Embed Size (px)
Citation preview

1
Post Hartree-Fock local-correlation method forcrystals
S. Casassa
C. Pisani, L. Maschio, M. Halo
Theoretical Chemistry Group, Dipartimento di Chimica I.F.M., Università di Torino
Excellence Center: Nanostructured Interfaces and Surfaces (NIS)
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

2
Electron Correlation (1)• Lowdin definition (1959):
? Ecorr = E0 − EHF
→ the difference between the exact energyof the fundamental state E0 and the Hartree-Fock energy EHF calculated by means of thevariational method
within the HF framework we can miss..
• STATIC correlation
? when the fundamental configuration is not a good approximation of theelectronic ground state of the system
• DYNAMIC correlation
? always: due to the lack of INSTANTANEOUS correlation in electrons motion
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

3
Electron Correlation (2)
♣ DENSITY FUNCTIONAL METHODS
• semi-empirical receipts
• need of parametrization by means of ab-initio results
• accurate but often not accurate enough:→ it can’t account for dispersive interactions between remote parts of the system
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
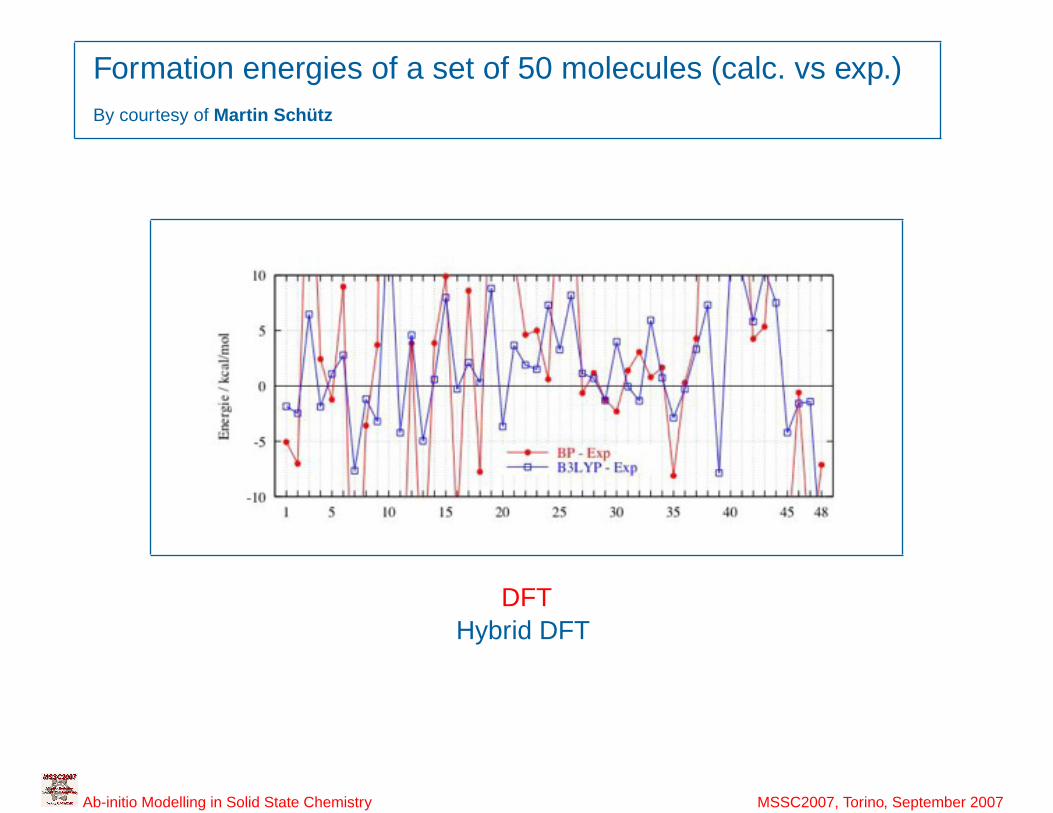
4
Formation energies of a set of 50 molecules (calc. vs exp.)By courtesy of Martin Schütz
DFTHybrid DFT
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

5
Electron Correlation (3)
♣ Møller-Plesset Perturbation theory, at second order (MP2)
♠ it is inadequate in many respects: in particular, it is non-variational
♥ is the simplest post-HF correlation technique,
♥ MP2 energy E(2) is size consistent;
♥ it provides an adequate treatment of long-range interactions;
♥ it permits the assessment of techniques, basis sets, etc., before introducing amore adequate treatment of short-range interactions (MP4, CCSD,..);
♥ the MP2 correlation energy estimates can be corrected using the simple GrimmeSCS (Spin-Component-Scaled) MP2 formula [Grimme, J. Chem. Phys. 118 (2003)9095], which has proved very efficient in a molecular context [Hill, Platts, Werner,PCCP 8 (2006) 4072].
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
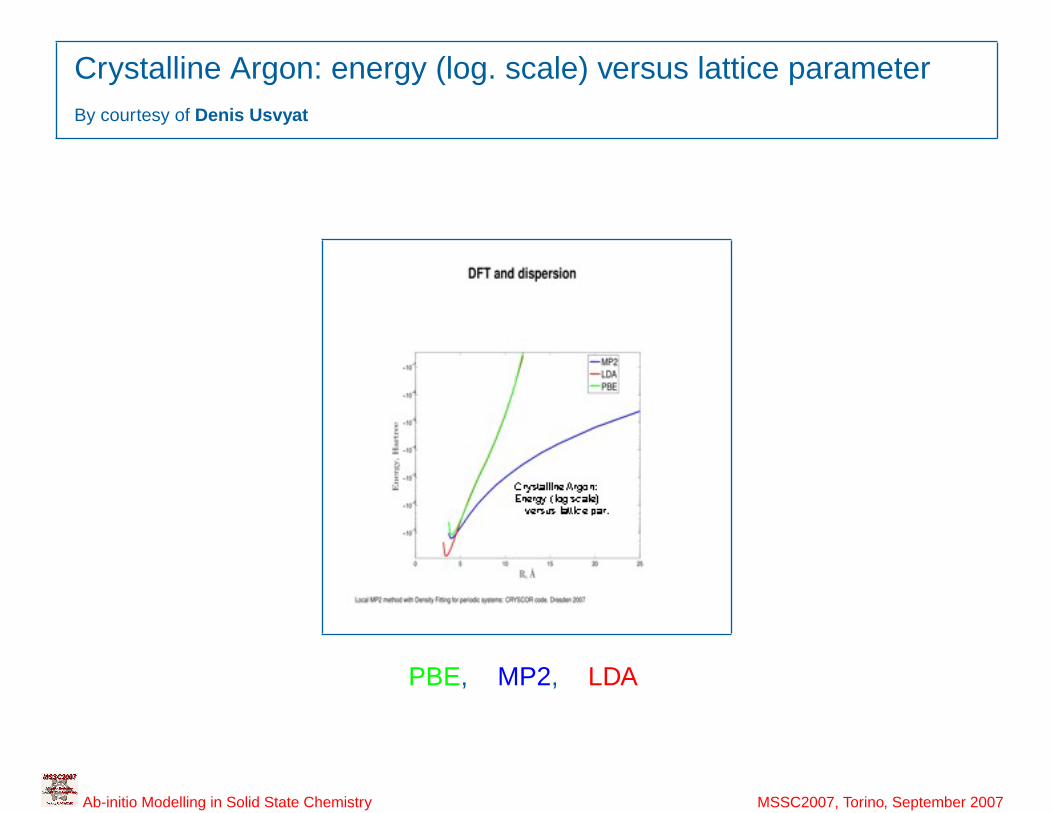
6
Crystalline Argon: energy (log. scale) versus lattice parameterBy courtesy of Denis Usvyat
PBE, MP2, LDA
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
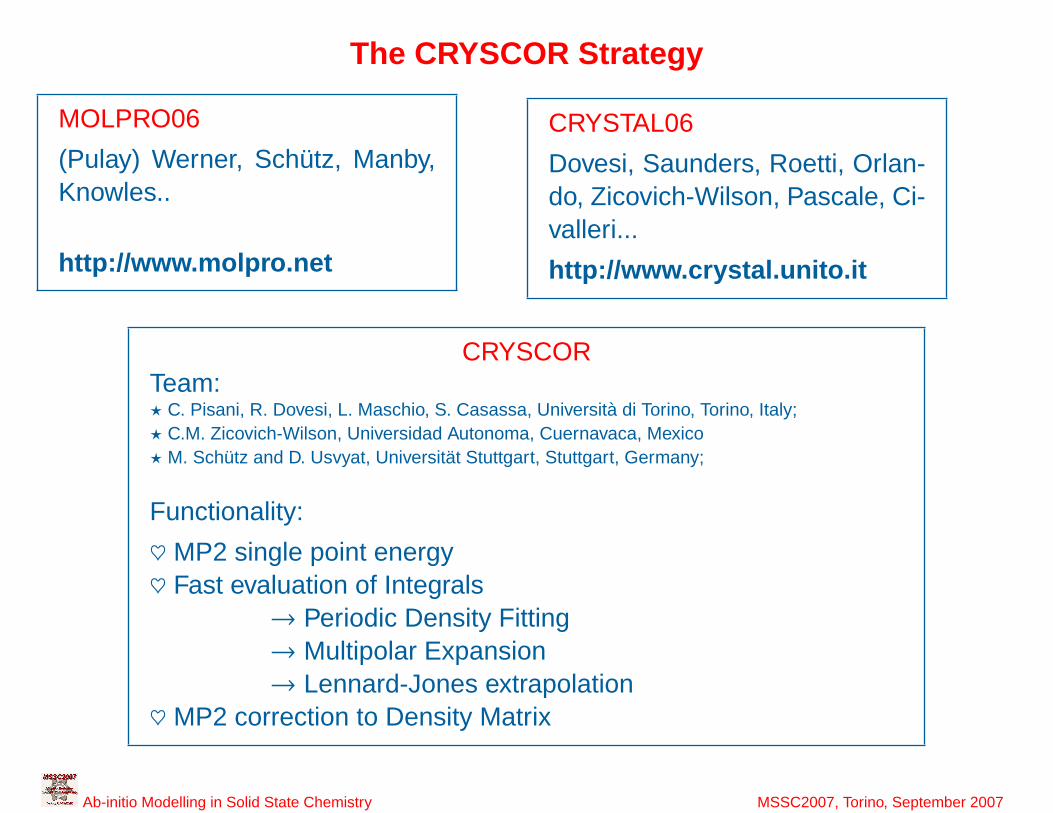
7
The CRYSCOR Strategy
MOLPRO06
(Pulay) Werner, Schütz, Manby,Knowles..
http://www.molpro.net
CRYSTAL06
Dovesi, Saunders, Roetti, Orlan-do, Zicovich-Wilson, Pascale, Ci-valleri...
http://www.crystal.unito.it
CRYSCORTeam:? C. Pisani, R. Dovesi, L. Maschio, S. Casassa, Università di Torino, Torino, Italy;? C.M. Zicovich-Wilson, Universidad Autonoma, Cuernavaca, Mexico? M. Schütz and D. Usvyat, Universität Stuttgart, Stuttgart, Germany;
Functionality:
♥ MP2 single point energy♥ Fast evaluation of Integrals
→ Periodic Density Fitting→ Multipolar Expansion→ Lennard-Jones extrapolation
♥ MP2 correction to Density Matrix
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

