Embed Size (px)
Citation preview

SHORT REPORT
A new syndrome, congenital extraocular muscle fibrosiswith ulnar hand anomalies, maps to chromosome 21qterT Tukel, A Uzumcu, A Gezer, H Kayserili, M Yuksel-Apak, O Uyguner, S H Gultekin, H-C Hennies,P Nurnberg, R J Desnick, B Wollnik. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
J Med Genet 2005;42:408–415. doi: 10.1136/jmg.2004.026138
Background: Congenital fibrosis of the extraocular muscles(CFEOM) is a heterogeneous group of disorders that may beassociated with other anomalies. The association of aCFEOM syndrome with ulnar hand abnormalities (CFEOM/U) has not been reported to date.Objective: To describe a new autosomal recessive syndromeof CFEOM and ulnar hand abnormalities, and localise thedisease causing gene.Methods: Clinical evaluation of the affected members andpositional mapping.Results: Six affected patients with CFEOM/U (aged 2 to 29years) from a large consanguineous Turkish family werestudied. Ophthalmological involvement was characterised bynon-progressive restrictive ophthalmoplegia with blepharop-tosis of the right eye. The postaxial oligodactyly/oligosyn-dactyly of the hands was more severe on the right side. Agenome-wide scan established linkage of this new autosomalrecessive syndrome to a locus on chromosome 21qter. Themultipoint LOD score was 4.53 at microsatellite markerD21S1259, and fine mapping defined a ,1.5 Mb criticalregion between microsatellite marker D21S1897 and thetelomere of the long arm.Conclusions: CFEOM/U maps to a 1.5 Mb region atchromosome 21qter. Future identification of the diseasecausing gene may provide insights into the development ofthe extraocular muscles and brain stem a motor neurones, aswell as anteroposterior limb development.
Heuk (1879) was the first to report the combination ofcongenital blepharoptosis and restricted eye move-ments.1 In 1950, Brown described this group of ocular
abnormalities in detail and classified the syndromes into fivedistinct phenotypes: horizontal retraction syndromes; stra-bismus fixus; vertical retraction syndromes; superior obliquetendon sheath syndromes; and a general fibrosis syndrome.2
Currently, the horizontal retraction syndromes are referred toas Duane syndrome, the superior oblique tendon sheathsyndromes as Brown syndrome, and the remaining syn-dromes as congenital fibrosis of the extraocular muscles(CFEOM).3 Recent neuropathological studies have shownthat some of the fibrosis syndromes result from develop-mental defects of particular brain stem a motor neurones andtheir corresponding axons.4 5
Duane syndrome, the most common of the CFEOMsyndromes, is characterised by limited abduction, variablylimited adduction, and globe retraction on attempted adduc-tion. Most cases are sporadic and only about 10% arefamilial.6 Cytogenetic analyses of sporadic cases revealeddeletions of 8q13. This locus was designated DURS1 (MIM
126800).7–9 Linkage analysis in autosomal dominant familiesmapped the familial disease to the DURS2 locus on 2q31(MIM 604356).10 To date, neither the DURS1 nor the DURS2gene has been identified.
Three other inherited CFEOM syndromes have beenmapped to different genetic loci:
N CFEOM1 (MIM 135700), an autosomal dominant dis-order, was mapped to 12q12,11 and the disease causinggene was recently identified as KIF21A.12 Affected indivi-duals have bilateral ptosis and restrictive ophthalmoplegia,and their eyes are fixed below the horizontally neutralposition with or without secondary esotropia or exotropia.CFEOM1 is phenotypically variable, with some patientshaving a milder expression which resembles CFEOM3.However, these families with the milder phenotypes havebeen linked to the CFEOM1 locus,13 leading to theirdesignation as CFEOM type 3A (MIM 607034).
N The CFEOM2 locus (MIM 602078), an autosomal recessivedisorder, was mapped to chromosome 11q13.3,14 andsubsequently mutations in the ARIX gene were described.15
Affected individuals with CFEOM2 have bilateral ptosis,with both eyes fixed in abduction.
N CFEOM3 (MIM 600638, formerly 604361), an autosomaldominant disorder, was mapped to chromosome 16q24.2.16
The phenotype of affected individuals in CFEOM3 familieswas variable and ranged from bilateral ptosis with fixedeyes in an infraducted and exotropic position to normallypositioned eyes with minimal limitation of vertical gazeand unilateral or absent ptosis. To date, the gene causingCFEOM3 has not been identified.
Several CFEOM syndromes occur in association with otheranomalies including the Duane radial ray syndrome (DRRS)(MIM 607323), the Wildervanck syndrome (MIM 314600),and familial horizontal gaze palsy with progressive scoliosis(MIM 607313).
DRRS is characterised by the Duane anomaly, radial rayabnormalities, and deafness. The DRRS syndrome—alsoknown as Okihiro syndrome17—is inherited as an autosomaldominant trait with variable expressivity. The DRRS locuswas mapped to 20q13 and subsequently SALL4 was identifiedas the disease causing gene.18 19
The features of the Wildervanck syndrome include theDuane anomaly, the Klippel-Feil anomaly (fused cervicalvertebrae), and congenital perceptive deafness. This disorderis mostly seen in females, suggesting that the syndrome is
Abbreviations: CFEOM, congenital fibrosis of extraocular muscles;CFEOM/U, congenital fibrosis of extraocular muscles with ulnar handabnormalities; DRRS, Duane radial ray syndrome; SNP, singlenucleotide polymorphism; OD, right eye; OS, left eye
408
www.jmedgenet.com
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from

inherited as a sex linked dominant with lethality in affectedmales.
Familial horizontal gaze palsy with progressive scoliosis isan autosomal recessive disorder characterised by progressiveexternal ophthalmoplegia and scoliosis. The disease causinggene has been mapped to chromosome 11q23–q25.20
Here, we describe a new autosomal recessive CFEOMsyndrome with prominent ulnar hand abnormalities in aconsanguineous Turkish family. The six affected individuals,aged 2 to 29 years, presented with right eye involvement andbilateral postaxial oligodactyly/oligosyndactyly of the hands,with the right more severely affected than the left. A genomescan of DNAs from family members mapped the disease locusto 21q with a multipoint LOD score of 4.525 at microsatellitemarker D21S1259. Further interrogation of the locus nar-rowed the critical region to ,1.5 Mb between D21S1897 andthe telomere of the long arm.
METHODSSubjects and medical evaluationSix affected individuals from the consanguineous Turkishfamily (fig 1) were clinically evaluated at the division of
medical genetics of the Child Health Institute and theorthoptics clinic of the department of ophthalmology,Istanbul Medical Faculty of Istanbul University, Turkey.The study was approved by the Institutional Review Board ofthe Child Health Institute of Istanbul University andinformed consent was obtained from each participant. Fiveof the six affected individuals had complete ophthalmologicalexaminations, skeletal x rays, abdominal ultrasound, echo-cardiography, and cranial magnetic resonance imaging(MRI).
Ophthalmological studiesVisual acuity was measured using a Snellen letter chartprojector transilluminated at approximately 100 cd/m2 andline acuity performances at 6 m were recorded. Non-cycloplegic refractive data were obtained using a retinoscopeor a Topcon KR-7000P autokeratorefractometer, and bi-nocular status was evaluated with a Clement–Clarke synop-tophore. Range of ocular movements was evaluated withHess screen tests in patients with binocular vision potential.Direct and indirect papillary reactions were recorded, andphotographic records of each patient were obtained. Duction
1 2 3 4
I
1 2 3 4 10 11 14 1513125-6-7
1stcousins
Distantly related
313226111
432123221
431117311
531117311
431117311
131117311
(1)(3)(1)(1)(1)(7)(3)(1)(1)
(5)(3)(3)(5)(3)(2)(3)(1)(1)
D21S1260D21S1890D21S1259D21S1912D21S171D21S1903D21S1897D21S1446rs881827
II
D21S1260D21S1890D21S1259D21S1912D21S171D21S1903D21S1897D21S1446rs881827
III
D21S1260D21S1890D21S1259D21S1912D21S171D21S1903D21S1897D21S1446rs881827
IV
D21S1260D21S1890D21S1259D21S1912D21S171D21S1903D21S1897D21S1446rs881827
143451212
431117311
443451212
131117311
143451212
533532311
431117311
131117311
8 9
313226111
133334221
313226111
123443121
313226111
431117311
313226111
531117311
10 11131117311
212535421
14
3
4 5 1312
2-3
2
313226111
313226111
1
6-7-8-9
4
1-2-3
3
Figure 1 Pedigree of the CFEOM/U family and 21qter haplotype data. The haplotypes of themost telomeric two markers, D21S1446 and rs881827, which segregated with the disease in thewhole family are shown in black. The inferred haplotypes of I-4 are indicated in parentheses.CFEOM/U, congenital fibrosis of extraocular muscles with ulnar hand abnormalities. (See onlinesupplemental table 1 for all marker data, obtainable from http://www.jmedgenet.com/supplemental/)
CFEOM with ulnar anomalies maps to 21qter 409
www.jmedgenet.com
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from

and versions in nine diagnostic gaze positions were evaluatedwith the cover test with special effort to hold the head in astraight position. Deviations in primary position when thehead was held straight were measured by the prism cover testand also during the synoptophore examination. Cycloverticaldeviations were evaluated with the three step approach,including the Bielschowsky head tilt test. Any aberrantmovements, globe retractions, or ptosis were also noted. Toassess ptosis, lid openings and levator functions weremeasured from the upper lid margin while attemptedsupraduction with frontalis function was controlled by theexaminer.
Photographs of the affected individuals were taken withtheir or their parents’ permission and blood samples wereobtained by venepuncture for DNA isolation and genemapping studies.
Histochemical and ultrastructural studiesSamples of the right superior rectus (SR) and inferior oblique(IO) muscles, obtained from patient III-11 at surgicalcorrection for strabismus, were frozen immediately in liquidnitrogen and kept at 280 C̊ until use. Fresh frozen muscletissue was processed for routine histology, histochemicalstaining (including haematoxylin and eosin, Gomori’smodified trichrome, NADH, alkaline phosphatase, acidphosphatase, ATPase at pH 9.4 and 4.2, and esterase), andelectron microscopy using standard techniques.21
DNA extraction and genotypingGenomic DNA was isolated from peripheral blood collected inEDTA from patients and family members using the DNAisolation kit for mammalian blood (Roche, Istanbul, Turkey).For the initial genome scan, DNAs were analysed using 422autosomal microsatellite markers from the genome-widehuman screening set (version 9) and single chromosome scanset (Invitrogen Life Technologies, Carlsbad, California, USA).As needed, additional microsatellite markers were obtainedfrom public databases (NCBI, Marshfield Institute, deCode),or new microsatellite markers were designed using thetandem repeat finder program.22 All new markers were onthe genomic contig NT_011515 and were named by theirpositions on the contig in kb (Human May 2004[hg17]assembly[NCBI Build 35] on the UCSC human genomebrowser). The new markers and their positions on chromo-some 21 were as follows: 1305K (44 748 500 base pairs (bp)),2044K (45 585 432 bp), 2849K (46 292 500 bp), 3086K(46 529 500 bp), 3258K (46 701 500 bp) (for primer infor-mation, see online supplemental table 2, obtainable fromhttp://www.jmedgenet.com/supplemental/). The primerswere designed using Primer3 software,23 and the fluorescentdye labelled forward primers were synthesised by InvitrogenLife Technologies. When no informative microsatellitemarkers were found in a particular region, single nucleo-tide polymorphisms (SNPs) were used for linkage analysis.SNP information was obtained from the UCSC GenomeBrowser and NCBI web sites, and primers were designedas above.
