Embed Size (px)
Citation preview

Bone 52 (2013) 366–371
Contents lists available at SciVerse ScienceDirect
Bone
j ourna l homepage: www.e lsev ie r .com/ locate /bone
Case Report
Severe osteoporosis and mutation in NOTCH2 gene in a woman withHajdu–Cheney syndrome
Ioannis P. Stathopoulos a,b,⁎, George Trovas a, Kalliopi Lampropoulou-Adamidou a,b, Theodora Koromila c,Panagoula Kollia c, Nikolaos A. Papaioannou a, George Lyritis a
a Laboratory for the Research of the Musculoskeletal System “Theodoros Garofalidis”, University of Athens, KAT hospital, Athens, Greeceb Third Orthopaedic Department, University of Athens, KAT hospital, Athens, Greecec Laboratory of Human Genetics, Department of Genetics & Biotechnology, Faculty of Biology, University of Athens, Athens, Greece
⁎ Corresponding author at: Laboratory for the ResearchGarofalidis”, University of Athens, KAT Hospital, 10, AthiGreece. Fax: +30 2108018122.
E-mail address: [email protected] (I.P. Stat
8756-3282/$ – see front matter © 2012 Elsevier Inc. Allhttp://dx.doi.org/10.1016/j.bone.2012.10.027
a b s t r a c t
a r t i c l e i n f oArticle history:Received 29 August 2012Revised 18 October 2012Accepted 19 October 2012Available online 29 October 2012
Edited by: Rene Rizzoli
Keywords:Hajdu–Cheney syndromeNOTCH2 mutationOsteoporosisAcro-osteolysisSkeletal dysplasia
Hajdu–Cheney syndrome (HCS) is a rare genetic disorder characterised by acro-osteolysis, skull deformationand generalised osteoporosis. Recently, truncating mutations in the last exon of NOTCH2, a protein-codinggene, were found to be responsible. We present the case of a young woman with HCS in whom clinical andradiologic diagnosis was confirmed with DNA tests.
© 2012 Elsevier Inc. All rights reserved.
Introduction
Hajdu–Cheney syndrome (HCS) is a rare disorder characterised byprominent skeletal dysplasia featured by craniofacial changes, dentalanomalies, short stature, acro-osteolysis and generalised osteoporosis[1–3]. Althoughmost of the reported cases are sporadic, thefirst case re-ports of HCS indicated that the syndrome is inheritable and the hereditypattern autosomal dominant. Recently, the genetic cause of HCS wasidentified to be mutations in the NOTCH2 gene, the coding gene forNotch2 receptor (Table 1) [4–6]. Notch receptors after binding to theirligands undergo changes that lead to translocation of their intracellulardomain to the nucleus, thus activating the transcription of Notch targetgenes that have among others a dominant role in skeletal developmentand bone remodeling [7]. Alterations of Notch signaling are associatedwith developmental disorders of the skeleton, such as recessivebrachydactyly, Alagille syndrome and spondylocostal dysostosis, andare also observed in osteosarcoma [7]. Here, we present the case of ayoung woman suffering from HCS for which we provide genetic confir-mation of the diagnosis.
of Musculoskeletal System “Th.nas str., Kifissia, 14561, Athens,
hopoulos).
rights reserved.
Patients and methods
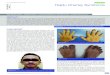
A 31-year-old woman presented in our outpatient clinic with ahistory of occasional back pain and multiple dental defects. She was139 cm tall and weighed 43 kg. Upon inspection, she appeared tohave many facial, head and hand abnormalities (Figs. 1 and 2A); shehad small face, with telecanthus and downslated palpebral fissures,micrognathia, small mouth, thin lips, long philtrum and full cheeks.Her ears were positioned low, her neck was short, her hair was coarseand many of her fingers were thick with pseudo-clubbing.
She was the first child of two healthy, non-consanguineous parents,delivered vaginally after a 37-week uncomplicated gestation. Accordingto her mother, her development was uneventful until the age of six,when it was noticed that the child was not growing in height. She hadrelatively retarded puberty (at the age of fifteen), but no menstrualdisorders. At the age of 16, she suffered an undisplaced forearm fractureafter a fall from a 2-m height, which was treated conservativelywith cast immobilisation for 6 weeks and healed well. At the age ofnineteen, dental hypermobility occurred and led to the loss of sixteeth. The patient's general health was good and her intelligencewas normal. Both of her parents were alive. However, both sufferedfrom arterial hypertension and her father had type II diabetes. Her27-year-old brother had no history of medical problems. The entirefamily provided informed consent to undergo laboratory investigation

Table 1NOTCH2 gene mutations associated with HCS.