8
The CRYSCOR Strategy
Main ideas
MOLPRO06
(Pulay) Werner, Schütz, Manby,Knowles..
http://www.molpro.net
♥ local correlation
♥ non canonical MP2 equations
♥ density fitting technique
♥ various levels of sophisticati-on: MP2, CCSD(T), ..
Background
CRYSTAL06
Dovesi, Saunders, Roetti, Or-lando, Zicovich-Wilson, Pascale,Civalleri...
http://www.crystal.unito.it
♥ geometrical and structuralanalysis of periodic systems
♥ accurate HF solution
♥ full use of symmetry
♥ local representation of occu-pied manifold (Wannier Func-tion)
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

9
The CRYSCOR Strategy
Main ideas
MOLPRO06
(Pulay) Werner, Schütz, Manby,Knowles..
http://www.molpro.net
♥ local correlation
♥ non canonical MP2 equations
♥ density fitting technique
♥ various levels of sophisticati-on: MP2, CCSD(T), ..
Background
CRYSTAL06
Dovesi, Saunders, Roetti, Or-lando, Zicovich-Wilson, Pascale,Civalleri...
http://www.crystal.unito.it
♥ geometrical and structuralanalysis of periodic systems
♥ accurate HF solution
♥ full use of symmetry
♥ local representation of occu-pied manifold (Wannier Func-tion)
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

10
Local-correlation approach for MOLECULESTheory: Meyer, Pulay, Saebø, (1976). MOLRPO: Werner, Knowles, Hetzer, Manby, Schütz...
exploitation of the short-range character of the dynamic correlation
1. BASIS SETboth occupied and virtual spaces must be described by a set of localizedfunction
→ ♠ orthonormality can be lost
2. TRUNCATION STRATEGY
• the number of excited configurations is limited by adopting a distance criterium• for each excited configuration the virtual space is reduced by adopting a
domain criterium
3. non-canonical MP2 equations:K i j
ab+∑
cd[ facTi jcdSdb+ SacT
i jcd fdc] −
∑cd[Sac
∑k( facT
k jcd + T ik
cd fk j)Scb] = 0
→ ♠ solution achieved by minimization of the Hylleraas functional
→ ♥ the problem scales with N, the basis set dimension!
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
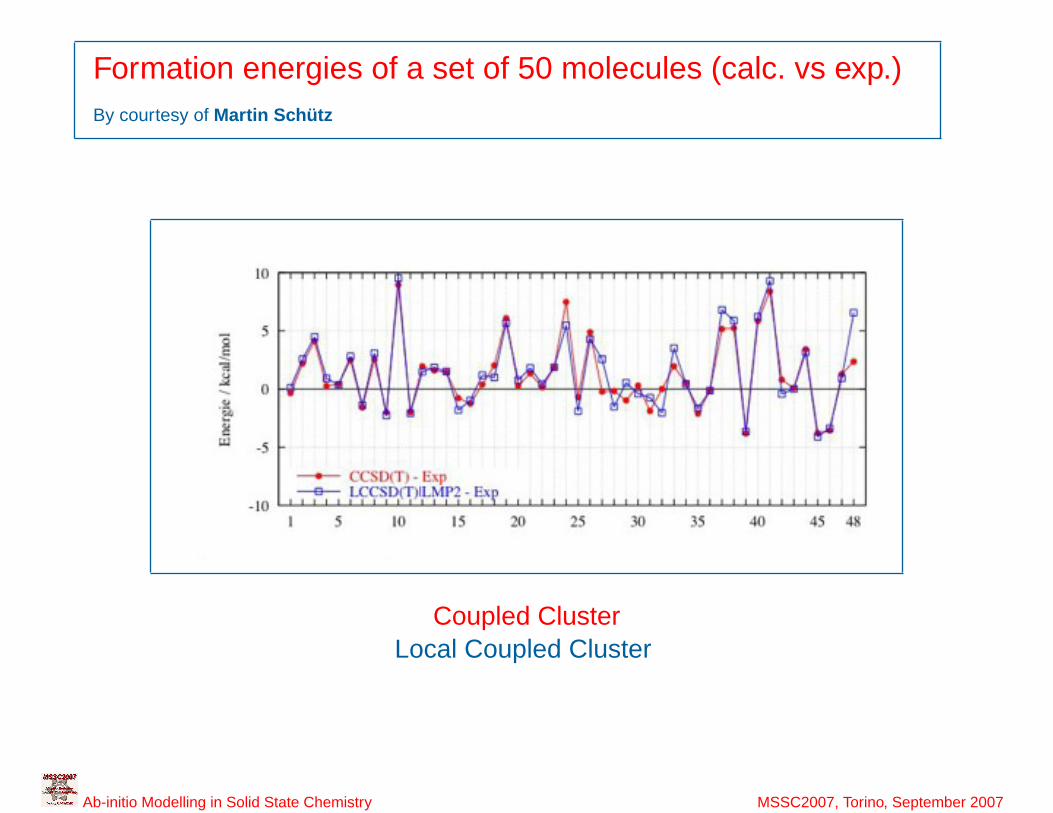
11
Formation energies of a set of 50 molecules (calc. vs exp.)By courtesy of Martin Schütz
Coupled ClusterLocal Coupled Cluster
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
12
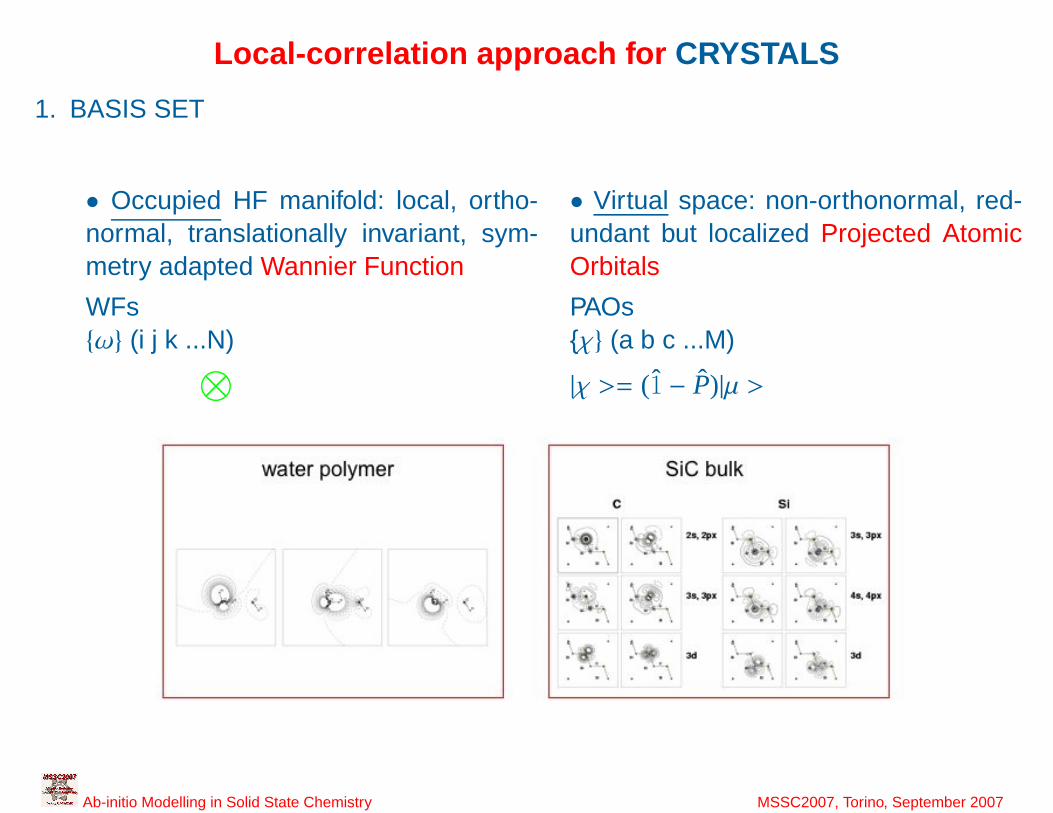
Local-correlation approach for CRYSTALS
1. BASIS SET
• Occupied HF manifold: local, ortho-normal, translationally invariant, sym-metry adapted Wannier Function
• Virtual space: non-orthonormal, red-undant but localized Projected AtomicOrbitals
WFs PAOsω (i j k ...N) χ (a b c ...M)⊗
|χ >= (1 − P)|µ >
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

