Embed Size (px)
Citation preview

Perioperative management of MYH9 hereditarymacrothrombocytopenia (Fechtner syndrome)Kathleen Selleng1, Lena E. Lubenow1, Andreas Greinacher1, Theodore E. Warkentin2,3
1Department of Transfusion Medicine and Immunology, Ernst-Moritz-Arndt University, Greifswald, Germany; 2Department of Pathology and
Molecular Medicine; 3Department of Medicine, Michael G. DeGroote School of Medicine, McMaster University, Hamilton, ON, Canada
Mutations of MYH9 – the gene for non-muscle myo-
sin heavy chain IIA (NMMHC-IIA) – are associated
with a heterogeneous group of hereditary macro-
thrombocytopenias (May–Hegglin anomaly, Sebastian
platelet syndrome, Fechtner syndrome, Epstein syn-
drome) characterized by thrombocytopenia with giant
platelets (‘macrothrombocytopenia’) and leukocyte
inclusions, often accompanied by abnormalities in renal
tubules (proteinuric glomerulonephritis), lenses (cata-
racts), or cochlea (sensorineural deafness) (1–3).
Despite thrombocytopenia, most patients either do not
bleed or have minor hemorrhagic tendencies. Indeed,
the major risk to the patient can be inappropriate
treatment with long-term corticosteroids or splenectomy
for misdiagnosed chronic autoimmune thrombocyto-
penia (4).
MYH9-related diseases exhibit autosomal dominant
inheritance, typically with a heterozygous MYH9 gene
mutation. Approximately 20% of cases are sporadic (2,
5, 6), and somatic mosaicism has been reported (7).
Abstract
Objective: Hereditary thrombocytopenias characterized by mutations in the gene for non-muscle myosin
heavy chain IIA (NMMHC-IIA) are known as MYH9-related hereditary macrothrombocytopenia, and include
the May–Hegglin anomaly, Sebastian platelet syndrome, Fechtner syndrome, and Epstein syndrome.
Despite the presence of thrombocytopenia, these patients often have only mild or non-bleeding pheno-
types. A major risk for these patients can be inappropriate treatment with long-term corticosteroids or
splenectomy for misdiagnosed chronic autoimmune thrombocytopenia, as well as inadequate peri- and
postoperative management. Methods: Using the case of a 44-yr-old male with Fechtner syndrome (macro-
thrombocytopenia, leukocyte inclusions, sensorineural deafness, glomerulonephritis) who underwent
neurosurgery for an intracerebral arteriovenous malformation, we describe current methods to diagnose
hereditary MYH9-related macrothombocytopenia by analysis of the blood smear, immunofluorescence
staining of the NMMHC-IIA in leucocytes, and by MYH9-gene sequencing. Results: Clusters of NMMHC-
IIA in granulocytes and a R1165C mutation in the MYH9-gene in two macrothrombocytopenic family mem-
bers confirmed the diagnosis of a MYH9-related disease. The patient had no bleeding diathesis by history
or physical examination. Thus no perioperative prohemostatic pharmacologic therapies or transfusions
were given, with only minimal bleeding observed. Postoperative antithrombotic thromboprophylaxis was
not given because of anticipated enhanced risk for bleeding. However, the patient developed symptomatic
pulmonary embolism on postoperative day 6, which was successfully managed with 8 months of
anticoagulation. Conclusion: MYH9-related hereditary macrothrombocytopenia does not necessarily protect
against postoperative venous thromboembolism, and affected patients who do not evince bleeding diathe-
sis should be considered for routine postoperative pharmacologic thromboprophylaxis.
