Embed Size (px)
Citation preview

Physikalische Chemie
für Mikrosystemtechnik
WS 2010/2011
P. Gräber e-mail: [email protected]
Lehrbücher:
• P.W. Atkins: Physikalische Chemie, Wiley – VCH • G. Wedler: Grundzüge der Physikalischen Chemie, Wiley – VCH • Engel/Reid: Physikalische Chemie, Pearson-Studium • P.W. Atkins, L. Jones: Chemie, einfach alles, Wiley – VCH
Textverarbeitung: B. Jöck Zeichnungen: R. Lehmann

Inhaltsverzeichnis: Seite
Anmerkungen zur Schreibweise bei Rechnungen, Diagrammen und Tabellen 1
1. Einleitung 5
2. Grundbegriffe
2.1. Systeme 6
2.2 SI Einheiten 6
2.3 Stoffmenge 8
2.4 Konzentrationsangaben 9
2.5 Temperaturmessung 11
2.6 Energieaustausch System – Umgebung 12
2.6.1 Elektrische Arbeit 16
2.6.2. Arbeit, kinetische Energie, potentielle Energie und innere Energie 17
2.6.3. Wärme und Wärmekapazität 19
3. Ideale Gase 21
3.1 Experimentelle Befunde über Gase 21
3.2. Kinetische Gastheorie (Modell) 23
3.3 Mischungen idealer Gase 27
3.4 Stoßzahlen und mittlere freie Weglänge 27
3.5 Die Geschwindigkeitsverteilung der Teilchen im Gas 38
3.6 Extensive und intensive Zustandsgrößen 44
4. Reale Gase 46
5. Flüssige und feste Stoffe, Aggregatzustände, Phasen 51
6. Energieerhaltung - Der erste Hauptsatz 54
6.1 Die Temperaturabhängigkeit von U und H 56
6.2.1 Expansion eines idealen Gases ins Vakuum 59
6.2.2 Adiabatische Expansion eines idealen Gases 60
6.2.3 Expansion eines realen Gases (Joule-Thomson-Effekt) 62
6.2.4 Innere Energie und Wärmekapazität idealer Gase 63
6.3 Enthalpieänderung bei Phasenumwandlungen 67

7. Die Enthalpieänderung bei chemischen Reaktionen 69
7.1 Temperaturabhängigkeit von rHΔ 70
7.2 Die Reaktionsenthalpie als Zustandsfunktion: Der Heß’sche Satz 71
7.3 Bildungsenthalpien 72
7.4 Standardzustände 73
7.5 Kalorimetrie 75
8. Die Richtung natürlicher Prozesse. Der 2. Hauptsatz 81
8.1 Reversible und irreversible Prozesse 81
8.2 Die Richtung natürlicher Prozesse 86
8.3 Die Entropie 86
8.4 Druck- und Temperaturabhängigkeit der Entropie 88
8.5 Umwandlungsentropie 89
8.6 Die Reaktionsentropie 90
8.7 Die Mischungsentropie 90
8.8 Der Absolutwert der Entropie 92
8.9 Wärmekraftmaschinen und Wärmepumpen 93
8.10 Anschauliche Bedeutung der Entropie 100
9. Die Freie Energie und die Freie Enthalpie 103
9.1 Die Richtung natürlicher Prozesse bei konstantem p und T 103
9.2 Die Temperatur- und Druckabhängigkeit von G 104
9.3 Das Chemische Potential 106
9.4 Die Temperaturabhängigkeit des chemischen Potentials 111
9.5 Das chemische Gleichgewicht 112
9.6 Berechnung der Freien Standardreaktionsenthalpie 115
9.7 Die Umrechnung von θGΔ r 298 und der Gleichgewichtskonstanten auf andere Temperaturen 116
9.8 Die Druckabhängigkeit von K 119
9.9 Das chemische Gleichgewicht und GΔr 121

10. Phasengleichgewichte 124
10.1. Dampfdruck von Flüssigkeiten 124
10.2 Gibbs’sches Phasengesetz 128
10.3. Zweikomponentige Systeme 128
10.3.1. Dampfdruckerniedrigung 128
10.3.2. Siedepunkterhöhung 131
10.3.3. Gefrierpunktserniedrigung 132
10.3.4. Osmotischer Druck 133
11. Reale Systeme
136
11.1. Molare und partielle molare Größen 136
11.2 Experimentelle Bestimmung der partiellen molaren Größen 138
11.3 Gibbs-Duhem-Gleichung 139
11.4 Die Thermodynamische Beschreibung realer Mischphasen 141
11.5 Der Dampfdruck über Mischungen 144
11.6 Aktivität und Aktivitätskoeffizient 147
11.7 Dampfdruckdiagramme 153
11.8 Schmelzdiagramme 159
Definitionen 163

PC MST Kap. 1-7_2010_11 1
Anmerkungen zur Schreibweise bei Rechnungen, Diagrammen und Tabellen: Physikalische Größen haben einen Zahlenwert und eine Einheit. Bei Unklarheiten über die Verwendung von Symbolen und Einheiten kann man nachsehen in dem Buch: "Größen, Einheiten und Symbole in der Physikalischen Chemie" herausgegeben von K.-H. Homann, Verlag Wiley - VCH 1. Physikalische Größe = Zahlenwert Einheit
Bsp.: Druck = p = 3500 Pa = 3,5 . 103 Pa = 3,5 kPa 2. Bei Umrechnungen von Einheiten gelten die gleichen algebraischen Regeln wie für
Zahlen. Man kann Einheiten mit negativen Exponenten oder als Brüche schreiben. Benutzt man negative Exponenten, lässt man Multiplikationszeichen weg.
Vorgehensweise: Man setzt in die Gleichung die Zahlenwerte mit den gegebenen Einheiten ein und rechnet dann schrittweise in die gewünschten Einheiten um.
Beispiele: Die Geschwindigkeit ist in km h−1 angegeben. Sie soll in m s−1 umgerechnet werden. v = 50 km h-1, 1 h = 3600 s, 1 km = 103 m
11 m50 13,9 m s3,6 s
−= = 3km 10 m 1 m
h 3600 s 3,6 s⎡ ⎤
= =⎢ ⎥⎣ ⎦
Berechnung der Gaskonstante:
1 1
1 1
p VRn T101325 Pa 22,4 L
1 mol 273 K8314 Pa L mol K8,314 J mol K
− −
− −
=
⋅=
⋅
=
=
p = 101325 Pa n = 1 mol T = 273 K V = 22,4 L
Einheiten:
Pa Lmol K
2 3 3 31N m 10 m 1N m10mol K mol K
− − −⋅= =
3 1 110 J mol K− − −=
1 Pa = 1 N m−2
1 L = 10−3m3 1 N m = 1 J
PC MST Kap. 1-7_2010_11 2
Ausdrücke hinter einem ln (...) oder exp (...) (Bsp. ⎟⎟⎠
⎞⎜⎜⎝
⎛−
Tk2vm
exp2
) müssen immer
einheitenlos sein. Das gleiche gilt für Glieder einer Summe, in der eine einheitenlose Zahl auftritt (Bsp: 1 + ....).
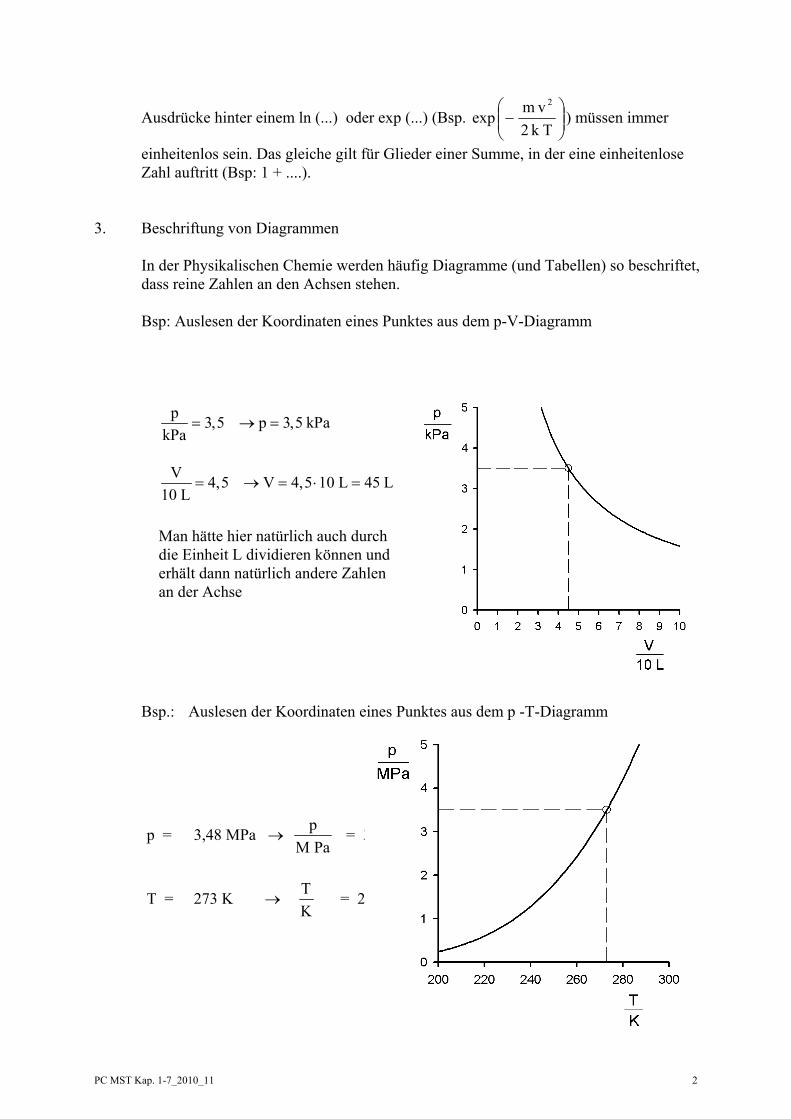
3. Beschriftung von Diagrammen
In der Physikalischen Chemie werden häufig Diagramme (und Tabellen) so beschriftet, dass reine Zahlen an den Achsen stehen. Bsp: Auslesen der Koordinaten eines Punktes aus dem p-V-Diagramm
Bsp.: Auslesen der Koordinaten eines Punktes aus dem p -T-Diagramm
p 3,5 p 3,5 kPakPa
= → =
V 4,5 V 4,5 10 L 45 L
10 L= → = ⋅ =
Man hätte hier natürlich auch durch die Einheit L dividieren können und erhält dann natürlich andere Zahlen an der Achse
p = 3,48 MPa → PaM
p = 3,48
T = 273 K → KT = 273

PC MST Kap. 1-7_2010_11 3
Bsp: Auslesen der Koordinaten eines Punktes aus einem ln p- 1T
-Diagramm
( )p pln 1,25 exp 1,25 3,49MPa MPa
= → = =
p 3,49 MPa=
33 110 K 13,66 3,66 10 K
T T− −= → = ⋅
T 273 K=
4. Beschriftung von Tabellen
KT
PaMp
TK103
PaMpln
216 0,52 4,62 - 0,66 273 3,49 3,66 1,75 304 7,38 3,29 1,99
Diese Übereinkunft bedeutet, dass die Beschriftung der Tabelle (oder des Diagramms) und die zugehörigen Zahlen als Gleichung zu lesen sind:
Bsp: p3,49 p 3,49 MPa,MPa
= → =
3
3 110 K 14,62 4,62 10 KT T
− −= → = ⋅
In der Biochemie/Biologie hat sich diese Schreibweise noch nicht durchgesetzt. Dort wird die Einheit so angegeben, dass sie durch ein Komma vom Symbol der Größe getrennt wird.
Sehr verbreitet ist auch die Angabe der Einheiten in eckigen Klammern
PC MST Kap. 1-7_2010_11 4
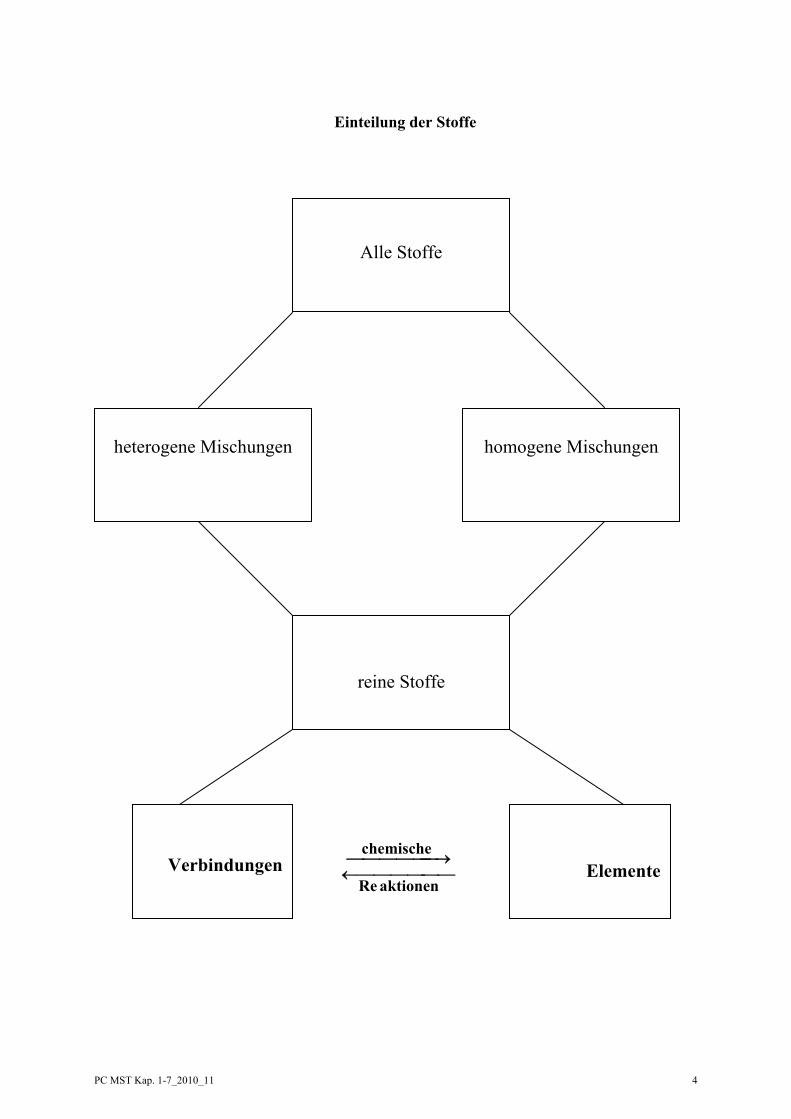
Einteilung der Stoffe
Alle Stoffe
heterogene Mischungen
homogene Mischungen
reine Stoffe
Verbindungen
Elemente⎯⎯⎯⎯⎯ ⎯←
⎯⎯⎯⎯⎯ →⎯
aktionenRe
chemische
PC MST Kap. 1-7_2010_11 5
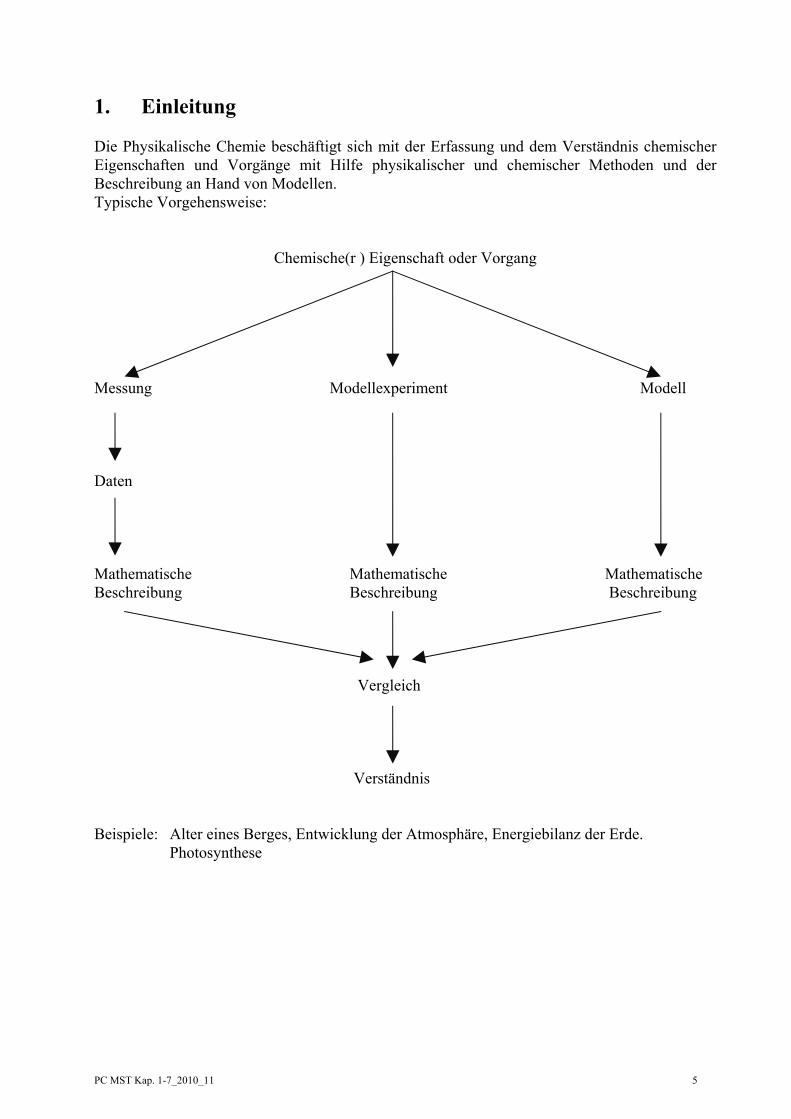
1. Einleitung Die Physikalische Chemie beschäftigt sich mit der Erfassung und dem Verständnis chemischer Eigenschaften und Vorgänge mit Hilfe physikalischer und chemischer Methoden und der Beschreibung an Hand von Modellen. Typische Vorgehensweise:
Chemische(r ) Eigenschaft oder Vorgang
Messung Modellexperiment Modell Daten Mathematische Mathematische Mathematische Beschreibung Beschreibung Beschreibung
Vergleich
Verständnis Beispiele: Alter eines Berges, Entwicklung der Atmosphäre, Energiebilanz der Erde.
Photosynthese
PC MST Kap. 1-7_2010_11 6
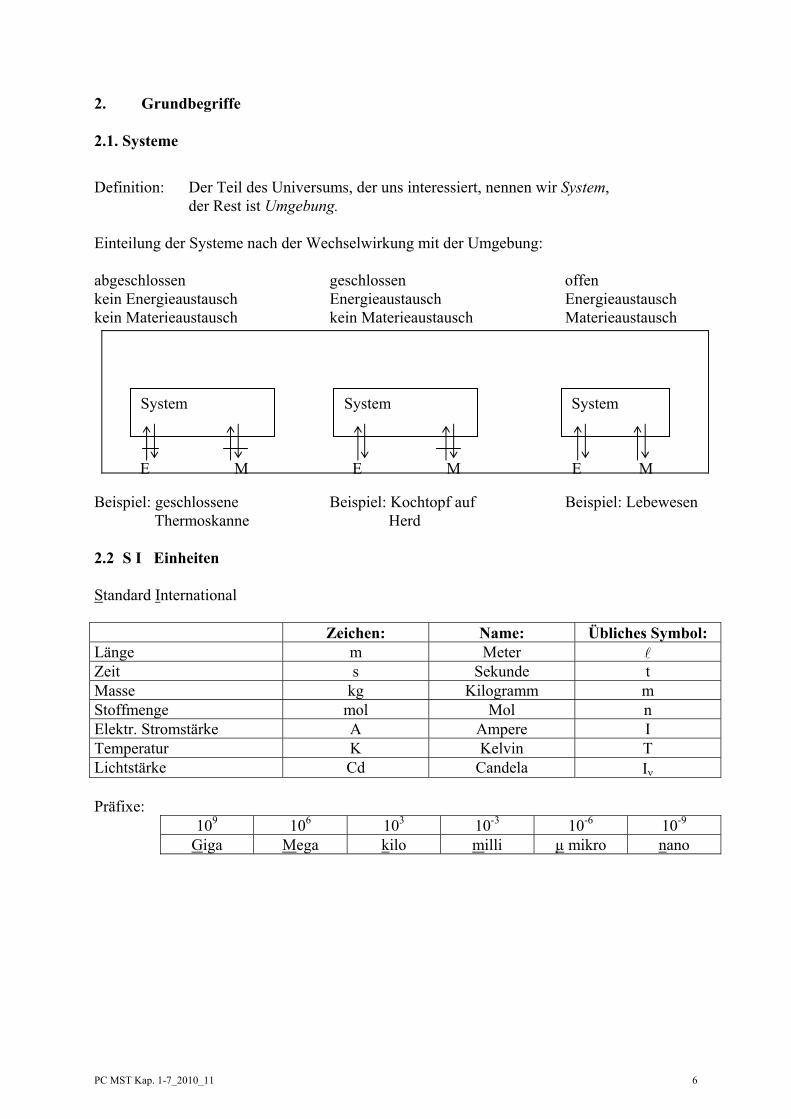
2. Grundbegriffe 2.1. Systeme
Definition: Der Teil des Universums, der uns interessiert, nennen wir System, der Rest ist Umgebung. Einteilung der Systeme nach der Wechselwirkung mit der Umgebung: abgeschlossen geschlossen offen kein Energieaustausch Energieaustausch Energieaustausch kein Materieaustausch kein Materieaustausch Materieaustausch
Beispiel: geschlossene Beispiel: Kochtopf auf Beispiel: Lebewesen Thermoskanne Herd 2.2 S I Einheiten Standard International Zeichen: Name: Übliches Symbol: Länge m Meter Zeit s Sekunde t Masse kg Kilogramm m Stoffmenge mol Mol n Elektr. Stromstärke A Ampere I Temperatur K Kelvin T Lichtstärke Cd Candela Iν Präfixe:
109 106 103 10-3 10-6 10-9 Giga Mega kilo milli µ mikro nano
System System System
E M E M E M

