Embed Size (px)
Citation preview

FRANCISCO ORTEGA
NEUROLOGÍA HOSPITAL ROOSEVELT
MAYO 2014

Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy: a heterogeneous degeneration involving brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964;10:333-359.
• Primera descripción que afecta principalmente mirada vertical, parálisispseudo bulbar, disartria, rigidez distónica del cuello y tronco superior, y otros síntomas piramidales y cerebelares menos constantes• Esta enfermedad consta de degeneración de las células del tronco cerebral predominantemente
Existian para la época algunas descripciones del sindrome piramidopalido yde casos con rigidez y demencia llamados enfermedad de Jackob. Sin embargo ninguna de estas descripciones incluian la descripción de unacombinación de distonia, oculomotor, pseudobulbar y mental

• Descrita por 1ª vez en 1964 por Steele, Richardson y Olszewski
como una “enfermedad cerebral progresiva”
• Caracterizado por oftalmoplejia supranuclear, parálisis pseudo bulbar, disartria, rigidez distonica de la nuca y tronco superior, demencia leve.
Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy: a heterogeneous degeneration involving brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964;10:333-359.

• Paciente hombre de 56 años, inicio con dificultad para tocar el piano con mano izquierda, inestable y torpe al ir al trabajo, dificultad inespecífica para ver objetos, síntomas que fueron empeorando. 56 empeoro con el trastorno de la marcha y paralasis de la mirada hacia abajo. Extensión de cuello con rigidez de este, pérdida de fenómeno de Bell. Dificultad para la fijación y para movimientos oculares. Un año después disartria, discurso lento y susurrado. Desorientación leve pero puede llevar una conversación. Caídas constantes y apraxia del tronco vertebral, facie rígida pero sin parkinsonismo. Presentó una convulsión y fue encontrado muerto en su cama boca abajo sofocado.
• Tiempo de seguimiento 4 años.


• Cerebro de 1,500 gramos, sin cambios macoscópicos de importancia, con sistema vascular con poca ateroesclerosis through the cerebral hemispheres
• La sección transversal del cerebelo y tronco cerebral evidenció dilatación de el acueducto, coliculosuperior y apariencia gelatinosa del nucleo dentado y pedúnculos cerebelares
• Microscópico:
• Severa pérdida neuronal y gliosis en el núcleo subtalámico, sustancia negra, coliculo superior, sustancia gris y nucleo dentado
• Enredos neurofibrilares estaban presentes en las regiones afectadas degeneración granulovacuolar en las neuronas del nucleo rojo y puente

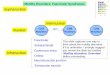
• Hombre de 50 años con trastornos intelectuales, trastornos visuales y depresión así como pérdida de la mirada hacia abajo. Postura distónica del cuello e hipertonicidad de la cadera. Disartria con espasticidad facial, apraxia del tronco al girar y al sentarse. La muerte por sofocación llego a los 56 años.
• Hallazgos neuropatológicos consistieron en pérdida neuronal,
enredos neurofibrilares, degeneración granulovacuolar, gliosis y
demielinización en núcleos de los ganglios de la base, tronco cerebral y cerebelo

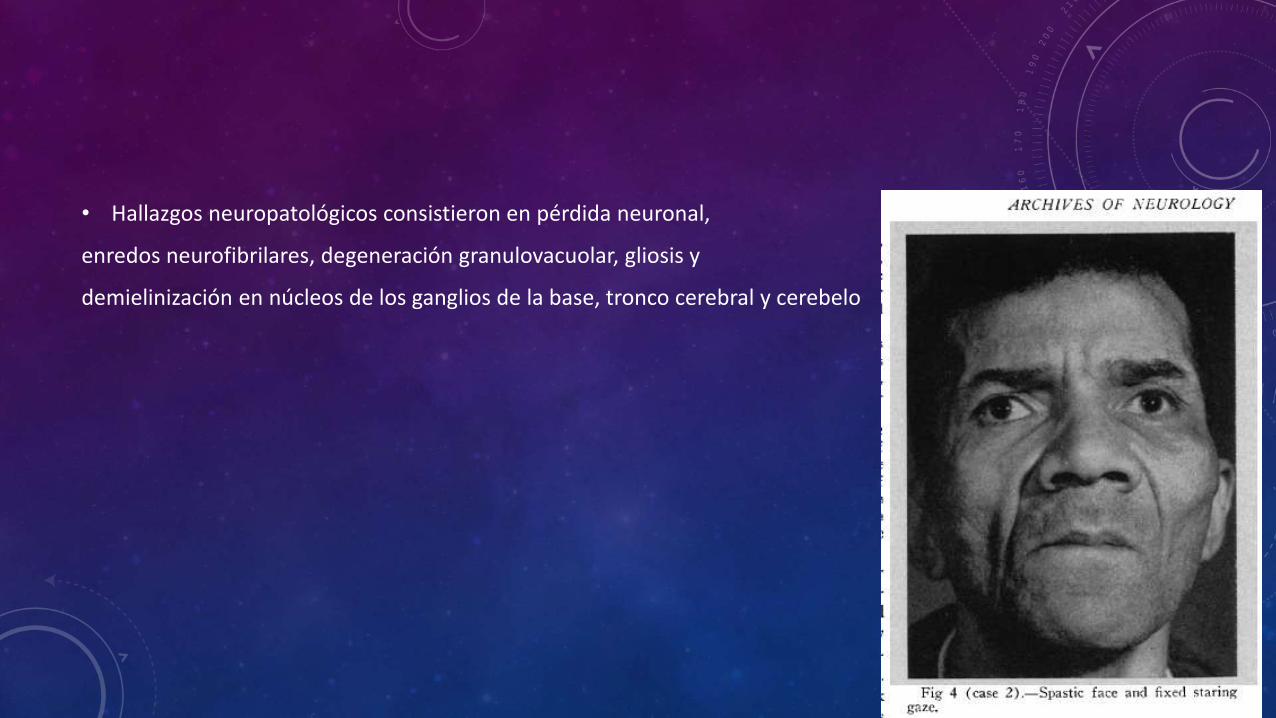
• 4 años antes de su muerte un paciente hombre de 59 años inició con dificultad para la visión, discurso en susurros, y problemas para deglutir liquidos. Presentó caídas frecuentes, reacciones explosivas emocionales, modera restricción de la marcha principalmente hacia los lados, pérdida de movimientos oculares de seguimiento, fenómeno de cabeza de muñeca, pérdida de la convergencia, facie fija y cuello extendido, con restricción al movimiento lateral, fenómeno de Bell perdido, movimientos de las manos torpes e inadecuados, movimientos atetoides en la prueba dedo nariz. Falleció al presentar convulsiones y aspiración bronquial.
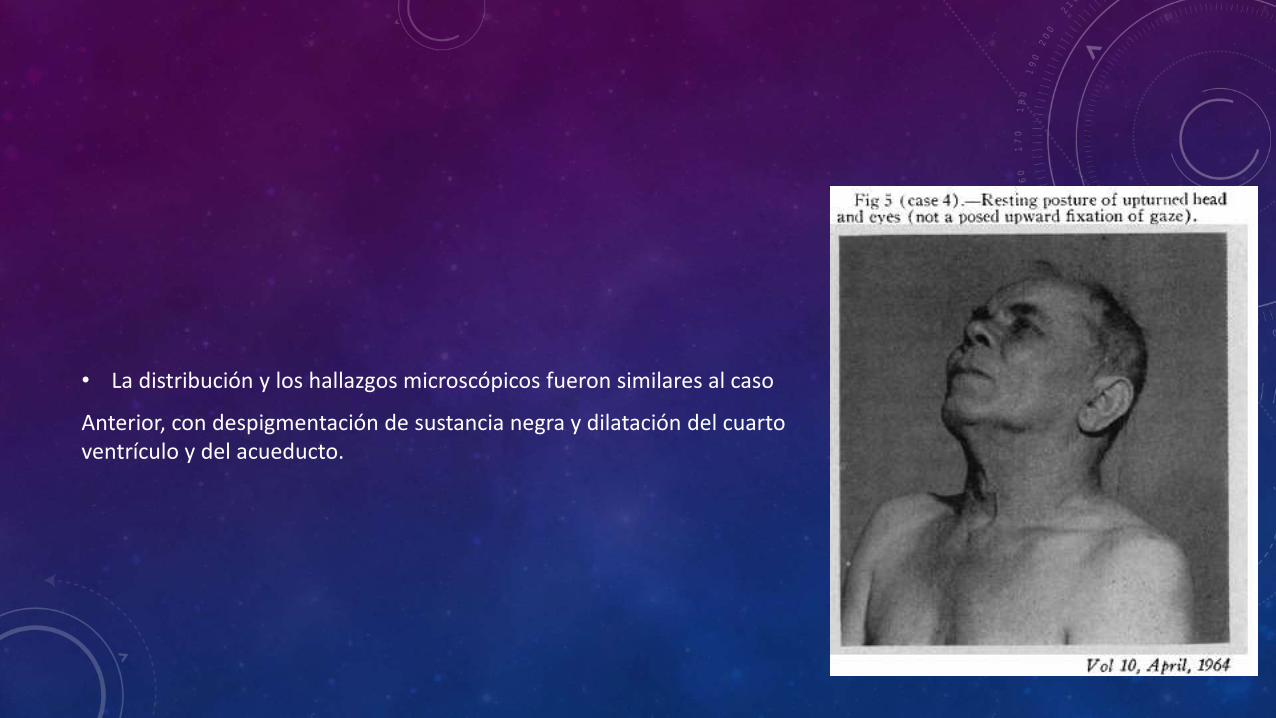
• La distribución y los hallazgos microscópicos fueron similares al caso
Anterior, con despigmentación de sustancia negra y dilatación del cuarto ventrículo y del acueducto.

