Embed Size (px)
Citation preview

Clinica e LaboratorioClinica e Laboratorio
Un percorso comune tra appropriatezza e Un percorso comune tra appropriatezza e laboratoriolaboratorio
‘’‘’LE CONNETTIVOPATIE”LE CONNETTIVOPATIE”
Dott.ssa Elisabetta BattagliaDott.ssa Elisabetta BattagliaResp. U.O.D. ReumatologiaResp. U.O.D. Reumatologia
A.R.N.A.S. A.R.N.A.S. Garibaldi-CTGaribaldi-CT

Gruppo di malattie infiammatorie croniche classificate tra le malattie Reumatiche
caratterizzate da
distribuzione sistemica
carattere autoimmune
tendenza alla formazione di autoanticorpi
LE CONNETTIVITILE CONNETTIVITI

Gli aspetti molto simili fra loro suggeriscono una comune origine patogenetica
• Elevato rapporto femmine/maschi• Maggiore prevalenza di malattia in età giovane• Presenza di alterazioni immunologiche• Associazione con specifici geni coinvolti nella regolazione immunitaria
(HLA I-II classe) ”molecola presentante” complessata con peptide antigenico e molecole costimolatorie
• Presenza di più patologie in seno ai componenti della stessa famiglia.
LELE CONNETTIVITICONNETTIVITI

Gli aspetti simili sul piano immunologico e genetico spiegano le loro Gli aspetti simili sul piano immunologico e genetico spiegano le loro similitudini sul piano dell’espressione fenotipicasimilitudini sul piano dell’espressione fenotipica
Le manifestazioni cliniche più frequenti sono rappresentate da:Le manifestazioni cliniche più frequenti sono rappresentate da:
Fenomeno di Raynaud Fenomeno di Raynaud Malessere generale Malessere generale Facile affaticabilità Facile affaticabilità Febbre Febbre Perdita di peso Perdita di peso Poliartrite spesso migrante Poliartrite spesso migrante Impegno cutaneo (chiazze, papule, porpora,alopecia).Impegno cutaneo (chiazze, papule, porpora,alopecia).
LE CONNETTIVITILE CONNETTIVITI

Connettiviti IndifferenziateConnettiviti IndifferenziateUCTD (Undifferentiated Connective Tissue Disease)UCTD (Undifferentiated Connective Tissue Disease)
Proprio in virtù della patogenesi poligenica si Proprio in virtù della patogenesi poligenica si manifestano forme manifestano forme
““INDIFFERENZIATE” INDIFFERENZIATE”
definite tali perché non soddisfano i criteri minimi di definite tali perché non soddisfano i criteri minimi di classificazione di una connettivite specificaclassificazione di una connettivite specifica
Mosca M. et all J.Rheumatol. 2002Mosca M. et all J.Rheumatol. 2002

Forme tutt’altro che rare (20-50 %) possono evolvere verso forme definite (Les, Sjogren, ecc) specialmente nei primi anni.
In molti casi le manifestazioni cliniche rimangono stabili nel tempo Esiste una buona possibilità, in una piccola parte di pazienti, di ottenere la remissione completa e prolungata.
Mosca M. et all J.Rheumatol. 2002Mosca M. et all J.Rheumatol. 2002
Connettiviti IndifferenziateConnettiviti IndifferenziateUCTD (Undifferentiated Connective Tissue Disease)UCTD (Undifferentiated Connective Tissue Disease)

Esistono solo criteri classificativi e non diagnostici. Il loro riconoscimento è molto importante perché non esiste
una definizione accettata all’unanimità. La positività degli ANA non è ritenuta da tutti necessaria Quadri clinici di solito paucisintomatici Esistono discordanze anche sulla durata dei sintomi (dai 12 ai
24 mesi) Evoluzione più frequente verso il LES, overlap syndrome, PDM.
Connettiviti IndifferenziateUCTD (Undifferentiated Connective Tissue Disease)UCTD (Undifferentiated Connective Tissue Disease)

OVERLAP SINDROME
In molti casi il quadro dei pazienti, presenta anomalie cliniche e laboratoristiche tipiche di connettiviti sistemiche ma con aspetti differenti fra loro, soddisfacendoCriteri classificativi di Inclusione di due o più connettiviti definite
In questi casi si definisce “Connettivite da sovrapposizione“
(Overlap syndrome)

LE CONNETTIVITILE CONNETTIVITI
Le principali sono rappresentate da :
• LUPUS ERITEMATOSO SISTEMICO• Sclerosi Sistemica• Polimiosite-Dermatomiosite• Malattia di Sĵogren• Connettivite Mista• Vasculiti

LUPUS ERITEMATOSO SISTEMICOLUPUS ERITEMATOSO SISTEMICO• Malattia infiammatoria cronica “sistemica” • Eziologia multifattoriale: UVA, virus, batteri, fattori ormonali,
ecc…• Presenza di Anticorpi Antinucleo (ANA) rivolti verso diverse
specificità antigeniche ad alto titolo.• Coinvolgimento sistemico e multiorgano che determina quadri
clinici diversi per sintomatologia,gravità e prognosi• Prognosi varia da una patologia cronica con decorso lento e
complicanze contenute a condizioni cliniche rapidamente evolutive fino alla mortalità precoce.

EPIDEMIOLOGIAEPIDEMIOLOGIA
• Età di insorgenza: esordio solitamente tra II e IV decennio di vita,qualsiasi fascia di
età, picco massimo in età fertile. • Rapporto M/F: 1/10• Razza: tutti i gruppi etnici e razziali, è più presente nelle popolazioni Afro
Caraibiche e la sua incidenza e gravità sono maggiori nei neri ed ispanici (207/100.000 abitanti)
• Incidenza: 1,8-7.6/100.000 Anno.

QUADRO CLINICO CLINICOEstremamente polimorfo all’esordio
Manifestazioni generali Astenia (>80%) Febbre
Manifestazioni frequenti:• Artralgie• Rash malare• Sierositi (pleuriti e pericarditi)• Perdita di peso• Eritema vasculitico della cute all’esposizione solare• F. Di Raynaud
Alterazioni ematologiche: leucopenia, anemia, piastrinopenia

MANIFESTAZIONI CUTANEECUTANEE
LE cutaneo cronico (LED)LE cutaneo cronico (LED)Lesioni papulose evolventi in cicatrici.Lesioni papulose evolventi in cicatrici.Nel 10% evolve in LESNel 10% evolve in LES
LE: lesioni edematose superficiali LE: lesioni edematose superficiali Non evolventi in cicatrici.Non evolventi in cicatrici.
LE acuto-classico: eritema a farfallaLE acuto-classico: eritema a farfalla 40%-eritema fisso40%-eritema fisso
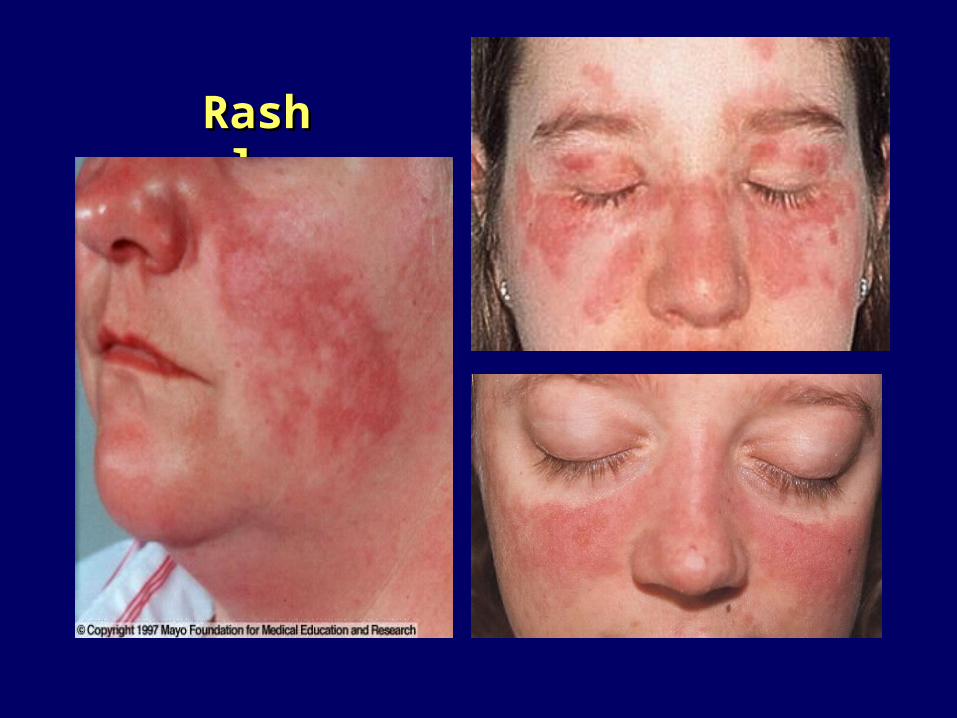
Rash malareRash malare
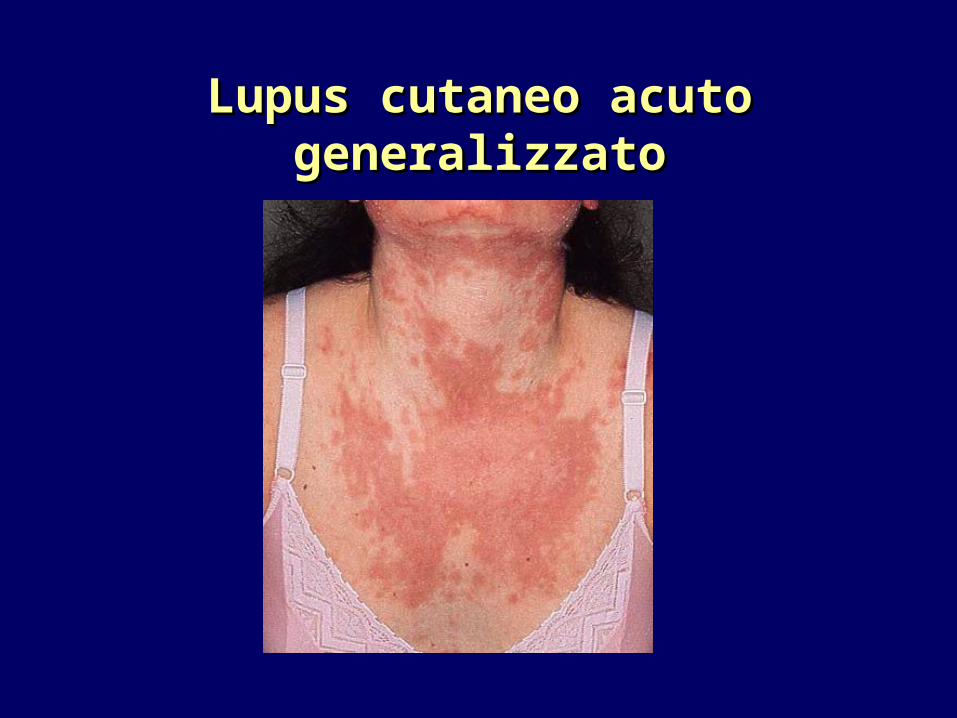
Lupus cutaneo acuto generalizzatoLupus cutaneo acuto generalizzato
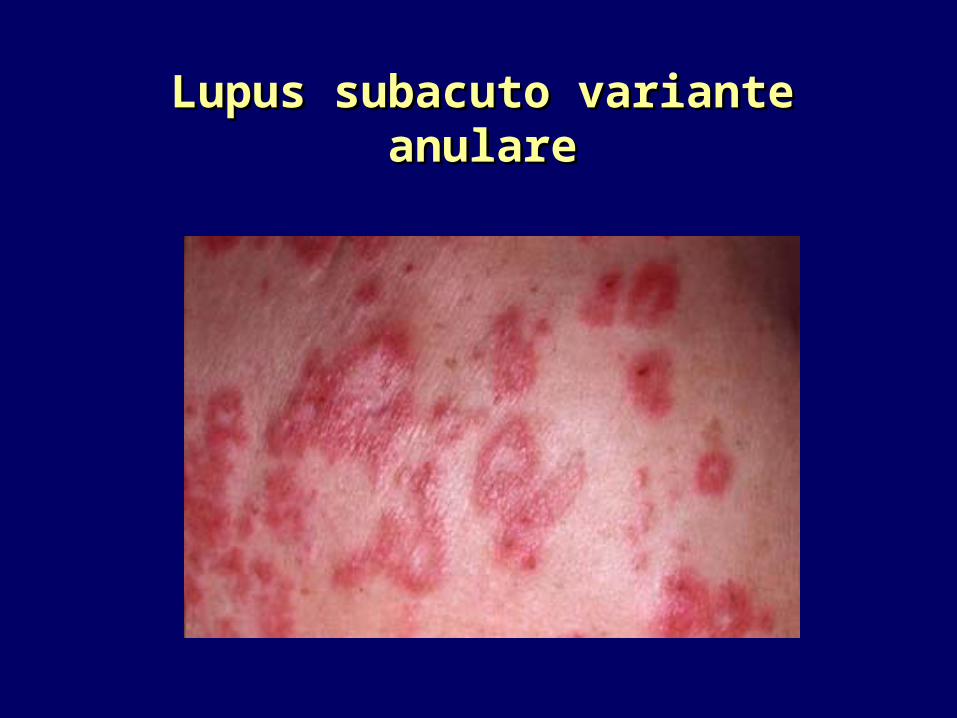
Lupus subacuto variante anulareLupus subacuto variante anulare
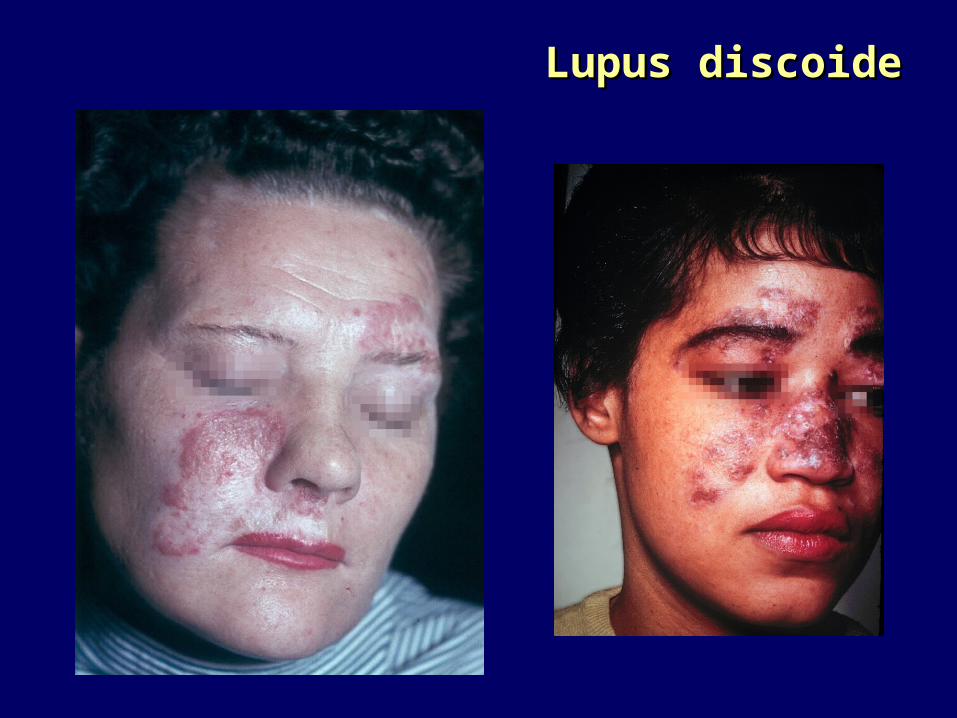
Lupus discoideLupus discoide