PC MST Kap. 1-7_2010_11 7

PC MST Kap. 1-7_2010_11 8
2.3 Stoffmenge Bei der Aufstellung chemischer Reaktionsgleichungen arbeitet man mit den Teilchenzahlen der beteiligten Stoffe. Die üblichen Stoffmengen, mit denen man umgeht, enthalten eine sehr große Zahl von Molekülen oder Atomen. Man führt daher eine Abkürzung für eine große Teilchenzahl ein. Def.: 1 Mol ist die Zahl von Teilchen in 12 g des Kohlenstoffisotops 12 C Die Teilchenzahl wird experimentell bestimmt. Man findet: 231 Mol 6 10 = ⋅ Teilchen. Diese Größe nennt man die Avogadrokonstante, NA, (früher Loschmidtzahl NL). NA = 23 16,022 10 mol−⋅ (exp. Bestimmung PC II) Eigentlich hat NA die Einheit „Teilchen pro mol“, man lässt aber das Wort „Teilchen“ weg. Aus der Definition und dem experimentellen Wert von NA ergibt sich sofort die Masse eines C-atoms. Masse ( )12
AC-atom N Masse⋅ = von 1 mol 12C-atomen ( ) ( )12
Am C N M C⋅ = Man nennt ( )M C die molare Masse oder Molmasse des Kohlenstoffs (der Ausdruck Molekulargewicht ist veraltet und wird nicht mehr verwendet). Auf Grund unserer Definition oben gilt: Definition: ( ) ⋅ -1M C = 12 g mol Damit können wir jetzt die Masse eines 12C-atoms ausrechnen:
m (12C) = ( ) -1
A
M C 12 g mol=N
⋅23 -16 10 mol⋅
-23=2 10 g⋅
Häufig verwendet man die sog. atomare Masseneinheit (mu)
Def.: 1 mu =1
12Masse eines 12C-atoms
= ( )112
12⋅ m C
Gleichung für m(12C) einsetzen:
1 mu = ( ) -1 -1
A A
M C1 12 g mol 1g mol= =12 N 12 N
⋅⋅ 23 -16 10 mol⋅
24u1 m = 1,66 10 g = 1 Da −⋅
In der Biochemie nennt man diese Größe ‘Dalton’ u1 m = 1 Da

PC MST Kap. 1-7_2010_11 9
Im Periodensystem werden die sog. relativen Molmassen angegeben
Mm Atom
mMasse des Atoms
Masse der Masseneinheit
m Atomm
NN
Molmasse des AtomsMolmasse der Masseneinheit
M AtomM Masseneinheit
ru
u
A
A
= =
= ⋅ =
=
( )
( )
( )( )
→ gleicher Zahlenwert wie die Molmasse aber ohne Einheit. Üblicherweise rechnet man nicht mit Teilchenzahlen, sondern mit Stoffmengen (Molzahlen).
Def.: Zahl der TeilchenStoffmenge = Zahl der Teilchen pro Mol
i Index für Stoff=ii
A
Nn =N
Die Masse einer Substanz, mi ist proportional der Zahl der Teilchen, Ni, d.h. mi = mi (1 Teilchen) . Ni Entsprechend gilt für die Molmasse Dividieren Mi = mi (1 Teilchen) . NA
m Ni i= = niM Ni A
Berechnung der Stoffmenge (Molzahl)
2.4 Konzentrationsangaben
Wenn man Stoffgemische hat, kann man deren Zusammensetzung auf unterschiedliche Art und Weise angeben: Massenanteil Massenbruch Volumenanteil Volumenbruch
Massenkonzentration: Yi = m
V gesamti
( )
i ii
i
m mW = =m m(gesamt)∑
i ii
i
V VV V(gesamt)
φ = =∑

PC MST Kap. 1-7_2010_11 10
Anteile werden häufig auch angegeben in %, ‰, ppm, ppb, ppt. In Zehnerpotenzen ausgedrückt bedeutet dies: 10-2 10-3 10-6 10-9 10-12
Stoffmengenanteil (Molenbruch): xn
nii
i
=∑
, x ii∑ = 1 , ni = Molzahl
wird häufig bei Mischungen benutzt. Stoffmengenkonzentration (kurz: Konzentration) Einheit: mol . m-3 oder mol . dm-3 V = Volumen der Lösung = n Vi m i
i,∑ wobei Vm,i das Molvolumen des
Stoffes i ist.
Abkürzung: „Molar“ M = moldm 3
Molalität: mnm
i
L= , ni = Molzahl
mL = Masse Lösungsmittel Einheit: mol . kg-1
Abkürzung: „Molal“ M* = molkg
Dichte: ρ = = = =Masse
VolumenmV
MolmasseMolvolumen
MVm
Das Molvolumen, Vm , ist das Volumen von einem Mol Teilchen. Massenanteil von Atomen in Molekülen: νi = stöchiometrischer Faktor von i
YM
Mii i=
ν Mi = Molmasse von Atom i
M = Molmasse des Moleküls Bsp. : Molvolumen von Fe
Molvolumen = Molmasse
Dichte, n
55,85 gMV =ρ=
1mol7,87 g
−⋅ 3 13 7,1cm mol
cm−
−=
ii
nc =V
ni = Molzahl

PC MST Kap. 1-7_2010_11 11
Bsp.: Volumen eines Atoms (Fe) Molvolumen = Volumen eines Teilchens . Avogadrokonstante
Vm = V (1 Teilchen) . NA
V (1 Teilchen) = 3 -1
m
A
V 7,1cm molN
⋅=
23 -16 10 mol⋅-23 31,18 10 cm= ⋅
Bei kugelförmigen Teilchen ist das Volumen V =43
3π r
rV cm
cm=⋅
=⋅ ⋅
⋅= ⋅
−−3
4118 10 3
41 4 103
23 33 8
π π,
, (Radius eines Eisenatoms)
2.5 Temperaturmessung a) Messverfahren: Erfahrung V = f( ) mit = Temperatur wenn p = const b) Kalibrierung = Definition von Fixpunkten:
1) Siedepunkt H2O bei p = 1.013 . 105 Pa = 100° C 2) Schmelzpunkt H2O bei p = 1.013 . 105 Pa = 0° C = V - V0 100 = V100 - V0
Temperaturskala:
= 100 (V - V0)/(V100 - V0) Die Größe (V100 - V0) ist für jeden Stoff, den man im Thermometer einsetzt, verschieden. Umgeformt ergibt sich:
= 100 V/(V100 - Vo) - 100 V0/(V100 - Vo) Man findet experimentell, dass für alle idealen Gase gilt: 100 V0/(V100 - V0) = 273,15 Setzen wir dies in Gleichung ein, ergibt sich
+ 273,15 = T = 100 V/(V100 - V0) oder V = V V
T100 0
100−
Man nennt T die absolute Temperatur, gemessen in Kelvin, K. Die Einteilung der Kelvinskala ist die Gleiche wie bei der Celsiusskala, der Zahlenwert des Schmelzpunktes des Eises ist jetzt aber 273,15 K, Vorteil: die absolute Skala ist unabhängig vom Stoff.
dividieren /100 = (V - V0)/(V100 - V0)
V100
V0
Celsiusskala
Kelvinskala 0 273,15 373,15 T/K
/100 C° -273,15 0
V A= ⋅ Länge = Fläche = A
V100 V V0

PC MST Kap. 1-7_2010_11 12
2.6 Energieaustausch System - Umgebung
Geschlossene und offene Systeme können mit der Umgebung Energie austauschen in Form von Arbeit W und Wärme Q. 2.6.1 Arbeit
Definition der Arbeit: Arbeit = Kraft . Weg dW = F ds⋅ = F ds cosα
= FFF
dxdydz
x
y
z
⎛
⎝
⎜⎜⎜
⎞
⎠
⎟⎟⎟⋅
⎛
⎝
⎜⎜⎜
⎞
⎠
⎟⎟⎟
Wenn Kraft und Weg gleiche Richtung z.B. in z-Richtung und Kraft konstant, so kann man die Gleichung einfach integrieren. Einschub Newton: Def.: Kraft ist eine Einwirkung auf einen Körper die
a) eine Bewegung oder b) eine Bewegungsänderung oder c) eine Deformation hervorruft.
⇒ hat Angriffspunkt, Richtung und Größe = Vektor Newton’sche Gesetze: 1) Trägheitsprinzip Jeder Körper verharrt im Zustand der Ruhe oder der gleichförmigen geradlinigen
Bewegung, wenn keine Kräfte auf ihn wirken. Beschleunigung = a = 0, wenn Kraft = F = 0 2) Aktionsprinzip a) Bei Einwirkung einer konstanten Kraft wird ein Körper gleichmäßig beschleunigt. b) Beschleunigung ist proportional der wirkenden Kraft (a ~F) c) Kraft (für eine bestimmte Beschleunigung) ist proportional zur Masse (F~m), Kraft = Masse . Beschleunigung F = ma (F, force; a, acceleration)
F α ds

PC MST Kap. 1-7_2010_11 13
Mit der Definition der Kraft ergibt sich für die Arbeit W = F . s
Einheit: = 1 kg m s-2 m = 1 kg m2 s-2 ≡def .
1 J
Das Vorzeichen der Arbeit wird folgendermaßen definiert:
Arbeit, die dem System zugeführt wird, ist positiv. Arbeit, die vom System geleistet wird, ist negativ. Herleitung verschiedener Arbeiten aus der Definitionsgleichung: Hubarbeit: W Fds= ∫
Wir betrachten nur eine Komponente des Vektors d.h. F hat immer die gleiche Richtung z.B. z-Richtung und F soll konstant sein.
→ W F z= ⋅ Masse m wird entgegen der Erdanziehung (= Gewicht = G = mg) von
der Kraft F von der Höhe z1 nach z2 gehoben
Man kann diese Gleichung auch anders formulieren:
mit a = dvdt
ergibt sich F = mdvdt
Mit v = dsdt
und a = d sdt
2
2 ergibt sich F = md sdt
2
2
Einheit: 1 Newton = 1 N = 1 kg . m . s-2 Häufig verwendet man auch den Impuls eines Teilchens: p = mV
Mit dieser Größe ergibt sich für die Kraft ( )d mvF =dt
3) Reaktionsprinzip Die Kräfte, die zwei Körper aufeinander ausüben, sind gleich groß aber entgegengerichtet. F12 = -F21
Einheiten: m: Masse, kg s: Weg, m t: Zeit, s v: Geschwindigkeit
v = dsdt
, ms
a: Beschleunigung
a = dvdt
d sdt
=2
2 , ms2
F: Kraft, F = a . m
ms
kg N2 1⋅ =
N = Newton
Anschaulich: Ein Newton ist die Kraft mit dem eine Tafel Schokolade (100 g) von der Erde angezogen wird. F = m . g = 0,1 kg . 9,81 ms-2 = 0,981 kg m s-2 ≈ 1 N, g = Erdbeschleunigung = 9,81 ms-2
Anschaulich: Ein Joule ist die Arbeit, die man braucht, um eine Tafel Schokolade vom Boden auf den Tisch (1 m Höhe) zu heben. W = 0,1 kg 9,81 m s-2 1 m = 0,981 kg m2 s-2 ≈1 N m ≈ 1 J

PC MST Kap. 1-7_2010_11 14
Dabei soll die Kraft F gerade gleich groß wie G sein (aber entgegen gesetzte Richtung). Eigentlich muss sie ein klein wenig größer als G sein, sonst könnten wird die Masse, m, nicht anheben. Dies wollen wir hier vernachlässigen.
Whub = = = =∫ ∫ ∫Fdz mgdz mg dz mgzz
z
z
z
z
z
z
z
1
2
1
2
1
2
1
2
= mg (z2 - z1) m in kg g = 9,8 ms-2 z2 - z1 = h = Höhe, m Ein Mensch, 70 kg, 1 Stockwerk (h = 3 m) Whub = 70 kg 9,8 m s-2 3 m = 2058 kg m2 s-2 = 2058 J Beschleunigungsarbeit: Ein Körper der Masse m wird reibungsfrei von der Geschwindigkeit v = 0 auf die Erdgeschwindigkeit ve beschleunigt.
Wbeschl.
=
= =
=
∫
∫ ∫
∫
Fds
mdvdt
ds mdsdt
dv
mvdv
s
s
s
s
s
s
s
s
1
2
1
2
1
2
1
2
Transformation der Grenzen des Integrals: Bei s1 soll v = 0 sein bei s2 soll v = ve sein
Wbeschl. = = =∫mvdv mv mvv V
e
e e
0
2
0
212
12
Beschleunigungsarbeit eines 100 m Läufers:
Endgeschwindigkeit ve = 10010
10 1ms
ms= −
( )Wbeschl. = ⋅
= =
−
−
12
70 10
3500 3500
1 2
2 2
kg ms
kg m s J
F = ma = mdvdt
mit dsdt
v= ergibt
sich
Einheiten: 1 kg m2 s-2 = 1 Nm = 1 J J = Joule

PC MST Kap. 1-7_2010_11 15
Volumenarbeit: Ein Körper mit dem Volumen V1 wird durch eine Kraft auf das Volumen V2 komprimiert (Luftpumpe).
Wvol = ∫ Fdss
s
1
2
Man definiert den Druck:
Druck =Kraft
Fläche, p = → = ⋅
FA
F p A
Das Volumen des Körpers ist gegeben durch V = A . s bzw. dV = A . ds (wenn A = konstant)
Wir ersetzen also in der Gleichung für die Arbeit ds = 1A
dV und F = pA
und erhalten
2
1
S
vols
1W p A dVA
= ⋅ ⋅∫
Transformation der Integrationsgrenzen: bei s1 haben wir das Volumen V1 bei s2 haben wir das Volumen V2
2
1
v
volv
W pdV= − ∫
Vorzeichen: Wenn man dem System Arbeit zuführt (zusammendrückt) wird dV negativ →Wvol negativ. Zugeführte Arbeit soll aber immer positiv gerechnet werden, daher negatives Vorzeichen
2
1
v
volv
W pdV= −∫ , wenn p = konstant → 2
1
v
volv
W pV= −
2 1= p(V V )− −
Einheiten
p = Kraft
FlächeN
mPa= =2
Pa = Pascal Luftdruck auf Meereshöhe ist P0 = 101325 Pa Dieser Druck hieß früher 1 atm. Da die Druckeinheit Pa sehr klein ist, verwendet man häufig 105Pa = 1 bar und 102Pa = 1 hPa = 1mbar = 1 hekto Pascal
Stempel F=pA A = Fläche
s1 s2
V1 V2
Einheiten:
W = 1 Pa . m3 = 1
23N
mm = 1N . m = 1 J

PC MST Kap. 1-7_2010_11 16
Elektrische Arbeit
Wel = ∫ Fdss
s
1
2
Elektrische Kraft F = Ladung . elektrische Feldstärke
= Q E
Zusammenhang Feldstärke - Potential (φ ): E = ddsφ
Setzen wir beide Gleichungen ein, so erhalten wir:
Wel 2 2 2
1 1 1
s s s
s s s
dQ E ds Q ds Q ddsφ
= = = φ∫ ∫ ∫
Transformation Integrationsgrenzen: Am Ort s1 haben wir das elektrische Potential 1φ Am Ort s2 haben wir das elektrische Potential 2φ
Wel ( )2 2 2
11 1
2 1Q d Q d Q Qφ φ φ
φφ φ
= φ = φ = φ = φ −φ∫ ∫
Man nennt die elektrische Potentialdifferenz 2 1 Uφ −φ = = Spannung. Damit ergibt sich: Wel = Q U Diese Gleichung kann man auch mit Hilfe der elektrischen Stromstärke ausdrücken:
I = Qt
LadungZeit
= , Q = I t
Damit ergibt sich für die Arbeit Wel = U I t Beispiel: Eine Glühbirne (100 W) brennt 5 Stunden. Wie groß ist die elektrische Arbeit? U = 220V, I = 0,45 A, t = 5h Wel = 220 V . 0,45 A . 5 h = 495 Wh = 0,495 kWh (Kilowattstunden). (Vom Elektrizitätswerk wird eine Spannung von 220 V geliefert, der Stromzähler im Haus misst die geflossene Ladung Q = I t. Daraus wird die gelieferte elektrische Arbeit berechnet, üblicherweise in kWh. (Preis 1 kWh ca. € 0,20).
Einheiten: U = Spannung, Volt, V I = Stromstärke, Ampere, A t = Zeit, Sekunde, s Q = Ladung, Coulomb, C 1V 1A 1 s = 1 Ws = 1 J⋅ ⋅ 1 W = 1 V 1 A W = Watt W = 1 As

PC MST Kap. 1-7_2010_11 17
2.6.2. Arbeit, kinetische Energie, potentielle Energie und innere Energie
Durch Zufuhr von Arbeit können wir die Energie eines Systems erhöhen (Energie = im System gespeicherte Arbeit). Die beschriebenen Arbeiten wirken auf das System unterschiedlich: 1. Arbeiten, die eine Beschleunigung des Systems hervorrufen dW = mvdv
W = 12
2mv E kin=
erhöhen die Bewegungsenergie (= kinetische Energie) des Systems. 2. Arbeiten gegen Kräfte, die nur eine Funktion des Ortes sind (konservative Kräfte, z.B.
Schwerkraft) und eine Änderung der Lage des Systems hervorrufen
W = Fds1
2
∫
Epot = − ∫ Fds1
2
erhöhen die potentielle Energie des Systems. (Minuszeichen drückt aus, dass Arbeit gegen die Kraft F verrichtet wird.
3. Arbeiten, die eine Erwärmung und/oder Verformung des Systems hervorrufen dW = dU W = ΔU (ohne dass die Lage und/oder Geschwindigkeit des Systems verändert wird), erhöhen die Innere Energie des Systems. Verallgemeinerung des Energieerhaltungssatzes: Abgeschlossene Systeme : dEkin + dEpot + dU = 0
Einschub In einem abgeschlossenen, reibungsfreien System gilt Ekin + Epot = E = konst. (‘Mechanische Energieerhaltung’) dEkin + dEpot = dE = 0
U = Innere Energie

PC MST Kap. 1-7_2010_11 18
Beispiel: Freier Fall aus der Höhe von 10 m. Wie groß ist die Geschwindigkeit beim Aufprall?
-dEpot = dEkin
− ∫mg dhh
h
A
E
= mvdvv
v
A
E
=∫
0
-mg(hE - hA) = ( )12
2 2m v vE A−
mghA = 12
2mv E
vE = 2gh A = 2 9 81 102⋅ ⋅−, ms m = 14 ms-1 ≈50 km . h-1
Einschub
Leistung = Energie
Zeit
P = dEdt
Beispiel: Mensch: Grundumsatzleistung: Energieverbrauch pro Zeit in Ruhezustand ist etwa 70 W. Bei Arbeitsleistung wird der Energieverbrauch pro Zeit etwa verdoppelt, d.h.
140 W. Die nach außen abgegebene Dauerleistung eines Menschen ist also etwa 70 W.
→ Arbeit an einem Tag: W = P . t = 70 W . 8h = 560 Wh Kosten elektrischer Arbeit: 1 kWh = € 0,20. Daraus folgt, dass der Energiewert der Arbeit eines Menschen pro Tag ca. 10 Cent beträgt.
Einheit:
J s-1 = 1 W
W = Watt Arbeit: P =
dWdt
Wärme: P = dQdt
Körper mit Masse m 10 m
hA vA= 0
hE vE

PC MST Kap. 1-7_2010_11 19
2.6.3. Wärme und Wärmekapazität Bringt man einen heißen Körper mit einem kälteren in Kontakt, so erwärmt sich der eine und der andere kühlt sich ab. Es fließt Energie (Wärme) vom heißeren zum kälteren Körper. Man beobachtet eine Temperaturerniedrigung des heißen Körpers, eine Temperaturerhöhung des kalten Körpers. Die Temperaturerhöhung dT ist proportional zur zugeführten Wärme. dQ = C dT C = Proportionalitätsfaktor = ‘Wärmekapazität’ in JK-1 C = dQ/dT Ist die Wärmekapazität konstant, lässt sich die Gleichung einfach integrieren:
dQ C dTT
TQ
= ∫∫1
2
0
daraus folgt Q = C (T2 - T1 ) = C Δ T
Die Wärmekapazität ist proportional zur Stoffmenge (wird gemessen als Masse m oder als Molzahl n). Man erhält: c = C/m ‘spezifische Wärmekapazität’ in J K-1 kg-1
c = C/n ‘molare Wärmekapazität in J K-1 mol-1 Man definiert: Die dem System zugeführte Wärmemenge ist positiv Die vom System abgeführte Wärmemenge ist negativ. Wärmeaustausch (Wärmebilanz) Ein Körper A mit Temperatur T1 wird mit einem Körper B der Temperatur T2 in Kontakt gebracht. Wie groß ist die Endtemperatur T3? Wie groß ist die geflossene Wärme?
Anfangszustand Endzustand
Abgeschlossenes System: kein Wärmeaustausch mit der Umgebung
A B T1 T1 > T2 T2 CA CB ↓ A B T3 T3 CA CB

PC MST Kap. 1-7_2010_11 20
Wärme fließt von A nach B bis sich eine gemeinsame Endtemperatur eingestellt hat (‘thermisches Gleichgewicht’). Da nach außen keine Wärme abgegeben wird, muss die von A abgegebene Wärme gleich der von B aufgenommenen sein, d.h. QA + QB = 0 also ist QA = - QB mit QA = CA (T3 - T1) und QB = CB (T3 - T2) Eingesetzt und aufgelöst nach der Endtemperatur T3 ergibt sich:
T3 = C T C T
C CA B
A B
1 2++
= m c T m c T
m c m cA A B B
A A B B
1 2++

