Embed Size (px)
Citation preview

MUSCLE
& NERVE
Invited Review
Pathophysiology of Immune-Mediated Demyelinating
Neuropathies—Part I: Neuroscience
Hessel Franssen, MD, PhD, and Dirk C.G. Straver, MD
This Reprint is Provided as a Member Service by the American Association of Neuromuscular & Electrodiagnostic Medicine
CME Credit AvailableFree to AANEM members
See instructions on the Journal
CME Guide page.

Pathophysiology of Immune-Mediated Demyelinating Neuropathies
—Part I: Neuroscience Hessel Franssen, MD, PhD, and Dirk C.G. Straver, MD
Department of Neurology, Section Neuromuscular Disorders, F02.230, Rudolf Magnus Institute for Neuroscience, University Medical Center Utrecht,
Heidelberglaan 100, 3584 CX Utrecht, The Netherlands
No one involved in the planning of this CME activity had any relevant financial relationships to disclose. (Authors/Faculty had nothing to disclose)
Reviewed and accepted by the 2013-2014 Monograph/Issues and Opinion Committee of the American Association of Neuromuscular & Electrodiagnostic Medicine
Certified for CME credit 01/2014 – 01/2017
Copyright© December 2013
AMERICAN ASSOCIATION OF NEUROMUSCULAR & ELECTRODIOGNOSTIC MEDICINE 2621 Super ior Dr NW Rochester , MN 55901
The ideas and op in ions in th is monograph a re so le ly those o f the author and do no t necessar i l y
represent those o f the AANEM.

AANEM Invited Review #45
CME STUDY GUIDE
Pathophysiology of Immune-Mediated Demyelinating Neuropathies
—Part I: Neuroscience
Hessel Franssen, MD, PhD, and Dirk C.G. Straver, MD Department of Neurology, Section Neuromuscular Disorders, F02.230, Rudolf
Magnus Institute for Neuroscience, University Medical Center Utrecht, Heidelberglaan 100, 3584 CX Utrecht, The Netherlands
EDUCATIONAL OBJECTIVES Upon completion of this monograph, the reader will acquire skills to: (1) review the
anatomy and physiology of normal myelinated axons and the methods of studying peripheral nerve physiology, and (2) recognize the pathophysiologic mechanisms and consequences of primary demyelination and secondary axonal degeneration.
CERTIFYING ORGANIZATION The American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM) is
accredited by the Accreditation Council for Continuing Medical Education (ACCME) to sponsor continuing medical education (CME) for physicians and certifies that this CME activity was planned and produced in accordance with ACCME Essentials.
CME CREDIT The AANEM designates this enduring material for a maximum of 2 AMA PRA Category 1 Credit(s)TM
.
Physicians should claim only the credit commensurate with the extent of their participation in the activity. Monographs published by the AANEM are reviewed every 3 years by the AAEM Education Committee for their scientific relevance. CME credit is granted for 3 years from the date of publish, review, or revision date. Individuals requesting credit for monographs that have been discontinued will be notified that CME credit is no longer available.
INSTRUCTIONS
The reader should carefully and thoroughly study the monograph. If further clarification is needed, the references should be consulted. Do not neglect illustrative material. To obtain CME: 1. Go to www.aanem.org/Marketplace. 2. Add specific Journal Review to cart. 3. Checkout
- Upon checkout an email will be sent directly to you with a CME survey link. - Click on the link; complete the survey; and print your transcript. - AANEM’s CME transcripts will update automatically.

INVITED REVIEW
PATHOPHYSIOLOGY OF IMMUNE-MEDIATED DEMYELINATINGNEUROPATHIES—PART I: NEUROSCIENCEHESSEL FRANSSEN, MD, PhD, and DIRK C.G. STRAVER, MD
Department of Neurology, Section Neuromuscular Disorders, F02.230, Rudolf Magnus Institute for Neuroscience, University MedicalCenter Utrecht, Heidelberglaan 100, 3584 CX Utrecht, The Netherlands
Accepted 26 August 2013
ABSTRACT: This first article of this review deals with neuro-scientific aspects of immune-mediated demyelinating neuropa-thies. It describes the anatomy and physiology of normalmyelinated axons, methods of studying peripheral nerve physi-ology, pathophysiological consequences of demyelination ordamage at the node of Ranvier, and the mechanisms that maylead to impaired axonal membrane dysfunction or axonaldegeneration. This article (part I) will be followed by a second(part II) dealing with clinical aspects of these neuropathies.
Muscle Nerve 48: 851–864, 2013
Immune-mediated demyelinating neuropathiesinvolve the myelin sheath, the node of Ranvier,the molecules that connect the Schwann cell mem-brane to the axolemma, and the axon itself. Theseneuropathies include Guillain–Barr�e syndrome(GBS), chronic inflammatory demyelinating poly-neuropathy (CIDP), multifocal motor neuropathy(MMN), anti–myelin-associated glycoprotein (MAG)neuropathy, and the syndrome of polyneuropathy,organomegaly, endocrinopathy, M-protein, and skinchanges (POEMS syndrome). The GBS subtypesacute inflammatory demyelinating polyneuropathy(AIDP) and acute motor axonal neuropathy(AMAN) will both be discussed. The emphasis inthis article will be on the physiological consequen-ces of demyelination or damage at the node ofRanvier for impulse transmission and associatedmechanisms that may lead to axonal membrane dys-function. For proper understanding of these mecha-nisms the anatomy and physiology of the normalmyelinated axon and studies of experimental demy-
elination will be discussed first. Ion channels arenamed by the channel name (not the gene name)as given in the International Union of Pharmacol-ogy (IUPHAR) Compendium of Voltage-Gated IonChannels.1
THE NORMAL MYELINATED AXON
Axon Geometry and Axolemmal Proteins. A myelin-ated axon consists of a series of successive node–internode configurations. At an internode (thepart of the axon between 2 adjacent nodes), theaxon is surrounded by a myelinating Schwann cell,whereas no myelin sheath is present at the node.The internode between 2 successive nodes can besubdivided into paranode, juxtaparanode, standardinternode, juxtaparanode, and paranode (Fig. 1).The myelinated axon is not a uniform tube,because the diameter and surface area of the nodeare much smaller than those of the internode. Thenode occupies only 0.1% of the total surface areaof a node and internode together. The juxtapara-node has a very large surface area, because it hasnot only the widest diameter but its surface isfluted as well.2 These differences in geometry arerelevant for rapid impulse propagation.
The node has a high density of voltage-gatedNa1 channels (1000–2000/lm2) and slow K1
channels (Fig. 1) (see review by Ritchie3). The jux-taparanode contains a high density of voltage-gated fast K1 channels. The standard internodehas the largest absolute number of Na1 channels,fast K1 channels, and slow K1 channels, but theirdensity is relatively low. The internodal Na1 chan-nel density of <25/lm2 is therefore not sufficientto sustain action potential propagation. The loca-tion of Na1/K1 pumps is controversial. Early stud-ies favored a nodal location, but advancedelectrophysiological and immunostaining techni-ques suggest localization in the internodal ratherthan the nodal membrane.4,5 The internodal axo-lemma also contains several types of glutamatereceptors, which employ signaling by second mes-sengers or Ca11 ions to activate ryanodine or IP3receptors on the axoplasmic reticulum; activationof these receptors releases Ca11 into the axon.Although the physiological role of these functional
Abbreviations: AIDP, acute inflammatory demyelinating polyneuropathy;AMAN, acute motor axonal neuropathy; CAP, compound action potential;Caspr-2, contactin-associated protein-2; ChAT, cholinacetyltransferase;CIDP, chronic inflammatory demyelinating polyneuropathy; CMAP, com-pound muscle action potential; Cx32, connexin-32; EAN, experimentalallergic neuritis; GBS, Guillain–Barr�e syndrome; HCN, hyperpolarization-activated cyclic nucleotide-gated; IUPHAR, International Union of Pharma-cology; MAC, membrane attack complex; MAG, myelin-associated glyco-protein; MBP, myelin basic protein; MMN, multifocal motor neuropathy;NCS, nerve conduction studies; NO, nitric oxide; P0, protein zero;PMP22, peripheral myelin protein-22; POEMS, polyneuropathy, organome-galy, endocrinopathy, M-protein, skin changes; SDTC, strength–durationtime constant; TAG-1, transient axonal glycoprotein-13Key words: demyelination; excitability; immune-mediated neuropathy;ion channels; safety factorThis study was supported by a grant from the Prinses Beatrix Spierfonds(to D.C.G.S.).Correspondence to: H. Franssen; e-mail: [email protected]
VC 2013 Wiley Periodicals, Inc.Published online 4 September 2013 in Wiley Online Library(wileyonlinelibrary.com). DOI 10.1002/mus.24070
Demyelinating Neuropathies MUSCLE & NERVE December 2013 851