SIEROSITI• Versamenti generalmente modesti. Versamenti generalmente modesti.
• Possono presentarsiPossono presentarsi: pleuriti, pericardite e ascite: pleuriti, pericardite e ascite
• Pleurite bilateralePleurite bilaterale ( 50% dei casi)( 50% dei casi)
• Ascite+ peritonite (11% dei casi)Ascite+ peritonite (11% dei casi); ;
• TTalvolta può divenire cronicaalvolta può divenire cronica.
• Pericardite: manifestazione cardiaca più frequente nel LES (48%)
• Può precedere i segni clinici del LES • Frequentemente asintomatica e decorso benigno.• La complicanza più grave è una pericardite purulenta (paziente
immunosoppresso)• Versamenti cospicui con tamponamento cardiaco e pericardite restrittiva sono
rari
• Frequente il dolore toracico

aterosclerosi accelerata
in parte correlabile alla terapia farmacologica e in parte alla stessa patologia
(Ab anti-fosfolipidi e anti-endotelio)
MIOCARDITErara, spesso asintomatica secondaria a
vasculite dei piccoli rami coronarici, si associa nel 25% a pericardite
ENDOCARDITE VERRUCOSA ATIPICA
(Libman Sacks)
non batterica, raramente produce alterazioni emodinamicamente
significative
CORONAROPATIE

MANIFESTAZIONI POLMONARI
• Dolore toracico pleuritico anche in assenza di pleurite• Polmonite acuta batterica,virale• Polmonite interstiziale• Polmonite lupica acuta• Polmonite cronica: 9% dei pazienti, tosse, frequente la presenza di
anticorpi anti RO• Sindrome “Shrinking Lung”• Ipertensione polmonare• Emorragia polmonare

POLMONITE LUPICA ACUTA
Non è possibile isolare dei patogeni
RaraRaraincidenza 1-12% 1-12%
•Tosse•Emottisi•Dispnea•Febbre•Pleurite•50% versamento pleurico
Terapia corticosteroidea ad alto dosaggio e antibiotica in base agli
esami colturali
Prognosi generalmente scadente: nel post-partum mortalità a breve termine del 50%
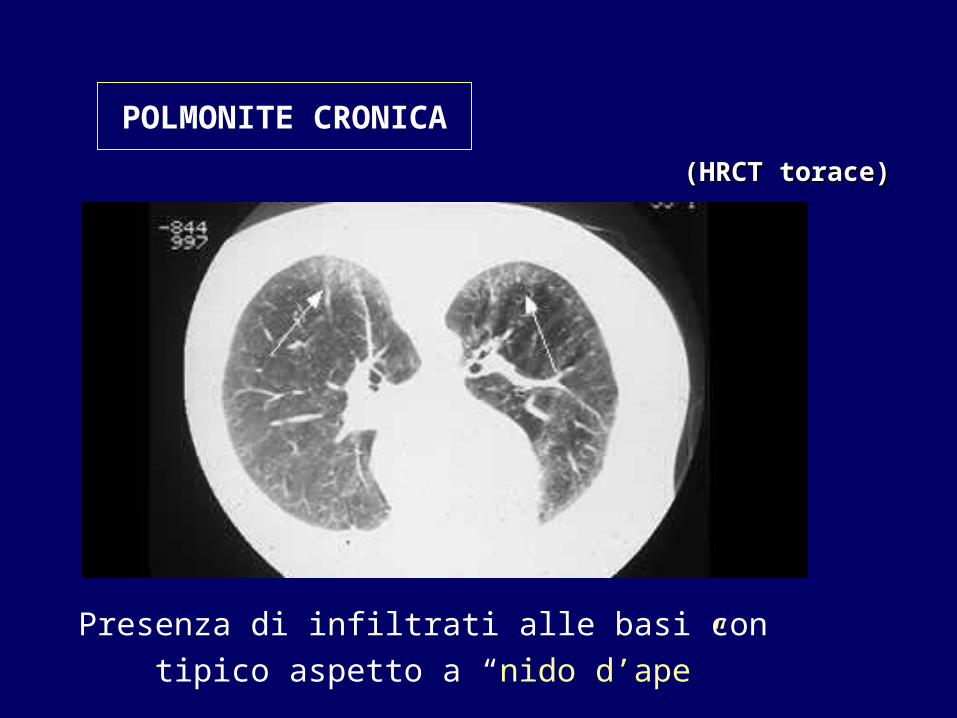
Presenza di infiltrati alle basi con tipico aspetto a “nido d’ape”
((HRCTHRCT torace)torace)
POLMONITE CRONICA
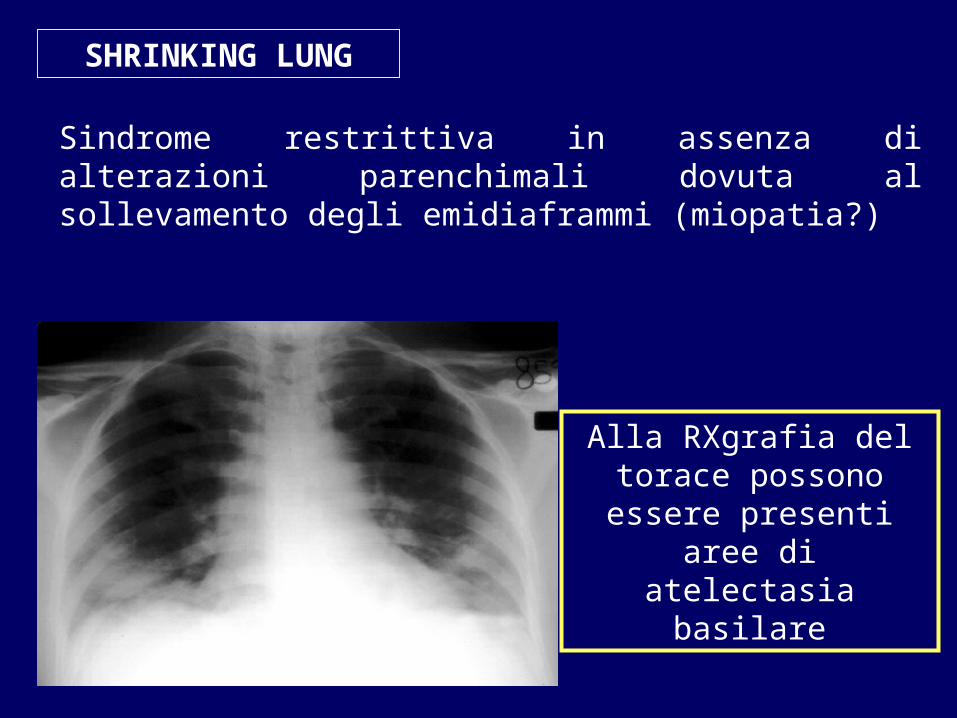
SHRINKING LUNG
Sindrome restrittiva in assenza di alterazioni parenchimali dovuta al sollevamento degli emidiaframmi (miopatia?)
Alla RXgrafia del torace possono essere presenti
aree di atelectasia basilare

MANIFESTAZIONI GASTROINTESTINALI
• Dolore addominale da Vasculite mesenterica• Sierosite e Pancretite• Epatomegalia (30%)• Epatite Lupica con ipertransaminasemia• Epatite da farmaci• Malattie infiammatorie dell’intestino• Morbo celiaco.

Manifestazioni Neurologiche• Lesioni frequenti e molteplici • 50% con prognosi sfavorevole ad eziopatogenesi vasculitica o immunologica• Anticorpi anti Proteina P-Ribosomiale e anti Membrana cellule nervose
presenti nel liquor• Cefalea non trattabile, responsiva agli steroidi• Convulsioni, accessi epilettici• Accidenti cerebrovascolari, ictus• Mielite trasversa• Multinevriti• Psicosi• Meningite da Fans• Organic brain syndrome: sindrome caratterizzata da delirio, instabilità
emotiva, riduzione della memoria• Disturbi neurocognitivi

IMPEGNO RENALE
• Incidenza: 30-80% (nelle diverse casistiche)
• Glomerulonefrite
• 25% dei pazienti con impegno renale sviluppa Insufficienza Renale Cronica
Importante fattore prognostico negativo in termini di morbilità e qualità di vita

MANIFESTAZIONI MUSCOLOSCHELETRICHE MUSCOLOSCHELETRICHE
Sindrome dolorosa da stress secondaria a molte malattie reumatiche.
i
ARTRITEsimmetrica, non erosiva (mani, polsi e piedi) tumefazione modesta a carico dei tessuti molli. Nel 10% possono svilupparsi deformità (A. Jaccoud)
MIOSITEMiopatia secondaria flogosi articolare, alla terapia steroidea o dovuta ad una infiammazione delle fibre muscolari
FIBROMIALGIA

MANIFESTAZIONI EMATOLOGICHE EMATOLOGICHE
• Anemia: normocitica e normocromica secondaria a malattia cronica, emolitica a patogenesi autoimmune
• Leucopenia: da Ab anti-leucociti
• Linfopenia: da Ab linfocitotossici
• Piastrinopenia: da Ab anti-piastrine
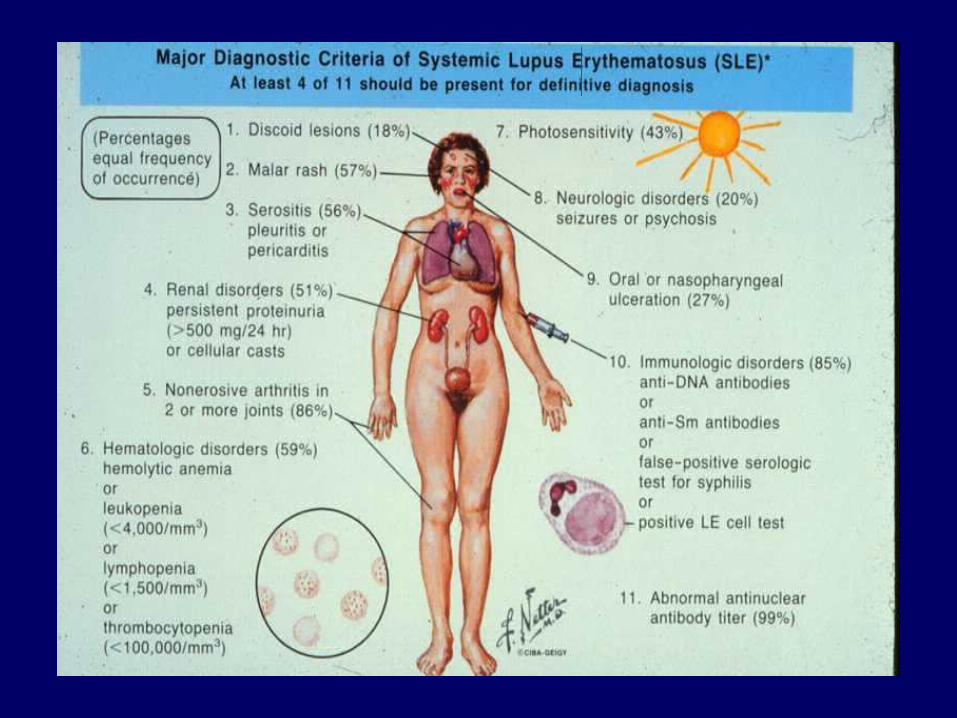
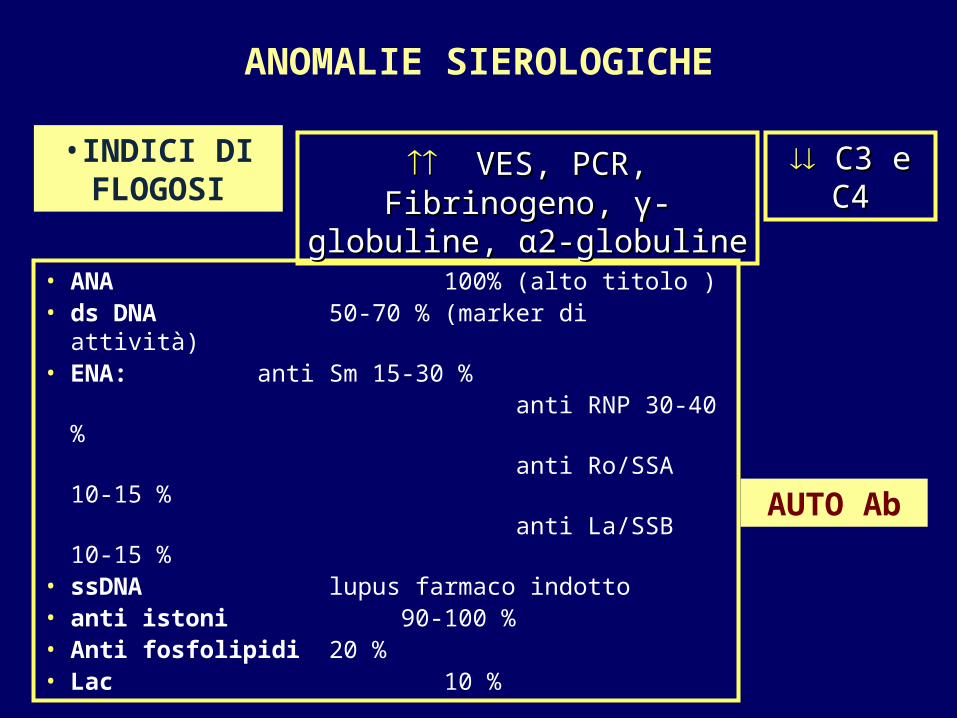
ANOMALIE SIEROLOGICHE
• ANA 100% (alto titolo )• ds DNA 50-70 % (marker di attività)• ENA: anti Sm 15-30 % anti RNP 30-40 % anti Ro/SSA 10-15 % anti La/SSB 10-15 %• ssDNA lupus farmaco indotto• anti istoni 90-100 %• Anti fosfolipidi 20 %• Lac 10 %
•INDICI DI FLOGOSI
VES, PCR, Fibrinogeno, VES, PCR, Fibrinogeno, γγ--globuline, globuline, αα2-globuline2-globuline
AUTO Ab
C3 e C4C3 e C4