Genomic DNAs were polymerase chain reaction (PCR)amplified in 96-well microtitre plates in an oil-free systemusing a DNA Engine PTC-200 thermal cycler (MJ Research,Waltham, Massachusetts, USA). Reaction mixtures (10 ml)contained 10 ng of genomic DNA, 2 mM MgCl2, 10 mM Tris-HCl, pH 8.3, 50 mM KCl, 200 nM of each primer, 0.2 mMdNTPs, and 0.5 U of Taq DNA polymerase (AmpliTaq Gold,Applied Biosystems, Foster City, California, USA). For PCR,the reaction mixtures were initially incubated at 95 C̊ for 10minutes, and amplified for 27 cycles with denaturation at94 C̊ for 30 seconds, annealing at 56 C̊ for 30 seconds,extension at 72 C̊ for 30 seconds, and a final extension step at
Table
1Su
mm
ary
ofcl
inic
alfin
ding
sin
patie
nts
with
cong
enita
lfib
rosi
sof
extr
aocu
lar
mus
cles
with
ulna
rha
ndab
norm
aliti
es(C
FEO
M/U
)sy
ndro
me
Featu
re
Fam
ilym
ember
II-9
II-14
III-1
0III
-11
IV-1
Rig
htLe
ftRig
htLe
ftRig
htLe
ftRig
htLe
ftRig
htLe
ft
Vis
uala
cuity
20/2
020/2
020/2
00
20/2
020/2
020/2
020/2
00
20/2
05/2
00
20/2
0A
nter
ior
segm
ent
Nor
mal
Nor
mal
Nor
mal
Nor
mal
Nor
mal
Nor
mal
Nor
mal
Nor
mal
Mic
roco
rnea
Nor
mal
Post
erio
rse
gmen
tN
orm
alN
orm
alN
orm
alN
orm
alN
orm
alN
orm
alN
orm
alN
orm
alTi
lted
disc
Nor
mal
Ocu
lar
mot
ility
Dev
iatio
n14
PDXT
12
PDXT
16
PDXT
18
PDXT
25
PDhT
25
PDhT
20
PDhT
Dys
func
tioni
ngm
uscl
e(s)
SR,IO
Lev,
SR,IO
SR,
IOIO
SR,
IOA
llPt
osis
Non
eN
one
Mar
ked
Non
eN
one
Non
eM
ildN
one
Mar
ked
Non
eA
nom
alou
she
adpo
stur
eC
hin
elev
atio
nC
hin
elev
atio
nSl
ight
head
tilt
Slig
hthe
adtil
tH
ead
turn
Forc
eddu
ctio
n(2
)(2
)(2
)(2
)(2
)(2
)(+
)(2
)H
and
abno
rmal
ities
Ab
5;
ClH
y4
Ab
5;
smal
lfift
hfin
ger
bud
Ab
5A
b5
Ab
5H
y4,5
;Sy
4–5
;A
bna
il5
Ab
5;
Cl4
Ab
5A
b3,4
,5A
b4,5
Han
dra
diog
raph
yA
bM
P5;
Hy
MP
4;
Hy
ulna
rst
yloi
dpr
oces
s;A
btr
ique
trum
and
pisi
form
;fu
sed
capi
tate
and
ham
ate
Ab
MP
5;
bipa
rtite
scap
hoid
Ab
MP
5;
Hy
ulna
rst
yloi
dpr
oces
s;fu
sed
triq
uetr
uman
dlu
nate
Ab
MP
5;
Hy
ulna
rst
yloi
dpr
oces
s;fu
sed
triq
uetr
uman
dlu
nate
Ab
MP
5A
bM
P5;
Hy
MP
4A
bM
P5
Ab
MP
5A
bM
P3,4
,5A
bM
P4,5
Ab,
abse
nt;
Cl,
clin
odac
tyly
;hT
,hy
potr
opia
;H
y,hy
popl
astic
;IO
,in
feri
orob
lique
mus
cle;
IR,
infe
rior
rect
usm
uscl
e;Le
v,le
vato
rpa
lpeb
rae
supe
rior
ism
uscl
e;M
,m
etac
arpa
lbon
e;P,
phal
ange
albo
ne;
PD,
pris
mdi
opte
rs;
SR,
supe
rior
rect
usm
uscl
e;Sy
,sy
ndac
tyly
;XT,
exot
ropi
a.N
umbe
rsfo
llow
ing
abbr
evia
tions
repr
esen
tth
ere
leva
ntfin
ger
num
bers
.
410 Tukel, Uzumcu, Gezer, et al
www.jmedgenet.com
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from

72 C̊ for seven minutes. PCR products were analysed witheither an ABI Prism 3100 genetic analyser or on an ABI Prism377 DNA sequencer using GeneScan analysis software(version 3.1.2) and Genotyper software (version 2.5)(Perkin-Elmer-Cetus, Norwalk, Connecticut, USA).
PCRs for SNPs were undertaken in a final volume of 50 mland at specific annealing temperatures for each fragment for35 cycles. Amplicons were sequenced on an ABI Prism 3700capillary array sequencer using the ABI Prism BigDyeTerminator ready reaction mix (Perkin-Elmer-Cetus).Electropherograms were inspected using ABI Prism sequen-cing analysis software (version 3.4.1).
Linkage analysisThe multipoint LOD score calculations for each chromosomewere individually carried out with the SimWalk2 program(version 2.86)24 under the assumption of autosomal recessiveinheritance with full penetrance. The Mega2 program(version 2.5)25 was used to create input files for theSimWalk2 program. As data on the population incidence ofthis unique disorder were unavailable, we used 0.001 as thedisease allele frequency. Loci with suggestive LOD scoreswere genotyped with a denser marker set. The highest LODscore obtained with SimWalk2 was confirmed with theLinkmap program (Fastlink package version 4.1).26
RESULTSClinical evaluationsFigure 1 shows the pedigree of the consanguineous Turkishfamily with six affected members in three sibships, of which
four (II-9, II-10, II-14, and IV-1) were offspring of first cousinmarriages, suggesting autosomal recessive inheritance. Ofnote, two affected siblings (III-10 and III-11) were theoffspring of an affected father (II-9) and a healthy mother(II-8). Although consanguinity between the parents couldnot clearly be documented, both originated from the samesmall village and thought they were distantly related. Thephysical and neurological examinations of all affectedmembers were normal with the exception of the ophthalmo-logical and hand abnormalities. The feet were normal andthere were no abdominal ultrasound or echocardiographicabnormalities in the affected individuals. The relevantfindings in each patient are described below and summarisedin table 1.
Patient 1 (II-9)This was a 29 year old man who had a right divergentstrabismus with hypotropia and 20/20 visual acuity bilater-ally, with no refractive error. He had 14 DD exotropia and 25DD hypotropia OD, with no torsional component and a slight‘‘chin up’’ anomalous head posture. He had a markedlyrestricted elevation OD, as well as secondary overaction of theinferior oblique muscle OS. Hess screen test results showedrestriction of the entire right superior gaze field. Cranial MRIshowed volume loss of the right superior rectus muscle andhypertrophy of the inferior rectus muscle. A forced ductiontest done under general anaesthesia showed no passiverestriction of eye movements, and the patient was diagnosedas having right double elevator palsy. He had absent fifthfingers bilaterally, and the proximal part of his right hand
A
B
C
D
II-9 II-14 III-10 III-11 IV-1
222
44
444
444
44
444
210
44
444
444
44
444
033
44
444
442
44
444
011
44
444
444
44
444
000
00
000
444
44
444
Figure 2 Clinical findings of the patients with congenital fibrosis of extraocular muscles with ulnar hand abnormalities (CFEOM/U). (A) facialphotographs. (B) Centre position of the eyes and eyelids (bar) if ptosis was present, and range of eye movement; a score of 4 indicates normalunrestricted movement; 3, 2, and 1 denote decreasing movement in the indicated direction. (C) and (D) Hand photographs and x rays. See text andtable 1 for details. Permission was obtained from the subjects or their parents for the photographs to be reproduced here.
CFEOM with ulnar anomalies maps to 21qter 411
www.jmedgenet.com
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from
was markedly hypoplastic compared with the left. Whilethe fourth finger on the right hand was incurved andhypoplastic (clinodactyly), the fourth finger of the left handwas normally developed and had a very small (,3–4 mm)fifth finger bud emerging from the ulnar side at themetacarpophalangeal level. On x ray, the metacarpal andphalangeal bones of the fifth fingers were absent bilaterally,and were hypoplastic for the right fourth finger. The rightulnar styloid process was also hypoplastic. In addition, thecarpal bones of the right hand were abnormal, the triquetrumand pisiform bones were absent, and the capitate and hamatebones were fused. The scaphoid bone of the left hand wasbipartite (fig 2).
Patient 2 (I I-14)This was a 26 year old man who had right hemiptosis anddivergent strabismus. He had 12 DD of exotropia and 25 DDof hypotropia OD without measurable torsional deviation.Elevation of the right eye was restricted, and this was moresignificant in abduction. The Bielschowsky head tilt test wasslightly positive on the right and he had a chin up headposture. His visual acuity was 20/200 (OD) and 20/20 (OS).Cranial MRI was similar to that of patient II-9, with volumeloss of the superior rectus muscle and hypertrophy of theinferior rectus muscle of the right eye. Under generalanaesthesia, the forced duction test showed no passiverestriction in ocular movements. The fifth fingers of bothhands were absent, and the fourth finger of the right handwas slightly thinner than on the left, although the patientwas right handed. On x ray, both hands were symmetrical,and the metacarpal and phalangeal bones of the fifth fingerswere absent. The ulnar styloid processes were hypoplastic,and the triquetrum and lunate bones were fused bilaterally(fig 2).
Patient 3 (I I I-10)This was a four year old girl who had 20/20 visual acuitybilaterally, 16 DD exotropia OD, and restricted elevation onadduction of both eyes, which was more marked on the right.Head posture appeared normal, but a slight head tilt to theright shoulder occurred occasionally. Cranial MRI was
normal. The forced duction test was normal, and the patientwas diagnosed as having pseudo-Brown syndrome because ofbilateral inferior oblique muscle dysfunction. The fifth fingerof the right hand was absent, and she had syndactyly of thefourth and fifth fingers on the left, both of which werehypoplastic, and the fifth finger had no nail. On x ray, themetacarpal and phalangeal bones of the fifth fingers wereabsent bilaterally, and the metacarpal and phalangeal bonesof the left fourth finger were hypoplastic (fig 2).