• La hipótesis establecida en ese entonces fue, degeneración neuronal primaria, que afectaría principalmente al tronco encéfalo o una degeneración de posible origen vírico.
• Steele describió, respecto a la neuropatología, ovillos neurofibrilares(cuerpos de hirano), degeneración granulovacuolar, perdida neuronal en los GB, tronco encéfalo y cerebelo.
Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy: a heterogeneous degeneration involving brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964;10:333-359.

• Es un cuadro degenerativo del SNC• Presentación esporádica• Comienza después de los cuarenta años• Los síntomas mas característicos considerados cardinales son:
• Parálisis supranuclear de la mirada
• Parálisis pseudobulbar
• Disartria
• Distonia axial
• Demencia
Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy: a heterogeneous degeneration involving brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964;10:333-359.

• Se describe como una mirada de “Mona Lisa” con muy poco parpadeo (0-4 por minuto)
• Discurso que arrastra palabras y cabeza retraída
• Marcha inestable y tambaleante “marcha de marinero ebrio”
• Imprudencia motora que se manifiesta al levantarse de la silla “signo de cohete”
• La ropa de mancha de comida por parálisis de la mirada hacia abajo y dificultad para deglutir
David J Burn and Andrew J Lees. Progressive supranuclear palsy: where are we now? Lancet Neurology2002; 1: 359–69

• Estudios genéticos sugieren un desorden neurodegenerativo, con presentación clínica diferente pero con hallazgos neuropatológicos similares
• Charcot en 1889 en Nouvelle Iconographie de la Salpêtrièredescribió “parálisis hemipléjica agitante con postura inusual del tronco y la cabeza, retrocolis y trastorno del movimiento ocular”
David J Burn and Andrew J Lees. Progressive supranuclear palsy: where are we now? Lancet Neurology2002; 1: 359–69

• Prevalencia de 5.3 por 100,000 habitantes mayores de 50 años muy similar en EEUU y UK
• La edad de desarrollo es de 60-65 años
• La vida media de los pacientes desde el inicio de los síntomas es de 5.8-5.9 años
• El rango que dura para ser diagnosticada la enfermedad es de 3.6 a 4.9 años
David J Burn and Andrew J Lees. Progressive supranuclear palsy: where are we now? Lancet Neurology2002; 1: 359–69

• Enfermedad de Parkinson (30%) trastornos del movimiento (20%) isquemia (10%) depresión (7%)
• En estudios patológicos 1/3 de pacientes con diagnóstico de Parkinson o demencia de cuerpos de Lewyson en realidad PSP
David J Burn and Andrew J Lees. Progressive supranuclear palsy: where are we now? Lancet Neurology2002; 1: 359–69