PC MST Kap. 1-7_2010_11 21
3. Ideale Gase In unserer Umgebung kommen Stoffe in verschiedenen Formen (‘Aggregatzuständen’) vor. Wir unterscheiden • gasförmige Stoffe verteilen sich gleichmäßig über den zur Verfügung stehenden Raum. • flüssige Stoffe im schwerelosen Zustand nehmen sie kugelförmige Gestalt an, auf der Erde sammeln sie sich an der tiefsten Stelle des Gefäßes und nehmen dessen Gestalt an. • feste Stoffe verändern ihre Form nicht. Alle Stoffe können durch Zufuhr von Energie aus dem festen in den gasförmigen Zustand gebracht werden. Sublimieren schmelzen verdampfen fest flüssig gasförmig
Das Verhalten eines Systems, in dem die Teilchen keine Wechselwirkungen zeigen, lässt sich am einfachsten beschreiben. Dies wird daher als erstes behandelt. 3.1 Experimentelle Befunde über Gase Allgemein beobachtet man, dass das Volumen V, eines Stoffes abhängt von der Stoffmenge n, dem Druck p, und der Temperatur T, d.h.. es gibt einen Zusammenhang V = f (n, p, T) wobei f eine Funktion ist, die wir im folgenden experimentell bestimmen wollen.
Fläche
AT
starke Wechselwirkung der Teilchen
mittelstarke Wechselwirkung der Teilchen
schwache oder keine Wechselwirkung der Teilchen
Zur Messung benutzen wir einen Zylinder, der mit einem reibungsfrei beweglichen, aber dicht schließenden Stempel versehen ist. Im Zylinder befindet sich das Gas, der Stempel kann mit verschiedenen Gewichten belastet werden und übt
dann einen genau bekannten Druck pa = FA
aus.
(Index a: Druck von außen)
Thermometer
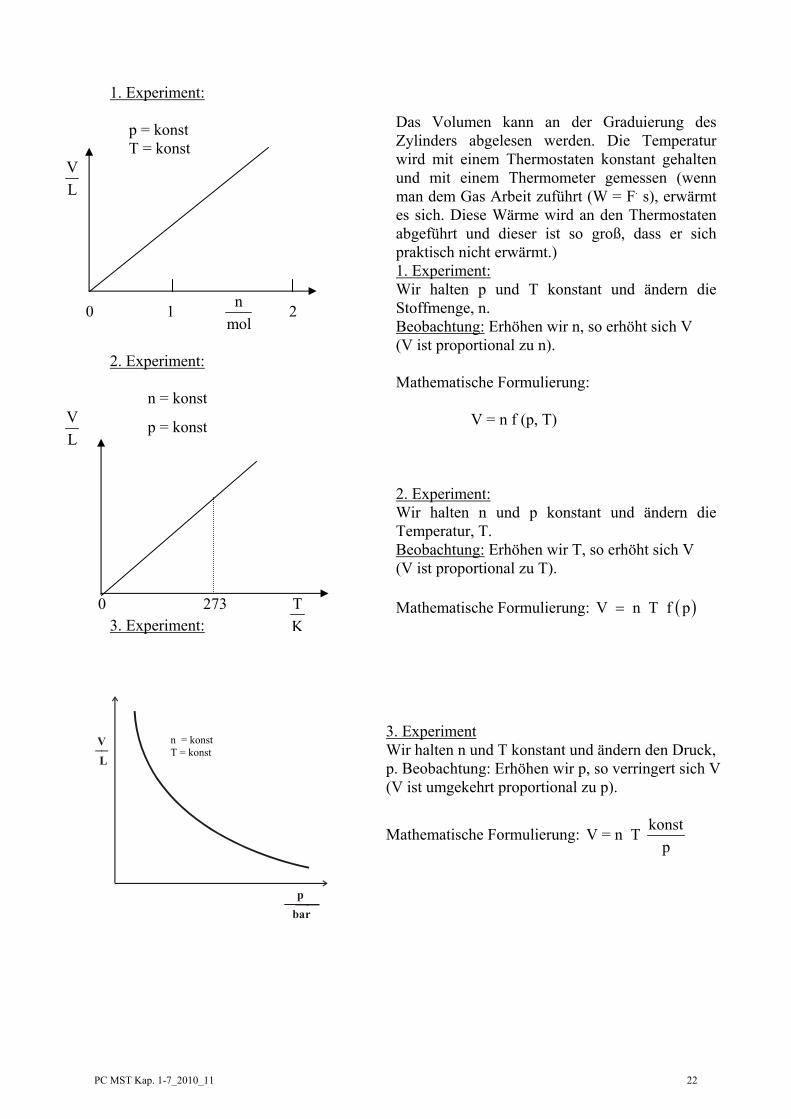
PC MST Kap. 1-7_2010_11 22
Das Volumen kann an der Graduierung des Zylinders abgelesen werden. Die Temperatur wird mit einem Thermostaten konstant gehalten und mit einem Thermometer gemessen (wenn man dem Gas Arbeit zuführt (W = F. s), erwärmt es sich. Diese Wärme wird an den Thermostaten abgeführt und dieser ist so groß, dass er sich praktisch nicht erwärmt.)
1. Experiment: Wir halten p und T konstant und ändern die
Stoffmenge, n. Beobachtung: Erhöhen wir n, so erhöht sich V (V ist proportional zu n). Mathematische Formulierung: V = n f (p, T)
2. Experiment: 2. Experiment: Wir halten n und p konstant und ändern die
Temperatur, T. Beobachtung: Erhöhen wir T, so erhöht sich V (V ist proportional zu T). Mathematische Formulierung: ( )V n T f p=
V
p
__L
bar___
1. Experiment: p = konst T = konst VL
0 1 nmol
2
2. Experiment:
n = konst VL
p = konst
3. Experiment:
0 273 TK
3. Experiment Wir halten n und T konstant und ändern den Druck, p. Beobachtung: Erhöhen wir p, so verringert sich V (V ist umgekehrt proportional zu p).
Mathematische Formulierung: konstV = n T p
n = konst T = konst

PC MST Kap. 1-7_2010_11 23
Die Konstante kann aus einem Satz (V, n, T, p) zusammengehörenden Daten bestimmt werden.
Konst = pVnT
= 101325 Pa2N m
322,4 dm−
⋅( )3110 m
273,15 K 1 mol
−
⋅
= 8,314 N m mol-1K-1 = 8,314 J mol-1 K-1 = R = ‘allgemeine Gaskonstante’ Da man für jedes Gas die gleiche Konstante erhält, nennt man sie die allgemeine Gaskonstante, R. Damit ergibt sich schließlich als Endgleichung pV = nRT Man nennt Gase, deren Verhalten sich mit dieser Gleichung exakt beschreiben lässt, ‘ideale Gase’, entsprechend ist dies die ‘ideale Gasgleichung’. (Gase, deren Verhalten sich nur angenähert damit beschreiben lässt, heißen ‘reale Gase’).
Manchmal benutzt man auch das Molvolumen in der idealen Gasgleichung. Mit mVV n
= erhält
man dann pVm = RT 3.2. Kinetische Gastheorie (Modell) Annahmen: 1) Gas besteht aus einzelnen Teilchen (Atome oder Moleküle)
2) Eigenvolumen der Teilchen << gegen Gefäßdimension 3) Teilchen in ungeordneter Bewegung 4) Teilchen verhalten sich wie starre Kugeln, d.h. sie üben keine anziehenden
oder abstoßenden Wechselwirkungen aufeinander aus. 5) Energie- und Impulserhaltungssatz gilt für Stöße der Teilchen untereinander
und mit der Wand. Berechnung des Druckes durch die Stöße der Teilchen auf die Wand nach diesem Modell
1) Aus der Mechanik: pFA
KraftFläche
= =
( )dv d mvF m a m
dt dt= ⋅ = = , mv ist der Impuls eines Teilchens.
Normalbedingungen: p0 = 101325 Pa n = 1 mol T = 273,15 K V = 22,4 dm3 (1 L = 1 dm3)

PC MST Kap. 1-7_2010_11 24
2) Die Kraft auf die Wand lässt sich also errechnen nach
Kraft = Im pulsänderungZeit
⋅ Zahl der auftreffenden Teilchen
d(mv)F Zdt
= ⋅
3) Berechnung Impulsänderung: Z Ein Teilchen fliegt mit dem Impuls xmv auf die Wand zu, wird reflektiert (dabei dreht sich das
Vorzeichen der Geschwindigkeit um) und fliegt mit dem Impuls xmv− davon. Impulsänderung des Teilchens |2 m v x|. Der auf die Wand übertragene Impuls ist entsprechend auch |2 m v x|.
( ) xd mv 2 mv→ = . 4) Berechnung Zahl der auftreffenden Teilchen.
Im Behälter sind NV
Teilchen pro Volumen enthalten. Zu jedem Zeitpunkt bewegt sich die
Hälfte in positiver, die andere Hälft in negativer x-Richtung, d.h. 12
NV
Teilchen
bewegen sich auf die Fläche A zu. Wie viele treffen in der Zeit dt auf?
Die Teilchen haben die mittlere Geschwindigkeit v xdxdt
= . Daraus folgt: Alle Teilchen, die
näher als xdx v dt= entfernt sind, treffen in dt auf, alle Teilchen, die weiter als xdx v= dt entfernt sind, treffen nicht auf
→ Zahl der auftreffenden Teilchen: x1 NZ v dtA2 V
=
5) Gesamtgleichung:
Kraft = ( ) xx
d mv 2 mv 1 NF Z v dt Adt dt 2 V
= =
und für den Druck erhalten wir:
2x
F Np mvA V= =
Fläche A

PC MST Kap. 1-7_2010_11 25
Wie hängt vx mit der Gesamtgeschwindigkeit v zusammen? Alle Geschwindigkeitskomponenten sind gleich groß, d.h. v v vx y z
2 2 2= =
v2 = v v vx y z2 2 2+ +
= 313
2 2 2v v vx x→ =
Im folgenden betrachten wir die sog. „mittlere quadratische Geschwindigkeit“ v 2 , d.h. erst quadrieren, dann mitteln. Es ergibt sich dann
21pV Nmv3
=
Umrechnung von Teilchenzahlen auf Molzahlen: Setzen wir die Definition der Molzahl
nN
N A
= ein, erhalten wir als Ergebnis der Modellbetrachtung:
2A
1pV nN mv3
→ =
Für die kinetische Energie eines Teilchens hatten wir erhalten 2kin
1 mv2
=ε (siehe Kap. 2.6.1).
Betrachten wir die Mittelwerte und berücksichtigen wir, dass es sich um eine linear
fortschreitende (translatorische) Bewegung handelt, so erhalten wir
ε εkin trans mv= =12
2 oder 2 2ε trans mv=
ε trans = mittlere kinetische Energie der Translation eines Teilchens. Entsprechend erhalten wir für die mittlere kinetische Energie von einem Mol Teilchen:
trans A transN Eε = Setzen wir dies noch ein, ergibt sich
pV = n23
Etrans → Ergebnis der Modellbetrachtung
pV = nRT → Ergebnis der Experimente Daraus erhalten wir schließlich
RT = 23
32
E E RTtrans trans→ =
d.h. die mittlere kinetische Energie der Translation der einzelnen Teilchen ist mit der Temperatur verknüpft. Der Vergleich zwischen Experiment und Modell ergibt die quantitative Verknüpfung thermodynamischer Größen (z.B. T) mit mechanischen Größen (z.B. Geschwindigkeit).
Vergleich

PC MST Kap. 1-7_2010_11 26
Rechnet man nicht auf molare Größen um, so erhält man
A trans
transA
2pV n N n RT3
3 R T2 N
= ε =
ε = transε = mittlere kinetische Energie pro Teilchen
Man nennt BA
R kN
= ‘Boltzmann-Konstante’,
Dies ist also eine Gaskonstante pro Molekül. Bislang hatten wir die Temperatur empirisch über das Volumen unseres Thermometers gemessen. Unsere Herleitung zeigt, dass die Temperatur mit der mittleren kinetischen Energie von Teilchen verknüpft ist. Der Umrechnungsfaktor von Temperatur in mittlere Energie ist die ‘Boltzmann-Konstante’ (für ein Teilchen) oder die ‘Gaskonstante’ (für ein Mol Teilchen). (Wenn man heute die Temperatur neu einführen würde, würde man vermutlich die Größe ‘RT’ als ‘Temperatur’ definieren. In der Thermodynamik taucht T auch immer verknüpft mit R als die Größe RT auf). Die Geschwindigkeit eines Teilchens lässt sich wie folgt errechnen:
2trans B
1 3m v k T2 2
ε = =
2 B B A
A
3 k T 3 k TN 3 RTvm mN M
→ = = = Bk NA = Rm NA = M
⎧⎨⎩
↑ Erweiterung mit NA „Wurzel aus dem mittleren Geschwindigkeitsquadrat“ Beispiel: O2
1 12
1
3 8,314 J mol K 300 Kv32 g mol
− −
−
⋅=
1 1 2 2
1 3 3 2 3 2
J mol K K Nm kg m mg mol 10 kg 10 s kg 10 s
− −
− − − −
⎡ ⎤= = =⎢ ⎥
⎣ ⎦
m484=
310 km 3600s
−⋅ ⋅
1
h
1741 km h−=
Für die mittlere Geschwindigkeit in einer Richtung, z.B. x-Richtung gilt:
2 2 Bx x B x
k T1 1mv k T v2 2 m
ε = = → =
pV =

PC MST Kap. 1-7_2010_11 27
3.3 Mischungen idealer Gase Mischt man verschiedene, ideale Gase, so verhält sich jedes Gas so, als ob es alleine vorhanden wäre. Dies muss so sein, da bei einem idealen Gas keine Wechselwirkungen zwischen den Teilchen auftreten. Entsprechend definiert man einen Partialdruck, pi. Def.: Der Partialdruck des Gases i ist der Druck, den man messen würde, wenn dieses Gas im betrachteten Volumen alleine vorhanden wäre. Der Partialdruck hängt mit dem Molenbruch zusammen. Dies ergibt sich aus folgender Überlegung: p V = n RT (1) Das Gas 2 wird jetzt vollständig entfernt, dabei nimmt der Druck ab von p auf p1 (da nur noch Gas 1 vorhanden ist. p1V = n1RT (2) Umgekehrt gilt, wenn Gas 1 vollständig entfernt wird p2V = n2RT (3) Addieren wir beide Gleichungen ergibt sich: (p1 + p2) V = (n1 + n2) RT (4) mit p1 + p2 = p und n1 + n2 = n Dividieren wir Gleichung (2) durch (4) ergibt sich
1p V
1 2(p p ) V+1n RT
=1 2(n n ) RT+ 1x= , mit x1 =
nn n
1
1 2+( = Molenbruch)
Allgemein gilt also: p pii∑ = (= Gesamtdruck) und
pp
nn
xi
i
i
ii∑ ∑
= =
3.4 Stoßzahlen und mittlere freie Weglänge Wir schätzen zunächst den mittleren Abstand der Teilchen ab. Hierzu betrachten wir die Zahl der Teilchen pro Volumen in einem Gas.
Aus pV = n R T = N
NRT
A
erhalten wir: NV
= p N
R TA⋅
Normalbedingungen: p0 = 101325 Pa, T0 = 273 K
= 101523 Pa 23 16 10 mol−⋅
8,314 Pa 3 1m mol− 1K− 273 K
25 3 19 32,65 10 m 2,65 10 cm− −= ⋅ =

PC MST Kap. 1-7_2010_11 28
Wir denken uns die Moleküle in einem Würfel von = 1cm Kantenlänge gleichmäßig verteilt, d.h. jedes Molekül hat ein bestimmtes Volumen a3 zur Verfügung. Dann gilt: Volumen des Würfels = Volumen für ein Teilchen ⋅ Zahl der Teilchen 3 = a3 ⋅ N
Abschätzung der Durchmesser der Teilchen (siehe z.B. Kap. 2.5) ergibt: d (Fe) ≈ 2,8 . 10-8 cm, für N2 erhält man d (N2) ≈ 3,2 . 10-8 cm
Die Teilchen fliegen mit großer Geschwindigkeit ( v ≈ 500 ms-1) umher, dabei stoßen sie zusammen.
Flugbahn eines Teilchens. Mittelwert über alle Strecken isΔ isΔ = λ = mittlere freie Weglänge
Def.: Man nennt die Zahl der Zusammenstöße, die ein Teilchen pro s erfährt die ‘Stoßzahl’ (Z). Def.: Der Weg, den das Teilchen zwischen zwei Stößen zurücklegt, ist die ‘mittlere freie Weglänge’ (λ)
a3 = 3 1
NVN N
V= =
a = 1
13
NV
⎛⎝⎜
⎞⎠⎟
a = 1
2 65 1019
13
, ⋅⎛⎝⎜
⎞⎠⎟
cm = 3,3 . 10-7 cm
Teilchen

PC MST Kap. 1-7_2010_11 29
Berechnung der Stoßzahl
Wir stellen uns vor, dass das Molekül A sich bewegt, die anderen Moleküle (B, C, D usw.) sind fixiert. Das Molekül A fliegt mit der Geschwindigkeit v und legt dabei in der Zeit dt den Weg
( vdxdt
= ) → dx = vdt zurück.
⇒ Alle Moleküle, deren Mittelpunkte sich innerhalb des Zylinders befinden, werden gestoßen ⇒ C und D nicht, B wird gestoßen. ⇒ Die Zahl der Zusammenstöße = Zahl der Moleküle im Zylindervolumen Der Zylinder hat einen Radius von rA + rB rA = Radius von Molekül A rB = Radius von Molekül B Die Grundfläche des Zylinders ist dann: π (rA + rB)2. Diese Größe nennt man auch den Stoßquerschnitt, σ σ = π(rA + rB)2 Das Volumen des Zylinders ist dann Vzyl = σ dx = σ vdt
Die Zahl der Teilchen (A) pro Volumeneinheit ist N A
V( )
. Wir erhalten damit für die Zahl der
Teilchen im Zylinder ( = Zahl der Zusammenstöße, d ZS) :
d ZS = zylN(A) N(A)V vdt
V V= σ
Falls A und B gleichartige Moleküle, dann ist Zylinderradius = 2 rA

PC MST Kap. 1-7_2010_11 30
Wir nennen d ZS N(A)Z 'Stoßzahl ' v
dt V= = = σ
Eigentlich ist Z keine Zahl, sondern diese Größe müsste Stoßhäufigkeit oder Stoßfrequenz heißen. Für v müssen wir eine mittlere Geschwindigkeit v A einsetzen und wir erhalten schließlich
1 AN(A)Z v
V= σ
= Zahl der Zusammenstöße, die ein A-Teilchen erfährt, wenn alle anderen Teilchen sich nicht bewegen. Einschub
Bisher (Kap. 3.2) haben wir mit der Größe vRT
MAA
2 3= = Wurzel aus dem mittleren
Geschwindigkeitsquadrat gearbeitet. Wir müssen hier aber die mittlere Geschwindigkeit
vRTMA
A=
8π
benutzen. Diese wird in Kap. 3.8 hergeleitet.
Da die anderen Teilchen sich aber auch bewegen, müssen wir die Relativgeschwindigkeit zweier Teilchen betrachten. Anschaulich:
|vrel| = 0 |vrel| = |vA| + |vB| v v vrel A B
2 2 2= + Situation, die im Mittel auftritt. Die Beziehung ( ) ( ) ( )v v vrel A B
2 2 2= + ergibt sich auch, wenn man ein korrekte Mittelung
(Integration über alle Orientierungen) durchführt. Sind die Moleküle A und B gleich, so ergibt sich
( ) ( ) ( ) ( ) ( ) ( ) ( )v v v v v v vAA A A A AA A A2 2 2 2 2 2
2 2 2= + = → = = Für die mittlere Relativgeschwindigkeit zwischen den Molekülen A und B ergibt sich:
A
vA
vB
A B vA vB
vrel
vA
vB
A B

PC MST Kap. 1-7_2010_11 31
( ) ( ) ( )2 2 2 B AAB A B
A B A B
8 RT 8 RT 8 RT M Mv v vM M M M
⎛ ⎞+= + = + = ⎜ ⎟π π π ⎝ ⎠
Man nennt die Größe B A
A B
M MM M
⎛ ⎞⋅= μ =⎜ ⎟+⎝ ⎠
reduzierte Masse und erhält damit
vRT
AB =8πμ
‘Mittlere Relativgeschwindigkeit von zwei verschiedenen Teilchen’
Falls die beiden Moleküle gleich sind (MA = MB), ergibt sich:
AMμ =2
und AA AA
8 RTv 2 v 2M
= =π
‘Mittlere Relativgeschwindigkeit von zwei gleichen Teilchen’
Beispiel: Berechnung der Stoßzahl für N2: ( )N AV
cm= ⋅ −2 65 1019 3, (wie vorher berechnet)
σA = π(rA + rB)2 = π(2rA)2 = π (d (N2))2 = 3,14 (0,32 . 10-7cm)2 = 0,32 . 10-14 cm2
2 2 1 1
A 1 3A
8 RT 8 8,314 kg m s mol K 273 KvM 3,14 28 mol 10 kg
− − −
− −
⋅ ⋅= =
π ⋅ ⋅
= 454 m s-1 (Vergleich vRT
Mm sA
A
2 13493= = − )
Z1A = ( )N AV
vA A⋅ ⋅σ 2
19 3 14 2 12,65 10 cm 0,32 10 cm 454 m s 2− − −= ⋅ ⋅ ⋅ ⋅
9 1 5,4 10 s −= ⋅ = Zahl der Zusammenstöße, die ein A-Teilchen erfährt, wenn alle anderen sich auch bewegen Die Gesamtzahl der Zusammenstöße aller A-Teilchen mit allen anderen A-Teilchen , wenn alle sich bewegen, ist dann ZA :
( ) ( )AA A A
N A N A 1Z v 2V V 2
σ= ⋅
9 1 19 3 28 3 115,4 10 s 2,65 10 cm 7,1 10 cm s2
− − − −= ⋅ ⋅ ⋅ ⋅ = ⋅
Der Faktor 12
taucht hier auf um die Stöße zwischen Teilchen gleicher Nummerierung nicht
doppelt zu zählen, d.h. Stoß 1→ 2 ist identisch mit Stoß 2 → 1.