Nucleotide mutation Protein alteration Reference
1) c.6208-6209delAGinsTCAACAC p.Pro2071ThrfsX8 [5]2) c.6272delT p.Phe2091SerfsX4 [5]3) c.6387delT p.Ser2129ArgfsX7 [5]4) c.6418C>T p.Gln2140X [5]5) c.6424-6427delTCTG p.Ser2142ArgfsX4 [5]6) c.6427-6428insT p.Glu2143X [4]7) c.6449-6450delCT p.Pro2149ArgfsX [4,5]8) c.6450delT p.Pro2149Profs5X [5]9) c.6460delT p.Val2151LeufsX4 [5]10) c.6622C>T p.Gln2208X [5,6]11) c.6853C>T p.Gln2285X [4]12) c.6949C>T p.Gln2317X [4]13) c.6973C>T p.Gln2324X [5]14) c.6986 G>T p.Glu2298X [5]15) c.7119 T>G p.Tyr2373X [4]16) c.7198C>T p.Arg2400X [5]17) c.7249-7250delCC p.Pro2417IlefsX6 [5]18) c.6667C>T p.Gln2223X [6]19) c.6403-6404delCT p.Leu2135GlufsX2 [6]20) c.6655-6840delinsG p.Pro2219GlyfsX10 [6]21) c.6457delT p.Ser2153ProfsX2 [6]22) c.7078C>T p.Gln2360X [6]
367I.P. Stathopoulos et al. / Bone 52 (2013) 366–371
and the studywas reviewed and approved by KATHospital InstitutionalReview Board.
Skull radiographs (Fig. 3) showed bathrocephaly, Wormian bonesof the lamboid suture, platybasia, absence of the frontal sinuses andlarge sella turcica. The anteroposterior projection of the spinal corddemonstrated scoliosis (double major — with a right thoracic and aleft thoracolumbar curve). At the lateral view, many of the vertebraspresented biconcave deformities of the upper and lower endplates(fishbone deformity) and decreased density (Fig. 4). The most im-pressive findings were observed at the anteroposterior view of thehands, where osteolysis of the distal phalanges was detected inmost of the fingers. In all lesions, osteolysis was found in a transversepattern across the width of the terminal phalanx (Fig. 2B).
Bone densitometry with dual-energy X-ray absorptiometry (DXA)(Lunar Prodigy Advance) showed a Z-score of −2.2 in total hip andfemoral neck and a Z-score of−5.4 in lumbar spine (L1–L4), suggestingthat her bonemineral density (BMD)was far below the expected valuesfor her age.We also assessed separately the trabecular and cortical bone
Fig. 1. A) Photograph of the patient's face. B) Lateral photograph of the patient's head and famicrognathia, small mouth, thin lips, long philtrum and full cheeks, low positioned ears, w
of the left tibia by peripheral quantitative computed tomography(pQCT) (Stratec XCT-3000 pQCT). Trabecular volumetric BMD (vBMD)at 4% of the tibia's length (counting from its distal end) was117.29 mg/cm3, cortical vBMD at 38% of the tibia's length was984.41 mg/cm3 and polar stress strain index (SSIPOL) at 14% of thetibia's length was 1131.73 mm3. In comparison to the values of healthywomen of the same age obtained from our laboratory's registry(unpublished data), the above-mentioned values were found to below (mean pQCT values of healthy women aged 25 to 35 years old: tra-becular vBMD at 4% of the tibia's length 229.66 mg/cm3; cortical vBMDat 38% of the tibia's length 1174.78 mg/cm3; SSIPOL at 14% of the tibia'slength 1236.63 mm3). Further laboratory examination using bloodand urinary tests was negative for bone metabolism disorders andendocrinopathies. Biochemical markers of bone turnover were withinnormal reference values (rf), except for high bone alkaline phosphatase(16.0 μg/l; rf below 14.3 μg/l).