Key words hereditary macrothrombocytopenia; MYH9-related disorder; Fechtner syndrome; pulmonary embolism; thromboprophylaxis
Correspondence Dr Theodore E. Warkentin, Hamilton Regional Laboratory Medicine Program, Room 1-180A, Hamilton Health
Sciences (General Site), 237 Barton St. E., Hamilton, ON, Canada L8L2X2. Tel: 905 527 0271 (ext. 46139); Fax: 905 577 1421; e-mail:
Accepted for publication 1 June 2007 doi:10.1111/j.1600-0609.2007.00913.x
CASE REPORT
European Journal of Haematology ISSN 0902-4441
ª 2007 The Authors
Journal compilation 79 (263–268) ª 2007 Blackwell Munksgaard 263

Genotype–phenotype correlation studies (8) suggest that
mutations in the C-terminal coiled coil region or trunca-
tion of the tailpiece is preferentially associated with
hematologic-only phenotypes (May–Hegglin anomaly,
Sebastian platelet syndrome), while mutations of the
head ATPase domain frequently are associated addition-
ally with nephropathy and ⁄or hearing loss (Fechtner syn-
drome, Epstein syndrome). However, this genotype–
phenotype pattern is not strictly applicable; a family car-
rying the same mutation in the MYH9 gene can show a
broad variety in the manifestations of non-hematologic
features. Furthermore, renal impairment, hearing loss
and cataracts rarely manifest during childhood, and at
widely differing ages during adulthood. There is evidence
that other protein abnormalities could explain some of
these phenotypic variations (9).
For patients with hereditary macrothrombocytopenia
requiring major surgery, there is uncertainty regarding
appropriate perioperative strategies to optimize hemosta-
sis (10) but also to prevent venous thrombosis. We
encountered a patient with MYH9-related macrothromb-
ocytopenia, leukocyte inclusions, sensorineural hearing
loss, and glomerulonephritis (Fechtner syndrome) requi-
ring neurosurgical resection of an arteriovenous malfor-
mation (AVM), whose postoperative course was
complicated not by bleeding but by pulmonary embol-
ism. This illustrates the potential for serious venous
thromboembolism in these patients despite their thromb-
ocytopenia.
Case report
Patient hemostatic history and laboratory investiga-tions
A 44-yr-old male of middle eastern descent, scheduled to
undergo neurosurgery for a posterior parietal AVM, was
referred for perioperative management of presumed chro-
nic autoimmune thrombocytopenia of at least 15 yr dur-
ation. He had previously undergone appendectomy and
tonsillectomy, and had sustained rib fractures, without
any significant bleeding or need for blood transfusions.
He also had progressive sensorineural hearing impair-
ment (confirmed by audiometry) and renal biopsy (with-
out bleeding) performed 2 yr earlier for proteinuria,
which was reported as showing ‘focal segmental
sclerosing glomerulonephritis’ without inflammation,
vasculitis, thrombosis, or deposition of immunoglobulin
or complement.
Hemoglobin was 124 g ⁄L, white blood cell count
8.4 · 109 ⁄L with normal leukocyte differential, and
platelet counts ranged from 40 to 90 · 109 ⁄L by the
electronic particle counter, with a mean platelet volume
(MPV) of 12.1 fL (normal, 7.4–10.4 fL). The patient’s
peripheral blood film revealed a platelet count estimated
to be about 100–120 · 109 ⁄L, giant platelets with
normal granulation, and faint blue-staining small
inclusion bodies within the granulocytes; the bleeding
time was 9 min (Ivy method, normal, 3–8 min). Inter-
national normalized ratio (1.0) and aPTT (25 s) were
normal. The serum creatinine was normal (73 lmol ⁄L).The patient exhibited proteinuria (1.28 g ⁄L; normal
<0.15 g ⁄L). Bone marrow aspiration and biopsy
revealed normal cellularity and normal trilineage
hematopoiesis; megakaryocytes were adequate, marrow
cytogenetics revealed no clonal chromosomal
abnormalities.
