, Bronowicki JP; CUPIC Study Group (2014a). Effectiveness of telaprevir or boceprevir in treatment-experienced patients with HCV genotype 1 infection and cirrhosis. Gastroenterology. 147 (1): 132-142.e4.
166. Hézode C, Forestier N, Dusheiko G, Ferenci P, Pol S, Goeser T, Bronowicki JP, Bourlière M, Gharakhanian S, Bengtsson L, McNair L, George S, Kieffer T, Kwong A, Kauffman RS, Alam J, Pawlotsky JM, Zeuzem S; PROVE2 Study Team (2009). Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N Engl J Med. 360 (18): 1839-50.
167. Hézode C, Hirschfield GM, Ghesquiere W, Sievert W, Rodriguez-Torres M, Shafran SD, Thuluvath PJ, Tatum HA, Waked I, Esmat G, Lawitz EJ, Rustgi VK, Pol S, Weis N, Pockros PJ, Bourlière M, Serfaty L, Vierling JM, Fried MW, Weiland O, Brunetto MR, Everson GT, Zeuzem S, Kwo PY, Sulkowski M, Bräu N, Hernandez D, McPhee F, Wind-Rotolo M, Liu Z, Noviello S, Hughes EA, Yin PD, Schnittman S (2014b).
-naive chronic hepatitis C genotype 1 or 4 infection: a randomised study. Gut. pii: gutjnl-2014-307498. doi: 10.1136/gutjnl-2014-307498.
168. Hill P (2005). Off licence and off label prescribing in children: litigation fears for physicians. Arch Dis Child. 90 Suppl 1:i17-8.
169. Hoffmann F (2008). (Arzneimittel)Routinedaten als Datenbasis für die Versorgungsforschung und Pharmakoepidemiologie._ Universität Bremen, Dissertation.
170. Hoffmann F, Meinecke P, Freitag MH, Glaeske G, Schulze J, Schmiemann G (2015). Who gets dipyrone (metamizole) in Germany? Prescribing by age, sex and region. J Clin Pharm Ther. 40 (3): 285-8.
171. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Paré PD (2004). The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 350 (26): 2645-53.
172. Huster S & Bohmeier A (2012). [The 'Myozyme' decision of the Federal Supreme Court of Switzerland and German Law: a constitutional rights and health insurance law perspective]. Z Evid Fortbild Qual Gesundhwes. 106 (6): 443-8.
173. ICH E9 (1998). International Conference on Harmonization – Statistical Principles for Clinical Trials. London UK. CPMP/ICH/363/96.
207

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
174. IQWiG (2012). Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen – IQWiG-Berichte Nr. 115. Telaprevir – Nutzenbewertung gemäß § 35a SGB V. Dossierbewertung Auftrag: A11-25, Version: 1.0, Stand der Information: 12.01.2012.
175. IQWiG (2013). Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen – Vorbericht (vorläufige Bewertung) V12-01: Leitlinienrecherche und –bewertung für das DMP COPD. Version 1.0. Stand der Information: 08.05.2013.
176. IQWiG (2013a). Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen – IQWiG-Berichte Nr. 196. Systematische Leitlinienrecherche und –bewertung sowie Extraktion relevanter Empfehlungen für das DMP Asthma bronchiale. Abschlussbericht. Auftrag: V12-03. Version: 1.0. Stand der Information: 26.11.2013.
177. IQWiG (2013b). Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen – IQWiG-Berichte Nr. 194. Systematische Leitlinienrecherche und –bewertung sowie Extraktion relevanter Empfehlungen für das DMP chronisch obstruktive Lungenerkrankung. Abschlussbericht. Auftrag: V12-01.Version:1.0 . Stand der Information: 05.11.2013.
178. IQWiG (2014). Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen – IQWiG-Berichte Nr. 239. Simeprevir – Nutzenbewertung gemäß § 35a SGB V. Dossierbewertung Auftrag: A14-18, Version: 1.0, Stand der Information: 28.08.2014.
179. IQWiG (2014a). Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen – IQWiG-Berichte Nr. 261. Daclatasvir – Nutzenbewertung gemäß § 35a SGB V. Dossierbewertung Auftrag: A14-31, Version: 1.0, Stand der Information: 27.11.2014.
180. IQWiG (2014b). Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen – IQWiG-Berichte Nr. 219. Sofosbuvir – Nutzenbewertung gemäß § 35a SGB V. Dossierbewertung Auftrag: A14-05, Version: 1.0, Stand der Information: 29.04.2014.
181. IQWiG (2015). Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen – IQWiG-Berichte Nr. 282. Ledipasvir/Sofosbuvir – Nutzenbewertung gemäß § 35a SGB V. Dossierbewertung Auftrag: A14-44, Version: 1.0, Stand der Information: 26.02.2015.
182. IQWiG (2015a). Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen – Allgemeine Methoden. Version 4.2. Stand der Information: 22.04.2015.
183. ISDB (2001): ISDB Declaration on therapeutic advance in the use of medicines. http://www.icf.uab.es/informacion/boletines/ISDB/ISDB.pdf. Letzter Zugriff: 01.04.2013.
184. Iwata R, Baur K, Stieger B, Mertens JC, Daly AK, Frei P, Braun J, Vergopoulos A, Stickel F, Sabrane K, Martin IV, Schmitt J, Goetze O, Day CP, Muellhaupt B, Geier A (2011). A common polymorphism in the ABCB11 gene is associated with advanced fibrosis in hepatitis C but not in non-alcoholic fatty liver disease. Clin Sci (Lond). 120 (7): 287-96.
185. Izumi N, Hayashi N, Kumada H, Okanoue T, Tsubouchi H, Yatsuhashi H, Kato M, Ki R, Komada Y, Seto C, Goto S (2014). Once-daily simeprevir with peginterferon and ribavirin for treatment-experienced HCV genotype 1-infected patients in Japan: the CONCERTO-2 and CONCERTO-3 studies. J Gastroenterol. 49 (5): 941-53.
208

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
186. Jacobson IM, Dore GJ, Foster GR, Fried MW, Radu M, Rafalsky VV, Moroz L, Craxi A, Peeters M, Lenz O, Ouwerkerk-Mahadevan S, De La Rosa G, Kalmeijer R, Scott J, Sinha R, Beumont-Mauviel M.
-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-1) (2014): A Phase III, randomised, double-blind, placebo-controlled trial. Lancet. 384: 403-13.
187. Jacobson IM, Gordon SC, Kowdley KV, Yoshida EM, Rodriguez-Torres M, Sulkowski MS, Shiffman ML, Lawitz E, Everson G, Bennett M, Schiff E, Al-Assi MT, Subramanian GM, An D, Lin M, McNally J, Brainard D, Symonds WT, McHutchison JG, Patel K, Feld J, Pianko S, Nelson DR; POSITRON Study; FUSION Study (2013). Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med. 368 (20): 1867-77.
188. Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S; ADVANCE Study Team (2011). Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 364 (25): 2405-16.
189. Janda C (2010). Medizinrecht. Konstanz: UVK Lucius, 1. Auflage.
190. Janssen (2013). Rote-Hand-Brief – Vorgehensweise bei Auftreten schwerer Hautreaktionen in Verbindung mit einer INCIVO®-Therapie. Stand der Information: 8. April 2013.
191. Janssen (2014). Fachinformation – Zusammenfassung der Merkmale des Arzneimittels Olysio® 150 mg Hartkapseln (Simeprevir). Stand der Information: Juni 2014.
192. Janzen RW & Ludwig WD (2012). Arzneimittelkommission der deutschen Ärzteschaft, Berlin. [Off-label therapy: current problems from the perspective of the Pharmaceutical Commission of the German Medical Profession]. Z Rheumatol. 71 (2): 108-10, 112-8.
193. Joerger M, Schaer-Thuer C, Koeberle D, Matter-Walstra K, Gibbons-Marsico J, Diem S, Thuerlimann B, Cerny T (2014). Off-label use of anticancer drugs in eastern Switzerland: a population-based prospective cohort study. Eur J Clin Pharmacol. 70 (6): 719-25.
194. Jones PW, Singh D, Bateman ED, Agusti A, Lamarca R, de Miquel G, Segarra R, Caracta C, Garcia Gil E (2012). Efficacy and safety of twice-daily aclidinium bromide in COPD patients: the ATTAIN study. Eur Respir J. 40 (4): 830-6.
195. Jonville-Béra AP, Béra F, Autret-Leca E (2005). Are incorrectly used drugs more frequently involved in adverse drug reactions? A prospective study. Eur J Clin Pharmacol. 61 (3): 231-6.
196. Kabisch M, Ruckes C, Seibert-Grafe M, Blettner M (2011). Randomized controlled trials: part 17 of a series on evaluation of scientific publications. Dtsch Arztebl Int. 108 (39): 663-8.
197. Kanniess F, Boulet LP, Pierzchala W, Cameron R, Owen R, Higgins M (2008). Efficacy and safety of indacaterol, a new 24-hour beta2-agonist, in patients with asthma: a dose-ranging study. J Asthma. 45 (10): 887-92.
198. Karabis A, Lindner L, Mocarski M, Huisman E, Greening A (2013). Comparative efficacy of aclidinium versus glycopyrronium and tiotropium, as maintenance treatment of moderate to severe COPD patients: a systematic review and network meta-analysis. Int J Chron Obstruct Pulmon Dis. 8: 405-23.
209