SINDROME DA ANTICORPI ANTI-FOSFOLIPIDI
Trombosi vascolari arteriose e/o venose ricorrenti Aborti ripetuti Trombocitopenia Anticorpi anti-fosfolipidi a titoli medio-alti

Sintomi Criterio1. Trombosi vascolari : uno o più episodi di trombosi arteriose,
venose o dei piccoli vasi, in qualsiasi organo o tessuto, confermate da tecniche di imaging, doppler o dall’istopatologia
2. Patologia ostetrica:• Una o più morti fetali oltre la 10^ settimana;• Uno o più parti prima della 34^ settimana, accompagnati
da preeclampsia o severa insufficienza placentare;• Tre o più aborti prima della 10^ settimana.
•

Sintomi non criterio MANIFESTAZIONI NEUROLOGICHE• Legate essenzialmente all’ ostruzione di arteriole cerebrali• Corea• Epilessia• TIA/ ictus• Emicrania/cefalea intrattabile• Mielite trasversa (rari casi) MANIFESTAZIONI CUTANEE • Livaedo reticularis: caratteristica della malattia• Ulcere cutanee secondarie a ischemia/tromboflebiti/occlusione
arteriole cutanee MALATTIA VALVOLARE CARDIACA NEFROPATIA MICROANGIOPATICA O ISCHEMICA
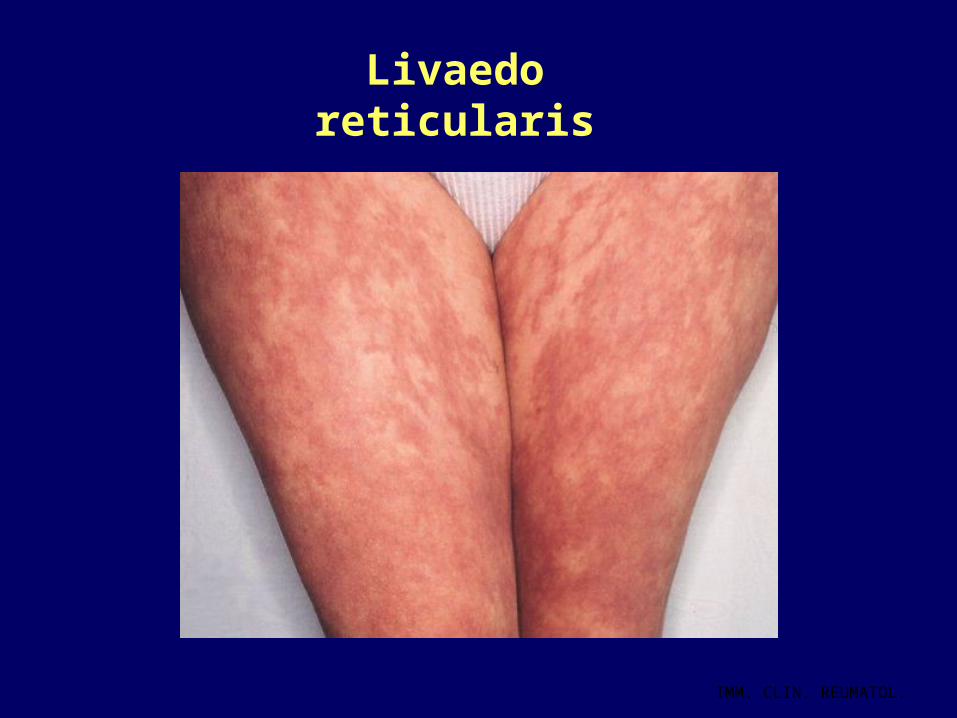
Livaedo reticularis
IMM. CLIN. REUMATOL.
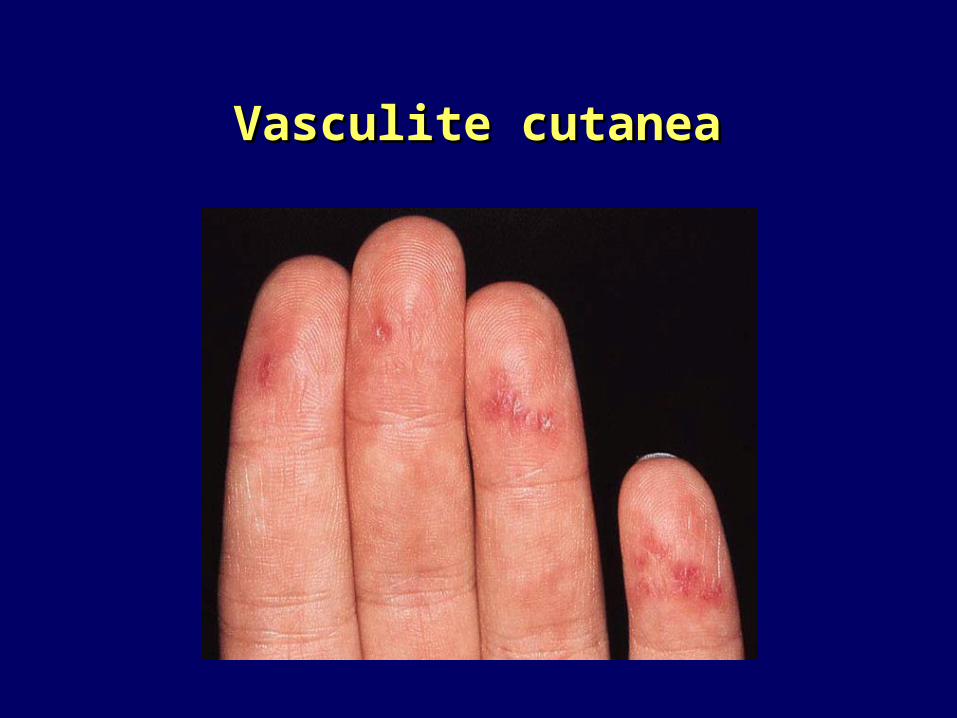
Vasculite cutaneaVasculite cutanea
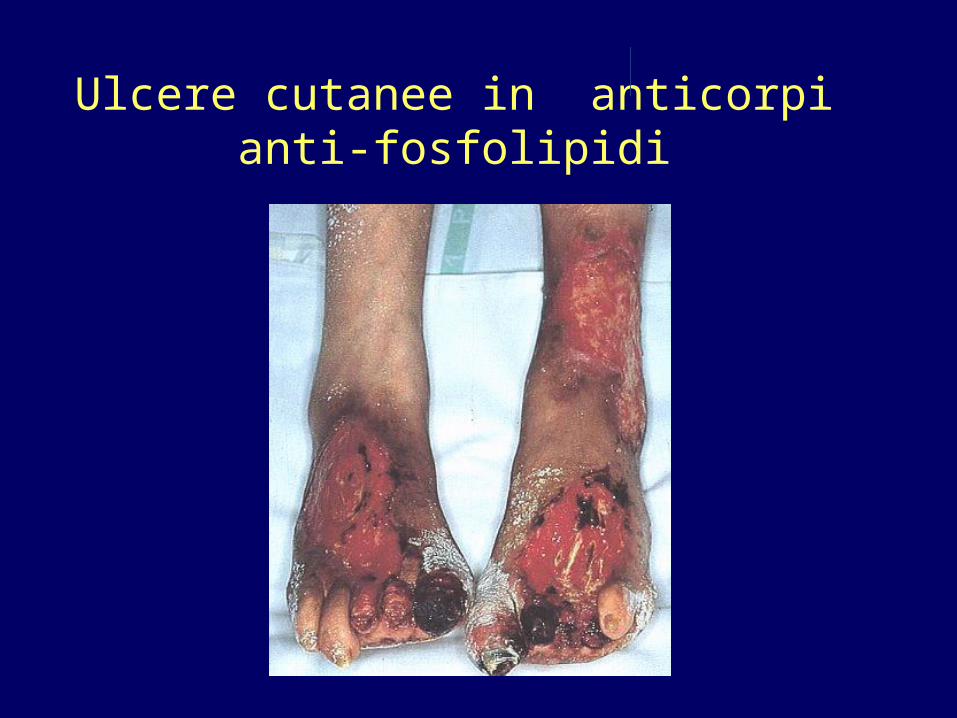
Ulcere cutanee in anticorpi anti-fosfolipidi

Criteri laboratoristici• Lupus Anticoagulant positivo
secondo le linee guida raccomandate dal Sottocomitato del Lupus Anticoagulante /Phosfolipd Dependent Antibodies della società Internazionale di Trombosied Emostasi
• Positività a titolo medio-alto anticorpi Anticardiolipina (aCL) di isotipo Ig G o Ig M (ELISA standardizzato)
• Riscontro di anticorpi anti- B2GPI di isotipo IgG e/o IgM
nel siero o plasma, ottenuta con metodica ELISA standardizzata
NB: Diagnosi di APL quando risulti positivo per uno dei 3 test-criterio in 2 successive determinazioni ripetute a distanza di almeno 12 settimane

TEST NON CRITERIO
• Anticorpi IgA Anticardiolipina• Anticorpi IgA Anti B2GPI• Anticorpi Antifosfatidilserina• Anticorpi Antifosfatidiletanolamina• Anticorpi Anti protrombina isolata (aPT-A)

Sclerodermia
Identifica alcune forme morbose caratterizzate da:
• Fibrosi progressiva del connettivo della cute, di organi o di interi apparati
• Lesioni caratteristiche a carico del microcircolo (vasculite)
• Anomalie della funzione immunitaria
secondarie alle secondarie alle arteritiarteriti responsabili di:responsabili di:
• Ischemie digitaliIschemie digitali• Malattia renaleMalattia renale• Ipertensione polmonareIpertensione polmonare
FIBROSI il il difetto centraledifetto centrale della della sclerodermiasclerodermia
rappresentarappresenta
coinvolge la cute, il polmone, l’apparato cardiovascolare, coinvolge la cute, il polmone, l’apparato cardiovascolare, l’apparato gastrointestinale e urinario.l’apparato gastrointestinale e urinario.
ALTERAZIONI DELMICROCIRCOLO

Sclerosi sistemicaCutanea diffusaCutanea limitata (CREST)
Scl circoscrittaLineareMorfea
Sclerosi sistemica sine scleroderma
Scl associata ad altre connettiviti (overlap syndromes)
Scl indotta da agenti
ambientali e chimiciCloruro di vinileSolventi organici (benzene toluene)Sindrome da olio tossicoFarmaci (bleomicina, pentazocina)
ClassificazioneClassificazione
Sclerosi sistemicaSclerosi sistemica
•Forma classica
•Estesa anche al tronco
•Decorso rapido
•Coinvolgimento multiorgano precoce
•Prognosi sfavorevole
Cutanea diffusa

Sclerosi sistemicaSclerosi sistemica
•Preceduta anche da molti anni dal F. di Raynaud
•Interessa il volto e la parte distale degli arti
•Impegno viscerale più tardivo o assente
•Prognosi più favorevole
Cutanea limitata
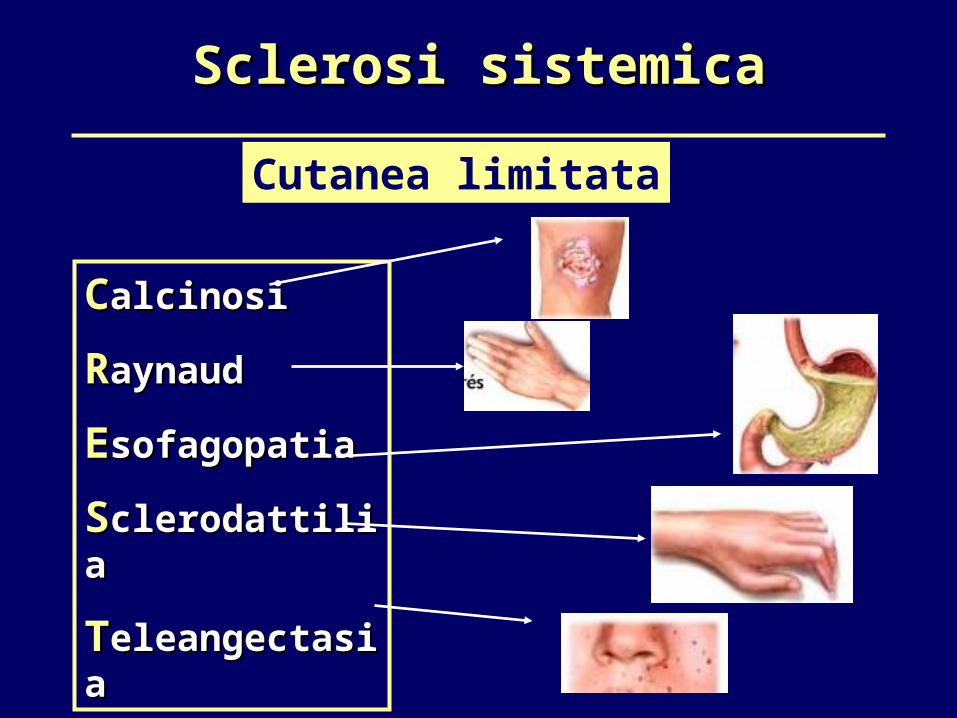
Sclerosi sistemicaSclerosi sistemica
Cutanea limitata
CCalcinosialcinosi
RRaynaudaynaud
EEsofagopatiasofagopatia
SSclerodattiliaclerodattilia
TTeleangectasiaeleangectasia

Sclerosi sistemicaSclerosi sistemica
SS sine scleroderma
non c’è evidenza di sclerosi cutanea ma è presente coinvolgimento sistemico.

