Embed Size (px)
Citation preview

MOLECULAR AND CELLULAR BIOLOGY, Nov. 2009, p. 5696–5709 Vol. 29, No. 210270-7306/09/$12.00 doi:10.1128/MCB.00191-09Copyright © 2009, American Society for Microbiology. All Rights Reserved.
Protein Phosphatase 2A-Dependent Dephosphorylation of ReplicationProtein A Is Required for the Repair of DNA Breaks Induced by
Replication Stress�†Junjie Feng,1 Timothy Wakeman,1 Sheila Yong,1 Xiaohua Wu,2
Sally Kornbluth,1 and Xiao-Fan Wang1*Department of Pharmacology and Cancer Biology, Duke University Medical Center, Durham, North Carolina 27710,1 and
Department of Molecular and Experimental Medicine, The Scripps Research Institute, La Jolla, California 920372
Received 10 February 2009/Returned for modification 13 March 2009/Accepted 14 August 2009
Eukaryotic genomic integrity is safeguarded by cell cycle checkpoints and DNA repair pathways, collectivelyknown as the DNA damage response, wherein replication protein A (RPA) is a key regulator playing multiplecritical roles. The genotoxic insult-induced phosphorylation of the 32-kDa subunit of human RPA (RPA32),most notably the ATM/ATR-dependent phosphorylation at T21 and S33, acts to suppress DNA replication andrecruit other checkpoint/repair proteins to the DNA lesions. It is not clear, however, how the DNA damage-responsive function of phosphorylated RPA is attenuated and how the replication-associated activity of theunphosphorylated form of RPA is restored when cells start to resume the normal cell cycle. We report here thatin cells recovering from hydroxyurea (HU)-induced genotoxic stress, RPA32 is dephosphorylated by theserine/threonine protein phosphatase 2A (PP2A). Interference with PP2A catalytic activity causes persistentRPA32 phosphorylation and increased HU sensitivity. The PP2A catalytic subunit binds to RPA following DNAdamage and can dephosphorylate RPA32 in vitro. Cells expressing a RPA32 persistent phosphorylationmimetic exhibit normal checkpoint activation and reenter the cell cycle normally after recovery but display apronounced defect in the repair of DNA breaks. These data indicate that PP2A-mediated RPA32 dephosphor-ylation is required for the efficient DNA damage repair.
The genomes of all living cells are under constant attack byexogenous DNA-damaging agents, as well as the intracellularby-products of normal metabolism. To cope with this chal-lenge, eukaryotic cells have evolved an elaborate surveillanceand maintenance system termed the DNA damage response,which is composed of a set of signal transduction and executionpathways that can detect DNA lesions, delay cell cycle pro-gression, facilitate repair processes, and induce apoptosis orsenescence if the level of DNA damage is beyond repair (re-viewed in references 18, 40, and 45).
The extensive phosphorylation of many checkpoint andDNA repair proteins by two phosphatidylinositol 3-kinase-re-lated kinases (PIKKs) ATM and ATR, along with their respec-tive preferred downstream kinases Chk2 and Chk1, appears toplay a major theme in the transduction and execution of theDNA damage response. Once these kinases are stimulated,they phosphorylate and activate important regulators such asRad17, Nbs1, BRCA1, H2AX, the 32-kDa subunit of replica-tion protein A (RPA32), Cdc25, and p53 to facilitate assemblyof DNA repair centers (foci) at the sites of DNA damage orcause alteration of their enzymatic or transcriptional activitiesleading to cell cycle arrest, apoptosis, or senescence (reviewedin references 1 and 26). Despite their overall similarity, these
two pivotal pathways differ in the types of DNA damage towhich they respond. While the ATM-Chk2 pathway respondsprimarily to DNA-damaging reagents that induce DNA dou-ble-stranded breaks (DSBs), the ATR-Chk1 pathway plays apredominant role in the cellular responses to UV and hy-droxyurea (HU), which induce base damage or replication forkstalling (27, 43).
Although much is known about the role of protein phosphor-ylation in the DNA damage response, there is relatively littleknowledge available concerning the function of protein de-phosphorylation in this process. Recently, mounting evidencehas prompted an emerging view that dephosphorylation ofthese phosphorylated checkpoint and repair proteins may alsoserve an important role in the DNA damage response, possiblyby allowing cells to recover from checkpoint arrest or by facil-itating the repair of DNA damage (reviewed in references 18and 34). In Schizosaccharomyces pombe and Saccharomycescerevisiae, recovery from checkpoint arrest following repair ofDNA lesions may require dephosphorylation and inactivationof Chk1 or Rad53 (a yeast Chk2 orthologue), which is medi-ated by Dis2 (S. pombe), a member of the protein phosphatase1 (PP1) phosphatase family, or Ptc2 and Ptc3 (the Wip1/PPM1D homologues) (S. cerevisiae), members of the PP2Cfamily of phosphatases (14, 28). In humans, it appears thatreentry into the cell cycle after the DNA damage response maydepend heavily on Wip1/PPM1D, which can reportedly de-phosphorylate Chk1, Chk2, and p53 (15, 31). In addition,BRCA1-dependent DNA DSB repair requires PP1-dependentdephosphorylation of BRCA1 (21, 57). Removal of �-H2AX,the phosphorylated histone H2AX generated at the site ofDNA DSBs whose function is to stabilize DNA repair foci, is
* Corresponding author. Mailing address: Department of Pharma-cology and Cancer Biology, Duke University Medical Center, C218LSRC Bldg., La Salle Street Extension, Durham, NC 27710. Phone:(919) 681-4861. Fax: (919) 681-7152. E-mail: [email protected].
† Supplemental material for this article may be found at http://mcb.asm.org/.
� Published ahead of print on 24 August 2009.
5696
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.

mediated by PP2A or PP4 and functions to facilitate repair ofDNA DSBs and allows subsequent resumption of DNA repli-cation (9, 10).
Replication protein A (RPA) is a heterotrimeric proteincomplex composed of the 70-kDa, 32-kDa, and 14-kDa sub-units (hereafter referred to RPA70, RPA32, and RPA14,respectively). RPA is a major single-stranded DNA(ssDNA)-binding protein in eukaryotic cells and is essentialfor all types of DNA metabolism, including DNA replica-tion, DNA recombination, and DNA damage repair (53).Besides its protective role in covering the ssDNA exposed byDNA damage, RPA has also been shown to be critical inactivation of the ATR pathway, likely by mediating therecruitment of ATR to the sites of DNA damage throughthe interaction between ATRIP and the RPA-ssDNA com-plexes (60). In addition, translocation of other importantcheckpoint/repair protein complexes, such as Rad17–Rfc2-5, Rad9-Hus1-Rad1, and Rad51/Rad52, to the sites ofDNA lesions may also rely on RPA (16, 38, 54, 61).
RPA is regulated through genotoxic stress-induced phos-phorylation. Upon DNA damage or replication stress, theSQ/TQ motif-containing threonine 21 (T21) and serine 33(S33) of the RPA32 subunit are phosphorylated in a processmediated by PIKKs ATM, ATR, and DNA-dependent proteinkinase (DNA-PK). In addition, phosphorylation of RPA32 isalso seen on at least five other serine residues (S4, S8, S11, S12,and S13), but the identities of the responsible kinases remainto be determined (reviewed in references 6 and 62). It has beendemonstrated that RPA32 phosphorylation plays an importantrole in the DNA damage response. Phosphorylation of RPA32stimulates genotoxic stress-induced interaction with two criti-cal checkpoint/repair complexes, Mre11-Rad50-Nbs1 andRad9-Hus1-Rad1 and may also promote the recruitment of theDSB repair proteins Rad51/Rad52 to sites of DNA damage(39, 54, 55). While phosphorylated RPA is competent to trans-locate to locations of DNA lesions, it is unable to associatewith replication centers and thus may function to mediate theS-phase checkpoint by suppressing DNA replication directly(36, 37, 48). The inability of phosphorylated RPA to supportDNA replication may be due to its altered duplex DNA bind-ing/denaturation ability and decreased interaction with DNApolymerase � (Pol �) (5, 35, 37). It is noteworthy that duringthis process, ATM/ATR-dependent phosphorylation of RPAat T21 and S33 is critical, whereas phosphorylation at othersites appears to be dispensable, indicating that distinct RPAfunctions may be differentially regulated by phosphorylation atdifferent sites (36).
Given the essential roles that unphosphorylated RPA playsin unperturbed cell growth and division, it is conceivable thatDNA damage-induced RPA32 phosphorylation needs to beattenuated when cells are recovering. Here we report that theATM/ATR-dependent phosphorylation of RPA32 at T21 andS33 is reversed by PP2A-mediated dephosphorylation. Inter-ference with PP2A activity causes persistent RPA32 phosphor-ylation and increased sensitivity to a replication stress inducerHU. The PP2A catalytic subunit associates with and dephos-phorylates RPA32 following HU-induced stress. Through amodel that mimics persistent phosphorylation of RPA32, weshow that cells substituted with T21 and S33 (T21/S33)-phos-phomimetic RPA32 exhibit increased HU/UV sensitivity but
nonetheless possess normal checkpoint activation and indistin-guishable resumption of DNA replication and progressionthrough mitosis after HU release compared with wild-type(WT) control cells. Further investigation demonstrates thatfollowing release from HU treatment, cells with mutantRPA32 display persistent DNA damage foci containing RPAand �-H2AX and exhibit a pronounced defect in the repair ofHU-induced DNA breaks, suggesting that PP2A-mediatedRPA32 dephosphorylation is required for the efficient repair ofDNA lesions.
MATERIALS AND METHODS
Plasmids and mutagenesis. C-terminally Flag-tagged RPA32 was generated bysubcloning into the pcDNA3 vector a PCR product containing the RPA32 codingsequence from p3a-RPA32, a generous gift from Marc Wold (University ofIowa). The pcDNA3-RPA32-Flag was further used as template to introduce theT21V and S33A (T21V/S33A) and T21D and S33D (T21D/S33D) substitutionsafter several rounds of mutagenesis following the QuikChange site-directedmutagenesis protocol (Stratagene). The mutagenesis primers are shown in TableS1 in the supplemental material. The Flag-tagged RPA32 variants were subse-quently cloned into pQCXIP (Clontech) vector for generating retroviruses. Theretroviral RPA32 short hairpin RNA expression vector was described previously(36). The expression constructs of the Flag-tagged PP1, PP2A, PP4, and PP6catalytic subunits were generously provided by Xin-Hua Feng (Baylor College ofMedicine). The Flag-tagged PP5 expression construct was described previously(2).
Cell culture and retroviral infection. HeLa and A549 cells were cultured inDulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine se-rum. U2OS cells were maintained in McCoy’s 5A medium with 10% fetal bovineserum The culture of HaCaT and HepG2 cells has been described previously(49). Various Flag-tagged RPA32 variants were introduced into HeLa cells byretroviral infection, followed by selection of puromycin-resistant cells. The sub-stitution lineages were further generated by silencing the endogenous RPA32 inthese cell lines that ectopically express each of the mutant RPA32 proteins,which was accomplished by two rounds of infection with retroviruses expressingthe RPA32 short hairpin RNA target sequence, followed by G418 selection. Theretroviral infection protocol was described previously (17).