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
199. Kerwin EM, D'Urzo AD, Gelb AF, Lakkis H, Garcia Gil E, Caracta CF; ACCORD I study investigators (2012). Efficacy and safety of a 12-week treatment with twice-daily aclidinium bromide in COPD patients (ACCORD COPD I). COPD. 9 (2): 90-101.
200. Kesselheim AS, Mello MM, Studdert DM (2011). Strategies and practices in off-label marketing of pharmaceuticals: a retrospective analysis of whistleblower complaints. PLoS Med. 8(4):e1000431.
201. Kesselheim AS, Myers JA, Avorn J (2011a). Characteristics of clinical trials to support approval of orphan vs nonorphan drugs for cancer. JAMA. 305 (22): 2320-6.
202. Khdour MR, Hallak HO, Alayasa KS, AlShahed QN, Hawwa AF, McElnay JC (2011). Extent and nature of unlicensed and off-label medicine use in hospitalised children in Palestine. Int J Clin Pharm. 33(4): 650-5.
203. Kim V & Criner GJ (2013). Chronic bronchitis and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 187 (3): 228–237.
204. Kirchhof B, Lehmacher W, Thomas S (2013). Bevacizumab versus Ranibizumab: Ist off-label use geboten? Dtsch Arztebl. 110 (15): A 708-13.
205. Kirsch F, Teuner CM, Menn P, Leidl R (2013). [Costs of illness for asthma and COPD in adults in Germany]. Gesundheitswesen. 75(7): 413-23.
206. Knöppel C, Klinger O, Soergel M, Seyberth HW, Leonhardt A (2000). Anwendung von Medikamenten außerhalb der Zulassung oder ohne Zulassung bei Kindern. Monatsschrift Kinderheilkunde. 148, 904-8.
207. Kölch M, Allroggen M, Fegert JM (2009): Off-Label-Use von Psychopharmaka in der Kinder- und Jugendpsychiatrie. Ein andauerndes ethisches, medizinisches und rechtliches Problem. Der Nervenarzt. VOL: 80 (7); p. 789-96.
208. Konietzko N & Fabel H (2000). Weißbuch Lunge 2000. Georg Thieme Verlag, Stuttgart.
209. Konietzko N & Fabel H (2005). Weißbuch Lunge 2005. Georg Thieme Verlag, Stuttgart
210. Korn S, Kerwin E, Atis S, Amos C, Owen R, Lassen C (2011). Indacaterol once-daily provides superior efficacy to salmeterol twice-daily in COPD: a 12-week study. Respir Med. 105 (5): 719-26.
211. Kornmann O, Dahl R, Centanni S, Dogra A, Owen R, Lassen C, Kramer B; INLIGHT-2 (Indacaterol Efficacy Evaluation Using 150-μg Doses with COPD Patients) study investigators (2011). Once-daily indacaterol versus twice-daily salmeterol for COPD: a placebo-controlled comparison. Eur Respir J. 37 (2): 273-9.
212. Kowdley KV, Gordon SC, Reddy KR, Rossaro L, Bernstein DE, Lawitz E, Shiffman ML, Schiff E, Ghalib R, Ryan M, Rustgi V, Chojkier M, Herring R, Di Bisceglie AM, Pockros PJ, Subramanian GM, An D, Svarovskaia E, Hyland RH, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Pound D, Fried MW; ION-3 Investigators (2014a). Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med. 370 (20): 1879-88.
210

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
213. Kowdley KV, Lawitz E, Crespo I, Hassanein T, Davis MN, DeMicco M, Bernstein DE, Afdhal N, Vierling JM, Gordon SC, Anderson JK, Hyland RH, Dvory-Sobol H, An D, Hindes RG, Albanis E, Symonds WT, Berrey MM, Nelson DR, Jacobson IM (2013). Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet. 381 (9883): 2100-7.
214. Kowdley KV, Lawitz E, Poordad F, Cohen DE, Nelson DR, Zeuzem S, Everson GT, Kwo P, Foster GR, Sulkowski MS, Xie W, Pilot-Matias T, Liossis G, Larsen L, Khatri A, Podsadecki T, Bernstein B (2014b). Phase 2b trial of interferon-free therapy for hepatitis C virus genotype 1. N Engl J Med. 370 (3): 222-32.
215. Kuebler KK, Buchsel PC, Balkstra CR (2008). Differentiating chronic obstructive pulmonary disease from asthma. J Am Acad Nurse Pract. 20 (9): 445-54.
216. Kuehn BM (2009). FDA panel: ban 2 popular asthma drugs. JAMA. 301 (4): 365-6.
217. Kunz R, Ollenschläger G, Raspe H, Jonitz G, Donner-Banzhoff U (2007). Lehrbuch evidenzbasierte Medizin. Köln: Deutscher Ärzteverlag. 2. Auflage.
218. KVNO (2008). Kassenärztliche Vereinigung Nordrhein. ADHS-Behandlung im Erwachsenenalter mit Methylphenidat: Gericht verneint Off-Label-Use. https://www.kvno.de/10praxis/40verordnungen/20heilmittel/a_z/adhs/index.html. Stand der Information: Juni 2008. Letzter Zugriff: 03.03.2015.
219. KVNO (2008a). Kassenärztliche Vereinigung Nordrhein – Off-label-Einsatz von Avastin ist rechtens. Arzneimittelinfo aktuell 8/2008.
220. Kvitkina T, ten Haaf A, Reken S, McGauran N, Wieseler B (2014). [Patient-relevant outcomes and surrogates in the early benefit assessment of drugs: first experiences]. Z Evid Fortbild Qual Gesundhwes. 108 (8-9): 528-38.
221. Kwo PY, Lawitz EJ, McCone J, Schiff ER, Vierling JM, Pound D, Davis MN, Galati JS, Gordon SC, Ravendhran N, Rossaro L, Anderson FH, Jacobson IM, Rubin R, Koury K, Pedicone LD, Brass CA, Chaudhri E, Albrecht JK; SPRINT-1 investigators (2010). Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, multicentre phase 2 trial. Lancet. 376 (9742): 705-16.
222. Kwo PY, Mantry PS, Coakley E, Te HS, Vargas HE, Brown R Jr, Gordon F, Levitsky J, Terrault NA, Burton JR Jr, Xie W, Setze C, Badri P, Pilot-Matias T, Vilchez RA, Forns X (2014). An interferon-free antiviral regimen for HCV after liver transplantation. N Engl J Med. 371(25): 2375-82.
223. Langen U, Schmitz R, Steppuhn H (2013) Häufigkeit allergischer Erkrankungen in Deutschland. Ergebnisse der Studie zur Gesundheit Erwachsener in Deutschland (DEGS1). Bundesgesundheitsbl–Gesundheitsforsch–Gesundheitsschutz. 56 (5/6): 698–706.
224. Lau S, Illi S, Sommerfeld C, Niggemann B, Bergmann R, von ME, Wahn U (2000). Early exposure to house-dust mite and cat allergens and development of childhood asthma: a cohort study. Multicentre Allergy Study Group. Lancet. 356 (9239): 1392-7.
225. Laufs A & Uhlenbrock W (2001). Handbuch des Arztrechts – Zivilrecht, Öffentliches Recht, Vertragsarztrecht, Krankenhausrecht, Strafrecht. München: C. H. Beck Verlag.
211

