Embed Size (px)
Citation preview

ORIGINAL PAPER
Recessive Spinocerebellar Ataxia with Paroxysmal CoughAttacks: A Report of Five Cases
Luis Velázquez-Pérez & Rigoberto González-Piña & Roberto Rodríguez-Labrada &
Raul Aguilera-Rodríguez & Lourdes Galicia-Polo & Yaimeé Vázquez-Mojena &
Ana M. Cortés-Rubio & Marla R. Trujillo-Bracamontes & Cesar M. Cerecedo-Zapata &
Oscar Hernández-Hernández & Bulmaro Cisneros & Jonathan J. Magaña
Published online: 6 October 2013# Springer Science+Business Media New York 2013
Abstract Hereditary ataxias are a heterogeneous group ofneurological diseases characterized by progressive cerebellarsyndrome and numerous other features, which result in greatdiversity of ataxia subtypes. Despite the characterization of anumber of both autosomal dominant and autosomal recessiveataxias, it is thought that a large group of these conditionsremains to be identified. In this study, we report the charac-terization of five patients (three Mexicans and two Italians)who exhibit a peculiar form of recessive ataxia associated withcoughing. The main clinical and neurophysiological featuresof these patients include cerebellar ataxia, paroxysmal cough,restless legs syndrome (RLS), choreic movements, atrophy ofdistal muscles, and oculomotor disorders. Brain magneticresonance imaging (MRI) revealed cerebellar atrophy, whilevideo polysomnography (VPSG) studies showed a severepattern of breathing-related sleep disorder, including sleep
apnea, snoring, and significant oxygen saturation in theabsence of risk factors. All patients share clinical featuresin the peripheral nervous system, including reduction ofamplitude and prolonged latency of sensory potentials inmedian and sural nerves. Altogether, clinical criteria as wellas molecular genetic testing that was negative for differentautosomal dominant and autosomal recessive ataxias suggestthe presence of a new form of recessive ataxia. This ataxia,in which cerebellar signs are preceded by paroxysmal cough,affects not only the cerebellum and its fiber connections, butalso the sensory peripheral nervous system and extracerebellarcentral pathways.
Keywords Ataxia . Autosomal recessive ataxia . Autosomaldominant ataxia . Cerebellar atrophy . Sensory neuropathy .
Sleep disorder paroxysmal cough
L. Velázquez-Pérez (*) :R. Rodríguez-Labrada :R. Aguilera-Rodríguez :Y. Vázquez-MojenaCenter for the Research and Rehabilitation of the Hereditary Ataxias(CIRAH), Carretera Central Km 5 ½ Reparto Edecío Pérez,80100 Holguín, Cubae-mail: [email protected]
L. Velázquez-Péreze-mail: [email protected]
R. González-PiñaDepartment of Brain Plasticity, National Rehabilitation Institute(INR), Mexico City, Mexico
L. Galicia-PoloClinic of Sleep Disorders, School of Medicine, UNAM,Mexico City, Mexico
A. M. Cortés-RubioMagnetic Resonance Service, INR, Mexico City, Mexico
M. R. Trujillo-BracamontesDepartment of Audiology and Otoneurology, INR,Mexico City, Mexico
C. M. Cerecedo-Zapata :O. Hernández-Hernández :J. J. Magaña (*)Laboratory of Genomic Medicine, Department of Genetics, INR,Calzada México-Xochimilco No. 289, Colonia Arenal Guadalupe,14389 Mexico City, Mexicoe-mail: [email protected]
B. CisnerosDepartament of Genetics andMolecular Biology, CINVESTAV-IPN,Mexico City, Mexico
Cerebellum (2014) 13:215–221DOI 10.1007/s12311-013-0526-3

Introduction
The term ataxia means absence of order, denotes a clinicalsyndrome of incoordination, and is used to describe neurologicaldiseases in which progressive ataxia is the most importantclinical presentation [1]. The classification of ataxias, accordingto current cause, can be subdivided into the following threemajor groups: hereditary ataxias, non-hereditary degenerativeataxias, and acquired ataxias [2].