Based on the clinical, radiologic and laboratory findings the predom-inant diagnosis was that of HCS. Recently, the genetic cause of HCS hasbeen attributed to truncating mutations in the NOTCH2 gene [4–6]. Inorder to examine whether such mutations were responsible for thehere-presented HCS incidence and to confirm the diagnosis, weperformed DNA sequencing analysis not only in the affected individual,but also in each available family member (parents and brother). Geno-mic DNA from peripheral blood was isolated using the QIAamp DNABloodMini Kit (QIAGEN). The entireNOTCH2 gene sequencewas ampli-fied by polymerase chain reaction (PCR) in a DNA Thermocycler(Mastercycler epgradient, Eppendorf) and sequenced. Two pairs ofprimers were designed in order to cover the seventeen mutations ofthe exon 34 that at the time of our study had been linked to HCS [4,5](Table 1, 1–17). Sanger sequencing was performed in triplicates persample in both forward and reverse directions. Genomic DNA wildtype sequences were obtained from GenBank [NOTCH2: Chromosome:1; NC_000001.10 (120454176..120612317), Location: 1p13-p11, ID:4853]. The p.Pro2149Profs5Xmutation in the HCS patient was detectedby sequences multiple alignments using Chromas software (ChromasLite 2.01, ChromasPro 1.5) and BLAST. Wild-type alleles of all studiedmutationswere found in the parental and brother samples (Fig. 5). Spe-cifically, themutation identified in the affected individual was shown tohave arisen de novo, confirming Simpson et al.'s previously reporteddata [5].
Additional examination did not reveal any non-skeletal defects,such as renal cysts and skin disorders.
ce. The following are noted: small face, telecanthus and downslated palpebral fissures,ith a crease in the lobules, short neck and coarse hair.

Fig. 2. A) Photograph of the patient's hands. Many of her fingers are thick (predominately the right thumb) with pseudo-clubbing. B) Anteroposterior radiograph of the patient'shands. Osteolysis of the distal phalanges is found in most of the fingers (only the left thumb, the left fourth and the right third fingers have normal appearance). In all lesionsosteolysis has a transverse pattern across the width of the terminal phalanx (white arrows).
Fig. 3. Radiographs of the patient's skull. A) Antero-posterior view. B) Lateral view. The following are noted: deep occipital fossa with protuberance of the squamous portion of theoccipital bone (bathrocephaly — large white arrows), Wormian bones of the lamboid suture (small white arrows), platybasia (angle a—Welcher basal angle >140°), absence of thefrontal sinuses and large sella turcica (black arrow).
368 I.P. Stathopoulos et al. / Bone 52 (2013) 366–371

Fig. 4. Radiographs of the patient's spine. A) Anteroposterior view. B) Lateral view. The following are noted: scoliosis (double major—with a right thoracic and a left thoracolumbarcurve — white arrows), biconcave deformities of the upper and lower endplates (fishbone deformity — black arrows) of many vertebras and decreased bone density.
Fig. 5. Sequence chromatograms covering the p.Pro2149Profs5X mutation, identified in pedigrees HCS patient (a), as well as the parental (b, c) and brother (d) wild type sequences,amplified from genomic DNA. The red arrow indicates the p.Pro2149Profs5X mutation's site detected in the HCS patient.
369I.P. Stathopoulos et al. / Bone 52 (2013) 366–371

370 I.P. Stathopoulos et al. / Bone 52 (2013) 366–371
Discussion
Hajdu–Cheney syndrome was first described by Hajdu andKauntze in 1948 under the term “cranio-skeletal dysplasia” [8]. In1965, Cheney gave a more precise radiologic description of the dis-ease and attributed it the name “acro-osteolysis” [9]. Followingthose two initial reports, more terms were introduced to refer tothe syndrome, such as “arthro-dento-osteo dysplasia” [10], “heredi-tary osteodysplasia with acro-osteolysis” [11] and “idiopathic familialacro-osteolysis” [12].
HCS presents mainly craniofacial and musculoskeletal clinicalfeatures (Table 2). Over 50% of the patients have short stature, char-acteristic coarse face with micrognathia, full cheeks, bushy eyebrows,telecanthus, abnormal dentition, short neck, digital anomalies(clubbed/stubby fingers/toes) and joint hypermobility [1–3]. Furtherto those features, renal, intestinal, cardiac, dermatological, neurologi-cal, and visual defects can also be observed. It is noteworthy that, over10% of the affected people appear to additionally have polycystic dis-ease of the kidneys [13].
