Embed Size (px)
Citation preview

Skript zur Vorlesung :
Theoretische Physik IV Statistische Physik
Vorlesung WS 17/18
Prof. Dr. Jens Timmer
9. Februar 2018
Ernst gemeint bis zum bitteren Ende
1

Inhaltsverzeichnis1 Einleitung 5
I Thermodynamik 9
2 Thermodynamische Systeme 9
3 Der 1. Hauptsatz der Thermodynamik 103.1 Thermodynamische Zustandsgleichungen . . . . . . . . . . . . . . . . 103.2 Mathematisches Vorspiel . . . . . . . . . . . . . . . . . . . . . . . . . 143.3 Innere Energie und Temperatur, 0. Hauptsatz . . . . . . . . . . . . . 183.4 Arbeit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 183.5 Wärme . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 223.6 Der 1. Hauptsatz . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
4 Der 2. Hauptsatz der Thermodynamik 294.1 Reversible und irreversible Prozesse . . . . . . . . . . . . . . . . . . . 304.2 Carnot’scher Kreisprozess . . . . . . . . . . . . . . . . . . . . . . . . 334.3 Thermodynamische Definition der Entropie . . . . . . . . . . . . . . . 364.4 Der 2. Hauptsatz . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 394.5 Fundamentalrelationen . . . . . . . . . . . . . . . . . . . . . . . . . . 434.6 Euler-Gleichung und Gibbs-Duhem-Relation . . . . . . . . . . . . . . 46
5 Der 3. Hauptsatz der Thermodynamik 49
6 Thermodynamische Potentiale 536.1 Prinzip der maximalen Entropie – Prinzip der minimalen Energie . . 546.2 Freie Energie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 566.3 Legendre-Transformation . . . . . . . . . . . . . . . . . . . . . . . . . 596.4 Enthalpie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 626.5 Freie Enthalpie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 656.6 Großkanonisches Potential . . . . . . . . . . . . . . . . . . . . . . . . 676.7 Maxwell-Relationen . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
7 Zusammenfassung Thermodynamik 71
II Statistische Mechanik 72
2

8 Ein bißchen Statistik 728.1 Verteilungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 728.2 Beispiele für Verteilungen . . . . . . . . . . . . . . . . . . . . . . . . 77
9 Entropie revisited 929.1 Vorspiel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 939.2 Formalisierung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 979.3 Entropie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1009.4 Zustandssumme und Entropie des idealen Gases . . . . . . . . . . . . 1019.5 Shannon-Information . . . . . . . . . . . . . . . . . . . . . . . . . . . 1129.6 Prinzip der maximalen Entropie . . . . . . . . . . . . . . . . . . . . . 1189.7 3. Hauptsatz revisited . . . . . . . . . . . . . . . . . . . . . . . . . . 1219.8 Entropische Kräfte revisited . . . . . . . . . . . . . . . . . . . . . . . 123
10 Gesamtheiten 12510.1 Ergodenhypothese . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12610.2 Mikrokanonische Gesamtheit . . . . . . . . . . . . . . . . . . . . . . . 12910.3 Kanonische Gesamtheit . . . . . . . . . . . . . . . . . . . . . . . . . . 13010.4 Großkanonische Gesamtheit . . . . . . . . . . . . . . . . . . . . . . . 13610.5 Äquivalenz der Gesamtheiten . . . . . . . . . . . . . . . . . . . . . . 14010.6 Fluktuationen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 141
11 Anwendungen 14311.1 Maxwell’sche Geschwindigkeitsverteilung . . . . . . . . . . . . . . . . 14311.2 Barometrische Höhenformel . . . . . . . . . . . . . . . . . . . . . . . 14411.3 Gleichverteilungs- und Virialsatz . . . . . . . . . . . . . . . . . . . . . 14511.4 Thermodynamische Freiheitsgrade in Quantensystemen . . . . . . . . 14811.5 Zwei-Niveau Systeme . . . . . . . . . . . . . . . . . . . . . . . . . . . 15111.6 Negative Temperaturen . . . . . . . . . . . . . . . . . . . . . . . . . . 15311.7 Relativistisches ideales Gas . . . . . . . . . . . . . . . . . . . . . . . . 156
12 Quantengase 16012.1 Symmetrie der Vielteilchenwellenfunktionen . . . . . . . . . . . . . . 16012.2 Besetzungszahl-Darstellung . . . . . . . . . . . . . . . . . . . . . . . 16412.3 Ideales Fermi-Gas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16612.4 Ideales Bose-Gas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16812.5 Das Planck’sche Strahlungsgesetz . . . . . . . . . . . . . . . . . . . . 17112.6 Phononen und spezifische Wärme von Festkörpern . . . . . . . . . . . 177
3