• Causas potenciales de anormalidad en movimientos oculares, inestabilidad postural y parkinsonismo que no son PSP
David R Williams, Andrew J Lees. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol 2009; 8: 270–79
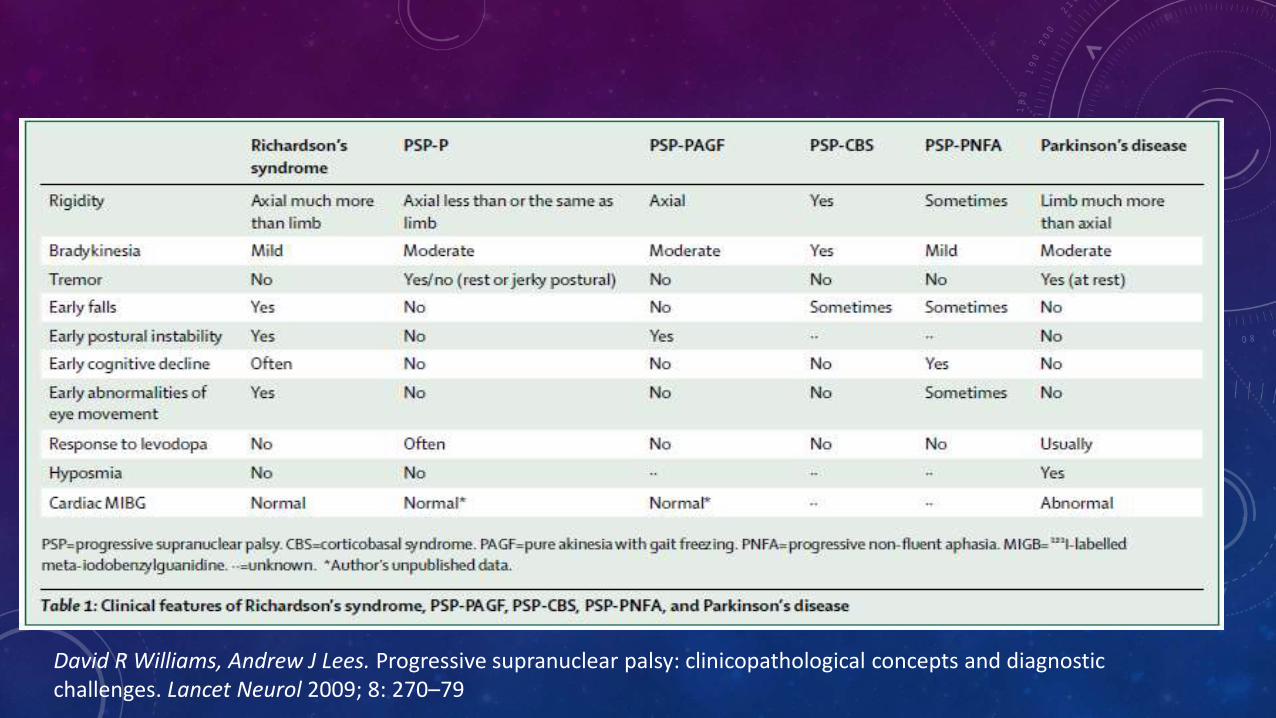
David R Williams, Andrew J Lees. Progressive supranuclear palsy: clinicopathological concepts and diagnosticchallenges. Lancet Neurol 2009; 8: 270–79

• Patologicamente la PSP se define con la acumulación de proteína tau y enredos neurofibrillas, mayormente en el pallidum, nucleo subtalamico, nucleo rojo, sustancia negra, tegmento pontino, striatum, nucleos oculomotores, medulla y nucleo dentado
David R Williams, Andrew J Lees. Progressive supranuclear palsy: clinicopathological concepts and diagnosticchallenges. Lancet Neurol 2009; 8: 270–79
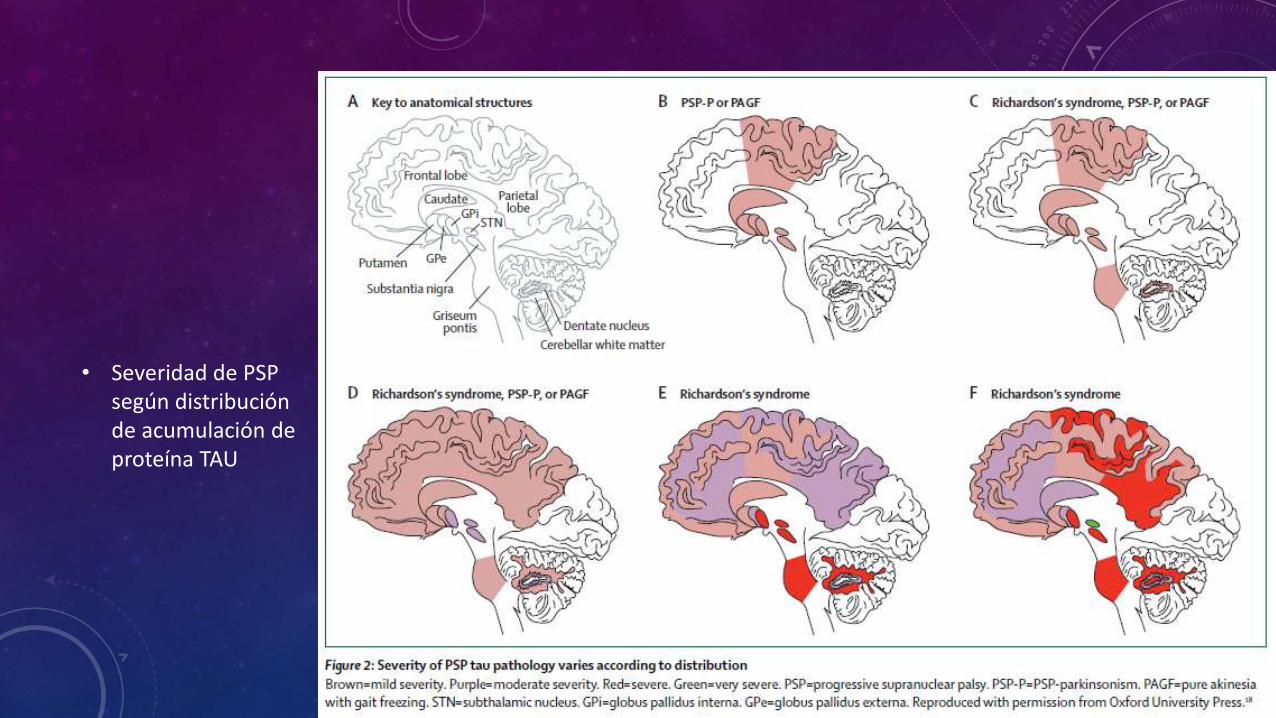
• Severidad de PSP según distribución de acumulación de proteína TAU

• Inestabilidad postural, caídas frecuentes (principalmente hacia atrás), paresia de la mirada vertical supranuclear son síntomas discriminatorios con otros síndromes Parkinsonianos
• Otro dato importante no ocurre en pacientes menores de 40 años
• Es más difícil de diagnosticar al principio de los síntomas
• Presentan mas apatía y deshinibición que los pacientes con Parkinson
• Mayor disminución en la atención, bradrifrenia, abilidades de categorización