PC MST Kap. 1-7_2010_11 32
Die Gesamtzahl der Zusammenstöße aller Moleküle A mit allen Molekülen B ist schließlich
( ) ( )AB AB AB
N A N BZ v
V Vσ= (wichtige Größe für chemische Reaktionen)
Mittlere Freie Weglänge Die mittlere freie Weglänge λ ist der Weg, den 1 Teilchen zwischen 2 Zusammenstößen zurücklegt. Wenn es mit der Geschwindigkeit vA fliegt, so ist der zurückgelegte Weg: dx = vAdt Mit der mittleren Geschwindigkeit ergibt sich entsprechend für den mittleren Weg: λ = v A dt Für die Zahl der Zusammenstöße eines Teilchens hatten wir erhalten
dZ1A = ( ) ( )
AA AN A N Av dt v 2 dt
V Vσ σ=
Wir lösen nach v A dt auf und setzen in die Gleichung für λ ein: ( )λσ
=dZ A
N AV
1
2
Da nach einem Zusammenstoß die freie Weglänge beendet ist(dZ1A = 1), ergibt sich:
( )λσ
=1
2N AV
Bsp. N2: λ =⋅ ⋅ ⋅− −
12 65 10 0 32 10 219 3 14 2, ,cm cm
= 8,3 . 10-6 cm
Vergleich Durchmesser mittlerer Teilchenabstand mittlere freie Weglänge 3,2 . 10-8cm 33 . 10-8 cm 830 . 10-8 cm Faktor ≈ 10 Faktor ≈ 20 Beispiel: Fußballspiel, Größe des Feldes: 105 m . 70 m, 23 Personen. Wir nehmen eine ideale, kugelförmige Person an, die einen Durchmesser von ca. 2 m (mit ausgestreckten Armen) haben soll. Wenn wir die 23 Personen gleichmäßig auf dem Feld verteilen, so wird ihr mittlerer Abstand (gleiche Überlegung wie der mittlere Abstand der Teilchen, Kap. 3.4)
a ≈ ≈105 70
2318
m mm .

PC MST Kap. 1-7_2010_11 33
Wenn diese Personen mit verbundenen Augen (ohne Wechselwirkungen aufeinander) auf dem Feld herumlaufen, dann verhalten sie sich wie Teilchen in einem idealen Gas. Wir nehmen für die Fußballspieler die gleichen Faktoren an wie beim idealen Gas zwischen Durchmesser der Spieler, ihrem mittleren Abstand und ihrer mittleren freien Weglänge. Wir erhalten dann für die mittlere freie Weglänge eines Spielers 18 m 20 360 m⋅ =
Einschub: Mittelwerte und Verteilungen 1) Mittelwertbildung bei einer geringen Zahl von Messungen. Wir führen die gleiche Messung mehrere Male (insgesamt Nges) durch und erhalten als
Ergebnis die Messwerte x1, x2, x3 usw. Daraus berechnen wir den Mittelwert der Messungen
folgendermaßen: xx
N
kk
ges
=∑
(1)
Zahl aller Messwerte = Anzahl der Summanden: Nges 2) Mittelwertbildung bei einer großen Zahl von Messungen Wir haben sehr viele Messwerte gemessen, von denen einige gleich sind: x1 wurde N1 mal gemessen x2 wurde N2 mal gemessen usw. Wir bilden den Mittelwert nach der folgenden Gleichung:
xN x N x
N NN xN
i i
i
=++
=∑∑
1 1 2 2
1 2
...................
(2)
Wir erhalten durch die Bildung der Gruppen i eine drastische Verringerung der Zahl der
Summanden. Der Index i zählt die Zahl der Gruppen, d.h. der Summanden. Mit N Ni
iges∑ = = Gesamtzahl der Messungen ergibt sich dann
xN x
NN
Nx
i ii
ges
i
gesi= =
∑∑ = i i
ih x∑ mit i
iges
NhN
= (3)
Spieler (Durchmesser): 2 m Abstand der Spieler: 18 m mittlere freie Weglänge der Spieler: 360 m Der Stoß mit der Spielfeldbegrenzung wird nicht gerechnet. Nach ca. 360 m stößt in diesem Fall Spieler Nr. 19 auf Nr. 16.
Faktor ca. 10
Faktor ca. 20

PC MST Kap. 1-7_2010_11 34
Wir nennen ii
ges
N hN
= die Häufigkeit für das Auftreten des Messwertes xi, und es gilt
ii
ii ges
Nh 1
N= =∑
∑
Wenn Nges sehr groß wird, so wird die Häufigkeit unabhängig von der Zahl der Messwerte Ni
und man nennt dann diese Größe die Wahrscheinlichkeit für das Auftreten des Messwertes xi
i
i
Nges
ges
Nlim pN
→∞
= (4)
Dabei ist ii
p 1=∑ und i ix p x=∑ (5)
Beispiele: a) Die Ergebnisse sind diskrete Zahlen wie z.B. beim Würfeln Wir berechnen die mittlere Punktzahl beim Würfeln nach Gleichung (5). Die Wahrscheinlichkeit für das Würfeln einer bestimmten Zahl ist gleich, d.h.
1 2 3 4 5 616
p = p = p = p = p = p =
Damit erhalten wir für den Mittelwert der Punktzahl bei einer großen Zahl von Würfen.
x = ⋅ + ⋅ + ⋅ + ⋅ + ⋅ + ⋅ =16
116
216
316
416
516
6 3 5,
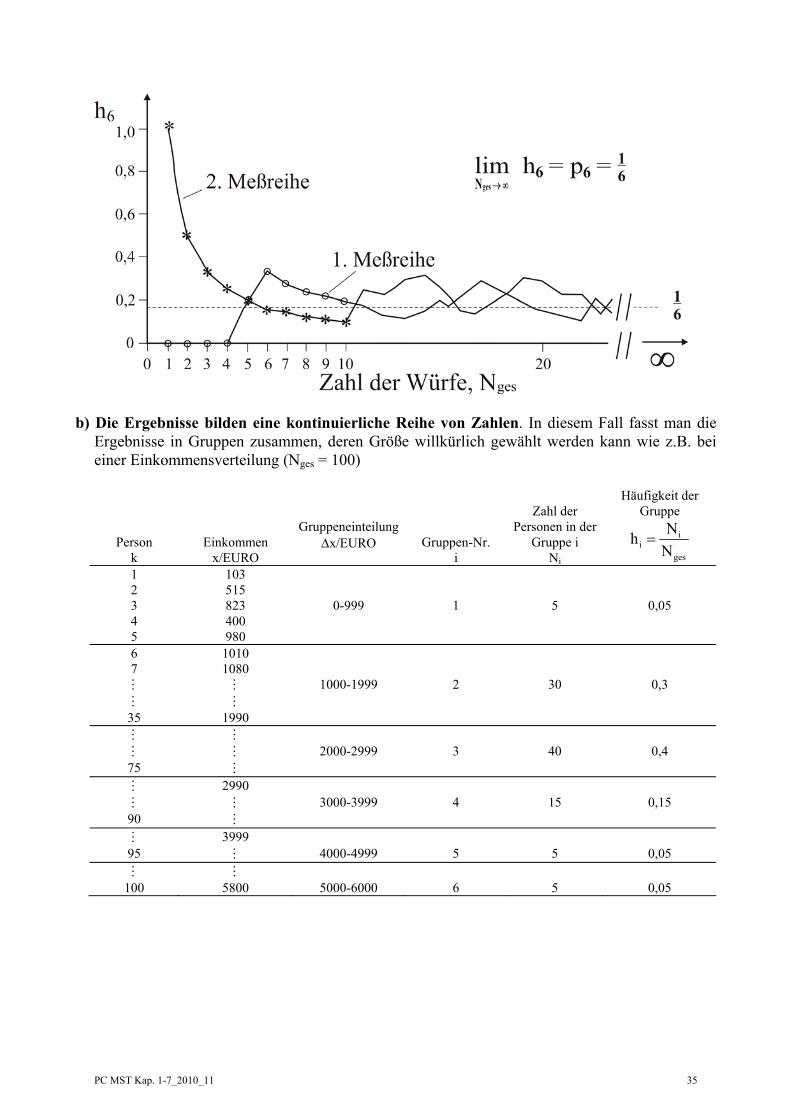
Zusammenhang zwischen Häufigkeit hi und Wahrscheinlichkeit pi: Wir würfeln mit einem Würfel insgesamt Nges mal, notieren die Ergebnisse und berechnen die Häufigkeit des Auftretens z.B. einer 6 1. Messreihe Ergebnis des Würfelns: 2, 3, 5, 1, 6, 6, 4, 3, 1, 2, ....................usw.
Häufigkeit: 66
ges
NhN
= : ,102,
92,
82,
72,
62,
51,
40,
30,
20,
10 ....................usw.
2. Messreihe Ergebnis des Würfelns: 6, 1, 2, 4, 5, 3, 3, 2, 1, 5, ........................usw.
Häufigkeit: 66
ges
NhN
= : ,101,
91,
81,
71,
61,
51,
41,
31,
21,
11 .......................usw.
PC MST Kap. 1-7_2010_11 35
b) Die Ergebnisse bilden eine kontinuierliche Reihe von Zahlen. In diesem Fall fasst man die Ergebnisse in Gruppen zusammen, deren Größe willkürlich gewählt werden kann wie z.B. bei einer Einkommensverteilung (Nges = 100)
Person k
Einkommen x/EURO
Gruppeneinteilung Δx/EURO
Gruppen-Nr. i
Zahl der
Personen in der Gruppe i
Ni
Häufigkeit der Gruppe
ii
ges
NhN
=
1 103 2 515 3 823 0-999 1 5 0,05 4 400 5 980 6 1010 7 1080
1000-1999 2 30 0,3
35 1990 2000-2999 3 40 0,4
75 2990 3000-3999 4 15 0,15
90 3999
95 4000-4999 5 5 0,05
100 5800 5000-6000 6 5 0,05

PC MST Kap. 1-7_2010_11 36
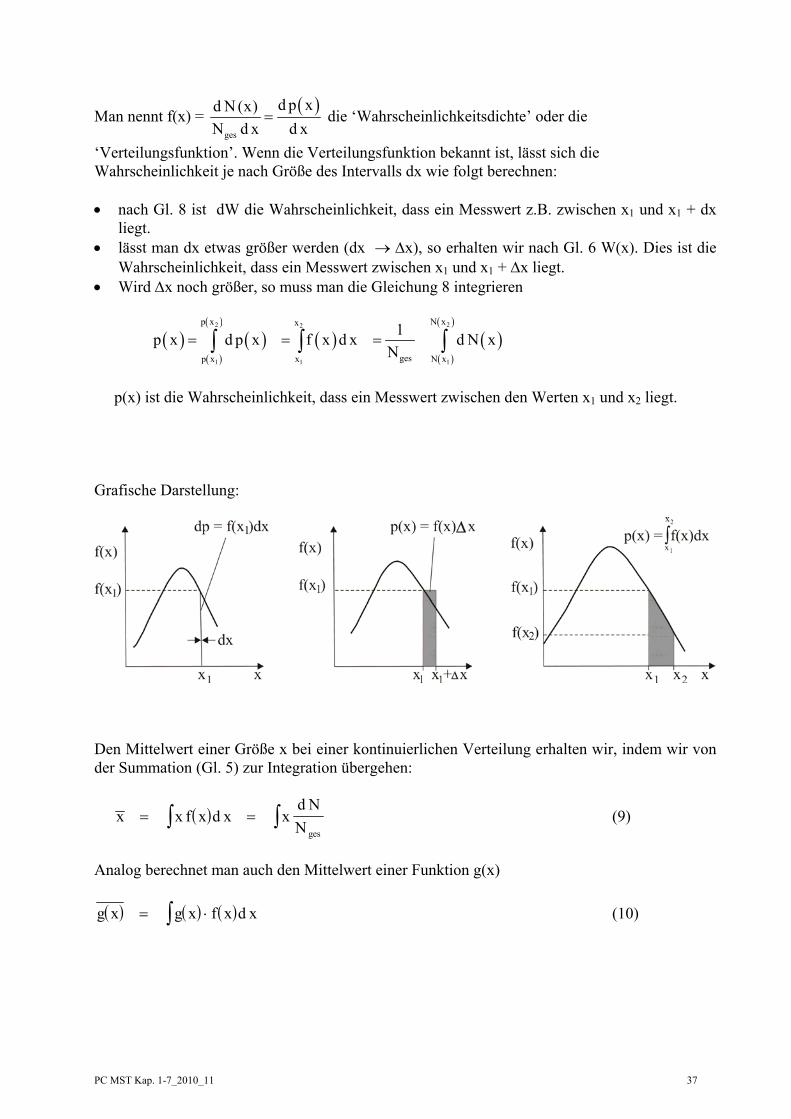
Berechnung des mittleren Einkommens:
Wir hätten die Gruppenbildung auch anders vornehmen können. Statt Δx = 1000, hätten wir z.B. Δx = 500 oder Δx = 100 wählen können. Dabei ändert sich natürlich die Zahl der Personen in der Gruppe und damit auch die Häufigkeit, d.h. dass die Häufigkeit von der Größe des Intervalls Δx abhängt. Wenn die Messgröße keine diskreten Werte (wie beim Würfeln) besitzt, so ist die Wahrscheinlichkeit, dass die Messgröße einen ganz bestimmten Wert hat, praktisch null d.h.
ges
N (x)p (x) 0N
= = (Bsp: Wie groß ist die Häufigkeit (Wahrscheinlichkeit) eines Einkommens von
exakt 2527,13 EURO) Es macht dann nur Sinn, nach der Wahrscheinlichkeit p(x) zu fragen, dass die Messgröße in einem bestimmten Intervall Δx liegt. Dies bedeutet, dass die Wahrscheinlichkeit für das Auftreten eines Messwertes, x, sowohl von der Größe von x (z.B. Höhe des Einkommens) als auch von der Größe von Δx (z.B. Breite der Gruppeneinteilung) abhängt. In erster Näherung wird p(x) proportional Δx sein, d.h. p(x) = f(x) Δx (6) Wenn wir den Übergang zu kleinen Δx machen, ergibt sich dp(x) = f(x) dx (7)
Setzen wir noch ges
NpN
= ein (Gl. 3 bzw. 4), so erhalten wir
dp(x) = ( )d N x
N ges
= f(x) dx (8)
N ii∑ = 100, ih 1=∑
i ix x h 0,05 500 € 0,3 1500 €= = ⋅ + ⋅∑ 0, 4 2500 € 0,15 3500 €+ ⋅ + ⋅ 0,05 4500 € 0,05 600 €+ ⋅ + ⋅ 2.525 €=
Häufigkeit
PC MST Kap. 1-7_2010_11 37
Man nennt f(x) = ( )ges
d p xd N (x)N d x d x
= die ‘Wahrscheinlichkeitsdichte’ oder die
‘Verteilungsfunktion’. Wenn die Verteilungsfunktion bekannt ist, lässt sich die Wahrscheinlichkeit je nach Größe des Intervalls dx wie folgt berechnen: • nach Gl. 8 ist dW die Wahrscheinlichkeit, dass ein Messwert z.B. zwischen x1 und x1 + dx
liegt. • lässt man dx etwas größer werden (dx → Δx), so erhalten wir nach Gl. 6 W(x). Dies ist die
Wahrscheinlichkeit, dass ein Messwert zwischen x1 und x1 + Δx liegt. • Wird Δx noch größer, so muss man die Gleichung 8 integrieren
( ) ( )( )
( )
( ) ( )( )
( )2 22
1 1 1
p x N xx
gesp x x N x
1p x d p x f x d x d N xN
= = =∫ ∫ ∫
p(x) ist die Wahrscheinlichkeit, dass ein Messwert zwischen den Werten x1 und x2 liegt. Grafische Darstellung:
Den Mittelwert einer Größe x bei einer kontinuierlichen Verteilung erhalten wir, indem wir von der Summation (Gl. 5) zur Integration übergehen:
( )x x f x d x xd NN ges
= = ∫∫ (9)
Analog berechnet man auch den Mittelwert einer Funktion g(x) ( ) ( ) ( )g x g x f x d x= ⋅∫ (10)

PC MST Kap. 1-7_2010_11 38
3.5 Die Geschwindigkeitsverteilung der Teilchen im Gas Die Teilchen im Gas haben nicht alle die gleiche (mittlere) Geschwindigkeit. Die Geschwindigkeitsverteilung kann man folgendermaßen herleiten. Die Gesamtenergie (= innere Energie) eines Gases verteilt sich auf die einzelnen Moleküle nach der Boltzmannverteilung (Herleitung: PC(IV)
N
NkT
Qi
ges
i
=−⎛⎝⎜
⎞⎠⎟
expε
,
Wir betrachten die Geschwindigkeitskomponente in x-Richtung, dann ergibt sich (εi = kinetische Energie der Teilchen, εpot = 0 (weil id. Gas)):
( )ε kin x m v x, =12
2
Die Laufzahl i ersetzen wir durch die kontinuierliche Variable vx
N
Nd N
N d vi
ges ges x
→
Wir erhalten dann:
d NN Q
m vk T
d vges
xx= −
⎛
⎝⎜
⎞
⎠⎟
12
2
exp
Die Konstante Q erhält man aus der Normierungsbedingung.
Es gilt: d N N gesO
Nges
=∫ oder d NN gesO
N
=∫ 1
Also gilt auch
12
1
2
2
2
Qm v
k Tdv
Qm v
k Tdv
xx
xx
exp
exp
−⎛
⎝⎜
⎞
⎠⎟ =
= −⎛
⎝⎜
⎞
⎠⎟
−∞
+∞
−∞
+∞
∫
∫
Lösung des Integrals: Substitution mk T
v z
mk T
dv dz
x
x
2
2
=
=
εi = εi (pot) + εi (kin), Energie der Teilchen Ni = Zahl der Teilchen mit Energie εi Nges = Gesamtzahl der Teilchen Q = Konstante
PC MST Kap. 1-7_2010_11 39
( )
( )
Q zmk T
dz
k Tm
z dz
= −
= −
−∞
+∞
−∞
+∞
∫
∫
exp
exp
2
2
1
2
2
Tabelle (oder Math. I - Vorlesung): ( )exp − =−∞
+∞
∫ z dz2 π
Qk T
m=
2 π
Einsetzen in Ausgangsgleichung
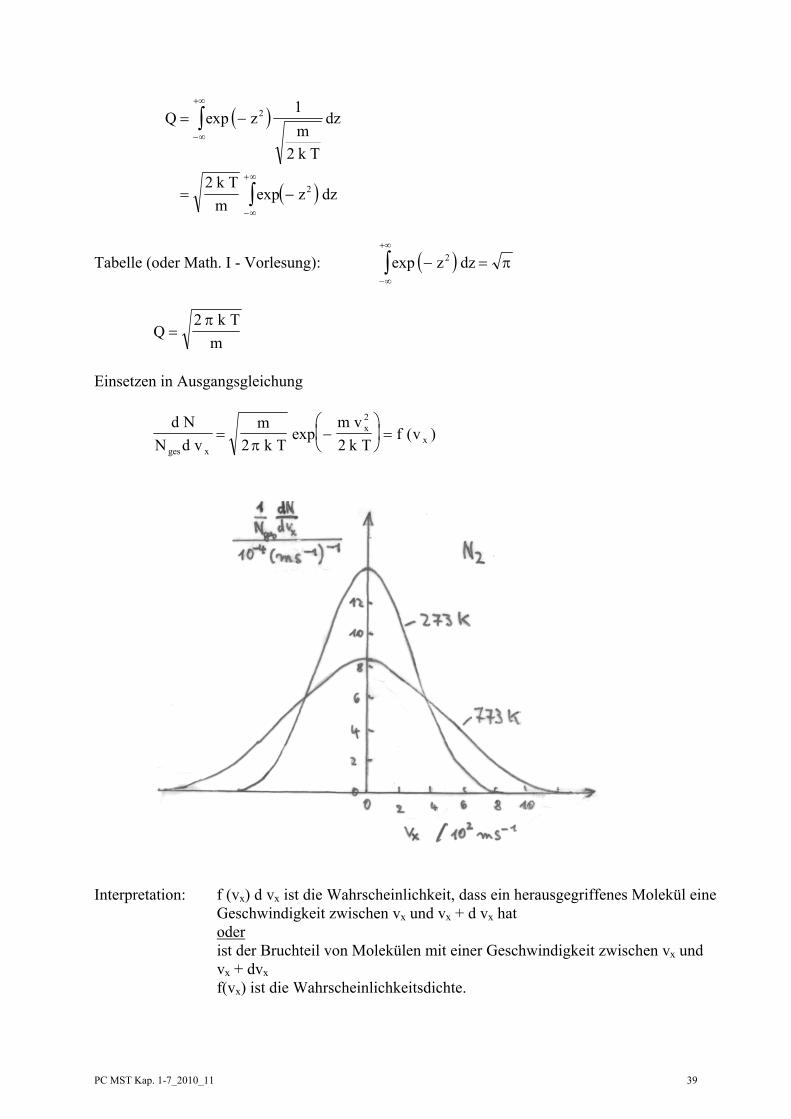
d N
N d vmk T
m vk T
f vges x
xx= −
⎛
⎝⎜
⎞
⎠⎟ =
2 2
2
πexp ( )
Interpretation: f (vx) d vx ist die Wahrscheinlichkeit, dass ein herausgegriffenes Molekül eine Geschwindigkeit zwischen vx und vx + d vx hat oder ist der Bruchteil von Molekülen mit einer Geschwindigkeit zwischen vx und vx + dvx f(vx) ist die Wahrscheinlichkeitsdichte.