Sclerodermia circoscrittaSclerodermia circoscritta
Soggetti giovani
bande sclerotiche a carico della fronte (deformità facciale), del tronco e delle estremità.
Lineare
Morfea
Bambini e giovani
placche sclerotiche singole o confluenti (morfea generalizzata), colorito cereo avorio. Prognosi buona.
Impegno viscerale assente. Possibilità di ANA positivi.

EpidemiologiaEpidemiologia
• Età di insorgenza : 30 e 50 anni ( qualsiasi età della vita)
Rapporto femmine/maschi variabile da 3:1 a 7:1 a seconda dell’età
• Incidenza media tra i 10 e 20 nuovi casi all’anno per milione di abitanti
• Differenze della prevalenza in seno alle diverse razze
• Associazione con antigeni di istocompatibilità
HLA A1, B8, DR1, DR3, DR5

EtiopatogenesiEtiopatogenesi
Il Il Sistema ImmunitarioSistema Immunitario gioca un ruolo essenziale nella gioca un ruolo essenziale nella patogenesi della malattia e nella sua progressionepatogenesi della malattia e nella sua progressione
• Attivazione dei linfociti B Attivazione dei linfociti B • Autoanticorpi circolantiAutoanticorpi circolanti• Prevalenza di risposta di tipo Th2Prevalenza di risposta di tipo Th2
ANOMALIEIMMUNOLOGICHE

Quadro clinicoQuadro clinico
ESORDIOESORDIO
Fenomeno di RaynaudFenomeno di Raynaud
In molti casi rappresenta il primo segno della malattiaPuò precedere di mesi la comparsa della sclerosi cutanea..
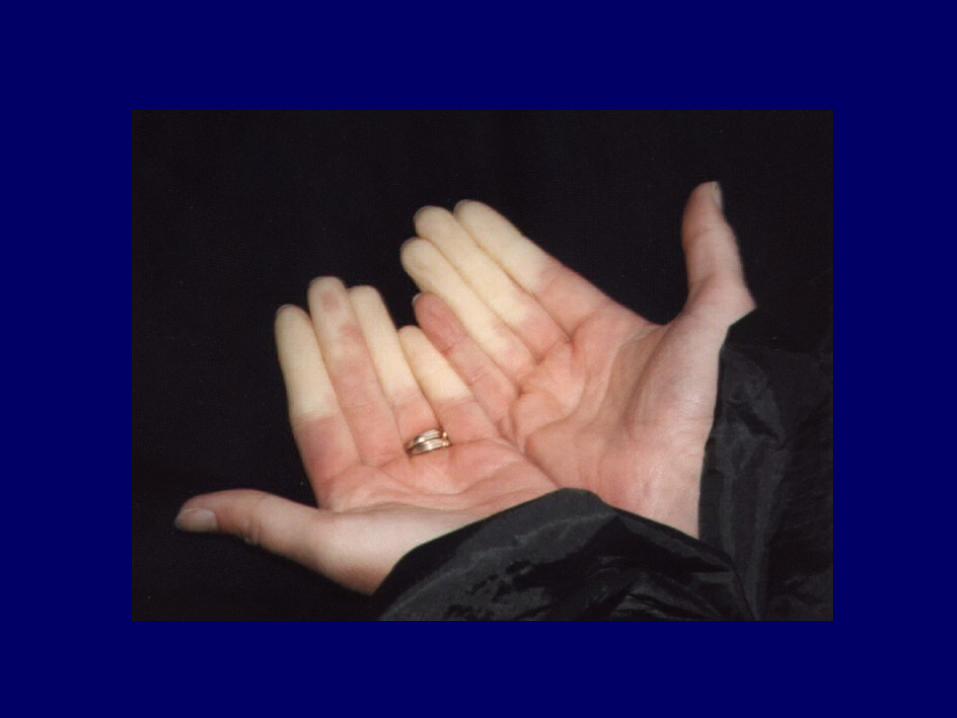
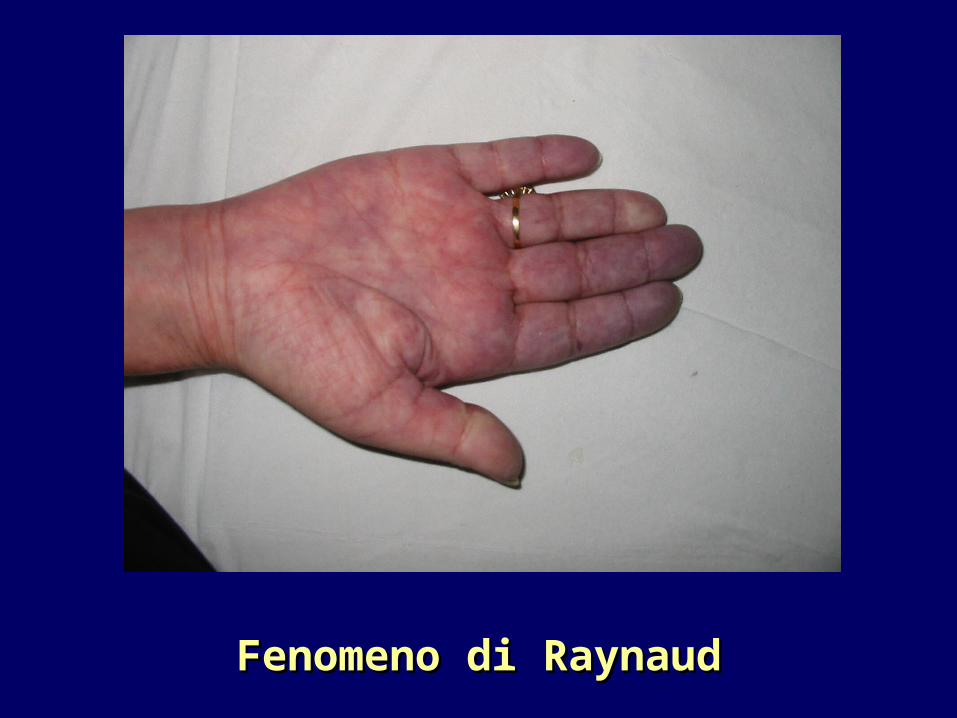
Fenomeno di RaynaudFenomeno di Raynaud
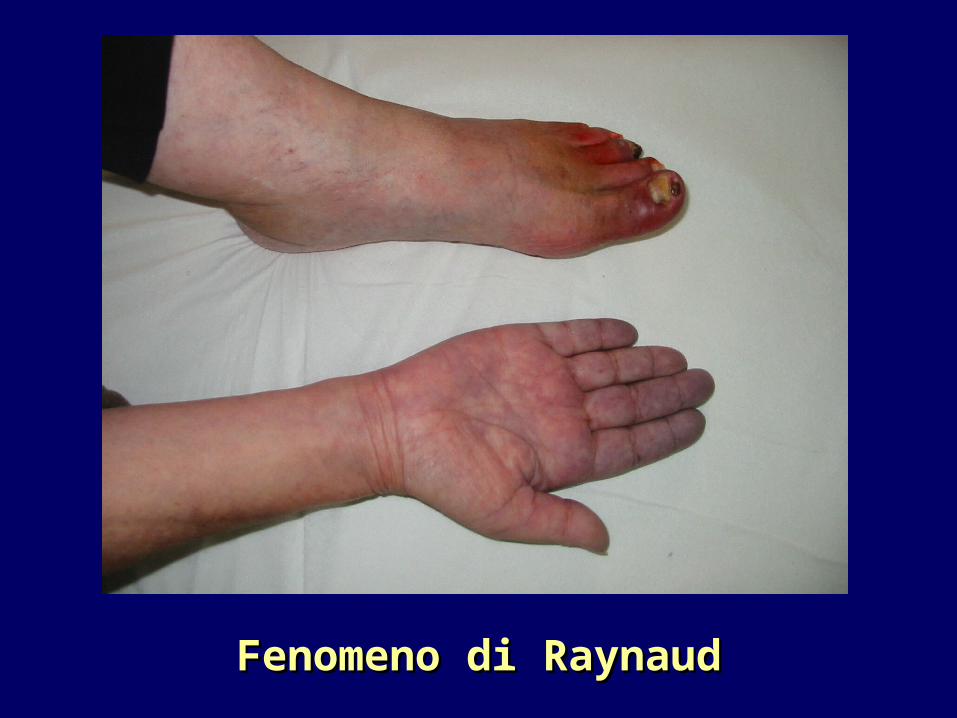
Fenomeno di RaynaudFenomeno di Raynaud

Quadro clinicoQuadro clinico
Astenia (80% dei pazienti)
Facile affaticabilità
Crampi muscolari
Artralgie
INIZIALE SPICCATO EDEMA DELLA CUTE
•Non improntabile
•Localizzato prevalentemente alle dita e al dorso delle mani talvolta agli avambracci
•Intensamente pruriginoso
•Determinante l’incapacità alla flessione delle dita.
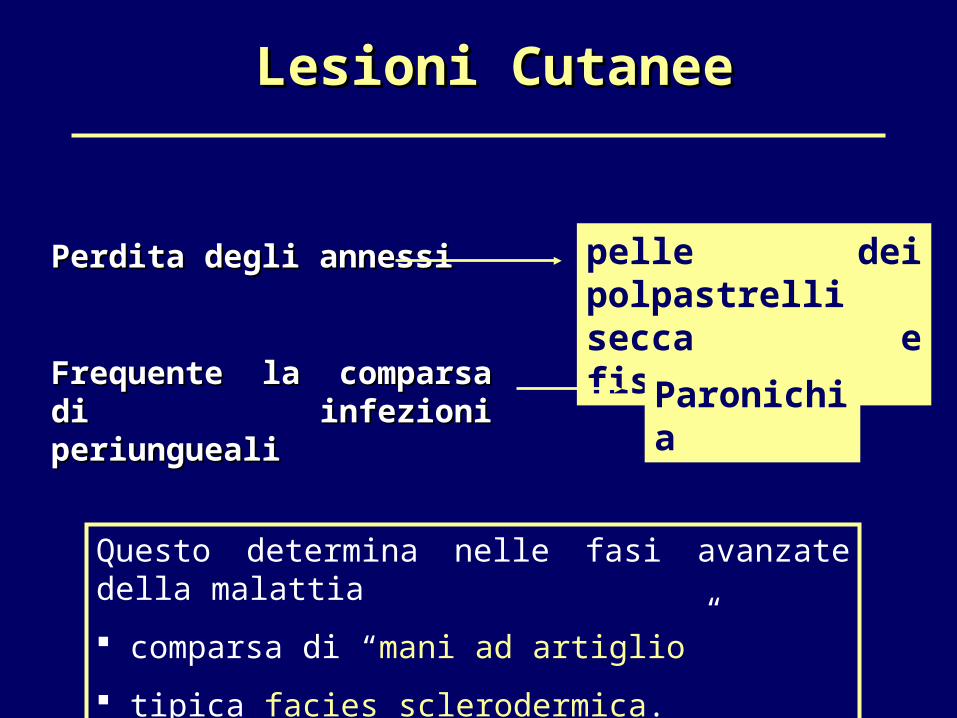
Lesioni CutaneeLesioni Cutanee
Questo determina nelle fasi avanzate della malattia
comparsa di “mani ad artiglio”
tipica facies sclerodermica.
pelle dei polpastrelli secca e fissurata
Perdita degli annessiPerdita degli annessi
Frequente la comparsa di Frequente la comparsa di infezioni periunguealiinfezioni periungueali Paronichia
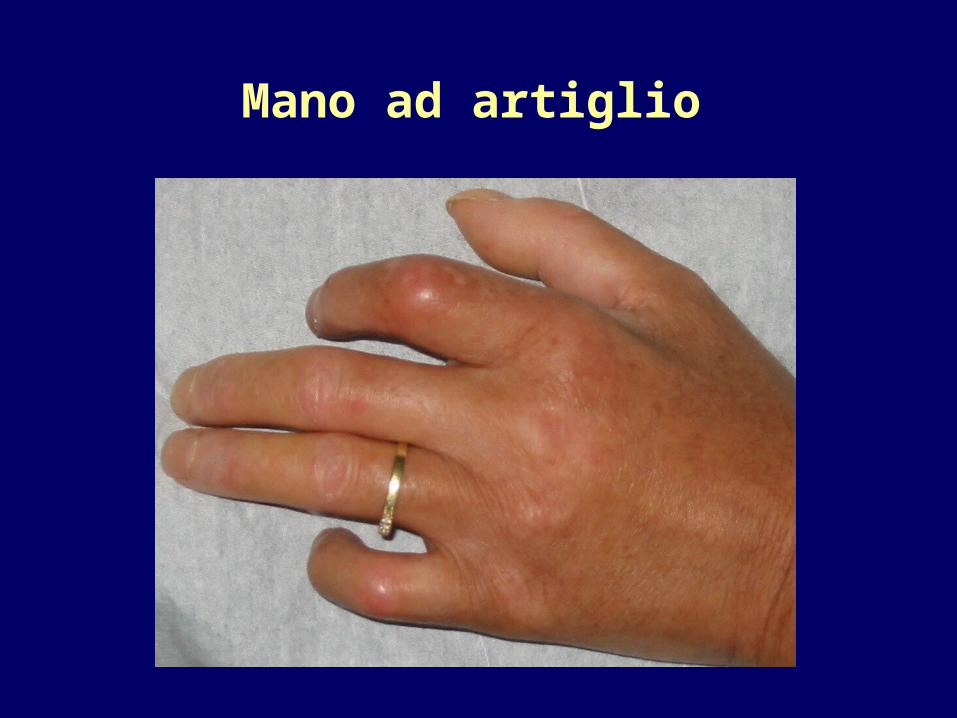
Mano ad artiglio