nanocomplexes is unknown, they may be involvedin intra-axonal Ca11 homeostasis.6
The nodal Na1 channels are anchored to spec-trin of the axonal cytoskeleton via ankyrin-G andto gliomedin of the Schwann cell microvilli via Nr-CAM and neurofascin-186.7,8 The juxtaparanodalK1 channels are anchored by contactin-associatedprotein-2 (Caspr-2) and transient axonal glycopro-tein 1 (TAG-1) according to a similar principle.TAG-1 is an adhesion molecule that is expressedon juxtaparanodal axolemma and apposingSchwann cell membrane. The interaction betweenTAG-1 on the axolemma with Caspr and TAG-1 onthe Schwann cell membrane is crucial for cluster-ing of juxtaparanodal Kv1.1 and 1.2 channels.9 Atthe paranode, axonal Caspr and contactin are con-nected to neurofascin-155 of paranodal Schwanncell loops; this complex forms septate-like junc-tions, which separate the nodal Na1 channel clus-ters from the juxtaparanodal K1 channel clusters.Gangliosides are also essential for the integrity ofthese junctions and ion channels, because, in GM1and GD1a knockout mice, paranodal loops do notattach to the axolemma, Na1 channels are dis-rupted, and K1 channels are mislocated to theparanode.10
Myelin Sheath Geometry and Schwann Cell
Molecules. The myelin sheath between 2 adjacentnodes is formed by a Schwann cell that, during
development, has rotated tens of times around theaxon. At regions of compact myelin the opposingmembranes of 1 Schwann cell are closely apposed.At regions of non-compact myelin the membranesare not apposed, resulting in the appearance ofcytoplasm. Non-compact myelin occurs around thenucleus in paranodal loops and at Schmidt–Lanter-man incisures. If a myelinating Schwann cell couldbe unrolled, it would form a flat trapezoid cellwith an apical edge of 1–2 mm that is attached tothe axon between 2 successive nodes and a parallellarger basal edge. Between these edges run broadstrands of compact myelin that are separated bysmaller strands of non-compact myelin. Whenrolled up, the lateral borders of the trapezoidform the paranodal Schwann cell loops that areattached firmly to the paranodal or myelin attach-ment section of the axolemma. Compact myelincontains the transmembrane molecules proteinzero (P0), peripheral myelin protein-22 (PMP22),and the intracellular myelin basic protein (MBP).Non-compact myelin contains a limited amount ofgangliosides in the membrane of paranodal loopsand the transmembrane molecules MAG, E-cadherin, and connexin-32 (Cx32) (reviewed byScherer and Arroyo7 and Willison11). P0, MAG, E-cadherin, and Cx32 play a role in adhesionbetween myelin lamellae, but the function ofPMP22 is unknown. The Cx32 molecules of adja-cent myelin layers form gap junctions and, thereby,a radial pathway through the myelin lamellae,which allows passage of molecules <1000 Da,including ions and second messengers.12 The lowelectrical resistance of this pathway may contributeto termination of the action potential (see nextsection).
The Schwann cell membrane expresses severaltypes of ion channels, including different types ofK1 channels which may pass outward or inward K1
currents (Fig. 1).13 These K1 channels are foundon the outer Schwann cell surface and in theSchwann cell microvilli in the myelin attachmentsection close to the fast K1 channels in the juxta-paranodal region of the axon. It is likely that theSchwann cell K1 channels maintain the K1 ionconcentration around the node and in the narrowperiaxonal space of the internode within certainlimits. This buffering is necessary, because, other-wise, prolonged firing (causing an increased flowof K1 ions out of the axon) would lead to extracel-lular accumulation of K1 ions, decreased potas-sium equilibrium potential (EK), and spontaneousgeneration of action potentials. It is likely thatNa1/K1-ATPase is also expressed on mature mye-lin sheath membranes, because Na1/K1-ATPase asubunits are part of the myelin proteome, ultracy-tochemistry showed ATPase activity that was most
FIGURE 1. Diagram of a myelinated axon. Top: subdivision into
sections with different diameters. Bottom: distribution of cur-
rents sustained by voltage-gated ion-channels which differ for
each section; Nat, transient Na1 current; Nap, persistent Na1
current; Ks, slow K1 current; Kf, fast K1 current; Ih,
hyperpolarization-activated cation current; the Na1/K1 pump
removes 3 Na1 ions from the axon in exchange for 2 K1 ions;
BB, Barrett and Barrett resistance, which is a low-resistance
pathway through the myelin sheath. [Color figure can be viewed
in the online issue, which is available at wileyonlinelibrary.com.]
852 Demyelinating Neuropathies MUSCLE & NERVE December 2013

prominent in paranodal loops, and immunohisto-chemistry shows Na1/K1-ATPase in isolated myelinfractions (reviewed by Stys et al.14).
Electrical Properties of the Axon and Myelin
Sheath. Experimental studies have shown thatlarge-diameter fibers have greater myelin thickness,axon diameter, and internodal distance. Simula-tions predict that an increase in each of theseparameters independently decreases internodalconduction time.15 This was proven experimentallyin periaxin-null mice which have normal myelinthickness, decreased internodal distance, andslower internodal conduction.16
Current is defined here as a net flow of positivecharges, although in axons it is actually carried bypositive and negative ions. Current through Na1
and K1 channels is carried by positively chargedions, but longitudinal current inside the axon orthrough the myelin sheath is carried by positivepotassium ions, negative chloride ions, and, to alesser extent, by positive sodium ions.17 Currentsin axon physiology have many different labels, butare primarily subdivided into ionic and capacitive.Ionic currents pass through structures with a con-ductance, such as ion channels. For instance, aninward ionic current causes positive charges topass through ion channels into the axon, yieldinga positive shift in membrane potential. Capacitivecurrents are defined as “passing through” the bili-pid axon membrane. Because the membrane isimpenetrable, however, ions do not actually passthe membrane, but accumulate on 1 side of it andleave it on the other side. For instance, an outwardcapacitive current causes positive charges to accu-mulate at the inside of the axon membrane anddisappear at its outside, resulting in a positive shiftin membrane potential. Current is also subdividedinto action current (the inward ionic current consist-ing of Na1 ions passing through voltage gated Na1
channels during the action potential) and drivingcurrent (the outward capacitive current that depo-larizes the node of Ranvier prior to an actionpotential).
The resting membrane potential is determinedlargely by the equilibrium potential for K1 ions(EK) and to a lesser extent by the Na1/K1 pumpand the equilibrium potential for sodium ions(ENa). It is unknown whether the 2-pore K1 leak-age channels, TREK and TRAAK, which werefound to be transported in peripheral axons,determine resting membrane potential in myelin-ated axons.18 EK arises because [Ki] is higher than[Ko] and because many K1 channels are still openat resting membrane potential. Due to the chemicalforce, K1 ions flow outward along their concentra-tion gradient to produce a positively charged layer
on the outside of the axolemma. This layer gener-ates an opposite electrostatic force that repels positiveK1 ions back into the axon. At EK, these oppositeforces are in equilibrium, so that there is no netflow of K1 ions. EK is slightly more negative thanresting membrane potential. For Na1 ions, boththe chemical and electrical forces are inward:[Na1
o] exceeds [Na1i], and the negative resting
membrane potential attracts the positive sodiumions. Therefore, ENa has a positive value. TheNa1/K1 pump contributes to the negative mem-brane potential because it is electrogenic; at eachcycle, it expels 3 Na1 ions for every 2 K1 ionsbrought into the axon, thereby generating a netoutward ionic current that removes positivecharges from the inside of the axon.19 In humanaxons, the resting membrane potential is 275 to280 mV.20
The classical model of myelin function is basedon early measurements showing that the sheathhas a high resistance and low capacitance, so thatit should be impenetrable for current and unableto store electrical charges.21 This model entailssome unrealistic assumptions, because it requiresthat both the nodal and internodal resting mem-brane potential be determined solely by the tinynode. It also requires a considerable outwardnodal leakage current that has to compensate forthe sodium influx during the action potential,although specific leakage channels have neverbeen discovered. Furthermore, it cannot explainthe slow potential changes associated with thedepolarizing afterpotential and threshold electroto-nus (see Excitability Studies subsection). Investiga-tions into the mechanisms of the depolarizingafterpotential showed that current may passthrough relatively low resistance pathways of themyelin sheath, such as the Schmidt–Lantermanincisures and gap junctions between myelin lamel-lae.12,22 These findings were incorporated into newmodels in which the myelin sheath still has a lowcapacitance but a low, instead of high, resistance,and in which internodal ion channels (mainly K1
channels) are important in the setting of interno-dal and nodal resting membrane potential. Theaforementioned slow potential changes could nowbe explained by currents through the low-resistance myelin pathways that slowly change thevoltage across the large capacitance of the interno-dal axolemma.
The fact that the surface of the node is muchsmaller than that of the internode is highly rele-vant for fast impulse propagation, as it entails thatthe capacitance (i.e., the ability to store electriccharges) of the nodal membrane is considerablysmaller than the capacitance of the internodalmembrane. Thus, when a capacitive current passes
Demyelinating Neuropathies MUSCLE & NERVE December 2013 853