Patient 4 (II I-11)This was a three year old boy who had a visual acuity of 20/200 (OD) and 20/20 (OS) with no significant refractive error.He had 18 DD exotropia and 20 DD hypotropia OD. Restrictedelevation during adduction was more marked than onabduction. He had a normal head posture, but a slight headtilt toward the right occurred intermittently. He had apositive forced duction test, showing a markedly restrictedelevation during adduction resembling that found in Brownsyndrome. Cranial MRI was normal. His fifth fingers wereabsent bilaterally, and the right fourth finger was slightlyincurved. On x ray, the metacarpal and phalangeal bones ofthe fifth fingers were absent bilaterally (fig 2).
Patient 5 (IV-1)This 2K year old girl was the most severely affected. She hadtotal ptosis and enophthalmia OD, and the right cornea wassmaller (radius, R = 11 mm) than the left (R = 12 mm). Shecould not fixate and follow objects with her right eye owingto profound amblyopia. Ocular movements of the right eyewere restricted in all directions, while they were normal onthe left. Examination of the fundus revealed a tilted disc OD.The MRI confirmed enophthalmia but did not reveal anyadditional abnormalities. The right third, fourth, and fifthfingers and the left fourth and fifth fingers were absent. Onx ray, the relevant metacarpal and phalangeal bones wereabsent, while the other bony structures were normal (fig 2).
Histopathological studiesHistological examination of the right superior rectus frompatient III-11 showed primarily fibroadipose tissue and nomuscle cells. In contrast, the skeletal muscle from the rightinferior oblique biopsy had mild non-specific variation infibre size on light microscopy, but no abnormalities wereobserved on histochemical analysis or electron microscopy(data not shown).
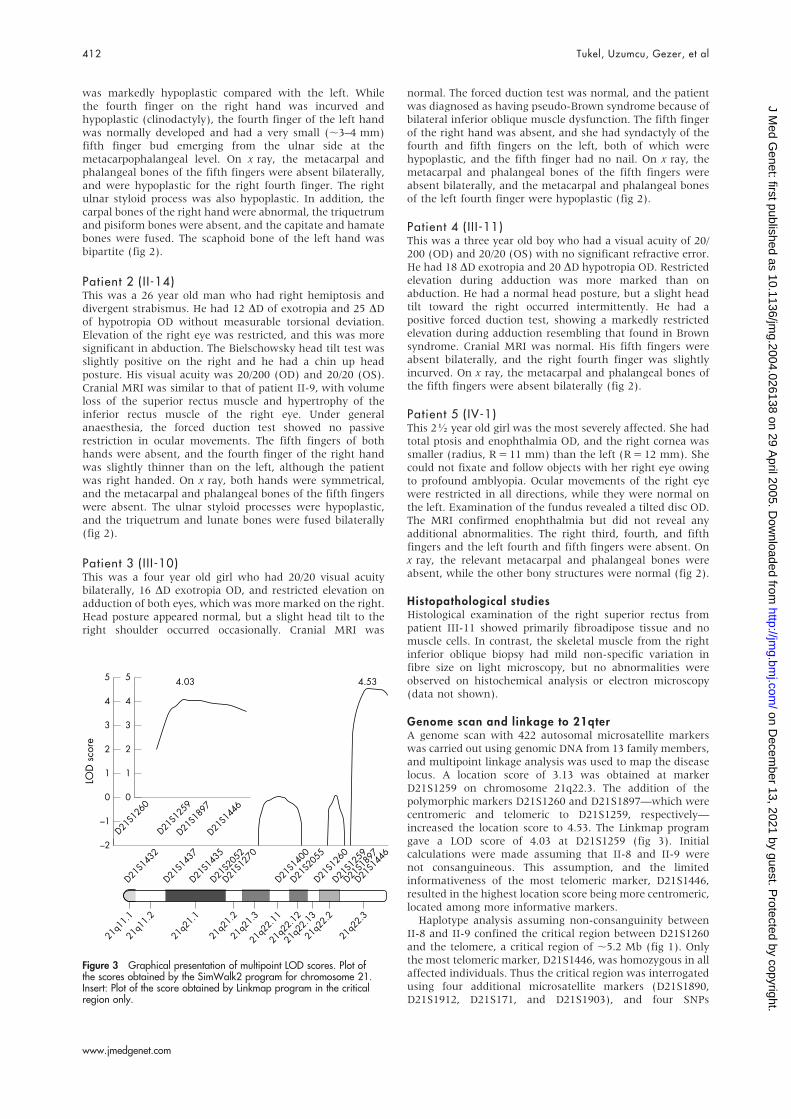
Genome scan and linkage to 21qterA genome scan with 422 autosomal microsatellite markerswas carried out using genomic DNA from 13 family members,and multipoint linkage analysis was used to map the diseaselocus. A location score of 3.13 was obtained at markerD21S1259 on chromosome 21q22.3. The addition of thepolymorphic markers D21S1260 and D21S1897—which werecentromeric and telomeric to D21S1259, respectively—increased the location score to 4.53. The Linkmap programgave a LOD score of 4.03 at D21S1259 (fig 3). Initialcalculations were made assuming that II-8 and II-9 werenot consanguineous. This assumption, and the limitedinformativeness of the most telomeric marker, D21S1446,resulted in the highest location score being more centromeric,located among more informative markers.
Haplotype analysis assuming non-consanguinity betweenII-8 and II-9 confined the critical region between D21S1260and the telomere, a critical region of ,5.2 Mb (fig 1). Onlythe most telomeric marker, D21S1446, was homozygous in allaffected individuals. Thus the critical region was interrogatedusing four additional microsatellite markers (D21S1890,D21S1912, D21S171, and D21S1903), and four SNPs
5
4
2
3
1
0
4.03 4.53
–2
–1
LOD
sco
re
D21S1
432
D21S1
437
D21S1
435
D21S2
052
D21S1
270
D21S1
400
D21S2
055
D21S1
260
D21S1
259
D21S1
897
D21S1
446
5
4
2
3
1
0
D21S1
260
D21S1
259
D21S1
897
D21S1
446
21q1
1.2
21q2
1.1
21q2
1.2
21q2
1.3
21q2
2.11
21q2
2.12
21q2
2.13
21q2
2.2
21q2
2.3
21q1
1.1
Figure 3 Graphical presentation of multipoint LOD scores. Plot ofthe scores obtained by the SimWalk2 program for chromosome 21.Insert: Plot of the score obtained by Linkmap program in the criticalregion only.
412 Tukel, Uzumcu, Gezer, et al
www.jmedgenet.com
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from

(rs234781, rs2839627, rs170916, and rs234728) whichspanned the region. Only the SNP data further narrowedthe centromeric boundary of the critical region by 2 Mb to,3.5 Mb from rs234728 to the telomere (online supplemen-tal table 1, obtainable from http://www.jmedgenet.com/supplemental/).
As additional microsatellite markers in the region (avail-able in public databases) were not informative, several newmarkers were identified and tested (online supplementaltables 1 and 2). Of these, 2044K was the most telomericheterozygous marker in patients III-10 and III-11, therebyfurther narrowing the critical region to ,1.5 Mb. Allmicrosatellite markers and SNPs telomeric to 2044K(rs2838917, rs725358, 2849K, rs2839168, rs1060609, 3086K,rs2839235, 3258K, rs2839281, D2151446, rs9722, andrs881827) were homozygous for the same allele in allpatients. Although these markers were not very informativein II-9 to II-14, SNP rs881827 clearly showed that affectedand unaffected individuals inherited different alleles. Thusthe CFEOM/U1 gene locus was localised to a critical region of,1.5 Mb from the new microsatellite marker, 2044K, to qter.Despite the fact that the new markers in the homozygousregion were not very informative, the location and LODscores were still significant when calculations were repeatedassuming consanguinity between II-8 and II-9, and with theinclusion of the new microsatellite markers and SNPs. Usingthe Simwalk2 program, a multipoint location score of 3.28was obtained at microsatellite D21S1446 through SNPrs881827. This result was confirmed with the Linkmapprogram, which gave a multipoint LOD score of 3.12 atD21S1446.
DISCUSSIONThe consanguineous Turkish patients described here define anew autosomal recessive syndrome of CFEOM with ulnarhand abnormalities (designated CFEOM/U). The affectedindividuals all had CFEOM of varying severity, which wasmainly confined to the right eye, and bilateral postaxialoligodactyly/oligosyndactyly of the hands, which was moresevere on the right. Although the clinical findings werevariable between affected individuals, penetrance was com-plete. Interestingly, the severity of the eye and handinvolvement was consistent in the same individual. Thiswas clearly observed in the most and least severely affectedindividuals, IV-1 and III-10, respectively.
All five patients had superior rectus and inferior obliquemuscle involvement, and three (II-14, III-11, and IV-1) alsohad levator palpebralis dysfunction. Phenotypically, threepatients (II-9, II-14, and III-11) had a double elevator palsy,perhaps caused by a superior rectus muscle paresis.27 PatientIII-11 was distinctive in having a more marked mechanicalrestriction of elevation during adduction, resulting in apositive forced duction test, as observed in patients withBrown syndrome; and patient III-10 had bilateral restrictedelevation during adduction with a negative forced ductiontest, which was interpreted as a pseudo-Brown syndromebecause of bilateral inferior oblique dysfunction. The fifthpatient had a more generalised and severe pattern ofextraocular muscle involvement with markedly restrictedeye movements in all directions and a total ptosis resemblinga generalised fibrosis syndrome.
Individuals affected with classical CFEOM (that is,CFEOM1) usually have bilateral ptosis and restrictiveophthalmoplegia, with their eyes fixed below the horizontalneutral position with or without secondary esotropia orexotropia. Necropsy examinations of individuals affected byclassical CFEOM revealed the absence of the superior divisionof the oculomotor nerve, which normally innervates thesuperior rectus and levator palpebrae superioris muscles.5 In
contrast, atypical patients with CFEOM2 and CFEOM3 havea restrictive ophthalmoplegia, caused by deficient function ofthe muscles innerved by the third or fourth cranial nerves.Unlike the classical type, they can raise their eyes above thehorizontal line or have unilateral involvement. For atypicalCFEOM, marked phenotypic variability has been reported.4
The patients described here can be classified as atypicalCFEOM as they have involvement of the superior and inferiordivisions of the third cranial nerve, although patient IV-1may also have involvement of the fourth and sixth cranialnerves, indicating the variability in the ocular phenotype. Inaddition to the ocular abnormalities, these patients hadoligodactyly/oligosyndactyly of the hands. The only ocularmotility disorder with upper limb defects is the Duane-radialray syndrome with radial ray abnormalities ranging fromhypoplasia of the thenar eminence to absence of the radialbone or forearm. In the family presented here, neither theocular nor the skeletal findings resembled the clinicalfindings of DRRS. Moebius syndrome, which is characterisedby congenital paresis or paralysis of the seventh (facial)cranial nerve frequently accompanied by dysfunction of othercranial nerves, may also be associated with arthrogryposisand hand abnormalities but is quite distinctive from thesyndrome we present here.