Microcheilia e microstomiaMicrocheilia e microstomia
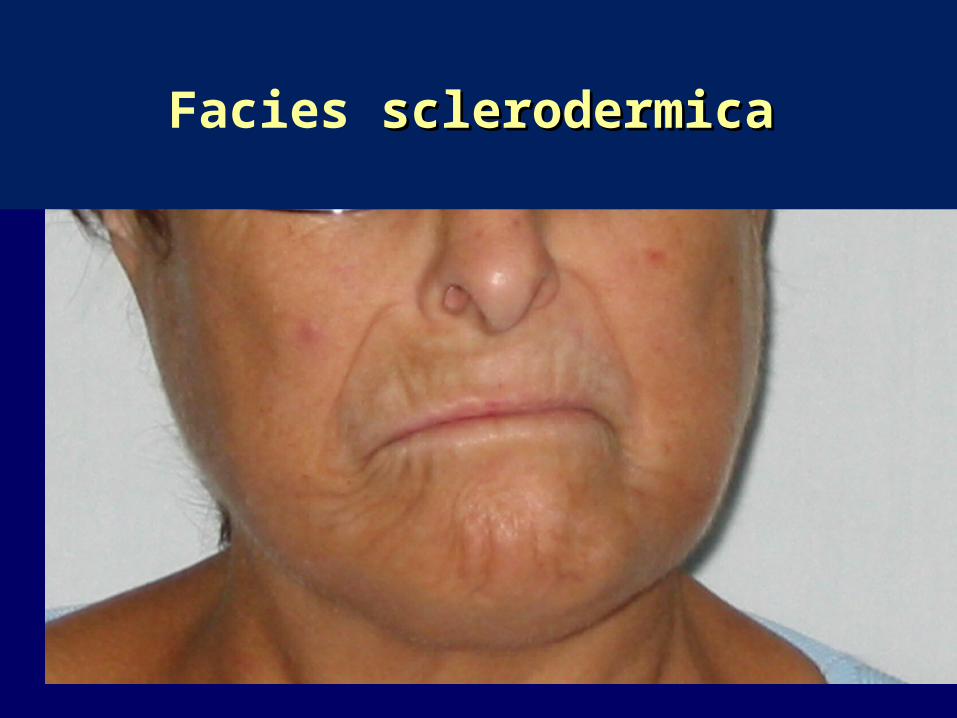
Facies sclerodermicasclerodermica

Lesioni CutaneeLesioni Cutanee
•Discromie melanodermia melanodermia falsa vitiligofalsa vitiligo
•CalcinosiCalcinosi (depositi di fosfato e (depositi di fosfato e carbonato di calcio che spesso affiorano carbonato di calcio che spesso affiorano alla cute)alla cute)
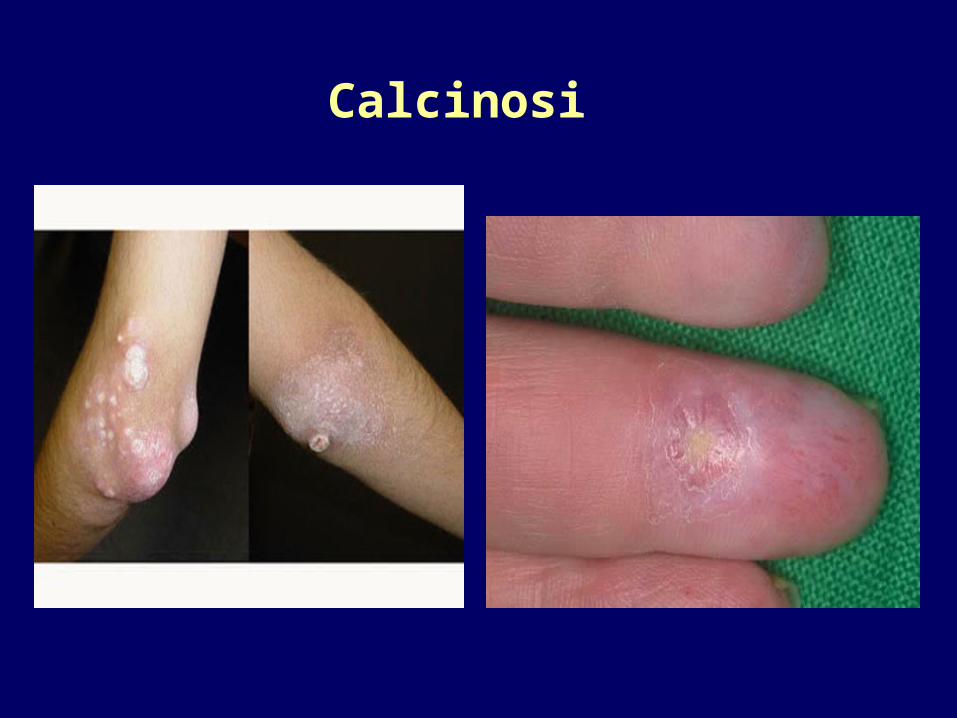
Calcinosi
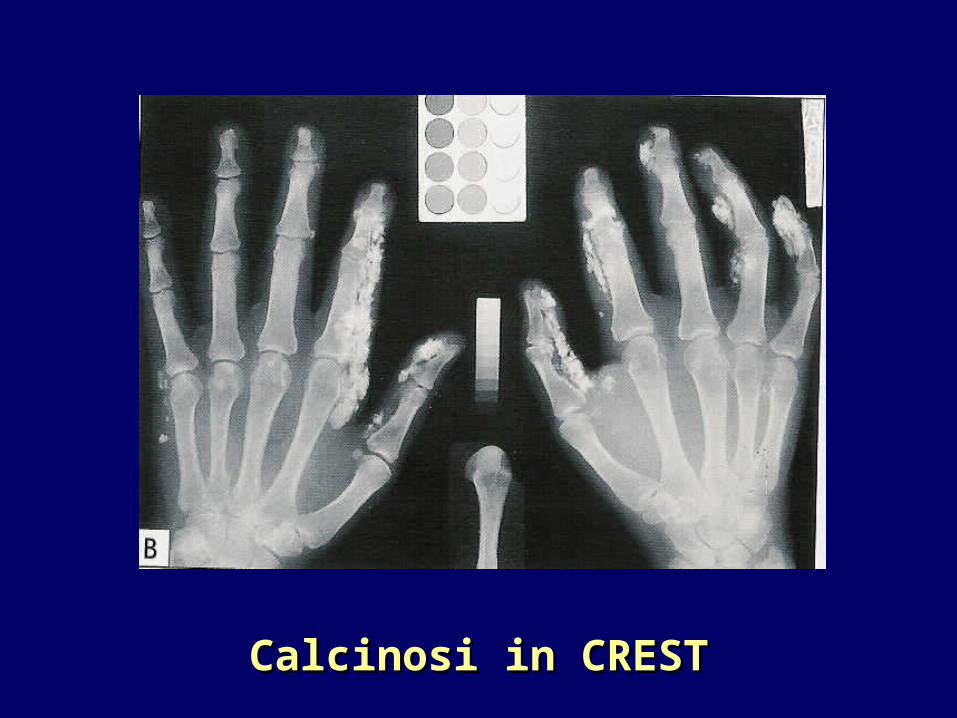
Calcinosi in CRESTCalcinosi in CREST

Dita delle mani e dei piedi
Osteolisi sottostante
Alterazioni VascolariAlterazioni Vascolari
F. DI RAYNAUD
TELEANGECTASIE
ULCERE ISCHEMICHE
Periferico: mani, piedi, punta del naso
Viscerale
Volto, palmo delle mani, dita, torace.(il numero aumenta con la durata della malattia)
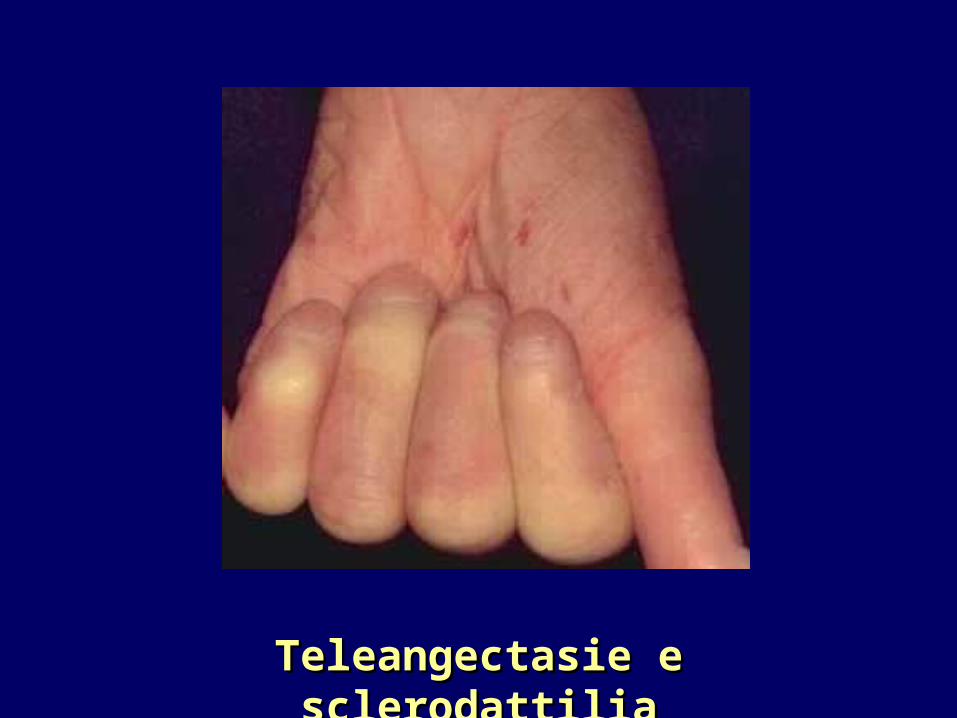
Teleangectasie e sclerodattiliaTeleangectasie e sclerodattilia
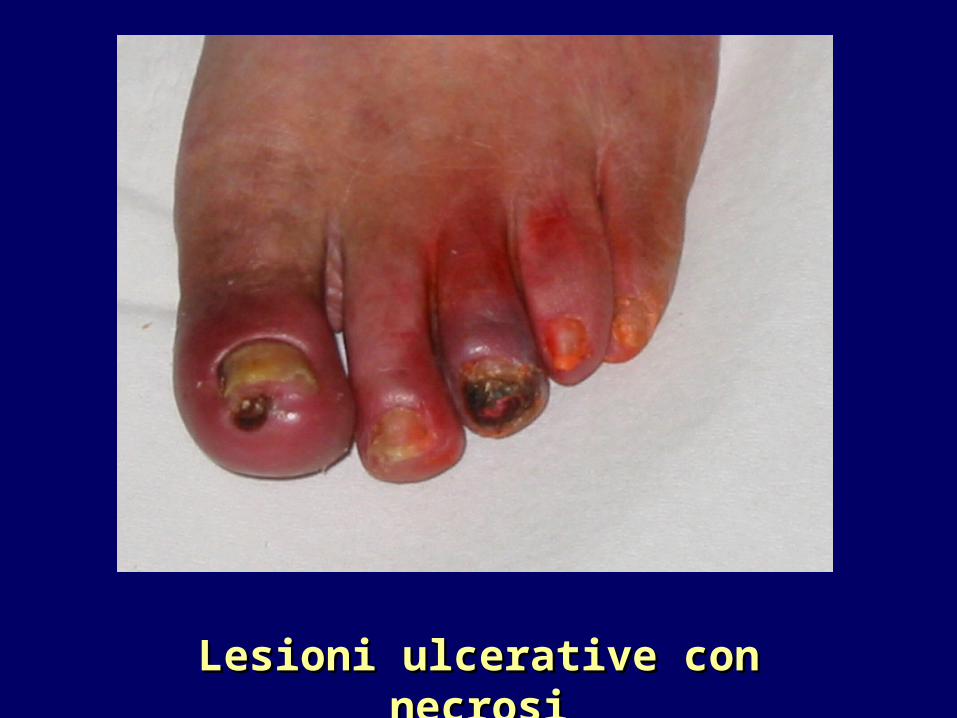
Lesioni ulcerative con necrosiLesioni ulcerative con necrosi

Impegno osteo-articolare
• Molto simile a quello che si osserva nell’ A . Reumatoide •Nelle fasi iniziali può manifestarsi poliartrite simmetrica• Artralgie: frequenti nella Scl diffusa•Artrite: 20-30%: mani e polsi
•Lesioni ossee: osteolisi da ischemia e da continua trazione tendinea sull’osso sottostante con riassorbimento delle falangi distali (autoamputazione). •Artropatia sclerodermica•Osteoporosi.