• Signos atípicos son distonia unilateral de una extremidad, levitación de brazo, apraxia ideomotora, mioclonus paladar (triangulo de Mollaret rojo, dentado, oliva)
• Neurosifilis, Whipple, atrofia multisistemas, Alzheimer, Parkinson, degeneración corticobasal.
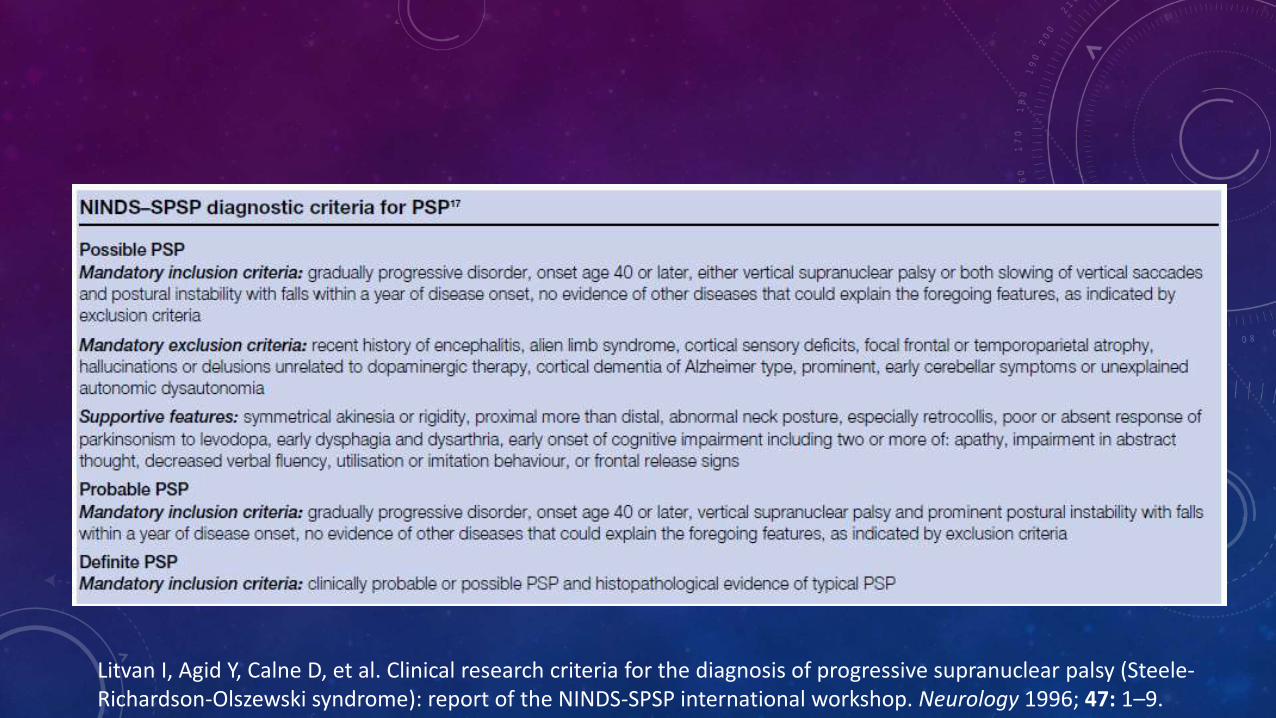
Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 1996; 47: 1–9.

• Concentración de proteína Tau en liquido cefalorraquídeo (aunque si es significante en degeneración corticobasal)
• Uso de levodopa y medición de proteínas de neurofilamentos puede dividir parkinsonismos atípicos
• Electro oculografia puede hacer diagnóstico temprano en PSP, disminución en velocidad de movimientos sacadicos
• Hipotensión ortostatica es rara por conservación simpática y parasimpática
• Anormalidades en potenciales visuales (P300 tiempos de amplitud y reacción) potenciales somatosensioriales, reflejos faciales, posturografia
• Ninguno tiene sensibilidad y especifidad adecuadas para diagnóstico