13 Näherungsverfahren 18113.1 Virialentwicklung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18213.2 Molekularfeldnäherung . . . . . . . . . . . . . . . . . . . . . . . . . . 18613.3 Renormierungsgruppentheorie . . . . . . . . . . . . . . . . . . . . . . 192
14 Phasenübergänge 19714.1 Klassifikation von Phasenübergängen . . . . . . . . . . . . . . . . . . 19714.2 Skalierungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19914.3 Landau-Theorie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 200
15 Danksagung 203
4

1 EinleitungTechnicalities:
• Welche Semester ?
• Skript, Übungsanmeldung und Kommunikation über homepage
• Skript ist dünn, ersetzt nicht das Studium von Lehrbüchern !
Hierarchie
– Vorlesung: Konzepte– Bücher: Konzepte und Details– Übungen rechnen: Verständnis
• Was schon in der Experimental-Physik gehört ?
• Inhaltsverzeichnis
• Wenn etwas unklar: Fragen !
• Kurzklausuren
• Münsteraufgaben, Münsterführung
Literatur:
• W. Greiner, L. Neise, H. Stöcker. Thermodynamik und Statistische Mechanik
Englische Version liegt kostenfrei auf dem Netz
• T. Fließbach. Statistische Physik
• J. Honerkamp. Statistical Physics
• W. Nolting. Theoretische Physik 6. Statistische Physik
• F. Schwabl. Statistische Mechanik
Unterschiede der Bücher:
• Verhältnis Text zu Gleichungen
• Systematik der Trennung zwischen Thermodynamik und Statistischer Physik
Diese Vorlesung: Fast komplette Trennung
5

Was bisher geschah:
• Klassische Mechanik
Newton: F = ma, z.B. F = −Gm1m2
r3~r
Lagrange:d
dt
∂
∂qL(q, q)− ∂
∂qL(q, q) = 0
Hamilton: q =∂
∂pH(q, p), p = − ∂
∂qH(q, p)
• Elektrodynamik
Maxwell’sche Gleichungen
∇E =1
ε0ρ, ∇B = 0, ∇× E = − ∂
∂tB, ∇×B = µ0j + ε0µ0
∂
∂tE
Ergeben z.B.:
1
c2
∂2
∂t2E =
∂2
∂x2E
• Quantenmechanik
Schrödinger-Gleichung
i~∂
∂tψ =
(− ~2
2m
∂2
∂x2+ V
)ψ
Das sind alles deterministische Bewegungsgleichungen. Machen die Physik quanti-tativ und prädiktiv.
And now for something completely different ...
• In der Statistischen Physik werden keine Bewegungsgleichungen, z.B. für dieWärme, abgeleitet
• Stattdessen: Verständnis des kollektiven Verhaltens vieler Teilchen
Beispiele:
• Neue Begriffe wie Temperatur
6

• Wirkungsgrad von Wärmemaschinen
• Eis, Wasser, Gas: Phasenübergänge
• Fermionische und bosonische Systeme
• ...
Statistische Physik besteht aus
• Thermodynamik, allerdings nicht sehr dynamisch
• Statistische Mechanik
• Nicht-Gleichgewichtssysteme
• Stochastische Prozesse
Thermodynamik und Statistische Mechanik
• Gleichgewichtszustände
• Anzahl Teilchen makroskopischer Systeme ist in der Größenordnung 1023
• Orte und Impulse definieren Mikrozustand
• Mikroskopische Dynamik ist chaotisch, i.e. nicht periodisch.
Frage: Chaos klar ?
• Detailierte Lösung der Bewegungsgleichungen hoffungslos ...
• ... und auch sinnlos, Anspielung auf Human Brain Project
• Beobachtung: Makrozustand beschreibbar durch wenige Zustandsvariablen wieVolumen, Druck, Temperatur, Energie
Thermodynamik:
• Theorie von Makrosystemen
• Beschreibt Abhängigkeiten der Zustandsvariablen
• Beschreibt mögliche Zustandsänderungen
7