Artropatia SclerodermicaArtropatia Sclerodermica
Anchilosi in flessioneAnchilosi in flessione

Alterazioni VisceraliAlterazioni Viscerali
•Apparato respiratorio•Apparato cardiovascolare•Apparato nefrovascolare•Apparato gastrointestinale•Sistema nervoso centrale e periferico•Apparato visivo
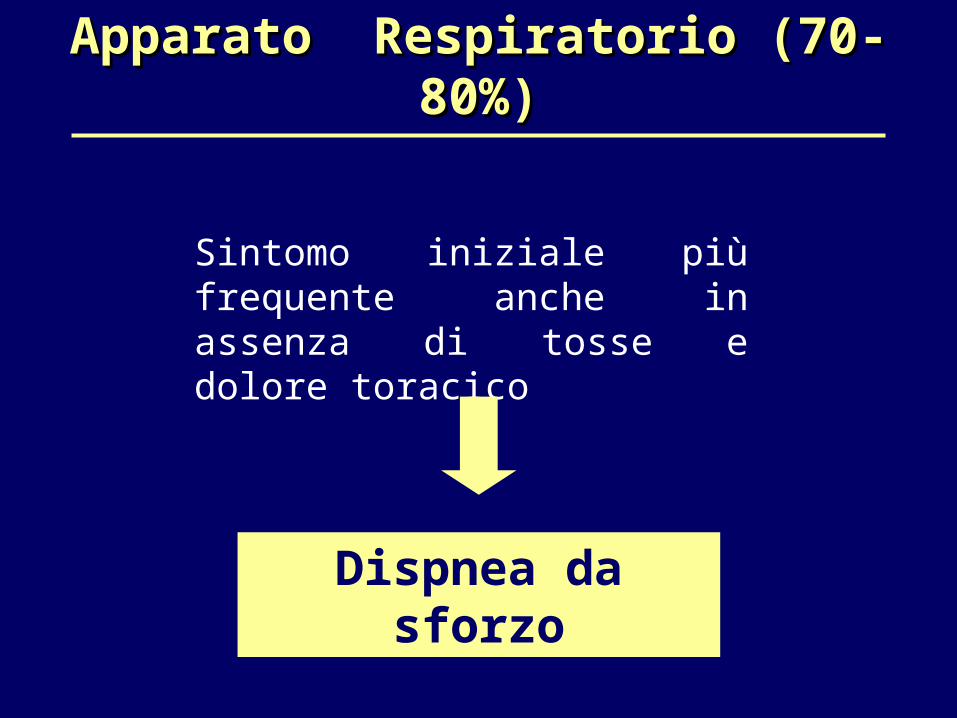
Apparato Respiratorio (70-80%)Apparato Respiratorio (70-80%)
Sintomo iniziale più frequente anche in assenza di tosse e dolore toracico
Dispnea da sforzo

•Fibrosi polmonare interstiziale: preceduta da alveolite basale. Progressiva compromissione della funzionalità respiratoria
•Ipertensione polmonare: cuore polmonare cronico Scompenso cardiaco congestizio
•Pleuropatie: raro il versamento•Polmoniti ab ingestis: da esofagopatia•Associazione con silicosi (Sindr. di Erasmus)•Associazione con K polmonare a cellule alveolari

Interstiziopatia PolmonareInterstiziopatia Polmonare
• Quadro caratteristico • Rappresenta uno dei criteri diagnostici • Esordio precoce• Più severa nella forme di SSC diffusa
Nonostante i recenti progressi terapeutici l’insufficienza respiratoria rappresenta una delle principali cause di morte nei pazienti con SSC
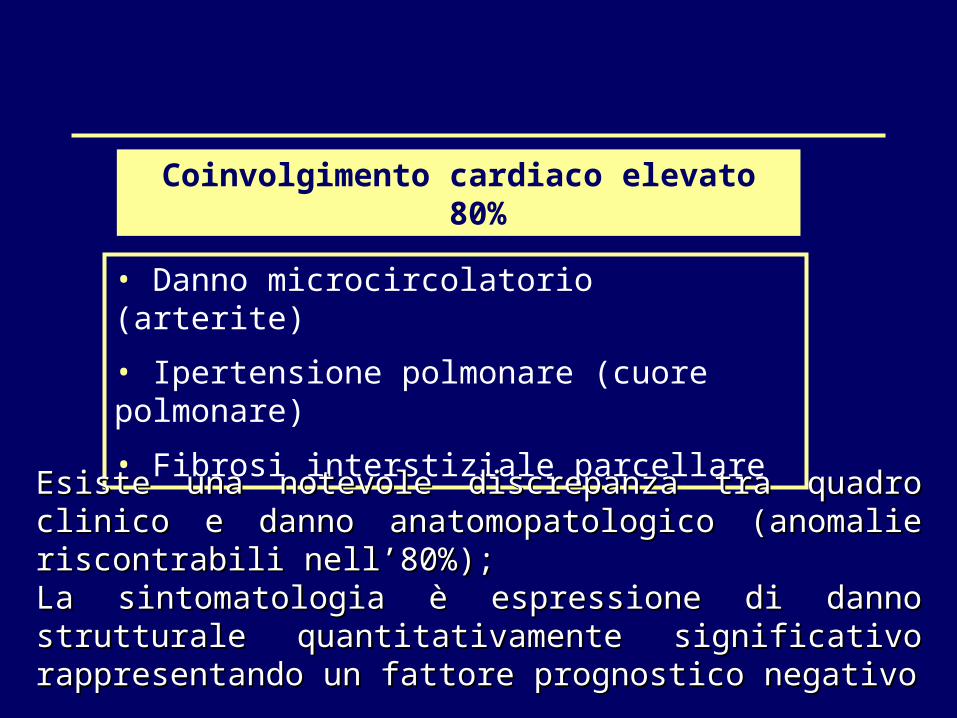
Coinvolgimento cardiaco elevato 80%
• Danno microcircolatorio (arterite) • Ipertensione polmonare (cuore polmonare)• Fibrosi interstiziale parcellare
Esiste una notevole discrepanza tra quadro clinico e danno Esiste una notevole discrepanza tra quadro clinico e danno anatomopatologico (anomalie riscontrabili nell’80%); anatomopatologico (anomalie riscontrabili nell’80%); La sintomatologia è espressione di danno strutturale La sintomatologia è espressione di danno strutturale quantitativamente significativo rappresentando un fattore quantitativamente significativo rappresentando un fattore prognostico negativoprognostico negativo

Apparato CardiovascolareApparato Cardiovascolare
• ArteriteArterite:: angor-infarto angor-infarto• Fibrosi muscolare parcellareFibrosi muscolare parcellare: evoluzione verso la : evoluzione verso la
miocardiopatia restrittivamiocardiopatia restrittiva• Fibrosi del tessuto di conduzione:Fibrosi del tessuto di conduzione:
tachicardia, aritmie ipercinetiche ventricolari-principale causa di tachicardia, aritmie ipercinetiche ventricolari-principale causa di morte)morte)
• Ipertensione polmonareIpertensione polmonare (cuore polmonare)(cuore polmonare)• Ipertensione arteriosaIpertensione arteriosa (nefrovascolare)(nefrovascolare)

Apparato NefrovascolareApparato Nefrovascolare
insuff. renale rapidamente progressiva
ipertensione arteriosa ”maligna”
anemia emolitica e piastrinopenia
Nefropatia sclerodermica
microematuria e proteinuria nel 50% dei pazienti. Lenta evoluzione verso l’insufficienza renale lieve-moderata
Crisi renale
(5-10%)(5-10%)
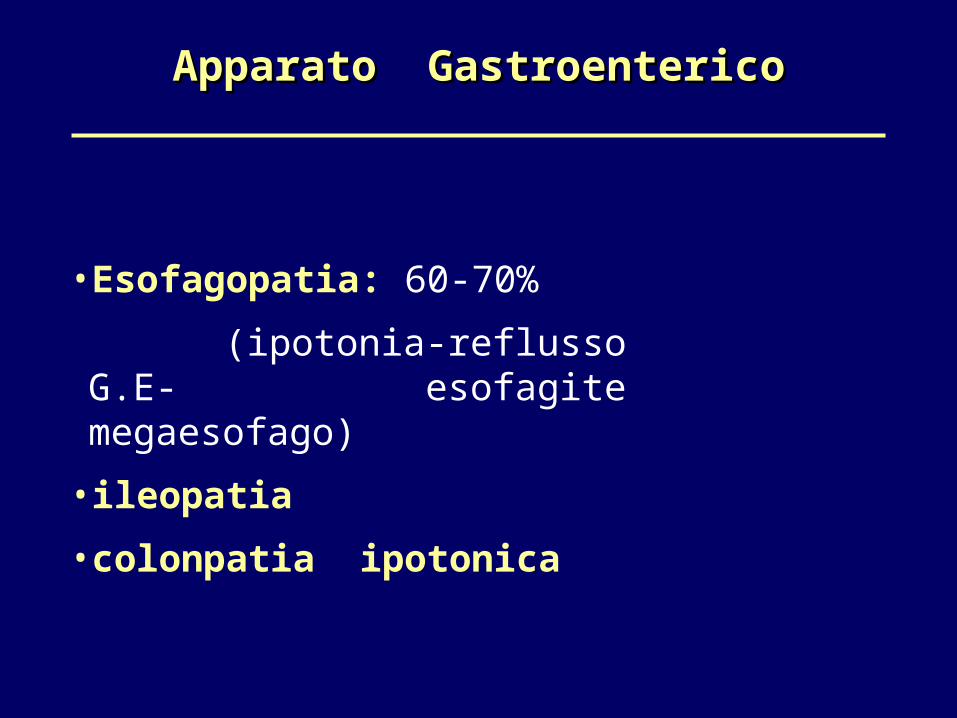
Apparato GastroentericoApparato Gastroenterico
•Esofagopatia: 60-70% (ipotonia-reflusso G.E- esofagite megaesofago)
•ileopatia •colonpatia ipotonica

• Disfagia più spiccata per i solidi
• Pirosi retrosternale• Senso di peso
postprandiale• Nausea e vomito
•Distensione addominale•Dolore crampiforme•Stipsi•Malnutrizione/
malassorbimento
Esofagopatia Ileocolopatia
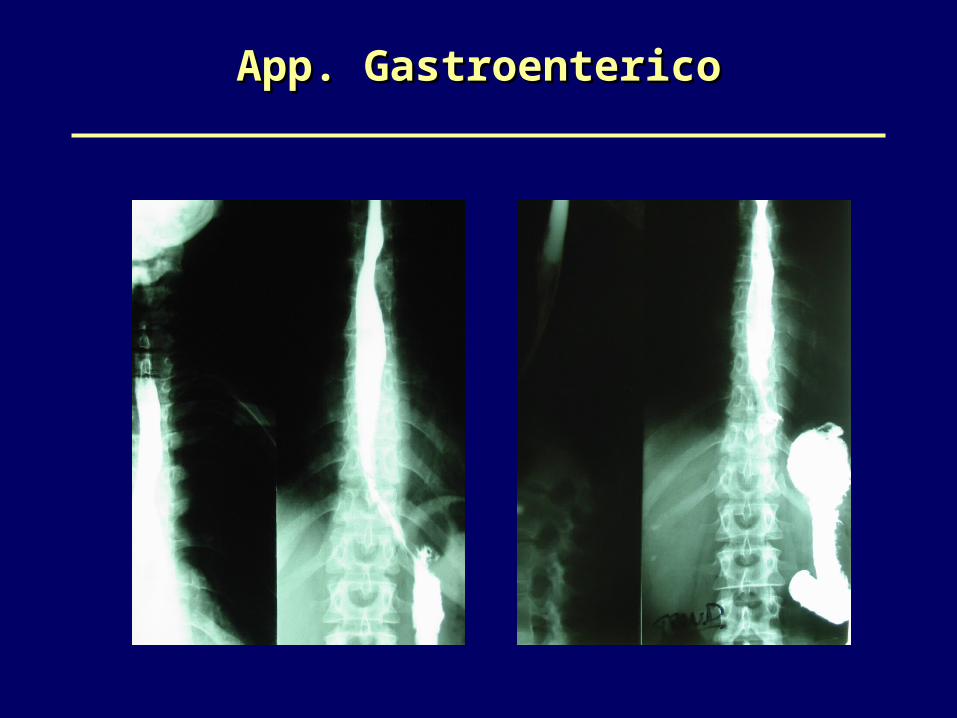
App. GastroentericoApp. Gastroenterico

Sistema NervosoSistema Nervoso
•Scarsamente suscettibile di lesioni per la minor presenza di tessuto connettivo
•Lesioni psico-organiche e ischemiche
•Neuropatie sensitive nn. cranici (trigemino, acustico, glosso-faringeo)
•S. da compressione (es. tunnel carpale)

Esami BioumoraliEsami Bioumorali• Indici di flogosi alterati nella fase edemigena in presenza di necrosi cutanee estese sierositi, miosite, artriti o infezioni
• Anemia micro/macrocitica (per alterato assorbimento intestinale di vit. B12 e folati) • Ipergammaglobulinemia di modesta entità
• Fattore reumatoide nel 20-30% dei casi

Esami BioumoraliEsami Bioumorali
•ANA positivi
(oltre il 95% dei pazienti solitamente con pattern granulare)
•Anti-centromero - ACA nel 70-90% dei pz con forma CREST
•Anti Scl-70 (anti Topoisomerasi-1) dimostrabili nel 30-35% dei pazienti “ marcatore della forma diffusa”