Hereditary ataxias are a heterogeneous group of neurologicaldiseases involving the Mendelian inheritance pattern. Thesegroups of neurodegenerative disorders are caused by degenera-tion of the cerebellum and its afferent and efferent connections,including the spinal cord, brainstem, and peripheral nerves[3, 4]. In general, the phenotypical presentation exhibits aprogressive cerebellar syndrome characterized by ataxic gait,cerebellar dysarthria, dysmetria, and dysdiadochokinesia.The hereditary ataxias are usually subdivided by their modeof inheritance into autosomal dominant, autosomal recessive,X-linked, or mitochondrial. Autosomal dominant formsmainly affect adults, and within this group of pathologies,spinocerebellar ataxias (SCA) represent the most frequent type.On the other hand, autosomal recessive ataxias usually havetheir onset in childhood or adolescence. This group encompassesfive main subgroups as follows: congenital, metabolic, DNArepair defects, degenerative, and ataxias with other features. Theclinical diagnosis of autosomal recessive ataxias must be con-firmed by ancillary tests such as magnetic resonance imaging(MRI), scanning, electrophysiological examination, and muta-tion analysis when the causative gene has been identified.
Despite progress in research on the different forms ofhereditary ataxias, it appears that a large group of these con-ditions remains to be identified. In 2006, Countinho et al. [5]reported six Portuguese families with autosomal dominantataxia that segregates with paroxysmal cough. However, therehave been no additional reports of ataxic kindreds exhibitingthis syndrome. Recently, we examined patients belonging totwo distinct families, a Mexican family and an Italian family,both of which exhibited this particular association betweenataxia and coughing; however, unlike the ataxic Portuguesekindred, the five patients described herein showed autosomalrecessive ataxia. We describe the main clinical and neuro-physiological characteristics of these patients which wereassessed following the ethical principles of the Declaration ofHelsinki. All subjects gave their informed consent.
Case Report
Mexican Kindred
A 67-year-old female patient (Mx-II-1; Table 1, Fig. 1a) beganat the age of 18 years to experience paroxysmal cough attacks
that never occurred during sleep, without any recognizableprecipitating factors. Duration of the coughing bouts was of afew minutes, and the episodes aborted when the patient drankwater. After 47 years of age, the patient was diagnosed as havingtype 2 diabetes mellitus (DM-2). At the age of 50 years, shenoticed gait instability and incoordination in the upper limbs,with the progressive increase of these clinical manifestations,confining the patient to a wheelchair at the age of 65 years. Theneurological examination demonstrated a progressive cerebellarsyndrome characterized by severe ataxia of the gait, dysarthria,dysmetria, dysdiadochokinesia, and intention tremor. The Scalefor the Assessment and Rating of Ataxia (SARA) [6] score was30. Sleep disturbances were frequent, with complaints such asinsomnia, muscle cramps, and restless legs syndrome (RLS).
