Embed Size (px)
Citation preview

American Journal of Medical Genetics 49:240-243 (1994)
Prenatal Diagnosis of Smith-Lemli-Opitz Syndrome, Type 11 Judith A. Johnson, David J. Aughton, Christine H. Comstock, Paul T. von Oeyen, James V. Higgins, and Rex Schulz Reproductive Genetics Program (J.A.J., D.J.A., P.T.u.O., C.H.C.), Departments of Pediatrics (D.J.A.) and Anatomic Pathology (R.S.), and Division of Fetal Imaging (C.H.C.), William Beaumont Hospital, Royal Oak, and Department of Pediatrics and Human Development (J.V.H.), Michigan State University, East Lansing, Michigan
Smith-Lemli-Opitz syndrome, type I1 (SLOS- 11) is a severe autosomal recessive disorder characterized by a distinctive face, unusual cleft palate, postaxial polydactyly, congenital heart defects, renal anomalies, and male pseudohermaphroditism. We present the first report of prenatal diagnosis of SLOS-11, as well as an additional report of prenatal detec- tion of multiple anomalies, in which a positive diagnosis of SLOS I1 was made postnatally. In neither case was the pregnancy known pro- spectively to be at risk for SLOS-11. In the former case, targeted sonographic examina- tion at 31 weeks of gestation showed intra- uterine growth retardation, atrioventricular septal defect, mesomelic shortening of the arms, small kidneys, overlapping fingers, and female external genitalia; a 46,XY chromo- some constitution had been ascertained pre- viously. A provisional diagnosis of SLOS-I1 was made prenatally. In the latter case, tar- geted sonographic examination at 18 weeks of gestation showed severe oligohydramnios, atrioventricular septal defect, and Dandy- Walker malformation. The kidneys and blad- der were not visualized. The chromosome constitution was 46,XX. The diagnosis of SLOS-I1 was made postnatally. In both cases, additional findings compatible with SLOS-I1 were noted postnatally. Prenatal detection of congenital heart defects and renal abnormal- ities, in combination with certain additional findings (most notably, female external geni- talia in the presence of a 46,XY karyotype, polydactyly, disproportionately short limbs, or intrauterine growth retardation) and a nor- mal karyotype, suggests the diagnosis of SLOS-11, and warrants further investigation. 0 1994 Wdey-Us$, Inc.
Received for publication June 1, 1993; revision received August 23, 1993.
Address reprint requests to Judith A. Johnson, M.S., Fkproduc- tive Genetics Program, William Beaumont Hospital, 3535 West Thirteen Mile Road, Suite 329, Royal Oak, MI 48073.
0 1994 Wiley-Liss, Inc.
KEY WORDS: Smith-Lemli-Opitz syndrome (Type 11), multiple abnormal- ities, prenatal diagnosis, ultra- sonography
INTRODUCTION Smith-Lemli-Opitz syndrome, type I1 (SLOS-11) is a
severe autosomal recessive disorder that is distin- guished from Smith-Lemli-Opitz syndrome, as origi- nally described [Smith et al., 19641, by the presence of major malformations, male pseudohermaphroditism, and early death [Curry et al., 19871. To date, at least 50 cases of SLOS-I1 have been reported. However, to our knowledge, prenatal diagnosis of the condition has not been reported.
We present 2 cases of SLOS-I1 that were ascertained prenatally through sonographic identification of multi- ple fetal abnormalities. Neither pregnancy was known prospectively to be at risk for the disorder. In one case, prenatal diagnosis of SLOS-I1 was possible through rec- ognition of malformations consistent with this diagnosis and of discordance between phenotypic and karyotypic sex. In the other case, the diagnosis of SLOS-I1 was made postnatally.
CLINICAL REPORTS Case 1
At 28 weeks of gestation, the consultand, a 25-year-old primigravida woman, underwent fetal sonography at another institution for postcoital spotting; this showed intrauterine growth retardation and an unspecified con- genital heart defect. Percutaneous umbilical blood sam- pling showed a 46,XY chromosome constitution. The consultand was referred to our institution where tar- geted fetal sonography confirmed severe intrauterine growth retardation, atrioventricular septal defect, small kidneys, mild mesomelic shortening of the upper limbs, overlapping fingers, and female external genitalia. A provisional diagnosis of SLOS-I1 was made.