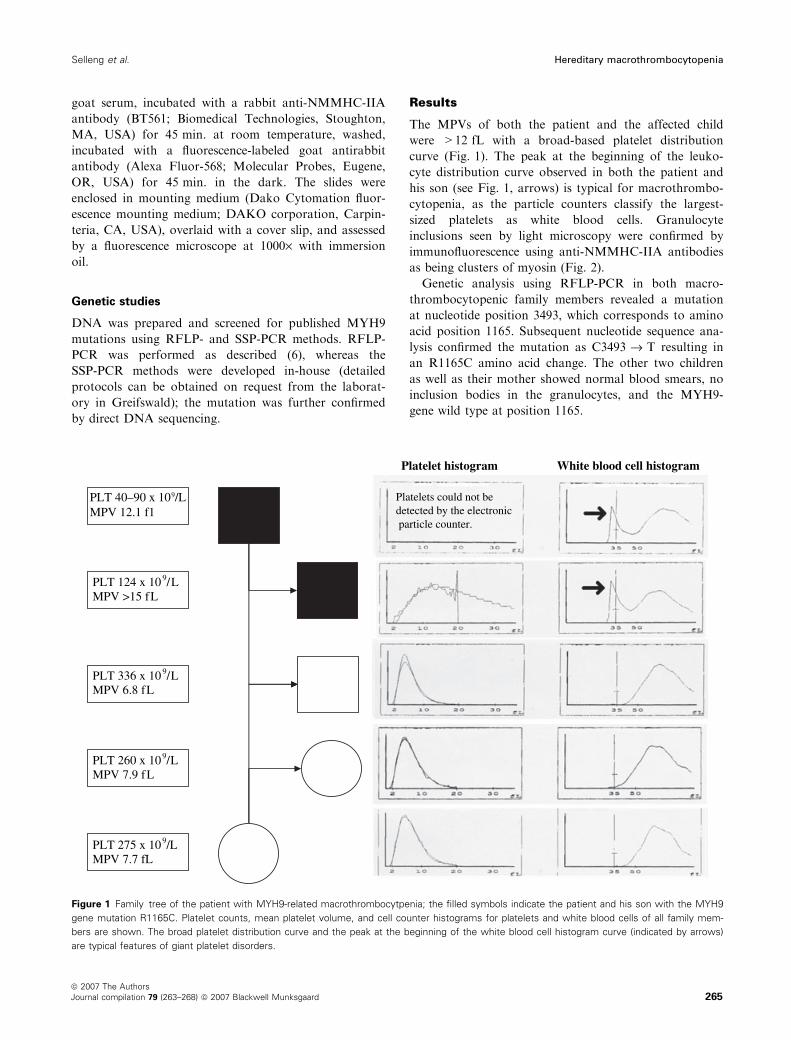
The patient’s 8-yr-old son also had macrothrombo-
cytopenia (124 · 109 ⁄L; MPV > 15 fL). However, the
other two children, and the patient’s wife, had normal
platelet counts (336, 260, and 275 · 109 ⁄L) and normal
MPVs (6.8, 7.9, and 7.7 fL) (Fig. 1).
Neurosurgery for AVM and postoperative pulmonaryembolism
The patient underwent image-guided left and right pari-
etal craniotomies to remove a parietal dural AVM
located near the superior sagittal sinus, with duraplasty.
No prophylactic or therapeutic administration of phar-
macologic prohemostatic therapy, such as tranexamic
acid or desmopressin, or blood products was given. The
estimated intraoperative blood loss was only 140 mL.
Also, no postoperative pharmacologic thromboprophy-
laxis was given, because of expected high risk for bleed-
ing and the patient was discharged on postoperative day
5. The next day the patient returned to hospital because
of abrupt-onset of dyspnea and left-sided pleuritic chest
pain. Spiral computed tomography (CT) confirmed pul-
monary embolism. The patient received therapeutic-dose
parenteral anticoagulation in-hospital, followed by
8 months of outpatient warfarin anticoagulation. No
bleeding complications were observed during anticoagu-
lant treatment.
Materials and methods
Informed written consent was obtained from the patient
and family members for performing the studies des-
cribed, and for preparing the case for publication.