Other clinical features included cognitive dysfunction,hyporeflexia of the upper and lower limbs, muscular hypotonia,decreased vibrational sense, and amyotrophy of the face and thedistal part of the upper and lower limbs, as well as choreiformmovements in hands and feet, with a score of 6 points on theInventory of Non-ataxia Symptoms (INAS) [7]. The score of theScale for Outcomes in PD-Autonomic (SCOPA-AUT) [8] was23, suggesting moderate autonomic dysfunctions. Other keycomplaints consisted of constipation and pollakiuria.
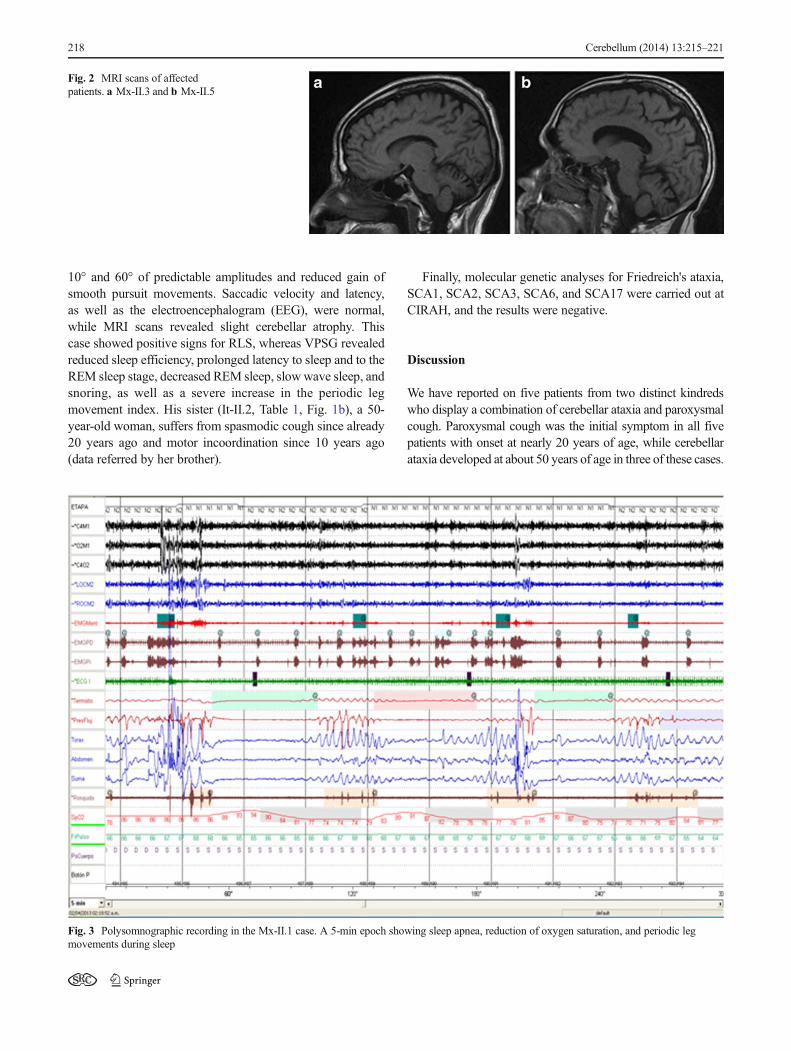
The family history revealed the following information: thepatient's parents (Mx-I.1 andMx-I.2; Fig. 1a) suffered fromDM-2 and both died at about 90 years of age. The patient has foursiblings; two of the brothers (Mx-II.3 and Mx-II.5; Table 1,Fig. 1) have ataxia, DM-2, allergies, and paroxysmal cough.However, there was no evidence of respiratory disease or gastriccomplaints in either, and both displayed the same neurologicalclinical features, including cerebellar syndrome paroxysmalcough, choreic movements in hands and feet, sleep abnormali-ties, sensory neuropathy, cognitive dysfunctions, andhyporeflexia in the lower limbs. SARA and INAS scores were12 and 5, respectively, whereas autonomic signs were slight(SCOPA-AUT scores, 8 points) in both cases. In the patientMx-II.3, the age of onset of cough and gait instability was at18 and 24 years, respectively, while in Mx-II.5, cough appearedat 27 years of age, while gait ataxia began at 50 years. Bothbrothers underwent neurophysiological assessment, and themost important findings in the peripheral nervous system werereduction of the amplitude and prolonged latency of the sensorypotentials in the median and sural nerves; however, motor nerveconduction velocities and latency and amplitude in the medianand peroneal nerves fell within the normal range in bothpatients. Brainstem auditory-evoked potential studies(BSAEP) showed abnormal morphology and reproducibilityof potentials, and the P40 components of the somatosensory-evoked potentials of the tibial nerves (Tn-SSEP) were absent.Oculomotor testing by video nystagmography revealed saccad-ic hypermetria, horizontal nystagmus, and impaired smoothpursuit in both cases. Furthermore, brain MRI revealed cerebel-lar atrophy in both of these individuals (Fig. 2a, b). With respect
216 Cerebellum (2014) 13:215–221
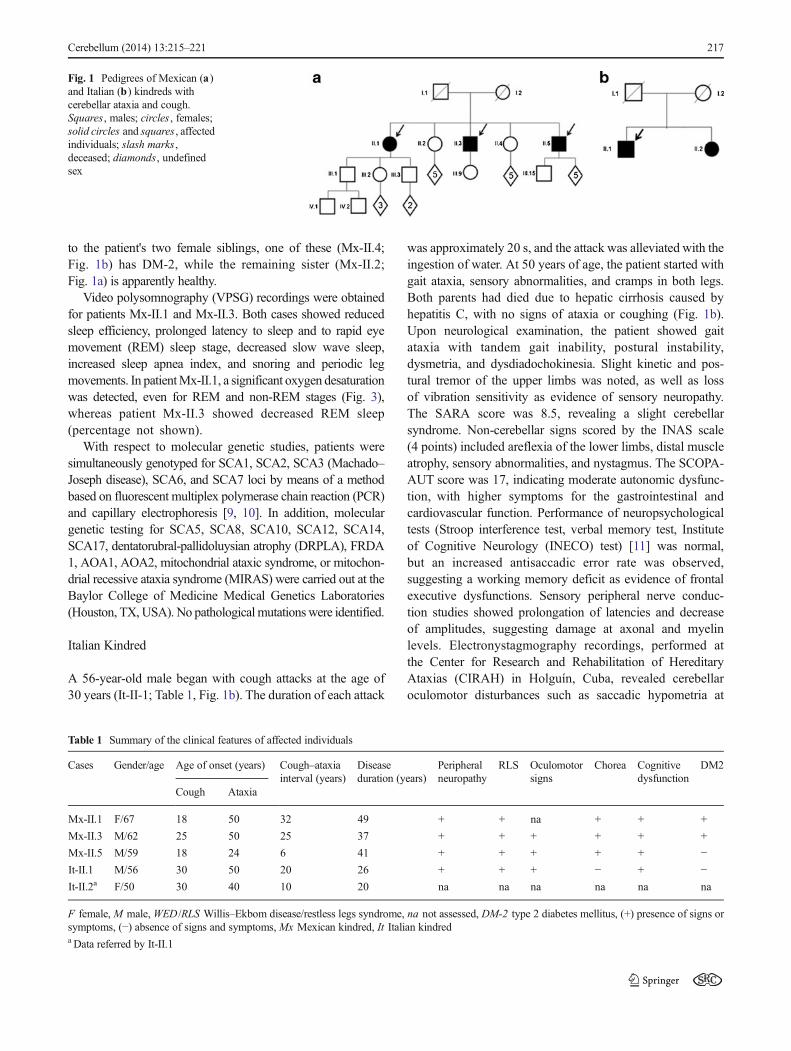
to the patient's two female siblings, one of these (Mx-II.4;Fig. 1b) has DM-2, while the remaining sister (Mx-II.2;Fig. 1a) is apparently healthy.
Video polysomnography (VPSG) recordings were obtainedfor patients Mx-II.1 and Mx-II.3. Both cases showed reducedsleep efficiency, prolonged latency to sleep and to rapid eyemovement (REM) sleep stage, decreased slow wave sleep,increased sleep apnea index, and snoring and periodic legmovements. In patientMx-II.1, a significant oxygen desaturationwas detected, even for REM and non-REM stages (Fig. 3),whereas patient Mx-II.3 showed decreased REM sleep(percentage not shown).
With respect to molecular genetic studies, patients weresimultaneously genotyped for SCA1, SCA2, SCA3 (Machado–Joseph disease), SCA6, and SCA7 loci by means of a methodbased on fluorescent multiplex polymerase chain reaction (PCR)and capillary electrophoresis [9, 10]. In addition, moleculargenetic testing for SCA5, SCA8, SCA10, SCA12, SCA14,SCA17, dentatorubral-pallidoluysian atrophy (DRPLA), FRDA1, AOA1, AOA2, mitochondrial ataxic syndrome, or mitochon-drial recessive ataxia syndrome (MIRAS) were carried out at theBaylor College of Medicine Medical Genetics Laboratories(Houston, TX,USA). No pathological mutationswere identified.