The prenatal history was remarkable for occasional alcohol use in the first trimester and for a urinary tract infection which was treated with Macrodantina. The consultand's mother and maternal grandmother each
Smith-Lemli-Opitz Syndrome, Type I1 241
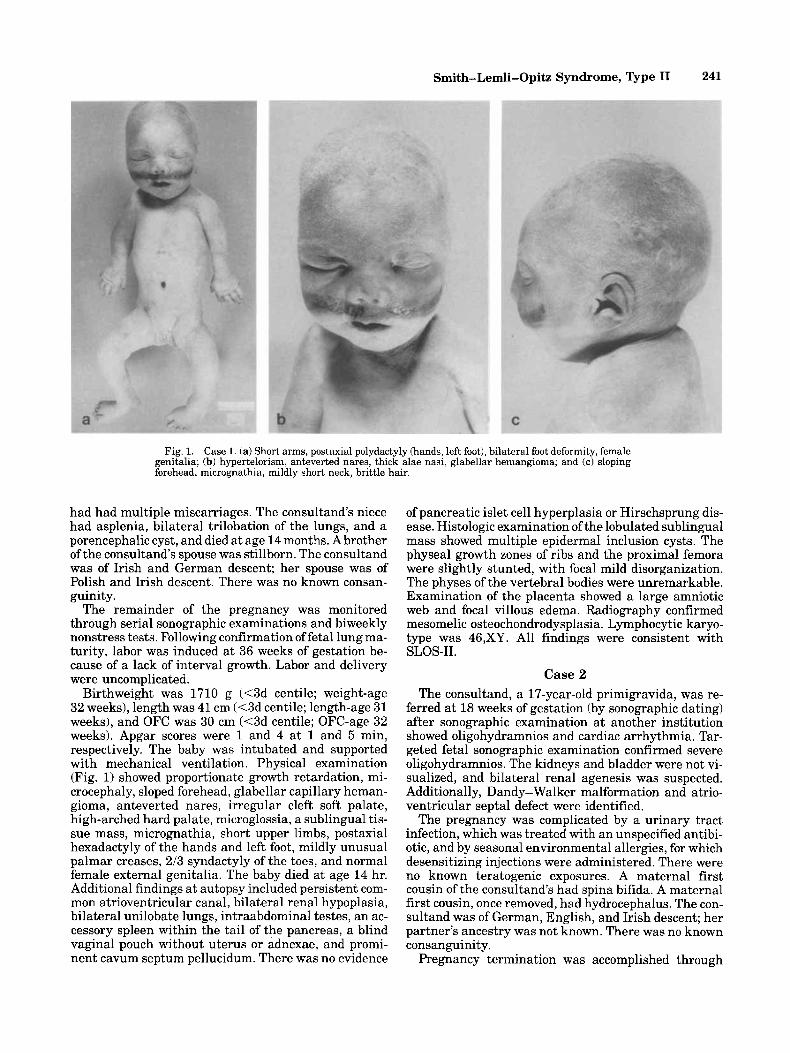
Fig. 1. Case 1. (a) Short arms, postaxial polydactyly (hands, left foot), bilateral foot deformity, female genitalia; (b) hypertelorism, anteverted nares, thick alae nasi, glabellar hemangioma; and (c) sloping forehead, micrognathia, mildly short neck, brittle hair.
had had multiple miscarriages. The consultand's niece had asplenia, bilateral trilobation of the lungs, and a porencephalic cyst, and died a t age 14 months. A brother of the consultand's spouse was stillborn. The consultand was of Irish and German descent; her spouse was of Polish and Irish descent. There was no known consan- guinity.
The remainder of the pregnancy was monitored through serial sonographic examinations and biweekly nonstress tests. Following confirmation of fetal lung ma- turity, labor was induced a t 36 weeks of gestation be- cause of a lack of interval growth. Labor and delivery were uncomplicated.
Birthweight was 1710 g (<3d centile; weight-age 32 weeks), length was 41 cm (<3d centile; length-age 31 weeks), and OFC was 30 cm (<3d centile; OFC-age 32 weeks). Apgar scores were 1 and 4 at 1 and 5 min, respectively. The baby was intubated and supported with mechanical ventilation. Physical examination (Fig. 1) showed proportionate growth retardation, mi- crocephaly, sloped forehead, glabellar capillary heman- gioma, anteverted nares, irregular cleft soft palate, high-arched hard palate, microglossia, a sublingual tis- sue mass, micrognathia, short upper limbs, postaxial hexadactyly of the hands and left foot, mildly unusual palmar creases, 213 syndactyly of the toes, and normal female external genitalia. The baby died at age 14 hr. Additional findings at autopsy included persistent com- mon atrioventricular canal, bilateral renal hypoplasia, bilateral unilobate lungs, intraabdominal testes, an ac- cessory spleen within the tail of the pancreas, a blind vaginal pouch without uterus or adnexae, and promi- nent cavum septum pellucidum. There was no evidence
of pancreatic islet cell hyperplasia or Hirschsprung dis- ease. Histologic examination of the lobulated sublingual mass showed multiple epidermal inclusion cysts. The physeal growth zones of ribs and the proximal femora were slightly stunted, with focal mild disorganization. The physes of the vertebral bodies were unremarkable. Examination of the placenta showed a large amniotic web and focal villous edema. Radiography confirmed mesomelic osteochondrodysplasia. Lymphocytic karyo- type was 46,XY. All findings were consistent with SLOS-11.