Antibodies and reagents. The antibodies used for immunoblotting were pur-chased from Abcam (rabbit polyclonal RPA32pT21 and PP4), BD Biosciences(rabbit polyclonal PP5), Bethyl Laboratories (rabbit polyclonal RPA32pS33,RPA32pS4/8, and Rad17pS645), Calbiochem (mouse monoclonal RPA32 andRPA70), Cell Signaling (rabbit polyclonal �-H2AX and Chk1 pSer317 and rabbitmonoclonal Chk1 pSer345 and p21 Waf1/Cip1), Millipore Chemicon (rabbitpolyclonal PP6), Upstate (rabbit polyclonal PP1 catalytic subunit and mousemonoclonal PP2A/C�), Santa Cruz (mouse monoclonal p53), and Sigma-Aldrich(mouse monoclonal Flag and �-tubulin). The reagents used in this study includeHU, okadaic acid (OA), DNase, and caffeine (all from Sigma-Aldrich) andMG132 (BioMol).
Immunoblotting and immunoprecipitation. For immunoblotting, cells werelysed in NETN (20 mM Tris-Cl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 0.5%Nonidet P-40) supplemented with protease inhibitors (20 �g/ml leupeptin, 10�g/ml pepstatin A, and 10 �g/ml aprotonin) and phosphatase inhibitors (20 mM�-glycerophosphate and 0.5 �M OA). Cell lysates were subsequently resolved bysodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblottedwith appropriate antibodies. For examination of the interaction between PP2A/Cand RPA32, HeLa cells were harvested with the previously described PP2Acoimmunoprecipitation buffer (3) (except that it contained 0.5% Nonidet P-40)containing protease and phosphatase inhibitors. Lysates were precleared andincubated with the anti-RPA32 antibody and protein A beads (Calbiochem).After extensive washing with lysis buffer, the immunoprecipitates were analyzedby immunoblotting with anti-RPA70 and anti-PP2A/C antibodies. The controlmouse immunoglobulin G was from Santa Cruz.
Immunofluorescence microscopy. Cells plated on glass coverslips were treatedwith various conditions before immunofluorescence analysis. In brief, cells werefixed and permeabilized with acetone-methanol (1:1) at �20°C for 10 min. Afterrehydration in phosphate-buffered saline (PBS) and incubation with blockingbuffer (3% bovine serum albumin [BSA] in PBS containing 0.1% Triton X-100)for 1 h, the cells were stained with primary antibodies in blocking buffer for 1 hat room temperature or overnight at 4°C, followed by three PBS washes andincubation with the appropriate secondary antibodies (Invitrogen) in blocking
VOL. 29, 2009 EFFICIENT DNA REPAIR REQUIRES RPA32 DEPHOSPHORYLATION 5697
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.

buffer for 2 h at room temperature. After the cells were washed with PBS, theywere mounted with fluorescence mounting medium containing 4�,6�-diamidino-2-phenylindole (DAPI) (Santa Cruz) and analyzed by fluorescence microscopy.For detection of incorporated bromodeoxyuridine (BrdU), cells were incubatedin medium containing 10 �M BrdU (BD Pharmingen) for 10 min prior to fixingand the DNA was denatured with 0.1 M HCl using standard procedures. Theprimary antibodies used were RPA32 and RPA70 (mouse monoclonal antibod-ies; Calbiochem), RPA32pS33 (rabbit polyclonal antibody; Bethyl), �-H2AX(rabbit polyclonal antibodies [Cell Signaling] and mouse monoclonal antibody[Upstate]), PP2A/C (rabbit polyclonal antibody; Santa Cruz), and anti-BrdU (ratmonoclonal antibody; Abcam). Experiments were performed three times, andcells detected with five or more discrete foci were regarded as focus positive.
Silencing of protein phosphatases. To knock down OA-sensitive protein phos-phatases (PP1, PP2A, PP4, PP5, and PP6), approximately 2 � 105 HeLa cellswere seeded per well in six-well plates. The next day, cells were transfected withsmall interfering RNA (siRNA) oligonucleotides against the catalytic subunit ofthe specific phosphatases (Santa Cruz) using Lipofectamine RNAiMAX reagent(Invitrogen) according to the manufacturer’s instructions. Cells were treated withthe indicated conditions and analyzed 48 to 72 h posttransfection.
In vitro phosphatase assay. HeLa cells were treated with 10 mM of HU for6 h, and the phosphorylated RPA32 substrate was prepared by immunoprecipi-tation using anti-RPA32 antibody and protein G beads. The phosphatase reac-tions were carried out in dephosphorylation buffer (20 mM HEPES [pH 7.0], 1mM dithiothreitol, 1 mM MnCl2, 100 mg/ml BSA, and 50 mM leupeptin) withpurified PP2A enzyme (Upstate) at 30°C for 30 min in the presence of differentconcentrations of OA.
Clonogenic survival assays. HeLa cells were plated at 500 cells per dish in60-mm dishes and exposed to pulses of the indicated doses of HU or UV 12 hpostseeding. After treatment, cells were rinsed twice with PBS and allowed torecover in drug-free medium. The cultures were then incubated for 10 to 14 dayswith the medium being changed every 3 days. Colonies were stained with crystalviolet (Sigma), counted, and normalized to untreated control. All survivingpoints were done in triplicate, and only colonies containing 50 or more cells werescored.
DNA synthesis assays. The DNA synthesis assays were performed as describedpreviously (11) to evaluate the UV-induced intra-S-phase checkpoint and theresumption of DNA synthesis following release from HU block. Specifically, cellswere incubated in medium containing 20 nCi/ml of [14C]thymidine (NEN) for24 h prior to UV treatment or exposure to pulses of HU. At the indicated timepoints after UV irradiation or release from HU block, cells were pulse labeledwith [3H]thymidine (2.5 �Ci/ml, 30 min; NEN) and harvested. The radioactivitywas determined by liquid scintillation counting, and the relative DNA synthesisrate was calculated by determining the ratio of 3H to 14C and normalization ofthe treated samples to the appropriate controls.
Cell cycle analysis. Cells pulse exposed to HU were harvested at variousrecovery time points. After fixation in 70% ethanol, cells were incubated withRNase A (100 �g/ml; Invitrogen) and propidium iodide (50 �g/ml; Sigma) for 30min at 37°C. The DNA content was then determined with a FACScan flowcytometer (BD Biosciences), and the cell cycle distributions were analyzed byCellQuest software.
Mitotic index assays. The mitotic index assays were carried out to examine theUV-induced G2/M checkpoint and the progression of mitosis following releasefrom HU block. Specifically, cells were irradiated with UV or pulse treated withHU, and at the indicated time points following treatment, cells were harvestedand fixed in 70% ethanol at �20°C. After permeabilization in 0.25% TritonX-100 in PBS, cells were incubated with anti-phospho-histone H3 antibody(pSer10; Upstate), followed by fluorescein isothiocyanate (FITC)-conjugatedsecondary antibody (Jackson ImmunoResearch). Cells were subsequently coun-terstained with propidium iodide, and the phospho-histone H3 fluorescence andDNA content were determined by flow cytometry. The percentage of mitoticcells was calculated as the mitotic index. For examination of the G2/M check-point, the mitotic index was normalized to that in the unperturbed controls.
Sequential labeling with IdU and CldU. The in vivo labeling of DNA withiododeoxyuridine (IdU) and chlorodeoxyuridine (CldU) was performed as pre-viously described (58). Briefly, cells were grown on slides, fixed with cold meth-anol, stored at 4°C for more than 30 min, and incubated in 1.5 N HCl in 0.5%Triton X-100 for 30 min at room temperature. After the cells were blocked in 3%BSA in PBS, the slides were incubated with rat anti-BrdU antibody BU1/75 (itcan detect CldU but does not bind IdU; AbCam) for 1 h at 37°C and thenincubated for a second time with mouse anti-BrdU monoclonal antibody (BDPharmingen) for 1 h at 37°C. After incubation with appropriate secondary anti-bodies, the slides were subjected to immunofluorescence.
Single-cell gel electrophoresis assay (comet assay). The repair kinetics ofHU-induced DNA breaks was evaluated by the alkaline comet assay according tothe manufacturer’s protocol (Trevigen). Briefly, cells were pulse exposed to HU(0.2 mM, 24 h) and harvested at various recovery time points for single-cell gelelectrophoresis. Nuclei were stained with Sybr green, and comets were visualizedby epifluorescence on a Zeiss microscope. Images were analyzed using the publicdomain software program ImageJ, and comets were evaluated by quantifying thetail moment of 75 cells using the comet-analyzing program CometScore (Tritek).
Senescence assay. Cells were cultured in six-well plates and underwent appro-priate treatments before they were evaluated for �-galactosidase activity accord-ing to the manufacturer’s protocol (Calbiochem). The percentage of �-galacto-sidase-positive cells was determined by bright-field microscopy after 200 cellswere scored for each sample.
Apoptosis assay. The percentage of apoptotic cells in the lethally treated cellswas analyzed by an apoptosis assay kit according to the manufacturer’s manual(Invitrogen). Briefly, cells were collected, washed once with cold PBS, andresuspended in the annexin-binding buffer before they were incubated withFITC, annexin V, and propidium iodide for 15 min at room temperature. Thestained cells were then analyzed by flow cytometry.
RESULTS
RPA32 undergoes dephosphorylation in cells released fromHU block. To investigate whether genotoxic insult-inducedRPA32 phosphorylation is attenuated during the recovery pro-cess, we examined the kinetics of RPA32 phosphorylation inHeLa cervical carcinoma cells released from the genotoxicstress induced by HU block. A potent inhibitor of the ribonu-cleotide reductase, HU causes ribonucleotide depletion andleads to inhibition of DNA replication and subsequent accu-mulation of DSBs as a result of replication fork collapse. Asshown in Fig. 1A, HeLa cells accumulated a high level ofphosphorylated RPA32 immediately before release from HUblock (0.2 mM, 24 h), as indicated by the mobility upshift ofRPA32 protein as well as an increase in the intensity of bandsdetected by the phospho-specific antibodies against RPA32(T21 and S33) in comparison with unperturbed cells. Followingrelease from HU treatment, the level of phosphorylatedRPA32 gradually reduced and dropped to near basal levels at12 h postrecovery, suggesting that RPA32 might undergo de-phosphorylation in this recovery process (Fig. 1A).