• Las anormalidades frecuentemente encontradas en RM son
• Dilatación del cuarto ventrículo
• Aumento del ángulo interpeduncular
• Hiperintensidad del pálido interno en T2
• Discreta hiperintensidad en el borde del putamen en T2
• Otros signos encontrados son
• Dilatación del acueducto de Silvio
• Dilatación del tercer ventrículo
• Hiperintensidad puntiforme en el mesencefalo
M. Vérin, L. Defebvre, C. Delmaire, Y. Rolland. Apport de l’IRM pour le diagnostic de paralysie supranucléaireprogressive. Rev Neurol (Paris) 2005 ; 161 : 2, 234-236
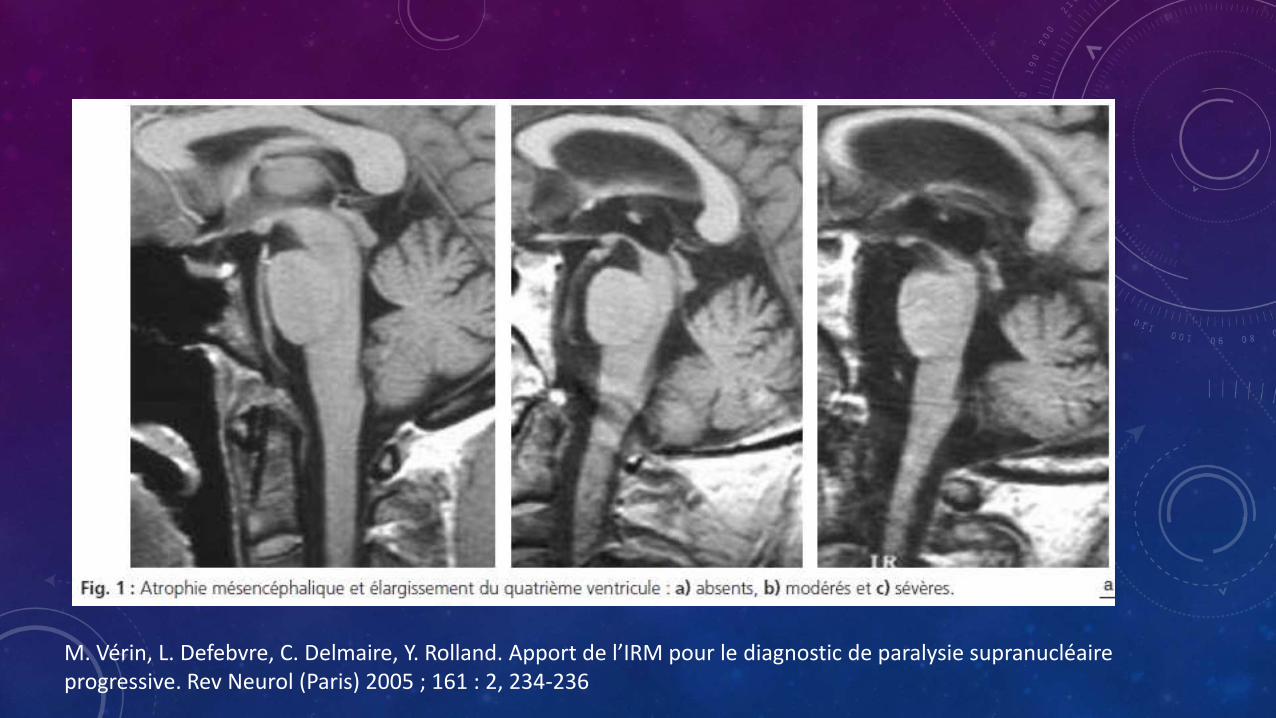
M. Vérin, L. Defebvre, C. Delmaire, Y. Rolland. Apport de l’IRM pour le diagnostic de paralysie supranucléaireprogressive. Rev Neurol (Paris) 2005 ; 161 : 2, 234-236
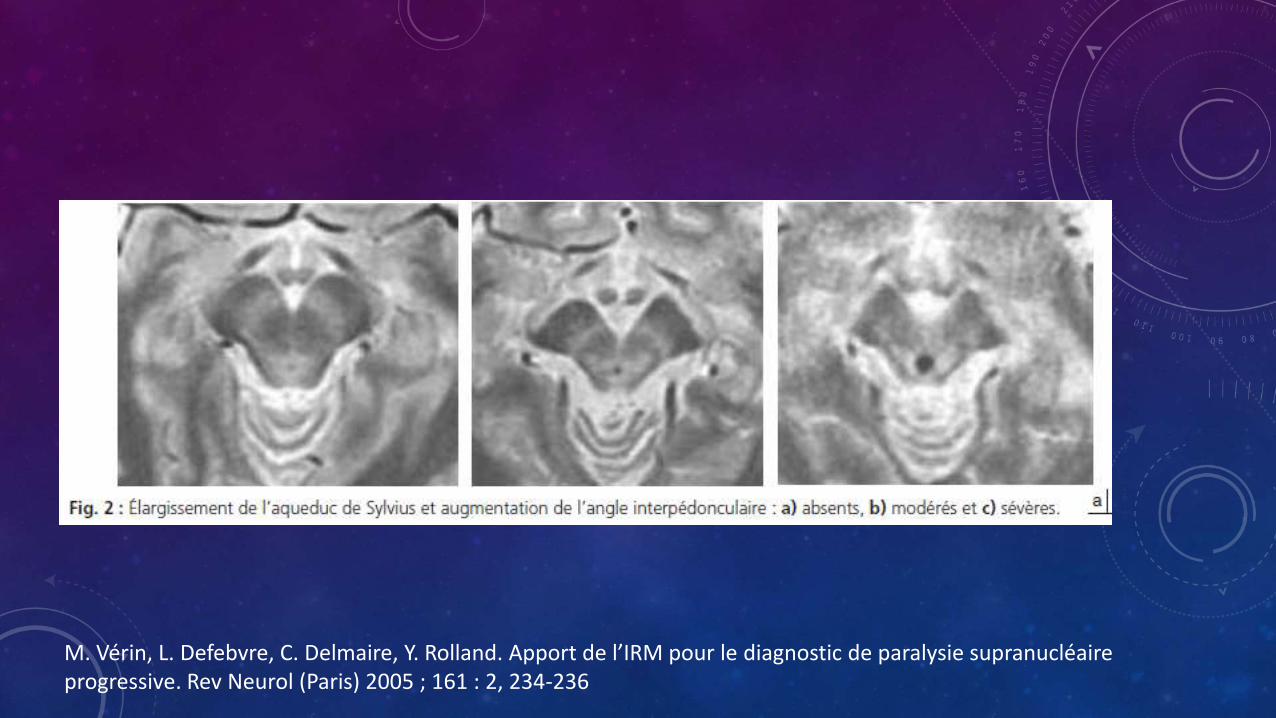
M. Vérin, L. Defebvre, C. Delmaire, Y. Rolland. Apport de l’IRM pour le diagnostic de paralysie supranucléaireprogressive. Rev Neurol (Paris) 2005 ; 161 : 2, 234-236