PC MST Kap. 1-7_2010_11 40
Die Verteilung der Teilchengeschwindigkeiten ohne Berücksichtigung der Richtung erhält man folgendermaßen: 1) Die Verteilungen in x-, y- und z-Richtung sind gleich und unabhängig von einander. 2) Die Wahrscheinlichkeit, dass ein Teilchen eine Geschwindigkeit zwischen vx und vx + dvx und v y und vy + dvy und vz und vz + dvz hat, ist dann gegeben durch F (vx, vy, vz) dvx, dvy, dvz = f (vx) . f (vy) . f (vz) dvx dvy dvz
= ( )mk T
mk T
v v v dv dv dvx y z x y z2 2
32
2 2 2
π⎛⎝⎜
⎞⎠⎟ ⋅ − + +
⎧⎨⎩
⎫⎬⎭
exp
Wir setzen ein: v2 = v v vx y z
2 2 2+ +
= mk T
m vk T
dv dv dvx y z2 2
32 2
π⎛⎝⎜
⎞⎠⎟ −
⎛
⎝⎜
⎞
⎠⎟exp
Anstelle des Volumenelementes im Geschwindigkeitsraum dvxdvydvz benötigen wir ein Volumenelement, das nur vom Betrag der Geschwindigkeit abhängt. Die Spitze aller Vektoren (von Nullpunkt aus gerechnet), die den gleichen Betrag der Geschwindigkeit haben, liegen auf einer Kugeloberfläche, (Radius der Kugel = v). Will man jetzt ein Volumenelement der Breite dv haben, so muss man das Volumen einer Kugelschale bestimmen
Vkugel = 43
3π v
Geschwindigkeit Kugelschale: dVKugel = 4 π v2 dv
Das Volumen der Kugelschale nimmt mit steigenden v zu ⇒ Gegensatz zu dvxdvydvz
Volumen Kugelschale = dV = 4 π v2 dv Wir erhalten dann
F (v) dv = 42 2
2
32 2
ππ
vmk T
m vk T
d v⎛⎝⎜
⎞⎠⎟ −
⎛
⎝⎜
⎞
⎠⎟exp
KugeldVdv
v

PC MST Kap. 1-7_2010_11 41
oder
F (v) =
322
2 M M v4 v exp2 R T 2 R T
⎛ ⎞⎛ ⎞−⎜ ⎟⎜ ⎟
⎝ ⎠ ⎝ ⎠π
π
Bsp. Wie groß ist die Wahrscheinlichkeitsdichte (= Anteil von Molekülen pro
Geschwindigkeitsintervall, dass N2-Moleküle bei 100 K eine Geschwindigkeit von 500 m s-1 haben?
M = 28 g mol-1, T = 100 K, v = 500 m s-1
( ) ( )⎟⎟⎠
⎞⎜⎜⎝
⎛
⋅⋅⋅
−⎟⎟⎠
⎞⎜⎜⎝
⎛⋅⋅π
⋅π=
−−
−−
−−−
−−−
K100KmolJ314.82sm500molg28exp
K100Kmolsmkg314,82molkg1028sm5004
vdNNd
11
21123
1122
1321
ges
= 5,8 . 10-4 s m-1 Wie groß ist der Anteil von N2-Molekülen, deren Geschwindigkeit bei 100 K zwischen 500 m s-1 und 510 m s-1 liegt? (v = 500 m s-1, v + d v = 510 m s -1, d v = 10 m s-1)
dNN
s m d v s m m sges
= ⋅ ⋅ = ⋅ ⋅ = ⋅− − − − − −5 8 10 5 8 10 10 5 8 104 1 4 1 1 3, , ,
Wenn wir dv = 1 m s-1 wählen, erhält man entsprechend d NN ges
= ⋅ −5 8 10 4,
1) Bestimmung der mittleren Geschwindigkeit aus der Verteilung:
( )v v F v d vmk T
vm v
k Td v= =
⎛⎝⎜
⎞⎠⎟ −
⎛
⎝⎜
⎞
⎠⎟
∞∞
∫∫ 42 2
32
32
00
ππ
exp
Ablesen der Daten aus Kurve bei 500 ms-1 (100 K):
( )ges
13 1
dNN dv
0, 610 ms
−− −=
( ) 13 1
ges
dN 0, 6 10 msN dv
−− −= ⋅
( ) 13 1
ges
dN 0, 6 10 ms dvN
−− −= ⋅
1dv 1 ms−=
( )3 1
ges
dN 0, 6 10 msN
− −= ⋅ ( )1
1ms−
−

PC MST Kap. 1-7_2010_11 42
Lösen des Integrals: Wir substituieren: m v
k Tz
22
2= ,
mk T
v z2
12⎛
⎝⎜
⎞⎠⎟ ⋅ =
zmk T
v332 3
2=⎛⎝⎜
⎞⎠⎟ ⋅ , d z
mk T
d v=⎛⎝⎜
⎞⎠⎟
2
12
( )vmk T
k Tm
z z d z=⎛⎝⎜
⎞⎠⎟ ⋅
⎛⎝⎜
⎞⎠⎟
−∞
∫42
232
42
3 2
0
ππ
exp
= 12
(siehe Tab., Math. I)
v
mk T
k Tm
k Tm
R TM
=⎛⎝⎜
⎞⎠⎟
⎛⎝⎜
⎞⎠⎟
⋅
= =
42
2 12
8 8
32
42
ππ
π π
2) Bestimmung der mittleren quadratischen Geschwindigkeit aus der Verteilung:
( )v v F v d v2 2
0
=∞
∫
=⎛⎝⎜
⎞⎠⎟ −
⎛⎝⎜
⎞⎠⎟
∞
∫42 2
32
42
0
ππmk T
vmv
k Td vexp
Lösen des Integrals: Substitution: mv
k Tz
22
2= ,
mk T
v z2
12⎛
⎝⎜
⎞⎠⎟ =
zmk T
v4 4
242
=⎛⎝⎜
⎞⎠⎟ , dz
mk T
d v=⎛⎝⎜
⎞⎠⎟
2
12
( )vmk T
k Tm
z z d z2
32
52
4 2
0
42
2=
⎛⎝⎜
⎞⎠⎟ ⋅
⎛⎝⎜
⎞⎠⎟
−∞
∫ππ
exp
=38
π (siehe Tab., Math.I)
vk Tm
R TM
2 3 3= =

PC MST Kap. 1-7_2010_11 43
3) Bestimmung der häufigsten (wahrscheinlichsten) Geschwindigkeit: Wir suchen das Maximum von F (v) → differenzieren,
( )d F v
d vmk T
vm v
k Tm vk T
vm v
k T=
⎛⎝⎜
⎞⎠⎟ −
⎛
⎝⎜
⎞
⎠⎟ −⎛⎝⎜
⎞⎠⎟ + −
⎛
⎝⎜
⎞
⎠⎟
⎧⎨⎪
⎩⎪
⎫⎬⎪
⎭⎪4
2 22
2
32
22 2
ππ
exp exp
Bei v = vW ist das Maximum von F (v), d. h. ( )d F v
d v⎛⎝⎜
⎞⎠⎟ = 0
42 2
2 0
32 2 2
ππmk T
vm v
k Tm v
k TWW W⎛
⎝⎜
⎞⎠⎟ −
⎛
⎝⎜
⎞
⎠⎟ − +
⎧⎨⎩
⎫⎬⎭=exp
vk Tm
R TMW = =
2 2
Für Integrale vom Typ In = x e dxn ax−∞
∫2
0
gilt:
n: 0 1 2 3 4 5
6
In: ( )12
1 2π / /a 1/(2a) ( )14
3 1 2π /
/a 1/(2a2) ( )3
85 1 2
π //
a 1/a3 ( )15
167 1 2
π //
a

PC MST Kap. 1-7_2010_11 44
3.6 Extensive und intensive Zustandsgrößen Zustandsvariable und Zustandsfunktionen beschreiben den Zustand eines Systems. Man unterscheidet zwei Typen von Zustandsgrößen. a) intensive: Unabhängig von der Masse des Systems. Beispiel: Druck p, Dichte ρ, Temperatur T Symbole: kleine Buchstaben, Ausnahme: T b) extensive: 1. setzen sich additiv aus den Zustandsgrößen der einzelnen Teilsysteme zusammen: V(gesamt) = V1 + V2 2. vergrößert man alle Massen des Systems um den Faktor a, so vergrößern sich auch die Extensivgrößen um diesen Faktor: aV(gesamt) = aV1 + aV2 Symbole: große Buchstaben, Ausnahme: m

PC MST Kap. 1-7_2010_11 45
Einschub: Zustandsfunktionen Man findet, dass V = f (n,p,T) Eine solche Gleichung nennt man eine Zustandsfunktion, da ihr Zahlenwert nur von den jeweiligen Werten der Größen n, p, T abhängt und nicht vom Weg, auf welchem dieser Zustand erreicht wird. Das Volumen kann also geändert werden durch Änderung von n,p und T. Mathematisch beschreibt man das so:
dV = p,T
V dnn
∂⎛ ⎞⎜ ⎟∂⎝ ⎠
+ n,T
V dpp
⎛ ⎞∂⎜ ⎟∂⎝ ⎠
+ n,p
V dTT∂⎛ ⎞
⎜ ⎟∂⎝ ⎠
totales partielle Differential Differentialquotienten Die Aussage, dass V eine Zustandsfunktion ist, lässt sich mathematisch verschieden ausdrücken: 1) dV = totales Differential 2) dV = unabhängig vom Weg 3) dV∫ = 0 Führt man Änderungen durch, die über verschiedene Zwischenzustände zum gleichen Anfangszustand zurücklaufen, so ist das Kreisintegral = 0.
( ) ( )VV
Tp pT
TP
4)T p
∂∂∂∂
⎛ ⎞ ⎛ ⎞∂∂⎜ ⎟ = ⎜ ⎟⎜ ⎟⎜ ⎟∂ ∂⎝ ⎠⎝ ⎠
Beispiel: Ist dV ein totales Differential? Noch mal differenzieren:
( )Vr h 2 rh
r
∂∂⎛ ⎞∂
= π⎜ ⎟⎜ ⎟∂⎝ ⎠,
( )Vh r 2 rr
h
∂∂⎛ ⎞∂
= π⎜ ⎟⎜ ⎟∂⎝ ⎠⇒ ja, Reihenfolge der Differentiationen ist vertauschbar.
4. Reale Gase
Größen, die jeweils beim Differenzieren konstant gehalten werden
Die Reihenfolge der Differentiationen ist vertauschbar (Schwartz’scher Satz).
V(Zylinder) = π r2 . h
dV = h r
V Vdr dhr h
∂ ∂⎛ ⎞ ⎛ ⎞+⎜ ⎟ ⎜ ⎟∂ ∂⎝ ⎠ ⎝ ⎠
h
V h2 rr
∂⎛ ⎞ = π⎜ ⎟∂⎝ ⎠
∂∂
πVh
rr
⎛⎝⎜
⎞⎠⎟ = 2
Eingesetzt: dV = 2πhrdr + πr2dh
PC MST Kap. 1-7_2010_11 46
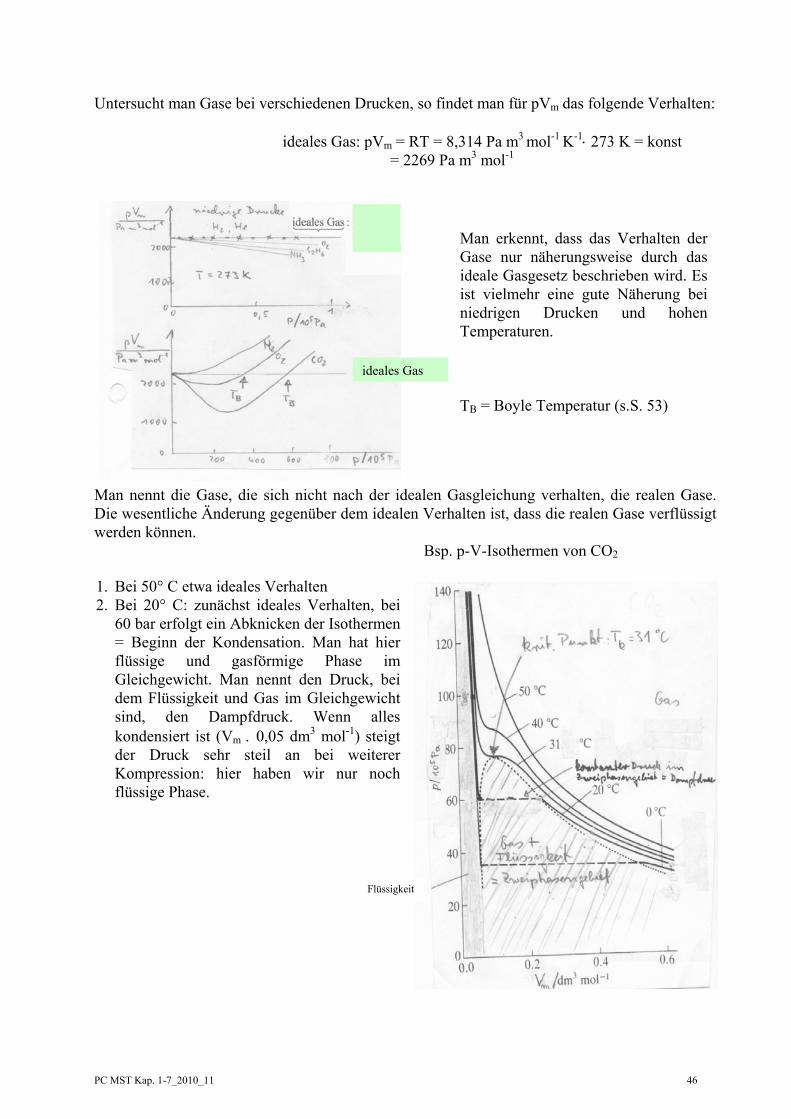
Untersucht man Gase bei verschiedenen Drucken, so findet man für pVm das folgende Verhalten:
ideales Gas: pVm = RT = 8,314 Pa m3 mol-1 K-1⋅ 273 K = konst = 2269 Pa m3 mol-1
Man nennt die Gase, die sich nicht nach der idealen Gasgleichung verhalten, die realen Gase. Die wesentliche Änderung gegenüber dem idealen Verhalten ist, dass die realen Gase verflüssigt werden können. Bsp. p-V-Isothermen von CO2
Man erkennt, dass das Verhalten der Gase nur näherungsweise durch das ideale Gasgesetz beschrieben wird. Es ist vielmehr eine gute Näherung bei niedrigen Drucken und hohen Temperaturen. TB = Boyle Temperatur (s.S. 53)
1. Bei 50° C etwa ideales Verhalten 2. Bei 20° C: zunächst ideales Verhalten, bei
60 bar erfolgt ein Abknicken der Isothermen = Beginn der Kondensation. Man hat hier flüssige und gasförmige Phase im Gleichgewicht. Man nennt den Druck, bei dem Flüssigkeit und Gas im Gleichgewicht sind, den Dampfdruck. Wenn alles kondensiert ist (Vm . 0,05 dm3 mol-1) steigt der Druck sehr steil an bei weiterer Kompression: hier haben wir nur noch flüssige Phase.
ideales Gas
Flüssigkeit

PC MST Kap. 1-7_2010_11 47
Eine der Isothermen besitzt einen Wendepunkt. Bei CO2 ist das die Isotherme bei 31° C. Diese Temperatur nennt man die kritische Temperatur Tk, die Werte von p und Vm am Wendepunkt nennt man den kritischen Druck, pk und das kritische Volumen, Vm, k. Oberhalb der kritischen Temperatur kann man keinen Phasenübergang zwischen Flüssigkeit und Gas mehr beobachten, d.h. es erfolgt dann eine kontinuierliche Änderung der Eigenschaften (‘fluide Phase’). Umgekehrt: Verflüssigung von Gasen nur unterhalb von Tk möglich. Die Ursache hierfür ist, dass bei höheren Temperaturen als der kritischen Temperatur die thermische Energie der Teilchen größer ist als deren Wechselwirkungsenergie und sie daher bei Bindung sofort wieder dissoziieren. Beschreibung der realen Gase durch verschiedene Gleichungen z.B. Potenzreihenansatz: p Vm = RT (ideales Gas) p Vm = RT + B p + C p2 + ............ ‘Virialgleichung’ Die van der Waals’sche Gleichung ist die erste Gleichung gewesen, mit der versucht wurde, das reale Verhalten zu beschreiben. Bei der van der Waals’schen Gleichung werden zwei Effekte berücksichtigt, die bei der idealen Gasgleichung vernachlässigt wurden. 1. Eigenvolumen der Teilchen: Das Volumen, in dem sich die Teilchen frei bewegen können, ist
um das Eigenvolumen (genau das 4-fache Eigenvolumen) vermindert. Das Molekül 2 kann nicht näher als 2 rA an das Molekül 1 heran, d.h. das schraffierte Volumen steht für die Bewegung von 2 nicht mehr zur Verfügung.
V(punktiert) = ( )3 3A A m o le k ü l
m o l e k ü l
4 4π 2 r = 8 π r = 8 V3 3
V
Das gleiche gilt umgekehrt für das Molekül 1, so dass für 1 und 2 zusammen dieses Volumen nicht mehr zur Verfügung steht.
→ V (ausgeschlossen, 1 Teilchen) = 4 molekülV → V (ausgeschlossen, NA Teilchen) = 4 Vm = b In Wirklichkeit wird b nicht aus VM bestimmt, sondern b ist ein experimenteller Parameter, der sich aus p, V, T-Werten des jeweiligen Gases ergibt.
2. Anziehungskräfte zwischen den Teilchen: Die Anziehung zwischen den Teilchen wirkt so, als
würde das Gas unter einem höheren äußeren Druck stehen, d.h. p(wirksam) = p + π. Dabei ist p der Außendruck und π der Binnendruck, der auf die Anziehungskräfte ( = van der Waals’sche Kräfte) zurückzuführen ist. Der Binnendruck ist proportional zur (Molekülzahldichte)2, d.h.
π ∼ NV
aVm
⎛⎝⎜
⎞⎠⎟ =
2
2
rA rA 1 2

PC MST Kap. 1-7_2010_11 48
Man korrigiert also den Druck u. das Volumen folgendermaßen:
p Vm = RT (id. Gas)
( )pa
VV b
mm+
⎛
⎝⎜
⎞
⎠⎟ −2 = RT (van der Waals - Gas)
Ersetzt man Vm = Vn
ergibt sich:
( )2
2
n ap V n bV
⎛ ⎞+ −⎜ ⎟
⎝ ⎠ = nRT
Für kleine Drucke kann man eine vereinfachte Form der van-der-Waals-Gleichung verwenden.
Herleitung:
p = RT
V ba
VV
m mm−
− ⋅2
mm m
m m
R T a 1 apV R Tb bV R T V1 1V V
⎛ ⎞= − = −⎜ ⎟− −⎜ ⎟⎜ ⎟
⎝ ⎠
Für Vm>> b gilt 1
11
−≈ +b
V
bV
m
m
(Anwendung von 1
11
−≈ +
xx , für kleine x)
mm m m
b a 1 apV R T 1 R T 1 bV R T V V R T
⎡ ⎤⎛ ⎞ ⎛ ⎞= + − = + −⎢ ⎥⎜ ⎟ ⎜ ⎟⎝ ⎠⎝ ⎠ ⎣ ⎦
Zur Abkürzung nennen wir B = ba
R T−
mm
1pV R T 1 BV
⎛ ⎞= +⎜ ⎟⎝ ⎠
, für Vm setzen wir die ideale Gasgleichung ein : VR Tpm =
mBpV R T 1 p
R T⎛ ⎞= + ⋅⎜ ⎟⎝ ⎠
pV R T B pm = +
Stoff 2 2
aL bar mol−
1
bL mol−
He 0,03457 0,02370 N2 1,408 0,03913 H2O 5,545 0,03053 C2H6 5,562 0,0638
In dieser Näherung ist B der erste Virialkoeffizient der Reihenentwicklung: B → 0 dann ideales Verhalten

PC MST Kap. 1-7_2010_11 49
Es gibt eine Temperatur bei der ba
R T= ist. Dann ist B = 0 und ein reales Gas verhält sich bei
dieser Temperatur wie ein ideales Gas. Man nennt diese Temperatur die ‘Boyle’ Temperatur.
B = ba
R TT
aR bB− = → =⋅
0
Die Zahlenwerte von a und b werden experimentell aus den kritischen Daten ( )k k kT , V , p bestimmt: Betrachtet man die Isotherme bei der kritischen Temperatur, so findet man am
kritischen Punkt eine horizontale Tangente → =⎛⎝⎜
⎞⎠⎟
d pd V
0 und einen Wendepunkt der Kurve
(→ =d pd V
2
2 0 ) d.h. an diesem Punkt gelten drei Gleichungen:
1. pR T
V ba
Vm m
=−
− 2
2. ( )
k2 3
m m,kk m,k
d p R T 2a 0d V VV b
⎛ ⎞ = − + =⎜ ⎟ −⎝ ⎠
3. ( )
2k
32 4m m,kk m,k
d p 2 R T 6a 0d V VV b
⎛ ⎞= − =⎜ ⎟
−⎝ ⎠
Wir setzen den Wert von b in Gl. 2 ein:
k2 3
m,km,k m,k
k2 3
m,km,k
2k m,k3
m,k
k m,k
R T 2a 0V1V V
3R T 2a
V2 V3
2a 4R T VV 9
9a R T V8
+ =⎛ ⎞−⎜ ⎟⎝ ⎠
=⎛ ⎞⎜ ⎟⎝ ⎠
= ⋅
=
Division: GleichungGleichung
23
:
( )
( )
( )( )
k2 3
m,k m,k
k3 4
m,km,k
3 4k m,k m,k
2 3m,km,k k
m,k m,k
m,k
R T 2aV b V
2R T 6aVV b
R T V b 2a VV 6aV b 2R T
V b V2 3
1b V3
−=
−
−=
−
−=
=

PC MST Kap. 1-7_2010_11 50
Setzen wir a und b in Gleichung 1 ein, ergibt sich:
k m,kk m,k m,k k2
m,k
R T V9 1p V V R T8 V 3
2 Vm, k3
⎛ ⎞⎛ ⎞+ − =⎜ ⎟⎜ ⎟⎝ ⎠⎝ ⎠
=
k m,kk m,k
m,k
9 R T 2 V2 p V3 8 V 3
⋅+ = R Tk
23
p Vk m k, = 14
R Tk
pk = 38
R TV
k
m k,
oder:
Sp V
R Tk m k
k
≡ = =, ,
38
0 375 Man nennt S den ‘kritischen Koeffizienten’, der für alle Gase, die
der von der Waals’schen Gleichung gehorchen, gleich sein sollte. Experimentell findet man aber für verschiedene Gase die Werte S = 0,250 - 0,315.
Aus der Gleichung für pk kann man R bestimmen: Rp V
Tk m k
k
=83
, . Ersetzt man alle Konstanten
(a, b, R) der van der Waals Gleichung durch die kritischen Daten erhält man:
2k m,k k
m,k k m,k2k k
p V T9 8 1 8 Tp+ V V p V8 3 T V 3 3 T
⎛ ⎞⎛ ⎞⋅ − =⎜ ⎟⎜ ⎟⋅ ⎝ ⎠⎝ ⎠
oder
2k m,k k
m,k
p 1 V T3 1 8p V TV
V
⎛ ⎞⎛ ⎞+ − =⎜ ⎟⎜ ⎟⎛ ⎞ ⎝ ⎠⎜ ⎟⎜ ⎟⎜ ⎟⎝ ⎠⎝ ⎠
Dies ist die reduzierte van der Waals Gleichung. Sie sollte für alle Stoffe gültig sein. Man nennt dies auch das‘ Theorem der übereinstimmenden Zustände’ Auch diese Aussage ist nicht streng gültig. Für die Beschreibung experimenteller Daten benutzt man daher häufig einen Potenzreihenansatz (‘Virialgleichung’) mit so vielen Gliedern wie nötig, um die Daten richtig zu beschreiben.