It is possible that the observed decrease in the level ofphosphorylated RPA was the result of ubiquitin-dependentproteasomal degradation of the phosphorylated RPA32. Toinvestigate this possibility, the proteasome inhibitor MG132was applied to HeLa cells exposed to pulses of HU. As shownin Fig. 1B, while the presence of MG132 effectively abrogateddegradation of p53, a protein which is continuously targetedfor proteasomal degradation in HeLa cells due to the presenceof human papillomavirus E6 protein (41), the addition ofMG132 did not block the decrease in the phosphorylated formof RPA32 or have any noticeable effect on total RPA32 proteinlevels, suggesting that phospho-RPA32 is not targeted for pro-teasomal degradation. We next tested whether the reduction ofRPA32 phosphorylation at T21 and S33 could be inhibited byokadaic acid, a wide-spectrum inhibitor of serine/threonineprotein phosphatases. As indicated in Fig. 1C, the presence ofas low as 50 nM of OA showed a significant suppression effect,suggesting that dephosphorylation by an OA-sensitive proteinphosphatase may account for the declining RPA32 phosphor-ylation in cells recovering from HU block. However, it is pos-sible that OA exerted its effect not by inhibiting RPA32 phos-phatases but by activating the kinases that phosphorylate
5698 FENG ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.
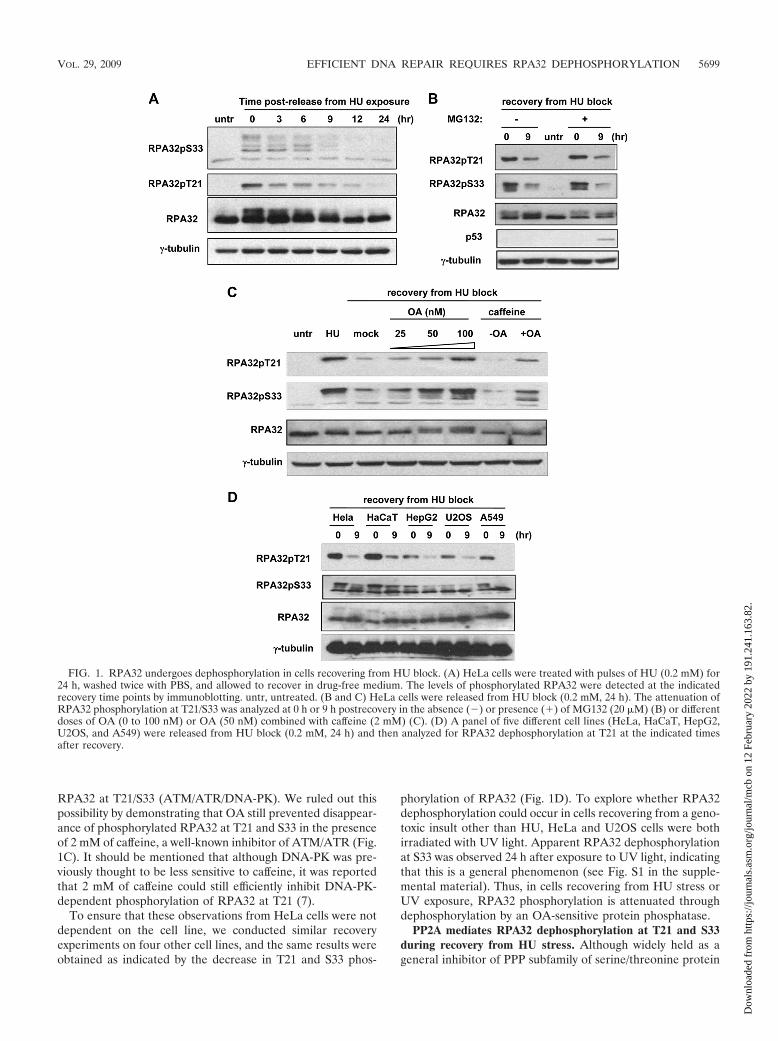
RPA32 at T21/S33 (ATM/ATR/DNA-PK). We ruled out thispossibility by demonstrating that OA still prevented disappear-ance of phosphorylated RPA32 at T21 and S33 in the presenceof 2 mM of caffeine, a well-known inhibitor of ATM/ATR (Fig.1C). It should be mentioned that although DNA-PK was pre-viously thought to be less sensitive to caffeine, it was reportedthat 2 mM of caffeine could still efficiently inhibit DNA-PK-dependent phosphorylation of RPA32 at T21 (7).
To ensure that these observations from HeLa cells were notdependent on the cell line, we conducted similar recoveryexperiments on four other cell lines, and the same results wereobtained as indicated by the decrease in T21 and S33 phos-
phorylation of RPA32 (Fig. 1D). To explore whether RPA32dephosphorylation could occur in cells recovering from a geno-toxic insult other than HU, HeLa and U2OS cells were bothirradiated with UV light. Apparent RPA32 dephosphorylationat S33 was observed 24 h after exposure to UV light, indicatingthat this is a general phenomenon (see Fig. S1 in the supple-mental material). Thus, in cells recovering from HU stress orUV exposure, RPA32 phosphorylation is attenuated throughdephosphorylation by an OA-sensitive protein phosphatase.
PP2A mediates RPA32 dephosphorylation at T21 and S33during recovery from HU stress. Although widely held as ageneral inhibitor of PPP subfamily of serine/threonine protein
FIG. 1. RPA32 undergoes dephosphorylation in cells recovering from HU block. (A) HeLa cells were treated with pulses of HU (0.2 mM) for24 h, washed twice with PBS, and allowed to recover in drug-free medium. The levels of phosphorylated RPA32 were detected at the indicatedrecovery time points by immunoblotting. untr, untreated. (B and C) HeLa cells were released from HU block (0.2 mM, 24 h). The attenuation ofRPA32 phosphorylation at T21/S33 was analyzed at 0 h or 9 h postrecovery in the absence (�) or presence (�) of MG132 (20 �M) (B) or differentdoses of OA (0 to 100 nM) or OA (50 nM) combined with caffeine (2 mM) (C). (D) A panel of five different cell lines (HeLa, HaCaT, HepG2,U2OS, and A549) were released from HU block (0.2 mM, 24 h) and then analyzed for RPA32 dephosphorylation at T21 at the indicated timesafter recovery.
VOL. 29, 2009 EFFICIENT DNA REPAIR REQUIRES RPA32 DEPHOSPHORYLATION 5699
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.
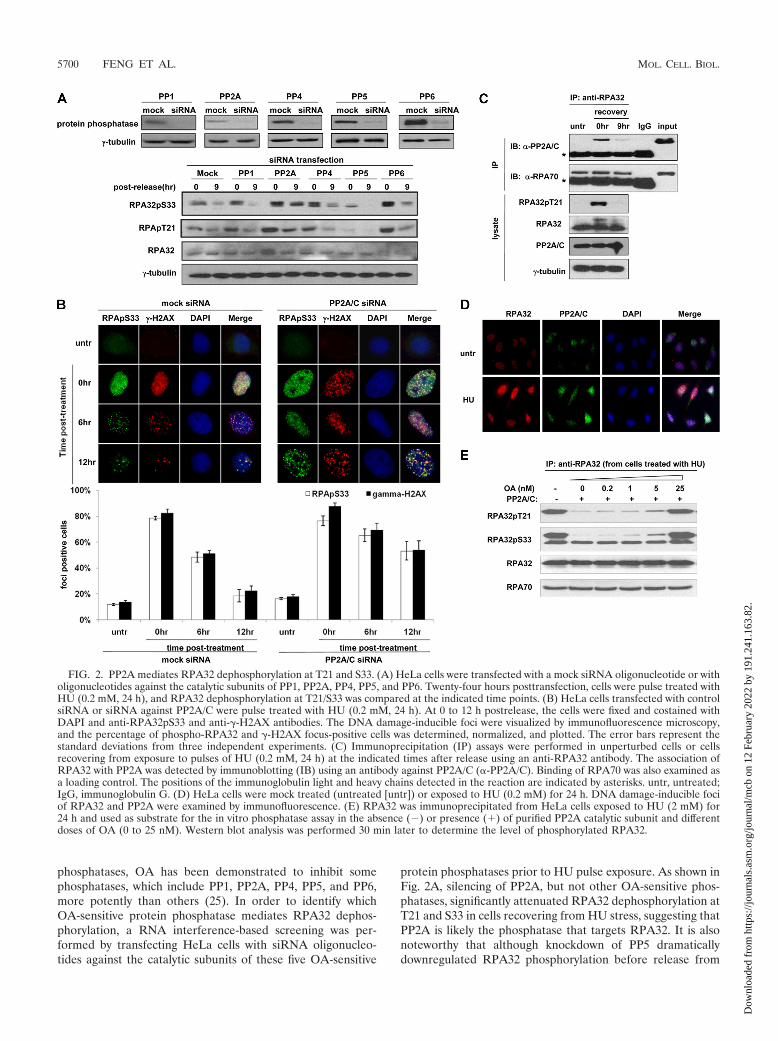
phosphatases, OA has been demonstrated to inhibit somephosphatases, which include PP1, PP2A, PP4, PP5, and PP6,more potently than others (25). In order to identify whichOA-sensitive protein phosphatase mediates RPA32 dephos-phorylation, a RNA interference-based screening was per-formed by transfecting HeLa cells with siRNA oligonucleo-tides against the catalytic subunits of these five OA-sensitive
protein phosphatases prior to HU pulse exposure. As shown inFig. 2A, silencing of PP2A, but not other OA-sensitive phos-phatases, significantly attenuated RPA32 dephosphorylation atT21 and S33 in cells recovering from HU stress, suggesting thatPP2A is likely the phosphatase that targets RPA32. It is alsonoteworthy that although knockdown of PP5 dramaticallydownregulated RPA32 phosphorylation before release from
FIG. 2. PP2A mediates RPA32 dephosphorylation at T21 and S33. (A) HeLa cells were transfected with a mock siRNA oligonucleotide or witholigonucleotides against the catalytic subunits of PP1, PP2A, PP4, PP5, and PP6. Twenty-four hours posttransfection, cells were pulse treated withHU (0.2 mM, 24 h), and RPA32 dephosphorylation at T21/S33 was compared at the indicated time points. (B) HeLa cells transfected with controlsiRNA or siRNA against PP2A/C were pulse treated with HU (0.2 mM, 24 h). At 0 to 12 h postrelease, the cells were fixed and costained withDAPI and anti-RPA32pS33 and anti-�-H2AX antibodies. The DNA damage-inducible foci were visualized by immunofluorescence microscopy,and the percentage of phospho-RPA32 and �-H2AX focus-positive cells was determined, normalized, and plotted. The error bars represent thestandard deviations from three independent experiments. (C) Immunoprecipitation (IP) assays were performed in unperturbed cells or cellsrecovering from exposure to pulses of HU (0.2 mM, 24 h) at the indicated times after release using an anti-RPA32 antibody. The association ofRPA32 with PP2A was detected by immunoblotting (IB) using an antibody against PP2A/C (�-PP2A/C). Binding of RPA70 was also examined asa loading control. The positions of the immunoglobulin light and heavy chains detected in the reaction are indicated by asterisks. untr, untreated;IgG, immunoglobulin G. (D) HeLa cells were mock treated (untreated [untr]) or exposed to HU (0.2 mM) for 24 h. DNA damage-inducible fociof RPA32 and PP2A were examined by immunofluorescence. (E) RPA32 was immunoprecipitated from HeLa cells exposed to HU (2 mM) for24 h and used as substrate for the in vitro phosphatase assay in the absence (�) or presence (�) of purified PP2A catalytic subunit and differentdoses of OA (0 to 25 nM). Western blot analysis was performed 30 min later to determine the level of phosphorylated RPA32.