Screening for MHY9 granulocyte inclusions
Immunofluorescence labeling was performed as described
(11), with minor modifications. In short, air-dried periph-
eral blood smears were fixed in ice-cold acetone (unfixed
blood smears can be sent within 1 wk by mail to the
laboratory in Greifswald, Germany), blocked with 10%
Hereditary macrothrombocytopenia Selleng et al.
264ª 2007 The Authors
Journal compilation 79 (263–268) ª 2007 Blackwell Munksgaard
goat serum, incubated with a rabbit anti-NMMHC-IIA
antibody (BT561; Biomedical Technologies, Stoughton,
MA, USA) for 45 min. at room temperature, washed,
incubated with a fluorescence-labeled goat antirabbit
antibody (Alexa Fluor-568; Molecular Probes, Eugene,
OR, USA) for 45 min. in the dark. The slides were
enclosed in mounting medium (Dako Cytomation fluor-
escence mounting medium; DAKO corporation, Carpin-
teria, CA, USA), overlaid with a cover slip, and assessed
by a fluorescence microscope at 1000· with immersion
oil.
Genetic studies
DNA was prepared and screened for published MYH9
mutations using RFLP- and SSP-PCR methods. RFLP-
PCR was performed as described (6), whereas the
SSP-PCR methods were developed in-house (detailed
protocols can be obtained on request from the laborat-
ory in Greifswald); the mutation was further confirmed
by direct DNA sequencing.
Results
The MPVs of both the patient and the affected child
were >12 fL with a broad-based platelet distribution
curve (Fig. 1). The peak at the beginning of the leuko-
cyte distribution curve observed in both the patient and
his son (see Fig. 1, arrows) is typical for macrothrombo-
cytopenia, as the particle counters classify the largest-
sized platelets as white blood cells. Granulocyte
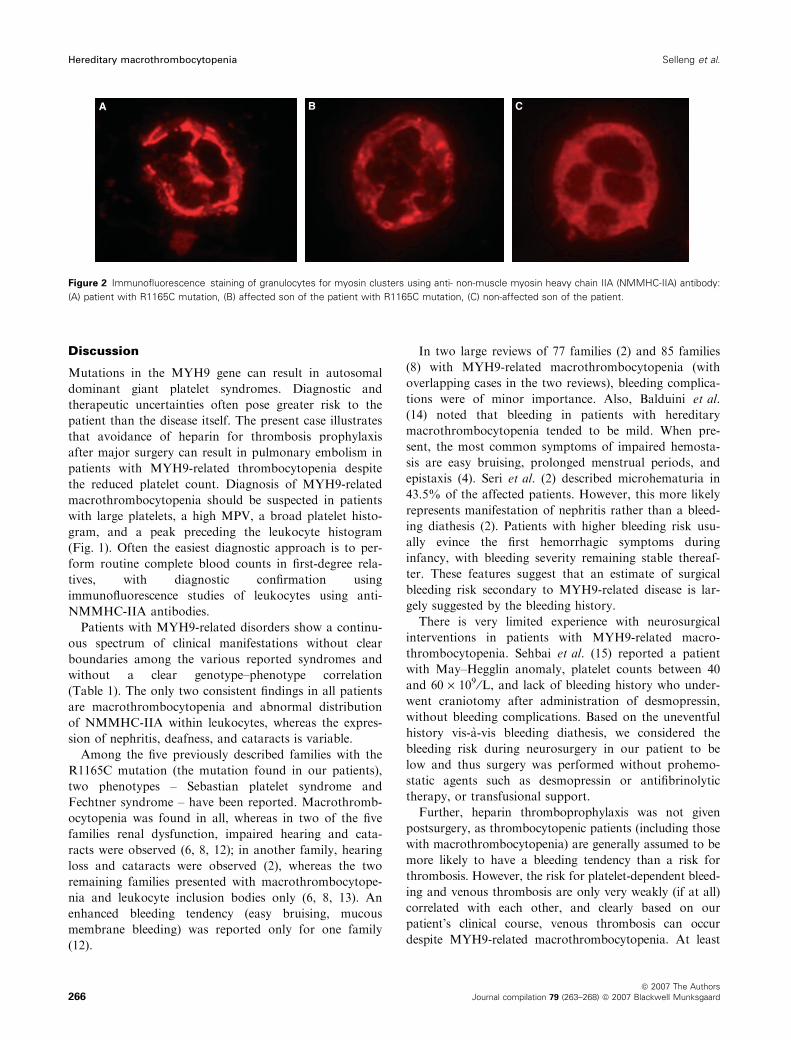
inclusions seen by light microscopy were confirmed by
immunofluorescence using anti-NMMHC-IIA antibodies
as being clusters of myosin (Fig. 2).