13
The CRYSCOR Strategy
Main ideas
MOLPRO06
(Pulay) Werner, Schütz, Manby,Knowles..
http://www.molpro.net
♥ local correlation
♥ non canonical MP2 equations
♥ density fitting technique
♥ various levels of sophisticati-on: MP2, CCSD(T), ..
Background
CRYSTAL06
Dovesi, Saunders, Roetti, Or-lando, Zicovich-Wilson, Pascale,Civalleri...
http://www.crystal.unito.it
♥ geometrical and structuralanalysis of periodic systems
♥ accurate HF solution
♥ full use of symmetry
♥ local representation of oc-cupied manifold (WannierFunction)
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
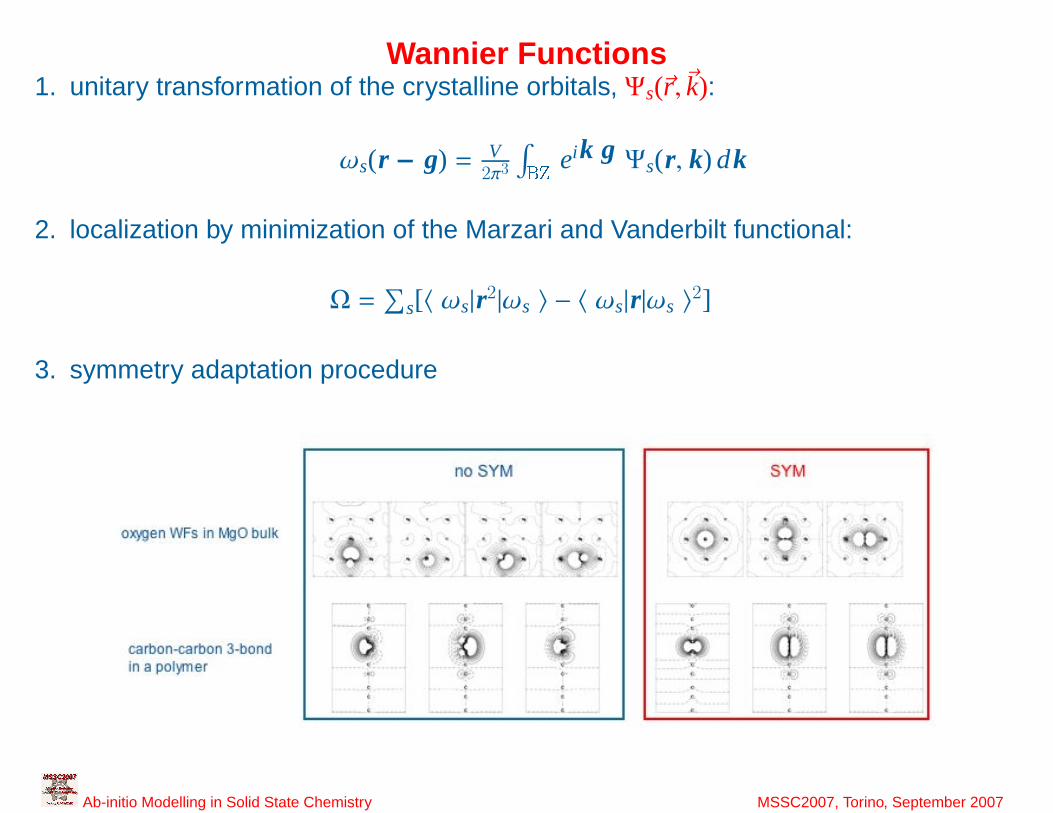
14
Wannier Functions1. unitary transformation of the crystalline orbitals, Ψs(~r ,~k):
ωs(r − g) = V2π3
∫BZ
eik g Ψs(r, k) dk
2. localization by minimization of the Marzari and Vanderbilt functional:
Ω =∑
s[〈 ωs|r2|ωs 〉 − 〈 ωs|r|ωs 〉2]
3. symmetry adaptation procedure
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
15
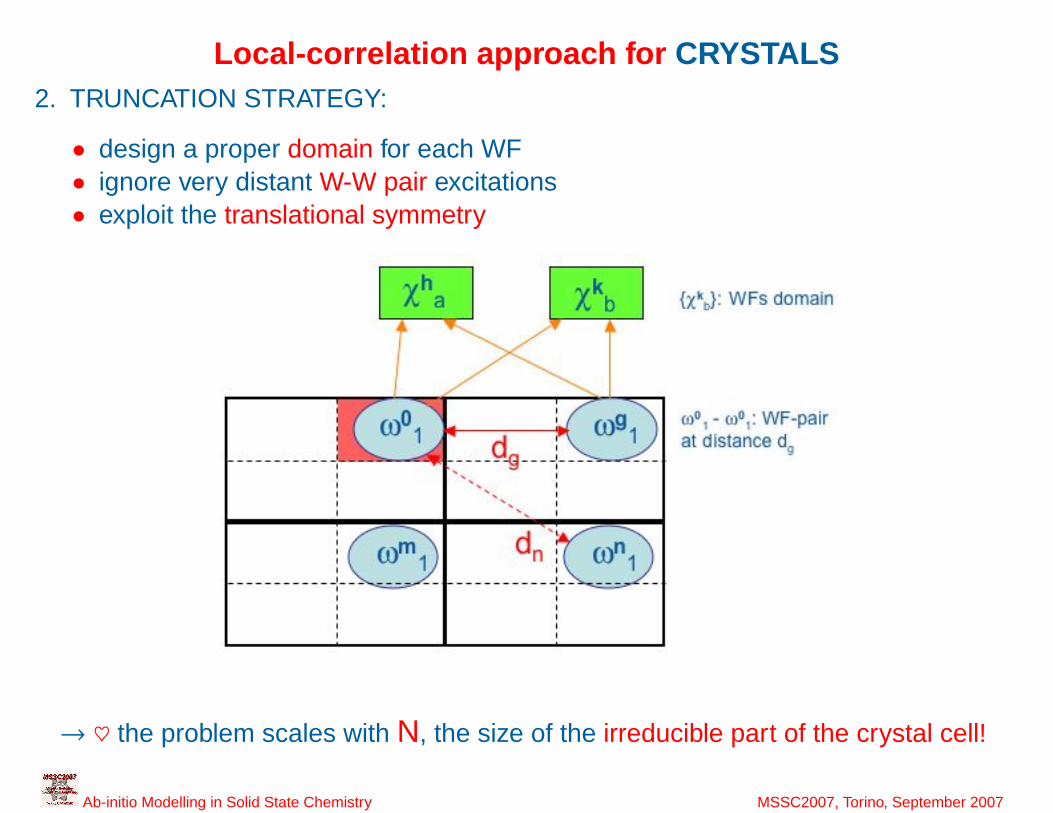
Local-correlation approach for CRYSTALS2. TRUNCATION STRATEGY:
• design a proper domain for each WF• ignore very distant W-W pair excitations• exploit the translational symmetry
→ ♥ the problem scales with N, the size of the irreducible part of the crystal cell!
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
16
The CRYSCOR Strategy
Main ideas
MOLPRO06
(Pulay) Werner, Schütz, Manby,Knowles..
http://www.molpro.net
♥ local correlation
♥ non canonical MP2 equati-ons
♥ density fitting technique
♥ various levels of sophisticati-on: MP2, CCSD(T), ..
Background
CRYSTAL06
Dovesi, Saunders, Roetti, Or-lando, Zicovich-Wilson, Pascale,Civalleri...
http://www.crystal.unito.it
♥ geometrical and structuralanalysis of periodic systems
♥ accurate HF solution
♥ full use of symmetry
♥ local representation of occu-pied manifold (Wannier Func-tion)
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