• Partiell prädiktiv, partiell beschreibend
Statistische Mechanik:
• Stellt Zusammenhang zwischen Mikro- und Makrozuständen her
• Leitet makroskopische Eigenschaften aus mikroskopischen Eigenschaf-ten/Wechselwirkungen ab, Anspielung auf emergentes Verhalten
• Prädiktiv, obwohl statistisch
Bonbon: Es wird sich zeigen, dass es eine konsistente konsequent klassische Statisti-sche Mechanik nicht geben kann, essentiell quantenmechanisch
Stichworte
• Gibbs’sches Paradoxon
• Spezifische Wärme großer Moleküle
• Normierung der Zustandssumme
• Planck’sches Strahlungsgesetz
8

Teil I
Thermodynamik2 Thermodynamische SystemeThermodynamisches System:Eine Menge von Materie - Gase, Fluide, Festkörper - deren physikalischen Ei-genschaften eindeutig und vollständig durch Angabe bestimmter makroskopischerVariablen beschrieben werden kann.
Ein thermodynamisches System heißt
• abgeschlossen oder isoliert, wenn es nichts mit Umwelt austauscht.
Gesamtenergie, Teilchenzahl und Volumen sind erhalten und damit zur Be-schreibung des Systems geeignet.
Beispiel: Perfekte Thermoskanne
• geschlossen, wenn es keine Materie mit Umwelt austauscht.
Energieaustausch ist erlaubt. Temperatur wird wichtige Grösse
Beispiel: Milchtüte
• offen, sonst. Austausch von Energie und Teilchen erlaubt.
Chemisches Potential wird wichtige Größe
• homogen, wenn es in allen Bereichen gleich ist
• inhomogen, wenn es verschiedene Phasen aufweist
Beispiel: flüssige und gasförmige Phase
• Workhorse der Thermodynamik: Das ideale Gas
Zustandsgrößen sind makroskopische Variablen, die den Zustand des Systems cha-rakterisieren.Beispiele: Volumen, Energie, Entropie, Temperatur, Druck
• Sie sind wegunabhängig, hängen nicht davon ab, wie das System in seinenZustand gekommen ist
9

• extensiv: proportional zur Systemgröße
Beispiele: Volumen, Energie
• intensiv: unabhängig von Systemgröße
Beispiele: Temperatur, Druck
Thermodynamische Zustandsgleichungen
• stellen Beziehungen zwischen Zustandsgrößen dar
• werden in der Thermodynamik meist empirisch bestimmt
• haben eingeschränkten Gültigkeitsbereich
• Systematische Ableitung in der Statistischen Mechanik
Thermodynamischer Limes:
N →∞, V →∞, N
V= ρ = const.
Thermodynamisches Gleichgewicht:
• Die (makroskopischen) Zustandsgrößen ändern sich nicht.
• Die Mikrozustände natürlich dauernd.
3 Der 1. Hauptsatz der Thermodynamik
3.1 Thermodynamische Zustandsgleichungen
Thermische Zustandsgleichung setzt p, V , T und N in Beziehung
• Beispiel: Ideales Gas
– Boyle (1664) und Mariotte (1676): pV = const. für T = const.
– Gay-Lussac: (1802): VT
= const. für p = const.
10

Zusammen, siehe Übung:
pV
T= const. ∝ N, da extensiv
ergibt Gasgleichung:
pV = NkT oder p = ρkT
mit Boltzmann-Konstante
k = 1.38 · 10−23 J
K
Gilt für geringe Drücke, geringe Dichte, hohe Temperaturen und im thermody-namischen Limes
• Beispiel: Reales Gas
Ideales Gas vernachlässigt:
– Eigenvolumen b der Teilchen
V → V −Nb
Kleiner Effekt
– Attraktive Wechselwirkung der Teilchen bewirkt Druckminderung pbinnenProportional zur quadrierten Dichte :
(NV
)2
pideal = preal + pbinnen, p→ p+
(N
V
)2
a
Relevanter Effekt
– Ergibt van der Waals Gleichung (1873), siehe Übung(p+
(N
V
)2
a
)(V −Nb) = NkT
Systematische Herleitung in Kap. 13.1.1
11