through the node, its tiny capacitance is chargedrapidly so that a membrane potential difference isreached quickly. On the contrary, it takes moretime for a capacitive current to reach a membranepotential difference across the large capacitance ofthe internode.
Depolarization is a shift in membrane potentialtoward values that are more positive than restingmembrane potential. Hyperpolarization is a shifttoward values that are more negative, and repolari-zation is a shift from depolarization or hyperpolar-ization toward resting membrane potential. Over awide range of membrane potential values, Na1
channel opening yields an inward ionic currentthat depolarizes membrane potential, whereas K1
channel opening yields an outward ionic currentthat repolarizes or hyperpolarizes membranepotential. Nodal Na1 channels generate transientand persistent currents, each of which is probablysustained by a different ortholog of the Nav1.6channel subtype.23,24 Transient Na1 channels areresponsible for the action potential, account for98% of Na1 currents, are closed at resting mem-brane potential, open for 1 to a few millisecondsduring depolarization, and inactivate thereafter.Recovery from this fast type of inactivation occursover tens of milliseconds by repolarization orhyperpolarization. Apart from this classical fastinactivation, Na1 channels in axons may alsoexhibit slow inactivation, the recovery of whichtakes tens of seconds.25,26 Persistent Na1 channelsaccount for 2% of sodium currents; they activate atmore negative membrane potentials and hardlyinactivate, causing a small persistent inward cur-rent at resting membrane potential that increasesexcitability. In myelinated mammalian axons, 3 K1
currents can be recorded, including 2 fast types(IKf1 and IKf2) and 1 slow type (IKs) (reviewed byReid et al.27). Each of these currents is activated bydepolarization. Patch clamping of human axonmembranes showed that the K1 currents are gen-erated by at least 5 different K1 channel types withdifferent but overlapping properties regarding con-ductance, membrane potential for activation, deac-tivation time, and inactivation time.27 Contrary toNa1 channels, inactivation of these K1 channels isslow (ranging from milliseconds to seconds) orabsent. IKf1 and IKf2 prevent the spread of excita-tion beyond the node after a single action poten-tial. Unlike in amphibian axons, they are notessential for membrane repolarization in mamma-lian axons.3 IKs activates slowly after an actionpotential, and during short-lasting repetitive firingits summated effect repolarizes membrane poten-tial so that extreme depolarization is prevented.IKf1 and IKs are important for determining restingmembrane potential and the type of accommoda-
tion observed in threshold electrotonus. Long-standing or extreme hyperpolarization activates alate inward current (Ih) that is generated byhyperpolarization-activated cyclic nucleotide-gatedchannels (HCN channels).28,29 Ih is carried by Na1
and K1 ions and drives membrane potential to lessnegative values, thereby preventing extremehyperpolarization.
Action Potential Propagation. At an active node, anaction potential is initiated by depolarization,which results in opening of transient Na1 channelsand influx of positively charged sodium ions(Fig. 2). This is a regenerative process, because theinflux depolarizes membrane potential and leadsto more Na1 channel openings, sodium influx,and more depolarization. The inward ionic currentat the active node forms part of a circuit thatdrops positive charges on the inside of the nodethat is next to be activated and draws positivecharges from the outside of this node, thereby cre-ating an outward capacitive current at the node-to-be-activated. This driving current is capacitive,because the Na1 channels at the node-to-be-activated are still closed. When this node is suffi-ciently depolarized by the driving current (thethreshold for an action potential is reached afterapproximately 20 ls of depolarization), its Na1
channels open, and the outward capacitive currentchanges into the inward ionic current of the actionpotential.30
The action potential is terminated by rapidNa1 inactivation, which stops the inward ionic cur-rent. Furthermore, the initial depolarization at thenode is reduced, because the inward current ofthe action potential not only returns through thenode to be activated but also through the interno-dal membrane and low-resistance pathwaysthrough the myelin sheath. This slow outwardcapacitive current smears the positive charges thathave entered during the action potential over thelarge internal surface of the internodal and, finally,the nodal membrane. The result is that both thenode and internode become slightly depolarized, aphenomenon known as the depolarizing afterpo-tential.22 The depolarizing afterpotential subsides,because its depolarization activates slow K1 chan-nels, which subsequently generate the hyperpola-rizing afterpotential.
PHYSIOLOGICAL METHODS
Experimental Methods. Electrophysiological meth-ods in animal studies that are comparable to nerveconduction studies (NCS) in humans includerecording of mixed compound action potentials(CAPs) of nerves that contain motor and sensoryaxons, pure motor or sensory CAPs of roots, andcompound muscle action potentials (CMAPs).
854 Demyelinating Neuropathies MUSCLE & NERVE December 2013

Several techniques have been developed tostudy electrical activity in single myelinated axons.These methods employ intra- or extracellular glasspipettes to record the membrane potential anddeliver electrical stimuli. In current clamping, a con-stant current stimulus changes the membranepotential to enable recording of subthresholdchanges in membrane potential (e.g., electroto-nus) or action potentials. In voltage clamping, themembrane potential is held at values set by theinvestigator, which enables recording of membranecurrent at different imposed membrane voltages.By selective pharmacological blockade of ion chan-nels, current and conductance of specific ion chan-nels can be determined. Macroscopic current orvoltage clamping has been employed to investigateevents at the node of an intact myelinated axon.Patch clamping employs extremely fine micropip-ettes with tips of about 1-lm diameter for currentor voltage clamping from an intact cell body, anexcised membrane patch containing many ionchannels, or a small membrane patch containingonly 1 ion channel.
Another approach is external longitudinal currentrecording, which records the extra-axonal part ofthe driving current loop by means of an electrodepair with a fixed distance of 70 lm placed at theoutside of a single axon in an intact nerve. Amicromanipulator allows recording from differentsites along the axon. When an action potential isevoked at a distant site and passes the recordingsite, the electrodes record the magnitude of the
external longitudinal current. Any difference inthis current between 2 recording sites reflects cur-rent that enters or leaves the axon. Comparing theexternal longitudinal current at a node and justbefore the node reveals a sudden decrease becauseof the strong inward current at the node duringan action potential. This method therefore allowsassessment of internodal conduction time andinternodal distance.
Some of the methods in single myelinatedaxons are considerably more difficult to performin mammalian and human fibers than in amphib-ian fibers, because the former are extremely fragileand are surrounded by large amounts of connec-tive tissue. Despite these difficulties, macroscopiccurrents in both normal human single myelinatedaxons and single mammalian axons, which weredemyelinated due to experimental allergic neuritisor toxic substances, have been characterized.31
This is of relevance because results in singlemyelinated amphibian or lower mammalian axonscannot always be extrapolated to human axonsdue to essential interspecies differences in ionchannel characteristics.
Excitability Studies. Excitability tests assess thejust-described passive and active properties at 1 siteof the axolemma. By adapting experimental proce-dures, these properties can now be recorded non-invasively and with only minor discomfort inhuman subjects.32 Conditioning and test stimuliare given at a point on a nerve. The conditioning
FIGURE 2. Saltatory action potential propagation in a normal myelinated axon. (A) At the active node (left), Na1 channels are opened,
inducing an inward ionic (action) current. This causes a current circuit that leads to an outward capacitive (driving) current at the node-
to-be-activated (middle). (B) Positive charges accumulate at the inside of the node-to-be-activated and are withdrawn at its outside;
this leads to depolarization of this node. (C) As soon as the node-to-be-activated is depolarized to threshold, its Na1 channels open,
and the outward capacitive current changes into an inward ionic (action) current. [Color figure can be viewed in the online issue, which
is available at wileyonlinelibrary.com.]
Demyelinating Neuropathies MUSCLE & NERVE December 2013 855
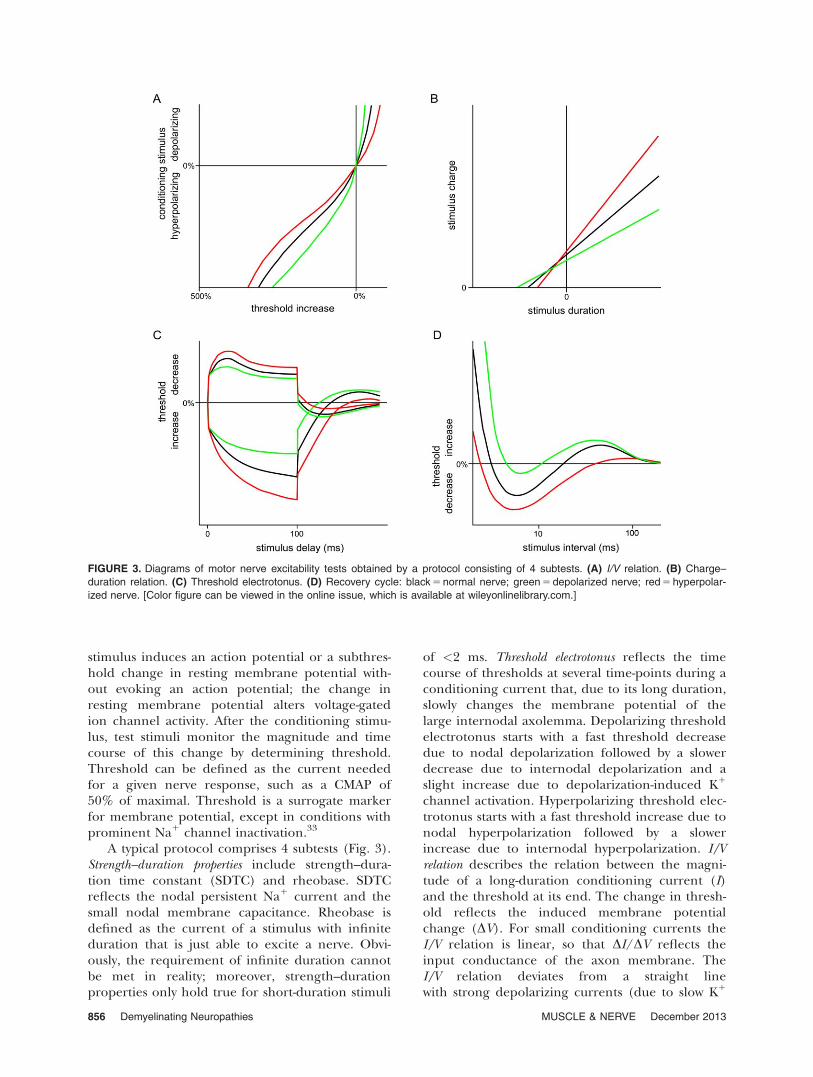
stimulus induces an action potential or a subthres-hold change in resting membrane potential with-out evoking an action potential; the change inresting membrane potential alters voltage-gatedion channel activity. After the conditioning stimu-lus, test stimuli monitor the magnitude and timecourse of this change by determining threshold.Threshold can be defined as the current neededfor a given nerve response, such as a CMAP of50% of maximal. Threshold is a surrogate markerfor membrane potential, except in conditions withprominent Na1 channel inactivation.33
A typical protocol comprises 4 subtests (Fig. 3).Strength–duration properties include strength–dura-tion time constant (SDTC) and rheobase. SDTCreflects the nodal persistent Na1 current and thesmall nodal membrane capacitance. Rheobase isdefined as the current of a stimulus with infiniteduration that is just able to excite a nerve. Obvi-ously, the requirement of infinite duration cannotbe met in reality; moreover, strength–durationproperties only hold true for short-duration stimuli
of <2 ms. Threshold electrotonus reflects the timecourse of thresholds at several time-points during aconditioning current that, due to its long duration,slowly changes the membrane potential of thelarge internodal axolemma. Depolarizing thresholdelectrotonus starts with a fast threshold decreasedue to nodal depolarization followed by a slowerdecrease due to internodal depolarization and aslight increase due to depolarization-induced K1
channel activation. Hyperpolarizing threshold elec-trotonus starts with a fast threshold increase due tonodal hyperpolarization followed by a slowerincrease due to internodal hyperpolarization. I/Vrelation describes the relation between the magni-tude of a long-duration conditioning current (I)and the threshold at its end. The change in thresh-old reflects the induced membrane potentialchange (DV). For small conditioning currents theI/V relation is linear, so that DI/DV reflects theinput conductance of the axon membrane. TheI/V relation deviates from a straight linewith strong depolarizing currents (due to slow K1
FIGURE 3. Diagrams of motor nerve excitability tests obtained by a protocol consisting of 4 subtests. (A) I/V relation. (B) Charge–
duration relation. (C) Threshold electrotonus. (D) Recovery cycle: black 5 normal nerve; green 5 depolarized nerve; red 5 hyperpolar-
ized nerve. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
856 Demyelinating Neuropathies MUSCLE & NERVE December 2013