Italian Kindred
A 56-year-old male began with cough attacks at the age of30 years (It-II-1; Table 1, Fig. 1b). The duration of each attack
was approximately 20 s, and the attack was alleviated with theingestion of water. At 50 years of age, the patient started withgait ataxia, sensory abnormalities, and cramps in both legs.Both parents had died due to hepatic cirrhosis caused byhepatitis C, with no signs of ataxia or coughing (Fig. 1b).Upon neurological examination, the patient showed gaitataxia with tandem gait inability, postural instability,dysmetria, and dysdiadochokinesia. Slight kinetic and pos-tural tremor of the upper limbs was noted, as well as lossof vibration sensitivity as evidence of sensory neuropathy.The SARA score was 8.5, revealing a slight cerebellarsyndrome. Non-cerebellar signs scored by the INAS scale(4 points) included areflexia of the lower limbs, distal muscleatrophy, sensory abnormalities, and nystagmus. The SCOPA-AUT score was 17, indicating moderate autonomic dysfunc-tion, with higher symptoms for the gastrointestinal andcardiovascular function. Performance of neuropsychologicaltests (Stroop interference test, verbal memory test, Instituteof Cognitive Neurology (INECO) test) [11] was normal,but an increased antisaccadic error rate was observed,suggesting a working memory deficit as evidence of frontalexecutive dysfunctions. Sensory peripheral nerve conduc-tion studies showed prolongation of latencies and decreaseof amplitudes, suggesting damage at axonal and myelinlevels. Electronystagmography recordings, performed atthe Center for Research and Rehabilitation of HereditaryAtaxias (CIRAH) in Holguín, Cuba, revealed cerebellaroculomotor disturbances such as saccadic hypometria at
Fig. 1 Pedigrees of Mexican (a)and Italian (b) kindreds withcerebellar ataxia and cough.Squares , males; circles , females;solid circles and squares , affectedindividuals; slash marks ,deceased; diamonds, undefinedsex
Table 1 Summary of the clinical features of affected individuals
Cases Gender/age Age of onset (years) Cough–ataxiainterval (years)
Diseaseduration (years)
Peripheralneuropathy
RLS Oculomotorsigns
Chorea Cognitivedysfunction
DM2
Cough Ataxia
Mx-II.1 F/67 18 50 32 49 + + na + + +
Mx-II.3 M/62 25 50 25 37 + + + + + +
Mx-II.5 M/59 18 24 6 41 + + + + + −It-II.1 M/56 30 50 20 26 + + + − + −It-II.2a F/50 30 40 10 20 na na na na na na
F female, M male,WED /RLS Willis–Ekbom disease/restless legs syndrome, na not assessed, DM-2 type 2 diabetes mellitus, (+) presence of signs orsymptoms, (−) absence of signs and symptoms, Mx Mexican kindred, It Italian kindreda Data referred by It-II.1
Cerebellum (2014) 13:215–221 217
10° and 60° of predictable amplitudes and reduced gain ofsmooth pursuit movements. Saccadic velocity and latency,as well as the electroencephalogram (EEG), were normal,while MRI scans revealed slight cerebellar atrophy. Thiscase showed positive signs for RLS, whereas VPSG revealedreduced sleep efficiency, prolonged latency to sleep and to theREM sleep stage, decreased REM sleep, slow wave sleep, andsnoring, as well as a severe increase in the periodic legmovement index. His sister (It-II.2, Table 1, Fig. 1b), a 50-year-old woman, suffers from spasmodic cough since already20 years ago and motor incoordination since 10 years ago(data referred by her brother).