Kalorische Zustandsgleichung: Eine Energiegleichung
Betrachte Ideales Gas in abgeschlossenem System:
• Im thermischen Gleichgewicht ändern sich die Geschwindigkeiten der Teilchenständig
• Aber im Mittel werden immer gleich viele Teilchen in einem Geschwindigkeits-kubus d3v vorliegen
• Geschwindigkeits-Verteilung f(~v) ändert sich nicht
Es gilt∫∞−∞ f(~v)d3v = 1 und Anzahl dN von Teilchen im Kubus d3v
dN = Nf(~v)d3v
• Druck entsteht durch Impulsübertrag bei Reflexion an Wänden
Sei x−Achse senkrecht zu Wand-Flächenelement O, Impulsübertrag: 2mvx
• Frage: Wieviele Teilchen mit Geschwindigkeit ~v treffen in dt auf O ?
Anzahl der Teilchen in Parallelepiped
dN = NdV
Vf(~v)d3v
mit dV/V Bruchteil des Volumens des Epipeds vom Gesamtvolumen mit dV =Ovxdt
12

• Damit Kraftstoß dFO auf Fläche O
dFOdt = 2mvxdN = 2Nmv2xf(~v)d3v
Odt
V
”Kürze” dt und sammele alle Teilchen mit positiver Geschwindigkeit vx auf:
p =1
O
∫dF0 =
N
V
∫ ∞0
dvx
∫ ∞−∞
dvy
∫ ∞−∞
dvzf(~v)2mv2x
Beachte: f(~v) kann nicht von Richtung von ~v abhängen, nur von |~v|Folge: ∫ ∞
0
dvx . . . =1
2
∫ ∞−∞
dvx . . .
• Damit
pV = mN
∫ ∞−∞
d3vf(~v)v2x
Integral: Mittlere quadradratische Geschwindigkeit senkrecht zur Fläche
• Wegen Isotropie ∫ ∞−∞
d3vf(~v)v2x = 〈v2
x〉 = 〈v2y〉 = 〈v2
z〉
Erwartungswert klar ?
Wegen~v2 = v2
x + v2y + v2
z
gilt
〈v2x〉 =
1
3〈~v2〉
und
pV = mN1
3〈~v2〉 =
2
3N〈εkin〉, 〈εkin〉 : mittlere kinetische Energie eines Teilchens
13

• Mit pV = NkT und E = N〈εkin〉 erhalten wir
E =3
2NkT
• Beachte: f(~v) muss nicht bekannt sein
• Schönere Herleitung in Kap. 9.4
3.2 Mathematisches Vorspiel
• Erinnere Taylorentwicklung
f(x) = f(x0) + f ′(x0)(x− x0) + ...
Taylor-Entwicklung 1. Ordnung
– gültig für kleine ∆x = x− x0
– ∆f = f(x0 + ∆x)− f(x0) ≈ f ′(x0)∆x
– beste lineare Näherung
– Infinitesimal: df = f ′(x)dx
• Mehr-dimensionaler Fall
∆f(x, y) = f(x0 + ∆x, y0 + ∆y)− f(x0, y0)
df(x, y) =∂f
∂x
∣∣∣∣y
dx+∂f
∂y
∣∣∣∣x
dy
Beschreibt Tangentialebene
• Definition: Das totale (oder vollständige oder exakte) Differential einer diffe-renzierbaren Funktion ist
df =N∑i=1
∂f
∂xidxi
14

• Erinnere 1. Semester
Kurvenintegration
f = f(x, y) = f(~x), ~x =
(xy
)df =
∂f
∂xdx+
∂f
∂ydy
= ~∇f · d~x
mit
~∇ =
(∂∂x∂∂y
)und d~x =
(dxdy
)• Für vollständiges Differential
df = ~F (~x) · d~x
gilt
~F (~x) = ~∇f(~x)
f ist Stammfunktion von ~F
• Wegintegral über ~F hängt nur von Anfangs- und Endpunkt ab, aber nicht vomIntegrationsweg.
• Beispiel: Arbeit W an Teilchen entlang einer Kurve C von ~x1 nach ~x2 in kon-servativem Kraftfeld ~F (~x) mit Potential V (~x)
W =
∫C
~F (~x) · d~x = −∫C
~∇V (~x) · d~x = −V (~x2) + V (~x1)
15

• Test auf Exaktheit:
Sei f(x, y) zweimal differenzierbare Funktion mit
df(x, y) =∂f
∂xdx+
∂f
∂ydy =
(FxFy
)·(dxdy
)= ~F · d~x
Es gilt der Satz von Schwarz
∂2f
∂x∂y=
∂2f
∂y∂x
oder∂Fy∂x
=∂Fx∂y
Damit: Drehe es herum. Gehe von beliebigem Differential und damit ~F (~x) ausund teste, ob es exakt ist.
• Beispiele:
– In 1-D ist jedes Differential trivial exakt
– Betrachte
~F · d~x = ydx+ xdy
Dann gilt
16