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
226. Lawitz E, Lalezari JP, Hassanein T, Kowdley KV, Poordad FF, Sheikh AM, Afdhal NH, Bernstein DE, Dejesus E, Freilich B, Nelson DR, Dieterich DT, Jacobson IM, Jensen D, Abrams GA, Darling JM, Rodriguez-Torres M, Reddy KR, Sulkowski MS, Bzowej NH, Hyland RH, Mo H, Lin M, Mader M, Hindes R, Albanis E, Symonds WT, Berrey MM, Muir A (2013b). Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis. 13 (5): 401-8.
227. Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC, Schultz M, Davis MN, Kayali Z, Reddy KR, Jacobson IM, Kowdley KV, Nyberg L, Subramanian GM, Hyland RH, Arterburn S, Jiang D, McNally J, Brainard D, Symonds WT, McHutchison JG, Sheikh AM, Younossi Z, Gane EJ (2013a). Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 368 (20): 1878-87.
228. Lawitz E, Poordad FF, Pang PS, Hyland RH, Ding X, Mo H, Symonds WT, McHutchison JG, Membreno FE. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR) (2014b): an open-label, randomised, phase 2 trial. Lancet. 383 (9916): 515-23.
229. Lawitz E, Sulkowski MS, Ghalib R, Rodriguez-Torres M, Younossi ZM, Corregidor A, DeJesus E, Pearlman B, Rabinovitz M, Gitlin N, Lim JK, Pockros PJ, Scott JD, Fevery B, Lambrecht T, Ouwerkerk-Mahadevan S, Callewaert K, Symonds WT, Picchio G, Lindsay KL, Beumont M, Jacobson IM (2014a). Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naïve patients: the COSMOS randomised study. Lancet. 384 (9956): 1756-65.
230. LG Ellwangen (2007). Landgericht Ellwangen. Urteil vom 18.07.2007, Az.: 2KLs 11 Js 21209/02.
231. Lipworth BJ (2005). Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet. 365 (9454): 167-75.
232. Lorenz J, Bals R, Magnussen H, Pfeifer M, Randerath W, Steinkamp G, Taube C, Teschler H, Vogelmeier C, Welte T, Worth H (2013). [Expert meeting chronic obstructive airway disease--cardiovascular aspects of COPD]. Pneumologie. 67 (12): 663-75.
233. Louw C, Williams Z, Venter L, Leichtl S, Schmid-Wirlitsch C, Bredenbroker D, Bardin PG (2007). Roflumilast, a phosphodiesterase 4 inhibitor, reduces airway hyperresponsiveness after allergen challenge. Respiration. 74 (4): 411-7.
234. Lynch SS & Cheng CM (2007). Bevacizumab for neovascular ocular diseases. Ann Pharmacother. 41 (4): 614-25.
235. Maasoumy B, Port K, Calle Serrano B, Markova AA, Sollik L, Manns MP, Cornberg M, Wedemeyer H (2013a). The clinical significance of drug-drug interactions in the era of direct-acting anti-viral agents against chronic hepatitis C. Aliment Pharmacol Ther. 38 (11-12): 1365-72.
236. Maasoumy B, Port K, Markova AA, Serrano BC, Rogalska-Taranta M, Sollik L, Mix C, Kirschner J, Manns MP, Wedemeyer H, Cornberg M (2013b). Eligibility and safety of triple therapy for hepatitis C: lessons learned from the first experience in a real world setting. PLoS One. 8(2): e55285.
237. Mahler DA, D'Urzo A, Bateman ED, Ozkan SA, White T, Peckitt C, Lassen C, Kramer B (2012). Concurrent use of indacaterol plus tiotropium in patients with COPD provides superior bronchodilation compared with tiotropium alone: a randomised, double-blind comparison. Thorax 9. (67): 781-8.
212

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
238. Manns M, Marcellin P, Poordad F, de Araujo ES, Buti M, Horsmans Y, Janczewska E, Villamil F, Scott J, Peeters M, Lenz O, Ouwerkerk-Mahadevan S, De La Rosa G, Kalmeijer R, Sinha R, Beumont-Mauviel M
-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-2): a randomised, double-blind, placebo-controlled Phase III trial. Lancet. 384: 414-26.
239. Manns M, Reesink H, Berg T, Dusheiko G, Flisiak R, Marcellin P, Moreno C, Lenz O, Meyvisch P, Peeters M, Sekar V, Simmen K, Verloes R (2011). Rapid viral response of once-daily TMC435 plus pegylated interferon/ribavirin in hepatitis C genotype-1 patients: a randomized trial. Antivir Ther.; 16 (7): 1021-33.
240. Manns MP, McCone J Jr, Davis MN, Rossaro L, Schiff E, Shiffman ML, Bacon B, Bourliere M, Sulkowski MS, Bruno S, Balart L, Bronowicki JP, Kwo P, Poordad F, Felizarta F, Reddy KR, Helmond FA, Sings HL, Pedicone LD, Burroughs M, Brass CA, Albrecht JK, Vierling JM (2014b). Overall safety profile of boceprevir plus peginterferon alfa-2b and ribavirin in patients with chronic hepatitis C genotype 1: a combined analysis of 3 phase 2/3 clinical trials. Liver Int. 34 (5): 707-19.
241. Marcellin P, Forns X, Goeser T, Ferenci P, Nevens F, Carosi G, Drenth JP, Serfaty L, De Backer K, Van Heeswijk R, Luo D, Picchio G, Beumont M (2011). Telaprevir is effective given every 8 or 12 hours with ribavirin and peginterferon alfa-2a or -2b to patients with chronic hepatitis C. Gastroenterology. 140 (2): 459-468.e1.
242. Marklund B, Tunsater A, Bengtsson C (1999). How often is the diagnosis bronchial asthma correct? FamPract. 16 (2): 112-6.
243. McHutchison JG, Everson GT, Gordon SC, Jacobson IM, Sulkowski M, Kauffman R, McNair L, Alam J, Muir AJ; PROVE1 Study Team (2009). Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med. 360 (18): 1827-38.
244. McHutchison JG, Manns MP, Muir AJ, Terrault NA, Jacobson IM, Afdhal NH, Heathcote EJ, Zeuzem S, Reesink HW, Garg J, Bsharat M, George S, Kauffman RS, Adda N, Di Bisceglie AM; PROVE3 Study Team (2010). Telaprevir for previously treated chronic HCV infection. N Engl J Med. 362(14): 1292-303.
245. McPhee F, Hernandez D, Zhou N, Yu F, Ueland J, Monikowski A, Chayama K, Toyota J, Izumi N, Yokosuka O, Kawada N, Osaki Y, Hughes EA, Watanabe H, Ishikawa H, Kumada H (2014). Virological escape in HCV genotype-1-infected patients receiving daclatasvir + ribavirin and peginterfer -2a or
19: 479-490.
246. MDS (2014). Medizinischer Dienst des Spitzenverbandes – Jahresstatistik 2013 zur Behandlungsfehler-Begutachtung der MDK Gemeinschaft.
247. Meier CR & Jick H (1997). Drug use and pulmonary death rates in increasingly symptomatic asthma patients in the UK. Thorax. 52: 612–617.
248. Mellor JD, Van Koeverden P, Yip SW, Thakerar A, Kirsa SW, Michael M (2012). Access to anticancer drugs: many evidence-based treatments are off-label and unfunded by the Pharmaceutical Benefits Scheme. Intern Med J. 42(11): 1224-9.
249. Meteran H, Thomsen SF, Harmsen L, Kyvik KO, Skytthe A, Backer V (2012). Risk of chronic bronchitis in twin pairs discordant for smoking. Lung. 190 (5): 557-61.
213