17
Local-correlation approach for CRYSTALS
3. EQUATIONS
Ri j
ab= K i j
ab+∑cd
[ facTi jcdSdb+ SacT
i jcd fdc] −
∑cd
[Sac
∑k
( facTk jcd + T ik
cd fk j)Scb] = 0
E2 =∑
i j
∑ab∈(i j )
K i j
ab(2 T i j
ab− T i jba)
K i j
ab= (i a| j b) =
∑µρνσ
cWFiµ cPAO
aρ cWFjν cPAO
bσ (µρ|νσ)
∑i j →
∑i0 jJ
∑ab→
∑aA bB
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

18
Periodic Local MP2 equations
Ri j
ab= K i j
ab+∑cd
[ facTi jcdSdb+ SacT
i jcd fdc] −
∑cd
[Sac
∑k
( fikTk jcd + T ik
cd fk j)Scb] = 0
E2 =∑
i j
∑ab∈(i j )
K i j
ab(2 T i j
ab− T i jba)
K i j
ab= (i a| j b) =
∑µρνσ
cWFiµ cPAO
aρ cWFjν cPAO
bσ (µρ|νσ)
• WFs Fock matrix
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

19
Periodic Local MP2 equations
Ri j
ab= K i j
ab+∑cd
[ facTi jcdSdb+ SacT
i jcd fdc] −
∑cd
[Sac
∑k
( fikTk jcd + T ik
cd fk j)Scb] = 0
E2 =∑
i j
∑ab∈(i j )
K i j
ab(2 T i j
ab− T i jba)
K i j
ab= (i a| j b) =
∑µρνσ
cWFiµ cPAO
aρ cWFjν cPAO
bσ (µρ|νσ)
• PAOs Overlap and Fock matrices
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

20
Periodic Local MP2 equations
Ri j
ab= K i j
ab+∑cd
[ facTi jcdSdb+ SacT
i jcd fdc] −
∑cd
[Sac
∑k
( facTk jcd + T ik
cd fk j)Scb] = 0
E2 =∑
i j
∑ab∈(i j )
K i j
ab(2 T i j
ab− T i jba)
K i j
ab= (i a| j b) =
∑µρνσ
cWFiµ cPAO
aρ cWFjν cPAO
bσ (µρ|νσ)
• Amplitudes.. which are the unknowns
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

21
Periodic Local MP2 equations
Ri j
ab= K i j
ab+∑cd
[ facTi jcdSdb+ SacT
i jcd fdc] −
∑cd
[Sac
∑k
( fikTk jcd + T ik
cd fk j)Scb] = 0
E2 =∑
i j
∑ab∈(i j )
K i j
ab(2 T i j
ab− T i jba)
K i j
ab= (i a| j b) =
∑µρνσ
cWFiµ cPAO
aρ cWFjν cPAO
bσ (µρ|νσ)
• Maximum WF-WF distance
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

22
Periodic Local MP2 equations
Ri j
ab= K i j
ab+∑cd
[ facTi jcdSdb+ SacT
i jcd fdc] −
∑cd
[Sac
∑k
( fikTk jcd + T ik
cd fk j)Scb] = 0
E2 =∑
i j
∑ab∈(i j )
K i j
ab(2 T i j
ab− T i jba)
K i j
ab= (i a| j b) =
∑µρνσ
cWFiµ cPAO
aρ cWFjν cPAO
bσ (µρ|νσ)
• Maximum WF-WF distance (range of∑
i j )
• Size of WF domains
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

23
Periodic Local MP2 equations
Ri j
ab= K i j
ab+∑cd
[ facTi jcdSdb+ SacT
i jcd fdc] −
∑cd
[Sac
∑k
( fikTk jcd + T ik
cd fk j)Scb] = 0
E2 =∑
i j
∑ab∈(i j )
K i j
ab(2 T i j
ab− T i jba)
K i j
ab= (i a| j b) =
∑µρνσ
cWFiµ cPAO
aρ cWFjν cPAO
bσ (µρ|νσ)
• Maximum WF-WF distance
• Size of WF domains
• Basis set µ (representation of WFs and PAOs in terms of AOs)
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

24
Periodic Local MP2 equations
Ri j
ab= K i j
ab+∑cd
[ facTi jcdSdb+ SacT
i jcd fdc] −
∑cd
[Sac
∑k
( fikTk jcd + T ik
cd fk j)Scb] = 0
E2 =∑
i j
∑ab∈(i j )
K i j
ab(2 T i j
ab− T i jba)
K i j
ab= (i a| j b) =
∑µρνσ
cWFiµ cPAO
aρ cWFjν cPAO
bσ (µρ|νσ)
• Maximum WF-WF distance
• Size of WF domains
• Basis set µ (representation of WFs and PAOs in terms of AOs)
• Truncation of WF and PAO tails (|cWFiµ | > tow; |cPAO
aν | > toq)
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