channel activation) and with strong hyperpolariz-ing currents (due to HCN channel activation).Recovery cycle reflects the membrane potential fluc-tuations that follow an action potential. It isassessed by giving a short-duration supramaximalconditioning stimulus (evoking action potentials)and recording thresholds at different time intervalsafter the conditioning stimulus. It comprises suc-cessively: refractoriness (threshold increase due totransient Na1 channel inactivation); superexcitabil-ity (threshold decrease due to the depolarizingafterpotential); and subexcitability (thresholdincrease due to slow K1 channel activation).
The effects of a permanently changed restingmembrane potential due to nerve pathology weremimicked by applying a continuous DC currentduring the entire excitability protocol.34 Persistentdepolarization of resting membrane potential by aDC current resulted in several changes. First,SDTC increased due to increased persistentsodium current. Second, threshold electrotonusshowed fanning-in. This is because massive interno-dal K1 channel opening yields an increased inputconductance through the axon membrane and lessmembrane potential change when a constantdepolarizing or hyperpolarizing current is passed
through it. Third, I/V slope became steeper due tothe increased input conductance. Fourth, refracto-riness increased due to increased transient Na1
channel inactivation. Fifth, superexcitabilitydecreased because the decreased potential differ-ence over the axon membrane decreases Na1
influx during an action potential, thereby decreas-ing the depolarizing afterpotential. Persistenthyperpolarization of resting membrane potentialby a DC current–induced changes in the oppositedirection (Fig. 3).
EXPERIMENTAL DEMYELINATION
Safety Factor. The physiological consequences ofdemyelination of a single axon are slowing of inter-nodal conduction time, persistent conductionblock, increased refractory period, rate-dependentblock, and warm block.35 Each of these phenom-ena can be explained by a reduced safety factor oftransmission (Fig. 4). Safety factor is the ratio:(available driving current) / (required driving cur-rent) for excitation of a node.36 In normal axons,it has a value of about 5–7, which is more than suf-ficient to sustain conduction in unfavorable physio-logical conditions. In axons affected by disease,the safety factor may be reduced by a decrease in
FIGURE 4. Different types of demyelination leading to reduced safety factor. (A) Segmental demyelination. The driving current leaks
through the damaged myelin sheath and impairs depolarization of the node-to-be-activated. (B) Paranodal demyelination. The driving
current is dissipated over an area consisting of the node and the denuded paranode; the large capacitance of this area impairs depola-
rization of the former nodal area. (C) Juxtaparanodal demyelination. The driving current is dissipated over a larger area consisting of
the node, paranode, and juxtaparanode; the very large capacitance of this area impairs depolarization to a greater extent than in (B);
depolarization is additionally impaired by activation of exposed juxtaparanodal K1 channels. [Color figure can be viewed in the online
issue, which is available at wileyonlinelibrary.com.]
Demyelinating Neuropathies MUSCLE & NERVE December 2013 857

available driving current, an increase in requireddriving current, or both. One of the factors con-tributing to the safety factor is the high density ofvoltage-gated Na1 channels at the node. If thesafety factor is reduced but above unity, conduc-tion is possible, albeit with slowed internodal con-duction time. If it falls below unity, internodalconduction is blocked. At critically demyelinatedinternodes with a safety factor of just above 1, con-duction slowing may change into conduction blockif physiological factors cause an additional reduc-tion in safety factor. This mechanism underliesrate-dependent block, warm block, and cold block,
which is discussed in part II of this review.37
Causes of reduced safety factor are demyelination,nodal Na1 channel damage or dysfunction, depo-larization, and hyperpolarization (Figs. 4 and 5).
Paranodal and Segmental Demyelination. Experimentaldemyelination has been induced by chemical sub-stances such as lysolecithin, immunization withdiphtheria toxin, injection with specific myelinpeptides, or injection with peripheral nerve myelinhomogenate, which yields the most prominentdemyelination.38 The latter 2 models are known asexperimental allergic neuritis (EAN). Pathologicalstudies in EAN show a T-cell–mediated response toP2, P0, or PMP22, with activated macrophagesinvading the myelin sheath and inducing demye-lination. EAN is regarded by some investigators asa model for AIDP, because both disorders sharesimilar pathological features.39
Segmental demyelination leads to a decreaseddriving current at the node-to-be-activated, becausepart of the driving current flows outward acrossthe damaged myelin sheath (Fig. 4A). Comparisonof CMAPs with histopathology findings in an EANmodel with predictable time course showed thatprogressive disappearance of myelin layers wasassociated with progressive conduction slowinguntil conduction became blocked.40,41 Computersimulations of segmental demyelination showedthat conduction is just possible if myelin thicknessis 2.7% of normal at 1 internode, or 4% of normalat 2 adjacent internodes.42 In the acute phase ofcomplete segmental demyelination, conduction isblocked in most axons, because the Na1 channeldensity of the denuded internodal membrane isnot sufficient to sustain continuous conduction.Only in very small demyelinated axons may the lowinternodal Na1 channel density be sufficient tosupport continuous conduction (reviewed by Wax-man et al.43). Computer simulations have shownthat the increased internodal Na1 channel densityin demyelinated regions is not sufficient to sustainconduction if membrane capacity at the denudedpart of the axon is increased too much, especiallywhere membrane capacity between myelinated anddemyelinated regions suddenly changes; this isknown as impedance mismatch.43
Paranodal demyelination also leads to dissipa-tion of the driving current over an area comprisingthe former nodal area and the demyelinated partof the paranodal axolemma (Fig. 4B). It thus takesmore time for the driving current to depolarizethe increased capacity of this enlarged area to thethreshold for an action potential; as a result, inter-nodal conduction time increases from its normalvalue of 20 ls to as much as 600 ls.30 If paranodaldemyelination becomes more extensive, driving
FIGURE 5. Nodal causes of reduced safety factor. (A) Damage
to or dysfunction of Na1 channels causing impaired inward ionic
(action) current. (B) Inactivation of Na1 channels in a perma-
nently depolarized axon causing impaired inward ionic current.
(C) increased membrane potential difference in a permanently
hyperpolarized axon; more driving current is needed to reach
threshold for activation of Na1 channels at the node-to-be-acti-
vated. [Color figure can be viewed in the online issue, which is
available at wileyonlinelibrary.com.]
858 Demyelinating Neuropathies MUSCLE & NERVE December 2013