It is estimated that approximately 1/600 newborn infantshave a congenital abnormality of the upper limb.28 Postaxiallimb deficiencies are most often unilateral and sporadic. Theyalso occur as a feature of various syndromes. An autosomaldominant, non-syndromic postaxial oligodactyly whichaffects all four extremities has also been described (MIM176240).29 However, the association of a congenital fibrosissyndrome with postaxial oligodactyly/oligosyndactyly is noveland has not been reported to date. Of note, only a fewcausative genes for this group of disorders have been mappedor identified.30
By multipoint linkage analysis, the disease locus forCFEOM/U1 was mapped to chromosome 21 between thenew microsatellite marker 2044K and the chromosome 21telomer, a critical region spanning ,1.5 Mb. Initial calcula-tions were made assuming that II-8 and II-9 were notconsanguineous, in order to prevent the lower location scoresthat would have been obtained because of the limited densityand informability of the marker grid in the genome-widemarker sets. In fact, the markers initially available lacked theability to detect the very small homozygous region shared bysiblings III-10 and III-11 and all other affected individuals.The homozygous region between 2044K and the telomer wasfurther refined by identifying several additional microsatel-lites and SNPs in the region, which were less than 500 kbapart. Although the telomeric markers were not veryinformative, recalculating the location score assuming con-sanguinity gave a score of 3.28 at microsatellite markerD21S1446 through SNP rs881827. This result was confirmedwith the Linkmap program which gave a LOD score of 3.12 atmarker D21S1446.
Based on the current Human May 2004 (hg17) Assembly(NCBI Build 35) on the UCSC human genome browser, thisregion of ,1.5 Mb contains 17 genes (C21orf123, COL18A1,SLC19A1, PCBP3, COL6A1, COL6A2, FTCD, C21orf56, LSS,MCM3APAS, AF426262, C21ofr57, C21orf58, PCNT2,C21orf106, S100B, and HRMT1L1), which have correspond-ing entries in PDB or SWISS-PROT, or are NCBI referencesequence mRNAs with a ‘‘reviewed’’ status. As there may beunrecognised genes in this ,1.5 Mb region, it may containabout 20 genes. However, there were no obvious candidategenes, and no obvious motifs. Efforts are under way tofurther refine the region of homozygosity and to identify thedisease causing gene.
CFEOM with ulnar anomalies maps to 21qter 413
www.jmedgenet.com
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from

ConclusionsA new autosomal recessive ocular motility syndrome withpostaxial oligodactyly and syndactyly (designated CFEOM/U1) was identified and its locus mapped to the mosttelomeric 1.5 Mb of chromosome 21. Future identificationand functional studies of the gene causing this newsyndrome may provide insights into the development of theextraocular muscles and their cranial motor nuclei, as well asantero-posterior limb development.
ELECTRONIC DATABASE INFORMATION
N Center for Medical Genetics, Marshfield Clinic ResearchFoundation, http://research.marshfieldclinic.org/genetics/Map_Markers/maps/IndexMapFrames.html
N National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/
N Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
N Primer3 primer design program, http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi
N Tandem Repeat Finder Program, http://c3.biomath.mssm.edu/trf.html
N University of California Santa Cruz (UCSC), HumanGenome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
ACKNOWLEDGEMENTSWe wish to thank the patients for participating in this study. Weacknowledge Monica Erazo for her excellent technical assistance.This work was supported in part by the Turkish Academy of Sciences,in the framework of the young scientist award program (BW/TUBA-GEBIP/2002-1-20), National Institutes of Health grants R37 DK34045(merit award), R01 DK026824 and M01 RR00071 for the MountSinai General Clinical Research Center from the National Institute forResearch Resources. TT is the recipient of an NIH postdoctoralfellowship in Mental Retardation and Developmental Disabilities(T32 HD7105).
Supplementary tables 1 and 2 can be found on ourweb site, www.jmedgenet.com/supplemental
Authors’ affiliations. . . . . . . . . . . . . . . . . . . . .
T Tukel, R J Desnick*, Department of Human Genetics, Istanbul Faculty ofMedicine of Istanbul University, Istanbul, TurkeyA Uzumcu, H Kayserili, M Yuksel-Apak, O Uyguner, B Wollnik*, ChildHealth Institute, Division of Medical Genetics, Istanbul UniversityA Gezer, Department of Ophthalmology, Istanbul Faculty of Medicine,Istanbul UniversityS H Gultekin, Department of Pathology, Section of Neuropathology,Istanbul UniversityH-C Hennies, P Nurnberg, Gene Mapping Centre and Department ofMolecular Genetics, Max Delbruck Centre for Molecular Genetics,Berlin, Germany
*Co-senior authors who contributed equally to this research
Competing interests: none declared
Correspondence to: Professor Robert J Desnick, Department of HumanGenetics, Mount Sinai School of Medicine, 1425 Madison Avenue, Box1498, New York, NY 10029, USA; [email protected] or Dr BerndWollnik, Institute of Child Health, Division of Medical Genetics, IstanbulUniversity, Millet Caddesi, Capa, 34390 Istanbul, Turkey; [email protected]
Revised version received 26 September 2004Accepted for publication 27 September 2004
REFERENCES1 Heuk G. Ueber angeborenen vererbtenBeweglichkeitsdefekts der Augen. Klin
Monatsbl Augenheilkd 1879;17:3253.2 Brown HW. Congenital structural muscle anomalies. In: Allen JH, ed.
Strabismus ophthalmic symposium. St Louis: CV Mosby Co,1950:205–36.
3 Traboulsi EI, Lee BA, Mousawi A, Khamis AR, Engle EC. Evidence of geneticheterogeneity in autosomal recessive congenital fibrosis of the extraocularmuscles. Am J Ophthalmol 2000;129:658–62.
4 Engle EC. Applications of molecular genetics to the understanding ofcongenital ocular motility disorders. Ann NY Acad Sci 2002;956:55–63.
5 Engle EC, Goumnerov BC, McKeown CA, Schatz M, Johns DR, Porter JD,Beggs AH. Oculomotor nerve and muscle abnormalities in congenital fibrosisof the extraocular muscles. Ann Neurol 1997;41:314–25.
6 Gutowski NJ. Duane’s syndrome. Eur J Neurol 2000;7:145–9.7 Vincent C, Kalatzis V, Compain S, Levilliers J, Slim R, Graia F, Pereira ML,
Nivelon A, Croquette MF, Lacombe D, et al. A proposed new contiguous genesyndrome on 8q consists of Branchio-Oto-Renal (BOR) syndrome, Duanesyndrome, a dominant form of hydrocephalus and trapeze aplasia;implications for the mapping of the BOR gene. Hum Mol Genet1994;3:1859–66.
8 Calabrese G, Stuppia L, Morizio E, Guanciali Franchi P, Pompetti F,Mingarelli R, Marsilio T, Rocchi M, Gallenga PE, Palka G, Dallapiccola B.Detection of an insertion deletion of region 8q13–q21. 2 in a patient withDuane syndrome: implications for mapping and cloning a Duane gene,Eur J Hum Genet 1998;6:187–93.
9 Calabrese G, Telvi L, Capodiferro F, Morizio E, Pizzuti A, Stuppia L, Bordoni R,Ion A, Fantasia D, Mingarelli R, Palka G. Narrowing the Duane syndromecritical region at chromosome 8q13 down to 40 kb. Eur J Hum Genet2000;8:319–24.
10 Appukuttan B, Gillanders E, Juo SH, Freas-Lutz D, Ott S, Sood R, VanAuken A, Bailey-Wilson J, Wang X, Patel RJ, Robbins CM, Chung M,Annett G, Weinberg K, Borchert MS, Trent JM, Brownstein MJ, Stout JT.Localization of a gene for Duane retraction syndrome to chromosome 2q31.Am J Hum Genet 1999;65:1639–46.
11 Engle EC, Kunkel LM, Specht LA, Beggs AH. Mapping a gene for congenitalfibrosis of the extraocular muscles to the centromeric region of chromosome12. Nat Genet 1994;7:69–73.
12 Yamada K, Andrews C, Chan WM, McKeown CA, Magli A, de Berardinis T,Loewenstein A, Lazar M, O’Keefe M, Letson R, London A, Ruttum M,Matsumoto N, Saito N, Morris L, Del Monte M, Johnson RH, Uyama E,Houtman WA, de Vries B, Carlow TJ, Hart BL, Krawiecki N, Shoffner J,Vogel MC, Katowitz J, Goldstein SM, Levin AV, Sener EC, Ozturk BT,Akarsu AN, Brodsky MC, Hanisch F, Cruse RP, Zubcov AA, Robb RM,Roggenkaemper P, Gottlob I, Kowal L, Battu R, Traboulsi EI, Franceschini P,Newlin A, Demer JL, Engle EC. Heterozygous mutations of the kinesin KIF21Ain congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nat Genet2003;35:318–21.
13 Sener EC, Lee BA, Turgut B, Akarsu AN, Engle EC. A clinically variant fibrosissyndrome in a Turkish family maps to the CFEOM1 locus on chromosome 12.Arch Ophthalmol 2000;118:1090–7.
14 Wang SM, Zwaan J, Mullaney PB, Jabak MH, Al-Awad A, Beggs AH,Engle EC. Congenital fibrosis of the extraocular muscles type 2, an inheritedexotropic strabismus fixus, maps to distal 11q13. Am J Hum Genet1998;63:517–25.
15 Nakano M, Yamada K, Fain J, Sener EC, Selleck CJ, Awad AH, Zwaan J,Mullaney PB, Bosley TM, Engle EC. Homozygous mutations in ARIX(PHOX2A)result in congenital fibrosis of the extraocular muscles type 2. Nat Genet2001;29:315–20.
16 Doherty EJ, Macy ME, Wang SM, Dykeman CP, Melanson MT, Engle EC.CFEOM3: a new extraocular congenital fibrosis syndrome that maps to16q24.2–q24.3. Invest Ophthalmol Vis Sci 1999;40:1687–94.
17 Okihiro MM, Tasaki T, Nakano KK, Bennett BK. Duane syndrome andcongenital upper-limb anomalies. A familial occurrence. Arch Neurol1977;34:174–9.
18 Kohlhase J, Heinrich M, Schubert L, Liebers M, Kispert A, Laccone F,Turnpenny P, Winter RM, Reardon W. Okihiro syndrome is caused by SALL4mutations. Hum Mol Genet 2002;11:2979–87.
19 Al-Baradie R, Yamada K, St Hilaire C, Chan WM, Andrews C, McIntosh N,Nakano M, Martonyi EJ, Raymond WR, Okumura S, Okihiro MM, Engle EC.Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and resultsfrom mutations in SALL4, a new member of the SAL family. Am J Hum Genet2002;71:1195–9.
20 Jen J, Coulin CJ, Bosley TM, Salih MA, Sabatti C, Nelson SF, Baloh RW.Familial horizontal gaze palsy with progressive scoliosis maps to chromosome11q23–25. Neurology 2002;59:432–5.
21 Carpenter S, Carpati G. Pathology of skeletal muscle, 2nd ed. New York:Oxford University Press, 2001.
22 Benson G. Tandem repeats finder: a program to analyze DNA sequences.Nucleic Acids Res 1999;27:573–80.
23 Rozen S, Skaletsky H. Primer3 on the WWW for general users and forbiologist programmers. Methods Mol Biol 2000;132:365–86.