5700 FENG ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.

HU (Fig. 2A), which was consistent with our previous reportthat PP5 plays a positive role in the ATR activation (59),dephosphorylation of RPA32 was nonetheless apparent (Fig.2A), indicating that PP5 is unlikely to be involved in thisprocess.
In agreement with this result, phosphorylated RPA32 focipersisted significantly longer in the PP2A/C-silenced cells,with 53.2% of cells being focus positive 12 h after releasefrom HU block. This was in sharp contrast with the mockcontrol cells that showed a significantly reduced number offoci and had only 18.5% of focus-positive cells at the 12-htime point (Fig. 2B). It is also noteworthy that phosphory-lated RPA32 foci colocalized with �-H2AX foci, which dis-played a similar kinetics as its number decreased over timeduring unperturbed recovery and also exhibited longer per-sistency in the PP2A-silenced cells. These data are consis-tent with a previous report that PP2A mediates �-H2AXdephosphorylation following pulse exposure to camptoth-ecin, a topoisomerase I inhibitor (9).
It is possible that PP2A does not mediate RPA32 dephos-phorylation directly but is somehow involved in this processindirectly. To assess this possibility, a coimmunoprecipitationassay was performed to examine whether PP2A could interactwith RPA32 at endogenous levels. As shown in Fig. 2C, thePP2A catalytic subunit did not associate with RPA32 in un-perturbed cells, but strong binding was observed when cellswere exposed to HU stress. This binding decreased signifi-cantly after 9 h of recovery. In addition, DNase treatment didnot abrogate the association between RPA32 and PP2A cata-lytic subunit, ruling out the possibility that their interaction ismediated by the chromatin bridge (see Fig. S2 in the supple-mental material). Therefore, PP2A likely plays a direct role inthe process of RPA32 dephosphorylation due to its inducedassociation with RPA32 following genotoxic exposure. In sup-port of this notion, immunofluorescence analysis also showedthat the catalytic subunit of PP2A colocalized with RPA32 fociin an HU stress-inducible manner (Fig. 2D). To examinewhether PP2A could dephosphorylate RPA32 directly, phos-phorylated RPA32 immunoprecipitated from HU-stressedcells was incubated with recombinant PP2A catalytic subunit.PP2A readily dephosphorylated RPA32 in vitro, reducing lev-els of phosphorylated RPA32 within 30 min, and this processwas markedly inhibited by the addition of 5 nM of OA (Fig.2E), a concentration known to block PP2A activity under theassay condition. Taken together, these data indicate that PP2Amediates RPA32 dephosphorylation at T21 and S33 in cellsrecovering from HU-induced genotoxic stress.
RPA32 dephosphorylation at T21/S33 is dispensable for thecheckpoint activation but is required for recovery from HUstress or UV irradiation. As previously mentioned, the un-phosphorylated form of RPA plays an essential role in sup-porting DNA replication and other DNA metabolism undernormal cellular conditions. It is therefore plausible that whencells are recovering from genotoxic stress, PP2A-dependentRPA32 dephosphorylation may be required to reverse geno-toxic stress-induced RPA32 phosphorylation, consequently re-storing RPA32 to its normal condition. In support of thishypothesis, PP2A-silenced HeLa cells exhibit deficient RPA32dephosphorylation at T21 and S33 after HU pulse exposure(Fig. 2A and B) and more importantly displayed significantly
reduced cell viability following pulse exposure to HU stress (24h) compared to cells treated with mock oligonucleotides (seeFig. S3A in the supplemental material).
Since PP2A is a versatile protein phosphatase targeting myr-iad substrates in numerous cellular processes, it is possible thatthe increased sensitivity to HU induced by PP2A knockdown iscaused by PP2A-dependent processes other than attenuatedRPA32 dephosphorylation. To better address whether RPA32dephosphorylation is required for this process, we created aRPA32-T21D/S33D mutant (called DD hereafter) to mimicthe persistent phosphorylation state of RPA32 at T21 and S33.The serine/threonine-to-aspartate conversion has been exten-sively utilized as phosphomimetic to study protein functions,and in many cases, the structures and activities of these phos-phorylation-mimicking proteins are identical to those of theactual phosphoproteins (22, 48, 52). In order to avoid potentialinterference by the presence of endogenous WT RPA32, HeLasubstitution cells were created in which the endogenousRPA32 was stably replaced with a nontargetable WT RPA32,the RPA32-DD mutant, or phospho-deficient RPA-T21V/S33A (VA) mutant. As shown in Fig. 3A, endogenous RPA32expression was effectively silenced by retrovirus-mediatedsiRNA, and the levels of the exogenously expressed RPA32variants were comparable to the level of endogenous RPA inthe vector control cells. Cells expressing the DD mutant ofRPA displayed normal morphology (data not shown) and alightly slower growth rate than that of control cells (see Fig.S3B in the supplemental material). The ability of RPA32-DDto complex with other RPA subunits was also indistinguishablefrom other RPA variants (data not shown), consistent with aprevious report on a RPA32 phosphomimetic which containedas many as eight substitutions (48). However, compared withthis report (48), which showed incompetent association of theextensively substituted RPA32 mutants with the DNA replica-tion centers, the RPA32-DD described here displayed no ap-parent deficiency in its colocalization with the DNA replicationcenters in the unperturbed cells (see Fig. S3C in the supple-mental material), indicating that the RPA32-DD is sufficient tosupport DNA replication, a result that is not surprising giventhe relatively normal cytology exhibited by the DD cells. Noother discernible phenotypes were found after 50 passages.
Since RPA translocation to sites of DNA damage has beendemonstrated to be an early event in the DNA damage re-sponse and plays an important role in activating ATR-depen-dent checkpoint pathways, we examined whether cells with theRPA32-DD phosphomimetic mutant exhibited defective up-stream checkpoint activation following genotoxic stress. AfterHU treatment, RPA32-DD formed punctuate foci in the nu-cleus that increased over time at a rate similar to that of theRPA32-WT, indicating that the RPA32-DD mutant translo-cates to chromatin normally upon DNA damage (Fig. 3B). Inaddition, ATR-dependent phosphorylation of two importantcheckpoint regulators, Chk1 and Rad17, displayed no distin-guishable difference in the DD cells compared with other cells,suggesting that the RPA32-DD substitution does not affectATR activation (Fig. 3C). The DD cells have an intact intra-Scheckpoint as evidenced by a reduction in DNA synthesis fol-lowing UV irradiation to an extent comparable to that of theWT and vector control lines, but in contrast with the phospho-deficient AA cells which maintained a relatively higher level of
VOL. 29, 2009 EFFICIENT DNA REPAIR REQUIRES RPA32 DEPHOSPHORYLATION 5701
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.
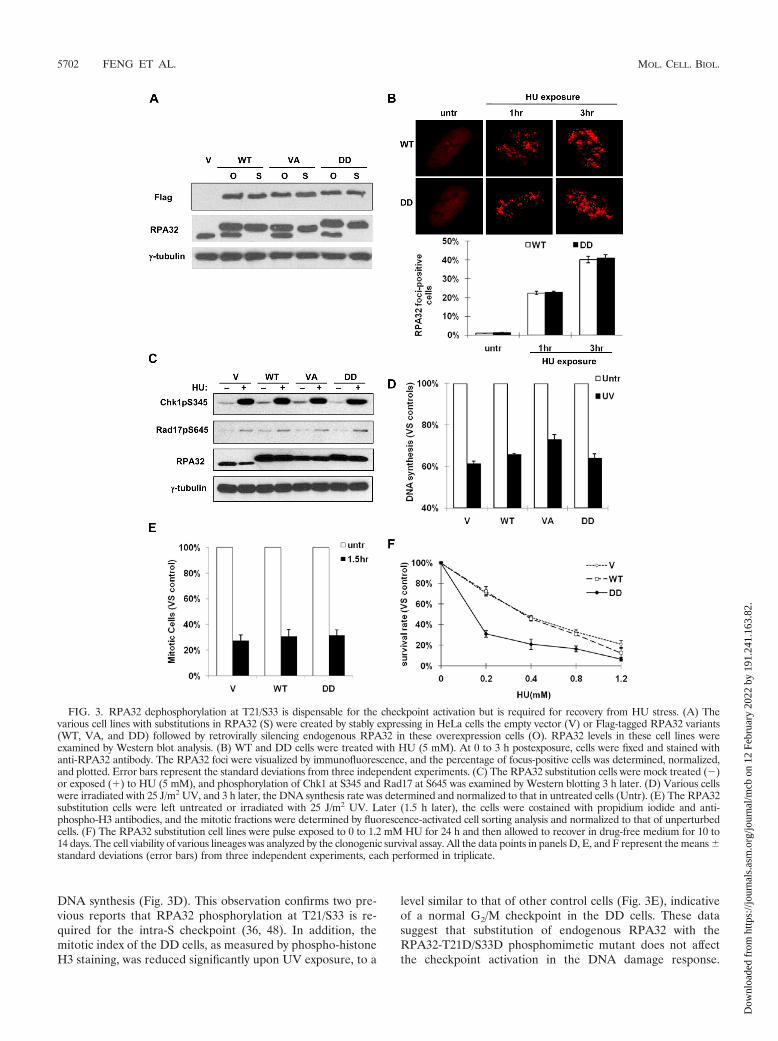
DNA synthesis (Fig. 3D). This observation confirms two pre-vious reports that RPA32 phosphorylation at T21/S33 is re-quired for the intra-S checkpoint (36, 48). In addition, themitotic index of the DD cells, as measured by phospho-histoneH3 staining, was reduced significantly upon UV exposure, to a
level similar to that of other control cells (Fig. 3E), indicativeof a normal G2/M checkpoint in the DD cells. These datasuggest that substitution of endogenous RPA32 with theRPA32-T21D/S33D phosphomimetic mutant does not affectthe checkpoint activation in the DNA damage response.
FIG. 3. RPA32 dephosphorylation at T21/S33 is dispensable for the checkpoint activation but is required for recovery from HU stress. (A) Thevarious cell lines with substitutions in RPA32 (S) were created by stably expressing in HeLa cells the empty vector (V) or Flag-tagged RPA32 variants(WT, VA, and DD) followed by retrovirally silencing endogenous RPA32 in these overexpression cells (O). RPA32 levels in these cell lines wereexamined by Western blot analysis. (B) WT and DD cells were treated with HU (5 mM). At 0 to 3 h postexposure, cells were fixed and stained withanti-RPA32 antibody. The RPA32 foci were visualized by immunofluorescence, and the percentage of focus-positive cells was determined, normalized,and plotted. Error bars represent the standard deviations from three independent experiments. (C) The RPA32 substitution cells were mock treated (�)or exposed (�) to HU (5 mM), and phosphorylation of Chk1 at S345 and Rad17 at S645 was examined by Western blotting 3 h later. (D) Various cellswere irradiated with 25 J/m2 UV, and 3 h later, the DNA synthesis rate was determined and normalized to that in untreated cells (Untr). (E) The RPA32substitution cells were left untreated or irradiated with 25 J/m2 UV. Later (1.5 h later), the cells were costained with propidium iodide and anti-phospho-H3 antibodies, and the mitotic fractions were determined by fluorescence-activated cell sorting analysis and normalized to that of unperturbedcells. (F) The RPA32 substitution cell lines were pulse exposed to 0 to 1.2 mM HU for 24 h and then allowed to recover in drug-free medium for 10 to14 days. The cell viability of various lineages was analyzed by the clonogenic survival assay. All the data points in panels D, E, and F represent the means standard deviations (error bars) from three independent experiments, each performed in triplicate.