PC MST Kap. 1-7_2010_11 51
5. Flüssige und feste Stoffe, Aggregatzustände, Phasen Materie kann in drei verschiedenen Aggregatzuständen vorkommen: Fest, flüssig, gasförmig. Die Unterschiede sind auf Unterschiede in den Wechselwirkungsenergien zurückzuführen. Fest: Starke Wechselwirkungsenergie Flüssig: Mittlere WW Gasförmig (real): Schwache WW Gasförmig (ideal): keine WW Bringt man 2 Teilchen (Atome oder Moleküle) aus großer Entfernung immer näher und betrachtet ihre potentielle Energie, so sieht das folgendermaßen aus:
- üblicherweise setzt man die Energie bei r → ∞, εpot = 0 - Nähert man Teilchen an, so ziehen sie sich gegenseitig an, εpot nimmt ab - Bei einem Abstand r0 erreicht εpot einen Minimalwert → Gleichgewichtsabstand - Bei kleinen Abständen stoßen die Teilchen sich ab, εpot nimmt stark zu. Diesen Typ von Kurve beobachtet man
eigentlich immer, sowohl wenn die Teilchen eine Bindung eingehen (z.B. HCl, H2) als auch bei ganz schwacher WW (z.B. He⋅⋅⋅He). Es unterscheiden sich jeweils die Abstandsabhängigkeiten von εpot (hängt von der jeweiligen Kraft ab) und die Größe der Bindungsenergie. Wenn die Bindungsenergie nach außen abgegeben werden kann, kommt es zur chemischen Bindung oder Aggregation (d.h. Bildung der flüssigen oder festen Phase). Umgekehrt kann jeder Stoff durch Zufuhr von thermischer Energie in den flüssigen bzw. gasförmigen Zustand überführt werden. Dann ist εtherm >> ε (Wechselwirkung). Die Theorien der Festkörper und Flüssigkeiten sind sehr viel schwieriger als die der Gase. Daher beschäftigen wir uns hier nur mit der empirischen Beschreibung: Es gilt bei konstanter Masse bzw. Stoffmenge (geschlossene Systeme) für das Volumen eines Stoffes: V = f (p, T) Für die Änderung des Volumens ergibt sich:
dV = ∂∂
∂∂
Vp
d pVT
d TT p
⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟
jeweils verglichen mit der thermischen Energie der Moleküle. Für die thermische Energie eines Atoms (sowohl eines isolierten als auch eines Atoms im Molekül) kann man im Mittel ε therm ≈ kT annehmen (genaue Berechnung siehe 6.24). Für ein Mol ergibt sich entsprechend Etherm ≈ RT

PC MST Kap. 1-7_2010_11 52
Führen wir eine Änderung durch, bei der das Volumen konstant bleibt (z.B. erst Volumenvergrößerung durch T-Erhöhung, dann Volumenverminderung durch p-Erhöhung) so ergibt sich: dV = 0 Daraus erhalten wir schließlich:
∂∂
pT
V
⎛⎝⎜
⎞⎠⎟ = p
T
VT
Vp
⎛ ⎞∂⎜ ⎟∂⎝ ⎠
−⎛ ⎞∂⎜ ⎟∂⎝ ⎠
Man definiert: Ausdehnungskoeffizient: α∂∂
=⎛⎝⎜
⎞⎠⎟
1V
VTO p
Kompressibilität: TO p
VV1
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
−=κ κ = Kappa
Spannungskoeffizient: β∂∂
=⎛⎝⎜
⎞⎠⎟
1p
pTO V
Nach unserer obigen Gleichung erhält man zwischen diesen Koeffizienten die Beziehung:
p O βακ
=
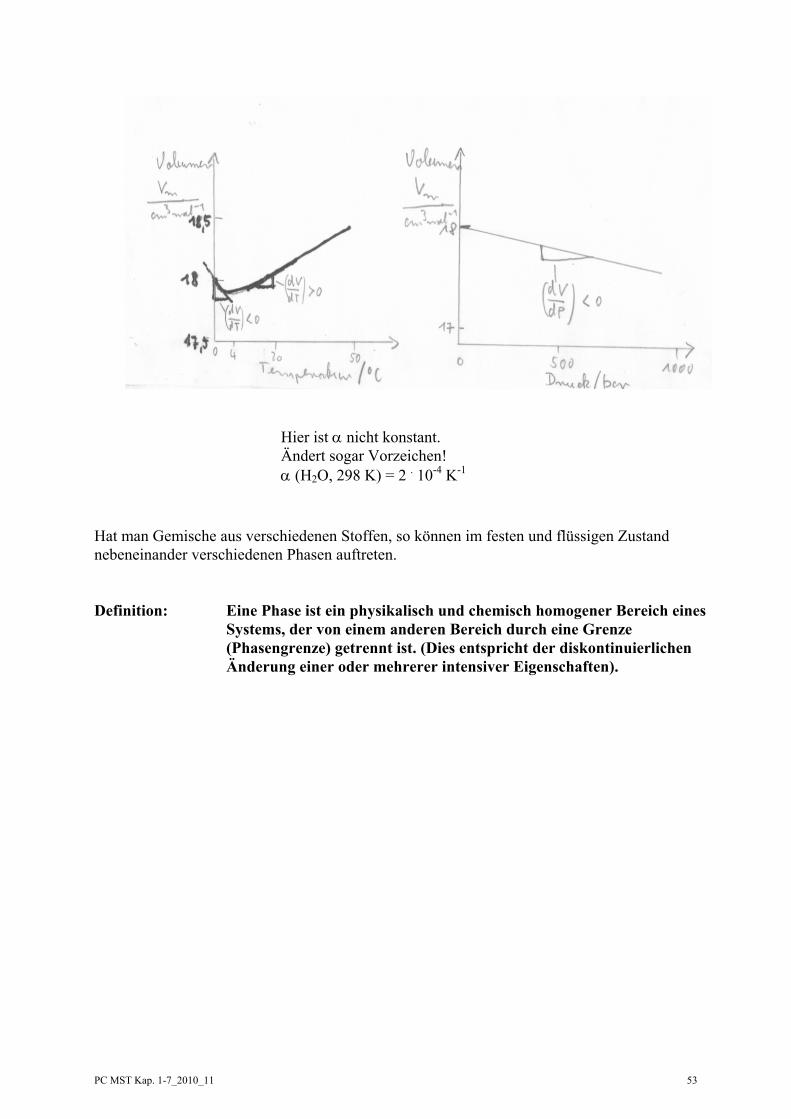
Diese Definitionen und Gleichungen gelten für Gase, Flüssigkeiten und Festkörper. Für Flüssigkeiten und Festkörper sind die Ausdehnungskoeffizienten und Kompressibilitäten sehr viel kleiner als für Gase und sie sind in erster Näherung konstant. Man kann dann einfach integrieren und erhält dann:
0
V
p0 V
1 V d VV T
α ∂⎛ ⎞= − → =⎜ ⎟∂⎝ ⎠ ∫ ( )0
T
0 0 0 0T T
V d T V V V T Tα αΔ
→ = + −∫
Konstanten (das muss aber experimentell für jeden Stoff geprüft werden!)
0
V
p0 V
1 V d VV P
κ ∂⎛ ⎞= − → =⎜ ⎟∂⎝ ⎠ ∫ ( )0
p
0 0 0 0p p
V d p V V V p pκ κΔ
− → = − −∫
PC MST Kap. 1-7_2010_11 53
Hat man Gemische aus verschiedenen Stoffen, so können im festen und flüssigen Zustand nebeneinander verschiedenen Phasen auftreten. Definition: Eine Phase ist ein physikalisch und chemisch homogener Bereich eines
Systems, der von einem anderen Bereich durch eine Grenze (Phasengrenze) getrennt ist. (Dies entspricht der diskontinuierlichen Änderung einer oder mehrerer intensiver Eigenschaften).
Hier ist α nicht konstant. Ändert sogar Vorzeichen! α (H2O, 298 K) = 2 . 10-4 K-1

PC MST Kap. 1-7_2010_11 54
6. Energieerhaltung - Der erste Hauptsatz Der erste Hauptsatz ist eine Erweiterung der mechanischen Energieerhaltung, wobei zusätzlich die Wärme berücksichtigt wird. Für ein abgeschlossenes System gilt: U = konstant, d U = O Für ein geschlossenes System gilt: d U = d Q + d W Für ein offenes System gilt: d U = d Q + d W + U d ni a i
i∑
Darin ist: U = Innere Energie = Summe der kinetischen und potentiellen Energie aller Moleküle
im System d W = Arbeitsaustausch mit der Umgebung = Austausch geordneter Bewegungsenergie
(d.h. alle Teilchen, die beteiligt sind, bewegen sich in gleicher Richtung). d Q = Wärmeaustausch mit der Umgebung = Austausch ungeordneter Bewegungsenergie (d.h. die Teilchen bewegen sich in beliebigen Richtungen).
i a ii
U d n =∑
Ob man einem System Arbeit oder Wärme zuführt, wird in der Umgebung definiert. Die Wirkung auf das System kann in beiden Fällen gleich sein.
d W = U . I . d t
System Adiabatische Wand d Q = 0 Flüssigkeit
Dem System wird Arbeit zugeführt (alle Elektronen bewegen sich in der gleichen Richtung). Ein Teil ihrer Bewegungsenergie wird auf die Atome des Drahts übertragen. Diese bewegen sich dann schneller aber ihre Bewegung ist ungeordnet. Diese Bewegungsenergie wird auf die Flüssigkeit übertragen. Folge: Die Flüssigkeit wird erwärmt.
Term, der den Energieaustausch mit der Umgebung beschreibt, der mit einem Materieaustausch verknüpft ist (z.B. Trinken von heißem Wasser: Dann ist Ui die Innere Energie pro Mol Wasser, ad n ist die von außen zugeführte Stoffmenge von Wasser)

PC MST Kap. 1-7_2010_11 55
d W = U . I d t Wir betrachten im Folgenden geschlossene Systeme. Die wichtigste Arbeit, die bei chemischen Reaktionen immer auftritt, ist die Druck-Volumen - Arbeit. → d U = d Q + d W = d Q - p d V (nur Druck-Volumen-Arbeit) 1. Im System sei das Volumen konstant: V = konst. → d V = 0
→ (d U)V = d Q → d U d Q U U QI
II Q
II I∫ ∫= → − =0
d.h. die Änderung der Inneren Energie ist gleich der zugeführten Wärme, wenn V = konstant. 2. Im System sei der Druck konstant: p = konstant
→ (d U)p = d Q - p d V → d U d Q p d VI
II Q
I
II
∫ ∫ ∫= −0
konst. ( )U U Q p V V Q p V p VII I II I II I− = − − = − + Umordnen: (UII + p VII) - (UI + p V1) = Q HII HI Wir nennen zur Abkürzung U + p V ≡ H die ‘Enthalpie’ oder ‘Wärmeinhalt’ des Systems, d.h. die Änderung der Enthalpie ist gleich der zugeführten Wärme, wenn p = konst. H = Enthalpie = Summe der kinetischen und potentiellen Energie aller Moleküle im
System und der Energie p V Wenn p = 0 (Vakuum), dann ist H = U Für Änderungen bei konstantem Druck (das sind z.B. alle Vorgänge, die bei Atmosphärendrucke ablaufen), muss nur die Änderung des Volumens beachtet werden: p Δ V
Der Heizplatte wird Arbeit zugeführt. Über die diathermane (wärmeleit-fähige) Wand wird der Flüssigkeit Wärme zugeführt (in der Wand und in der Heizplatte befinden sich die Teilchen in ungeordneter Bewegung). Folge: Die Flüssigkeit wird erwärmt.
System Adiabatische Wand Flüssigkeit Diathermane Wand
Heizplatte

PC MST Kap. 1-7_2010_11 56
6.1 Die Temperaturabhängigkeit von U und H Die Enthalpie ist folgendermaßen definiert H ≡ U + p V Wir differenzieren und erhalten d H = d U + d (p V) = d U + p d V + V d p Wir setzen d U = d Q - p d V ein und erhalten für dH d H = d Q + V d p Die folgenden Ableitungen sind für U und H völlig analog und die entsprechenden Schritte sind nebeneinander dargestellt.

PC MST Kap. 1-7_2010_11 57
U = f (V, T)
U Ud U d V d T d Q p d V
V T∂ ∂
= + = −∂ ∂
⎛ ⎞ ⎛ ⎞⎜ ⎟ ⎜ ⎟⎝ ⎠ ⎝ ⎠
beschreibt Eigenschaften des Systems
Auflösen nach d Q:
d QUT
d TUV
p d VV T
=⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟ +
⎡
⎣⎢⎢
⎤
⎦⎥⎥
∂∂
∂∂
V = konst. → d V = 0
d QUT
dTV
=⎛⎝⎜
⎞⎠⎟
∂∂
d Qd T
UT
CV V
V
⎛⎝⎜
⎞⎠⎟ =
⎛⎝⎜
⎞⎠⎟ ≡
∂∂
→ Die Änderung der Inneren Energie mit der Temperatur bei V = konst. ist die Wärmekapazität des Systems.
d U C d T d U C d TVU
U
VT
T
= → =∫ ∫1
2
1
2
a) CV = konst. → U2 - U1 = CV (T2 - T1)
b) CV = a + b T + .....→ U2 - U1 = ( )a b T d TT
T
+∫1
2
= a ( T2 - T1) + ( )12 2
212b T T−
H = f (p, T)
T P
H Hd H d p d T d Q V d p
p T∂ ∂
= + = +∂ ∂
⎛ ⎞ ⎛ ⎞⎜ ⎟ ⎜ ⎟⎝ ⎠ ⎝ ⎠
Auflösen nach d Q:
d QHT
d THp
V d pp T
=⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟ −
⎡
⎣⎢⎢
⎤
⎦⎥⎥
∂∂
∂∂
p = konst. → d p = 0
d QHT
d Tp
=⎛⎝⎜
⎞⎠⎟
∂∂
d Qd T
HT
Cp p
p
⎛⎝⎜
⎞⎠⎟ =
⎛⎝⎜
⎞⎠⎟ ≡
∂∂
→ die Änderung der Enthalpie mit der Temperatur bei p = konst. ist die Wärmekapazität des Systems.
d H = Cp d T → d H C d TH
H
pT
T
1
2
1
∫ ∫=
a) C p = konst. → H2 - H1 = Cp (T2 - T1)
b) C p = a’ + b’ T + ... → H2 - H1 = ( )a b T d TT
T
' '+∫1
2
= a’ (T2 - T1) + ( )12 2
212b T T' −
beschreibt
WW mit der
Umgebung

PC MST Kap. 1-7_2010_11 58
Häufig verwendet man ‘intensive’ Größen, die entweder auf ein mol (oder ein g) bezogen sind.
Symbole: Vm = Vn
, Um = Un
usw. (manchmal werden diese Größen auch durch Fettdruck
oder durch Überstreichen kenntlich gemacht (V oder V ). Nicht einheitlich in der Literatur. Für Größen, die auf die Masseneinheit bezogen sind, werden im Allgemeinen kleine Buchstaben
verwendet: cv = Cm
V (Wärmekapazität pro g oder spezifische Wärme).
Umrechnung cp in cV: Üblicherweise misst man cp, die Größe cV kann man leichter theoretisch berechnen. Zum Vergleich muss man beide Größen ineinander umrechnen. Es ergibt sich:
cp - cV = ∂∂
∂∂
HT
UT
p v
⎛⎝⎜
⎞⎠⎟ −
⎛⎝⎜
⎞⎠⎟
Einsetzen der Definitionsgleichung: H = U + p V
= ∂∂
∂∂
∂∂
UT
pVT
UT
p p v
⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟ −
⎛⎝⎜
⎞⎠⎟
= ∂∂
∂∂
∂∂
∂∂
∂∂
UT
UV
VT
pVT
UT
V T p p v
⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟ −
⎛⎝⎜
⎞⎠⎟
= pUV
VT
T p
+⎛⎝⎜
⎞⎠⎟
⎡
⎣⎢⎢
⎤
⎦⎥⎥
⎛⎝⎜
⎞⎠⎟
∂∂
∂∂
Für das totale Differential von U gilt:
d UUT
d TUV
d VV T
=⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟
∂∂
∂∂
Division durch d T bei p = konst. führt zu:
∂∂
∂∂
∂∂
∂∂
UT
UT
UV
VT
p V T p
⎛⎝⎜
⎞⎠⎟ =
⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟⎛⎝⎜
⎞⎠⎟ →Einsetzen
Wir benutzen die folgende Beziehung (Herleitung siehe Wedler):
∂∂
∂∂
UV
TpT
pT v
⎛⎝⎜
⎞⎠⎟ =
⎛⎝⎜
⎞⎠⎟ − → Einsetzen
p v
p
p Vc c p T pT Tv
⎡ ⎤∂ ∂⎛ ⎞ ⎛ ⎞− = + −⎢ ⎥⎜ ⎟ ⎜ ⎟∂ ∂⎢ ⎥⎝ ⎠ ⎝ ⎠⎣ ⎦

PC MST Kap. 1-7_2010_11 59
Ersetzen wir den Spannungskoeffizient ∂∂
pT
V
⎛⎝⎜
⎞⎠⎟ noch durch Ausdehnungskoeffizienten
∂∂
VT
p
⎛⎝⎜
⎞⎠⎟ und die Kompressibilität
T
Vp
∂∂
⎛ ⎞⎜ ⎟⎝ ⎠
, so ergibt sich:
cp - cv =
2
p
V p
T
VTp VT T
T T Vp
∂∂∂ ∂
∂ ∂ ∂∂
⎛ ⎞⎜ ⎟
⎛ ⎞ ⎛ ⎞ ⎝ ⎠= −⎜ ⎟ ⎜ ⎟ ⎛ ⎞⎝ ⎠ ⎝ ⎠
⎜ ⎟⎝ ⎠
= 2
0VT ακ
Diese Gleichung gilt für feste, flüssige und gasförmige Stoffe; für ideale Gase wird die Beziehung besonders einfach. Es gilt:
V p
p R V RundT V T p
∂ ∂∂ ∂
⎛ ⎞ ⎛ ⎞= =⎜ ⎟ ⎜ ⎟
⎝ ⎠ ⎝ ⎠
Setzen wir dies ein, erhalten wir:
cp - cv = R R T R RT RV p R T
⋅ ⋅⋅ = =
6.2.1 Expansion eines idealen Gases ins Vakuum Thermometer Wasserbad Gas (p, V) Vakuum
Kolben 1 wird mit Gas gefüllt. Kolben 2 ist evakuiert. Dann wird Hahn 3 geöffnet: Gas strömt in den Kolben 2. Beobachtung: T ändert sich nicht, d.h. d T = 0

PC MST Kap. 1-7_2010_11 60
Thermodynamische Beschreibung:
dU d Q + d W= = v
U Ud V d TV T
⎛ ⎞ ⎛ ⎞∂ ∂+⎜ ⎟ ⎜ ⎟∂ ∂⎝ ⎠ ⎝ ⎠
WW mit Umgebung Systemeigenschaften 1 d Q = 0 da adiabatisch 2 d W = 0 keine Arbeit wird nach außen abgeben 3 d T = 0 experimenteller Befund
4 d V ≠ 0 → ∂∂
UV
T
⎛⎝⎜
⎞⎠⎟ = 0
→ d.h. die Innere Energie eines idealen Gases hängt nicht vom Volumen ab. Dies ist auch anschaulich klar, da die Teilchen keine WW aufeinander ausüben, d.h. der mittlere Abstand der Teilchen spielt keine Rolle (Volumen ∼ (mittlerer Teilabstand)3).
6.2.2 Adiabatische Expansion eines idealen Gases Thermodynamische Beschreibung:
d U = d Q + d W = ∂∂
∂∂
UV
d VUT
d TT V
⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟
WW (Umgebung) System (= Gas.) 1 d Q = 0, da adiabatisch 2 d W = -p d V nur Druck - Volumen - Arbeit.
3 ∂∂
UV
T
⎛⎝⎜
⎞⎠⎟ = 0 da ideales Gas
4 ∂∂
UT
V
⎛⎝⎜
⎞⎠⎟ = n cV
Gas wird expandiert und leistet dabei Arbeit. Dabei ist das Gas gegen die Umgebung wärmeisoliert a) Wie groß ist die Arbeit? b) Wie lautet die Zustandsgleichung?
beweglicher adiabatische Wand Stempel (adiabatisch)
Gas (p0, V0)

PC MST Kap. 1-7_2010_11 61
Man erhält damit:
d U = d W = - p d V = n cV d T
a) Volumenarbeit: d W n c d TW
VT
T
0 1
2
∫ ∫=
W = n cV (T2 - T1) wenn cV = konst. b) Zustandsgleichung: d W = - p d V = n cV d T ideales Gasgesetz einsetzen
pn R T
V=
- n R Td VV
= n cV d T, Variablentrennung
− = ∫∫Rd VV
cd TTV
T
T
V
V
00
− = → =RVV
cTT
Rc
VV
TTV
V
ln ln ln ln0 0
0
0
VV
TT
RcV0
0
⎛⎝⎜
⎞⎠⎟
=
VV
TT
01
0
⎛⎝⎜
⎞⎠⎟
=−κ
Wir setzen die ideale Gasgleichung ein Tp Vn R
= und erhalten
p Vn R
V T Vκ κ− −=10 0
1
oder
Diesen beiden Gleichungen nennt man ‘Poisson’-Gleichung oder Adiabaten-Gleichung.
Wir setzen ein: R = cp - cV
→ Rc
c cc
ccV
p V
V
p
V=
−= −1
mit κ =cc
p
V
RcV
= −κ 1
T0 V01κ− = T Vκ-1 = konst.
p Vκ = T0 V n R p V konsto01
0κ κ− = = .