24 Sobel E, Lange K. Descent graphs in pedigree analysis: applications tohaplotyping, location scores, and marker-sharing statistics. Am J Hum Genet1996;58:1323–37.
414 Tukel, Uzumcu, Gezer, et al
www.jmedgenet.com
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from

25 Mukhopadhyay N, Almasy L, Schroeder M, Mulvihill WP, Weeks DE. Mega2,a data-handling program for facilitating genetic linkage and associationanalyses [abstract]. Am J Hum Genet 1999;65:A436.
26 Lathrop GM, Lalouel JM, Julier C, Ott J. Strategies for multilocus linkageanalysis in humans. Proc Natl Acad Sci USA 1984;81:3443–6.
27 von Noorden GK, Campos EC. Paralytic Strabismus. Binocular vision andocular motility, 6th ed. St Louis: CV Mosby, 2001:442.
28 Flatt AE. Genetics and inheritance. In: The care of congenital hand anomalies.St Louis: Quality Medical Publishing, 1994.
29 Wulfsberg EA, Mirkinson LJ, Meister SJ. Autosomal dominanttetramelic postaxial oligodactyly. Am J Med Genet1993;46:579–83.
30 Manouvrier-Hanu S, Holder-Espinasse M, Lyonnet S. Genetics of limbanomalies in humans. Trends Genet 1999;15:409–17.
Get published within days of acceptance with JMG
We are delighted to announce that the Journal of Medical Genetics launched a ‘‘publishahead of print’’ programme in March 2005. Selected papers are fast tracked and publishedonline months before they appear in the print journal.Papers of more significance to the international ophthalmology community are publishedwithin days of acceptance. The first published article is the raw accepted manuscript; editedand typeset versions are also published as soon as they are available.In addition to being available on JMG Online, the publish ahead of print articles aresearchable through PubMed/Medline – establishing primacy for your work. They are linkedfrom the JMG Online home page.The JMG’s publish ahead of print programme is unique among the major clinical geneticsjournals – to take advantage of this service submit your papers to Journal of Medical Geneticsusing our online submission and review system Bench.Press (http://submit-jmg.bmjjournals.com). For further information contact [email protected].
CFEOM with ulnar anomalies maps to 21qter 415
www.jmedgenet.com
on Decem
ber 13, 2021 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.2004.026138 on 29 A
pril 2005. Dow
nloaded from

SHORT REPORT
Further evidence for LBP-1c/CP2/LSF association inAlzheimer’s disease familiesL Bertram, M Parkinson, M B McQueen, K Mullin, M Hsiao, R Menon, T J Moscarillo, D Blacker,R E Tanzi. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
J Med Genet 2005;42:857–862. doi: 10.1136/jmg.2004.024596
Objectives: Several studies suggested chromosome 12 har-bours an Alzheimer’s disease (AD) risk factor gene.Significant association of a single nucleotide polymorphism(SNP) in the 39 UTR of transcription factor CP2 (LBP-1c/CP2/LSF or TFCP2) at 12q13 was reported in three independentcase-control studies, but no family based analyses have beenperformed to date.Methods: Genotypes for three SNPs were generated in twoindependent AD family samples. A meta-analysis on allpublished case-control studies was also performed.Results: The A allele of the 39 UTR SNP was associated withincreased risk for AD in one sample (odds ratio (OR) 2.1,95% confidence interval (95% CI) 1.1 to 4.3), but not in theother, possibly due to low power. Haplotype analysesshowed that this allele is part of a putative risk-haplotypeovertransmitted to affected individuals in one sample and inboth samples combined. Meta-analysis of the previouslyassociated 39 UTR SNP showed a trend towards a protectiveeffect of the A allele in AD (OR 0.73, 95% CI 0.5 to 1.1).Conclusions: This is the first study to examine LBP-1c/CP2/LSF in AD families, and the fifth to independently showsignificant association. While our results support a role of thisgene in AD pathogenesis, the direction of the effect remainsuncertain, possibly indicating linkage disequilibrium withanother variant nearby.
Alzheimer’s disease (AD) is a neurodegenerative dis-order with a complex genetic background. The rare,early onset autosomal dominant forms of AD are
caused by mutations in three genes (APP, PSEN1, andPSEN2), all of which lead to an increase in b amyloid protein(Ab) levels in brain.1 Disease onset is typically before thesixth decade of life, and pathogenic mutations displayvirtually 100% penetrance. The more common late onsetform of AD (that is, with disease onset usually between 60and 80 years of age) is likely governed by genetic suscept-ibility factors of smaller effect and greatly reduced pene-trance, which are transmitted in a non-Mendelian fashion.2
The only established risk factor to date is the e4 allele ofapolipoprotein E (APOE) on chromosome 19q13, which isinvolved in the accumulation and/or clearance of Ab in thebrain of AD patients.3 While several dozen papers arepublished each year claiming or refuting association withadditional candidate genes on just about every chromosome,none of these has been unequivocally confirmed.4
Since the discovery of APOE e4, numerous laboratoriesworldwide have performed either linkage, or associationbased, full genome screens in search of other AD predispos-ing variants. Linkage to chromosome 12 was one of the firstmajor signals to emerge from these efforts. However, while
some studies predominantly observed the strongest findingson the short arm of this chromosome (on 12p13, near10 Mb)5–7, other groups detected more pronounced linkageapproximately 40 Mb distal (on 12q13, near 50 Mb).8 9 Todate, it remains unclear whether these signals are caused bythe same underlying locus, or are actually the result of twodistinct genes. The latter hypothesis is supported by the factthat there is at least one candidate gene in each region thathas received independent confirmation—albeit not unequi-vocally—in at least ten studies: a2-macroglobulin (A2M) on12p13, and the lipoprotein receptor related protein-1 (LRP1)on 12q13 (reviewed in Bertram and Tanzi10 and in Saunders etal11). Other candidates on chromosome 12 reported to beassociated with AD include: on 12p13, oxidised lipoproteinreceptor-1 (OLR1), and on 12q13, transcription factor CP2(LBP-1c/CP2/LSF) and neurotrophin-3 (NTF3). Of these, LBP-1c/CP2/LSF (also known as TFCP2) has received the mostconsistent support from independent groups with four papersreporting significant association, and thus far no publishednegative study.12–15
LBP-1c/CP/LSF encodes a nuclear transcription factor thatregulates the expression of A2M and glycogen synthasekinase-3b (GSK3b),16 17 and also interacts with Fe65,18 whichserves as an adapter molecule for the cytoplasmic domain ofAb precursor protein (APP), and may also be involved in theregulation of gene expression via interaction with the APPintracellular domain (AICD).19 The original paper associatingLBP-1c/CP/LSF with AD examined three independent case-control series from France, the US, and the UK (table 1).12
While two of these (France and UK) showed a significantprotective effect in carriers of the A allele at a singlenucleotide polymorphism (SNP) in the 39 UTR of the gene,the authors detected no significant association in the USsample. Combining all three populations revealed a signifi-cantly decreased risk of developing AD in carriers of the Aallele v the G/G genotype (odds ratio (OR) 0.58, 95%confidence intervals (95% CI) 0.44 to 0.75; table 1). Twosubsequent independent case-control studies by Taylor et al13
and Luedecking-Zimmer et al14 replicated the protective effectof the A allele with similar effect sizes (table 1). Finally, afourth case-control study from Italy also detected a sig-nificant association between this SNP and AD.15 In contrastto the other papers, however, the data of this reportsuggested an over-representation of the A allele in AD casesv controls. To date, there are no published reports investigat-ing the potential role of the LBP-1c/CP2/LSF gene in familybased AD datasets, which have the advantage of beingunbiased in the presence of population admixture. In thisstudy we have examined a total of three SNPs in LBP-1c/CP2/
Abbreviations: AD, Alzheimer’s disease; CAG, Consortium onAlzheimer’s Genetics; CLR, conditional logistic regression; LD, linkagedisequilibrium; OR, odds ratio; PDT, pedigree disequilibrium test; SNP,single nucleotide polymorphism; 95% CI, 95% confidence interval
857
www.jmedgenet.com
LSF in two independent and carefully ascertained andevaluated AD family samples, and provide further supportfor a significant role of this gene in contributing to overall ADrisk.
METHODSSamplesThe NIMH AD genetics init iative study sampleSubjects were collected following a standardised protocolapplying NINCDS/ADRDA criteria for the diagnosis of AD.20
Over the 10 years that the participating families have beenfollowed, a clinical diagnosis of AD has been confirmed atautopsy in 94% of the cases.21 The NIMH sample includes1439 individuals (69% female) from 437 families with at leasttwo affected individuals (994 affected individuals (mean(SD) age of onset 72.4 (7.7) years, range 50–97 years), 411unaffected individuals, and 34 with unknown phenotype.
Consortium on Alzheimer’s Genetics (CAG) studysampleSubjects for this second, independently ascertained, ADfamily sample were collected under the auspices of theConsortium on Alzheimer’s Genetics, a collaborative effort ofthe Massachusetts AD Research Center, the University ofCalifornia, Los Angeles, the University of California, SanDiego, and the University of Rochester Medical Center.22
NINCDS/ADRDA criteria were used for a clinical diagnosisof AD, and probands were included only if they had at leastone unaffected living sibling willing to participate in thisstudy. Unlike the NIMH sample, no affected individualbeyond the proband was required; thus, the vast majority offamilies are not multiplex. Currently, data and specimencollection is completed for 489 individuals (62.6% female)from 217 sibships in which all affected individuals displayedan onset age >50 years (n=224 affected individuals (mean(SD) age of onset 71.2 (9.1) years, range 50–89 years),n=265 unaffected individuals). Most sibships consisted ofjust one discordant sibpair, but in 41 families there weremore than two siblings available.
GenotypingGenotypes for a total of three polymorphisms (that is, theoriginal 39 UTR SNP, rs4438107 (,10 kb proximal), andrs10876135 (773 bp distal)) in LBP-1c/CP/LSF were generatedusing fluorescent polarisation detected single base extension(FP-SBE) on a Criterion Analyst AD (Molecular Devices,Sunnyvale, CA). PCR primers were designed to yield aproduct of approximately 250 bp in length and added to
,10 ng of genomic DNA using individually optimised PCRconditions (sequences available on request). PCR primers andunincorporated dNTPs were degraded by the direct additionof exonuclease I (0.1–0.15 U/rxn) and shrimp alkalinephosphatase (1 U/rxn). The single base extension step wascarried out using Thermosequenase (0.4 U/rxn) and theappropriate mix of R110-ddNTP, TAMRA-ddNTP (3 mM),and all four unlabeled ddNTPs (22 or 25 mM) to the Exo1/SAP treated PCR product. To assess genotyping quality andensure consistency of the genotyping calls, ,10% of thesamples were randomly duplicated and genotyped twice. Forall three SNPs combined, average genotyping efficiency was97.8%, and the discrepancy rate (based on comparison toblinded duplicated samples) was below 0.3% in both samples.