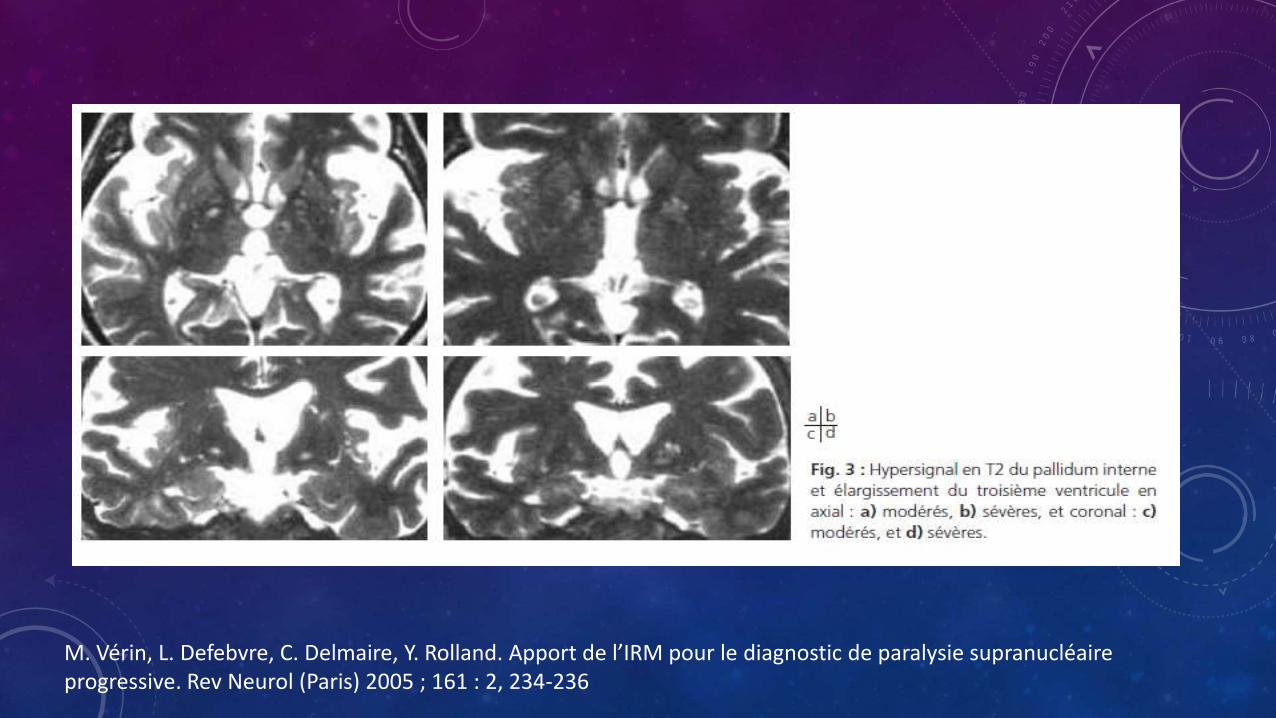
M. Vérin, L. Defebvre, C. Delmaire, Y. Rolland. Apport de l’IRM pour le diagnostic de paralysie supranucléaireprogressive. Rev Neurol (Paris) 2005 ; 161 : 2, 234-236
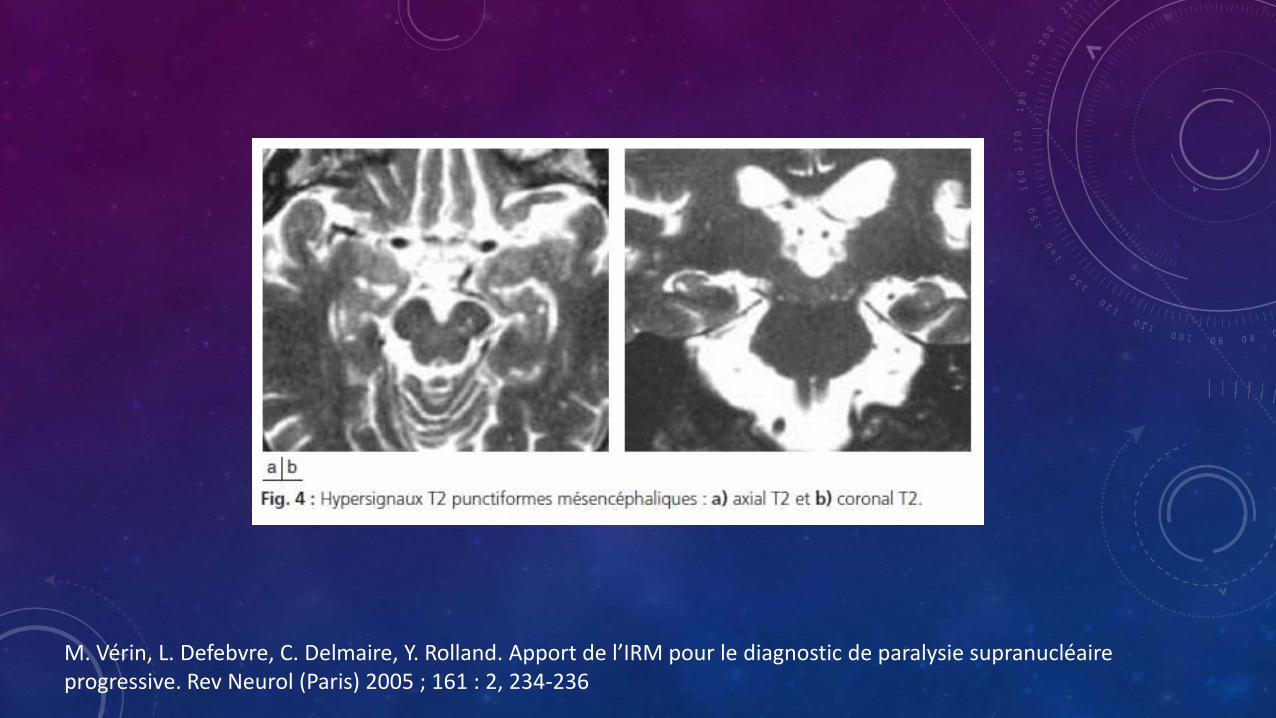
M. Vérin, L. Defebvre, C. Delmaire, Y. Rolland. Apport de l’IRM pour le diagnostic de paralysie supranucléaireprogressive. Rev Neurol (Paris) 2005 ; 161 : 2, 234-236
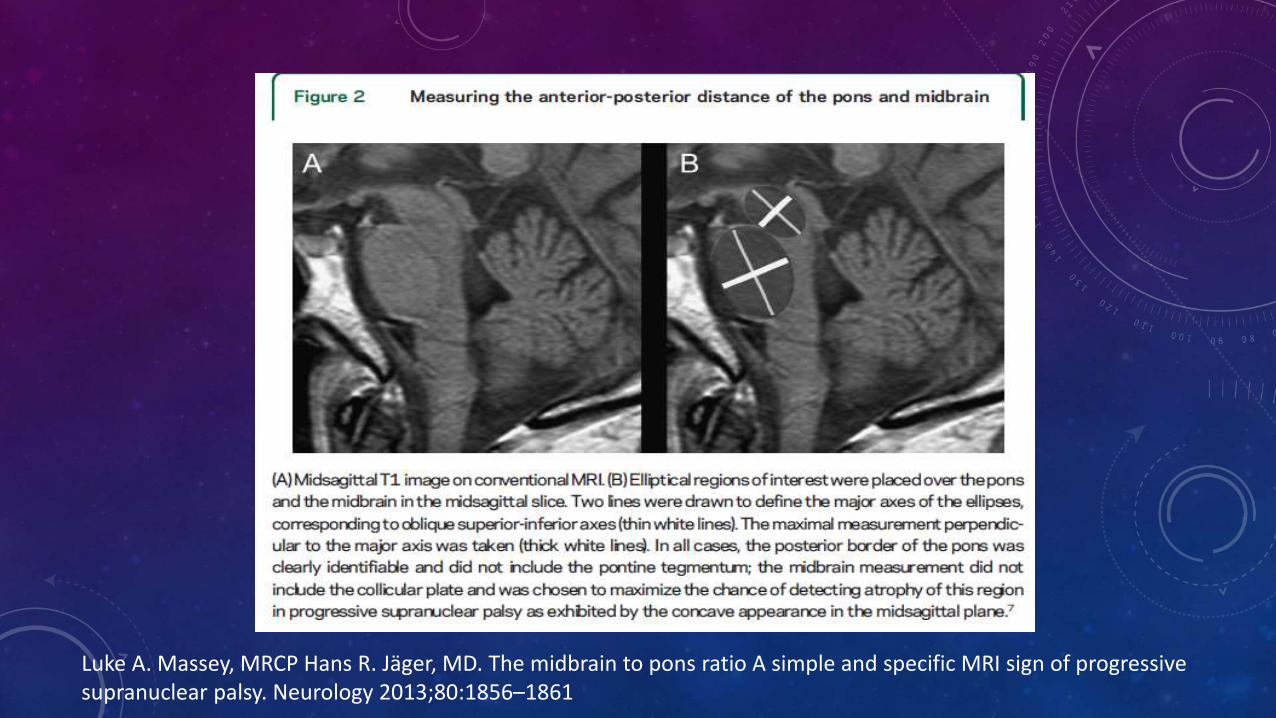
Luke A. Massey, MRCP Hans R. Jäger, MD. The midbrain to pons ratio A simple and specific MRI sign of progressive supranuclear palsy. Neurology 2013;80:1856–1861
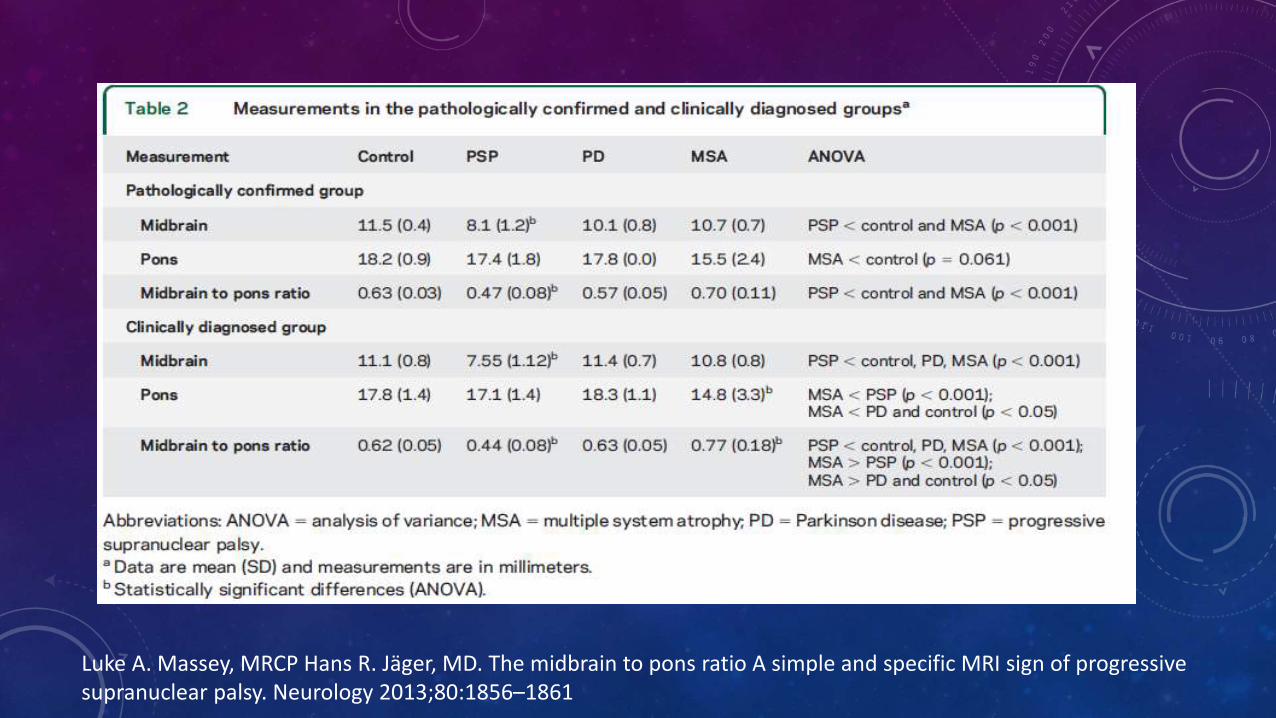
Luke A. Massey, MRCP Hans R. Jäger, MD. The midbrain to pons ratio A simple and specific MRI sign of progressive supranuclear palsy. Neurology 2013;80:1856–1861