current is insufficient to depolarize the enlargednode to threshold, so that action potential propa-gation is blocked. If demyelination also involvesthe juxtaparanode, fast K1 channels in that regionwill be exposed or dispersed to the node (Figs. 1and 4C).38,44 Activation of these channels by thedriving current may then shorten the duration ofthe action potential or prevent reaching thethreshold for an action potential so that conduc-tion is blocked. Consistent with this, inhibition offast K1 channels by application of 4-aminopyridineon single demyelinated mammalian motor axonsresolved conduction block or increased the dura-tion of action potentials in intra- or extra-axonalrecordings.38,45 Unfortunately, neither resolutionof conduction block on NCS nor clinical improve-ment were observed in human demyelinating neu-ropathies after administration of 4-aminopyridine.46
Demyelination may affect refractoriness. Therefractory period is caused mainly by transientnodal Na1 channel inactivation after an actionpotential. In the absolute refractory period, notenough non-inactivated Na1 channels are availableto generate an action potential. In the relativerefractory period, an action potential may be gen-erated, but the action and driving currents aresmaller than normal because many Na1 channelsare still inactivated. With demyelination, the abso-lute refractory period may be prolonged, becausemore driving current is needed to depolarize thedriven node due to current leakage and increasednodal capacity. The refractory period was investi-gated in demyelinated single axons by externallongitudinal current recording.30 Progressivelydecreasing the interval between 2 closely spacedstimuli resulted in an increased internodal conduc-tion time for the second stimulus until conductionbecame blocked; the interstimulus interval atwhich this occurred was defined as the refractoryperiod. This was increased from a normal value ofabout 1.0 ms to values ranging from 1.8 to 4.3 msin demyelinated axons.
In single axons that were slightly demyelinateddue to EAN, decreased nodal peak sodium cur-rents are associated with increased membranecapacitance, suggesting dispersion of Na1 channelsafter myelin loosening (Fig. 4).44 Another study inEAN showed that, prior to the appearance of clini-cal deficits and demyelination, immunostaining ofnodal Na1 channel clusters in spinal roots becameweaker, and Na1 channel labeling became diffusedinto the paranodal membrane; whole nerve record-ings showed signs of conduction block, warmblock, and slowing.47 Thereafter Na1 channelstaining became undetectable, indicating that <50channels/lm2 were present; this was associatedwith severe clinical signs and myelin damage vary-
ing from paranodal to total segmental demyelin-ation. Computer simulations could explain theelectrophysiological findings by diffusion of Na1
channel clusters combined with loss of paranodalseals between axon and Schwann cell.47
In EAN induced by whole peripheral nervemyelin, the density of the nodal adhesion mole-cules gliomedin and neurofascin became dimin-ished, and autoantibodies to these molecules werefound in the earliest stage.38 This was followed byparanodal retraction leading to widened nodes,demyelination, disruption of nodal Na1 channeland Kv7.2 channel clusters, and mislocalization ofjuxtaparanodal Kv1.2 channels to the paranodeand node (Figs. 4 and 5). Nodal complement dep-ositions were not detected. Clinical signs startedwhen the adhesion molecules disappeared, andtheir severity correlated with the number of dis-rupted nodes. Whole nerve recordings showedconduction block, slowing, and an increasedrefractory period. Signs of conduction block dimin-ished after application of the Kv1 channel blocker4-aminopyridine, but not after a Kv7.2 channelblocker, suggesting that exposure of juxtaparano-dal Kv1 channels contributed to the block. Thus,the nodal abnormalities in EAN induced byperipheral nerve myelin may cause a conductiondeficit on top of that caused by paranodal demye-lination, because the action current is impaired bydisruption of Na1 channel clusters. The remainingaction current is counteracted by Kv1 channelsthat are mislocalized at the node.
Restoration of Conduction. Restoration of conduc-tion after demyelination may occur by diffuseexpression of Na1 channels on the denuded inter-node, formation of new Na1 channel clusters, andremyelination.
External longitudinal current recording hasshown continuous conduction along totally demye-linated axons 4–6 days after demyelination bydiphtheria toxin, suggesting that conduction wasrestored by a diffuse increase in Na1 channel den-sity on the internodal membrane (Fig. 5).48 Inter-nodal foci of increased inward current were alsoobserved, suggesting clustering of Na1 channels.These results were supported by the finding of anincreased density of particles on freeze fractureelectron microscopy (likely representing Na1 chan-nels) and increased Na1 channel immunoreactivityon demyelinated internodal membranes.43 Theincreased expression of voltage-gated Na1 chan-nels along denuded internodes may, however, beunfavorable for axon survival (see Axonal Degener-ation subsection).
After chemical demyelination, external longitu-dinal current recordings show foci of inward
Demyelinating Neuropathies MUSCLE & NERVE December 2013 859

current (phi-nodes) separated by 100–200 lm.Because this is the shortened internodal distanceafter remyelination, phi-nodes may represent smallNa1 channel clusters out of which nodes willdevelop later.49 During recovery in EAN, broadNa1 channel aggregates appear adjacent to theedges of remyelinating Schwann cells (Fig. 5).47
When these move laterally, the Na1 channel clus-ters fuse to form new nodes with shortened inter-nodal distance; the latter is associated with clinicalrecovery.
Restoration of CMAPs evoked proximal to thelesion was associated with appearance of smallnumbers of axons encircled with 2–8 turns of mye-lin lamellae (8–20% of normal).40 When myelinthickness was one-third of normal, maximal con-duction velocity became normal, which indicatesthat even moderate demyelination may go unde-tected by NCS.
Effects of Anti-Ganglioside Antibodies. The findingof antibodies against gangliosides in severalimmune-mediated neuropathies promptedresearch into pathological mechanisms affectingthe complicated fine structures at and around thenode where gangliosides are mainly located. Gan-gliosides are glycolipids consisting of a lipid cer-amide component located in the bilipidmembrane and an extracellular sugar portion towhich 0, 1, or more sialic acid residues may beattached. High-resolution light and electronmicroscopy of teased fibers from rat spinal rootsand sciatic nerves has shown gangliosides on thefollowing structures: GM1 on nodal axolemma,paranodal axolemma, abaxonal Schwann cell mem-brane, and Schwann cell microvilli; GT1b on nodalaxolemma, internodal axolemma, and abaxonalSchwann cell membrane; and GD1a on nodes andabaxonal Schwann cell membrane.50,51 Theamount of gangliosides GM1, GD1a, and GD1b inhuman roots was greater in axons than in myelinin 1 study,52 but greater in myelin in anotherstudy.53
Early animal studies suggested that anti-GM1antibodies may bind to Na1 channels, resulting inblocking of nodal Na1 currents. Injection of anti-GM1 serum from a patient with MMN into rat sci-atic nerve induced conduction block and increasedtemporal dispersion on motor NCS and nodalimmunoglobulin deposits on immunopathologicalstudies; however, demyelination was observed inonly 6.5% of axons.54 Because the amount ofCMAP reduction on proximal versus distal stimula-tion was considerable and the percentage ofdemyelinated axons small, the block was possiblynot caused by demyelination but by binding ofGM1 antibodies to nodes of Ranvier. Voltage
clamping of rat myelinated single axons showedthat application of high-concentration anti-GM1antisera without complement had little effect onNa1 currents but increased delayed fast outwardK1 currents elicited by depolarization. Anti-GM1antibodies plus active complement, however,caused both suppression of Na1 current andincrease of K1 and non-specific leakage currents.55
The effects on K1 currents were consistent withexposure of K1 channels due to juxtaparanodaldemyelination. In neuron-like cells, application ofIgG anti-GM1 raised in rabbits caused a reversible,substantial voltage-independent reduction in Na1
currents, but only in high concentrations and inthe presence of complement.56 Complementalone, or complement and anti-GM2 or anti-GM4,had no effect. It was suggested that both GM1 andthe Na1 channel protein contain sialic acid resi-dues so that anti-GM1 with complement mayimpair nodal Na1 channel function, leading toblocking of impulse propagation.57
Later studies, however, did not support the pos-sibility of immune attacks on the Na1 channel pro-tein itself. External longitudinal current recordingsalong single motor axons of intact rat ventral rootsincubated with high-titer anti-GM1-sera obtainedfrom patients with AMAN or MMN showed block-ing in only 2% of fibers. In these fibers the currentpatterns at the site of block resemble those of atypical demyelinative block, characterized byintense passive outward currents; the current pat-terns did not resemble those induced by the Na1
channel blocker tetrodotoxin.58 Also, applicationof sera from 10 patients with AIDP to rat sciaticnerve induced conduction block in only 2 of the10.59 Up to 6 hours of incubation of desheathedsciatic nerve with anti-GM1 (IgM or IgG) or antiGQ1b (IgG) and complement resulted in nodalantibody depositions, but not in conduction blockon whole nerve recordings.60 Finally, the GM1 moi-ety Gal(b1–3)GalNAc was found on human axo-lemma, but was not obligatory colocalized withvoltage-gated Na1 or K1 channels.50 These find-ings suggest that anti-GM1 in the presence of com-plement may cause demyelination or secondaryreduction in Na1 currents by damage to the axo-lemma in which the Na1 channels are embedded,but it is unlikely to be directed against the Na1
channel protein itself.Damage to nodal structures of axons in roots
was observed in rabbits where AMAN was inducedby subcutaneous injection of GM1 ganglioside.61
The acute phase showed disruption of several mol-ecules, including nodal Na1 channel clusters, bIVspectrin in the axonal cytoskeleton, and axoglialjunction molecules involved in stabilizing ion-channel clusters, such as moesin in Schwann cell
860 Demyelinating Neuropathies MUSCLE & NERVE December 2013