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
250. Meyer F & Grunert G (2005). „Off-Label-Use“: Haftungs- und Regressrisiken für Ärzte, Apotheker und Pharmaunternehmen. PharmR. 205ff.
251. Molina JM, Orkin C, Iser DM, Zamora FX, Nelson M, Stephan C, Massetto B, Gaggar A, Ni L, Svarovskaia E, Brainard D, Subramanian GM, McHutchison JG, Puoti M, Rockstroh JK; PHOTON-2 study team (2015). Sofosbuvir plus ribavirin for treatment of hepatitis C virus in patients co-infected with HIV (PHOTON-2): a multicentre, open-label, non-randomised, phase 3 study. Lancet. 385 (9973): 1098-106.
252. Montnémery P, Hansson L, Lanke J, Lindholm LH, Nyberg P, Löfdahl CG, Adelroth E (2002). Accuracy of a first diagnosis of asthma in primary health care. Fam Pract. 19 (4): 365-8.
253. Moreno C, Berg T, Tanwandee T, Thongsawat S, Van Vlierberghe H, Zeuzem S, Lenz O, Peeters M, Sekar V, De Smedt G (2012). Antiviral activity of TMC435 monotherapy in patients infected with HCV genotypes 2-6: TMC435-C202, a phase IIa, open-label study. J Hepatol. 56(6): 1247-53.
254. Morgan RL, Baack B, Smith BD, Yartel A, Pitasi M, Falck-Ytter Y (2013). Eradication of hepatitis C virus infection and the development of hepatocellular carcinoma: a meta-analysis of observational studies. Ann Intern Med. 158 (5 Pt 1): 329-37.
255. MSD (2012): Rote-Hand-Brief –Information für Angehörige der medizinischen Fachkreise über Arzneimittelinteraktionen zwischen Victrelis® (Boceprevir) und Ritonavir-geboosterten HIV-Proteaseinhibitoren. Stand der Information: 23.02.2012.
256. MSD (2014). Fachinformation – Zusammenfassung der Merkmale des Arzneimittels Incivo® 375 mg Filmtabletten (Telaprevir). Stand der Information: Juli 2014.
257. MSD (2015a). Fachinformation – Zusammenfassung der Merkmale des Arzneimittels Victrelis® 200 mg Hartkapseln (Boceprevir). Stand der Information: Februar 2015.
258. Mühlbauer B, Janhsen K, Pichler J, Schoettler P (2009). Off-label use of prescription drugs in childhood and adolescence: an analysis of prescription patterns in Germany. Dtsch Arztebl Int. 106 (3): 25-31.
259. Muir AJ, Poordad FF, McHutchison JG, Shiffman ML, Berg T, Ferenci P, Heathcote EJ, Pawlotsky JM, Zeuzem S, Reesink HW, Dusheiko G, Martin EC, George S, Kauffman RS, Adda N (2011). Retreatment with telaprevir combination therapy in hepatitis C patients with well-characterized prior treatment response. Hepatology. 54 (5): 1538-46.
260. Müller H (2009). Die Rechtsproblematik des Off-Label-Use: Das Spannungsfeld zwischen Haftungs-, Versicherungs- und Werberecht. Berlin: LIT; 1. Auflage.
261. Murakami E, Imamura M, Hayes CN, Abe H, Hiraga N, Honda Y, Ono A, Kosaka K, Kawaoka T, Tsuge M, Aikata H, Takahashi S, Miki D, Ochi H, Matsui H, Kanai A, Inaba T, McPhee F, Chayama K (2014). Ultradeep sequencing study of chronic hepatitis C virus genotype 1 infection in patients treated with daclatasvir, peginterferon, and ribavirin. Antimicrob Agents Chemother. 58: 2105-2112.
262. Mutschler E, Geisslinger G, Kroemer HK, Schäfer-Korting M (2008): Mutschler Arzneimittelwirkungen – Lehrbuch der Pharmakologie und Toxikologie. Stuttgart: Wissenschaftliche Verlagsgesellschaft Stuttgart. 9. Auflage.
263. Neumann-Haefelin C, Blum HE, Thimme R (2012). Direct antiviral treatment strategies in chronic hepatitis C. Dtsch Med Wochenschr. 137 (25-26): 1360-5.
214

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
264. Novartis Pharma (2014). Fachinformation – Zusammenfassung der Merkmale des Arzneimittels Onbrez® Breezhaler®. Stand der Information: Dezember 2014.
265. Novartis Pharma (2014). Fachinformation – Zusammenfassung der Merkmale des Arzneimittels Ultibro® Breezhaler®. Stand der Information: Januar 2015.
266. Nölling T (2014). Es bleibt dabei: Leitlinien sind nicht rechtlich verbindlich. GMS Mitt AWMF 2014;11:Doc6. http://dx.doi.org/10.3205/awmf000295. Letzter Zugriff: 10.05.2015.
267. NVL (2006). Nationale VersorgungsLeitlinie COPD. Langfassung Version 1.9. Januar 2012, basierend auf der Fassung vom Februar 2006. Derzeit in Überarbeitung.
268. NVL (2009). Nationale VersorgungsLeitlinie Asthma. Langfassung. Version 5, 2. Auflage, August 2013, basierend auf der Fassung vom Dezember 2009. Derzeit in Überarbeitung.
269. Oba Y & Lone NA (2013). Efficacy and safety of roflumilast in patients with chronic obstructive pulmonary disease: a systematic review and meta-analysis. Ther Adv Respir Dis. 7 (1): 13-24.
270. OLG Köln (1990). Oberlandesgericht Köln – Urteil vom 20.05.1990. Az.: 27 U 169/87.
271. Ollenschläger G, Oesingmann U, Kolkmann FW (2000). Eignen sich Leitlinien als Steuerungsinstrumente im Gesundheitswesen? Forum für Gesundheitspolitik. 255-257. Zusammenfassung.
272. Ortho Biotech (2002). Wichtige Information zur Arzneimittelsicherheit ERYPO®/EPREX® (Epoetin alfa). Art der Anwendung – nur intravenöse Applikation bei Patienten mit Nierenversagen & Erinnerungshinweis betreffend Lagerung bei 2-8°C aufbewahren. Stand der Information: 11.12.2002.
273. Osinusi A, Kohli A, Marti MM, Nelson A, Zhang X, Meissner EG, Silk R, Townsend K, Pang PS, Subramanian GM, McHutchison JG, Fauci AS, Masur H, Kottilil S (2014). Re-treatment of chronic hepatitis C virus genotype 1 infection after relapse: an open-label pilot study. Ann Intern Med. 161 (9): 634-8.
274. Osinusi A, Meissner EG, Lee YJ, Bon D, Heytens L, Nelson A, Sneller M, Kohli A, Barrett L, Proschan M, Herrmann E, Shivakumar B, Gu W, Kwan R, Teferi G, Talwani R, Silk R, Kotb C, Wroblewski S, Fishbein D, Dewar R, Highbarger H, Zhang X, Kleiner D, Wood BJ, Chavez J, Symonds WT, Subramanian M, McHutchison J, Polis MA, Fauci AS, Masur H, Kottilil S (2013). Sofosbuvir and ribavirin for hepatitis C genotype 1 in patients with unfavorable treatment characteristics: a randomized clinical trial. JAMA. 310 (8): 804-11.
275. Osinusi A, Townsend K, Kohli A, Nelson A, Seamon C, Meissner EG, Bon D, Silk R, Gross C, Price A, Sajadi M, Sidharthan S, Sims Z, Herrmann E, Hogan J, Teferi G, Talwani R, Proschan M, Jenkins V, Kleiner DE, Wood BJ, Subramanian GM, Pang PS, McHutchison JG, Polis MA, Fauci AS, Masur H, Kottilil S (2015). Virologic response following combined ledipasvir and sofosbuvir administration in patients with HCV genotype 1 and HIV co-infection. JAMA. 313 (12): 1232-9.
276. Owens J (2007). 2006 drug approvals: finding the niche. Nat Rev Drug Discov. 6 (2): 99-101.
277. Page CP (2014). Phosphodiesterase inhibitors for the treatment of asthma and chronic obstructive pulmonary disease. Int Arch Allergy Immunol. 165 (3): 152-64.
215