Statistical analysesSingle locus and haplotype based tests of association weredone in FBAT (v.1.5.3). FBAT uses a generalised scorestatistic to perform a variety of TDT type tests and despitethe true underlying genetic model, FBAT performs bestassuming an additive genetic model, which was used here.23
We used the empirical variance function of the program toaccount for the presence of linkage in the area24 (as wassuggested by previous studies), and an equal weight offsetcorrection to incorporate genotypes from both affected andunaffected individuals. All analyses were performed on thefull NIMH sample, the CAG sample, and on the two samplescombined. All single locus analyses were repeated using thepedigree disequilibrium test (PDT)25 26 to confirm resultsobtained with FBAT (note that the PDT currently does notaccommodate haplotype tests). While both tests computevalid p values under the null hypothesis of linkage but noassociation, the PDT statistic can be less efficient under somecircumstances as it only includes discordant sibships andignores families where only affected siblings are available.Further, to assess the magnitude of any potential effect ondisease risk for the 39 UTR SNP, we performed conditionallogistic regression (CLR) stratified on family,27 comparingcarriers of the A allele to carriers of the GG genotype. All ORare adjusted for age, gender, and APOE e4 allele status. Notethat confidence intervals may be too narrow because CLRmay slightly underestimate the standard errors when multi-ple affected and unaffected subjects are included in eachfamily. However, the magnitude of this effect is expected tobe small unless genetic effects are very large.28 Finally, toassess whether families showing association with the 39 UTRSNP in LBP-1c/CP/LSF overlap with families associated withthe intron 18 deletion in A2M,11 we determined for bothpolymorphisms which of the NIMH pedigrees showed
Table 1 Summary of published case-control AD association studies for the 39 UTR SNP in LBP-1c/CP/LSF
Study
AD cases Normal controls Results
Subjects, n(% women)
Onset age,mean (SD)
Subjects, n(% women)
Age,mean (SD)
A allelefrequency(AD v CTRL) OR (95% CI)
Panza et al15
Italy 166 (63%) 69.4 (10.3) 225 (68%) 71.3 (10.4) 0.06 v 0.02 2.97 (1.33 to 6.66)Luedecking-Zimmer et al14
USA 564 (68%) 77.3 (6.4) 523 (66%) 76.8 (6.3) 0.05 v 0.07 0.65 (0.43 to 0.96)Taylor et al13
UK 239 (64%)* 81.2 (7.8) 342 (59%)* 82.1 (3.8) 0.05 v 0.08 0.59 (0.35 to 1.00)Lambert et al12
All combined 1139 (64.6%) 70.5 (6.6) 1317 (62.5%) 73.4 (9.2) 0.04 v 0.07 0.58 (0.44 to 0.75)UK 159 (67%) 65.7 (11.1) 205 (51%) 60.8 (11.3) 0.04 v 0.09 0.46 (0.23 to 0.93)USA 296 (67%) 75.7 (0.2) 462 (67%) 79.2 (9.3) 0.06 v 0.07 0.87 (0.56 to 1.38)France 684 (63%) 69.4 (8.4) 650 (63%) 73.2 (8.6) 0.04 v 0.07 0.48 (0.33 to 0.70)
Studies are shown in chronological order, with the most recent study listed first. Odds ratios (ORs) and 95% confidence intervals (CI) as reported by authors; someORs are adjusted for co-variables (like age, gender, and APOE e4 status) and might thus vary slightly from the crude ORs presented in fig 1, which were used tocalculate summary ORs.
858 Bertram, Parkinson, McQueen, et al
www.jmedgenet.com

transmission of at least two risk alleles to affected individuals(using the Viewstat option in FBAT).
Haplotype block predictionsHaplotype blocks were estimated using the programHaploview based on the four gamete rule (fourth gamete at0.02 frequency; see Haploview website for details at http://www.broad.mit.edu/personal/jcbarret/haploview/index.php).Haploview was also used for the calculation of pairwiselinkage disequilibrium (LD) measures across all three SNPs.
Meta analysisStudy specific crude ORs and 95% CIs were calculated fromthe raw data for each of the case-control studies investigatingthe association between the LBP-1c/CP/LSF 39 UTR SNP andAD. The Q statistic, a test for heterogeneity among the studyspecific ORs, that is distributed approximately as x2 with k21degrees of freedom (k=number of studies),29 resulted in a pvalue ,0.1, suggesting significant between-study heteroge-neity. Therefore, to calculate a summary OR for all studies,we used the DerSimonian and Laird30 random effects model,which utilises weights that incorporate both the within studyand between study variance. Note the ORs estimated fromthe CLR in our family based analyses are adjusted for anumber of co-variables (see above), which is why we electednot to combine them with the crude or differently adjustedORs from the case-control studies. Statistical Analysis System(SAS) was used for the statistical analyses and resultinggraphs.
RESULTSThe results of the single locus and haplotype associationanalyses are shown in table 2. Allele frequencies, asestimated by FBAT, were very similar for all three SNPs inboth samples, and for the 39 UTR SNP are comparable to theprevious reports (see legend to table 2). Genotype frequenciesfor all SNPs were in Hardy-Weinberg equilibrium (p.0.90).Testing the 39 UTR SNP in FBAT revealed significantovertransmission of the A allele to affected individuals inthe NIMH families (p=0.05). This was confirmed using thePDT (p=0.04), and resulted in a significant risk increase incarriers of the A allele v non-carriers (OR 2.1, 95% CI 1.1 to4.3) using CLR stratified on family. Due to the low numbers,heterozygous and homozygous A allele carriers could not beexamined separately, but all three subjects carrying the A/Agenotype were affected (two autopsy confirmed, one clinicalAD diagnosis), in accordance with the observation that the Aallele confers risk in this sample. While the same allele wasalso overtransmitted to affected individuals in families of the
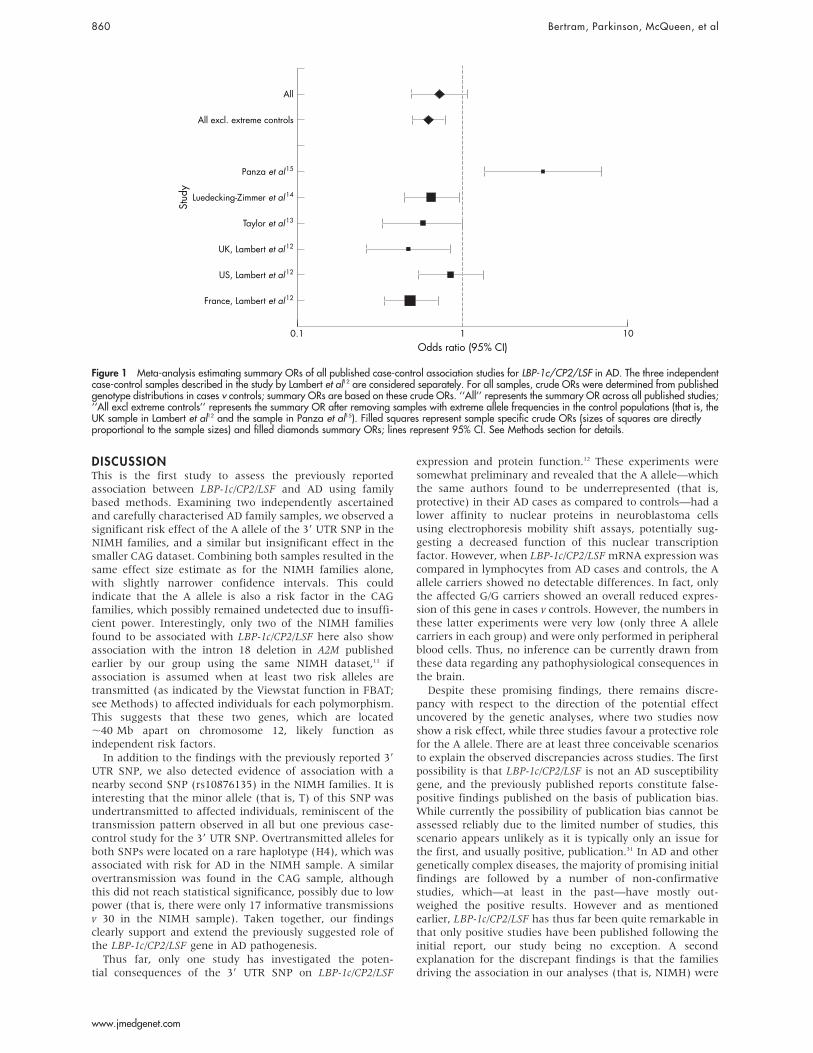
CAG sample resulting in a similar effect size estimate as forthe NIMH families, this did not reach statistical significancein any of the analyses (OR 2.1, 95% CI 0.5 to 8.5), possiblydue to low power in this overall smaller sample. Also, therewere no homozygous A allele carriers in these families.However, combining both samples yielded slightly decreasedp values (FBAT=0.03; PDT=0.02) and narrower confidenceintervals in the effect size estimates (OR 2.1, 95% CI 1.1 to3.7). In addition to these findings, one of the other two testedSNPs (that is, rs10876135, located 773 bp further 39) alsoshowed significant association in the NIMH but not in theCAG sample (p=0.04 and 0.2, respectively; table 2). All threeSNPs showed strong pairwise LD and were estimated toreside within the same haplotype block, which is in goodagreement with predictions from the International HapMapProject (http://www.hapmap.org/). Thus, all three SNPs werecombined in the haplotype analyses which showed evidencefor one rare haplotype (H4) being significantly overtrans-mitted to affected individuals in the NIMH sample (p=0.03;table 2). A similar frequency and transmission pattern forthis haplotype were observed in the CAG families, althoughthe overtransmission to affected individuals did not reachstatistical significance, again possibly due to low power.However, as for the 39 UTR SNP, the association signal of thishaplotype was strongest when both samples were combined(p=0.01). None of these SNPs showed a significantinteraction with APOE e4, gender, or onset age in our CLRanalyses (data not shown). This is noteworthy because mostof the previous studies demonstrating a protective role for theA allele of the 39 UTR SNP were comprised of late onset ADsamples. In this study, however, effect size estimates arequite comparable in families of late (OR 1.8 (1.0–3.2)) andearly/mixed onset (OR 2.2 (0.7–7.2); combined sample).Combining all four previously published case-control
studies into one meta-analysis revealed an overall protectiveeffect of the A allele, although this did not reach statisticalsignificance when all six independent case-control serieswere considered (0.73, 95% CI 0.5 to 1.1; fig 1). Interestingly,allele-frequency estimates across studies were quite similarfor the AD cases (ranging from 0.04 to 0.06), but wereconsiderably more variable in the control populations(ranging from 0.02 to 0.09; table 1). In an attempt to reducethis variability, we removed the two populations with themost extreme allele frequency estimates in healthy controls(that is, Panza et al15 and the UK sample from Lambert et al12)and repeated the analyses. As expected, the resultingsummary OR proved somewhat more stable, indicating asignificant protective effect across the remaining samples(OR 0.62, 95% CI 0.5 to 0.8; fig 1).