25
Periodic Local MP2 equations
Ri j
ab= K i j
ab+∑cd
[ facTi jcdSdb+ SacT
i jcd fdc] −
∑cd
[Sac
∑k
( fikTk jcd + T ik
cd fk j)Scb] = 0
E2 =∑
i j
∑ab∈(i j )
K i j
ab(2 T i j
ab− T i jba)
K i0 jJaA bB
= (i a| j b) =∑
µM ρR νNσScWF
i0 µM cPAOaA ρR cWF
jJ νN cPAObBσS(µMρR|νNσS)
• Maximum WF-WF distance
• Size of WF domains
• Basis set µ (representation of WFs and PAOs in terms of AOs)
• Truncation of WF and PAO tails (|cWFiµ | > tow; |cPAO
aν | > toq)
• Exact, density-fitting and multipolar treatment of K integrals andpre-screening of exact K integrals
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
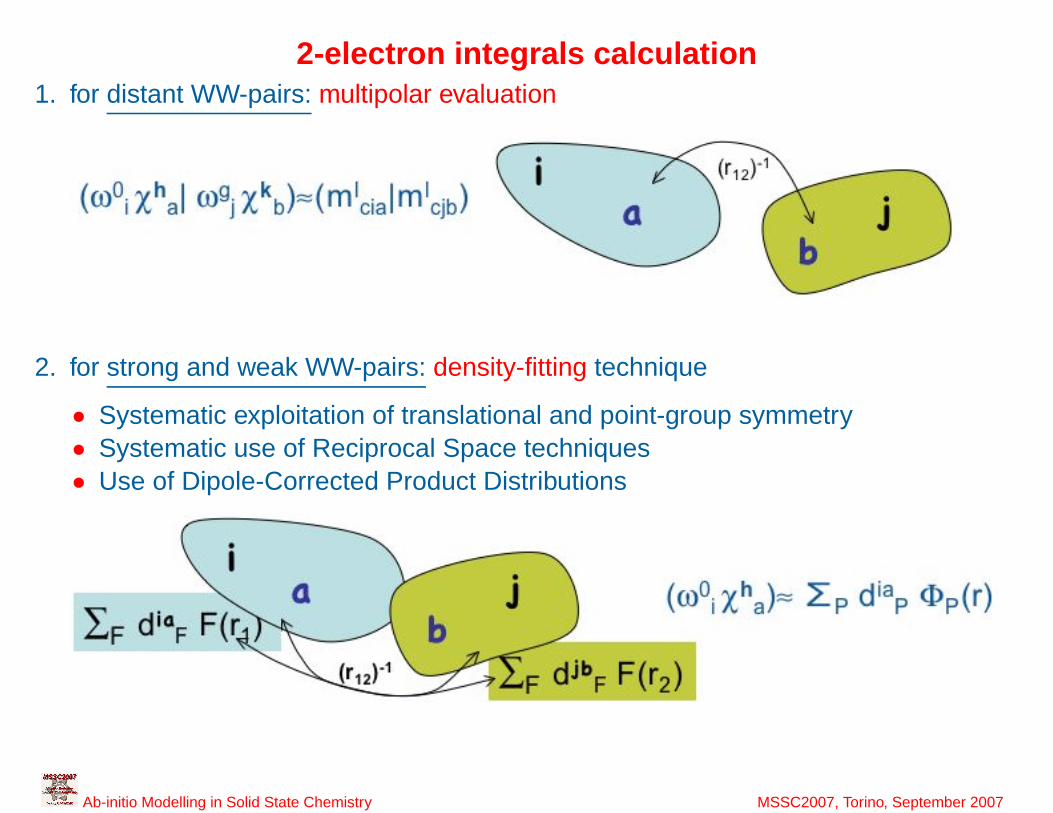
26
2-electron integrals calculation1. for distant WW-pairs: multipolar evaluation
2. for strong and weak WW-pairs: density-fitting technique
• Systematic exploitation of translational and point-group symmetry• Systematic use of Reciprocal Space techniques• Use of Dipole-Corrected Product Distributions
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
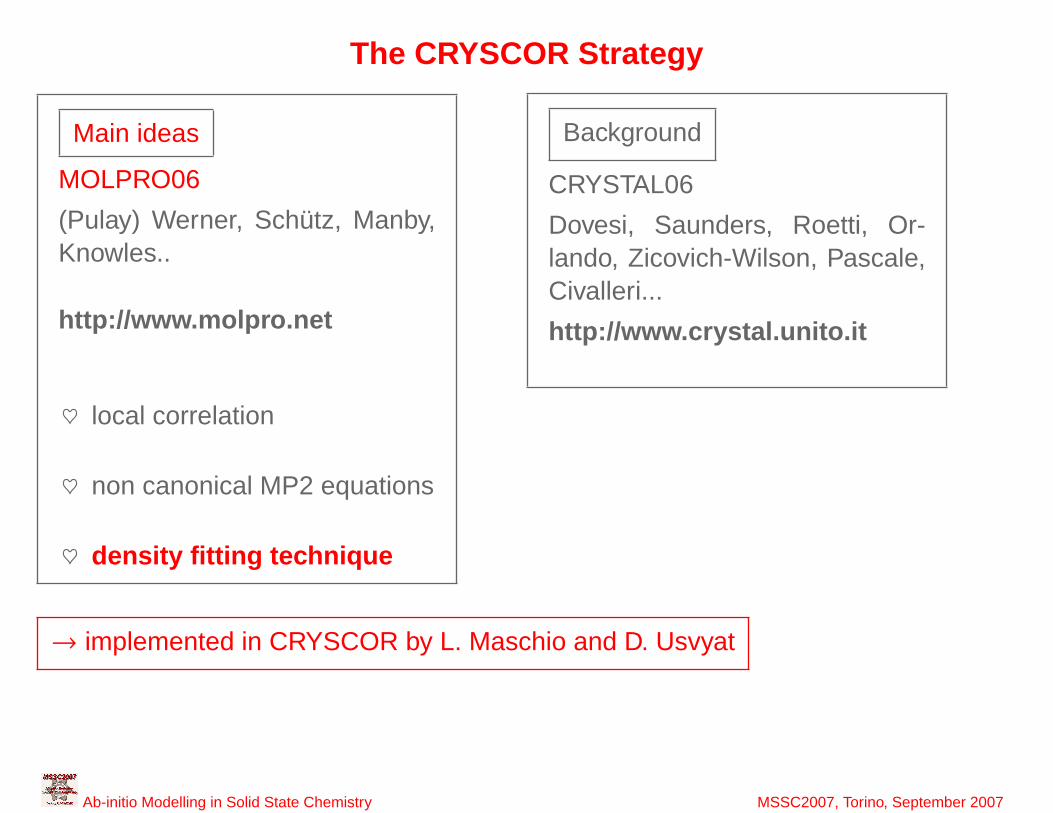
27
The CRYSCOR Strategy
Main ideas
MOLPRO06
(Pulay) Werner, Schütz, Manby,Knowles..
http://www.molpro.net
♥ local correlation
♥ non canonical MP2 equations
♥ density fitting technique
Background
CRYSTAL06
Dovesi, Saunders, Roetti, Or-lando, Zicovich-Wilson, Pascale,Civalleri...
http://www.crystal.unito.it
→ implemented in CRYSCOR by L. Maschio and D. Usvyat
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

28
Df-technique (1)
K i j
ab= (ωi χa|ω j χb)
ρia = (ωiχa)
ΦP(r) = fitting function
MOLECULAR CASE
ρia ≈ ρia =∑P dia
PΦP(r)
∆w = (ρia − ρia|w12|ρia − ρia)
w12 =1
r12
JPQ =∫
dr1∫ΦP(r1) 1
r12ΦQ(r2) dr2
JiaP=∫
dr1∫ΦP(r1) 1
r12ρia(r2) dr2
diaP=∑Q Jia
Q[J−1]QP
K i jab = (ρia|ρ jb) + (ρia|ρ jb) − (ρia|ρ jb)
K i jab =∑P dia
PJ jbP
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

29
Df-technique (1)
K i j
ab= (ωi χa|ω j χb)
ρia = (ωiχa)
ΦP(r) = fitting function
MOLECULAR CASE
ρia ≈ ρia =∑P dia
PΦP(r)
∆w = (ρia − ρia|w12|ρia − ρia)
w12 =1
r12
JPQ =∫
dr1∫ΦP(r1) 1
r12ΦQ(r2) dr2
JiaP=∫
dr1∫ΦP(r1) 1
r12ρia(r2) dr2
diaP=∑Q Jia
Q[J−1]QP
K i jab = (ρia|ρ jb) + (ρia|ρ jb) − (ρia|ρ jb)
K i jab =∑P dia
PJ jbP
FITTING DOMAIN for each different product distribution
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

30
Df-technique (2)
MOLECULAR CASE PERIODIC CASE
indices: i, j, a, b, P ... indices: i I, j J, a A, b B, P P ...
ρia ≈ ρia =∑P dia
PΦP(r) ρia A =
∑PP dia A
PP ΦPP(r)
diaP=∑Q Jia
Q[J−1]QP di aA
PP =∑QQ Ji aA
QQ [J−1]QQPP
K i jab =∑P dia
PJ jbP
˜K i0 jJ
aA bB =∑PP di aA
PP J jJ bBPP
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

31
Df-technique (2)
MOLECULAR CASE PERIODIC CASE
indices: i, j, a, b, P ... indices: i I, j J, a A, b B, P P ...
ρia ≈ ρia =∑P dia
PΦP(r) ρia A =
∑PP di aA
PP ΦPP(r)
diaP=∑Q Jia
Q[J−1]QP di aA
PP =∑QQ Ji aA
QQ [J−1]QQPP
infinite matrix ....?
K i jab =∑P dia
PJ jbP
˜K i0 jJ
aA bB =∑PP di aA
PP J jJ bBPP
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