5702 FENG ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.

Therefore, for studies of the recovery process from the DNAdamage response where RPA32 is dephosphorylated at T21and S33, the cells with the RPA32-DD substitution provide uswith an opportunity to investigate the events influenced byRPA32 dephosphorylation by serving as a persistent phosphor-ylation mimic model.
To examine whether RPA32 dephosphorylation is requiredfor efficient recovery from HU stress, the cells with substitu-tions in RPA32 were pulse treated to a range of doses of HUfor 24 h, and their viability after recovery was analyzed by theclonogenic survival assay. As shown in Fig. 3F, the survival rateof DD cells was significantly reduced compared to that of WTor vector control cells; only 30.9% of DD cells survived andformed colonies after recovery from pulse exposure to 0.2 mMof HU, which was in sharp contrast with WT cells (72.4%) andvector control cells (70.8%). Similarly, DD cells were also lesscapable to survive UV irradiation compared to control cells,albeit to a lesser extent than with HU treatment (see Fig. S3Din the supplemental material). Taken together, cells with theRPA32-DD mutant are defective in the recovery from HUstress or UV irradiation, suggesting that RPA32 dephosphor-ylation at T21 and S33 is required in this process.
RPA32 dephosphorylation is not required for the resump-tion of DNA replication or subsequent initiation of mitosisfollowing release from HU block. It has been reported thatgenotoxic stress-induced RPA32 phosphorylation prevents itsassociation with replication centers and thus may suppressDNA synthesis following DNA damage (48). Recent evidenceindicates that phosphorylation at the ATM/ATR-responsiveT21 and S33 sites is critical for this function (36). Given thesefindings, it is plausible that RPA32 dephosphorylation at T21/S33 might be required for the resumption of DNA replicationfollowing recovery from HU block. However, as the thymidineincorporation assay indicated, the DNA synthesis activity ofthe RPA32-DD substitution cells increased at a rate compara-ble to that of WT and vector control lines within 3 hours afterrelease from HU pulse exposure (0.2 mM, 24 h) (Fig. 4A),implying that RPA32 dephosphorylation is not required toresume the arrested DNA replication following release fromHU block.
HU is a reversible inhibitor of DNA replication, and cellssynchronized by HU exposure reenter the cell cycle if releasedfrom HU block. Since the resumption of DNA replicationseems to be unaffected, it is possible that progression throughthe downstream G2/M boundary requires RPA32 dephosphor-ylation at T21/S33. To test this possibility, the various substi-tution lines were released from pulse exposure to HU (0.2 mM,24 h), and their progression through mitosis was evaluated byphospho-histone-H3 staining at different time points. As indi-cated in Fig. 4B, the mitotic profile of the DD cells showed nonoticeable difference compared with that of other two lines(VA and WT); the mitotic indices of all three lines remainedlow until 4 h postrelease when cells started to divide, and thepercentage of the mitotic cells peaked at 8 h and dropped to alevel similar to that of the 0-h time point at 10 h. Thus, RPA32dephosphorylation at T21/S33 is not necessary for mitotic pro-gression in cells recovering from HU block.
The above results were supported by an independent exper-iment performed on HeLa cells pulse exposed to two differentlevels of HU for 24 h. In sharp contrast with cells pulse treated
with a sublethal dose (0.2 mM) of HU, where RPA32 wasapparently dephosphorylated gradually during recovery, cellstreated with a lethal dose (2 mM) exhibited persistent RPA32phosphorylation up to 24 h postrelease (Fig. 4C). The lethallytreated cells could resume DNA replication in S phase andreenter the cell cycle with no apparent defects, except that theydisplayed an approximately 3-h delay in their recovery com-pared with sublethally treated cells (Fig. 4D), indicating adispensable role of RPA32 dephosphorylation in the resump-tion of DNA replication and the subsequent mitosis followingrelease from HU block.
There is a possibility that DNA replication resumes only incells where RPA32 is dephosphorylated, or alternatively atsites where the bound RPA32 is dephosphorylated and not atsites where RPA32 remains phosphorylated. To test these pos-sibilities, HeLa cells were pulse-labeled with BrdU for 10 min6 hours after release from HU block (2 mM, 24 h) and thentested for colocalization between phosphorylated RPA32 andincorporated BrdU, which usually indicates where DNA rep-lication is occurring. As shown in Fig. 4E, phosphorylatedRPA32 and BrdU strongly colocalized, indicating again thatresumption of DNA replication is independent of RPA32 de-phosphorylation. However, it is possible that DNA replicationfollowing release from HU block may not be resumed at thestalled replication forks but restart at the later origins that havenot yet been fired, which could in theory allow for completionof DNA replication. To differentiate these two events, a pre-viously reported sequential labeling approach (24, 58) wasadopted such that cells were pulse-labeled with IdU before HUstress, followed by a second labeling with CldU at differenttime points after release from HU block. As indicated (see Fig.S4 in the supplemental material), all cells displayed a signifi-cant degree (95%) of coincident labeling (yellow) of CldU(red) with prelabeled IdU (green) at 3 h postrelease from 0.2mM of HU pulse treatment (HeLa and DD cells) or 6 hpostrelease from 2 mM of HU pulse treatment (HeLa cells). Inthe latter case, RPA32 still remains hyperphosphorylated (Fig.4C). In sharp contrast, only a negligible level (�5%) of post-labeled CldU (red alone) was observed. Thus, cells preferen-tially resume DNA replication at the previously stalled repli-cation forks regardless of the phosphorylation status or thephosphomimetic mutation of RPA32, corroborating thatRPA32 dephosphorylation is dispensable in resumption ofDNA replication.
To confirm that RPA32 dephosphorylation is not requiredfor resumption of mitosis following recovery from HU block,mitotic index analysis was carried out. As revealed in Fig. 4F,the lethally treated HeLa cells exhibit a lagging but nonethe-less apparent progression through mitosis in comparison withthe cells pulse exposed to a sublethal dose of HU, indicatingthat RPA32 dephosphorylation at T21/S33 is not required forthis process. Taken together, these data indicate that RPA32dephosphorylation is not required to resume DNA replicationor progress through the mitotic phase during recovery fromHU-induced genotoxic stress.
It is noteworthy that the kinetics of �-H2AX dephosphory-lation were similar to the kinetics of RPA32 dephosphorylationin cells pulse treated with sublethal doses of HU, and similar toRPA32, this dephosphorylation was significantly attenuated inlethally treated cells (Fig. 4C). In contrast, dephosphorylation
VOL. 29, 2009 EFFICIENT DNA REPAIR REQUIRES RPA32 DEPHOSPHORYLATION 5703
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.
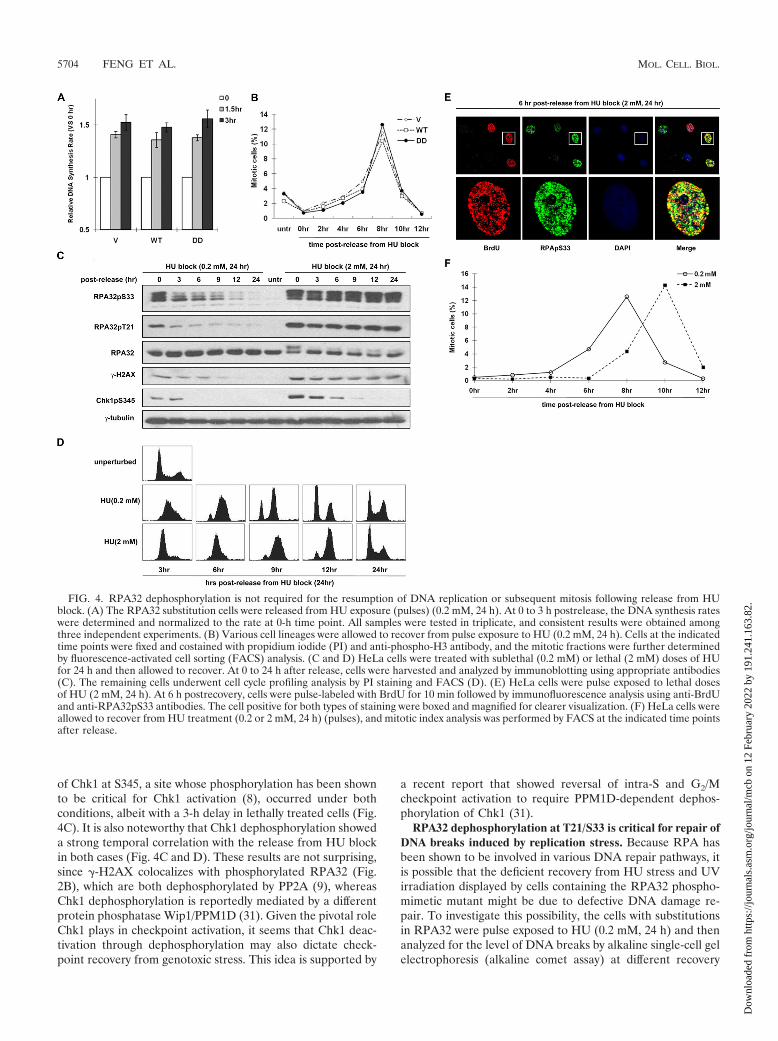
of Chk1 at S345, a site whose phosphorylation has been shownto be critical for Chk1 activation (8), occurred under bothconditions, albeit with a 3-h delay in lethally treated cells (Fig.4C). It is also noteworthy that Chk1 dephosphorylation showeda strong temporal correlation with the release from HU blockin both cases (Fig. 4C and D). These results are not surprising,since �-H2AX colocalizes with phosphorylated RPA32 (Fig.2B), which are both dephosphorylated by PP2A (9), whereasChk1 dephosphorylation is reportedly mediated by a differentprotein phosphatase Wip1/PPM1D (31). Given the pivotal roleChk1 plays in checkpoint activation, it seems that Chk1 deac-tivation through dephosphorylation may also dictate check-point recovery from genotoxic stress. This idea is supported by
a recent report that showed reversal of intra-S and G2/Mcheckpoint activation to require PPM1D-dependent dephos-phorylation of Chk1 (31).