•Radiologia tradizionale: aumento reticolare della trama polmonare (fibrosi) soprattutto alle basi, aumento dell’ombra cardiaca.
Microcalcificazioni sottocutanee, osteolisi delle falangi distali da necrosi ischemica.
•TC ad alta risoluzione: individuazione di lesioni polmonari precoci quali l’alveolite (aspetto a “vetro smerigliato”) che precede la fibrosi (aspetto a “nido d’ape”)
•Spirometria: insufficienza ventilatoria di tipo restrittivo, riduzione DLCO
Esami StrumentaliEsami Strumentali
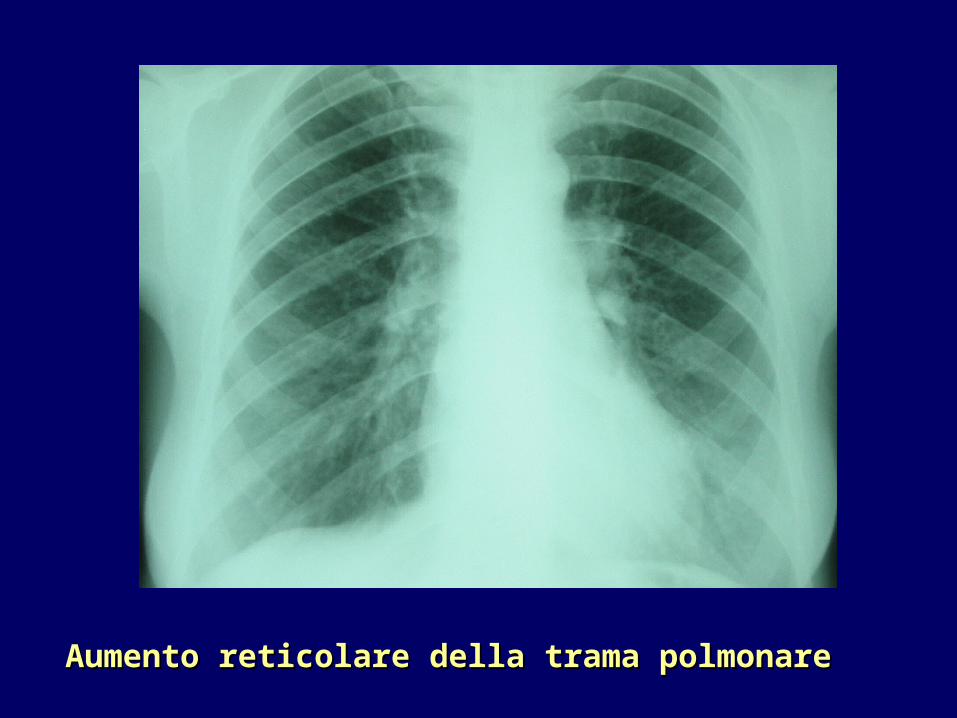
Aumento reticolare della trama polmonareAumento reticolare della trama polmonare
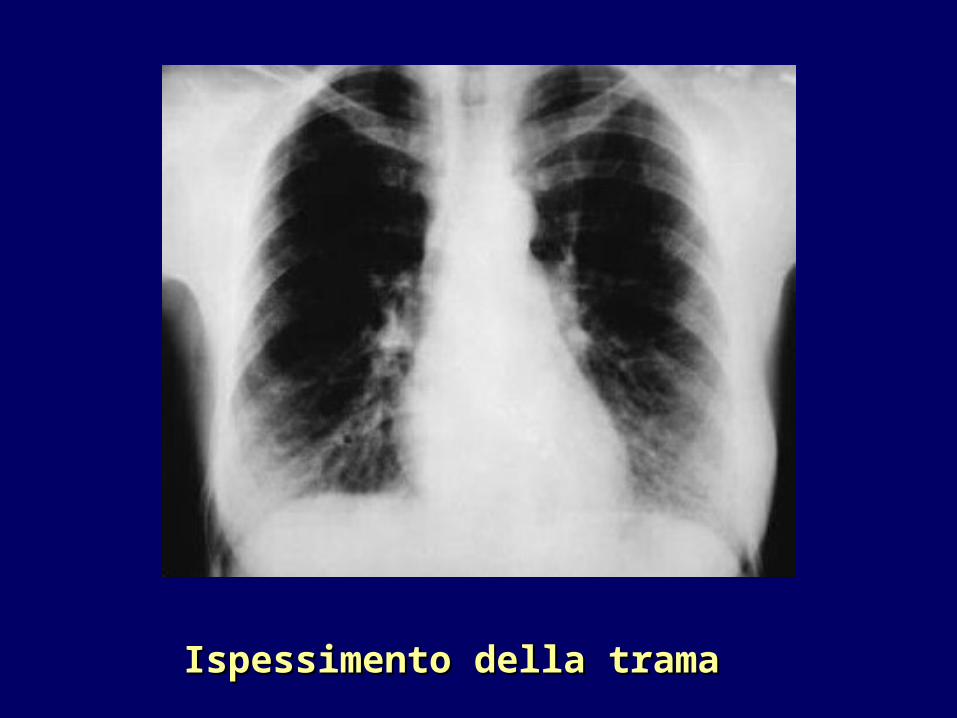
Ispessimento della trama Ispessimento della trama
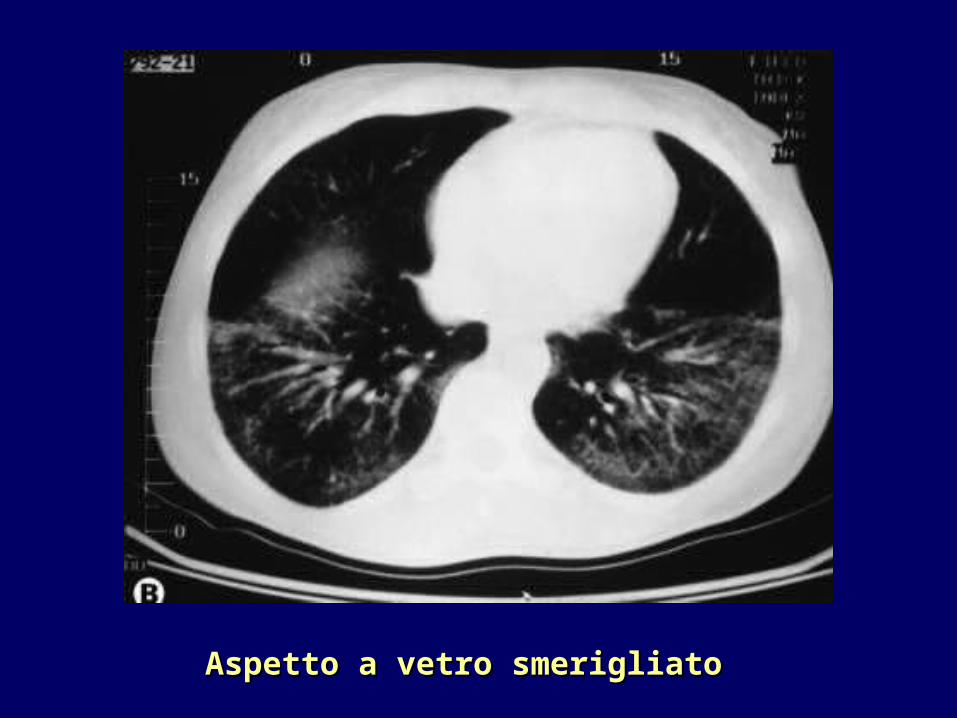
Aspetto a vetro smerigliatoAspetto a vetro smerigliato
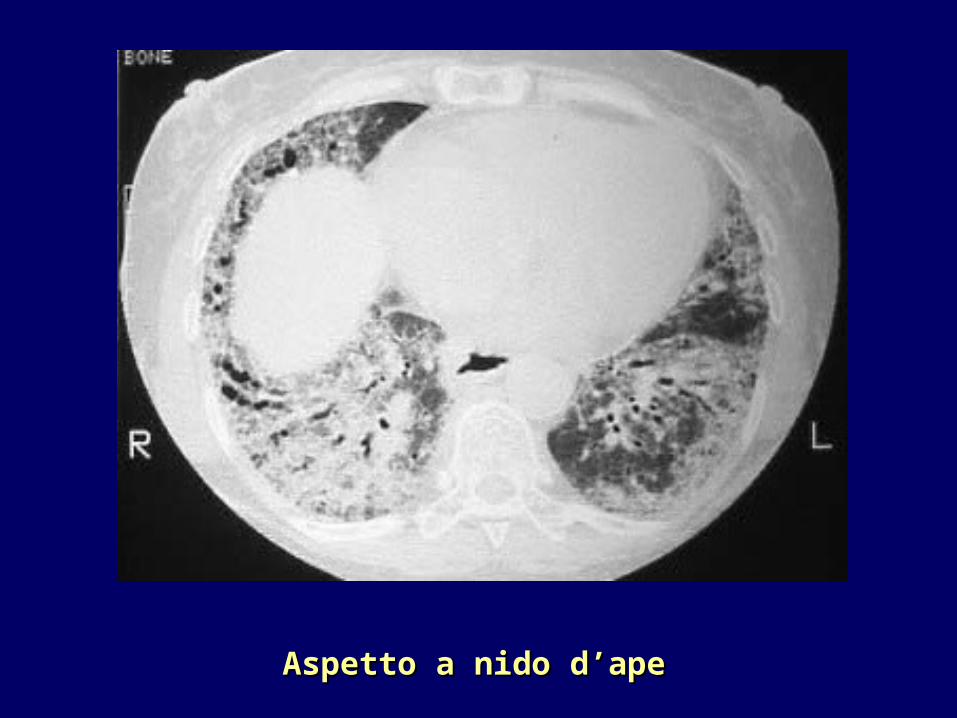
Aspetto a nido d’apeAspetto a nido d’ape

•ECG ed Ecocardio: valutazione di turbe di conduzione e aritmie; morfologia e cinetica cardiaca, pericardio, valvole e valutazione PAP
•Manometria e scintigrafia esofagea: studio della compromissione della peristalsi a livello esofageo
•Capillaroscopia: dilatazione delle anse con formazione di megacapillari tortuosi, distruzione dei capillari, aree avascolarizzate
Esami StrumentaliEsami Strumentali

Diagnosi differenzialeDiagnosi differenziale
• Sindromi similsclerodermicheSindromi similsclerodermiche• Scleredema di BuschkeScleredema di Buschke• ScleromixedemaScleromixedema• Reazione del trapianto verso l’ospiteReazione del trapianto verso l’ospite• AmiloidosiAmiloidosi• Porfiria cutanea tardaPorfiria cutanea tarda• Fascite eosinofila (sindr. di Shulman)Fascite eosinofila (sindr. di Shulman)• Sindrome eosinofilia-mialgiaSindrome eosinofilia-mialgia• Sindrome di WernerSindrome di Werner• Lichen scleroatroficoLichen scleroatrofico

Polimiosite /DermatomiositePolimiosite /Dermatomiosite
Gruppo eterogeneo di malattie muscolari acquisite caratterizzate da un processo infiammatorio a carico della muscolatura scheletrica

• Polimiosite (PM)• Dermatomiosite (DM)• Dermatomiosite dell’infanzia• Miosite nelle neoplasie• Miosite associata ad altre connettiviti • Miosite da corpi inclusi (MCI)• Dermatomiosite amiopatica
Classificazione

ASSOCIAZIONE CON ALTRE MALATTIE
1) Connettiviti (SSc, LES, MCTD, SS, AR)
2) Malattie autoimmuni (m. di Crohn,sarcoidosi, celiachia, miastenia, trombocitopenia
autoimmune, Hashimoto etc)
3) Neoplasie:- PM: polmone, mammella, linfoma, prostata- DM:ovaio, stomaco, mammella, La neoplasia può precedere, seguire o essere contemporanea alla miositeL’associazione è marcata soprattutto per la DMUno screening per neoplasia è fondamentale in tutti i pazienti

• Malattia ubiquitaria ma più frequente nella razza nera
• rapporto F/M variabile
• Età: PM >18 aa DM: tutte le età MCI>50 aa
• incidenza: 1-12 casi/anno/milione
• prevalenza: 4 casi/100 000
Epidemiologia

• Ipostenia o Astenia muscolare, simmetrica, a carico dei muscoli dei cingoli scapolare e pelvico, tronco e collo (interessamento dei muscoli distali nei casi gravi )
• Deficit muscolare:– salire le scale– alzarsi dalla sedia e dal letto– accovacciarsi– incrociare le gambe– deambulare– sollevare il capo dal cuscino– deglutire– voce nasale (incapacità di sollevare il palato, m. cricofaringeo)– disfagia (mm faringe 10-15% casi)
• Dolore spontaneo o provocato a carico delle masse muscolari
Manifestazioni muscolo-scheletriche
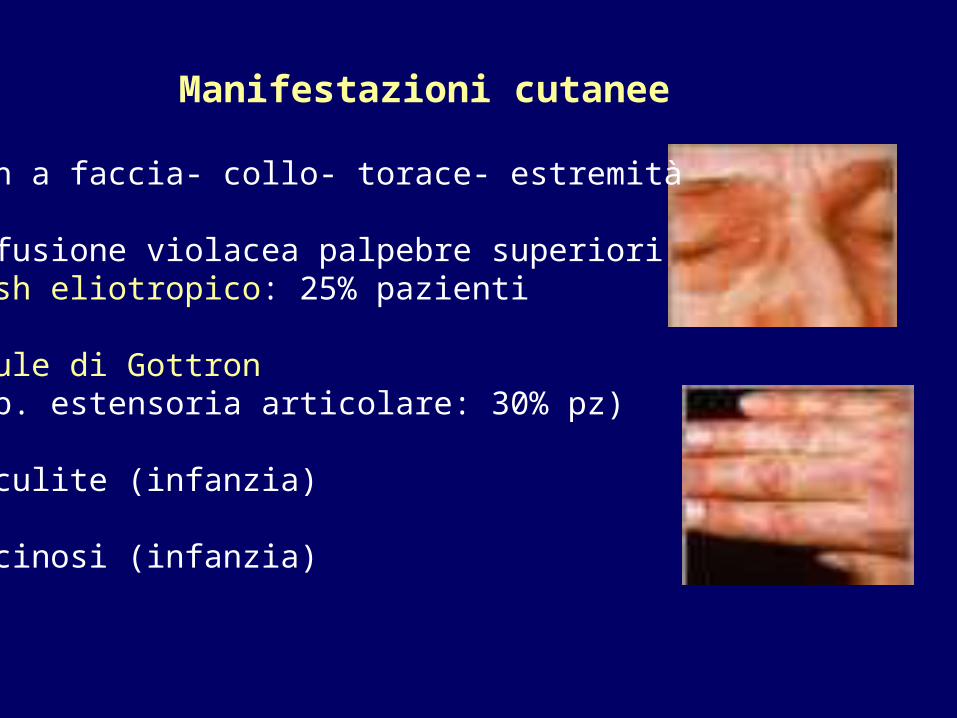
- Rash a faccia- collo- torace- estremità
- Soffusione violacea palpebre superiori Rash eliotropico: 25% pazienti
- Papule di Gottron (sup. estensoria articolare: 30% pz)
- Vasculite (infanzia)
- Calcinosi (infanzia)
Manifestazioni cutanee

• Manifestazioni polmonari:– ipoventilazione– polmonite ab ingestis (difetti della deglutizione)– interstiziopatia polmonare (Jo-1)– fibrosi polmonare 5-10%
• Manifestazioni cardiache:– miocardite con insufficienza congestizia– disturbi della conduzione AV– prolasso della mitrale
• Manifestazioni renali:– rare (solo da persistente mioglobinuria)
• Manifestazioni articolari:– Artralgie (artrite rara)

• Enzimi muscolari aumentati:CPK, LDH, aldolasi, SGOT– Isoenzimi CPK: CPK- MM (muscolo scheletrico) CPK- BB (encefalo e m.liscio)– CPK-MB (miocardio)
• Mioglobina– proteina respiratoria della cellula muscolare– aumentata nel 70-80% delle PM– la mioglobinuria è più rara
• VES elevata
Laboratorio