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
278. Pan L, Guo YZ, Zhang B, Yan JH (2013). Does roflumilast improve dyspnea in patients with chronic obstructive pulmonary disease? A meta-analysis. J Thorac Dis. 5 (4): 422-9.
279. Pearce N, Douwes J, Beasley R (2000). Is allergen exposure the major primary cause of asthma? Thorax. 55 (5): 424-31.
280. Pearlman DS, Greos L, LaForce C, Orevillo CJ, Owen R, Higgins M (2008). Bronchodilator efficacy of indacaterol, a novel once-daily beta2-agonist, in patients with persistent asthma. Ann Allergy Asthma Immunol. 101 (1): 90-5.
281. PEI (1998). Paul-Ehrlich-Institut – Pressemitteilung: Wichtiger Schritt für noch mehr Sicherheit im Blutspendewesen. http://www.pei.de/DE/infos/presse/pressemitteilungen/archiv-pressemitteilungen/1998/05-wichtiger-schritt-fuer-noch-mehr-sicherheit-im-blutspendewesen.html. Juli 1998. Letzter Zugriff: 09.04.015.
282. Penn JS, Madan A, Caldwell RB, Bartoli M, Caldwell RW, Hartnett ME (2008). Vascular endothelial growth factor in eye disease. Prog Retin Eye Res. 27 (4): 331-71.
283. Plate V, Nies P, Behles C, Schweim H (2008): Wohin treibt der Off-Label-Use?, A&R 6. 261-68.
284. Poethko-Müller C, Zimmermann R, Hamouda O, Faber M, Stark K, Ross RS, Thamm M (2013). Die Seroepidemiologie der Hepatitis A, B und C in Deutschland – Ergebnisse der Studie zur Gesundheit Erwachsener in Deutschland (DEGS1). Bundesgesundheitsblatt. 56 (5/6): 707-715.
285. Pol S, Ghalib RH, Rustgi VK, Martorell C, Everson GT, Tatum HA, Hézode C, Lim JK, Bronowicki JP, Abrams GA, Bräu N, Morris DW, Thuluvath PJ, Reindollar RW, Yin PD, Diva U, Hindes R, McPhee F, Hernandez D, Wind-Rotolo M, Hughes EA, Schnittman S (2012). Daclatasvir for previously untreated chronic hepatitis C genotype-1 infection: a randomised, parallel-group, double-blind, placebo-controlled, dose-finding, phase 2a trial. Lancet Infect Dis. 12 (9): 671-7.
286. Polosa R, Knoke JD, Russo C, Piccillo G, Caponnetto P, Sarva M, Proietti L, Al Delaimy WK (2008). Cigarette smoking is associated with a greater risk of incident asthma in allergic rhinitis. J Allergy Clin Immunol. 121 (6): 1428-34.
287. Poole SG & Dooley MJ (2004). Off-label prescribing in oncology. Support Care Cancer. 12 (5): 302-5.
288. Poordad F, Hezode C, Trinh R, Kowdley KV, Zeuzem S, Agarwal K, Shiffman ML, Wedemeyer H, Berg T, Yoshida EM, Forns X, Lovell SS, Da Silva-Tillmann B, Collins CA, Campbell AL, Podsadecki T, Bernstein B (2014). ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med. 370 (21): 1973-82.
289. Poordad F, McCone J Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP; SPRINT-2 Investigators (2011): Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 364 (13): 1195-206.
290. Postma DS, Bush A, van den Berge M (2015). Risk factors and early origins of chronic obstructive pulmonary disease. Lancet. 385 (9971): 899-909.
291. Prayle AP, Hurley MN, Smyth AR (2012). Compliance with mandatory reporting of clinical trial results on ClinicalTrials.gov: cross sectional study. BMJ. 344: d7373. doi: 10.1136/bmj.d7373.
216

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
292. Pritzkuleit R, Beske F, Katalinic A (2010). [Disease numbers in pneumology - a projection to 2060]. Pneumologie. 64 (9): 535-40.
293. Rabe KF, Vermeire PA, Soriano JB, Maier WC (2000). Clinical management of asthma in 1999: the Asthma Insights and Reality in Europe (AIRE) study. Eur Respir J. 16 (5): 802-7.
294. Ram FS & Sestini P (2003). Regular inhaled short acting beta2 agonists for the management of stable chronic obstructive pulmonary disease: cochrane systematic review and meta-analysis. Thorax. 58: 580-4.
295. Räpple T (1991). Das Verbot bedenklicher Arzneimittel: Eine Kommentierung zu § 5 AMG, Dissertation Baden-Baden.
296. Rasch A, Dintsios CM (2015). [Subgroups in the early benefit assessment of pharmaceuticals: a methodical review]. Z Evid Fortbild Qual Gesundhwes. 109 (1): 69-78.
297. Ray SM, McMillen JC, Treadway SA, Helmer RS, Franks AS (2012). Indacaterol: a novel long-acting (2)-agonist. Pharmacotherapy. 32 (5): 456-74.
298. Reddy KR, Zeuzem S, Zoulim F, Weiland O, Horban A, Stanciu C, Villamil FG, Andreone P, George J, Dammers E, Fu M, Kurland D, Lenz O, Ouwerkerk-Mahadevan S, Verbinnen T, Scott J, Jessner W (2015). Simeprevir versus telaprevir with peginterferon and ribavirin in previous null or partial responders with chronic hepatitis C virus genotype 1 infection (ATTAIN): a randomised, double-blind, non-inferiority phase 3 trial. Lancet Infect Dis. 15 (1): 27-35.
299. Reesink HW, Fanning GC, Farha KA, Weegink C, Van Vliet A, Van 't Klooster G, Lenz O, Aharchi F, Mariën K, Van Remoortere P, de Kock H, Broeckaert F, Meyvisch P, Van Beirendonck E, Simmen K, Verloes R (2010). Rapid HCV-RNA decline with once daily TMC435: a phase I study in healthy volunteers and hepatitis C patients. Gastroenterology. 138 (3): 913-21.
300. Restrepo RD (2007). Use of inhaled anticholinergic agents in obstructive airway disease. Respir Care. 52 (7): 833-51.
301. RKI (2005). Robert Koch-Institut – Gesundheitsberichterstattung des Bundes – Armut, soziale Ungleichheit und Gesundheit. Expertise des Robert Koch-Instituts zum 2. Armuts- und Reichtumsbericht der Bundesregierung.
302. RKI (2014a). Robert Koch-Institut: Virus-Hepatitis C: Situationsbericht Deutschland 2013. http://www.rki.de/DE/Content/Infekt/EpidBull/Archiv/2014/31/Art_01.html. Letzter Zugriff: 14.01.2015.
303. RKI (2014b). Robert Koch-Institut. Chronische Lungenerkrankungen. http://www.rki.de/DE/Content/Gesundheitsmonitoring/Themen/Chronische_Erkrankungen/lungenerkrankungen/lungenerkrankungen_tab.html;jsessionid=0F3A062733A30AB39DC93A14CF095465.2_cid363. Letzter Zugriff: 29.12.2014.
304. RKI (2014c). Robert Koch-Institut - Chronische Bronchitis. Faktenblatt zu GEDA 2012: Ergebnisse der Studie »Gesundheit in Deutschland aktuell 2012«. RKI, Berlin.
305. RKI (2014d). Robert Koch-Institut – Asthma bronchiale. Faktenblatt zu GEDA 2012: Ergebnisse der Studie „Gesundheit in Deutschland aktuell“. RKI, Berlin.
217