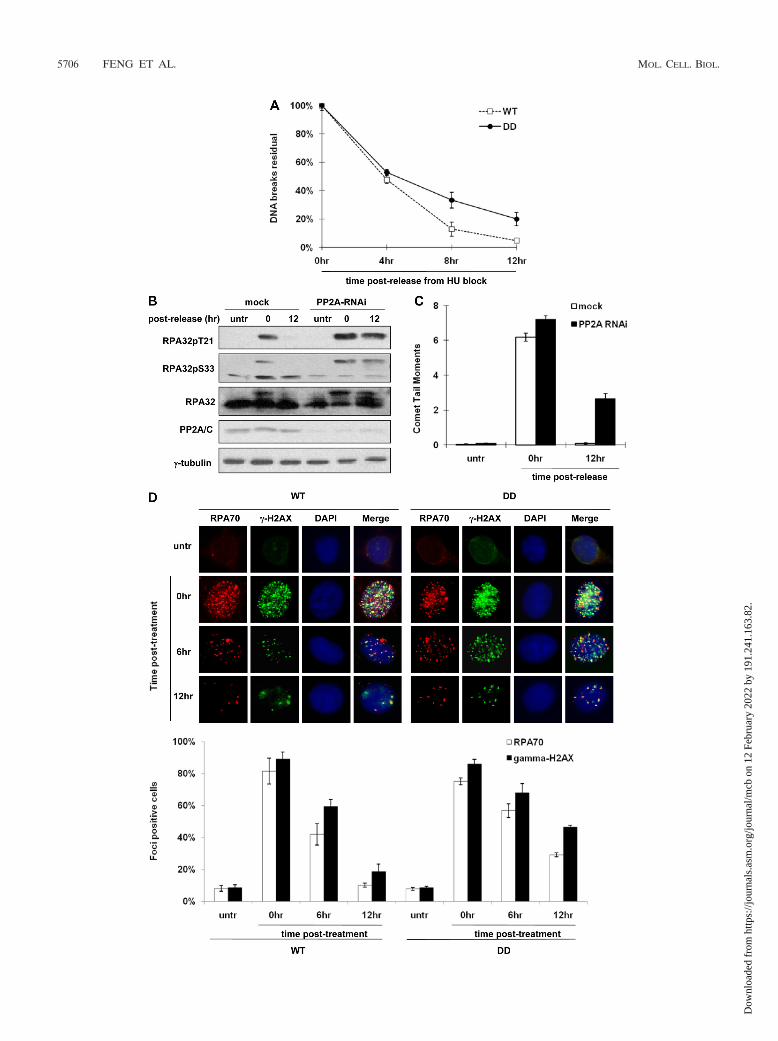
RPA32 dephosphorylation at T21/S33 is critical for repair ofDNA breaks induced by replication stress. Because RPA hasbeen shown to be involved in various DNA repair pathways, itis possible that the deficient recovery from HU stress and UVirradiation displayed by cells containing the RPA32 phospho-mimetic mutant might be due to defective DNA damage re-pair. To investigate this possibility, the cells with substitutionsin RPA32 were pulse exposed to HU (0.2 mM, 24 h) and thenanalyzed for the level of DNA breaks by alkaline single-cell gelelectrophoresis (alkaline comet assay) at different recovery
FIG. 4. RPA32 dephosphorylation is not required for the resumption of DNA replication or subsequent mitosis following release from HUblock. (A) The RPA32 substitution cells were released from HU exposure (pulses) (0.2 mM, 24 h). At 0 to 3 h postrelease, the DNA synthesis rateswere determined and normalized to the rate at 0-h time point. All samples were tested in triplicate, and consistent results were obtained amongthree independent experiments. (B) Various cell lineages were allowed to recover from pulse exposure to HU (0.2 mM, 24 h). Cells at the indicatedtime points were fixed and costained with propidium iodide (PI) and anti-phospho-H3 antibody, and the mitotic fractions were further determinedby fluorescence-activated cell sorting (FACS) analysis. (C and D) HeLa cells were treated with sublethal (0.2 mM) or lethal (2 mM) doses of HUfor 24 h and then allowed to recover. At 0 to 24 h after release, cells were harvested and analyzed by immunoblotting using appropriate antibodies(C). The remaining cells underwent cell cycle profiling analysis by PI staining and FACS (D). (E) HeLa cells were pulse exposed to lethal dosesof HU (2 mM, 24 h). At 6 h postrecovery, cells were pulse-labeled with BrdU for 10 min followed by immunofluorescence analysis using anti-BrdUand anti-RPA32pS33 antibodies. The cell positive for both types of staining were boxed and magnified for clearer visualization. (F) HeLa cells wereallowed to recover from HU treatment (0.2 or 2 mM, 24 h) (pulses), and mitotic index analysis was performed by FACS at the indicated time pointsafter release.
5704 FENG ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.

time points. Even though DD cells accumulated slightly lowerlevels of DNA breaks before HU release (data not shown),repair of the DNA breaks, as indicated by the decline of therelative comet tail moments, was significantly less efficientcompared to WT cells (Fig. 5A). At 12 h postrelease, the DDcells still contained a substantially higher level of unresolvedDNA breaks (22.4%) compared to the WT cells (3.3%). Thesedata indicate that cells expressing the RPA32 phosphomimeticmutant are less efficient in the repair of DNA breaks inducedby HU, suggesting that RPA32 dephosphorylation at T21/S33is required for this repair process. Consistent with this result,PP2A/C-silenced HeLa cells exhibit less RPA32 dephosphor-ylation and a significantly higher level of unresolved DNAbreaks 12 h after recovery from HU stress (36.7% relative tothe 0-h point, calculated by the ratio between comet tail mo-ments, Fig. 5B and C). This is in sharp contrast to the mockcontrol cells where RPA32 was fully dephosphorylated andDNA repair was essentially complete (1.4%) (Fig. 5B and C).
In response to genotoxic stress, a large number of check-point and repair proteins including the Mre11/Rad50/Nbs1complex, RPA, 53BP1, and �-H2AX accumulate at sites ofDNA damage and form huge protein complexes termed DNAdamage foci, which are essentially the DNA repair centers thatdo not resolve until the repair process is completed (13, 30).For this reason, these protein foci are regarded as markers forDNA damage. To confirm the above results through the cometassays, we examined the kinetics of RPA foci and �-H2AXfoci, two markers broadly used to indicate DNA damage, in thecells with substitutions in RPA32 recovering from HU block(0.2 mM, 24 h). As shown in Fig. 5D, both RPA70 and�-H2AX exhibited HU stress-induced punctate staining in WTand DD cells, which also displayed a significant level of colo-calization. In both RPA32 substitution cells, the number of thespeckles formed by these two proteins as well as the percentageof the focus-positive cells declined over time following recov-ery from HU pulse exposure. However, the rate of reduction inDD cells was significantly lower than that in WT cells; at 12 hpostrecovery, only 18.7% of WT cells but as many as 46.6% ofDD cells were �-H2AX focus positive, and the percentage ofDD cells that contained unresolved RPA70 foci was almostthree times that of WT cells (29.2% versus 10.2%), confirmingagain that DNA damage repair is defective in the DD cells.
Taken together, these results indicate that the phosphomi-metic DD mutant cells are deficient in the repair of DNAbreaks induced by replication stress, demonstrating that PP2A-dependent RPA32 dephosphorylation at T21 and S33 is nec-essary for efficient DNA damage repair.
DISCUSSION
In this report we show that in cells recovering from thegenotoxic stress induced by HU, RPA32 undergoes PP2A-dependent dephosphorylation at T21 and S33, a process dis-pensable for checkpoint activation or cell cycle reentry butcritical for efficient DNA damage repair. Our findings, com-bined with several previous studies, have established that de-phosphorylation of checkpoint/repair proteins is likely to be asimportant in the DNA damage response as the well-docu-mented upstream phosphorylation cascades known to initiateand propagate the DNA damage response.
RPA is a versatile protein playing numerous roles in theDNA damage response, including recognizing and stabilizingDNA breaks, activating/mediating checkpoint pathways, andassisting with the downstream repair of diverse types of DNAlesions. Although much is known about the functions of RPA,little is known about how RPA’s multiform activities are reg-ulated. On the basis of our study and others, it seems that thedynamic phosphorylation and dephosphorylation of theRPA32 subunit at T21 and S33 might provide a mechanismcontrolling the numerous functions of RPA in the DNA dam-age response. Upon DNA damage or replication stress, RPA32is phosphorylated by ATM/ATR at sites of DNA damage orstalled replication forks, and phosphorylated RPA suppressesongoing DNA replication and helps recruit other checkpoint/repair proteins. Later, during recovery from DNA damage-induced checkpoint arrest, phosphorylated RPA32 is removedthrough PP2A-dependent dephosphorylation, an event that isessential for efficient DNA repair. For this entire process,initial phosphorylation and subsequent dephosphorylation ofRPA32 need to be tightly regulated in order for a proper DNAdamage response to occur. Suppression or failure of initialRPA32 phosphorylation causes a defective S-phase checkpointleading to DNA damage-resistant DNA synthesis, and delay ordysfunction of subsequent RPA32 dephosphorylation results ininefficient repair of DNA lesions.
It is noteworthy that in addition to the ATM/ATR-respon-sive T21 and S33, several other serine residues (S4, S8, S11,S12, and S13) on the N terminus of RPA32 also undergo DNAdamage-inducible phosphorylation. Therefore, it is possiblethat dephosphorylation of these sites is also regulated by PP2Aand required for the DNA damage response. On the basis ofour observations, PP2A is seemingly responsible for dephos-phorylation of some, if not all, of these residues, since Westernblotting with an anti-RPA32 antibody showed an apparentPP2A-dependent reduction of multiple slower-migrating bandsin cells recovering from HU stress, which can be indicative ofa loss of protein phosphorylation (Fig. 1A and 5B). In supportof this, the results of Western blotting with an antibody thatrecognizes doubly phosphorylated RPA32 on both S4 and S8suggested at least one of these two sites may also be dephos-phorylated in a PP2A-dependent manner (see Fig. S5 in thesupplemental material). Due to lack of appropriate phospho-specific antibodies against all of these seven individual sites, wedid not address this question further. Regardless of whetherPP2A is involved, given the high level of cytotoxicity inducedby overexpression of RPA32 hyperphosphomimic mutant thathas substitutions of all seven serine/threonine sites (data notshown) and the essential role unphosphorylated RPA32 playsin supporting unperturbed DNA replication, it is conceivablethat cells would not survive persistent RPA32 hyperphosphor-ylation following genotoxic stress and that dephosphorylationof these DNA damage-inducible sites is likely a required eventduring recovery.
With the use of RPA32 phosphomimetic mutants, it hasbeen shown by two groups that PIKK-dependent RPA32 phos-phorylation may prevent RPA from associating with replica-tion machinery following genotoxic stress, because these ec-topically expressed RPA32 mutants exhibit defects in bindingwith replication centers and forming S-phase-specific foci (36,48). In our study, however, the cytology of HeLa cells with the
VOL. 29, 2009 EFFICIENT DNA REPAIR REQUIRES RPA32 DEPHOSPHORYLATION 5705
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.