32
Df-technique (3): how to overcome the ”infinite” problem
1. calculation of the JiaAPP and J
P0QQ integrals
2. skip to reciprocal space
→ for each k point of the Monkhorst set:
• Fourier Transform: JPQ(k) =∑
Q JP0QQ exp[ikRQ]
JiaAP
(k) =∑
P JiaAPP exp[ikRP]
• matrix inversion: [J−1]QP(k)• coefficients calculation: diaA
P(k) =
∑Q Ji aA
Q(k) [J−1]QP(k)
• integrals evaluation:˜
K i jJaA bB(k) =
∑P diaA
P(k) J jJ bB
P(k)
3. ANTI FOURIER TRANSFORM:˜
K i jJaA bB =
1Nk
∑k˜
K i jJaA bB(k) exp[−ikRJ]
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

33
Df-technique (3): how to overcome the ”infinite” problem
1. calculation of the JiaAPP and J
P0QQ integrals
2. skip to reciprocal space
→ for each k point of the Monkhorst set :
• Fourier Transform: JPQ(k) =∑
Q JP0QQ exp[ikRQ]
JiaAP
(k) =∑
P JiaAPP exp[ikRP]
• matrix inversion: [J−1]QP(k)• coefficients calculation: diaA
P(k) =
∑Q Ji aA
Q(k) [J−1]QP(k)
• integrals evaluation:˜
K i jJaA bB(k) =
∑P diaA
P(k) J jJ bB
P(k)
3. ANTI FOURIER TRANSFORM:˜
K i jJaA bB =
1Nk
∑k˜
K i jJaA bB(k) exp[−ikRJ]
good calibration of the Monkhorst net is needed!
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

34
Df-technique (3): how to overcome the ”infinite” problem
1. calculation of the JiaAPP and J
P0QQ integrals
2. skip to reciprocal space
→ for each k point of the Monkhorst set:
• Fourier Transform: JPQ(k) =∑
Q JP0QQ exp[ikRQ]
JiaAP
(k) =∑
P JiaAPP exp[ikRP]
• matrix inversion: [J−1]QP(k)• coefficients calculation: diaA
P(k) =
∑Q Ji aA
Q(k) [J−1]QP(k)
• integrals evaluation:˜
K i jJaA bB(k) =
∑P diaA
P(k) J j bL
P(k)
3. ANTI FOURIER TRANSFORM:˜
K i jJaA bB =
1Nk
∑k˜
K i jJaA bB(k) exp[−ikRJ]
L = B - J
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