Genetic analysis using RFLP-PCR in both macro-
thrombocytopenic family members revealed a mutation
at nucleotide position 3493, which corresponds to amino
acid position 1165. Subsequent nucleotide sequence ana-
lysis confirmed the mutation as C3493 fi T resulting in
an R1165C amino acid change. The other two children
as well as their mother showed normal blood smears, no
inclusion bodies in the granulocytes, and the MYH9-
gene wild type at position 1165.
PLT 40–90 x 109/LMPV 12.1 f1
01x421TLP 9 L/Lf51>VPM
01x633TLP 9 L/Lf8.6VPM
01x062TLP 9 L/Lf9.7VPM
01x572TLP 9 L/Lf7.7VPM
Platelet histogram White blood cell histogram
Platelets could not bedetected by the electronic particle counter.
Figure 1 Family tree of the patient with MYH9-related macrothrombocytpenia; the filled symbols indicate the patient and his son with the MYH9
gene mutation R1165C. Platelet counts, mean platelet volume, and cell counter histograms for platelets and white blood cells of all family mem-
bers are shown. The broad platelet distribution curve and the peak at the beginning of the white blood cell histogram curve (indicated by arrows)
are typical features of giant platelet disorders.
Selleng et al. Hereditary macrothrombocytopenia
ª 2007 The Authors
Journal compilation 79 (263–268) ª 2007 Blackwell Munksgaard 265
Discussion
Mutations in the MYH9 gene can result in autosomal
dominant giant platelet syndromes. Diagnostic and
therapeutic uncertainties often pose greater risk to the
patient than the disease itself. The present case illustrates
that avoidance of heparin for thrombosis prophylaxis
after major surgery can result in pulmonary embolism in
patients with MYH9-related thrombocytopenia despite
the reduced platelet count. Diagnosis of MYH9-related
macrothrombocytopenia should be suspected in patients
with large platelets, a high MPV, a broad platelet histo-
gram, and a peak preceding the leukocyte histogram
(Fig. 1). Often the easiest diagnostic approach is to per-
form routine complete blood counts in first-degree rela-
tives, with diagnostic confirmation using
immunofluorescence studies of leukocytes using anti-
NMMHC-IIA antibodies.
Patients with MYH9-related disorders show a continu-
ous spectrum of clinical manifestations without clear
boundaries among the various reported syndromes and
without a clear genotype–phenotype correlation
(Table 1). The only two consistent findings in all patients
are macrothrombocytopenia and abnormal distribution
of NMMHC-IIA within leukocytes, whereas the expres-
sion of nephritis, deafness, and cataracts is variable.
Among the five previously described families with the
R1165C mutation (the mutation found in our patients),
two phenotypes – Sebastian platelet syndrome and
Fechtner syndrome – have been reported. Macrothromb-
ocytopenia was found in all, whereas in two of the five
families renal dysfunction, impaired hearing and cata-
racts were observed (6, 8, 12); in another family, hearing
loss and cataracts were observed (2), whereas the two
remaining families presented with macrothrombocytope-
nia and leukocyte inclusion bodies only (6, 8, 13). An
enhanced bleeding tendency (easy bruising, mucous
membrane bleeding) was reported only for one family
(12).