Finally, molecular genetic analyses for Friedreich's ataxia,SCA1, SCA2, SCA3, SCA6, and SCA17 were carried out atCIRAH, and the results were negative.
Discussion
We have reported on five patients from two distinct kindredswho display a combination of cerebellar ataxia and paroxysmalcough. Paroxysmal cough was the initial symptom in all fivepatients with onset at nearly 20 years of age, while cerebellarataxia developed at about 50 years of age in three of these cases.
Fig. 2 MRI scans of affectedpatients. a Mx-II.3 and b Mx-II.5
Fig. 3 Polysomnographic recording in the Mx-II.1 case. A 5-min epoch showing sleep apnea, reduction of oxygen saturation, and periodic legmovements during sleep
218 Cerebellum (2014) 13:215–221

Additionally, these patients showed peripheral neuropathy,chorea, RLS, autonomic dysfunctions, cognitive deficits, oculo-motor disturbances, and sleep disorders, which were importantfeatures to understand the phenotype, as result of the multisys-tem involvement of peripheral and central nervous systems.This clinical profile is common in several molecular forms ofhereditary ataxia, supporting the heterogeneity of these entities[1–4]. Nevertheless, the cough represents a phenotypical hall-mark in these patients.
With regard to the inheritance pattern, genealogical analysessuggest autosomal recessive inheritance because the clinicalpicture appeared in both males and females, and this phenotypewas not observed in their parents or in their descendants.Moreover, the age of onset at about 20–30 years does notpreclude the inheritance pattern suggested for this new formof ataxia; although recessive ataxias are regularly known asearly-onset diseases, several authors have reported a largeheterogeneity in relation to age of onset, progression, andphenotypic manifestations [12]. For example, Friedreich'sataxia has been classified, according to age of onset, intoearly-onset (before 10 years) [13], late-onset (after 25 years)[14], and very late-onset types (after 40 years) [15].
The majority of early-onset autosomal recessive cerebellarataxias can be clinically discarded in both kindred becauseataxia usually appears prior to the age of 20 years in theseentities. Additionally, autosomal recessive cerebellar ataxiatype 1, autosomal recessive cerebellar ataxia type 2, andNiemann–Pick disease type C were excluded based on theabsence of the neuropathic criteria, because all patientsdisplayed clinical and electrophysiological signs of peripheralneuropathy. Likewise, molecular genetic testing allowed us toexclude Friedreich's ataxia in four patients. In the Mexicankindred, different molecular genetic tests were conducted formitochondrial ataxic syndrome, ataxia telangiectasia, andataxia with oculomotor apraxia types 1 and 2. The absenceof the main clinical hallmarks for ataxia telangiectasia andataxia with oculomotor apraxia types 1 and 2 (telangiectasiaand oculomotor apraxia, respectively), as well as the laterage of onset, helped us to exclude these pathologies fromthe Italian family. Moreover, molecular testing excludedthe presence of various autosomal dominant ataxias in thetwo genealogies.
