Embed Size (px)
Citation preview

American Journal of Medical Genetics 33:447-449 (1989)
Brief Clinical Report Postaxial Acrofacial Dysostosis: Report of a Brazilian Patient A. Richieri-Costa and M.L. Guion-Almeida Laboratorio de Genetica Humana, Hospital de Pesquisa e ReabilitaGao de Lesdes Labio-Palatais, USP, Bauru, Brazil
We report on a Brazilian child with postaxial acrofacial dysostosis (AFD)-type Genee-Wie- demann. Clinical and genetic aspects of the postaxial acrofacial dysostoses are discussed.
KEY WORDS: Genee-Wiedemann syndrome, Robin sequence, oligodactyly, cleft palate, mandibulofacial dysostosis, autosomal reces- sive inheritance
INTRODUCTION The postaxial acrofacial dysostoses are a rare type of
mandibulofacial dysostosis associated with ulnar and fib- ular defects. Most reported cases have been sporadic [Birch-Jensen, 1949; Genee, 1969; Wiedemann, 1973; Brunoni et al., 1987; Meinecke and Wiedemann, 19871, but autosomal recessive [Fineman, 1981; Opitz and Stick- ler, 19871 and autosomal dominant inheritance [Robinow et al., 19861 have been reported. Clinical manifestations include Robin sequence, oligodactyly, and multiple other anomalies such as vertebral and rib defects, congenital heart defect, extra nipples, and single umbilical artery [Opitz and Stickler, 19871. Here we report on a sporadic case of postaxial acrofacial dysostosis AFD-type Genke- Wiedemann.
CLINICAL REPORT Patient J.R.S. (Figs. 1, 2), a boy, was born in 1982 to
a G5P3 healthy 23 year-old mother and her unrelated 26 year-old husband. There were no similarly affected rela- tives. Pregnancy was normal with absence of exposure to toxins, infections, traumatic incidents, or radiation. De- livery was normal and at term. Birth weight, height, and OFC were not recorded. Cleft palate and limb anomalies
Received for publication August 23, 1988; revision received Feb- ruary 24, 1989.
Address reprints request to Dr. A. Richieri-Costa, Laboratorio de Genetica Humana, Hospital de Pesquisa e Reabilitacio de Lesaes Labio-Palatais, USP, CEP 17043, Bauru, SP, Brazil.
0 1989 Alan R. Liss, Inc.
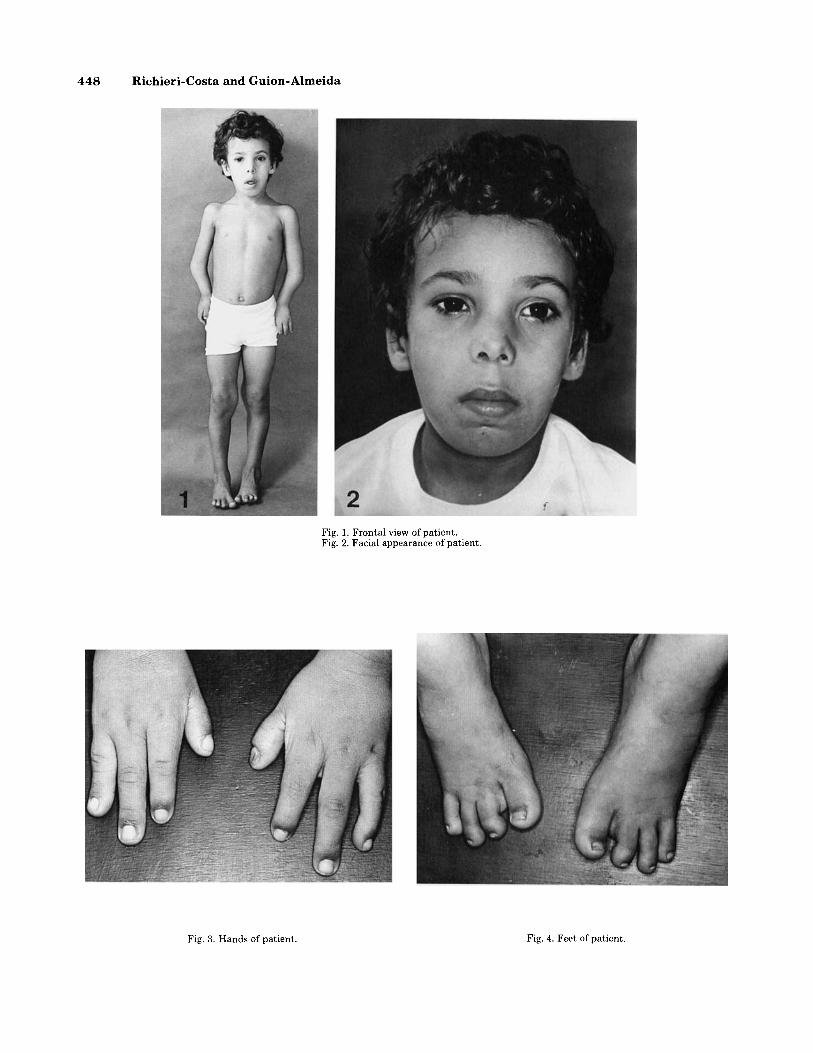
were noticed at birth. Neuropsychological development was normal. Shortly after birth, he was diagnosed as having the Pierre Robin “syndrome”; glossoptosis re- quired almost continuous medical care in the first few months of life. Clinical examination at age 5 years showed height: 105 cm; weight: 14,500 g; OFC: 50 cm; inner canthal distance: 2.8 cm; outer canthal distance: 7.9 cm (all normal measurements); median cleft of the soft pal- ate, micrognathia, mild malar hypoplasia, ulnar deviation of hands at wrists, and absence of the 5th rays; and hypoplastic and anteriorly placed thumbs (Fig. 3), genua valga and absence of the 5th toes (Fig. 4). Roentgeno- grams showed bilaterally bowed, hypoplastic, and abnor- mally modeled radii and ulnae, single carpal bone, ab- sence of the 5th ray, and marked hypoplasia of ray 1 (Fig. 5); and bilateral afhypoplastic tarsal bones, absence of the 5th ray (Fig. 6).