Table 2 Association analyses of three SNPs in LBP-1c/CP/LSF in two independent family samples
Single locus analyses*Haplotype analyses�
rs4438107 39 UTR Rs10876135
FBAT z score (p) z score (p) z score (p) Allele z score (p)
NIMH (n = 437) 20.4 (0.7) 1.9 (0.05) 22.0 (0.04) H4 2.1 (0.03)CAG (n = 217) 0.5 (0.6) 1.1 (0.3) 1.4 (0.2) H4 1.3 (0.2)Combined (n = 654) 0 (1) 2.2 (0.03) 20.9 (0.4) H4 2.5 (0.01)
PDT x2 (p) x2 (p) x2 (p)
NIMH (n = 437) 0.5 (0.5) 5.1 (0.02) 2.8 (0.09) – –CAG (n = 217) 1.2 (0.3) 0.5 (0.5) 1.5 (0.2) – –Combined (n = 654) 0 (0.9) 5.5 (0.02) 2.1 (0.15) – –
*Minor allele frequencies in the NIMH (CAG) sample are: rs4438107 (T) = 0.46 (0.47), 39UTR (A) = 0.06 (0.07), rs10876135 (T) = 0.08 (0.06). z score for minorallele or denoted haplotype allele (positive values indicate overtransmission to affected individuals).�Haplotype frequencies in the NIMH (CAG) sample are: H1 (C-G-C) = 0.51 (0.48), H2 (T-G-C) = 0.39 (0.43), H3 (T-G-T) = 0.06 (0.04), H4 (C-A-C) = 0.04 (0.04).Association statistics are presented for risk associated haplotype (H4) and 1 df.
LBP-1c/CP2/LSF association in AD families 859
www.jmedgenet.com
DISCUSSIONThis is the first study to assess the previously reportedassociation between LBP-1c/CP2/LSF and AD using familybased methods. Examining two independently ascertainedand carefully characterised AD family samples, we observed asignificant risk effect of the A allele of the 39 UTR SNP in theNIMH families, and a similar but insignificant effect in thesmaller CAG dataset. Combining both samples resulted in thesame effect size estimate as for the NIMH families alone,with slightly narrower confidence intervals. This couldindicate that the A allele is also a risk factor in the CAGfamilies, which possibly remained undetected due to insuffi-cient power. Interestingly, only two of the NIMH familiesfound to be associated with LBP-1c/CP2/LSF here also showassociation with the intron 18 deletion in A2M publishedearlier by our group using the same NIMH dataset,11 ifassociation is assumed when at least two risk alleles aretransmitted (as indicated by the Viewstat function in FBAT;see Methods) to affected individuals for each polymorphism.This suggests that these two genes, which are located,40 Mb apart on chromosome 12, likely function asindependent risk factors.In addition to the findings with the previously reported 39
UTR SNP, we also detected evidence of association with anearby second SNP (rs10876135) in the NIMH families. It isinteresting that the minor allele (that is, T) of this SNP wasundertransmitted to affected individuals, reminiscent of thetransmission pattern observed in all but one previous case-control study for the 39 UTR SNP. Overtransmitted alleles forboth SNPs were located on a rare haplotype (H4), which wasassociated with risk for AD in the NIMH sample. A similarovertransmission was found in the CAG sample, althoughthis did not reach statistical significance, possibly due to lowpower (that is, there were only 17 informative transmissionsv 30 in the NIMH sample). Taken together, our findingsclearly support and extend the previously suggested role ofthe LBP-1c/CP2/LSF gene in AD pathogenesis.Thus far, only one study has investigated the poten-
tial consequences of the 39 UTR SNP on LBP-1c/CP2/LSF
expression and protein function.12 These experiments weresomewhat preliminary and revealed that the A allele—whichthe same authors found to be underrepresented (that is,protective) in their AD cases as compared to controls—had alower affinity to nuclear proteins in neuroblastoma cellsusing electrophoresis mobility shift assays, potentially sug-gesting a decreased function of this nuclear transcriptionfactor. However, when LBP-1c/CP2/LSF mRNA expression wascompared in lymphocytes from AD cases and controls, the Aallele carriers showed no detectable differences. In fact, onlythe affected G/G carriers showed an overall reduced expres-sion of this gene in cases v controls. However, the numbers inthese latter experiments were very low (only three A allelecarriers in each group) and were only performed in peripheralblood cells. Thus, no inference can be currently drawn fromthese data regarding any pathophysiological consequences inthe brain.Despite these promising findings, there remains discre-
pancy with respect to the direction of the potential effectuncovered by the genetic analyses, where two studies nowshow a risk effect, while three studies favour a protective rolefor the A allele. There are at least three conceivable scenariosto explain the observed discrepancies across studies. The firstpossibility is that LBP-1c/CP2/LSF is not an AD susceptibilitygene, and the previously published reports constitute false-positive findings published on the basis of publication bias.While currently the possibility of publication bias cannot beassessed reliably due to the limited number of studies, thisscenario appears unlikely as it is typically only an issue forthe first, and usually positive, publication.31 In AD and othergenetically complex diseases, the majority of promising initialfindings are followed by a number of non-confirmativestudies, which—at least in the past—have mostly out-weighed the positive results. However and as mentionedearlier, LBP-1c/CP2/LSF has thus far been quite remarkable inthat only positive studies have been published following theinitial report, our study being no exception. A secondexplanation for the discrepant findings is that the familiesdriving the association in our analyses (that is, NIMH) were
10Odds ratio (95% CI)
Stud
y
0.1 1
All
All excl. extreme controls
Panza et al15
Luedecking-Zimmer et al14
Taylor et al13
UK, Lambert et al12
US, Lambert et al12
France, Lambert et al12
Figure 1 Meta-analysis estimating summary ORs of all published case-control association studies for LBP-1c/CP2/LSF in AD. The three independentcase-control samples described in the study by Lambert et al12 are considered separately. For all samples, crude ORs were determined from publishedgenotype distributions in cases v controls; summary ORs are based on these crude ORs. ‘‘All’’ represents the summary OR across all published studies;‘‘All excl extreme controls’’ represents the summary OR after removing samples with extreme allele frequencies in the control populations (that is, theUK sample in Lambert et al12 and the sample in Panza et al15). Filled squares represent sample specific crude ORs (sizes of squares are directlyproportional to the sample sizes) and filled diamonds summary ORs; lines represent 95% CI. See Methods section for details.
860 Bertram, Parkinson, McQueen, et al
www.jmedgenet.com

ascertained based on the presence of at least two AD cases infirst degree relatives of the same pedigree, while all previoussamples did not specifically consider family history. Thiscould potentially lead to the sampling of genetically distinctpopulations, that is, samples that are governed by differentgenetic risk factors and risk alleles. However, the observationthat at least one other investigation (by Panza et al) alsodescribed an over-representation of the A allele in their ADcases as compared to controls, suggests that differences inascertainment are probably not responsible for the observeddifferences in allele and genotype distributions acrossstudies. Finally, it is possible that the 39 UTR SNP is notactually pathogenic, but that the observed associationsmerely reflect LD with another genetic variant nearby. Inthis case, the—still elusive—true disease predisposing variantwould have independently occurred on the haplotype back-ground of the major allele in four of the examined case-control populations, while in the sample by Panza et al andour study it has arisen coupled with the minor allele. In theformer samples the A allele would thus appear as protective(since the actual risk allele is actually in LD with the G alleleat the 39 UTR SNP), while in the latter cases it would appearas a risk factor. On the other hand, there could be severalindependent and rare disease modifying variants within theLBP-1c gene, which would have also arisen on differenthaplotype backgrounds (for example, similar to what isobserved for PSEN1). Both alternatives are consistent withthe analyses provided in this study, which—at least in thefamilies analysed here—favour the existence of risk increas-ing variant(s) on the H4 background. It is noteworthy thatsimilar observations, that is, significant associations withopposite alleles across different samples and populations,have actually been reported with several other AD candidategenes in the past (for example, A2M (recently reviewed inSaunders et al11), LRP1,32 33 tumour necrosis factor a(TNFA),34 35 and butyrylcholinesterase K (BChE-K)36 37). Ifthey do not merely represent a collection of varying false-positive findings, these differences could be attributed to thedifferent patterns of LD across populations of different originand/or differing degrees of population heterogeneity. Whilewe favour this last alternative as the most likely explanationfor the observed differences with the 39 UTR SNP, clearlymore studies need to be performed on the potentialassociation of this and possibly other polymorphisms inLBP-1c/CP2/LSF and AD.In conclusion, we provide additional and independent
evidence suggesting that genetic variants in LBP-1c/CP2/LSFsignificantly alter the risk for developing AD. More studieswill need to be performed to further establish this associa-tion, and to more definitively assess which variant(s) areactually responsible for the observed effects and how theyaffect disease pathogenesis.
ACKNOWLEDGEMENTSThe authors wish to thank all families for participating in this study.
ELECTRONIC-DATABASE INFORMATION
The Haploview website can be found at http://www.broad.mit.edu/personal/jcbarret/haploview/index.php and International HapMaP Project can befound at http://www.hapmap.org/.
Authors’ affiliations. . . . . . . . . . . . . . . . . . . . .
L Bertram, M Parkinson, K Mullin, M Hsiao, R Menon, R E Tanzi,Genetics and Aging Research Unit, MassGeneral Institute forNeurodegenerative Diseases (MIND), Department of Neurology,Massachusetts General Hospital, Harvard Medical School, Charlestown,MA, USA
M B McQueen, Department of Biostatistics, Harvard School of PublicHealth, Boston, MA, USAT J Moscarillo, D Blacker, Gerontology Research Unit, Department ofPsychiatry, Massachusetts General Hospital, Harvard Medical School,Charlestown, MA, USA
This work was sponsored by grants from the NIMH, the NIA (ADRC),and the Alzheimer Association. LB was a fellow of the DeutscheForschungsgemeinschaft (DFG) and now receives a fellowship from theHarvard Center for Neurodegeneration and Repair (HCNR), Core A,and a stipend from the National Alliance for Research on Schizophreniaand Depression (NARSAD). MBM is supported by a National ResearchService Award, Training Program in Psychiatric Epidemiology andBiostatistics (T32 MH17119).
Competing interests: none declared
Correspondence to: Dr Rudolph E Tanzi, Genetics and Aging ResearchUnit, MGH-East (MIND), 114 16th St., Charlestown, MA 02129, USA;[email protected]
Revised version received 16 December 2004Accepted for publication 6 January 2005
REFERENCES1 Selkoe DJ. Deciphering the genesis and fate of amyloid beta-protein yields
novel therapies for Alzheimer disease. J Clin Invest 2002;110(10):1375–81.2 Tanzi RE, Bertram L. New frontiers in Alzheimer’s disease genetics. Neuron
2001;32(2):181–4.3 Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J,
Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familialAlzheimer disease. Proc Natl Acad Sci U S A 1993;90(5):1977–81.
4 Bertram L, Tanzi RE. Alzheimer’s disease: one disorder, too many genes?Hum Mol Genet 2004;13(Spec No 1):R135–41.
5 Wu WS, Holmans P, Wavrant-DeVrieze F, Shears S, Kehoe P, Crook R,Booth J, Williams N, Perez-Tur J, Roehl K, Fenton I, Chartier-Harlin MC,Lovestone S, Williams J, Hutton M, Hardy J, Owen MJ, Goate A. Geneticstudies on chromosome 12 in late-onset Alzheimer disease. JAMA1998;280(7):619–22.