In two large reviews of 77 families (2) and 85 families
(8) with MYH9-related macrothrombocytopenia (with
overlapping cases in the two reviews), bleeding complica-
tions were of minor importance. Also, Balduini et al.
(14) noted that bleeding in patients with hereditary
macrothrombocytopenia tended to be mild. When pre-
sent, the most common symptoms of impaired hemosta-
sis are easy bruising, prolonged menstrual periods, and
epistaxis (4). Seri et al. (2) described microhematuria in
43.5% of the affected patients. However, this more likely
represents manifestation of nephritis rather than a bleed-
ing diathesis (2). Patients with higher bleeding risk usu-
ally evince the first hemorrhagic symptoms during
infancy, with bleeding severity remaining stable thereaf-
ter. These features suggest that an estimate of surgical
bleeding risk secondary to MYH9-related disease is lar-
gely suggested by the bleeding history.
There is very limited experience with neurosurgical
interventions in patients with MYH9-related macro-
thrombocytopenia. Sehbai et al. (15) reported a patient
with May–Hegglin anomaly, platelet counts between 40
and 60 · 109 ⁄L, and lack of bleeding history who under-
went craniotomy after administration of desmopressin,
without bleeding complications. Based on the uneventful
history vis-a-vis bleeding diathesis, we considered the
bleeding risk during neurosurgery in our patient to be
low and thus surgery was performed without prohemo-
static agents such as desmopressin or antifibrinolytic
therapy, or transfusional support.
Further, heparin thromboprophylaxis was not given
postsurgery, as thrombocytopenic patients (including those
with macrothrombocytopenia) are generally assumed to be
more likely to have a bleeding tendency than a risk for
thrombosis. However, the risk for platelet-dependent bleed-
ing and venous thrombosis are only very weakly (if at all)
correlated with each other, and clearly based on our
patient’s clinical course, venous thrombosis can occur
despite MYH9-related macrothrombocytopenia. At least
A B C
Figure 2 Immunofluorescence staining of granulocytes for myosin clusters using anti- non-muscle myosin heavy chain IIA (NMMHC-IIA) antibody:
(A) patient with R1165C mutation, (B) affected son of the patient with R1165C mutation, (C) non-affected son of the patient.
Hereditary macrothrombocytopenia Selleng et al.
266ª 2007 The Authors
Journal compilation 79 (263–268) ª 2007 Blackwell Munksgaard
for one patient with MYH9-related disease recurrent
thrombosis has been described (16). Despite a history of
epistaxis and bleeding gums since childhood (platelet counts
between 18 and 56 · 109 ⁄L), this patient with Fechtner syn-
drome (MYH9-R702H) developed at least four episodes of
venous thrombosis, beginning at 23 yr of age (spontaneous
proximal deep-vein thrombosis and pulmonary embolism),
with recurrences of deep-vein thrombosis (4 months later),
caval vein thrombosis (12 yr later), and thrombotic occlu-
sions of the arteriovenous fistulae (because of hemodialysis
for Fechtner syndrome-related renal failure). No cause for
thrombophilia was identified in this patient (except hype-
rhomocysteinemia) despite an extensive investigation.
Neurosurgical procedures are a major risk factor for
thrombosis (17–21). It is a Grade 1A recommendation
of the American College of Chest Physicians (ACCP)
that thromboprophylaxis should be routinely used in
patients undergoing major neurosurgery (21). In our
patient, thrombocytopenia was judged as a risk factor
for bleeding complications and was the reason against
the use of pharmacologic thromboprophylaxis. How-
ever, when postoperative pulmonary embolism was
diagnosed, the patient required therapeutic-dose antico-
agulant therapy for an extended period. This arguably
was associated with a much higher bleeding risk than
short-term perioperative thromboprophylaxis with hep-
arin. The occurrence of pulmonary embolism in this
patient with MYH9-related macrothrombocytopenia
illustrates that protection against venous thromboembo-
lism is not necessarily afforded by this platelet
disorder. When a careful preoperative history and
physical examination suggest that there is no (or
minimal) bleeding diathesis despite MYH9-related
macrothrombocytopenia postoperative pharmacologic
thromboprophylaxis may well be appropriate.