microvilli and Caspr at paranodes. The severity ofthese abnormalities was associated with extensionof membrane attack complex and complementdeposits at the paranode. These features occurredmore frequently in nodes of motor rather thansensory roots.62 Recovery was associated with reap-pearance of Na1 channel clusters on each side ofthe former nodal area. These binary clusters fusedduring subsequent recovery (Fig. 5). Because axo-nal degeneration was mild, it is conceivable thatthe clinical signs were caused by conduction blockarising from Na1 channel disruption and associ-ated reduced nodal Na1 current density. Contraryto EAN, nodal disruption in AMAN rabbits wascomplement-dependent, as intravenous administra-tion of a complement inhibitor decreased thenumber of nodes with Na1 channel disruptionand complement deposits.63
Injection of IgG anti-GD1a or GT1b antibodiesinto rat sciatic nerve induced nodal IgG deposits,nodal complement deposits, and disruption ofnodal or paranodal structures.62 Cholinacetyltrans-ferase (ChAT) staining, which is used to distin-guish motor from sensory axons, showed that eachof these features occurred more often in motorthan in sensory nodes. The disruption was charac-terized by damage to nodal bIV spectrin, wideningof the nodal gap, or disappearance of paranodalCaspr clusters. Serial motor conduction studies ofthe sciatic nerve revealed a temporary decrease inthe amplitude of the proximally evoked CMAPwith preserved distally evoked CMAPs. Because thiswas not accompanied by conduction slowing ortemporal dispersion, it may have reflected conduc-tion block due to nodal dysfunction and not demy-elination. On the other hand, injection of IgGanti-GD1b antibodies caused nodal disruption, andcomplement deposits that were found more oftenin sensory than in motor nodes. Furthermore, inan acute sensory axonal neuropathy-like rabbitmodel induced by immunization with GD1b, nodesand paranodes showed disruption of Caspr andNa1 channel clusters together with membraneattack complex (MAC) deposits, and the disrup-tion was more frequent in dorsal than ventralroots.62 These findings indicate that: (i) nodal andparanodal disruption is a common mechanism inanimal models of axonal GBS subtypes; (ii) anti-bodies to GM1, GD1a, and GT1b affect motoraxons predominantly and sensory axons to a lesserdegree; and (iii) antibodies to GD1b affect sensoryaxons predominantly and motor axons to a lesserdegree.
The most distal part of axon branches at theneuromuscular synapse can also be affected byanti-ganglioside antibodies.64 Exposure of neuro-muscular synapses in ex vivo mouse diaphragms to
anti-GQ1b antibodies showed antibody binding toneuromuscular synapses and profound electrophys-iological effects consisting of a considerable rise inminiature endplate potential frequency duringtens of minutes and blocking of neuromusculartransmission. These effects were complement-dependent, and the blocking was probably theresult of structural damage to axon terminals dueto MAC formation and Ca11-mediated degenera-tion. Subsequent studies showed that antibodies togangliosides GD3, GD1a, GM1, and GD1b elicitedsimilar or partly similar effects, suggesting thatsome symptoms in AMAN may be caused by neuro-muscular synapse pathology. Antibodies to GQ1b,GM1, and GD1a also inhibited evoked acetylcho-line release that was not complement-dependent.65
Whether or not anti-ganglioside antibodies arepathogenic was shown to be dependent on accessi-bility and density of the antigenic gangliosides andon endocytic processing of antibodies.64,66
AxonalDegenerationinDemyelinatingNeuropathies. Axonaldegeneration in demyelinating neuropathies maycause permanent and severe clinical deficits.Unfortunately, its mechanisms in peripheral neuro-pathies are not well understood.14 Nevertheless,animal studies have revealed several putativemechanisms.
Direct injury of the nodal axolemma with sub-sequent axonal degeneration was suggested by thefinding of nodal IgG deposition, complement acti-vation, and MAC in AMAN rabbits and nodal IgGand complement deposits in fatal humanAMAN.61,67,68 MAC causes 5-nm-wide pores in theaxolemma that allow water and ions, includingmassive amounts of Ca11, to enter the axon. HighCa11 concentrations activate the calpain pathway,causing proteolytic cleavage of neurofilaments andthe intra-axonally located inactivation-gate of Na1
channels so that inactivation fails, although activa-tion is still possible.69 Ex vivo incubation of mouseintramuscular desheathed distal myelinated motoraxon bundles with anti-GD1a antibodies and com-plement resulted in nodal complement deposits,nodal MAC formation, loss of nodal Nav1.6,ankyrin-G, and neurofascin, and loss of paranodalCaspr; juxtaparanodal Kv1.1 staining remainedunaffected.70 Loss of Nav staining started between15 and 30 minutes after addition of complement.Perineural electrophysiological recording (a vari-ant of external longitudinal current recording)showed loss of inward Na1 currents and outwardK1 currents. The pathological abnormalities wereprevented by adding complement or calpain inhib-itors. Therefore, the damage to nodal proteins waslikely caused by a sequence of antibodies, comple-ment activation, MAC formation, Ca11 entry, and
Demyelinating Neuropathies MUSCLE & NERVE December 2013 861