PC MST Kap. 1-7_2010_11 62
6.2.3 Expansion eines realen Gases (Joule-Thomson-Effekt)
Bei Druck auf den Kolben links wird das Gas bei konstantem Druck p2 durch die Fritte hindurchgedrückt, der gesamte Druckabfall erfolgt in der Fritte. Auf der rechten Seite expandiert das Gas bei konstantem Druck p1 und schiebt dabei den Kolben rechts zurück. Thermodynamische Beschreibung: Wechselwirkung mit Umgebung links rechts d U2 = d Q2 - p2 d V2 d U1 = d Q - p1d V1
d U p d V d QAnf
Ende Q
Anf
Ende
2 2 2 20
2
+ =∫ ∫∫..
d U d V d QAnf
Ende
Anf
Ende Q
1 1 10
1
. .∫ ∫ ∫+ =
U2 (E) - U2 (A) + p2 (V2 (E) - V2 (A) ) = Q2 U1 (E) - U1 (A) + p1 (V1 (E) - V1 (A)) = Q1
Δ U2 + p2 Δ V2 = Q2 Δ U1 + p1 Δ V1 = Q1 Wenn das System adiabatisch ist, gilt Q2 = Q1 = 0 oder Δ U2 + p2 Δ V2 = Δ U1 + p1 Δ V1 Δ H2 = Δ H1 → die Enthalpie des Gases ändert sich bei der
Expansion nicht Was passiert im Gas?
d H =∂∂
∂∂
HT
d THp
d pp T
⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟
→ aus der Betrachtung der WW mit Umgebung ergab sich: d H = 0

PC MST Kap. 1-7_2010_11 63
Auflösen der Gleichung: Umrechnung auf molare Größen
m m
T T T T
p p pH
p
H n HH Hp p p pT
p C n c cHT
⎛ ⎞ ⎛ ⎞ ⎛ ⎞ ⎛ ⎞∂ ⋅ ∂∂ ∂⎜ ⎟ ⎜ ⎟ ⎜ ⎟ ⎜ ⎟∂ ∂ ∂ ∂⎛ ⎞∂ ⎝ ⎠ ⎝ ⎠ ⎝ ⎠ ⎝ ⎠= − = − = − = −⎜ ⎟∂ ⋅⎛ ⎞∂⎝ ⎠⎜ ⎟∂⎝ ⎠
p
pH
VT VTT
p c
∂∂∂
∂
⎛ ⎞−⎜ ⎟
⎛ ⎞ ⎝ ⎠=⎜ ⎟
⎝ ⎠= ‘Joule-Thomson-Koeffizient’
In der Abbildung sind Isenthalpen aufgetragen. Die Punkte geben das jeweilige Maximum dieser Kurven, d.h. die Inversionstemperatur an. Im schraffierten Bereich nimmt mit abnehmendem Druck (Entspannung) die Temperatur ab (Abkühlung). Nur in diesem Bereich lässt sich das Gas mit dem Joule-Thomson Effekt verflüssigen. Der Koeffizient ist temperaturabhängig und druckabhängig.
Die Temperatur, bei der ∂∂
Tp
H
⎛⎝⎜
⎞⎠⎟ = 0 nennt man die Inversionstemperatur. Will man ein Gas
durch diesen Effekt abkühlen (und schließlich verflüssigen) muss Druck und Temperatur des Gases unter der Inversionstemperatur Ti liegen, oberhalb dieser Temperatur erwärmt sich das Gas bei der Expansion. 6.2.4 Innere Energie und Wärmekapazität idealer Gase Die Atome in einem einatomigen Gas haben drei Translationsfreiheitsgrade der Bewegung: ( )x y zv , v , v . Entsprechend hatten wir für die gesamte kinetische Energie 3
trans kin 2 k Tε = ε =
gefunden. Die Energie für einen Freiheitsgrad ist entsprechend 1kin 2 k Tε = . Beispiel:
Edelgase, Hg-dampf. Zweiatomige Moleküle Bsp. O2, N2 3 Translationsfreiheitsgrade ( )x y zv , v , v , zusätzlich 2 Rotationsfreiheitsgrade
Wir benutzen die folgende Beziehung:∂∂
∂∂
Hp
V TVT
T p
⎛⎝⎜
⎞⎠⎟ = −
⎛⎝⎜
⎞⎠⎟
Man erhält für N2
1
H
T 0,142 K barp
∂∂
−⎛ ⎞ =⎜ ⎟⎝ ⎠
, wobei
dieser Wert natürlich von p und T
abhängt.
Diese Rotation geht nicht, weil sie nicht angeregt werden kann (Trägheitsmoment zu klein)

PC MST Kap. 1-7_2010_11 64
Eine Schwingung kann Energie in Form potentieller und kinetischer Energie aufnehmen. Gleichverteilungssatz: Die vorhandene Energie wird so verteilt, dass jeder Freiheitsgrad im Mittel gleich viel Energie erhält. Schwingungsfreiheitsgrade müssen dabei doppelt gerechnet werden wegen εkin und εpot. 3-atomige Moleküle, gewinkelt, Bsp. H2O
3 Translationsfreiheitsgrade
3 Rotationsfreiheitsgrade
3 Schwingungsfreiheitsgrade
3-atomige, gestreckte Moleküle, Bsp. CO2
+ − + wie oben, aber aus der Bildebene heraus
1 Schwingungsfreiheitsgrad
xF Dx= −
potx
dF Dx
dxε
= − = −
( )21pot 02 D x xε = −
pot
12 kTε =
1 2
kin 2 mvε =
kin12 kTε =
kin pot konstkT
ε + ε = ε =
=
3 Translationsfreiheitsgrade
2 Rotationsfreiheitsgrade (wie bei zweiatomigen Molekülen)
4 Schwingungsfreiheitsgrade

PC MST Kap. 1-7_2010_11 65
Vielatomige gewinkelte Moleküle Es gilt immer: 3 Translationsfreiheitsgrade
3 Rotationsfreiheitsgrade
Berechnung der Zahl der Schwingungsfreiheitsgrade: Wenn ein Molekül aus den Atomen aufgebaut wird, bleibt die Zahl der Freiheitsgrade der beteiligten Atome erhalten. Ein Atom hat immer nur 3 Translationsfreiheitsgrade. Ein N-atomiges Molekül hat also 3 N Freiheitsgrade. Ein Molekül hat immer 3 Translations-freiheitsgrade Z(T) und 2 (lineare Moleküle) oder 3 (alle anderen Moleküle) Rotations-freiheitsgrade, Z(R). Die Zahl der Schwingungsfreiheitsgrade Z(S) ist dann: Z(S) = 3 N–Z(T) – Z(R) → Bsp. Molekül mit N = 9 Atomen: C2H5OH Z(S) = 3 9 3 3 = 21⋅ − − Jetzt können wir die Energie der verschiedenen Moleküle berechnen:
12 k T Zε = ⋅ Z = Faktor für Berechnung der Energie des Moleküls
= Z(T) + Z(R) + 2 Z(S) Wir führen hier 2 Z(S)ein, weil ein Schwingungsfreiheitsgrad doppelt gezählt werden muss. Hg: Z = 3 nur T N2: Z = 3 + 2 + 2 H2O: Z = 3 + 3 + 6 Die Energie pro mol ist entsprechend
AE N= ε = 1 1A2 2k T Z N R T Z=
Bei idealen Gasen (keine WW zwischen den Molekülen) ist die so berechnete Energie gleich der Inneren Energie. Die Energie der Teilchen bei T = 0 bezeichnen wir mit U0 und wir erhalten dann für die thermische Energie E z.B.
30 2E U U R T= − = (einatomige Gase)
70 2E U U R T= − = (N2)
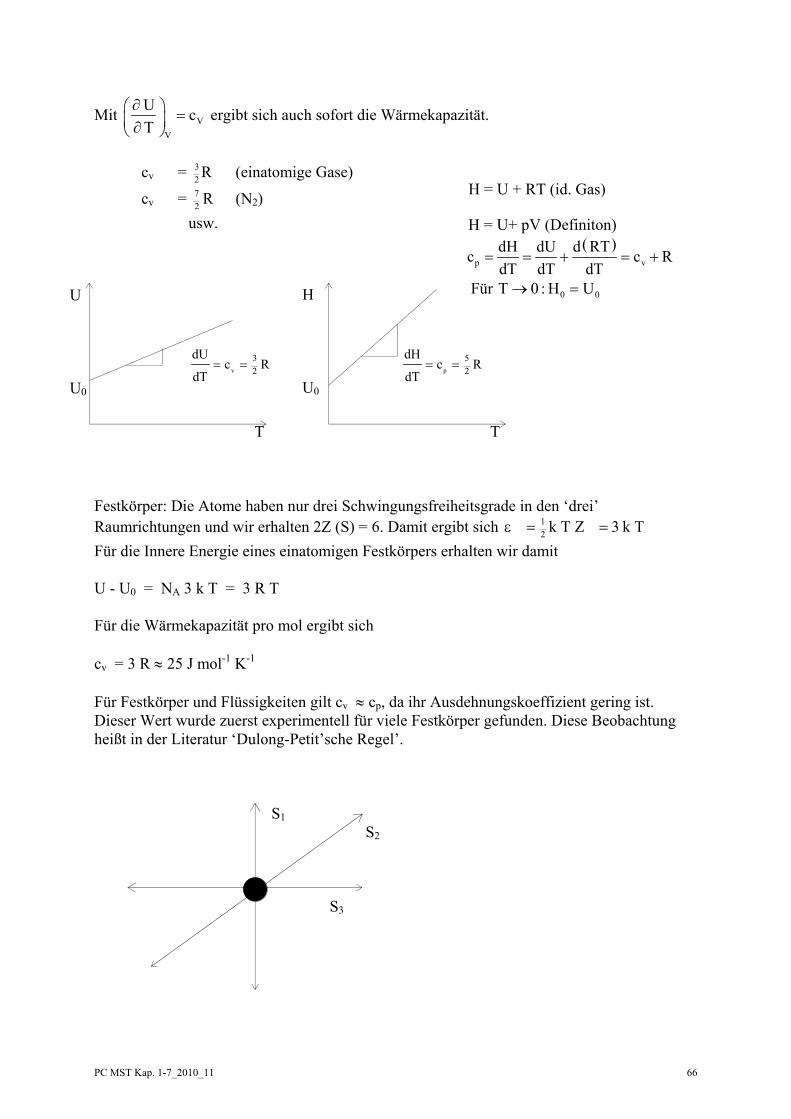
PC MST Kap. 1-7_2010_11 66
Mit VV
U cT
∂⎛ ⎞ =⎜ ⎟∂⎝ ⎠ ergibt sich auch sofort die Wärmekapazität.
cv = 3
2 R (einatomige Gase)
cv = 72 R (N2)
usw.
v
32
dUc R
dT= = p
52
dHc R
dT= =
T T Festkörper: Die Atome haben nur drei Schwingungsfreiheitsgrade in den ‘drei’ Raumrichtungen und wir erhalten 2Z (S) = 6. Damit ergibt sich 1
2 k T Z 3 k Tε = = Für die Innere Energie eines einatomigen Festkörpers erhalten wir damit U - U0 = NA 3 k T = 3 R T Für die Wärmekapazität pro mol ergibt sich cv = 3 R ≈ 25 J mol-1 K-1 Für Festkörper und Flüssigkeiten gilt cv ≈ cp, da ihr Ausdehnungskoeffizient gering ist. Dieser Wert wurde zuerst experimentell für viele Festkörper gefunden. Diese Beobachtung heißt in der Literatur ‘Dulong-Petit’sche Regel’. S1 S2 S3
H = U+ pV (Definiton)
H = U + RT (id. Gas)
( )p v
dH dU d RTc c RdT dT dT
= = + = +
0 0Für T 0 : H U→ = U U0
H U0

PC MST Kap. 1-7_2010_11 67
6.3 Enthalpieänderung bei Phasenumwandlungen Wir betrachten folgendes System p
Isotherme, T = konst flüss
Gas
H = f (p, T, ng, nfl) pD = Dampfdruck = konstant V ∧Gleichgewicht zwischen Gas und flüssig
dH = g fl g fl gfl
g flT,n n p,n ,n p,T,np,T,ng fl
H H H Hd p d T d n d np T n n
∂ ∂ ∂ ∂⎛ ⎞ ⎛ ⎞ ⎛ ⎞ ⎛ ⎞+ + +⎜ ⎟ ⎜ ⎟ ⎜ ⎟ ⎜ ⎟∂ ∂ ∂ ∂⎝ ⎠ ⎝ ⎠ ⎝ ⎠⎝ ⎠
1. d T = 0 2. d p = 0 3. kein Stoffaustausch mit Umgebung: ng + nfl = n (gesamt) = konst. (∧Massenerhaltung) → d ng + d nfl = 0 → d ng = - d nfl Zur Abkürzung nennt man
ii p, T
H Hn
∂⎛ ⎞ ≡⎜ ⎟∂⎝ ⎠ = ‘partielle molare Enthalpie’
Für reine Stoffe (wie bei dem hier betrachteten System) gilt:
ii ip, T
H H Hn n
∂⎛ ⎞ = =⎜ ⎟∂⎝ ⎠= ‘molare Enthalpie’
Hg = molare Enthalpie des Gases Hfl = molare Enthalpie der Flüssigkeit Hf = molare Enthalpie des festen Stoffes Setzt man diese Größen ein, so erhält man d H = (Hg - Hfl) d ng, Hg- Hfl = Änderung der Enthalpie, wenn bei p,
T = konst. 1 mol Flüssigkeit in den gasförmigen Zustand übergeht.
p
Gas
flüssig
z.B. H2O flüssig und gasförmig, kein Fremdgas! wie z.B. Luft
pD flüss + Gas

PC MST Kap. 1-7_2010_11 68
Man bezeichnet: Hg - Hfl ≡ ΔvapH ‘Verdampfungsenthalpie’ (‘vaporisation’) Hg - Hf ≡ ΔsubH ‘Sublimationsenthalpie’ (‘sublimation’) Hfl - Hf ≡ ΔfusH ‘Schmelzenthalpie’ (‘fusion’)
Analoge Beziehungen gelten für die Änderung der Inneren Energie bei der Phasenumwandlung. Man benutzt sie für die Nebenbedingung V = konst.
( )d H c gaspdT p
⎛ ⎞⎜ ⎟= =⎜ ⎟⎝ ⎠

PC MST Kap. 1-7_2010_11 69
7. Die Enthalpieänderung bei chemischen Reaktionen Wir betrachten ein System, das aus einer Phase besteht, in dem einen Gemisch verschiedener Stoffe vorhanden ist, zwischen denen eine chemische Reaktion abläuft. Reaktion: νA A + νB B→← νC C + νD D positiv für Produkte νi = stöchiometrische Koeffizienten negativ für Edukte i = Index für Stoff Die Enthalpie hängt jetzt von den Molzahlen aller vorhandenen Stoffe ab: H = f (p, T, ni)
d H = i i j j jj
A B C DT,n p,n p,T,n A p,T,n B p,T,n Dp,T,n CA B C D
H H H H H Hd p d T d n dn d n d np T n n n n
HKurzschreibweise: d nini i p,T,n ij
∂ ∂ ∂ ∂ ∂ ∂∂ ∂ ∂ ∂ ∂ ∂
∂∂
≠ ≠ ≠≠
⎛ ⎞⎛ ⎞ ⎛ ⎞ ⎛ ⎞⎛ ⎞ ⎛ ⎞+ + + + +⎜ ⎟⎜ ⎟ ⎜ ⎟ ⎜ ⎟⎜ ⎟ ⎜ ⎟
⎝ ⎠ ⎝ ⎠ ⎝ ⎠ ⎝ ⎠ ⎝ ⎠⎝ ⎠
⎛ ⎞⎜ ⎟∑⎜ ⎟⎝ ⎠ ≠
Die Änderungen der Stoffmengen (Molzahlen) beim Ablauf chemischer Reaktionen sind aber nicht unabhängig voneinander, sondern durch die stöchiometrische Gleichung miteinander verknüpft. Analog ist bei der Verdampfung (Kap. 6.3) die Änderung der Stoffmenge der gasförmigen Phase mit der der flüssigen Phase verknüpft. Bsp. CH4 + 2 O2 → CO2 + 2 H2O Verbrennt man nun 0,01 mol CH4 (d.h. d n molCH4
0 01= − , ) so gilt notwendigerweise → d n mol, d n mol, d n molO CO H O2 2 2
2 0 01 1 0 01 2 0 01= − ⋅ = ⋅ = ⋅, , , Daraus ergibt sich: Aus der Stöchiometrie der Reaktion kann man aus der Angabe der Stoffmengenänderung eines Stoffes die Stoffmengenänderungen aller anderen Stoffe berechnen. Diese Größe nennt man die "Reaktionsvariable". Definition: i io in n= + ν ξ
oder: i
i
d ndξ =ν
Einheit: mol (ξ (griech.) gesprochen: ksi)
Man erhält dann: jj
i ip,T,n ip,T,n ii i i
H Hd n dn n ≠≠
⎛ ⎞ ⎛ ⎞∂ ∂= ν ξ⎜ ⎟ ⎜ ⎟∂ ∂⎝ ⎠ ⎝ ⎠
∑ ∑ i ii
H d= ν ξ∑
(ν (griech.) gesprochen: nü)
in = Stoffmenge i
ion = Stoffmenge i am Anfang der Reaktion
iν = stöchiometrischer Faktor

PC MST Kap. 1-7_2010_11 70
j
ii p,T,n i
H Hn
≠
⎛ ⎞∂≡ =⎜ ⎟∂⎝ ⎠
partielle molare Enthalpie des Stoffes i
Wir nennen i ii p,T
rHH H
⎛ ⎞∂ν = Δ = ⎜ ⎟∂ ξ⎝ ⎠
∑ die ‘Reaktionsenthalpie’(Einheit: Jmol-1).
Für das obige Beispiel explizit geschrieben, ist die Reaktionsenthalpie
( ) ( ) ( ) ( )r i i 2 2 2 4i
H H 2 H H O 1 H CO 2 H O 1 H CHProdukte Edukte
Δ = ν = + − −∑
Interpretation: d Q V d p+
T, p, p,T
H H Hd H d p d T dp T
Systemeigenschaften ξ ξ
∂ ∂ ∂⎛ ⎞ ⎛ ⎞ ⎛ ⎞= + + ξ =⎜ ⎟ ⎜ ⎟ ⎜ ⎟∂ ∂ ∂ξ⎝ ⎠ ⎝ ⎠ ⎝ ⎠
Wir halten Druck und Temperatur konstant, d.h. dp = d T = 0. Daraus folgt
rp,T
Hd Q d H d⎛ ⎞∂
= ξ = Δ ξ⎜ ⎟∂ ξ⎝ ⎠
Def.: Die Reaktionsenthalpie ΔrH ist die Wärmemenge, die beim Ablauf der chemischen
Reaktion frei wird (genauer formuliert: wenn 1 mol chemische Umsetzungen der Art, wie sie in der stöchiometrischen Gleichung beschrieben sind, stattgefunden haben). Um die Temperatur konstant zu halten, muss die Reaktionsenthalpie abgeführt werden (oder zugeführt werden, wenn diese Wärme verbraucht wird).
Def.: Wenn Wärme abgegeben wird bei der chemischen Reaktion (ΔH<0), nennt man die Reaktion exotherm, wenn Wärme aufgenommen wird (ΔH>0) endotherm.
7.1 Temperaturabhängigkeit von ΔrH Die Enthalpie eines Stoffgemisches, in dem eine chemische Reaktion (beschrieben durch die Reaktionsvariableξ ) stattfindet, ist gegeben durch
d H = ,p T,p ,T
H H Hd T d d pT pξ ξ
∂ ∂ ∂⎛ ⎞ ⎛ ⎞ ⎛ ⎞+ ξ +⎜ ⎟ ⎜ ⎟ ⎜ ⎟∂ ∂ ξ ∂⎝ ⎠ ⎝ ⎠ ⎝ ⎠
↓ ↓ Cp ΔrH Darin ist: Cp = gesamte Wärmekapazität des Systems (Gemisch aller Stoffe) cpi = molare Wärmekapazität des Stoffes i Unter der Nebenbedingung p = konst. d p = 0 erhalten wir
WW mit Umgebung
d Q V d p+

PC MST Kap. 1-7_2010_11 71
d H = Cp d T + Δ rH d ξ H ist eine Zustandsfunktion. Dann gilt der Schwartz’sche Satz (siehe Kap. 3.6), d.h. die zweiten Ableitungen müssen gleich sein:
T
T
prp
T
HHT
T
CH CT
ξ
ξ
ξ
⎛ ⎞⎛ ⎞⎛ ⎞ ∂⎛ ⎞∂ ∂∂ ⎜ ⎟⎜ ⎟⎜ ⎟⎜ ⎟ ∂∂ ξ ⎝ ⎠⎝ ⎠ ⎜ ⎟⎜ ⎟ =⎜ ⎟ ⎜ ⎟∂ ∂ ξ⎝ ⎠ ⎝ ⎠
∂⎛ ⎞∂ Δ⎛ ⎞ = = Δ⎜ ⎟⎜ ⎟∂ ∂ ξ⎝ ⎠ ⎝ ⎠
Die Größe ∂
∂ξ
Cp ist analog zu ∂∂ξ
H definiert, d. h.
i
pi p p
iT
Cc C
∂⎛ ⎞= ν = Δ⎜ ⎟∂ ξ⎝ ⎠∑
Integration dieser Gleichung ergibt:
2 2 2
1 1 1
H T T
p r 2 r 1 pH T T
d H C d T H H C d TΔ
Δ
Δ = Δ → Δ = Δ + Δ∫ ∫ ∫ = Δ H c d Ti pT
T
i11
2
+ ∑∫ ν
Diese Gleichung nennt man den ‘Kirchhoff’schen Satz’. Dieser beschreibt die Temperatur-abhängigkeit der Reaktionsenthalpie. Man kann die Gleichung integrieren entweder unter der Voraussetzung: a) Δ Cp = konst. oder: b) Δ Cp = f (T) Bsp. Verbrennung von Methan:
( ) ( ) ( ) ( )ΔC c c H O c CO c O c CHp i p ii
p p p p= = + − −∑ν 2 22 2 2 4
7.2 Die Reaktionsenthalpie als Zustandsfunktion: Der Heß’sche Satz U und H sind Zustandsfunktionen, ebenso ΔU und ΔH. Das bedeutet, dass ihr Wert nur vom Anfangs- und Endzustand abhängt, nicht vom Weg. Für chemische Reaktionen, die über Zwischenstufen verlaufen, setzt sich die Reaktionsenthalpie dann additiv aus den ΔH’s der einzelnen Schritte zusammen.