Acknowledgements
We thank the patient and his family for permission to
publish this history.
Table 1 MYH9-related disease
Families with macrothrombocytopeniaand inclusion bodies only Exon Mutation
Families with additional symptoms;e.g. deafness and ⁄ or nephritis and ⁄ or cataract
Exons encoding the globular head
del N76-S81 1 (7)
2 1 N93K
1 A95T
1 S96L 3
1 10 K371N
16 R702C 7
R702H 4
R705H 11
1 (22) Q706E
Exons encoding the coiled-coil domain of NMMHC-IIA
24 del E1066-A1072 1 (2)
1 25 S1114P
1 T1155I
2 R1165C 42
26 R1165L 2
1 delL1205-Q1207
1 D1424H 2
5 30 D1424N 8
1 D1424Y 1
31 V1516L 1 (23)
37 I1816V 1
15 38 E1841K 5
1 5774delA preterm
1 5779delC preterm
13 40 R1933X
1 5828delG preterm
E1945X 1 (2)
To date, 26 MYH9 mutations have been identified in 91 unrelated families. Data are taken from Dong et al. 2005 (8) unless otherwise indicated.
Values in parenthesis indicate references.1Non-syndromic deafness without hematological symptoms.2Including the family of the present report.
Selleng et al. Hereditary macrothrombocytopenia
ª 2007 The Authors
Journal compilation 79 (263–268) ª 2007 Blackwell Munksgaard 267

Financial support
This study was funded by the Heart & Stroke Founda-
tion of Ontario (#T-5207 and #T-6157); Deutsche For-
schungsgemeinschaft (Gr 1096 ⁄2-4); German Federal
Ministry for Education and Research (NBL3 program,
reference 01-ZZ0403), Deutsche Forschungsgesellschaft
(Graduiertenkolleg GRK-840), Landesforderungs-
programm EFRE.
References
1. Toren A, Rozenfeld-Granot G, Rocca B, et al. Autosom-
al-dominant giant platelet syndromes: a hint of the same
genetic defect as in Fechtner syndrome owing to a similar
genetic linkage to chromosome 22q11-13. Blood
2000;96:3447–51.
2. Seri M, Pecci A, Di Bari F, et al. MYH9-related disease:
May-Hegglin anomaly, Sebastian syndrome, Fechtner syn-
drome, and Epstein syndrome are not distinct entities but
represent a variable expression of a single illness. Medicine
(Baltimore) 2003;82:203–15.
3. Greinacher A, Nieuwenhuis HK, White JG. Sebastian
platelet syndrome: a new variant of hereditary macro-
thrombocytopenia with leukocyte inclusions. Blut
1990;61:282–8.
4. Greinacher A, Mueller-Eckhardt C. Hereditary types of
thrombocytopenia with giant platelets and inclusion
bodies in the leukocytes. Blut 1990;60:53–60.
5. Heath KE, Campos-Barros A, Toren A, et al. Nonmuscle
myosin heavy chain IIa mutations define a spectrum of
autosomal dominant macrothrombocytopenias: may-
hegglin anomaly and fechtner, sebastian, epstein, and
alport-like syndromes. Am J Hum Genet 2001;69:1033–45.
6. Kunishima S, Matsushita T, Kojima T, et al. Identifica-
tion of six novel MYH9 mutations and genotype-pheno-
type relationships in autosomal dominant
macrothrombocytopenia with leukocyte inclusions. J Hum
Genet 2001;46:722–9.
7. Kunishima S, Matsushita T, Yoshihara T, Nakase Y,
Yokoi K, Hamaguchi M, Saito H. First description of
somatic mosaicism in MYH9 disorders. Br J Haematol
2005;128:360–5.
8. Dong F, Li S, Pujol-Moix N, Luban NL, Shin SW, Seo
JH, Ruiz-Saez A, Demeter J, Langdon S, Kelley MJ.