5706 FENG ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.

T21D/S33D mutant substitution was rather normal, and theDD mutant form of RPA32 displayed no apparent dysfunctionin its colocalization with BrdU-labeled replication centers (seeFig. S3C in the supplemental material). One possible explana-tion for this is that RPA32-DD could still associate with rep-lication centers but with slightly lower efficiency compared towild-type RPA32. This could result in insufficient binding ofRPA32-DD to replication centers if there is abundant endog-enous wild-type RPA32 to compete with, as was the case in thetwo previous reports which both used overexpression systems(27, 29). If the interference caused by endogenous RPA32 isremoved by RNA interference silencing, as in our substitutionmodel, mutant RPA32 is still able to support DNA replicationwith no apparent defects. In support of this hypothesis, the DDsubstitution cells exhibit relatively normal replication, and theRPA32-DD mutant protein migrates to sites of DNA breaksefficiently and supports ATR-dependent checkpoint activationwith no observable defects. Therefore, it is a useful model tostudy events downstream of checkpoint activation in the DNAdamage response that are related to RPA32 dephosphoryla-tion.
Surprisingly, our data indicate that RPA32 dephosphoryla-tion is not required for resumption of DNA replication afterrelease from HU block, as cells released from a lethal dose ofHU resumed DNA replication normally (Fig. 4D and E) (seeFig. S4 in the supplemental material) regardless of persistentRPA32 phosphorylation (Fig. 4C). This result differs from theconsensus view that phosphorylated RPA is unable to supportDNA replication due to its reduced capability to bind withDNA Pol � and to denature duplex DNA (5, 35, 37). Onepossibility is that DNA synthesis during unperturbed S-phasereplication and in the recovery from replication stress mayutilize different DNA polymerases and involve distinct mech-anisms. While the former process requires Pol � primase andunphosphorylated RPA-dependent initiation at replication or-igins and relies heavily on two other processive, high-fidelitypolymerases (Pol � and Pol ε) at replication elongation, DNAsynthesis in the recovering cells mainly resumes at the arrestedreplication forks or starts as part of a repair process and there-fore may not need the initiation step and may be mediated bynonreplicative but repair-specific polymerases (23, 42, 51). Insupport of this idea, several nonreplicative DNA polymerases,including X-family polymerases , �, TdT, Y-family polymer-ase �, B-family polymerase �, and Rev1, have been found to beinvolved in the nonhomologous end joining and homologousrecombination repair synthesis of DSBs, the major type ofDNA breaks induced by collapse of replication forks followingHU exposure (12, 19, 33, 42, 51). In addition, it has also been
shown that the Pol �-primase complex is not required forDSB-induced homologous recombination during the matingtype switch in budding yeast (50).
As discussed above, although resumption of DNA replica-tion does not require RPA32 dephosphorylation, there was aclose temporal correlation between resumption of DNA rep-lication and Chk1 dephosphorylation regardless of the level ofHU exposure (Fig. 4C and D). On the basis of a previousreport that PPM1D-dependent dephosphorylation of Chk1 ab-rogates checkpoints, we believe that Chk1 dephosphorylation-dependent deactivation may dictate resumption of DNA rep-lication as part of a checkpoint recovery process (31).Interestingly, while cells pulse treated with a lethal dose (2mM) of HU and cells with the phosphomimetic mutation ofRPA32 (DD) as well underwent apparent checkpoint recoveryby resuming cell cycle following release (Fig. 4D) (see Fig. S7in the supplemental material), these cells contained signifi-cantly high levels of unresolved DNA breaks during the recov-ery process, as indicated by the persistently high levels of phos-phorylated RPA32 and �-H2AX (Fig. 4C and 5D), as well asthe data from the comet assays (Fig. 5A) (see Fig. S6 in thesupplemental material). This is similar to an intriguing mech-anism termed “checkpoint adaptation” originally discovered inyeast. In this mechanism, cells reenter the cell cycle despite thepersistence of unrepaired DNA breaks, an advantage presum-ably conferred on unicellular yeast cells to allow them to sur-vive a long-term arrest when facing persistent genotoxicstresses (47). This mechanism had been believed to be absentin metazoans because of the possibility that it could promotegenomic instability, but recent studies have shown that such aphenomenon may exist in cells of higher organisms as well (18,46, 56). These data in our study evidently suggest that check-point adaptation also occurs in human cells. Thus far, it isunclear why adaptation exists in higher organisms. Although itwas speculated that this mechanism may facilitate the elimina-tion of cells containing irreparable DNA damage through mi-totic catastrophe (4), we observed a significant proportion ofthese adapted cells undergoing senescence, which was accom-panied by a increase of p21 protein levels (see Fig. S8A andS8B in the supplemental material). Therefore, checkpoint ad-aptation may also ultimately trigger senescence. Although wedid not see a significant contribution of apoptosis in this pro-cess (see Fig. S8C in the supplemental material), it is in theorypossible that checkpoint-adapted cells may also end up beingapoptotic. More in-depth studies are required to examine thisinteresting phenomenon.
This study has established a causal relationship betweenRPA32 dephosphorylation and repair of DNA breaks; how-
FIG. 5. RPA32 dephosphorylation is necessary for the efficient repair of DNA breaks induced by HU. (A) WT and DD cells were allowed torecover from HU exposure (0.2 mM, 24 h) (pulses). At 0 to 12 h postrecovery, cells were harvested and analyzed by alkaline comet assay. Repairof DNA breaks was evaluated by the level of the residual DNA breaks, which is calculated by the comet tail moments at various recovery timepoints relative to that at the 0-h time point. (B and C) HeLa cells were transfected with control siRNA or siRNA against the PP2A catalytic subunit.At 24 h posttransfection, the cells were pulse exposed to HU (0.2 mM, 24 h) and then allowed to recover. RPA32 dephosphorylation was analyzedby immunoblotting (B), and repair of DNA breaks was evaluated by alkaline comet assay (C). untr, untreated; RNAi, RNA interference. (D) WTand DD cells were allowed to recover from HU treatment (0.2 mM, 24 h) (pulses). At 0 to 12 h postrelease, cells were fixed and stained with DAPIand anti-RPA70 and anti-�-H2AX antibodies. Immunofluorescence was carried out to visualize the DNA damage foci formed by RPA70 and�-H2AX, and the percentages of the focus-positive cells were calculated, normalized, and plotted. All the data points in panels A, C, and Drepresent the means standard deviations (error bars) from three independent experiments.
VOL. 29, 2009 EFFICIENT DNA REPAIR REQUIRES RPA32 DEPHOSPHORYLATION 5707
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.

ever, the question of what specific role RPA32 dephosphory-lation plays in this process remains open. Presumably, RPA32dephosphorylation may play a role by recruiting certain repair-essential proteins to or removing other suppressive ones fromthe sites of DNA breaks where the DNA repair process isoccurring. Accumulating evidence has shown that the repair ofDNA breaks is a complicated multistep process orchestratedby a variety of DNA repair proteins that form dynamic buttightly regulated repair foci at sites of DNA damage (20, 29, 32,44). Alternatively, phosphorylated RPA32 may negatively reg-ulate certain downstream steps of DNA repair pathways thatdo not involve translocation of any repair proteins, and acti-vation of these steps may therefore necessitate prior PP2A-dependent dephosphorylation of RPA32. To understand whatspecific role RPA32 dephosphorylation plays in the DNA re-pair process, it would be inviting to temporally dissect theentire process, including recruitment/removal of the repairproteins and activation of the stepwise DNA repair pathways.
ACKNOWLEDGMENTS
We thank Mark Wold, Xin-Hua Feng, and David MacAlpine forproviding crucial reagents. We also thank Qinhong Wang and SamJohnson for technical help with immunofluorescence microscopy andXing Guo for assistance with the immunoprecipitation assay.
This work was supported by NIH grant (CA123250) to X.-F.W.
REFERENCES
1. Abraham, R. T. 2001. Cell cycle checkpoint signaling through the ATM andATR kinases. Genes Dev. 15:2177–2196.
2. Ali, A., J. Zhang, S. Bao, I. Liu, D. Otterness, N. M. Dean, R. T. Abraham,and X. F. Wang. 2004. Requirement of protein phosphatase 5 in DNA-damage-induced ATM activation. Genes Dev. 18:249–254.
3. Arnold, H. K., and R. C. Sears. 2006. Protein phosphatase 2A regulatorysubunit B56� associates with c-Myc and negatively regulates c-Myc accumu-lation. Mol. Cell. Biol. 26:2832–2844.
4. Bartek, J., and J. Lukas. 2007. DNA damage checkpoints: from initiation torecovery or adaptation. Curr. Opin. Cell Biol. 19:238–245.
5. Binz, S. K., Y. Lao, D. F. Lowry, and M. S. Wold. 2003. The phosphorylationdomain of the 32-kDa subunit of replication protein A (RPA) modulatesRPA-DNA interactions. Evidence for an intersubunit interaction. J. Biol.Chem. 278:35584–35591.
6. Binz, S. K., A. M. Sheehan, and M. S. Wold. 2004. Replication protein Aphosphorylation and the cellular response to DNA damage. DNA Repair(Amsterdam) 3:1015–1024.
7. Block, W. D., D. Merkle, K. Meek, and S. P. Lees-Miller. 2004. Selectiveinhibition of the DNA-dependent protein kinase (DNA-PK) by the radio-sensitizing agent caffeine. Nucleic Acids Res. 32:1967–1972.
8. Chen, Y., and Y. Sanchez. 2004. Chk1 in the DNA damage response: con-served roles from yeasts to mammals. DNA Repair (Amsterdam) 3:1025–1032.
9. Chowdhury, D., M. C. Keogh, H. Ishii, C. L. Peterson, S. Buratowski, and J.Lieberman. 2005. �-H2AX dephosphorylation by protein phosphatase 2Afacilitates DNA double-strand break repair. Mol. Cell 20:801–809.
10. Chowdhury, D., X. Xu, X. Zhong, F. Ahmed, J. Zhong, J. Liao, D. M.Dykxhoorn, D. M. Weinstock, G. P. Pfeifer, and J. Lieberman. 2008. APP4-phosphatase complex dephosphorylates gamma-H2AX generated dur-ing DNA replication. Mol. Cell 31:33–46.
11. Cliby, W. A., C. J. Roberts, K. A. Cimprich, C. M. Stringer, J. R. Lamb, S. L.Schreiber, and S. H. Friend. 1998. Overexpression of a kinase-inactive ATRprotein causes sensitivity to DNA-damaging agents and defects in cell cyclecheckpoints. EMBO J. 17:159–169.
12. de Feraudy, S., C. L. Limoli, E. Giedzinski, D. Karentz, T. M. Marti, L.Feeney, and J. E. Cleaver. 2007. Pol eta is required for DNA replicationduring nucleotide deprivation by hydroxyurea. Oncogene 26:5713–5721.
13. Dellaire, G., and D. P. Bazett-Jones. 2007. Beyond repair foci: subnucleardomains and the cellular response to DNA damage. Cell Cycle 6:1864–1872.