6 Mayeux R, Lee JH, Romas SN, Mayo D, Santana V, Williamson J, Ciappa A,Rondon HZ, Estevez P, Lantigua R, Medrano M, Torres M, Stern Y, Tycko B,Knowles JA. Chromosome-12 mapping of late-onset Alzheimer diseaseamong Caribbean Hispanics. Am J Hum Genet 2002;70(1):237–43.
7 Myers A, Wavrant De-Vrieze F, Holmans P, Hamshere M, Crook R,Compton D, Marshall H, Meyer D, Shears S, Booth J, Ramic D, Knowles H,Morris JC, Williams N, Norton N, Abraham R, Kehoe P, Williams H,Rudrasingham V, Rice F, Giles P, Tunstall N, Jones L, Lovestone S, Williams J,Owen MJ, Hardy J, Goate A. Full genome screen for Alzheimer disease: stageII analysis. Am J Med Genet 2002;114(2):235–44.
8 Pericak-Vance MA, Bass MP, Yamaoka LH, Gaskell PC, Scott WK,Terwedow HA, Menold MM, Conneally PM, Small GW, Vance JM,Saunders AM, Roses AD, Haines JL. Complete genomic screen in late-onsetfamilial Alzheimer disease. Evidence for a new locus on chromosome 12.JAMA 1997;278(15):1237–41.
9 Rogaeva E, Premkumar S, Song Y, Sorbi S, Brindle N, Paterson A, Duara R,Levesque G, Yu G, Nishimura M, Ikeda M, O’Toole C, Kawarai T, Jorge R,Vilarino D, Bruni AC, Farrer LA, St George-Hyslop PH. Evidence for anAlzheimer disease susceptibility locus on chromosome 12 and for further locusheterogeneity. JAMA 1998;280(7):614–8.
10 Bertram L, Tanzi RE. Dancing in the dark? The status of late-onset Alzheimer’sdisease genetics. J Mol Neurosci 2001;17(2):127–36.
11 Saunders AJ, Bertram L, Mullin K, Sampson AJ, Latifzai K, Basu S, Jones J,Kinney D, MacKenzie-Ingano L, Yu S, Albert MS, Moscarillo TJ, Go RC,Bassett SS, Daly MJ, Laird NM, Wang X, Velicelebi G, Wagner SL, Becker DK,Tanzi RE, Blacker D. Genetic association of Alzheimer’s disease with multiplepolymorphisms in alpha-2-macroglobulin. Hum Mol Genet2003;12(21):2765–76.
12 Lambert JC, Goumidi L, Vrieze FW, Frigard B, Harris JM, Cummings A,Coates J, Pasquier F, Cottel D, Gaillac M, St Clair D, Mann DM, Hardy J,Lendon CL, Amouyel P, Chartier-Harlin MC. The transcriptional factor LBP-1c/CP2/LSF gene on chromosome 12 is a genetic determinant of Alzheimer’sdisease. Hum Mol Genet 2000;9(15):2275–80.
13 Taylor AE, Yip A, Brayne C, Easton D, Evans JG, Xuereb J, Cairns N,Esiri MM, Rubinsztein DC. Genetic association of an LBP-1c/CP2/LSF genepolymorphism with late onset Alzheimer’s disease. J Med Genet2001;38(4):232–3.
14 Luedecking-Zimmer E, DeKosky ST, Nebes R, Kamboh MI. Association of the39 UTR transcription factor LBP-1c/CP2/LSF polymorphism with late-onsetAlzheimer’s disease. Am J Med Genet 2003;117B(1):114–7.
15 Panza F, D’Introno A, Colacicco AM, Capurso C, Basile AM, Torres F,Capurso A, Solfrizzi V. LBP-1c/CP2/LSF gene polymorphism and risk ofsporadic Alzheimer’s disease. J Neurol Neurosurg Psychiatry2004;75(1):166–8.
16 Bing Z, Reddy SA, Ren Y, Qin J, Liao WS. Purification and characterization ofthe serum amyloid A3 enhancer factor. J Biol Chem1999;274(35):24649–56.
LBP-1c/CP2/LSF association in AD families 861
www.jmedgenet.com

17 Lau KF, Miller CC, Anderton BH, Shaw PC. Molecular cloning andcharacterization of the human glycogen synthase kinase-3beta promoter.Genomics 1999;60(2):121–8.
18 Zambrano N, Minopoli G, de Candia P, Russo T. The Fe65 adaptor proteininteracts through its PID1 domain with the transcription factor CP2/LSF/LBP1.J Biol Chem 1998;273(32):20128–33.
19 Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] activecomplex of APP with Fe65 and histone acetyltransferase Tip60. Science2001;293(5527):115–20.
20 Blacker D, Albert MS, Bassett SS, Go RC, Harrell LE, Folstein MF. Reliabilityand validity of NINCDS-ADRDA criteria for Alzheimer’s disease. The NationalInstitute of Mental Health Genetics Initiative. Arch Neurol1994;51(12):1198–204.
21 Blacker D, Bertram L, Saunders AJ, Moscarillo TJ, Albert MS, Wiener H,Perry RT, Collins JS, Harrell LE, Go RC, Mahoney A, Beaty T, Fallin MD,Avramopoulos D, Chase GA, Folstein MF, McInnis MG, Bassett SS, Doheny KJ,Pugh EW, Tanzi RE. Results of a high-resolution genome screen of 437Alzheimer’s disease families. Hum Mol Genet 2003;12(1):23–32.
22 Mullin K, Bertram L, Moscarillo TJ, Becker KD, Wang C, Growdon J,Blacker D, Tanzi RE. Genetic association within the IDE region on chromosome10 - The Consortium on Alzheimer’s Genetics (CAG). Poster (abstract no.202.8) presented at the 33rd Annual Meeting of the Society for Neuroscience,New Orleans, LA, 2003.
23 Rabinowitz D, Laird N. A unified approach to adjusting association tests forpopulation admixture with arbitrary pedigree structure and arbitrary missingmarker information. Hum Hered 2000;50(4):211–23.
24 Lake SL, Blacker D, Laird NM. Family-based tests of association in thepresence of linkage. Am J Hum Genet 2000;67(6):1515–25.
25 Martin ER, Monks SA, Warren LL, Kaplan NL. A test for linkage andassociation in general pedigrees: the pedigree disequilibrium test. Am J HumGenet 2000;67(1):146–54.
26 Martin ER, Bass MP, Kaplan NL. Correcting for a potential bias in the pedigreedisequilibrium test. Am J Hum Genet 2001;68(4):1065–7.
27 Witte JS, Gauderman WJ, Thomas DC. Asymptotic bias and efficiency incase-control studies of candidate genes and gene-environment interactions:basic family designs. Am J Epidemiol 1999;149(8):693–705.
28 Siegmund KD, Langholz B, Kraft P, Thomas DC. Testing linkage disequilibriumin sibships. Am J Hum Genet 2000;67(1):244–8.
29 Laird NM, Mosteller F. Some statistical methods for combining experimentalresults. Int J Technol Assess Health Care 1990;6(1):5–30.
30 DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials1986;7(3):177–88.
31 Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replicationvalidity of genetic association studies. Nat Genet 2001;29(3):306–9.
32 Sanchez-Guerra M, Combarros O, Infante J, Llorca J, Berciano J, Fontalba A,Fernandez-Luna JL, Pena N, Fernandez-Viadero C. Case-control study andmeta-analysis of low density lipoprotein receptor-related protein gene exon 3polymorphism in Alzheimer’s disease. Neurosci Lett 2001;316(1):17–20.
33 Kolsch H, Ptok U, Mohamed I, Schmitz S, Rao ML, Maier W, Heun R.Association of the C766T polymorphism of the low-density lipoproteinreceptor-related protein gene with Alzheimer’s disease. Am J Med Genet2003;121B(1):128–30.
34 Perry RT, Collins JS, Wiener H, Acton R, Go RC. The role of TNF and itsreceptors in Alzheimer’s disease. Neurobiol Aging 2001;22(6):873–83.
35 Alvarez V, Mata IF, Gonzalez P, Lahoz CH, Martinez C, Pena J,Guisasola LM, Coto E. Association between the TNFalpha-308 A/Gpolymorphism and the onset-age of Alzheimer disease. Am J Med Genet2002;114(5):574–7.
36 Lehmann DJ, Johnston C, Smith AD. Synergy between the genes forbutyrylcholinesterase K variant and apolipoprotein E4 in late-onset confirmedAlzheimer’s disease. Hum Mol Genet 1997;6(11):1933–6.
37 Hiltunen M, Mannermaa A, Helisalmi S, Koivisto A, Lehtovirta M,Ryynanen M, Riekkinen P Sr, Soininen H. Butyrylcholinesterase K variant andapolipoprotein E4 genes do not act in synergy in Finnish late-onsetAlzheimer’s disease patients. Neurosci Lett 1998;250(1):69–71.
A Uzumcu, H Kayserili, M Y Apak, OUyguner, Bernd Wollnik*, Child HealthInstitute, Division of Medical Genetics,Istanbul University, Istanbul, TurkeyA Gezer, Department of Ophthalmology,
Istanbul Faculty of Medicine, IstanbulUniversityS H Gultekin, Department of Pathology,
Section of Neuropathology, Mount SinaiSchool of Medicine of New York UniversityH-C Hennies, P Nurnberg, Gene
Mapping Center and Department ofMolecular Genetics, Max Delbruck Centerfor Molecular Genetics, Berlin, Germany* Co-senior authors who contributed
equally to this research
Correspondence to:Professor Robert J Desnick, PhD, MD,
Department of Human Genetics, MountSinai School of Medicine, 1425 MadisonAvenue, Box 1498, New York, NY 10029,USA; [email protected] Wollnik, MD, Institute of Child
Health, Division of Medical Genetics,Istanbul University, Millet Caddesi, Capa,34390 Istanbul, Turkey; [email protected] figure 3 the marker D21S1400 should be
D21S1440.On page 413 in the second paragraph, the
marker D2151446 should be D21S1446.
CORRECTION
doi: 10.1136/jmg.2005.26138corr1
In the paper titled, A new syndrome, con-genital extraocular muscle fibrosis with ulnarhand anomalies, maps to chromosome 21qter(J Med Genet 2004;42:408-15) there are anumber of errors. The affiliations and corre-spondence details were incorrect, the correctdetails have been listed below:T Tukel, R J Desnick*, Department of
HumanGenetics, Mount Sinai School of Medi-cine of New York University, New York, USA
862 Bertram, Parkinson, McQueen, et al
www.jmedgenet.com























![Jacobo Grinberg.vision Extraocular [Articulo]](https://img.pdfslide.net/doc/110x75/577cde0c1a28ab9e78ae476f/jacobo-grinbergvision-extraocular-articulo.jpg)