35
Df-technique (3): how to overcome the ”infinite” problem
1. calculation of the JiaAPP and J
P0QQ integrals
2. skip to reciprocal space
→ for each k point of the Monkhorst set:
• Fourier Transform: JPQ(k) =∑
Q JP0QQ exp[ikRQ]
JiaAP
(k) =∑
P JiaAPP exp[ikRP]
• matrix inversion: [J−1]QP(k)• coefficients calculation: di aA
P(k) =
∑Q Ji aA
Q(k) [J−1]QP(k)
• integrals evaluation:˜
K i jJaA bB(k) =
∑P diaA
P(k) J j bL
P(k)
3. ANTI FOURIER TRANSFORM:˜
K i jJaA bB =
1Nk
∑k˜
K i jJaA bB(k) exp[−ikRJ]
convergence PROBLEMS in the summation over FF integrals!
→ use of the original dipole-corrected method→ fitting functions: Poisson (and few Gaussian)
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
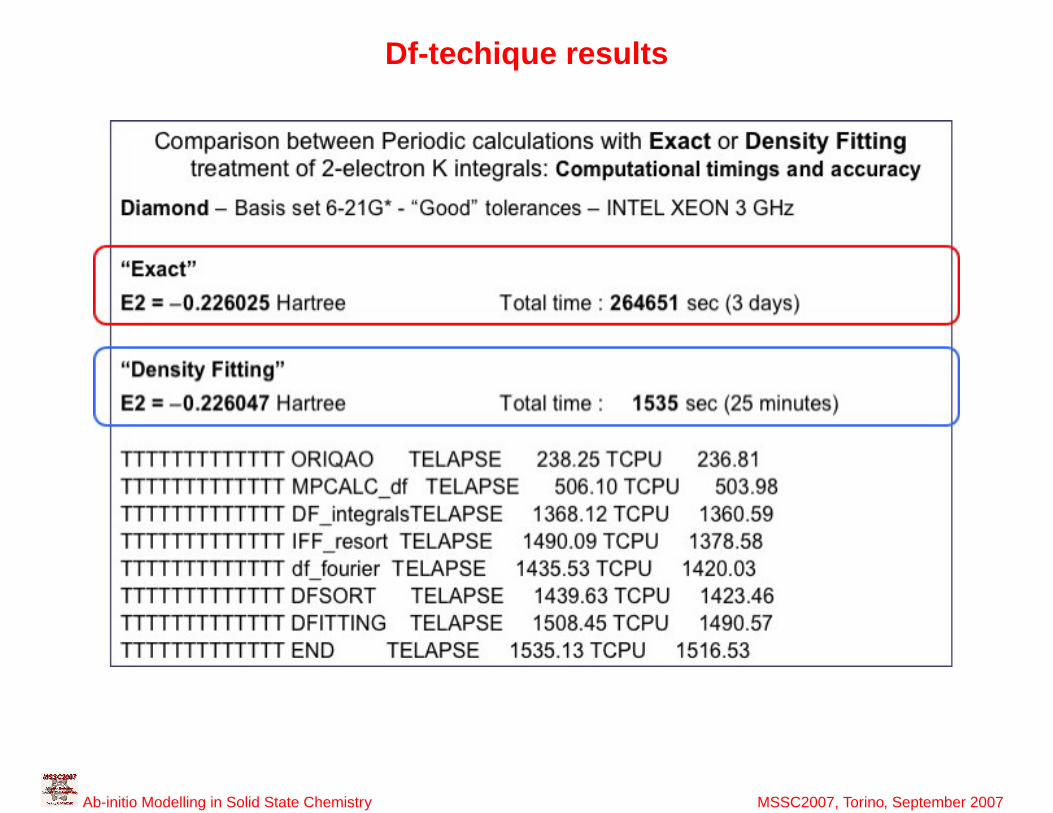
36
Df-techique results
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
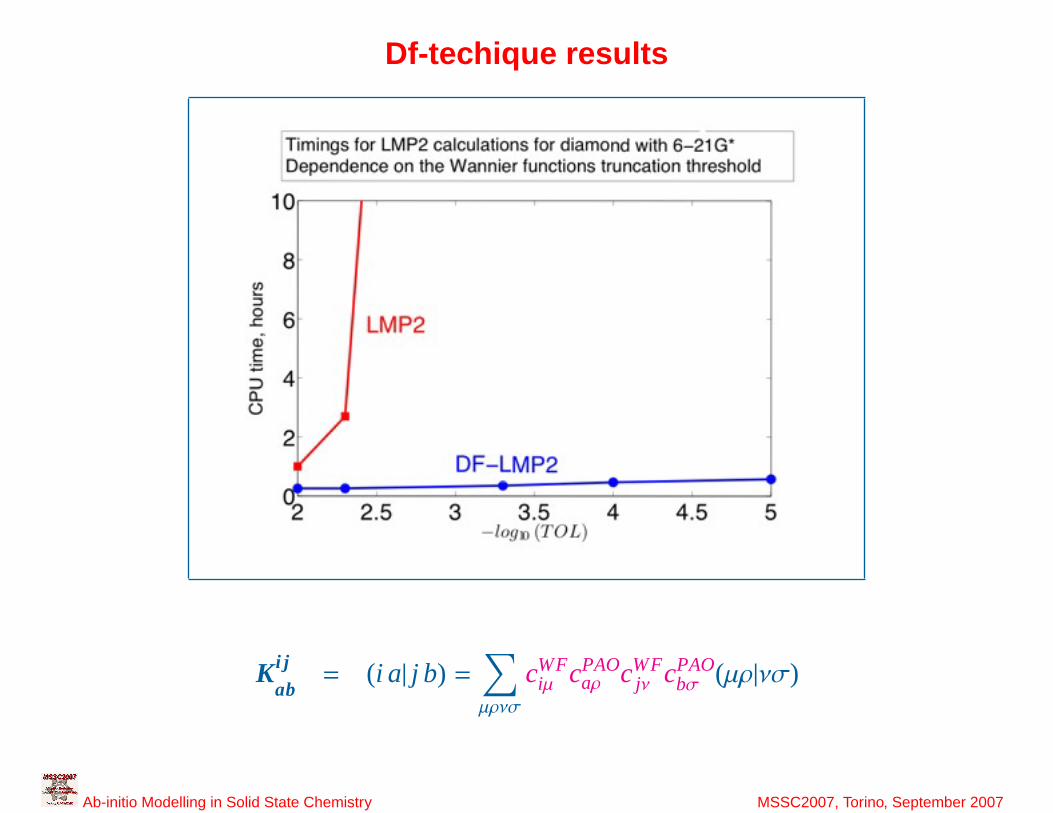
37
Df-techique results
K i j
ab= (i a| j b) =
∑µρνσ
cWFiµ cPAO
aρ cWFjν cPAO
bσ (µρ|νσ)
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
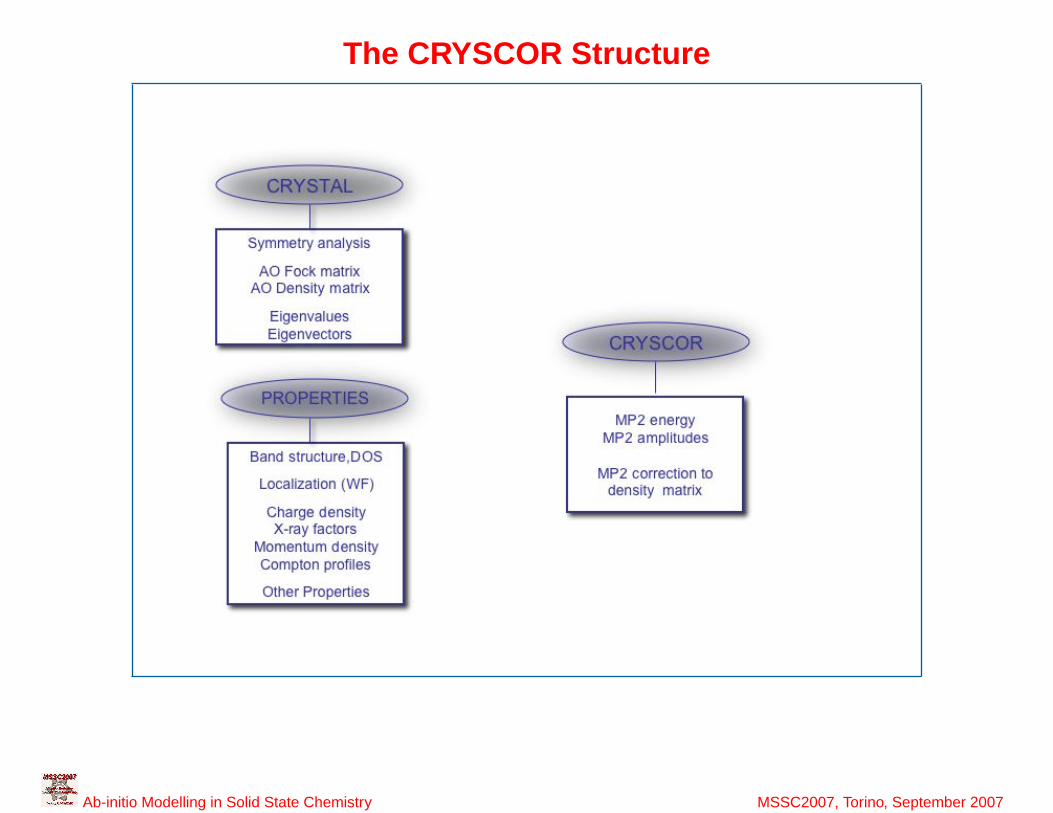
38
The CRYSCOR Structure
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
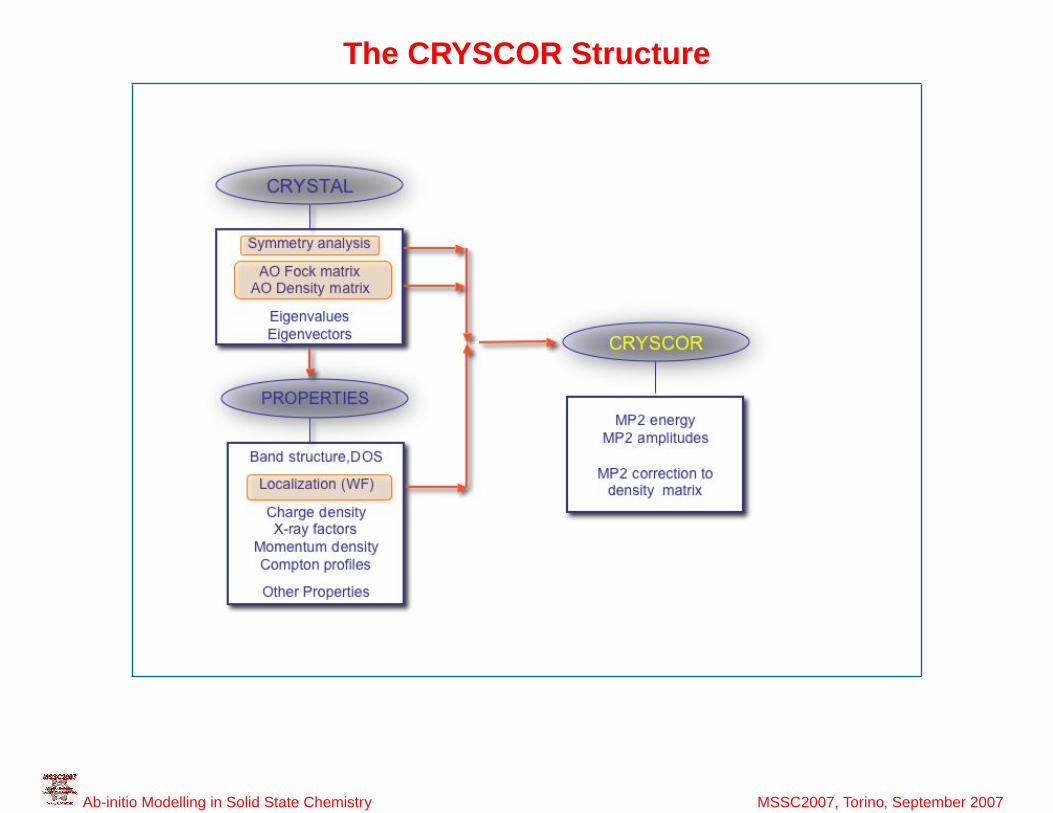
39
The CRYSCOR Structure
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
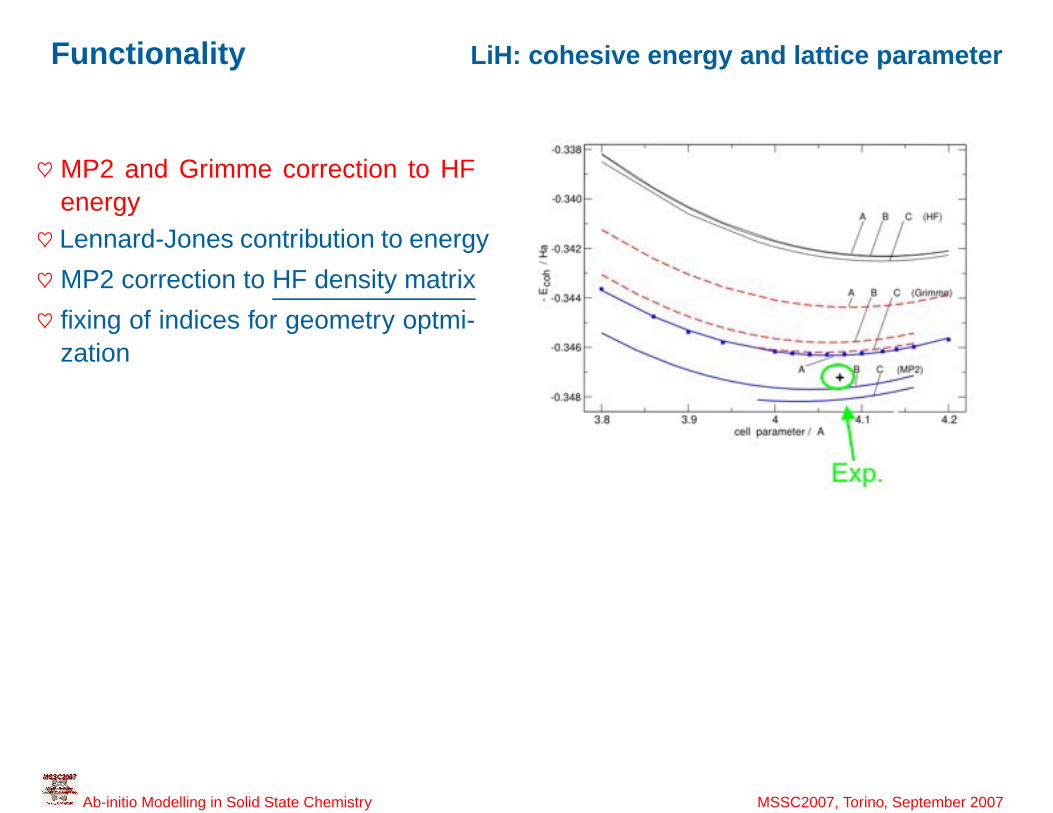
40
Functionality LiH: cohesive energy and lattice parameter
♥ MP2 and Grimme correction to HFenergy♥ Lennard-Jones contribution to energy
♥ MP2 correction to HF density matrix
♥ fixing of indices for geometry optmi-zation
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