• Tau es importante en la estabilización de los microtubulos en el citoesqueleto los hace resistentes a la proteólisis
• Glicación y transglutaminación están implicados en su acumulación lo que produce enredos neurofibrilares
• Hay muerte celular por lesión toxica y predisposición genética con disfunción mitocondrial
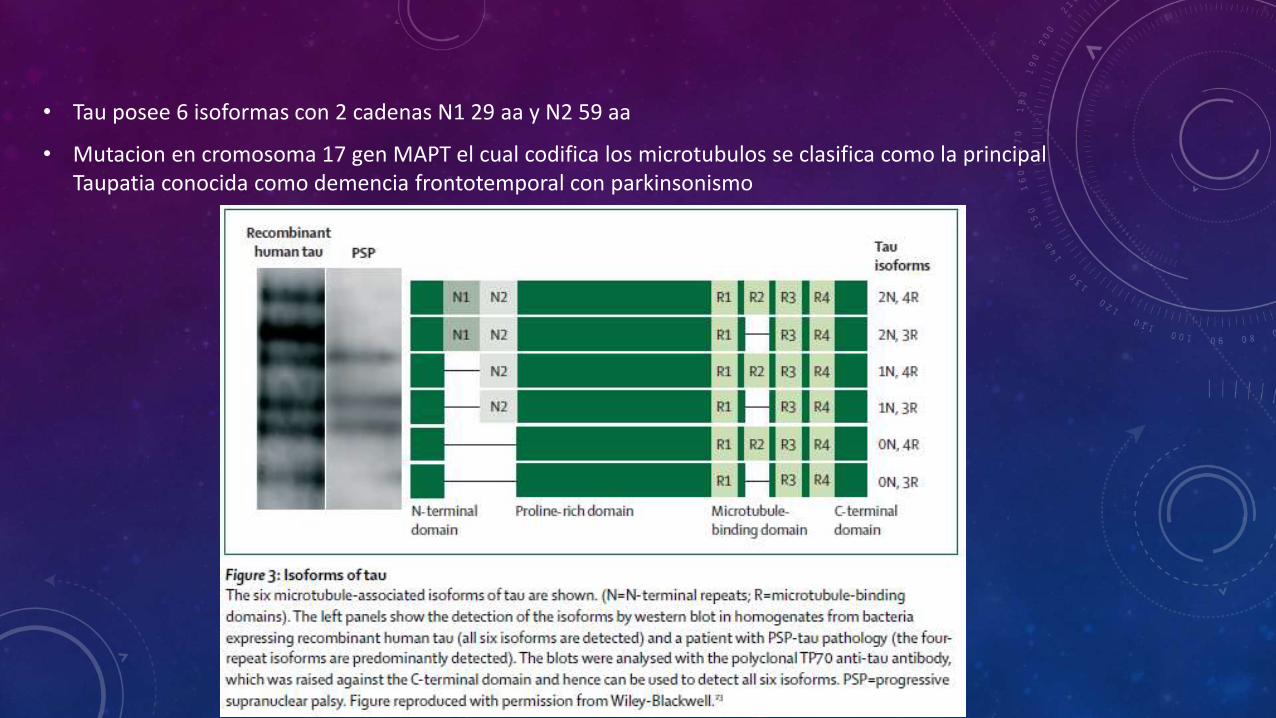
• Tau posee 6 isoformas con 2 cadenas N1 29 aa y N2 59 aa
• Mutacion en cromosoma 17 gen MAPT el cual codifica los microtubulos se clasifica como la principal Taupatia conocida como demencia frontotemporal con parkinsonismo