Patients with HCS have distinctive radiologic appearance(Table 2). Acro-osteolysis and skull defects are the most constantfindings in plain radiographs. Brennan and Pauli [2] estimated that84% of the patients they reviewed had acro-osteolysis (the remaining16% were at an age that this feature could not be identified). In addi-tion, Ramos et al. [1] reported that acro-osteolysis is present in more
Table 2Clinical and radiological features of HCS.
Clinical features Radiological features
Head/face/neck:– coarse facea
– micrognathiaa
– full cheeksa
– short necka
– bushy eyebrowsa
– telecanthusa
– coarse hair– low hairline in the forehead– facial hirsutism– down slanted palpebral fissures– synophrys– flat nasal bridge– small mouth– long philtrum– low set ears– high arched palate and cleft palate
Skull:– Wormian bones (>65% of patients)– open suturesa
– platybasia (with or without basilarinvagination) a
– abnormalities of the sella turcicaa
– absence or hypoplasia of the frontalsinuses– dolichocephaly– bathrocephaly– resorption of alveolar bone– thickened calvaris– obtuse mandibular angle– micrognathiaa
Digital anomalies (clubbed/stubbyfingers/toes) a
Acro-osteolysis (>90% of patients)
Short staturea Spine:– biconcave/compression deformitiesof the vertebraea
– scoliosis– kyphosis– spondylolisthesis– decreased bone densitya
Joint hypermobilitya
Abnormal dentitiona
Neurological defects:– headaches (mostly occipital or frontal)– backaches– conductive and sensorineuralhearing loss– optic nerve atrophy– optic disc swelling– paralysis of the facial nerve– hydrocephalus– syringomyelia
Other:– polycystic disease of the kidneys– intestinal malrotation– congenital heart disease
Other:– elongated/serpentine fibula– dislocation and hypoplasia of theradial head– bone cysts– genu valgum– coxa valga– bowing of the long bones– multiple fractures of the long bones– mesomelic shortening of the arms
a Observed in over 50% of the patients.
than 90% of the HCS patients. Acro-osteolysis consists of bone loss ofthe terminal phalanges of the fingers and, less often, the toes. Twoacro-osteolysis patterns have been reported: (a) distal to proximaland (b) transverse [2]. All fingers can be affected, but usually one ormore are normal. The bony defect is responsible for the clinical ap-pearance of the fingers (short and clubbed) and, in some cases, isaccompanied by pain and fingernail anomalies (discoloration anddystrophy).
Plain radiographs of the skull show Wormian bones in over 65% ofthe patients, while open sutures, platybasia (with or without basilar in-vagination) and abnormalities of the sella turcica are observed in over50% of the cases [2]. Radiographs of the spine showdecreasedboneden-sity in 60% of the patients, biconcave/compression deformities ofthe vertebrae in approximately 50%, scoliosis in over 35%, kyphosis inover 20% and spondylolisthesis in over 10% of the cases [1–3]. Some pa-tients that present bilateral or unilateral elongated/serpentine fibula,which in combination to polycystic kidney disease, are considered thepredominant manifestations of the “serpentine fibula polycystic kidneysyndrome” (SFPKS) [14]. Based on recent evidence, it seems that thetwo syndromes, HCS and SFPKS, represent variable expressions of thesame entity [15–17].
In agreement with the radiologically detected osteopenia, BMDpresents a marked decrease, particularly in spine, when comparedto the age-adjusted normal range values. High turnover osteoporosiswas observed in iliac crest bone biopsies of a few patients with HCS[12], whereas osteoclast activity has been reported to be increased[18–20], normal [20] or decreased [12]. Reduced bone formationrate has also been described [18,20]. Whether a primary osteoclasticor osteoblastic dysfunction is responsible for the generalised osteopo-rosis remains unclear. In the present case we detected an increase inbone alkaline phosphatase, but the other biochemical markers ofbone turnover were within normal reference values.