To date, there has been a unique report of cerebellar ataxiaassociated with paroxysmal cough, which was described insix Portuguese families [5]. In contrast with our cases, theinheritance pattern of the Portuguese kindred was autosomaldominant, and the onset of cough in such patients was atlater ages. Therefore, the kindreds in this report represent arare form of recessive ataxia associated with paroxysmalcough and other neurological manifestations. With respectto the Mexican kindred, we observed familial aggregationfor DM-2; however, the fact that this metabolic disease waspresent indiscriminately in cerebellar-affected and unaffected
siblings indicates that DM-2 did not segregate with cerebellarataxia in this family. This is distinct from Friedreich's ataxia,in which increased susceptibility to DM-2 exists in more than20 % of cases with DM-2 [16].
VPSG studies showed a severe pattern of breathing-relatedsleep disorders including sleep apnea, snoring, and significantoxygen desaturation in the absence of risk factors such asobesity or craniofacial dysmorphology. Therefore, these sleepdisorders could be another specific hallmark of this new typeof hereditary ataxias, contributing to its diagnosis.
Cough, Cerebellum, and Ataxia
Cough is a complex respiratory act characterized usually byenhanced inspiratory, compressive, and expulsive phases,which expels mucus and foreign irritants out of the respi-ratory tract. Physiologically, the activation of airway coughreceptors in the lower airway activates the afferent vagaltracts projecting to the pontine–medullary rhythm-generatingnetwork, which trigger an automatic sequence of events thatultimately activates the cough reflex [17]. The medullarycomponents of the network include three major regions—rostral ventral respiratory group (rVRG), pre-Botzinger Com-plex (pre-BotC), and Botzinger Complex (BotC)—whereasthe pontine component is subdivided into rostral and caudalparts [18]. Besides, previous functional MRI studies providedinsights into the role of supramedullary regions in generatingcoughing, such as the primary motor and sensory cortices,inferior frontal gyrus, operculum, insula, anterior and mid-cingulate cortex, ventral and dorsomedial thalamus, caudatenucleus, putamen, and cerebellum [19, 20].
The specific role of the cerebellum in the coughing controlis still unclear. Nevertheless, there are several evidences fromanimal models. Electrical stimulation of the cervical vagusafferents in anesthetized cats produced evoked potentials inthe cerebellar cortex and its deep structures through the acti-vation of climbing fibers [21]. Besides, afferent transmissionby mossy fiber has been also demonstrated in dogs [22].Autoradiographic studies demonstrated that some vagal fibersproject directly to the fastigial and anterior interpositecerebellar nuclei [23]. Moreover, cerebellar ablation andlesion of the rostral interposed nucleus reduced the coughfrequency in cats, suggesting that the cerebellum, specificallythe INr, is implicated inmodulation of the frequency of centrallygenerated coughing [24].
In addition, other evidences supporting the cerebellarinvolvement in cough control are the presence of cough andother central breathing disorders in patients with cerebellarneurodegeneration, such as SCA2, SCA3,multisystemic atrophy[25, 26], and the form of dominant ataxia with spasmodic coughdescribed in a Portuguese family, in which cerebellar signs arepreceded by spasmodic cough [5]. Integrating the evidencesgiven above, we could hypothesize that the spasmodic cough
Cerebellum (2014) 13:215–221 219

in these patients is produced by degeneration of the cerebellarinterpositus nucleus, climbing and mossy fibers, and vagalafferents to the cerebellum as well as the neuronal loss ofthe pontine–medullary rhythm-generating network. Therefore,these abnormalities may produce an increased mechanism ofcentral facilitation of the cough reflex.
The mechanism supporting the presence of cough as a signthat precedes cerebellar syndrome is unclear. Nevertheless, thepresence of a lobule-specific degeneration pattern in the cerebel-lum may explain it [27], whether the regions involved in thecontrol of the cough degenerate earlier than regions involved inthe coordination control. Another putative hypothesis may bethat the neurodegeneration of the pontine–medullary rhythm-generating network begins prior than that in the cerebellum. Thepresence of clinical manifestations preceding gait ataxia is notexclusive for this type of ataxia because these have been reportedin some recessive and dominant ataxias. For example, scoliosisor cardiomyopathy can precede ataxic gait until several years inFriedreich's ataxia [16]. In SCA2, the saccadic slowing [28],REM sleep disorders [29], and painful cramps appear morethan 15 years before ataxia onset [30, 31], whereas in SCA7,visual symptoms can be detected even for 12 years prior tocerebellar syndrome [32].