Ca11-mediated Na1 channel damage. Electrophysi-ological dysfunction was prevented by complementinhibition but not by calpain inhibition. Despiteprevention of Na1 channel damage by calpaininhibitors, MAC formation probably still occurredand caused loss of ionic concentration gradientsand inability to generate action potentials.
Inflammatory cells in demyelinating lesionsproduce nitric oxide (NO), a free radical andinflammatory mediator that may induce energydepletion of Na1/K1-ATPase by inhibiting mito-chondrial respiration. In vitro studies have shownthat, when rat dorsal root axons were exposed toNO and stimulated at 100 HZ, CAPs on wholenerve recording permanently disappeared, evenafter removal of NO.71 CAP disappearance was pre-vented by adding Na1 channel blockers to themedium. Pathological examination showed thatadministration of NO in vivo caused axonal degen-eration that could be prevented by adding Na1
channel blockers or Na1/Ca11 exchanger block-ers. These results suggest that Na1 influx (inducedby repetitive firing and permanent depolarizationdue to Na1/K1-ATPase failure) and activity of theNa1/Ca11 exchanger are key factors in the devel-opment of axonal degeneration. The continuedNa1 influx despite long-standing depolarizationsuggests opening of persistent Na1 channels,which, contrary to transient Na1 channels, do notinactivate during long-standing depolarization.Therefore, NO-induced axonal degeneration mayinitiate a cascade, which has been observed inanoxia of the rat optic nerve. This cascade startswith Na1/K1-ATPase failure due to energy deple-tion. Pump failure results in axoplasmic Na1 accu-mulation and depolarization. Depolarizationactivates nodal persistent Na1 channels, yieldingadditional Na1 accumulation and depolarization.Furthermore, the Na1 accumulation inducesreverse operation of the axolemmal Na1/Ca11
exchanger so that the excess Na1 is removed inexchange for Ca11. The resulting axoplasmicCa11 accumulation will induce Ca11-mediatedaxonal degeneration.72
Ca11-mediated axonal degeneration may alsobe the result of Ca11 release from intra-axonalstores in the axoplasmic reticulum or mitochon-dria.6 Activation of axolemmal glutamate receptorsinduces opening of ryanodine or IP3 receptors onthe axoplasmic reticulum by several mechanismsso that Ca11 ions are released into the axon.Ca11 release from mitochondria may be inducedby intra-axonal Na1 accumulation and reversal ofthe mitochondrial Na1/Ca11 exchanger.
Axonal survival may be at risk if remyelinationdoes not occur and impulse propagation isrestored by increased expression of Na1 channels
along the entire denuded part of the axon.40,73
The resulting continuous conduction may result inabnormally large intra-axonal Na1 accumulationand Ca11-mediated axonal degeneration asdescribed earlier.
Increased packing density of axonal neurofila-ments, as found in anti-MAG neuropathy, mayresult in impaired axonal transport leading to axo-nal degeneration, which is further described in thesecond article of this review.37 MyelinatingSchwann cells were shown to transfer polyribo-somes with mRNA to the underlying part of theaxon.74 This raises the possibility that at least someaxonal proteins are synthesized locally instead ofbeing transported from the soma by axonal trans-port. Impairment of this mechanism due to demye-lination may therefore contribute to axonaldegeneration.
Altered Expression of Na1 Channel Subtypes. Changesin expression of ion channel subtypes in demyeli-nated axons have been found in animal studiesand pathological studies in multiple sclerosispatients and are likely to contribute to axonal dys-function and axonal degeneration in immune-mediated neuropathies as well. In axons that weredenuded due to complete demyelination, Na1
channel expression was upregulated, resulting infacilitation of continuous conduction. Upregula-tion of the normal subtype Nav1.6 was associatedwith axonal degeneration, but that of subtypeNav1.2 was not.75 This is because, contrary toNav1.2, Nav1.6 produces a large persistent Na1
influx that may induce Ca11-mediated axonaldegeneration. In another animal study, upregula-tion of Nav1.3 and Nav1.7 expression in dorsalroot ganglia neurons was found in a model ofinflammatory pain. Gating properties of these sub-types will result in a hyperexcitable state thatcauses spontaneous generation of action potentialsand thus may contribute to pain in inflamma-tion.76 Altered Na1 channel subtype expressionhas not been investigated, to our knowledge, inhuman immune-mediated neuropathies but investi-gations in hereditary neuropathies have shown thathuman peripheral neurons are capable of express-ing subtypes other than Nav1.6.77
REFERENCES
1. IUPHAR compendium of voltage-gated ion channels. Pharmacol Rev2005;57:385–540.
2. Berthold CH, Rydmark M. Morphology of normal peripheral axons.In: Waxman SG, Kocsis JD, Stys PK, editors. The axon. New York:Oxford University Press; 1995. p 13–48.
3. Ritchie JM. Physiology of axons. In: Waxman SG, Kocsis JD, Stys PK,editors. The axon. New York: Oxford University Press; 1995. p 68–96.
4. Stys PK, Waxman SG, Ransom BR. Ion pumps and exchangers. In:Waxman SG, Kocsis JD, Stys PK, editors. The axon. New York: OxfordUniversity Press; 1995. p 296–310.
5. Kleinberg CS, Arroyo EJ, King C, Scherer SS. Na,K-ATPase isoformsin the PNS: cell-type specific expression and internodal localizationin myelinated axons. J Peripher Nerv Syst 2007;12(suppl):S45.
862 Demyelinating Neuropathies MUSCLE & NERVE December 2013