Case 2 The consultand, a 17-year-old primigravida, was re-
ferred a t 18 weeks of gestation (by sonographic dating) after sonographic examination at another institution showed oligohydramnios and cardiac arrhythmia. Tar- geted fetal sonographic examination confirmed severe oligohydramnios. The kidneys and bladder were not vi- sualized, and bilateral renal agenesis was suspected. Additionally, Dandy-Walker malformation and atrio- ventricular septa1 defect were identified.
The pregnancy was complicated by a urinary tract infection, which was treated with an unspecified antibi- otic, and by seasonal environmental allergies, for which desensitizing injections were administered. There were no known teratogenic exposures. A maternal first cousin of the consultand's had spina bifida. A maternal first cousin, once removed, had hydrocephalus. The con- sultand was of German, English, and Irish descent; her partner's ancestry was not known. There was no known consanguinity.
Pregnancy termination was accomplished through

242 Johnson et al.
urea infusion and prostaglandin induction of labor at 1g4/7 weeks of gestation. Placental karyotype was 46,XX. Birthweight was 360 g, crown-heel length was 24 cm, and birth OFC was 19 cm. Physical examination showed large, low-set, and floppy ears, and a small man- dible. The palate was intact, but of unusual contour, with broad alveolar ridges anteriorly and a high arch posteriorly. There was mesomelic shortening and post- axial hexadactyly of all limbs, bilateral foot deformity, 213 syndactyly of the right foot, and 213 and 516 syndac- tyly of the left foot. Additional autopsy findings included absence of the cerebellar vermis (maceration precluded confirmation of other findings of Dandy-Walker malfor- mation), persistent common atrioventricular canal, bi- lateral unilobate hypoplastic lungs, agenesis of the right kidney and ureter, severe hypoplasia of the left kidney, and absence of the gallbladder. Pancreatic islet cell hyperplasia was present. Microscopic examination of the proximal humeri, proximal femora, and ribs showed a mild stunting of the physes, with focal mild disorganization, as seen in Case 1. The vertebral bodies were unremarkable. Examination of the placenta showed a single umbilical artery, focal villous edema, and intervillositis secondary to urea installation. A di- agnosis of SLOS-I1 was made.
DISCUSSION SLOS-I1 was first described by Curry et al. [19871,
who reported 19 infants with a more severe phenotype than had been previously associated with SLOS. A sec- ond series of 8 severely affected infants was described by Le Merrer et al. [1988].The diagnosis of SLOS-I1 is based on the presence of 3 or more of the following: congenital heart defect, postaxial hexadactyly, cataracts, severe genital ambiguity or pseudohermaphroditism in a 46,XY male, cleft palate and small tongue, early death, and the presence of at least one other specific malforma- tion including renal abnormalities, large adrenals, Hirschsprung disease, unilobated lungs, pancreatic is- let cell hyperplasia, 213 syndactyly of the toes, redun- dant nuchal skin, short limbs, facial hemangiomata, andjoint contractures [Curry et al., 19871. Both infants described herein conform to these diagnostic guidelines. To our knowledge, Case 2 is the first in which Dandy- Walker malformation has been described in association with SLOS-11.
Prenatal diagnosis of SLOS-11, as described in Case 1, has not been reported previously. Prenatal recognition of malformations associated with SLOS-11, with postna- tal determination of the diagnosis (as in Case 2), has been described in several reports. Two cases described by Rutledge et al. [19841, one in which short humeri were noted on sonographic examination at 35 weeks of gesta- tion, and another in which intrauterine growth retarda- tion was noted in the third trimester, were later recog- nized as being consistent with SLOS-11. Abnormal prenatal sonographic findings were noted in 5 of the 19 cases of Curry et al. [1987l: one with marginally short limbs and intrauterine growth retardation, one with intrauterine growth retardation alone, and 3 with intra- uterine growth retardation and oligohydramnios. Bel- mont et al. [1987] described oligohydramnios and intra-
uterine growth retardation at 31 weeks gestation in an infant later identified as having SLOS-11. Donnai et al. [19871 described unspecified malformations and oli- gohydramnios in a fetus at 27 weeks in which the diag- nosis of SLOS-I1 was made postnatally. Abnormalities were noted prenatally in 3 cases reported by Le Merrer et al. [1988]: one with oligohydramnios and intrauterine growth retardation at 29 weeks of gestation, one with unspecified multiple malformations, and one with intra- uterine growth retardation ascertained at 20 weeks of gestation. In addition, Donnai et al. [19861, Curry et al. [19871, and McKeever et al. 119901 described decreased late third trimester maternal serum and/or urinary es- trio1 levels in pregnancies in which the neonate was subsequently found to have SLOS-11.