∂Fx∂y− ∂Fy
∂x= 1− 1 = 0
und das Differential ist exakt– Betrachte
~F · d~x = yx dx+ x2 dy
Es folgt
∂Fx∂y− ∂Fy
∂x= x− 2x = −x 6= 0
und das Differential ist nicht exakt.
• Nicht-exakte Differentiale:
– Wegintegral hängt vom Weg ab– werden mit δf bezeichnet
• In drei Dimensionen
df =
FxFyFz
· dx
dydz
= ~F · d~x
rot~F = 0 =⇒ df ist vollständiges Differential
• Integrierender Faktor
– Trick, aus einem nicht-exakten Differential ein exaktes zu machen– Betrachte zweites Beispiel und ergänze 1
x
1
x
(yx dx+ x2 dy
)– Es folgt
∂Fx∂y− ∂Fy
∂x= 1− 1 = 0
ein exaktes Differential.– 1
xist der integrierende Faktor für nicht exaktes Differential yx dx+ x2 dy
– Siehe Übung
17

3.3 Innere Energie und Temperatur, 0. Hauptsatz
Ein isoliertes System erhält die Gesamtenergie.Diese innere Energie U ist
• die Summe aller Energien
• eine Zustandsgröße
• extensiv
• im allgemeinen in Bezug auf ihre Änderung interessant
Beispiel: Ideales Gas
U =3
2NkT, mit Boltzmann-Konstante k = 1.38 · 10−23 J
K
• Der 0. Hauptsatz: Es gibt eine intensive Zustandsvariable Temperatur T , sodass sich Systeme genau dann miteinander im thermischen Gleichgewicht be-finden, wenn sie den selben Wert von T aufweisen.
• Bei einem isolierten System ist die innere Energie identisch mit der aus Me-chanik und Elektrodynamik bekannten Gesamtenergie.
• Für nicht-isolierte Systeme kommt noch Arbeit und/oder Wärmeaustausch hinzu.
3.4 Arbeit
• Die innere Energie U ist im isolierten System erhalten.
• U ändert sich aber bei Interaktion mit Umgebung.
Betrachte Kolben, der mit Kraft ~Fa auf System drückt
18

Im Gleichgewicht: ~Fa = −~FiVerschiebe Kolben um d~s
δW = ~Fa · d~s = −~Fi · d~s
Vorzeichenkonvention:
– System leistet Arbeit, Energie wird abgeführt: δW < 0
– Am System wird Arbeit geleistet, Energie wird zugeführt: δW > 0
Da ~Fa‖d~s und dV = Ads, folgt
~Fi · d~s =FiAAds = pdV
und somit:
δW = −pdV, beachte: dV < 0
Beachte: Die Arbeit ist keine Zustandsgröße
Beispiel: Kompression eines Gases
V1 → V2, V2 < V1
∆W = −∫ V2
V1
p(V )dV
Proof by examples:
19
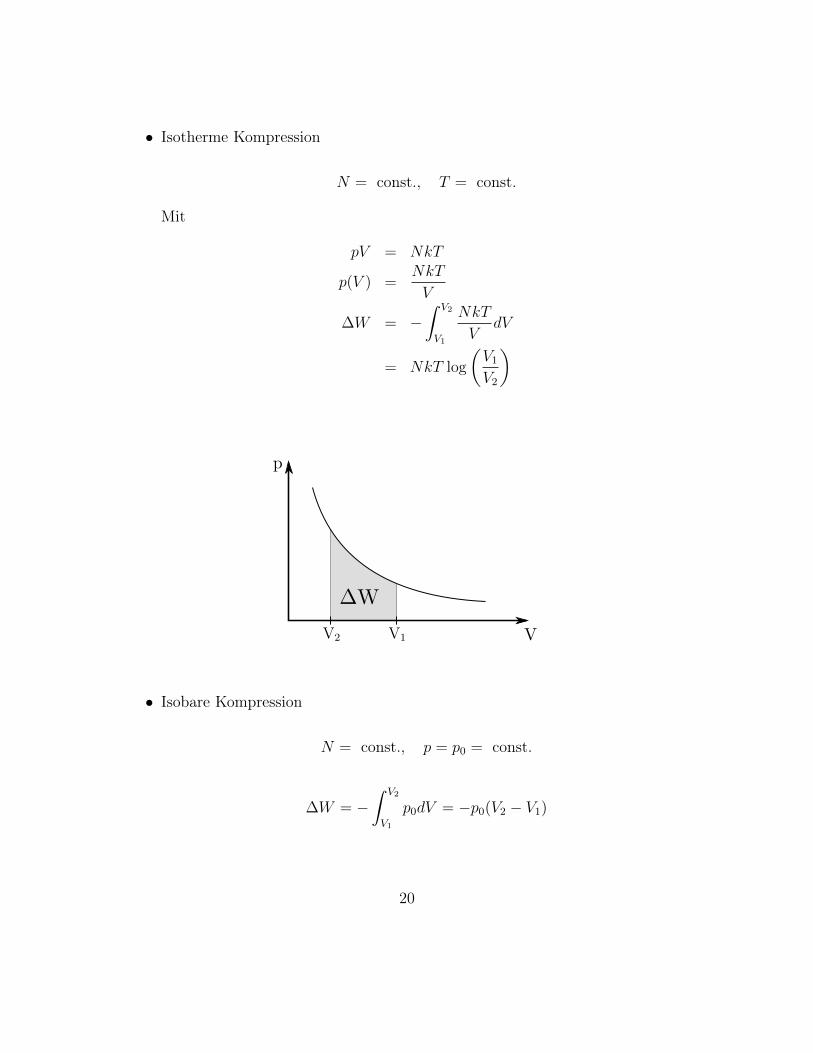
• Isotherme Kompression
N = const., T = const.
Mit
pV = NkT
p(V ) =NkT
V
∆W = −∫ V2
V1
NkT
VdV
= NkT log
(V1
V2
)
V1V2
• Isobare Kompression
N = const., p = p0 = const.
∆W = −∫ V2
V1
p0dV = −p0(V2 − V1)
20