14. den Elzen, N. R., and M. J. O’Connell. 2004. Recovery from DNA damagecheckpoint arrest by PP1-mediated inhibition of Chk1. EMBO J. 23:908–918.
15. Fujimoto, H., N. Onishi, N. Kato, M. Takekawa, X. Z. Xu, A. Kosugi, T.Kondo, M. Imamura, I. Oishi, A. Yoda, and Y. Minami. 2006. Regulation ofthe antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. CellDeath Differ. 13:1170–1180.
16. Golub, E. I., R. C. Gupta, T. Haaf, M. S. Wold, and C. M. Radding. 1998.
Interaction of human rad51 recombination protein with single-strandedDNA binding protein, RPA. Nucleic Acids Res. 26:5388–5393.
17. Guo, X., A. Ramirez, D. S. Waddell, Z. Li, X. Liu, and X. F. Wang. 2008. Axinand GSK3-� control Smad3 protein stability and modulate TGF-� signaling.Genes Dev. 22:106–120.
18. Harrison, J. C., and J. E. Haber. 2006. Surviving the breakup: the DNAdamage checkpoint. Annu. Rev. Genet. 40:209–235.
19. Hirano, Y., J. Reddy, and K. Sugimoto. 2009. Role of budding yeast Rad18in repair of HO-induced double-strand breaks. DNA Repair (Amsterdam)8:51–59.
20. Houtsmuller, A. B., S. Rademakers, A. L. Nigg, D. Hoogstraten, J. H. Hoei-jmakers, and W. Vermeulen. 1999. Action of DNA repair endonucleaseERCC1/XPF in living cells. Science 284:958–961.
21. Hsu, L. C. 2007. Identification and functional characterization of a PP1-binding site in BRCA1. Biochem. Biophys. Res. Commun. 360:507–512.
22. Huang, W., and R. L. Erikson. 1994. Constitutive activation of Mek1 bymutation of serine phosphorylation sites. Proc. Natl. Acad. Sci. USA 91:8960–8963.
23. Hubscher, U., G. Maga, and S. Spadari. 2002. Eukaryotic DNA polymerases.Annu. Rev. Biochem. 71:133–163.
24. Itakura, E., I. Sawada, and A. Matsuura. 2005. Dimerization of the ATRIPprotein through the coiled-coil motif and its implication to the maintenanceof stalled replication forks. Mol. Biol. Cell 16:5551–5562.
25. Janssens, V., and J. Goris. 2001. Protein phosphatase 2A: a highly regulatedfamily of serine/threonine phosphatases implicated in cell growth and sig-nalling. Biochem. J. 353:417–439.
26. Kastan, M. B., and J. Bartek. 2004. Cell-cycle checkpoints and cancer.Nature 432:316–323.
27. Kumagai, A., and W. G. Dunphy. 2006. How cells activate ATR. Cell Cycle5:1265–1268.
28. Leroy, C., S. E. Lee, M. B. Vaze, F. Ochsenbien, R. Guerois, J. E. Haber, andM. C. Marsolier-Kergoat. 2003. PP2C phosphatases Ptc2 and Ptc3 are re-quired for DNA checkpoint inactivation after a double-strand break. Mol.Cell 11:827–835.
29. Lisby, M., J. H. Barlow, R. C. Burgess, and R. Rothstein. 2004. Choreogra-phy of the DNA damage response: spatiotemporal relationships amongcheckpoint and repair proteins. Cell 118:699–713.
30. Lisby, M., and R. Rothstein. 2004. DNA damage checkpoint and repaircenters. Curr. Opin. Cell Biol. 16:328–334.
31. Lu, X., B. Nannenga, and L. A. Donehower. 2005. PPM1D dephosphorylatesChk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 19:1162–1174.
32. Lukas, C., J. Falck, J. Bartkova, J. Bartek, and J. Lukas. 2003. Distinctspatiotemporal dynamics of mammalian checkpoint regulators induced byDNA damage. Nat. Cell Biol. 5:255–260.
33. McIlwraith, M. J., A. Vaisman, Y. Liu, E. Fanning, R. Woodgate, and S. C.West. 2005. Human DNA polymerase eta promotes DNA synthesis fromstrand invasion intermediates of homologous recombination. Mol. Cell 20:783–792.
34. Nyberg, K. A., R. J. Michelson, C. W. Putnam, and T. A. Weinert. 2002.Toward maintaining the genome: DNA damage and replication checkpoints.Annu. Rev. Genet. 36:617–656.
35. Oakley, G. G., S. M. Patrick, J. Yao, M. P. Carty, J. J. Turchi, and K. Dixon.2003. RPA phosphorylation in mitosis alters DNA binding and protein-protein interactions. Biochemistry 42:3255–3264.
36. Olson, E., C. J. Nievera, V. Klimovich, E. Fanning, and X. Wu. 2006. RPA2is a direct downstream target for ATR to regulate the S-phase checkpoint.J. Biol. Chem. 281:39517–39533.
37. Patrick, S. M., G. G. Oakley, K. Dixon, and J. J. Turchi. 2005. DNA damageinduced hyperphosphorylation of replication protein A. 2. Characterizationof DNA binding activity, protein interactions, and activity in DNA replica-tion and repair. Biochemistry 44:8438–8448.
38. Plate, I., S. C. L. Hallwyl, I. Shi, L. Krejci, C. Muller, L. Albertsen, P. Sung,and U. H. Mortensen. 2008. Interaction with RPA is necessary for Rad52repair center formation and for its mediator activity. J. Biol. Chem. 283:29077–29085.
39. Robison, J. G., L. Lu, K. Dixon, and J. J. Bissler. 2005. DNA lesion-specificco-localization of the Mre11/Rad50/Nbs1 (MRN) complex and replicationprotein A (RPA) to repair foci. J. Biol. Chem. 280:12927–12934.
40. Sancar, A., L. A. Lindsey-Boltz, K. Unsal-Kacmaz, and S. Linn. 2004. Mo-lecular mechanisms of mammalian DNA repair and the DNA damage check-points. Annu. Rev. Biochem. 73:39–85.
41. Scheffner, M., B. A. Werness, J. M. Huibregtse, A. J. Levine, and P. M.Howley. 1990. The E6 oncoprotein encoded by human papillomavirus types16 and 18 promotes the degradation of p53. Cell 63:1129–1136.
42. Shcherbakova, P. V., K. Bebenek, and T. A. Kunkel. 2003. Functions ofeukaryotic DNA polymerases. Sci. Aging Knowledge Environ. 2003:RE3.
43. Shiloh, Y. 2006. The ATM-mediated DNA-damage response: taking shape.Trends Biochem. Sci. 31:402–410.
44. Solomon, D. A., M. C. Cardoso, and E. S. Knudsen. 2004. Dynamic targetingof the replication machinery to sites of DNA damage. J. Cell Biol. 166:455–463.
5708 FENG ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.

45. Su, T. T. 2006. Cellular responses to DNA damage: one signal, multiplechoices. Annu. Rev. Genet. 40:187–208.
46. Syljuasen, R. G., S. Jensen, J. Bartek, and J. Lukas. 2006. Adaptation to theionizing radiation-induced G2 checkpoint occurs in human cells and dependson checkpoint kinase 1 and Polo-like kinase 1 kinases. Cancer Res. 66:10253–10257.
47. Toczyski, D. P., D. J. Galgoczy, and L. H. Hartwell. 1997. CDC5 and CKIIcontrol adaptation to the yeast DNA damage checkpoint. Cell 90:1097–1106.
48. Vassin, V. M., M. S. Wold, and J. A. Borowiec. 2004. Replication protein A(RPA) phosphorylation prevents RPA association with replication centers.Mol. Cell. Biol. 24:1930–1943.
49. Waddell, D. S., N. T. Liberati, X. Guo, J. P. Frederick, and X. F. Wang. 2004.Casein kinase Iepsilon plays a functional role in the transforming growthfactor-beta signaling pathway. J. Biol. Chem. 279:29236–29246.
50. Wang, X., G. Ira, J. A. Tercero, A. M. Holmes, J. F. Diffley, and J. E. Haber.2004. Role of DNA replication proteins in double-strand break-inducedrecombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 24:6891–6899.
51. Weill, J. C., and C. A. Reynaud. 2008. DNA polymerases in adaptive immu-nity. Nat. Rev. Immunol. 8:302–312.
52. Wittekind, M., J. Reizer, J. Deutscher, M. H. Saier, and R. E. Klevit. 1989.Common structural changes accompany the functional inactivation of HPrby seryl phosphorylation or by serine to aspartate substitution. Biochemistry28:9908–9912.
53. Wold, M. S. 1997. Replication protein A: a heterotrimeric, single-strandedDNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev.Biochem. 66:61–92.
54. Wu, X., S. M. Shell, and Y. Zou. 2005. Interaction and colocalization ofRad9/Rad1/Hus1 checkpoint complex with replication protein A in humancells. Oncogene 24:4728–4735.
55. Wu, X., Z. Yang, Y. Liu, and Y. Zou. 2005. Preferential localization ofhyperphosphorylated replication protein A to double-strand break repairand checkpoint complexes upon DNA damage. Biochem. J. 391:473–480.
56. Yoo, H. Y., A. Kumagai, A. Shevchenko, and W. G. Dunphy. 2004. Adapta-tion of a DNA replication checkpoint response depends upon inactivation ofClaspin by the Polo-like kinase. Cell 117:575–588.
57. Yu, Y. M., S. M. Pace, S. R. Allen, C. X. Deng, and L. C. Hsu. 2008. APP1-binding motif present in BRCA1 plays a role in its DNA repair function.Int. J. Biol. Sci. 4:352–361.
58. Zachos, G., M. D. Rainey, and D. A. Gillespie. 2003. Chk1-deficient tumourcells are viable but exhibit multiple checkpoint and survival defects. EMBOJ. 22:713–723.
59. Zhang, J., S. Bao, R. Furumai, K. S. Kucera, A. Ali, N. M. Dean, and X. F.Wang. 2005. Protein phosphatase 5 is required for ATR-mediated check-point activation. Mol. Cell. Biol. 25:9910–9919.
60. Zou, L., and S. J. Elledge. 2003. Sensing DNA damage through ATRIPrecognition of RPA-ssDNA complexes. Science 300:1542–1548.
61. Zou, L., D. Liu, and S. J. Elledge. 2003. Replication protein A-mediatedrecruitment and activation of Rad17 complexes. Proc. Natl. Acad. Sci. USA100:13827–13832.
62. Zou, Y., Y. Liu, X. Wu, and S. M. Shell. 2006. Functions of human replicationprotein A (RPA): from DNA replication to DNA damage and stress re-sponses. J. Cell Physiol. 208:267–273.
VOL. 29, 2009 EFFICIENT DNA REPAIR REQUIRES RPA32 DEPHOSPHORYLATION 5709
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 12
Feb
ruar
y 20
22 b
y 19
1.24
1.16
3.82
.





