DISCUSSION
The published reports on Genee-Wiedemann syn- drome include 20 affected patients [Birch-Jensen, 1949; Genee, 1969; Wiedemann, 1973; Smith and Jones, 1975; Wildervanck, 1975; Miller et al., 1979; Fineman, 1981; Robinow et al., 1986; Brunoni et al., 1987; Meinecke and Wiedemann, 1987; Donnai e t al., 1987; Opitz and Stick- ler, 1987; Fryns and Van den Berghe, 1988; present case].
Clinical heterogeneity seems to occur in this type of AFD, since in the reported cases, the manifestations ranged from Robin sequence-oligodactyly [Robinow et al., 19861 to a typical multiple congenital anomalies syn- drome with additional involvement of the spine, heart, ribs with extra nipples, and single umbilical artery among other less common defects [Brunoni et al., 1987; Opitz and Stickler, 19871. Most reported patients have been isolated cases [Birch-Jensen, 1949; Genee, 1969; Wiede- mann, 1973; Brunoni et al., 1987; Donnai e t al., 1987; Meinecke and Wiedemann, 19871; however, recurrence in sibs [Fineman, 19811 and parental consanguinity [Wildervanck, 19751 have been reported. Robinow et al., [1986] reported on a woman and her equally affected son with the Robin sequence and postaxial oligodactyly very similar to the Genbe-Wiedemann syndrome, but they suggested that their observation concerned a new auto- soma1 dominant condition.
448 Richieri-Costa and Guion-Almeida
Fig. 1. Frontal view of patient. Fig. 2. Facial appearance of patient.
Fig. 3. Hands of patient. Fig. 4. Feet of patient.

Postaxial Acrofacial Dysostosis 449
Fig. 5 . Radiological aspects of patient’s upper limbs.
The pattern of anomalies presented by our patient characterizes the nosologic group of AFD, and the pos- taxial involvement observed strongly suggests the diag- nosis of the Genbe-Wiedemann syndrome in this patient.
REFERENCES Birch-Jensen A (1949): Congenital deformities of the upper extremities.
Domus Biologiae Humanae, Universitatis Hafniensis. Andelsbogi- rykkeriet i Odense and Det danske Forlag, 285 pp.
Brunoni D, Guidugli-Net0 J, Chedick ES, Borovik CL (1987): Acrofacial dysostosis: A new type? Rev Bras Genet 10:353-360.
Donnai D, Hughes HE, Winter RM (1987): Postaxial acrofacial dysos- tosis (Miller) syndrome. J Med Genet 24:422-425.
Fineman RM (1981): Recurrence of the postaxial acrofacial dysostosis- syndrome in a sibship: Implications for genetic counseling. J Pediatr 9887-88.
Fryns JP, Van den Berghe H (1988): Acrofacial dysostosis with postaxial limb deficiency. Am J Med Genet 29:205-208.
Genee E (1969): Une forme extensive de dysostose mandibulo-faciale. J Genet Hum 17:45-52.
Meinecke P, Wiedemann HR (1987): Robin sequence and oligodactyly in mother and son: Probably a further example of the postaxial acrofacial dysostosis syndrome. Am J Med Genet 27:953-956.
Miller M, Fineman R, Smith DW (1979): Postaxial acrofacial dysostosis syndrome. J Pediatr 95:970-975.
Opitz JM, Stickler GM (1987): The Genbe-Wiedemann syndrome, an acrofacial dysostosis: Further observation, Am J Med Genet 27971- 975.
Robinow M, Johnson GF, Apesos J (1986): Robin sequence and oligo- dactyly in mother and son. Am J Med Genet 25:293-297.
Smith DW, Jones KL (1975): Case report 28, patient 1. Synd Ident 3:7. Wiedemann HR (1973): Missbildungs-Retardierungs-Syndrom mit Feh-
len des 5 Strahls an Handen and Fiissen, Gaumenspalte, dysplas- tischen Ohren und Augenlidern und radioulnarer Synostose. Klin Padiatr 185:181-186.
Wildervanck LS (1975): Case report 28, patient 3. Synd Ident 3:8.
Fig. 6. Radiological aspects of patient’s feet.