PC MST Kap. 1-7_2010_11 72
Bsp.: Wir betrachten folgende Zustandsänderungen eines Systems:
Die Enthalpieänderung auf dem Weg I → II→ III muss gleich sein der Enthalpieänderung auf dem Weg I → III COgas + 1
2 O2 gas → CO2 gas Δ H2 = -283,1 kJ/mol
Cfest + O2 gas → CO2 gas Δ H3 = -393,7 kJ/mol
Cfest + 1
2O2 gas → COgas Δ H1 ist nicht messbar, da CO sofort weiter reagiert;
Δ H1 = Δ H3 - Δ H2 = −110,6 kJ/mol Man bezeichnet diesen Befund als den “Heß’schen Satz”. Er beschreibt die Anwendung des 1. Hauptsatzes auf den Energieumsatz bei chemischen Reaktionen und er wurde vor der Formulierung des 1. Hauptsatzes gefunden. 7.3 Bildungsenthalpien Es ist nicht notwendig, für alle möglichen chemischen Reaktionen Δ H zu messen. Auf Grund des 1. Hauptsatzes (bzw. des Heß’schen Satzes) kann man Δ H einer Reaktion berechnen, wenn man die jeweilige Reaktion durch eine Folge anderer Reaktionen ersetzen kann.
I C + O2 ΔH3 II III CO2
ΔH1 CO + 1
2O2
ΔH2

PC MST Kap. 1-7_2010_11 73
Bei einer chemischen Reaktion sind nun alle Elemente, die in den Edukten enthalten sind, auch in den Produkten enthalten. Daher können wir immer den folgenden Kreisprozess konstruieren, der hier am Beispiel der Verbrennung der Glukose gezeigt ist: Elemente: 6 C, 6 H2, 9 O2 H ΔBH (Glukose) Bildungsenthalpien ( )B 2H COΔ ΔBH (O2) ( )B 2H H OΔ Edukte: C6H1206 + 6 O2 Daraus ergibt sich sofort: ΔB H (Glu) + 6 ΔB H (O2) + Δr H1 = 6 ΔB H (CO2) + 6 ΔB H (H2O) Δr H1 = 6 ΔB H (CO2) + 6 ΔB H(H2O) - ΔB H (Glu) - 6 ΔB H (O2) H (Produkte) - H (Edukte) Def.: Die Bildung einer Verbindung aus den Elementen nennt man die ‘Bildungsreaktion’. Die
dazugehörende Reaktionsenthalpie ist die ‘Bildungsenthalpie’ (‘enthalpy of formation’). Index: B oder f (engl.).
Die Reaktionsenthalpie einer beliebigen chemischen Reaktion lässt sich aus der
Differenz der Bildungsenthalpien von Produkten und Edukten berechnen. Man tabelliert nun die Bildungsenthalpien aller chemischen Verbindungen. Dabei muss beachtet werden, dass die Zahlenwerte von den Zuständen der Stoffe, die beteiligt sind, abhängen (z.B. ob flüssiges oder gasförmiges Wasser, oder ob eine Verbindung in reiner oder gelöster Form entsteht). 7.4 Standardzustände 1. Der Standardzustand einer gasförmigen Substanz bei beliebiger Temperatur ist der
eines reinen idealen Gases bei p = 1 bar (Index: g (gasförmig). 2. Der Standardzustand einer flüssigen Substanz bei beliebiger Temperatur ist der einer reinen Flüssigkeit bei p = 1 bar (Index: l (liquid) oder fl (flüssig)). 3. Der Standardzustand einer festen Substanz bei beliebiger Temperatur ist der einer
reinen festen Substanz bei p = 1 bar (Index: s (solid) oder f (fest)). 4. Der Standardzustand einer Substanz in einer Lösung bei beliebiger Temperatur ist der
einer idealen Lösung mit der Standardkonzentration (m = 1 mol kg-1 oder c = 1 mol L-1) bei p = 1 bar (Index: aq)).
Bildungsenthalpien
Reaktionsenthalpie ΔrH1 Produkte: 6 CO2 + 6 H2O

PC MST Kap. 1-7_2010_11 74
Anmerkungen: • Die Wahl des Standardzustandes der Elemente spielt keine Rolle bei der Berechnung
der Reaktionsenthalpien, da für die Bildung der Edukte und die der Produkte der gleiche Standardzustand verwendet wird und dieser bei der Differenzbildung herausfällt.
• In der statistischen Thermodynamik wird als Standardzustand der der isolierten Atome in unendlicher Entfernung verwendet.
• Bei der Festlegung des Druckes hat man früher (vor 1982) die Festlegung p = 1 atm = 1,01325 bar getroffen. Heute wird p = 1 bar verwendet. Zahlenmäßig kann man diesen Unterschied vernachlässigen.
• Größen, die sich auf die Standardzustände beziehen, werden durch den IndexӨ gekennzeichnet, z.B. ΔHӨ. Da ΔHӨ von der Temperatur abhängt, wählt man als Temperatur für die Tabellierung der Daten T = 298 K. Nomenklatur: ΔH298
Ө oder ΔHӨ (298):
Die Innere Energie (und die Enthalpie) eines Stoffes kann nicht absolut bestimmt werden, sondern man misst immer nur Änderungen. Daher kann man (willkürlich) einen Nullpunkt wählen und alle Enthalpien relativ zu diesem Nullpunkt angehen (analog gibt man Höhen auf der Erdoberfläche relativ zur Meereshöhe an ( = NN)). Def.: Die Bildungsenthalpie aller Elemente in ihrem stabilsten Zustand
(= Standardzustand) ist Null. BΔ HӨ (Elemente) ≡ 0 Def.: Unter der Enthalpie einer Verbindung versteht man die Bildungsenthalpie
dieser Verbindung, wobei die Enthalpien der Elemente im Standardzustand und unter Standardbedingungen willkürlich auf Null gesetzt sind.
Bsp: ( ) ( ) ( )1
2 2 22H g O g H O fl+ → ) Enthalpie des Wassers≡Bildungsenthalpie des Wassers ( ) ( ) ( ) ( ) ( )1
2 B 2 B 2 B 2 B 22H H O H H O H H H O H H Oθ θ θ θ θ= − − = Δ
BH= Δ∑Ө (Edukte)
RΔ HӨ
B (Pr odukte)H= =Δ∑
Edukte
Produkte

PC MST Kap. 1-7_2010_11 75
Allgemein: rΔ HӨ = ν ii∑ H
iӨ = i B
iν Δ∑ Hi
Ө
Bsp: C6H12O6 (f)+ 6 O2 (g) → 6 CO2 (g) + 6 H2O (fl)
rΔ H Ө = 6 ΔB HӨ (CO2) + 6 ΔB HӨ (H2O) - 6 ΔB HӨ (O2) - ΔB HӨ (Glu)
1 1 16 ( 393,51) kJ mol +6 ( 285,83) kJ mol 6 (0) ( 1274) kJ mol− − −= − ⋅ − − − −
1= 2802,04 kJ mol−−
Diese Reaktionsenthalpien können dann auf beliebige andere Temperaturen und Drucke umgerechnet werden. Analoge Beziehungen wie für die Enthalpie kann man auch für die Innere Energie herleiten. Die Innere Energie benutzt man sinnvollerweise dann, wenn beim Experiment V = konst. ist. Zusammenstellung der Gleichungen für die Reaktionsenergie: U = f (V, T, ni)
d UUT
d TUV
d VU
d d Q p d VV T V T
=⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟ = −
∂∂
∂∂
∂∂ξ
ξξ ξ, , ,
rV,T
U U⎛ ⎞∂
= Δ⎜ ⎟∂ ξ⎝ ⎠ = Reaktionsenergie
2
i
1
Tr
V r 2 r 1 V V i ViT
U C U U C d T, C cT
ξ
⎛ ⎞∂ Δ= Δ → Δ = Δ + Δ Δ = ν⎜ ⎟∂⎝ ⎠
∑∫
7.5 Kalorimetrie
(= Wärme, die bei der chemischen Reaktion frei wird, wenn V = konst.)
Früher wurde als Einheit der Wärmemenge die Kalorie verwendet (1cal = 4,18 J). Die Kalorie ist die Wärmemenge, die man benötigt, um 1 g Wasser um 1 K zu erwärmen. Die Kalorie wird heute noch im nichtwissenschaftlichen Bereich, z.B. bei der Angabe des Energieinhaltes von Nahrungsmitteln verwendet. Schreibweise: 1 CAL = 1 kcal = 4184 J. Reaktionsenthalpien werden meistens kalori-metrisch bestimmt. Wir betrachten im Folgenden ein häufig verwendetes adiabatisches Kalorimeter, indem man (bei V = konst.) Verbrennungsenergien misst.

PC MST Kap. 1-7_2010_11 76
i i
iV,n T,n V,T,nj ii
U U Ud U d T d V d nT V n
System
∂ ∂ ∂∂ ∂ ∂ ≠
⎛ ⎞⎛ ⎞ ⎛ ⎞= + + ⎜ ⎟⎜ ⎟ ⎜ ⎟
⎝ ⎠ ⎝ ⎠ ⎝ ⎠∑ d Q d W
WW mit Umgebung= +
Wir unterscheiden Druck−Volumen−Arbeit von den anderen Arbeiten: d W p d V + d W'= −
i
V rT,n
Ud U C d T d V U d d Q pd V d W 'V
⎛ ⎞∂= + + Δ ξ = − +⎜ ⎟∂⎝ ⎠
CV = Wärmekapazität des Systems, rUΔ = Reaktionsenergie Dies ist die Grundgleichung der Kalorimetrie, aus der durch Einführung der jeweiligen experimentellen Bedingungen die benötigten Zusammenhänge hergeleitet werden können.
Dann wird aus der Grundgleichung: V rC d T U d 0+ Δ ξ = Wir integrieren von der Anfangs- bis zur Endtemperatur, entsprechend vom Anfangs- bis zum Endwert der Reaktionsvariablen.
E E
A A
T
r VT
U d C d Tξ
ξ
Δ ξ = −∫ ∫ (oder mit dd n i
iξ
ν= )
E
A
ni
rin
d nU= Δν∫
Wenn rUΔ und CV konstant, ergibt sich:
( ) ( )r E A V E AU C T TΔ ξ −ξ = − − oder
Beispiel: Messung der Wärmekapazität CV:
(U ist in dieser Gleichung die Spannung, I die elektrische Stromstärke, d t Zeit)
Beispiel: Messung von rUΔ : V = konst.: d V = 0 adiabatisch: d Q = 0 keine Arbeit: d W = 0
Setzen wir die Molzahl eines verschwindenden Stoffes ein, der den stöchiometrischen Faktor νi = -1 hat, so ergibt sich
( ) ( )r E A V E AU n n C T T−Δ − = − − Wenn die Reaktion vollständig ist, so ist
En 0= und man erhält
r VTU Cξ
ΔΔ = −
Δ r V
A
TU CnΔ
Δ = −
V = konst.: d V = 0 adiabatisch d Q = 0 keine chemische Reaktion: zugeführte elektrische Arbeit:
d ξ = 0 d W’ = U I d t

PC MST Kap. 1-7_2010_11 77
Wir erhalten aus der Grundgleichung:
CV d T = d W’
d W C d TW
VT
T
A
E
''
0∫ ∫=
CV = W
T TE A
'−
W’ = zugeführte elektrische Arbeit
Damit ist CV bekannt und rUΔ kann ermittelt werden. Umrechnung von rUΔ in rHΔ (analog der Umrechnung von cp in cV) d H = d U + p d V + V d p,
Wir setzen ,V T, V,T
U U Ud U d T d V dT Vξ ξ
∂ ∂ ∂ ξ∂ ∂ ∂ ξ
⎛ ⎞ ⎛ ⎞ ⎛ ⎞= + +⎜ ⎟ ⎜ ⎟ ⎜ ⎟⎝ ⎠ ⎝ ⎠ ⎝ ⎠
ein
und betrachten den Fall, dass d T = 0, d p = 0 und dividieren durch ∂ξ :
d Hd
U UV
Vp
V
p T V T T p T p Tξ
∂∂ξ
∂∂
∂∂ξ
∂∂ξ
ξ
⎛⎝⎜
⎞⎠⎟ =
⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟
⎛⎝⎜
⎞⎠⎟ +
⎛⎝⎜
⎞⎠⎟
, , , , ,
,
mit r i iip,T
V V V⎛ ⎞∂
≡ Δ = ν⎜ ⎟∂ ξ⎝ ⎠∑ = Reaktionsvolumen
νi = stöchiometrischer Koeffizient von i Vi = Molvolumen von i Def.: Das Reaktionsvolumen ist die Volumenveränderung, wenn 1 mol Umsetzungen der
Art, die durch die zugehörige stöchiometrische Gleichung beschrieben werden, stattfinden.
Damit ergibt sich:
r r rT,
U H U + +p VV
ξ
⎡ ⎤⎛ ⎞∂Δ = Δ Δ⎢ ⎥⎜ ⎟∂⎢ ⎥⎝ ⎠⎣ ⎦
mit ∂∂
∂∂
ξ
UV
TpT
pT V
⎛⎝⎜
⎞⎠⎟ =
⎛⎝⎜
⎞⎠⎟ −
,
Spezialfälle: 1) Nur flüssige und feste Stoffe sind beteiligt (z.B. Reaktionen in Lösungen). Dann ist das
auftretende ΔV sehr klein und es gilt ΔrH ≈ ΔrU (weil sich das Volumen praktisch nicht ändert ΔrV ≈ 0).

PC MST Kap. 1-7_2010_11 78
2) Für Reaktionen, bei denen nur ideale Gase beteiligt sind, gilt: ∂∂
UV
T
⎛⎝⎜
⎞⎠⎟ = 0 und wir erhalten
r r r r i i r ii i
H U p V U p V U p VΔ = Δ + Δ = Δ + ν = Δ + ν∑ ∑
Mit pV = R T ergibt sich ii
H U R TΔ = Δ + ν∑
Bsp.: 2 2 3 i r ri
N 3 H 2 NH 2 1 3 2 H U 2 RT+ → → ν = + − − = − → Δ = Δ −∑
→ Wenn i 0ν =∑ d.h. wenn sich die Molzahl der Gase bei der Reaktion nicht ändert, gilt r rH UΔ = Δ . 3) Bei Reaktionen, bei denen Gase und feste und flüssige Stoffe beteiligt sind, müssen nur die
Volumina der Gase berücksichtigt werden, d.h. in der Summe müssen nur die stöchiometrischen Koeffizienten der beteiligten gasförmigen Stoffe berücksichtigt werden.
r r i
iH U R TΔ = Δ + ν∑ (Gase)
Druckabhängigkeit von Δ H (analoge Herleitung wie T–abhängigkeit )
r rr
T p
H VV Tp T
⎛ ⎞ ⎛ ⎞∂ Δ ∂Δ= Δ −⎜ ⎟ ⎜ ⎟∂ ∂⎝ ⎠ ⎝ ⎠
(für ideale Gase gilt: rH 0p
⎛ ⎞∂ Δ=⎜ ⎟∂⎝ ⎠
)
Häufig benutzte Enthalpien: 1. Bildungsenthalpien
2. Verbrennungsenthalpien
3. Phasenumwandlungsenthalpien
4. Dissoziationsenthalpien von Bindungen: (AB (g) → A (g) + B (g))
Bindungsdissoziationsenthalpien ΔBDH (A-B)/kJ mol-1 bei 298 K
H - H 436 H - Cl 431 H - O 428 H - OH 492 H - CH3 435 H3C - CH3 368

PC MST Kap. 1-7_2010_11 79
5. Mittlere Dissoziationsenthalpie von Bindungen aus verschiedenen Verbindungen (Mittelwert): Mittlere Bindungdissoziationsenthalpien ΔBDH (A-B) /kJ mol-1
H C N O H 436 C 413 348 N 388 305 163 O 460 360 157 146
6. Atomisierungsenthalpie: ΔAH Zerlegung eines Moleküls in Atome. Dies ist im Fall H - H → gleich der
Bindungsdissoziationsenthalpie, im Fall von Na (fest) ist das gleich der Sublimationsenthalpie: Na (f) → Na (ges).
Standard–Atomisierungsenthalpien 1
AH / kJ mol−Δ
C (s) 716.68 Na (s) 108.4 K (s) 89.8 Cu (s) 339.3
Diese Größen sind sehr nützlich bei der Berechnung (Abschätzung) von Bildungsenthalpien
von Stoffen, deren Struktur bekannt ist, aber keine Daten über die Bildungsenthalpie vorliegen.
Beispiel: Abschätzung der Bildungsenthalpie von Methanol. Die Pfeilrichtungen im Diagramm zeigen, in welcher Richtung die jeweiligen Enthalpienänderungen definiert sind. H C(f)+2 H2(g)+ 1
2O2(g)
0
Bsp.:
H - O → H + O, Δ H = 428 kJ mol-1 H - OH → H + OH, Δ H = 492 kJ mol-1 Mittelwert
( ) 1BD
1H 428 492 460 kJ mol2
−Δ = + =
BDH= Δ∑ Θ
= ∑aller mittleren Bindungsdissoziations-enthalpien
CH3OH(g) vapΔ H Θ
C(g) + 4 H(g) + O(g)
AH= Δ∑ Θ
= ∑aller Atomisierungsenthalpien
BΔ H Θ Bildungsenthalpie CH3OH(fl)

PC MST Kap. 1-7_2010_11 80
B vap BD AH H H H 0Δ + Δ + Δ − Δ =∑ ∑
( ) ( ) ( ){ }( ) ( ) ( ){ }{ } { }
[ ]
( )
B vap BD BD BD
1A A A2
1
1 1
B
H H 3 H C H H C O H O H
H C 2 H H H H O O
= 38 3 413 + 1 360 + 1 463 + 717 + 2 436 + 497 kJ mol
38 2062 + 1838 kJ mol = 262 kJ mol
H experimentell 239 kJ m
−
− −
Δ = −Δ − Δ − + Δ − + Δ − +
Δ + Δ − + Δ −
⎡− − ⋅ ⋅ ⋅ ⋅ ⎤⎣ ⎦= − − −
↑
Δ = − 1ol−
7. Lösungsenthalpie Löst man eine Substanz in Wasser, so ist dies mit einer Enthalpieänderung verbunden. Wenn
man wenig Substanz in viel Wasser löst, nennt man diese Größe ‘Lösungsenthalpie bei unendlicher Verdünnung’ ΔLH. Anschaulich bedeutet dies, dass keine Wechselwirkungen zwischen den gelösten Molekülen stattfinden sondern nur Wechselwirkungen zwischen dem gelösten Stoff und dem Wasser (= ideale Lösung). Index = aq.
Bsp. ( ) ( )HCl g HCl aq→ 1H 74,85 kJmol−Δ = − Bestimmung der Bildungsenthalpie in Lösung:
1) Bildungsreaktion: ( ) ( ) ( )2 21 1H g Cl g HCl g2 2
+ → , ( ) 1BH g 92,3kJmol−Δ = −
2) Lösungsreaktion: ( ) ( )HCl g HCl aq→ , 1LH 74,9 kJmol−Δ = −
3) Bildungsreaktion in Lösung: ( ) ( ) ( )2 21 1H g Cl g HCl aq2 2
+ → , ( ) 1BH aq 167,2 kJ mol−Δ = −
Die Bildungsenthalpie in Lösung kann man als Summe der Bildungsenthalpien von H+ (aq) und Cl- (aq) auffassen. Man kann diese beiden Größen aber nicht getrennt messen. Daher setzt man
willkürlich ( ) ( )21 H g H aq ,2
+→ ( )( )BH H aq 0+Δ ≡
→ Daraus ergibt sich dann sofort BHΔ für Cl- und durch Messung der Bildungsenthalpie von NaCl (aq) die für Na+ (aq) usw.
Standard-Bildungsenthalpien von gelösten Ionen in unendlicher Verdünnung 1
BH / kJ mol−Δ .
Kationen Anionen H+ 0 OH- - 230.0 Na+ - 240.1 Cl- - 167.2 Cu2+ + 64.8 SO4
2− - 909.3 Al3+ - 524.7 PO4
3− - 1277.4
← Unterschied durch Verwendung mittlerer Bindungdissoziationsenthalpien





