6. Stirling DP, Stys PK. Mechanisms of axonal injury: internodal nano-complexes and calcium deregulation. Trends Mol Med 2010;16:160–170.
7. Scherer SS, Arroyo EJ. Recent progress on the molecular organiza-tion of myelinated axons. J Peripher Nerv Syst 2002;7:1–12.
8. Eshed Y, Feinberg K, Poliak S, Sabanay H, Sarig-Nadir O, Spiegel I, etal. Gliomedin mediates Schwann cell–axon interaction and the molec-ular assembly of the nodes of Ranvier. Neuron 2005;47:215–229.
9. Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, etal. Juxtaparanodal clustering of Shaker-like K1channels in myelin-ated axons depends on Caspr2 and TAG-1. J Cell Biol 2003;162:1149–1160.
10. Susuki K, Baba H, Tohyama K, Kanai K, Kuwabara S, Hirata K, et al.Gangliosides contribute to stability of paranodal junctions and ionchannel clusters in myelinated nerve fibers. Glia 2007;55:746–757.
11. Willison HJ. The immunobiology of Guillain–Barr�e syndromes.J Peripher Nerv Syst 2005;10:94–112.
12. Balice-Gordon RJ, Bone LJ, Scherer SS. Functional gap junctions inthe schwann cell myelin sheath. J Cell Biol 1998;142:1095–1104.
13. Baker MD. Electrophysiology of mammalian Schwann cells. Prog Bio-phys Mol Biol 2002;78:83–103.
14. Stys PK. The axo-myelinic synapse. Trends Neurosci 2011;34:393–400.15. Moore JW, Joyner RW, Brill MH, Waxman SD, Najar-Joa M. Simula-
tions of conduction in uniform myelinated fibers. Relative sensitivityto changes in nodal and internodal parameters. Biophys J 1978;21:147–160.
16. Court FA, Sherman DL, Pratt T, Garry EM, Ribchester RR,Cottrell DF, et al. Restricted growth of Schwann cells lacking Cajalbands slows conduction in myelinated nerves. Nature 2004;431:191–195.
17. Nygren A, Halter JA. A general approach to modeling conductionand concentration dynamics in excitable cells of concentric cylindri-cal geometry. J Theor Biol 1999;199:329–358.
18. Bearzatto B, Lesage F, Reyes R, Lazdunski M, Laduron PM. Axonaltransport of TREK and TRAAK potassium channels in rat sciaticnerves. Neuroreport 2000;11:927–930.
19. Rakowski RF, Gadsby DC, De WP. Stoichiometry and voltage depend-ence of the sodium pump in voltage-clamped, internally dialyzedsquid giant axon. J Gen Physiol 1989;93:903–941.
20. Schwarz JR, Reid G, Birch R, Bostock H. Action potentials and mem-brane currents in human node of Ranvier. Pflugers Arch 1993;422:R18.
21. Tasaki I. New measurements of the capacity and the resistance of themyelin sheath and the nodal membrane of the isolated frog nervefiber. Am J Physiol 1955;181:639–650.
22. Barrett EF, Barrett JN. Intracellular recording from vertebratemyelinated axons: mechanism of the depolarizing afterpotential. JPhysiol 1982;323:117–144.
23. Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodiumchannel Na(v)1.6 is localized at nodes of Ranvier, dendrites, and syn-apses. Proc Natl Acad Sci USA 2000;97:5616–5620.
24. Taddese A, Bean BP. Subthreshold sodium current from rapidly inac-tivating sodium channels drives spontaneous firing of tuberomammil-lary neurons. Neuron 2002;33:587–600.
25. Marban E, Yamagishi T, Tomaselli GF. Structure and function ofvoltage-gated sodium channels. J Physiol 1998;508:647–657.
26. Ruff RL. Effects of temperature on slow and fast inactivation of ratskeletal muscle Na(1) channels. Am J Physiol 1999;277:C937–C947.
27. Reid G, Scholz A, Bostock H, Vogel W. Human axons contain at leastfive types of voltage-dependent potassium channel. J Physiol 1999;518:681–696.
28. Herrmann S, Stieber J, Ludwig A. Pathophysiology of HCN channels.Pflugers Arch 2007;454:517–522.
29. Tomlinson S, Burke D, Hanna M, Koltzenburg M, Bostock H. In vivoassessment of HCN channel current (I(h)) in human motor axons.Muscle Nerve 2010;41:247–256.
30. Rasminsky M, Sears TA. Internodal conduction in undissecteddemyelinated nerve fibres. J Physiol 1972;227:323–350.
31. Schwarz JR, Reid G, Bostock H. Action potentials and membrane cur-rents in the human node of Ranvier. Pflugers Arch 1995;430:283–292.
32. Kiernan MC, Burke D, Andersen KV, Bostock H. Multiple measuresof axonal excitability: a new approach in clinical testing. MuscleNerve 2000;23:399–409.
33. Burke D, Kiernan MC, Bostock H. Excitability of human axons. ClinNeurophysiol 2001;112:1575–1585.
34. Kiernan MC, Bostock H. Effects of membrane polarization andischaemia on the excitability properties of human motor axons.Brain 2000;123:2542–2551.
35. Kaji R, Sumner AJ. Ouabain reverses conduction disturbances in sin-gle demyelinated nerve fibers. Neurology 1989;39:1364–1368.
36. Tasaki I. Nervous transmission. Springfield, IL: C.C. Thomas; 1953.37. Franssen H, Straver DC. Pathophysiology of immune-mediated
demyelinating neuropathies—part II: neurology. Muscle Nerve2013.
38. Lonigro A, Devaux JJ. Disruption of neurofascin and gliomedin atnodes of Ranvier precedes demyelination in experimental allergicneuritis. Brain 2009;132:260–273.
39. Hughes RA, Cornblath DR. Guillain–Barr�e syndrome. Lancet 2005;366:1653–1666.
40. Saida K, Sumner AJ, Saida T, Brown MJ, Silberberg DH. Antiserum-mediated demyelination: relationship between remyelination andfunctional recovery. Ann Neurol 1980;8:12–24.
41. Sumner AJ, Saida K, Saida T, Silberberg DH, Asbury AK. Acute con-duction block associated with experimental antiserum-mediateddemyelination of peripheral nerve. Ann Neurol 1982;11:469–477.
42. Koles ZJ, Rasminsky M. A computer simulation of conduction indemyelinated nerve fibres. J Physiol 1972;227:351–364.
43. Waxman, SG. Pathophysiology of demyelinated axons. In: WaxmanSG, Kocsis JD, Stys PK, editors. The axon. New York: Oxford Univer-sity Press; 1995. p 438–462.
44. Schwarz JR, Corrette BJ, Mann K, Wietholter H. Changes of ionicchannel distribution in myelinated nerve fibres from rats with experi-mental allergic neuritis. Neurosci Lett 1991;122:205–209.
45. Bostock H, Sears TA, Sherratt RM. The effects of 4-aminopyridineand tetraethylammonium ions on normal and demyelinated mamma-lian nerve fibres. J Physiol 1981;313:301–315.
46. Bergin PS, Miller DH, Hirsch NP, Murray NM. Failure of 3,4-diami-nopyridine to reverse conduction block in inflammatory demyelinat-ing neuropathies. Ann Neurol 1993;34:406–409.
47. Novakovic SD, Levinson SR, Schachner M, Shrager P. Disruption andreorganization of sodium channels in experimental allergic neuritis.Muscle Nerve 1998;21:1019–1032.
48. Bostock H, Sears TA. The internodal axon membrane: electricalexcitability and continuous conduction in segmental demyelination. JPhysiol 1978;280:273–301.
49. Smith KJ, Bostock H, Hall SM. Saltatory conduction precedes remye-lination in axons demyelinated with lysophosphatidyl choline. J Neu-rol Sci 1982;54:13–31.
50. Sheikh KA, Deerinck TJ, Ellisman MH, Griffin JW. The distributionof ganglioside-like moieties in peripheral nerves. Brain 1999;122:449–460.
51. Gong Y, Tagawa Y, Lunn MP, Laroy W, Heffer-Lauc M, Li CY, et al.Localization of major gangliosides in the PNS: implications forimmune neuropathies. Brain 2002;125:2491–2506.
52. Ogawa-Goto K, Funamoto N, Abe T, Nagashima K. Different cer-amide compositions of gangliosides between human motor and sen-sory nerves. J Neurochem 1990;55:1486–1493.
53. Svennerholm L, Bostrom K, Fredman P, Jungbjer B, Lekman A,Mansson JE, et al. Gangliosides and allied glycosphingolipids inhuman peripheral nerve and spinal cord. Biochim Biophys Acta1994;1214:115–123.
54. Santoro M, Uncini A, Corbo M, Staugaitis SM, Thomas FP, Hays AP,et al. Experimental conduction block induced by serum from apatient with anti-GM1 antibodies. Ann Neurol 1992;31:385–390.
55. Takigawa T, Yasuda H, Kikkawa R, Shigeta Y, Saida T, Kitasato H.Antibodies against GM1 ganglioside affect K1and Na1currents inisolated rat myelinated nerve fibers. Ann Neurol 1995;37:436–442.
56. Weber F, Rudel R, Aulkemeyer P, Brinkmeier H. Anti-GM1 antibod-ies can block neuronal voltage-gated sodium channels. Muscle Nerve2000;23:1414–1420.
57. Elmer LW, O’Brien BJ, Nutter TJ, Angelides KJ. Physicochemicalcharacterization of the alpha-peptide of the sodium channel from ratbrain. Biochemistry 1985;24:8128–8137.
58. Hirota N, Kaji R, Bostock H, Shindo K, Kawasaki T, Mizutani K, et al.The physiological effect of anti-GM1 antibodies on saltatory conduc-tion and transmembrane currents in single motor axons. Brain 1997;120:2159–2169.
59. Arasaki K, Kusunoki S, Kudo N, Tamaki M. The pattern of antigan-glioside antibody reactivities producing myelinated nerve conductionblock in vitro. J Neurol Sci 1998;161:163–168.
60. Paparounas K, O’Hanlon GM, O’Leary CP, Rowan EG, Willison HJ.Anti-ganglioside antibodies can bind peripheral nerve nodes of Ranv-ier and activate the complement cascade without inducing acute con-duction block in vitro. Brain 1999;122:807–816.
61. Susuki K, Rasband MN, Tohyama K, Koibuchi K, Okamoto S,Funakoshi K, et al. Anti-GM1 antibodies cause complement-mediateddisruption of sodium channel clusters in peripheral motor nervefibers. J Neurosci 2007;27:3956–3967.
62. Susuki K, Yuki N, Schafer DP, Hirata K, Zhang G, Funakoshi K, etal. Dysfunction of nodes of Ranvier: a mechanism for anti-ganglioside antibody-mediated neuropathies. Exp Neurol 2012;233:534–542.
63. Phongsisay V, Susuki K, Matsuno K, Yamahashi T, Okamoto S,Funakoshi K, et al. Complement inhibitor prevents disruption ofsodium channel clusters in a rabbit model of Guillain–Barr�e syn-drome. J Neuroimmunol 2008;205:101–104.
64. Plomp JJ, Willison HJ. Pathophysiological actions of neuropathy-related anti-ganglioside antibodies at the neuromuscular junction.J Physiol 2009;587:3979–3999.
Demyelinating Neuropathies MUSCLE & NERVE December 2013 863

65. Buchwald B, Zhang G, Vogt-Eisele AK, Zhang W, Ahangari R, GriffinJW, et al. Anti-ganglioside antibodies alter presynaptic release andcalcium influx. Neurobiol Dis 2007;28:113–121.
66. Fewou SN, Rupp A, Nickolay LE, Carrick K, Greenshields KN,Pediani J, et al. Anti-ganglioside antibody internalization attenuatesmotor nerve terminal injury in a mouse model of acute motor axonalneuropathy. J Clin Invest 2012;122:1037–1051.
67. Griffin JW, Li CY, Ho TW, Xue P, Macko C, Gao CY, et al. Guillain–Barr�e syndrome in northern China. The spectrum of neuropathologi-cal changes in clinically defined cases. Brain 1995;118:577–595.
68. Hafer-Macko CE, Sheikh KA, Li CY, Ho TW, Cornblath DR,McKhann GM, et al. Immune attack on the Schwann cell surface inacute inflammatory demyelinating polyneuropathy. Ann Neurol1996;39:625–635.
69. von Reyn CR, Spaethling JM, Mesfin MN, Ma M, Neumar RW, SmithDH, et al. Calpain mediates proteolysis of the voltage-gated sodiumchannel alpha-subunit. J Neurosci 2009;29:10350–10356.
70. McGonigal R, Rowan EG, Greenshields KN, Halstead SK, HumphreysPD, Rother RP, et al. Anti-GD1a antibodies activate complement andcalpain to injure distal motor nodes of Ranvier in mice. Brain 2010;133:1944–1960.
71. Kapoor R, Davies M, Blaker PA, Hall SM, Smith KJ. Blockers ofsodium and calcium entry protect axons from nitric oxide-mediateddegeneration. Ann Neurol 2003;53:174–180.
72. Waxman SG, Black JA, Ransom BR, Stys PK. Anoxic injury of ratoptic nerve: ultrastructural evidence for coupling between Na1influxand Ca21-mediated injury in myelinated CNS axons. Brain Res 1994;644:197–204.
73. Bostock H, Sears TA. Continuous conduction in demyelinated mam-malian nerve fibers. Nature 1976;263:786–787.
74. Court FA, Hendriks WT, MacGillavry HD, Alvarez J, van Minnen J.Schwann cell to axon transfer of ribosomes: toward a novel under-standing of the role of glia in the nervous system. J Neurosci 2008;28:11024–11029.
75. Waxman SG. Axonal conduction and injury in multiple sclerosis: therole of sodium channels. Nat Rev Neurosci 2006;7:932–941.
76. Black JA, Liu S, Tanaka M, Cummins TR, Waxman SG. Changes inthe expression of tetrodotoxin-sensitive sodium channels within dorsalroot ganglia neurons in inflammatory pain. Pain 2004;108:237–247.
77. Bai Y, Ianokova E, Pu Q, Ghandour K, Levinson R, Martin JJ, et al.Effect of an R69C mutation in the myelin protein zero gene on mye-lination and ion channel subtypes. Arch Neurol 2006;63:1787–1794.
864 Demyelinating Neuropathies MUSCLE & NERVE December 2013






