After the first reports of HCS, it became apparent that the diseasehas a genetic background, since many patients had positive familyhistory (although the majority were sporadic cases). Based on pheno-typic studies of the affected relatives, it was revealed that the syn-drome is a pleiotropic autosomal dominant disorder and sporadiccases should be considered as the result of de novo mutations. Recentgenetic studies confirmed that finding and explained part of the path-ophysiology of HCS. HCS is caused by mutations located within thelast exon of NOTCH2, a gene that encodes the Notch 2 receptor [4,5].Notch receptors are single-pass transmembrane proteins that medi-ate cell-to-cell interactions. They play a regulatory role in skeletal de-velopment and bone remodeling by suppressing chondrocyte andosteoblast differentiation andmodulating osteoclastogenesis [7]. Muta-tions of exon 34 of NOTCH2 lead to production of a protein that lacksthe complete PEST (proline-, glutamic acid-, serine-, threonine-rich)domain [4,5], which mediates the proteosomal degradation of Notch 2receptor. Deletion of the PEST domain results in persistence of Notch in-tracellular signal, which is proposed to account for many of the HCSskeletal defects (predominately the bone loss) [4,5,7].
So far, HCS diagnosis has been based on the clinical and radiologicfindings. However confirmation of the disease by genetic analysisshould be the gold standard, especially for the cases that differentialdiagnosis is difficult or when the patient has positive familial historybut clinical and radiologic appearance are not distinctive, as the char-acteristic features of HCS are recognisable after the first few years oflife [21,22]. Differential diagnosis should be made to distinguish HCSfrom other conditions that are characterised by acro-osteolysis, suchas scleroderma and Reynaud's disease, and diseases with multipleWormian bones, such as osteogenesis imperfecta, cleidocranial dys-plasia, and hypothyroidism [21,23,24].
Many of the defects are progressive and can lead to serious dis-comfort (acro-osteolysis usually evolves and results in destructionof the terminal phalanges, while progression of platybasia and basilarinvagination often leads to worsening of the neurologic state of the

371I.P. Stathopoulos et al. / Bone 52 (2013) 366–371
patients) [2,25]. However, lifespan seems to be unaffected, thoughthere are reports of deaths at a young age [2]. Treatment is most ofthe times symptomatic. Prevention of fractures is aimed by anti-osteoporotic therapy, but bisphosphonates administered orally [26],intravenously [27] or in combination with anabolic therapy [28]have not proved effective in all cases.
Conclusions
The present case displays most of the clinical and radiologic findingsof HCS. Genetic analysis of the patient confirmed recent reports aboutthe association of HCS with truncating mutations in the exon 34 ofNOTCH2 gene. Better understanding of the underlying pathophysiologicalmechanism will provide novel insights in the study of bone metabolismin this rare disease and eventually offer additional treatment options.
References
[1] Ramos FJ, Kaplan BS, Bellah RD, Zackai EH, Kaplan P. Further evidence that theHajdu–Cheney syndrome and the “serpentine fibula-polycystic kidney syndrome”are a single entity. Am J Med Genet 1998;78:474–81.
[2] Brennan AM, Pauli RM. Hajdu–Cheney syndrome: evolution of phenotype andclinical problems. Am J Med Genet 2001;100:292–310.
[3] Currarino G. Hajdu–Cheney syndrome associated with serpentine fibulae andpolycystic kidney disease. Pediatr Radiol 2009;39:47–52.
[4] Isidor B, Lindenbaum P, Pichon O, Bezieau S, Dina C, Jacquemont S, et al. Truncatingmutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporo-sis. Nat Genet 2011;43:306–8.
[5] Simpson MA, Irving MD, Asilmaz E, Gray MJ, Dafou D, Elmslie FV, et al. Mutationsin NOTCH2 cause Hajdu–Cheney syndrome, a disorder of severe and progressivebone loss. Nat Genet 2011;43:303–5.
[6] Majewski J, Schwartzentruber JA, Caqueret A, Patry L, Marcadier J, Fryns J-P, et al.Mutations in NOTCH2 in families with Hajdu–Cheney syndrome. Hum Mutat2011;32:1114–7.
[7] Zanotti S, Canalis E. Notch regulation of bone development and remodeling andrelated skeletal disorders. Calcif Tissue Int 2012;90:69–75.
[8] Hajdu N, Kauntze R. Cranio-skeletal dysplasia. Br J Radiol 1948;21:42–8.[9] Cheney WD. Acro-Osteolysis. Am J Roentgenol Radium Ther Nucl Med 1965;94:
595–607.