In conclusion, the association of paroxysmal coughingwithrecessive ataxia could represent a new syndrome. This form ofataxia is a neurodegenerative disease that affects not only thecerebellum and its fiber connections, but also the sensoryperipheral nervous system and the extracerebellar centralpathways. Further, molecular studies are necessary to confirmour hypothesis.
Acknowledgments We are grateful to the Mexican and Italian patientsfor the collaboration. This work was supported by the Cuban Ministry ofPublic Health and CONACyT fellowship 203861 to L-VP and by theSecretary of Science, Technology and Innovation of Distrito Federal grant(PICSA12-162) to JJ-M.
Conflicts of interest The authors declare that there are no conflicts ofinterest.
References
1. Klockgether T, Paulson H. Milestones in ataxia. Mov Disord.2011;26(6):1–8.
2. Klockgether T. Update on degenerative ataxias. Curr Opin Neurol.2011;24:339–45.
3. Velázquez-Pérez L, Rodríguez-Labrada R, García-Rodríguez J,Almaguer-Mederos L, Cruz-Mariño T, Laffita-Mesa JM. A compre-hensive review of spinocerebellar ataxia type 2 in Cuba. Cerebellum.2011;10:184–98.
4. Matilla-Dueñas A. The ever expanding spinocerebellar ataxias.(Editorial). Cerebellum. 2012;11:821–7.
5. Coutinho P, Cruz V, Tuna A, Silva E, Guimaraes J. Cerebellar ataxiawith paroxistic cough. A new form of dominant ataxia. Arch Neurol.2006;63:553–5.
6. Schmitz-Hubsch T, du Montcel ST, Baliko L, Berciano J, Boesch S,Depondt C, et al. Scale for the assessment and rating of ataxia:development of a new clinical scale. Neurology. 2006;66(11):1717–20.
7. Schmitz-Hubsch T, Coudert M, Bauer P, Giunti P, Globas C, Baliko L,et al. Spinocerebellar ataxia types 1, 2, 3, and 6: disease severity andnonataxia symptoms. Neurology. 2008;71(13):982–9.
8. Visser M, Marinus J, Stiggelbout AM, Van Hilten JJ. Assessment ofautonomic dysfunction in Parkinson's disease: the SCOPA-AUT.Mov Disord. 2004;11(19):1306–12.
9. DorschnerMO,BardenD, StephensK.Diagnosis of five spinocerebellarataxia disorders bymultiplex amplification and capillary electrophoresis.J Mol Diagn. 2002;4(2):108–13.
10. Magaña JJ, Tapia-Guerrero YS, Velázquez-Pérez L, Cerecedo-ZapataC, Maldonado-Rodríguez M, Jano-Ito J, et al. Analysis of CAGrepeats in five SCA loci in Mexican population: epidemiologicalevidence of a SCA7 founder effect. Clin Genet. 2013. doi:10.1111/cge.12114.
11. Torralva T, Roca M, Gleichgerrcht E, López P, Manes F. INECOFrontal Screening (IFS): a brief, sensitive, and specific tool toassess executive functions in dementia. J Int Neuropsychol Soc.15(5):777–786.
12. Anheim M, Tranchant C, Koenig M. The autosomal recessive cere-bellar ataxias. N Engl J Med. 2012;366(7):636–46.
13. Anheim M, Mariani LL, Calvas P, et al. Exonic deletions of FXNcause early-onset Friedreich's ataxia. Arch Neurol. 2012;69(7):912–6.
14. Schöls L, Amoiridis G, Przuntek H, Frank G, Epplen JT, Epplen C.Friedreich's ataxia: revision of the phenotype according to moleculargenetics. Brain. 1997;120:2131–40.
15. Berciano J, Infante J, García A, Polo JM, Volpini V, Combarros O.Very late-onset Friedreich's ataxia with minimal GAA1 expansionmimicking multiple system atrophy of cerebellar type. Mov Disord.2005;20:1643–5.