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
306. Rodrigo GJ & Neffen H (2012). Comparison of indacaterol with tiotropium or twice-daily long-acting beta -agonists for stable COPD: a systematic review. Chest. 142 (5): 1104-10.
307. Röhrig B, du Prel JB, Blettner M (2009). Study design in medical research: part 2 of a series on the evaluation of scientific publications. Dtsch Arztebl Int. 106 (11): 184-9.
308. Rosenfeld PJ (2011). Bevacizumab versus ranibizumab for AMD. N Engl J Med. 364 (20): 1966-7.
309. Rückeshäuser P (2011). Off-Label-Use: Die rechtlichen Probleme des zulassungsüberschreitenden Einsatzes von Arzneimitteln. Dr. Kovac, 1. Auflage.
310. Rzehak P, Schoefer Y, Wichmann HE, Heinrich J (2008). A prospective study on the association between hay fever among children and incidence of asthma in East Germany. Eur J Epidemiol. 23 (1): 17-22.
311. Sackett DL, Rosenberg WM, Gray JA, Haynes RB, Richardson WS (1996). Evidence based medicine: what it is and what it isn't. BMJ. 312 (7023): 71-2.
312. Salo PM, Arbes SJ, Jr., Crockett PW, Thorne PS, Cohn RD, Zeldin DC (2008). Exposure to multiple indoor allergens in US homes and its relationship to asthma. J Allergy Clin Immunol. 121 (3): 678-84.
313. Schäfer T, Heinrich J, Böhler E, Klemm E, Merkl J, Ruhdorfer S, Weigl L, Wessner D, Wichmann HE, Ring J; MONICA/KORA-Studiengruppe (2005). [Allergies in adults]. Gesundheitswesen. 67 Suppl 1: S187-92.
314. Schiedermair R & Pohl HU (2012). Gesetzeskunde für Apotheker. Eschborn: Govi, 17. Auflage.
315. Schimmelpfennig-Schütte R (2004) Recht auf Behandlung und Off-Label-Use in der Gesetzlichen Krankenversicherung (GKV). MedR. 22: 655-659.
316. Schmucker R (2005). Die deutsche Arzneimittelzulassung im europäischen Wettbewerb. Diskussionspapier 2005-1. Institut für Medizinische Soziologie – Fachbereich Humanmedizin der Johann Wolfgang Goethe-Universität. Frankfurt am Main.
317. Schneider A, Gantner L, Maag I, Borst MM, Wensing M, Szecsenyi J (2005). Are ICD-10 codes appropriate for performance assessment in asthma and COPD in general practice? Results of a cross sectional observational study. BMC Health Serv Res. 5 (1): 11.
318. Scholz H, Kuschinsky G, Böger RH (2005). Taschenbuch der Arzneibehandlung: Angewandte Pharmakologie. Berlin: Springer.
319. Schott G, Pachl H, Ludwig WD (2010). Publikationsbias in Abhängigkeit von der Art der Finanzierung bei klinischen Studien. Z Evid Fortbild Qual Gesundhwesen 104:314-322.
320. Schwabe U & Paffrath D (2011): Arzneiverordnungs-Report 2011. Berlin, Heidelberg: Springer-Verlag.
321. SG Düsseldorf (2008). Sozialgericht Düsseldorf – Urteil vom 02.07.2008. Az.: S2 KA 181/07. https://sozialgerichtsbarkeit.de/sgb/esgb/show.php?modul=esgb&id=80635. Letzter Zugriff: 01.04.2015.
322. Shaaban R, Zureik M, Soussan D, Neukirch C, Heinrich J, Sunyer J, Wjst M, Cerveri I, Pin I, Bousquet J, Jarvis D, Burney PG, Neukirch F, Leynaert B (2008). Rhinitis and onset of asthma: a longitudinal population based study. Lancet. 372 (9643): 1049-57.
218

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
323. Sherman KE, Flamm SL, Afdhal NH, Nelson DR, Sulkowski MS, Everson GT, Fried MW, Adler M, Reesink HW, Martin M, Sankoh AJ, Adda N, Kauffman RS, George S, Wright CI, Poordad F; ILLUMINATE Study Team (2011). Response-guided Telaprevir combination treatment for hepatitis C virus infection. N Engl J Med. 365 (11): 1014-24.
324. Sickmüller B & Lietz Chr (2011). Der Off-Label-Use aus Sicht der Pharmaindustrie aus Becker U, Wilman N (2011): Im Zweifel auf Privatrezept? Sozial- und haftungsrechtliche Aspekte des Off-Label-Use. Normos, 1. Auflage.
325. Simmonds P, Bukh J, Combet C, Deléage G, Enomoto N, Feinstone S, Halfon P, Inchauspé G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon E, Shin-I T, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ, Widell A (2005). Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 42 (4): 962-73.
326. Spring J, Clague J, Ind PW (1992). A comparison of the effect of salmeterol and salbutamol in normal subjects. Br J Clin Pharmacol. 33 (2): 139-41.
327. Stahmeyer JT, Rossol S, Bert F, Abdelfattah M, Wedemeyer H, Krauth C (2013). Die Kosten der Versorgung von Hepatitis C Patienten in Deutschland. Gesundheitswesen. 75: A274.
328. Stapff M (2008). Arzneimittelstudien - Eine Einführung in klinische Prüfungen für Ärzte, Studenten, medizinisches Assistenzpersonal und interessierte Laien, Kpt. A Klinische Studien in der Arzneimittelforschung und Kpt. B Einbindung in Rechtsnormen, München: Zuckschwerdt, 5. Auflage.
329. Steinbrook R (2002). Testing medications in children. N Engl J Med. 347: 1462 – 1470.
330. Steinman MA, Bero LA, Chren MM, Landefeld CS (2006). Narrative review: the promotion of gabapentin: an analysis of internal industry documents. Ann Intern Med. 145(4): 284-93.
331. Stock S, Redaelli M, Luengen M, Wendland G, Civello D, Lauterbach KW (2005). Asthma: prevalence and cost of illness. Eur Respir J. 25(1): 47-53.
332. Sturton RG, Trifilieff A, Nicholson AG, Barnes PJ (2008): Pharmacological characterization of indacaterol, a novel once daily inhaled 2 adrenoceptor agonist, on small airways in human and rat precision-cut lung slices. J Pharmacol Exp Ther. 324 (1): 270-5.
333. Sugihara N, Kanada S, Haida M, Ichinose M, Adachi M, Hosoe M, Emery C, Higgins M, Kramer B (2010). 24-h bronchodilator efficacy of single doses of indacaterol in Japanese patients with asthma: a comparison with placebo and salmeterol. Respir Med. 104 (11): 1629-37.
334. Sugihara N, Kanada S, Haida M, Ichinose M, Adachi M, Hosoe M, Emery C, Higgins M, Kramer B (2010). 24-h bronchodilator efficacy of single doses of indacaterol in Japanese patients with asthma: a comparison with placebo and salmeterol. Respir Med. 104 (11): 1629-37.
335. Sulkowski M, Pol S, Mallolas J, Fainboim H, Cooper C, Slim J, Rivero A, Mak C, Thompson S, Howe AY, Wenning L, Sklar P, Wahl J, Greaves W; P05411 study investigators (2013a). Boceprevir versus placebo with pegylated interferon alfa-2b and ribavirin for treatment of hepatitis C virus genotype 1 in patients with HIV: a randomised, double-blind, controlled phase 2 trial. Lancet Infect Dis. 13 (7): 597-605.
219

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
336. Sulkowski MS, Eron JJ, Wyles D, Trinh R, Lalezari J, Wang C, Slim J, Bhatti L, Gathe J, Ruane PJ, Elion R, Bredeek F, Brennan R, Blick G, Khatri A, Gibbons K, Hu YB, Fredrick L, Schnell G, Pilot-Matias T, Tripathi R, Da Silva-Tillmann B, McGovern B, Campbell AL, Podsadecki T (2015). Ombitasvir, paritaprevir co-dosed with ritonavir, dasabuvir, and ribavirin for hepatitis C in patients co-infected with HIV-1: a randomized trial. JAMA. 313 (12): 1223-31.
337. Sulkowski MS, Gardiner DF, Rodriguez-Torres M, Reddy KR, Hassanein T, Jacobson I, Lawitz E, Lok AS, Hinestrosa F, Thuluvath PJ, Schwartz H, Nelson DR, Everson GT, Eley T, Wind-Rotolo M, Huang SP, Gao M, Hernandez D, McPhee F, Sherman D, Hindes R, Symonds W, Pasquinelli C, Grasela DM; AI444040 Study Group (2014b). Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med. 370 (3): 211-21.
338. Sulkowski MS, Naggie S, Lalezari J, Fessel WJ, Mounzer K, Shuhart M, Luetkemeyer AF, Asmuth D, Gaggar A, Ni L, Svarovskaia E, Brainard DM, Symonds WT, Subramanian GM, McHutchison JG, Rodriguez-Torres M, Dieterich D; PHOTON-1 Investigators (2014a). Sofosbuvir and ribavirin for hepatitis C in patients with HIV coinfection. JAMA. 312(4): 353-61.
339. Sulkowski MS, Sherman KE, Dieterich DT, Bsharat M, Mahnke L, Rockstroh JK, Gharakhanian S, McCallister S, Henshaw J, Girard PM, Adiwijaya B, Garg V, Rubin RA, Adda N, Soriano V (2013b). Combination therapy with telaprevir for chronic hepatitis C virus genotype 1 infection in patients with HIV: a randomized trial. Ann Intern Med. 159 (2): 86-96.
340. Susser S, Vermehren J, Forestier N, Welker MW, Grigorian N, Füller C, Perner D, Zeuzem S, Sarrazin C (2011). Analysis of long-term persistence of resistance mutations within the hepatitis C virus NS3 protease after treatment with telaprevir or boceprevir. J Clin Virol. 52(4): 321-7.
341. Svartengren M, Engström G, Anderson M, Hallberg J, Edula G, de Verdier MG, Dahlbäck M, Lindberg CM, Forsman-Semb K, Nihlén U, Fehniger TE (2009). Twins studies as a model for studies on the interaction between smoking and genetic factors in the development of chronic bronchitis. Biochem Soc Trans. 37 (Pt 4): 814-8.
342. Svensson S, Menkes DB, Lexchin J (2013). Surrogate outcomes in clinical trials: a cautionary tale. JAMA Intern Med. 173 (8): 611-2.
343. SVR (2001). Sachverständigenrat zur Begutachtung der Entwicklung im Gesundheitswesen. Bedarfsgerechtigkeit und Wirtschaftlichkeit: Band III Über -, Unter- und Fehlversorgung. Gutachten 2000/2001 des Sachverständigenrates zur Begutachtung der Entwicklung im Gesundheitswesen. Bonn: Sachverständigenrat zur Begutachtung der Entwicklung im Gesundheitswesen.
344. Takeda (2013). Fachinformation – Zusammenfassung der Merkmale des Arzneimittels Daxas® 50 Mikrogramm Filmtabletten. Stand der Information: August 2013.
345. Thimme R, Heim M, Baumert TF, Nassal M, Moradpour D.(2014). Hepatitis B and C: from molecular virology to new antiviral therapies (part 2). Dtsch Med Wochenschr. 139 (15): 778-82.
346. Tinkelman DG, Price DB, Nordyke RJ, Halbert RJ (2006). Misdiagnosis of COPD and asthma in primary care patients 40 years of age and over. J Asthma. 43 (1): 75-80.
347. Torrent M, Sunyer J, Munoz L, Cullinan P, Iturriaga MV, Figueroa C, Vall O, Taylor AN, Anto JM (2006). Early life domestic aeroallergen exposure and IgE sensitization at age 4 years. J Allergy Clin Immunol. 118 (3): 742-8.
220