Genotype-phenotype correlation in MYH9-related
thrombocytopenia. Br J Haematol 2005;130:620–7.
9. Toren A, Rozenfeld-Granot G, Heath KE, et al. MYH9
spectrum of autosomal-dominant giant platelet syndromes:
unexpected association with fibulin-1 variant-D
inactivation. Am J Hematol 2003;74:254–62.
10. Matzdorff AC, White JG, Malzahn K, Greinacher A.
Perioperative management of a patient with Fechtner
syndrome. Ann Hematol 2001;80:436–9.
11. Kunishima S, Kojima T, Matsushita T, et al. Mutations
in the NMMHC-A gene cause autosomal dominant
macrothrombocytopenia with leukocyte inclusions
(May-Hegglin anomaly ⁄ Sebastian syndrome). Blood
2001;97:1147–9.
12. Pujol-Moix N, Kelley MJ, Hernandez A, Muniz-Diaz E,
Espanol I. Ultrastructural analysis of granulocyte
inclusions in genetically confirmed MYH9-related disor-
ders. Haematologica 2004;89:330–7.
13. Seri M, Cusano R, Gangarossa S, et al. Mutations in
MYH9 result in the May-Hegglin anomaly, and Fechtner
and Sebastian syndromes. The May-Heggllin ⁄FechtnerSyndrome Consortium. Nat Genet 2000;26:103–5.
14. Balduini CL, Iolascon A, Savoia A. Inherited thrombo-
cytopenias: from genes to therapy. Haematologica
2002;87:860–80.
15. Sehbai AS, Abraham J, Brown VK. Perioperative man-
agement of a patient with May-Hegglin anomaly requiring
craniotomy. Am J Hematol 2005;79:303–8.
16. Heller PG, Pecci A, Glembotsky AC, Savoia A, Negro
FD, Balduini CL, Molinas FC. Unexplained recurrent
venous thrombosis in a patient with MYH9-related
disease. Platelets 2006;17:274–5.
17. Black PM, Baker MF, Snook CP. Experience with
external pneumatic calf compression in neurology and
neurosurgery. Neurosurgery 1986;18:440–4.
18. Macdonald RL, Amidei C, Baron J, Weir B, Brown F,
Erickson RK, Hekmatpanah J, Frim D. Randomized,
pilot study of intermittent pneumatic compression devices
plus dalteparin versus intermittent pneumatic compression
devices plus heparin for prevention of venous thrombo-
embolism in patients undergoing craniotomy. Surg Neurol
2003;59:363–72.
19. Epstein NE. A review of the risks and benefits of differing
prophylaxis regimens for the treatment of deep venous
thrombosis and pulmonary embolism in neurosurgery.
Surg Neurol 2005;64:295–301.
20. Swann KW, Black PM. Deep vein thrombosis and
pulmonary emboli in neurosurgical patients: a review.
J Neurosurg 1984;61:1055–62.
21. Geerts WH, Pineo GF, Heit JA, Bergqvist D, Lassen MR,
Colwell CW, Ray JG. Prevention of venous thrombo-
embolism: the Seventh ACCP Conference on Antithrom-
botic and Thrombolytic Therapy. Chest 2004;126:338S–
400S.
22. Otsubo K, Kanegane H, Nomura K, Ogawa J, Miyawaki
T, Kunishima S. Identification of a novel MYH9 muta-
tion in a patient with May-Hegglin anomaly. Pediatr
Blood Cancer 2006;47:968–9.
23. Ma ES, Wong CL, Shek TW, Hui SP. Hematologic and
genetic characterization of an MYH9-related disorder in a
Chinese family. Haematologica 2006;91:1002–3.
Hereditary macrothrombocytopenia Selleng et al.
268ª 2007 The Authors
Journal compilation 79 (263–268) ª 2007 Blackwell Munksgaard





