16. Cruz Mariño T, González Zaldívar Y, Laffita JM, Almaguer LE,Aguilera R, Almaguer D, et al. Uncommon phenotypical featuresin Cuban families with Friedreich ataxia. Neurosci Lett. 2010;472:85–9.
17. Mazzone SB, McGovern AE, Yang SK, Woo A, Phipps S, Ando A,et al. Sensorimotor circuitry involved in the higher brain control ofcoughing. Cough. 2013;9(1):7.
18. Rybak IA, Shevtsova NA, Paton JF, Dick TE, St-John WM,Mörschel M, et al. Modeling the ponto-medullary respiratory net-work. Respir Physiol Neurobiol. 2004;143(2–3):307–19.
19. Simonyan K, Saad ZS, Loucks TM, Poletto CJ, Ludlow CL. Func-tional neuroanatomy of human voluntary cough and sniff production.Neuroimage. 2007;37:401–9.
20. Mazzone SB, Cole LJ, AndoA, EganGF, FarrellMJ. Investigation ofthe neural control of cough and cough suppression in humans usingfunctional brain imaging. J Neurosci. 2011;31(8):2948–58.
21. Hennemann HE, Rubia FJ. Vagal representation in the cerebellum ofthe cat. Pflugers Arch. 1978;375(2):119–23.
22. Sobusiak T, Zimny R, Matlosz Z. Primary glossopharyngeal andvagal afferent projection into the cerebellum in the dog. J Hirnforsch.1971;13:117–34.
23. Zheng Z, Dietrichs E, Walberg F. Cerebellar afferent fibres from thedorsal motor vagal nucleus in the cat. Neurosci Lett. 1982;32:113–8.
24. Xu F, Frazier DT, Zhang Z, Baekey DM, Shannon R. Cerebellarmodulation of cough motor pattern in cats. J Appl Physiol. 1997;83:391–7.
25. Sriranjini SJ, Pal PK, Krishna N, Sathyaprabha TN. Subclinicalpulmonary dysfunction in spinocerebellar ataxias 1, 2 and 3. ActaNeurol Scand. 2010;122(5):323–8.
26. Schwarzacher SW, Rüb U, Deller T. Neuroanatomical characteristicsof the human pre-Bötzinger complex and its involvement in neuro-degenerative brainstem diseases. Brain. 2011;134(Pt 1):24–35.
220 Cerebellum (2014) 13:215–221

27. Jung BC, Choi SI, Du AX, Cuzzocreo JL, Ying HS, Landman BA,et al. MRI shows a region-specific pattern of atrophy in spinocerebellarataxia type 2. Cerebellum. 2012;11(1):272–9.
28. Velázquez-Pérez L, Seifried C, Abele M, Wirjatijasa F, Rodríguez-Labrada R, Santos-Falcón N, et al. Saccade velocity is reduced inpresymptomatic spinocerebellar ataxia type 2. Clin Neurophysiol.2009;120(3):632–5.
29. Rodríguez-Labrada R, Velázquez-Perez L, Ochoa NC, Polo LG,Valencia RH, Cruz GS, et al. Subtle rapid eye movement sleepabnormalities in presymptomatic spinocerebellar ataxia type 2 genecarriers. Mov Disord. 2011;26(2):347–50.
30. Velázquez-Pérez Luis and Rodríguez Labrada R. Early manifesta-tions of spinocerebellar ataxias type 2. Holguín: Ediciones Holguín;2012. ISBN 978-959-221-353-1 [Book in Spanish].
31. Jacobi H, Reetz K, du Montcel ST, Bauer P, Mariotti C, Nanetti L,et al. Biological and clinical characteristics of individuals at risk forspinocerebellar ataxia types 1, 2, 3, and 6 in the longitudinalRISCA study: analysis of baseline data. Lancet Neurol. 2013;12(7):650–8.
32. Manrique RK, Noval S, Aguilar-Amat MJ, Arpa J, Rosa I, Contreras I.Ophthalmic features of spinocerebellar ataxia type 7. JNeuroophthalmol.2009;29(3):174–9.
Cerebellum (2014) 13:215–221 221







![Spinocerebellar ataxia: an update · ataxia with pigmentary macular degeneration and con-sists of only SCA 7 [20]. ADCA type 3 refers to ‘pure’ cerebellar ataxia, which includes](https://img.pdfslide.net/doc/110x75/5f60a23d2190f22226185a55/spinocerebellar-ataxia-an-update-ataxia-with-pigmentary-macular-degeneration-and.jpg)





