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
348. Toyota J, Ozeki I, Karino Y, Asahina Y, Izumi N, Takahashi S, Kawakami Y, Chayama K, Kamiya N, Aoki K, Yamada I, Suzuki Y, Suzuki F, Kumada H (2013). Virological response and safety of 24-week telaprevir alone in Japanese patients infected with hepatitis C virus subtype 1b. J Viral Hepat. 20 (3): 167-73.
349. Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R (2008). Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med. 358 (3): 252-60.
350. Upham JW & James AL (2011). Remission of asthma: The next therapeutic frontier? Pharmacol Ther. 130 (1): 38-45.
351. van Haren-Willems J & Heijdra Y (2010) Increasing evidence for gender differences in chronic obstructive pulmonary disease. Womens Health (Lond Engl). 6 (4): 595–600.
352. van Schalkwyk E, Strydom K, Williams Z, Venter L, Leichtl S, Schmid-Wirlitsch C, Bredenbröker D, Bardin PG (2005). Roflumilast, an oral, once-daily phosphodiesterase 4 inhibitor, attenuates allergen-induced asthmatic reactions. J Allergy Clin Immunol. 116 (2): 292-8.
353. van Schayck CP, Dompeling E, van Herwaarden CL, Folgering H, Verbeek AL, van der Hoogen HJ, van Weel C (1991). Bronchodilator treatment in moderate asthma or chronic bronchitis: continuous or on demand? A randomised controlled study. BMJ. 303 (6815): 1426-31.
354. Vedula SS, Bero L, Scherer RW, Dickersin K (2009). Outcome reporting in industry-sponsored trials of gabapentin for off-label use. N Engl J Med. 361 (20): 1963-71.
355. Veldt BJ, Heathcote EJ, Wedemeyer H, Reichen J, Hofmann WP, Zeuzem S, Manns MP, Hansen BE, Schalm SW, Janssen HL (2007). Sustained virologic response and clinical outcomes in patients with chronic hepatitis C and advanced fibrosis. Ann Intern Med. 147 (10): 677-84.
356. Vestbo J, Sorensen T, Lange P, Brix A, Torre P, Viskum K (1999). Long-term effect of inhaled budesonide in mild and moderate chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 353 (9167): 1819-23.
357. vfa (2013). vfa-Positionspapier „Orphan Drugs“. http://www.vfa-bio.de/vb-de/download-manager/_pos-orphandrugs.pdf. Letzter Zugriff: 03.12.2013.
358. vfa bio (2015). Die forschenden Pharma-Unternehmen. Immer mehr Patienten mit seltenen Erkrankungen behandelbar. April 2015. http://www.vfa-bio.de/vb-de/aktuelle-themen/orphans/immer-mehr-patienten-mit-seltenen-erkrankungen-behandelbar.html. Letzter Zugriff: 03.05.2015.
359. Vogelmeier C, Buhl R, Criee CP, Gillissen A, Kardos P, Köhler D, Magnussen H, Morr H, Nowak D, Pfeiffer-Kascha D, Petro W, Rabe K, Schultz K, Sitter H, Teschler H, Welte T, Wettengel R, Worth H (2007). Leitlinie der Deutschen Atemwegsliga und der Deutschen Gesellschaft für Pneumologie und Beatmungsmedizin zur Diagnostik und Therapie von Patienten mit chronisch obstruktiver Bronchitis und Lungenemphysem (COPD). Pneumologie. 61 (5): e1 40.
360. Vogelmeier CF, Bateman ED, Pallante J, Alagappan VK, D'Andrea P, Chen H, Banerji D (2013). Efficacy and safety of once-daily QVA149 compared with twice-daily salmeterol-fluticasone in patients with chronic obstructive pulmonary disease (ILLUMINATE): a randomised, double-blind, parallel group study. Lancet Respir Med. 1 (1): 51-60.
221

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
361. Walter U (2012) Ärzte und ihre Verordnung von Arzneimitteln im Off-Label-Use, 105-118, in: Becker U & Wilman N (Hrsg.) Im Zweifel auf Privatrezept. Sozial- und haftungsrechtliche Aspekte des Off-Label-Use. Normos. 1. Auflage.
362. Wedemeyer H, Hardtke S, Cornberg M (2012) Therapie der Hepatitis C – Aktuelle Standards und zukünftige Entwicklungen. Chemother J. 21: 1-7.
363. Wedemeyer H, Jensen DM, Godofsky E, Mani N, Pawlotsky JM, Miller V; on behalf of the Definitions/Nomenclature Working Group* of the HCV DrAG (HCV Drug Development Advisory Group), under the auspices of the Forum for Collaborative HIV Research (2012a). Recommendations for standardized nomenclature and definitions of viral response in trials of HCV investigational agents. Hepatology. [Epub ahead of print].
364. Wedzicha JA & Seemungal TA (2007). COPD exacerbations: defining their cause and prevention. Lancet. 370 (9589): 786-96.
365. Wedzicha JA, Decramer M, Ficker JH, Niewoehner DE, Sandström T, Taylor AF, D'Andrea P, Arrasate C, Chen H, Banerji D (2013). Analysis of chronic obstructive pulmonary disease exacerbations with the dual bronchodilator QVA149 compared with glycopyrronium and tiotropium (SPARK): a randomised, double-blind, parallel-group study. Lancet Respir Med. 1 (3): 199-209.
366. Wegner C (2013): Off-Label-Use – Eine sozialversicherungsrechtliche Betrachtung. AV Akademikerverlag.
367. Wegscheider K, Drabik A, Bleich C, Schulz H (2015). [Benefit assessment in health services research and epidemiology]. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 58 (3): 298-307.
368. Wetterling T (2004): Rechtsprechung zur Verabreichung von Medikamenten außerhalb der zugelassenen Indikationsgebiete („off-label-use“). Fortschritte der Neurologie Psychiatrie. VOL: 72(5); p 255-9.
369. White WB, Cooke GE, Kowey PR, Calverley PM, Bredenbröker D, Goehring UM, Zhu H, Lakkis H, Mosberg H, Rowe P, Rabe KF (2013). Cardiovascular safety in patients receiving roflumilast for the treatment of COPD. Chest. 144 (3): 758-65.
370. WHO (2012). World Health Organization . Prevention & Control of Viral Hepatitis Infection: Framework for global action. http://who.int/csr/disease/hepatitis/GHP_Framework_En.pdf. Letzter Zugriff: 16.04.2015.
371. WHO (2013). World Health Organization - Asthma. Fact sheet N°307. WHO, Genf www.who.int/mediacentre/factsheets. Updates 11/2013. Letzter Zugriff: 01.02.2015.
372. WHO (2013). World Health Organization - Asthma. Fact sheet N°307. WHO, Genf www.who.int/mediacentre/factsheets. Updates 11/2013. Letzter Zugriff: 01.02.2015.
373. Wiegard N (2014). Entscheidung des Monats 05/2014. Forschungsstelle Recht der Gesundheitswirtschaft Lehrstuhl für Bürgerliches Recht, Arbeitsrecht und Sozialrecht Fakultät für Rechtswissenschaft - Universität Bielefeld. Professor Dr. Oliver Ricken.
374. Wieseler B (2010). [Results registries for clinical trials--a milestone on the way to transparency in clinical research?]. Z Evid Fortbild Qual Gesundhwes. 104 (4): 298-305.
222