We conclude that prospective prenatal diagnosis of SLOS-I1 is sometimes possible in pregnancies not known to be at risk for the condition, and that careful postnatal documentation of anomalies facilitates retro- spective diagnosis of SLOS-I1 when the diagnosis cannot be affirmed prenatally. Intrauterine growth retarda- tion, congenital heart defects, and renal abnormalities are common, albeit nonspecific, prenatal findings asso- ciated with SLOS-11. Perhaps the most specific prenatal finding is the observation of female external genitalia in the presence of a 46,XY chromosome complement and associated with other malformations, as the differential diagnosis of multiple malformation syndromes with male pseudohermaphroditism is relatively limited. Pre- natal diagnosis of any of these findings suggests the possibility of SLOS-11, and should prompt targeted so- nographic examination for possible related abnormal- ities including postaxial polydactyly, mesomelic short- ening of the extremities, or micrognathia. Prenatal recognition of the possibility of SLOS-I1 is important so that distinctive, but subtle and potentially easily over- looked findings, such as sublingual tissue mass, micro- gnathia, pancreatic islet cell hyperplasia, unilobate lungs, and Hirschsprung disease may be specifically sought at autopsy, and so that couples may be counselled accurately regarding their recurrence risk in future pregnancies. Finally, these cases underscore the impor- tance of sonographic assessment of fetal genitalia and correlation with karyotype in the comprehensive prena- tal evaluation of fetal anomalies.
ACKNOWLEDGMENTS We thank Dr. Helga Toriello and Dr. Cynthia Curry
for generously bringing their clinical expertise to bear in the evaluation and diagnosis of Case 2, and Dr. S. Samuel Yang for his thorough anatomic examinations of both cases.
REFERENCES Belmont JW, Hawkins E, Hejtmancik JF, Greenberg F (1987): Two
cases of severe lethal Smith-Lemli-Opitz syndrome. Am J Med Genet 26:65-67.
Curry CJR, Carey JC, Holland JS, Chopra D; Fineman R, Golabi M, Sherman S, Pagon RA, Allanson J, Shulman S, Barr M, McGravey V, Dabiri C, Schimke N, Ives E, Hall BD (1987): Smith-Lemli-Opitz syndrome-type 11: Multiple congenital anomalies with male pseu- dohermaphroditism and frequent early lethality. Am J Med Genet 26:45-57.

Donnai D, Young ID, Owen WG, Clark SA, Miller PFW, Knox WF (1986): The lethal multiple congenital anomaly syndrome of poly- dactyly, sex reversal, renal hypoplasia, and unilobar lungs. J Med Genet 23:64-71.
Donnai D, Burn J, Hughes H (1987): Smith-Lemli-Opitz syndromes: Do they include the Pallister-Hall syndrome? Am J Med Genet 28:741-743.
Le Merrer M, Briard ML, Girard S, Mulliez Pi, Moraines C, Imbert MC (1988): Lethal acrodysgenital dwarfism: A severe lethal condition resembling Smith-Lemli-Opitz syndrome. J Med Genet 25:89-95.
Smith-Lemli-Opitz Syndrome, Type I1 243
McKeever PA, Young ID (1990): Smith-Lemli-Opitz syndrome I1 A disorder of the fetal adrenals? J Med Genet 27:465-466.
Rutledge JC, Friedman JM, Harrod W E , Currarion G, Wright CG, Pinckney L, Chen H (1984). “New” lethal multiple congenital anomaly syndrome Am J Med Genet 19.255-264.
Smith DW, Lemli L, Opitz JM (1964). A newly recognized syndrome of multiple congenital anomalies. J Pediatr 64210-217








![Genetics of atrioventricular canal defects...rib polydactyly, Smith-Lemli-Opitz, and oral-facial-digital type IV syndromes [22, 23, 49] while ciliary func-tion is directly involved](https://img.pdfslide.net/doc/110x75/60e702c468ac877b2d356a48/genetics-of-atrioventricular-canal-defects-rib-polydactyly-smith-lemli-opitz.jpg)