SINDROME DA ANTICORPI ANTI-Jo1
F > M Miopatia prossimale Raynaud: 60% Vasculite leucocitoclastica Artrite non erosiva delle mani Fibrosi interstiziale polmonare ANA positivo Positività Anti-Jo-1 (Istidil t-RNA sintetasi)

ELETTROMIOGRAFIA • Positiva nel 70-90% dei pz• La contrattura volontaria del muscolo produce:
– attività di breve durata– bassa ampiezza dei potenziali– potenziali polifasici
• A riposo:– potenziali di fibrillazione
BIOPSIA MUSCOLARE• utile come conferma• la sua negatività non esclude la diagnosi
Indagini diagnostiche

DIAGNOSI DIFFERENZIALE
• Distrofie muscolari
• Miopatia da danno neurogeno
• Miopatia iatrogena (corticosteroidi)
• Miopatie endocrine
• Polimialgia reumatica
• Miosite infettiva

PROGNOSI
• Malattia severa
• Fatale in una minoranza di casi
• Andamento cronico nel 30-40% dei casi
• Possibili recidive
• Più benigna mediamente nell’infanzia
• Prognosi più grave nell’adulto >60 anni

MALATTIA DI SJOGRENMalattia infiammatoria cronica, sistemica, eziologia multifattoriale
patogenesi autoimmune
• Forma primitiva e forma secondaria Overlap con altre connettiviti• Interessa prevalentemente le ghiandole esocrine (salivari, lacrimali, vaginali)• Solitamente decorso lento• Rapporto M/F di 1/9• Insorgenza tra IV–V decade di vita• Prevalenza da 6 a 27 /100.000 abitanti

Impegno Oculare• Spesso subclinico
• Cheratocongiuntivite secca (manifestazione tipica che comporta distruzione dell’epitelio congiuntivale)
• Le forme non trattate possono comportare gravi complicanze(congiuntivite secondaria, ulcerazione corneale e perforazione)
• La perforazione corneale può causare uveite, cataratta e glaucoma.
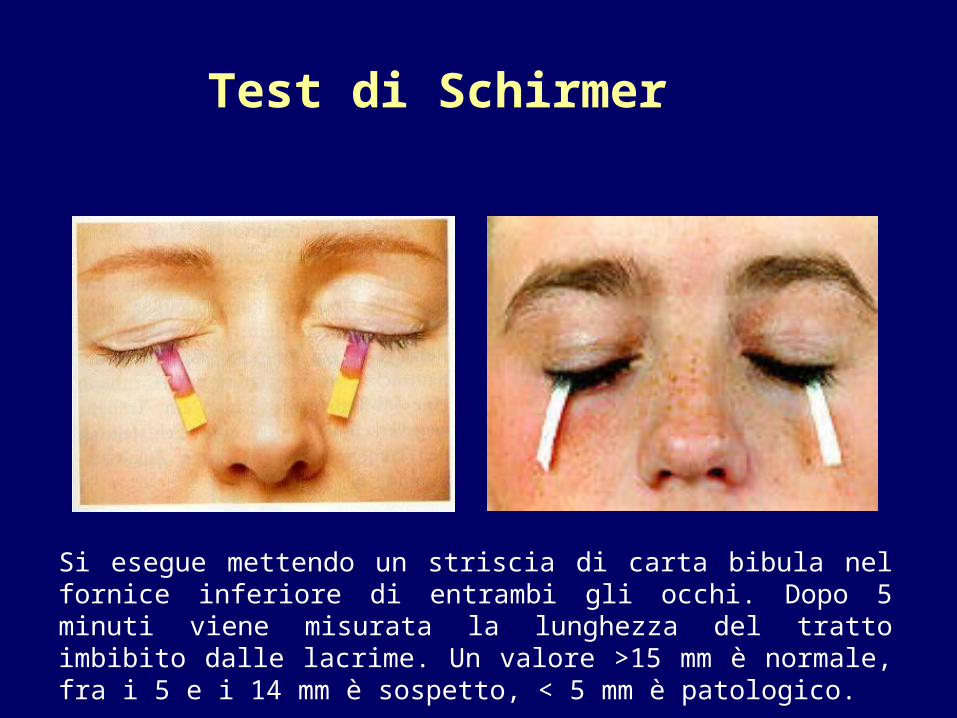
Test di Schirmer
Si esegue mettendo un striscia di carta bibula nel fornice inferiore di entrambi gli occhi. Dopo 5 minuti viene misurata la lunghezza del tratto imbibito dalle lacrime. Un valore >15 mm è normale, fra i 5 e i 14 mm è sospetto, < 5 mm è patologico.
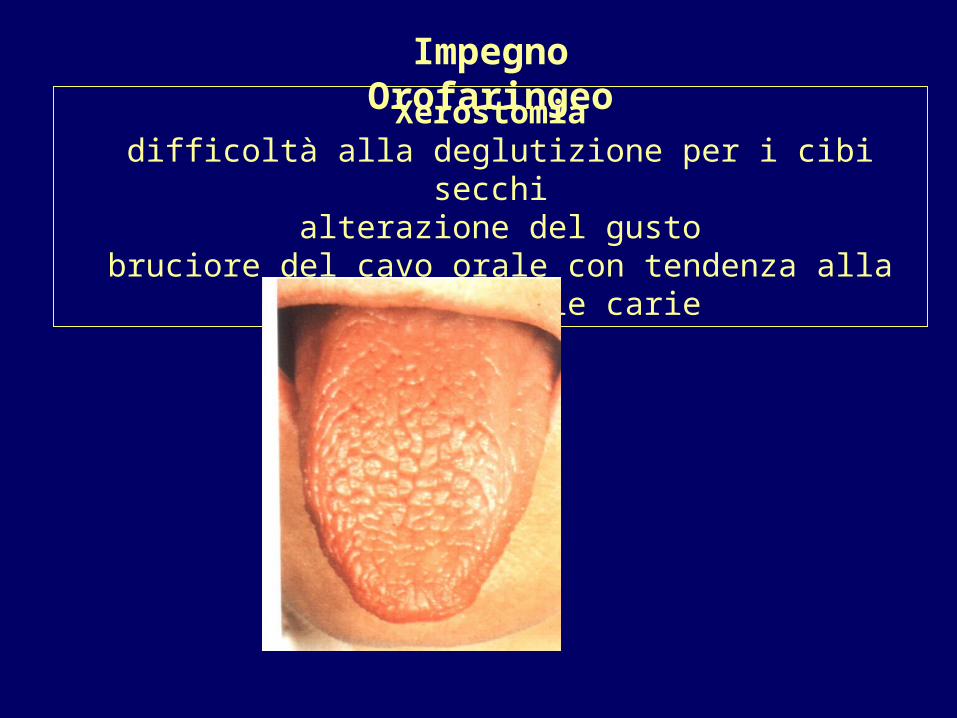
Xerostomia difficoltà alla deglutizione per i cibi secchi
alterazione del gusto bruciore del cavo orale con tendenza alla candidiasi e alle carie
Impegno Orofaringeo

Tumefazione parotidea
Tumefazione ghiandole salivari maggiori spesso bilaterale
presente nel 60% dei casi, episodica,può anche diventare persistente.

Valutazione dell’impegno orofaringeo
• Biopsia delle ghiandole salivari accessorie: valuta la presenza di infiltrato infiammatorio
• Scialografia: valuta i cambiamenti dei dotti delle ghiandole salivari.
• Scintigrafia parotidea: fornisce una valutazione funzionale delle ghiandole salivari attraverso la valutazione dell’uptake di tecnezio. Nei pazienti con SS tale uptake è ritardato o assente.

Impegno articolare
Impegno renale
Impegno polmonare
Impegno gastrointestinale
Gastrite atrofica pancreatite sub clinica
Cirrosi biliarecolangite sclerosante
Impegno del SNC

rischio di sviluppare linfomi risulta essere di 44 volte superiore rispetto alla popolazione generale.
La proliferazione dei linfociti B determina spesso gammopatie monoclonali o macroglobulinemia di
Waldenstrom
Malattie LinfoproliferativeMalattie Linfoproliferative

ANOMALIE SIEROLOGICHE
• Emocromo: anemia, leucopenia, linfopenia.• Indici di flogosi: Ves, PCR, ipergammaglobulinemia, fibrinogeno• Amilasemia: frequentemente aumentata.
• ANA: 95-100%• Anti RO-SSA: 50-90%• Anti LA-SSB: 50-90%• FR: 90%

CONNETTIVITE MISTA (MCTD)
• Sindrome overlap che unisce le manifestazioni del LES, sclerodermia, polimiosite ed è caratterizzata dalla presenza di anticorpi anti-U1-RNP
• Colpisce tipicamente il sesso femminile con un rapporto M:F = 1:9

CRITERI DIAGNOSTICI
1. Sierologici: positività di anti-RNP ad alto titolo
2. Clinici:• Edema delle mani• Sinovite• Miosite• Fenomeno di Raynaud• Acrosclerosi
Diagnosi= criteri sierologici + almeno 3 clinici

VASCULITI
Malattie eterogenee, sistemiche caratterizzate da flogosi o necrosi della parete dei vasi sanguigni
(capillari, arteriole e venule)
La loro espressione clinica è il risultato:
ischemia dei tessuti irrorati dai vasi sanguigni infiammati
sintomi secondari al processo infiammatorio sistemico
presente

PATOGENESI
• Vasculiti da immunità cellulo-mediata
• Vasculiti da immunocomplessi circolanti (CIC)
• Vasculiti ANCA associate
Nell’ambito della stessa forma di vasculite possono essere coinvolti meccanismi patogenetici diversi come nella Granulomatosi di Wegener
immunità cellulo-mediata (granuloma) + ANCA

• Vasculiti da immunità cellulo mediata: Vasculiti da immunità cellulo mediata: Arterite a cellule giganti, Arterite a cellule giganti, Arterite di Takayasu, Granulomatosi di WegenerArterite di Takayasu, Granulomatosi di Wegener
• Vasculiti da immunocomplessi: Vasculiti da immunocomplessi: Crioglobulinemia Mista, Crioglobulinemia Mista, Panarterite Nodosa, Vasculiti da ipersensibiltà, Overlap con altre Panarterite Nodosa, Vasculiti da ipersensibiltà, Overlap con altre connettiviti.connettiviti.
• Vasculiti Anca associate: Vasculiti Anca associate: Churg Strauss-G.di Wegener Churg Strauss-G.di Wegener
PatogenesiPatogenesi

VASCULITI ANCA ASSOCIATE
Vasculiti Sistemiche associate alla presenza in circolo di anticorpi diretti contro antigeni citoplasmatici dei neutrofili (ANCA)
•P Anca: P Anca: Anticorpi anti MieloperossidasiAnticorpi anti Mieloperossidasi
• C Anca:C Anca: Anticorpi Antiperossidasi 3 (PR3 )Anticorpi Antiperossidasi 3 (PR3 )
Sono un’entità clinico patologica caratterizzata da manifestazioni sistemiche che coinvolgono diversi organi e apparati
i
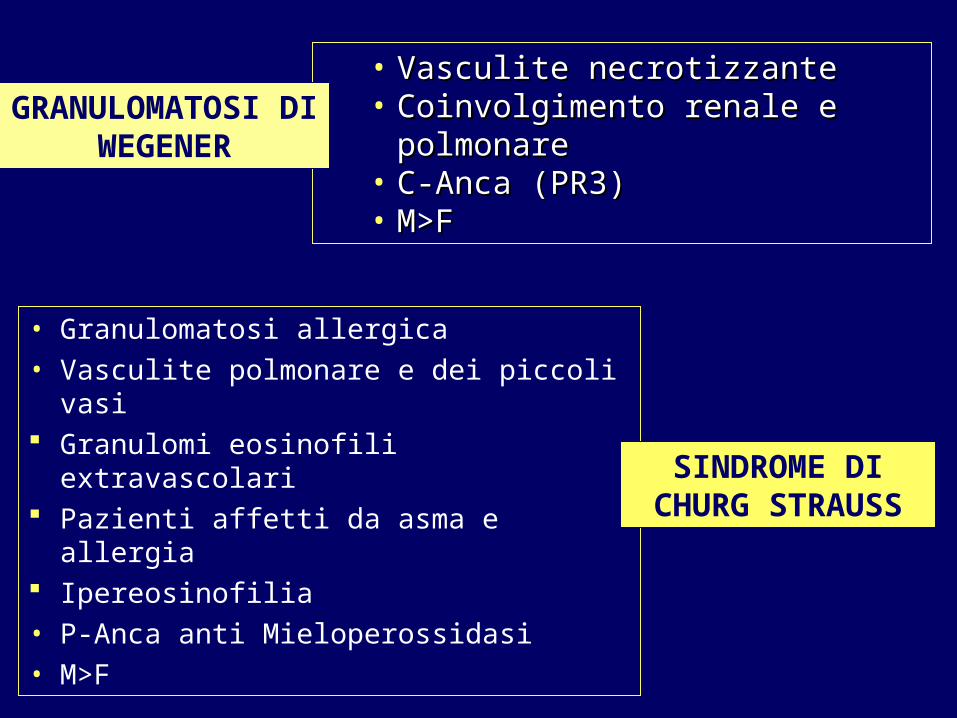
• Vasculite necrotizzanteVasculite necrotizzante• Coinvolgimento renale e polmonareCoinvolgimento renale e polmonare• C-Anca (PR3)C-Anca (PR3)• M>F M>F
• Granulomatosi allergica• Vasculite polmonare e dei piccoli vasi Granulomi eosinofili extravascolari Pazienti affetti da asma e allergia Ipereosinofilia• P-Anca anti Mieloperossidasi• M>F
GRANULOMATOSI DI WEGENER
SINDROME DI CHURG STRAUSS

CONCLUSIONI
L’esperienza di questi ultimi anni ha evidenziato che uno degli elementi fondamentali per la sopravvivenza dei pazienti affetti da Connettiviti Sistemiche è rappresentato dalla tempestività diagnostica .
E la molteplicità dei fattori eziologici e dei fattori patogenetici E la molteplicità dei fattori eziologici e dei fattori patogenetici rappresenta il presupposto fondamentale che impone unarappresenta il presupposto fondamentale che impone una
Appropiatezza terapeutica interdisciplinareAppropiatezza terapeutica interdisciplinare





