[10] Herrmann J, Zugibe FT, Gilbert EF, Opitz JM. Arthro-dento-osteo dysplasia (Hajdu–Cheney syndrome). Review of a genetic “acro-osteolysis” syndrome. Z Kinderheilkd1973;114:93–110.
[11] Elias AN, Pinals RS, Anderson HC, Gould LV, Streeten DH. Hereditary osteodysplasiawith acro-osteolysis. (The Hajdu–Cheney syndrome). Am J Med 1978;65:627–36.
[12] Udell J, Schumacher Jr HR, Kaplan F, Fallon MD. Idiopathic familial acroosteolysis:histomorphometric study of bone and literature review of the Hajdu–Cheney syn-drome. Arthritis Rheum 1986;29:1032–8.
[13] Kaplan P, Ramos F, Zackai EH, Bellah RD, Kaplan BS. Cystic kidney disease inHajdu–Cheney syndrome. Am J Med Genet 1995;56:25–30.
[14] Exner GU. Serpentine fibula–polycystic kidney syndrome. A variant of theMelnick–Needles syndrome or a distinct entity? Eur J Pediatr 1988;147:544–6.
[15] Isidor B, Le Merrer M, Exner GU, Pichon O, Thierry G, Guiochon-Mantel A, et al.Serpentine fibula–polycystic kidney syndrome caused by truncating mutationsin NOTCH2. Hum Mutat 2011;32:1239–42.
[16] Gray MJ, Kim CA, Bertola DR, Arantes PR, Stewart H, Simpson MA, et al. Serpentinefibula polycystic kidney syndrome is part of the phenotypic spectrum of Hajdu–Cheney syndrome. Eur J Hum Genet 2012;20:122–4.
[17] Albano LM, Bertola DR, Barba MF, Valente M, Robertson SP, Kim CA. Phenotypicoverlap in Melnick–Needles, serpentine fibula-polycystic kidney and Hajdu–Cheney syndromes: a clinical and molecular study in three patients. ClinDysmorphol 2007;16:27–33.
[18] Nunziata V, di Giovanni G, Ballanti P, Bonucci E. High turnover osteoporosis inacro-osteolysis (Hajdu–Cheney syndrome). J Endocrinol Invest 1990;13:251–5.
[19] Blumenauer BT, Cranney AB, Goldstein R. Acro-osteolysis and osteoporosis as mani-festations of the Hajdu–Cheney syndrome. Clin Exp Rheumatol 2002;20:574–5.
[20] Brown DM, Bradford DS, Gorlin RJ, Desnick RJ, Langer LO, Jowsey J, et al. The acro-osteolysis syndrome:morphologic and biochemical studies. J Pediatr 1976;88:573–80.
[21] Marik I, Kuklik M, Zemkowa D, Kozlowski K. Hajdu–Cheney syndrome: report of afamily and a short literature review. Australas Radiol 2006;50:534–8.
[22] Leidig-Bruckner G, Pfeilschifter J, Penning N, Limberg B, Priemel M, Delling G,et al. Severe osteoporosis in familial Hajdu–Cheney syndrome: progression ofacro-osteolysis and osteoporosis during long-term follow-up. J Bone Miner Res1999;14:2036–41.
[23] Kemp SS, Dalinka MK, Schumacher HR. Acro-osteolysis. Etiologic and radiologicalconsiderations. JAMA 1986;255:2058–61.
[24] O'Reilly MA, Shaw DG. Hajdu–Cheney syndrome. Ann Rheum Dis 1994;53:276–9.[25] Hoey H, Hinde F, Grant DB. Hajdu–Cheney syndrome associated with intrauterine
fractures and arachnoid cysts. J R Soc Med 1983;76:521–3.[26] Drake WM, Hiorns MP, Kendler DL. Hadju–Cheney syndrome: response to thera-
py with bisphosphonates in two patients. J Bone Miner Res 2003;18:131–3.[27] Hwang S, Shin DY, Moon SH, Lee EJ, Lim SK, Kim OH, et al. Effect of zoledronic acid
on acro-osteolysis and osteoporosis in a patient with Hajdu–Cheney syndrome.Yonsei Med J 2011;52:543–6.
[28] McKiernan F. Integrated anti-remodeling and anabolic therapy for the osteoporosisof Hajdu–Cheney syndrome: 2-year follow-up. Osteoporos Int 2008;19:379–80.