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
375. Windeler J, Koch K, Lange S, Ludwig WD (2010). Arzneimittelmarktneuordnungsgesetz: Zu guter Letzt ist alles selten. Dtsch Arztebl; 107 (42): A-2032.
376. Windt R (2010). Analyse der medikamentösen Versorgung von Asthmapatienten im Erwachsenenalter mit Routinedaten. Dissertation. Südwestdeutscher Verlag für Hochschulschriften.
377. Windt R, Boeschen D, Glaeske G (2013): Innovationsreport 2013 – Auswertungsergebnisse von Routinedaten der Techniker Krankenkasse aus den Jahren 2010 und 2011 (Langfassung). Zentrum für Sozialpolitik; Techniker Krankenkasse, Hamburg; Eigenverlag.
378. Windt R, Boeschen D, Glaeske G (2014): Innovationsreport 2014 – Auswertungsergebnisse von Routinedaten der Techniker Krankenkasse aus den Jahren 2011 und 2012 (Langfassung). Zentrum für Sozialpolitik; Techniker Krankenkasse, Hamburg; Eigenverlag.
379. Witmer AN, Vrensen GF, Van Noorden CJ, Schlingemann RO (2003). Vascular endothelial growth factors and angiogenesis in eye disease. Prog Retin Eye Res. 22( 1): 1-29.
380. Wong KA, Worth A, Martin R, Svarovskaia E, Brainard DM, Lawitz E, Miller MD, Mo H (2013). Characterization of Hepatitis C virus resistance from a multiple-dose clinical trial of the novel NS5A inhibitor GS-5885. Antimicrob Agents Chemother. 57 (12): 6333-40.
381. Yang WH, Martinot JB, Pohunek P, Beier J, Magula D, Cameron R, Owen R, Higgins M (2007). Tolerability of indacaterol, a novel once-daily beta2-agonist, in patients with asthma: a randomized, placebo-controlled, 28-day safety study. Ann Allergy Asthma Immunol. 99 (6): 555-61.
382. Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, Focaccia R, Younossi Z, Foster GR, Horban A, Ferenci P, Nevens F, Müllhaupt B, Pockros P, Terg R, Shouval D, van Hoek B, Weiland O, Van Heeswijk R, De Meyer S, Luo D, Boogaerts G, Polo R, Picchio G, Beumont M; REALIZE Study Team (2011). Telaprevir for retreatment of HCV infection. N Engl J Med. 3 64 (25): 2417-28.
383. Zeuzem S, Berg T, Gane E, Ferenci P, Foster GR, Fried MW, Hezode C, Hirschfield GM, Jacobson I, Nikitin I, Pockros PJ, Poordad F, Scott J, Lenz O, Peeters M, Sekar V, De Smedt G, Sinha R, Beumont-Mauviel M (2014b). Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology. 146 (2): 430-41.e6.
384. Zeuzem S, Dusheiko GM, Salupere R, Mangia A, Flisiak R, Hyland RH, Illeperuma A, Svarovskaia E, Brainard DM, Symonds WT, Subramanian GM, McHutchison JG, Weiland O, Reesink HW, Ferenci P, Hézode C, Esteban R; VALENCE Investigators (2014a). Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N Engl J Med. 370 (21): 1993-2001.
385. Zeuzem S, Jacobson IM, Baykal T, Marinho RT, Poordad F, Bourlière M, Sulkowski MS, Wedemeyer H, Tam E, Desmond P, Jensen DM, Di Bisceglie AM, Varunok P, Hassanein T, Xiong J, Pilot-Matias T, DaSilva-Tillmann B, Larsen L, Podsadecki T, Bernstein B (2014c). Retreatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med. 370 (17): 1604-14.
386. Zipprich A & Dollinger M (2010). Hepatitis C – Die vernachlässigten Genotypen. Hepatitis&more. 2/2010, 18-20.
223

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
11 Abkürzungen
μg Mikrogramm LDV Ledipasvir
Abs. Absatz LL Leitlinie
AMG Arzneimittelgesetz MDK Medizinischer Dienst der Krankenkassen
AMNOG Gesetz zur Neuordnung des Arzneimittelmark-tes
NVL Nationale VersorgungsLeitlinie
Az. Aktenzeichen OLG Oberlandesgericht
BEK Barmer Ersatzkasse Nr. Nummer
BfArM Bundesinstitut für Arzneimittel und Medizin-produkte
OLU Off-Label Use
BMG Bundesministerium für Gesundheit OMV Ombitasvir
BOC Boceprevir PEG- PEG-Interferon alpha
BSG Bundessozialgericht PEI Paul-Ehrlich-Institut
BSSichG Beitragssatzsicherungsgesetz PRV Paritaprevir
BVerfG Bundesverfassungsgericht pU pharmazeutischer Unternehmer
BW Behandlungswoche(n) RBV Ribavirin
cHC chronische Hepatitis C RTV Ritonavir
d.h. das heißt RNA Ribonukleinsäure
DAA Direkt antiviral wirkende Substanzen RKI Robert Koch-Institut
DCV Daclatasvir SG Sozialgericht
DGVS Deutsche Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten
SGB V 5. Sozialgesetzbuch
DPhG Deutsche Pharmazeutische Gesellschaft SMV Simeprevir
DSV Dasabuvir SOF Sofosbuvir
EbM Evidenzbasierte Medizin SVR Sustained Virological Response
EKG Elektrokardiogramm TK Techniker Krankenkasse
EMA European Medicines Agency TVR Telaprevir
etc. et cetera u.a. unter anderem
FI Fachinformation vs. versus
G-BA Gemeinsamer Bundesausschuss ZH Zirrhose
GEDA Gesundheit in Deutschland aktuell
gem. gemäß
GKV Gesetzliche Krankenversicherung
GKV-WSG GKV-Wirtschaftlichkeitsstärkungsgesetz
GMG GKV-Modernisierungsgesetz
GT Genotyp
HIV Humanes Immundefizienz Virus
i.v. intravenös
ILU In-Label Use
IQWiG Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen
224

Nutzen und Risiken der zulassungsüberschreitenden Arzneimittelanwendung in der ambulant-ärztlichen Versorgung
12 Abstract
Off-label use (OLU) is defined as the prescription of pharmaceutical drugs for an indication or other
application that has not been approved by the governing authorization body. Off-label uses are not
only common practice but are often the standard and relate to the need for a patient-oriented and
patient-specific therapy. The current thesis discusses off-label use from the perspective of health
services research. The question is of particular interest because civil and criminal requirements on
the one hand and the health insurance law on the other have a direct impact on the doctor as a
health care provider and the patient as a beneficiary.
The first part of this thesis (Chapters 1-6) briefly outlines the legal aspects, the second part (chapters
7-9) analyses the possible conditions that enable an off-label use in drug therapy of chronic diseases.
The focus is primarily on possible structural and administrative gaps as the cause of an OLU. The
present work integrates different areas of supply research.
This thesis considers the strengths and limitations of different databases and indicates more trans-
parency with regard to off-label use discussing the gap between the admission authorised usage and
patient-specific care. Finally it outlines the implications for better health care provision.
225





