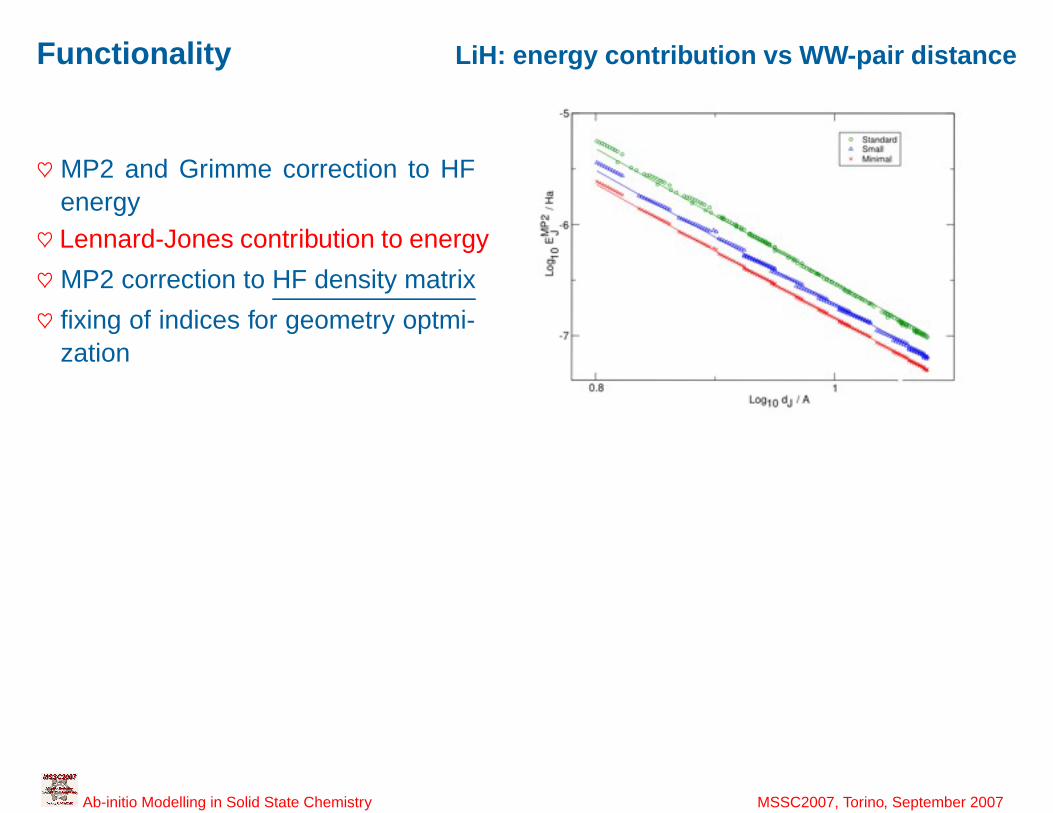
41
Functionality LiH: energy contribution vs WW-pair distance
♥ MP2 and Grimme correction to HFenergy♥ Lennard-Jones contribution to energy
♥ MP2 correction to HF density matrix
♥ fixing of indices for geometry optmi-zation
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007
42
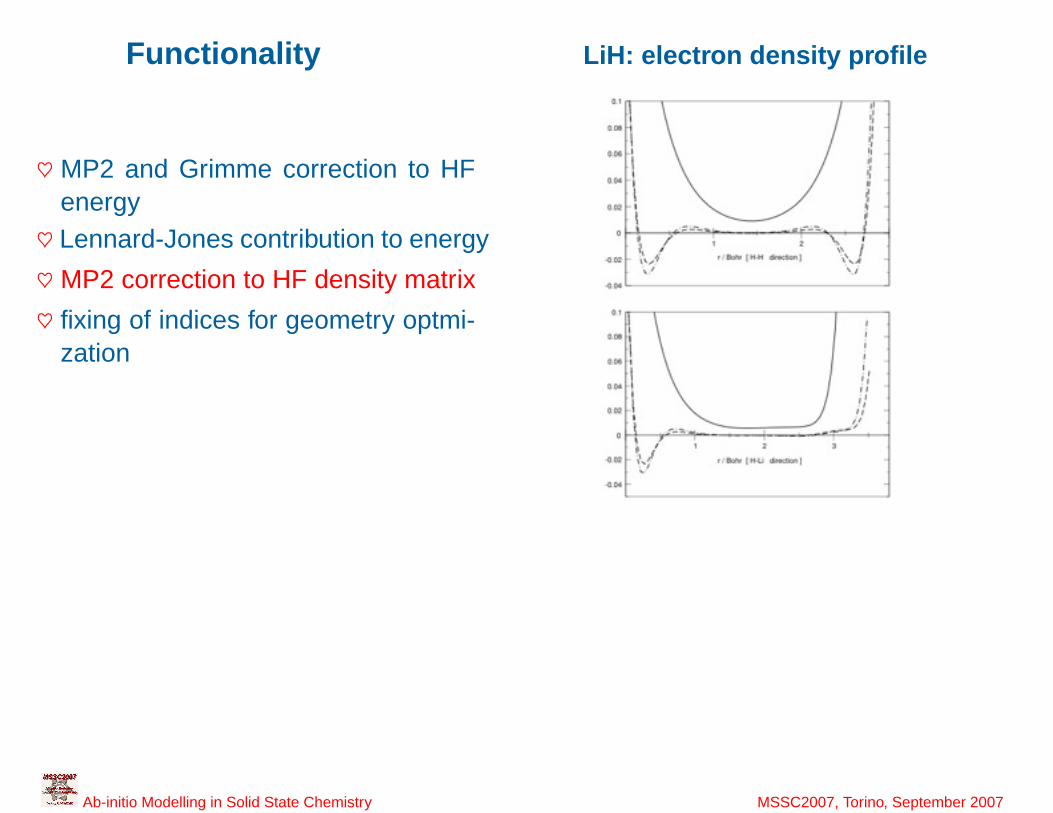
Functionality LiH: electron density profile
♥ MP2 and Grimme correction to HFenergy♥ Lennard-Jones contribution to energy
♥ MP2 correction to HF density matrix
♥ fixing of indices for geometry optmi-zation
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

43
Functionality and successfully tested systems
♥ MP2 and Grimme correction to HFenergy♥ Lennard-Jones contribution to energy
♥ MP2 correction to HF density matrix
♥ fixing of indices for geometry optmi-zation
• Some study cases: LiH [S. Casassa, M. Halo, L. Maschio, C. Roetti and C. Pisani, Theor.Chem. Acc., (2006)] and MgO bulk [poster: M. Halo et al.a]
• CRYSCOR TUTORIAL: LiH, H2O polymer, Argon adsorbed on MgO
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

44
The perfectTest
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007

45
Conclusion and Perspectives
♥ Refinement and standardization of the basic program
? Beta-version of the code in few months
♥ Tests
? different systems? assessing basis set quality
♥ Extension to other local correlation schemes (CCSD, MP4, ..)
♣ Acknowledgments
? all of you for your kind attention
?
Ab-initio Modelling in Solid State Chemistry MSSC2007, Torino, September 2007





