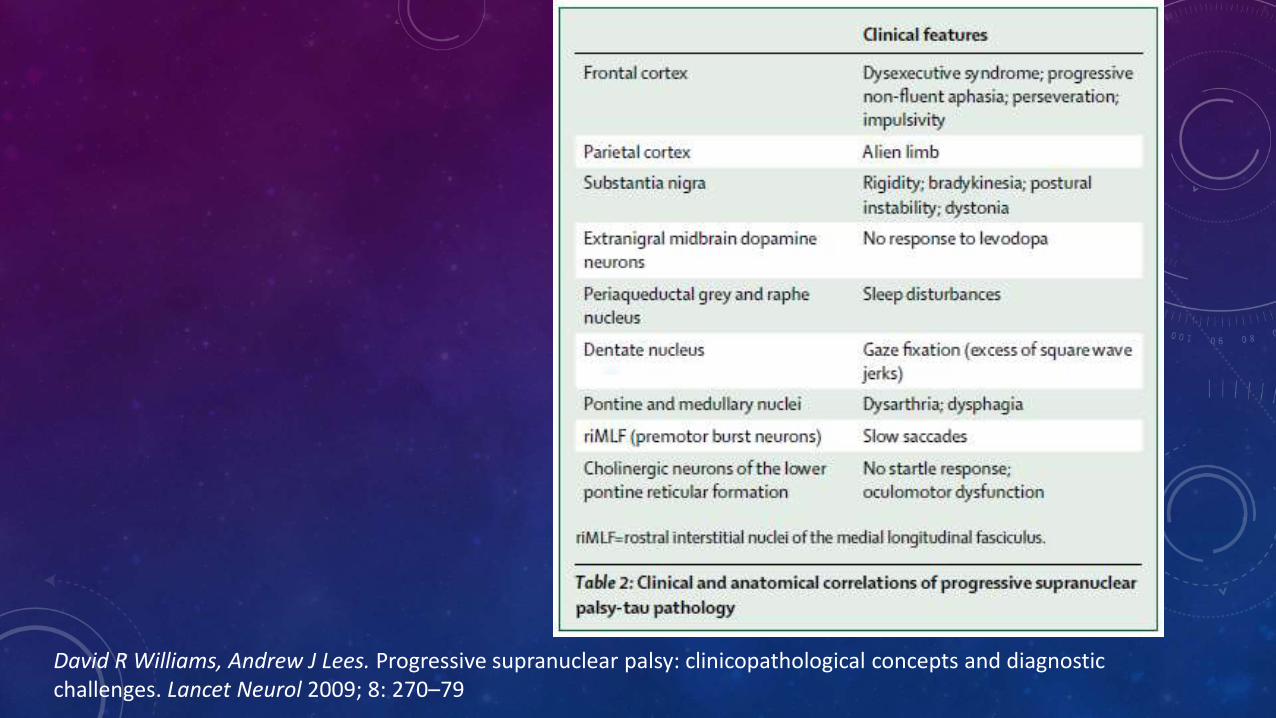
David R Williams, Andrew J Lees. Progressive supranuclear palsy: clinicopathological concepts and diagnosticchallenges. Lancet Neurol 2009; 8: 270–79
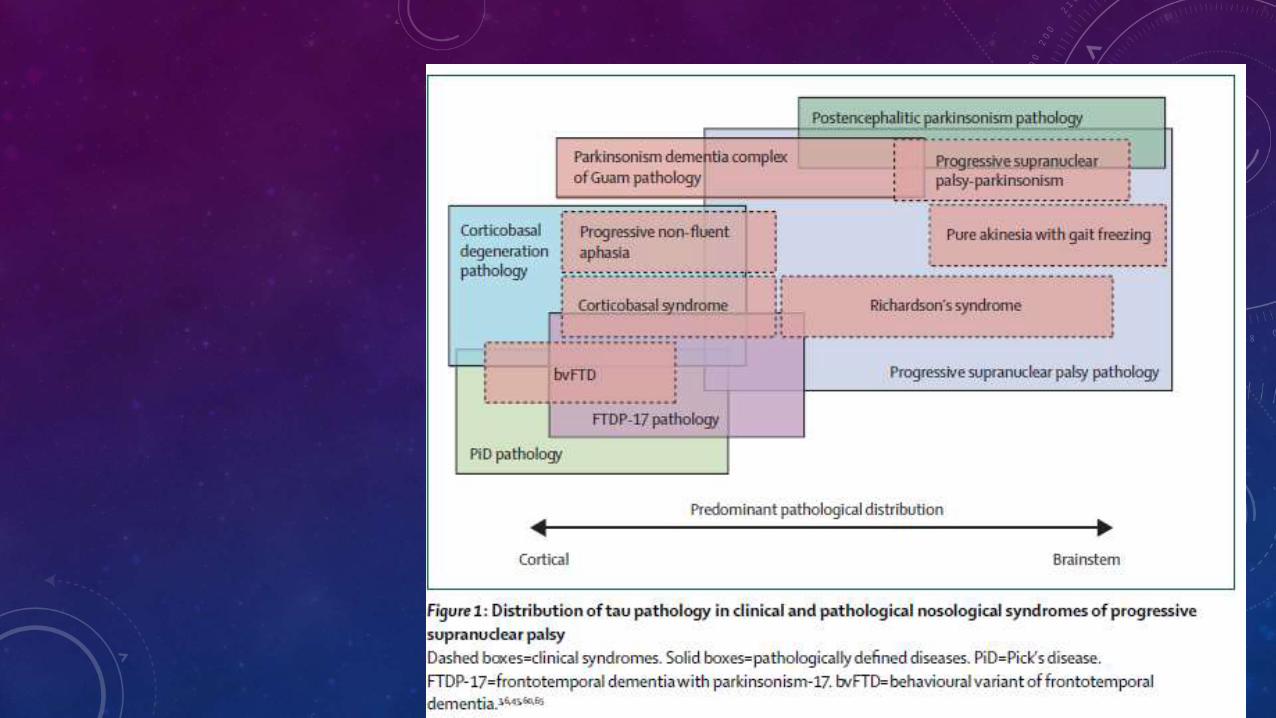
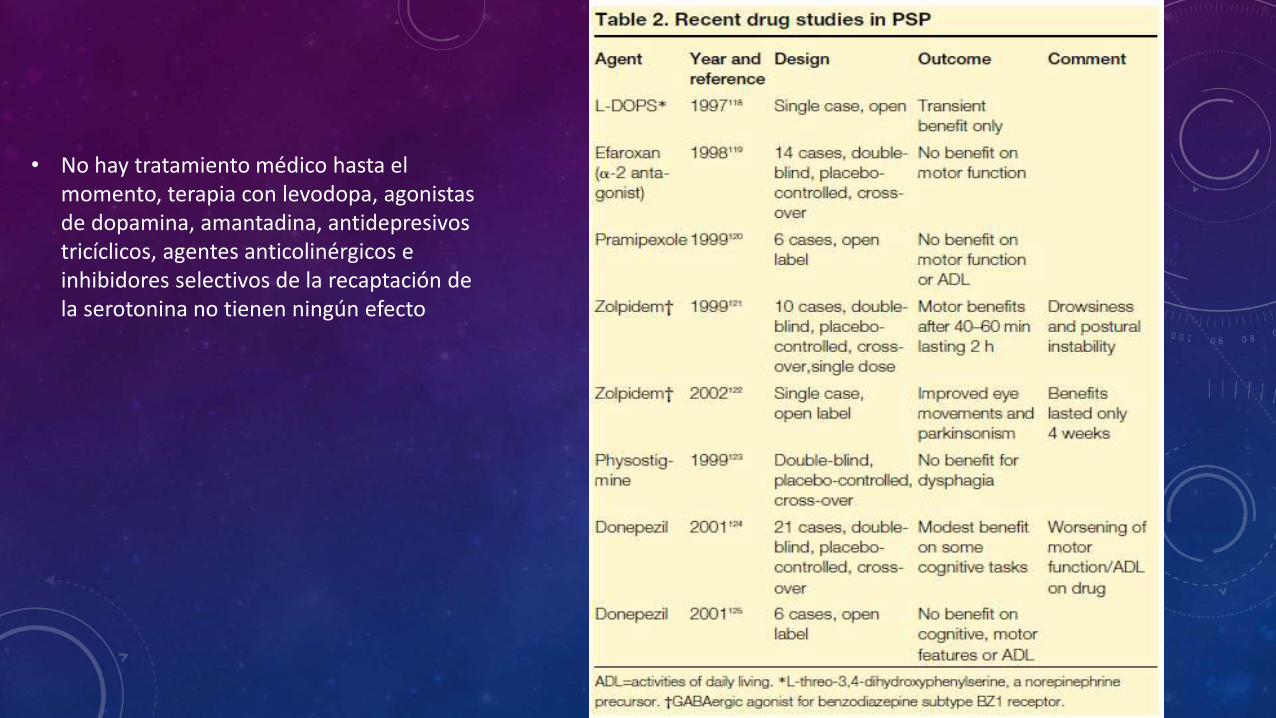
• No hay tratamiento médico hasta el momento, terapia con levodopa, agonistas de dopamina, amantadina, antidepresivos tricíclicos, agentes anticolinérgicos e inhibidores selectivos de la recaptación de la serotonina no tienen ningún efecto