V1V2
• Ergo: ∆W hängt vom Vorgehen ab.
Es gilt:
• Die Größe X ist genau dann eine Zustandsgröße, wenn sie ein vollständigesDifferential dX besitzt.
1. weekWeitere Beispiele für Arbeit
• Änderung der elektrischen Ladung q im elektrischen Potential φ
δW = φdq
Widerstand des Systems gegen Zuführung weiterer Ladung analog zu Druckals Widerstand gegen Kompression
• Änderung der Teilchenzahl N , mit chemischem Potential µ
δW = µdN
Widerstand gegen Zuführung von weiteren Teilchen
• δW ist immer ein Produkt aus einer intensiven und einer extensiven Zustands-größe, Energie-konjugierte Variablen
• Alle Arten von Arbeit können (im Prinzip) ineinander überführt werden
21

Integrierender Faktor revisited:
• δW kein exaktes Differential
δW = −pdV
• 1/p ist integrierender Faktor
dV = −1
pδW
3.5 Wärme
Antoine Laurent de Lavoisier (1787) Kalorische Theorie: Wärme als Substanz
• Ein weiteres Element, nicht zu erzeugen, nicht zu vernichten
• Unsichtbar
• Gewichtslos
• Selbstabstoßend, darum gleichen sich Temperaturunterschiede aus
• Lockerung der Moleküle durch Ansammlung der kalorischen Substanz führtzum Schmelzen/Verdampfen
• Widerlegung der Theorie: Sir Benjamin Thompson, Graf Rumford (1789) Ge-genbeispiel: Reibung (beim Bohren von Kanonen)
Mayer (1845):Wärme ist eine Form der Energie(änderung), keine Zustandsgröße.
Definition:δQ ist die Wärmemenge, die eine Temperaturänderung einer Substanz von dT bewirkt
δQ = C(T ) dT
mit C, der totalen Wärmekapazität der erwärmten Substanz
• C ist extensive Größe, da δQ extensiv und dT intensiv
22

• Zur Charakterisierung einer Substanz besser intensive Größe:
c =C
Nspezifische Wärme(kapazität)
odercM =
NA
NC molare spezifische Wärme(kapazität)
mit Avogadro-Konstante NA ≈ 6 · 1023 die Anzahl der Teilchen pro Mol (12 gC12)
CV und Cp
• Wärmekapazität hängt von Prozessführung ab, p = konstant (Cp) oder V =konstant (CV )
• Allgemeinen gilt Cp > CV , da bei p = const. auch Volumenausdehnungsarbeitgeleistet werden muss
• Betrachte ideales Gas bei V = const. und Temperaturänderung dT
dU = δQ = dQ = CV dT
Dann gilt
CV =
(∂U
∂T
)V
Ideales Gas:
U =3
2NkT
CV =
(∂U
∂T
)V
=3
2Nk
cV =CVN
=3
2k
23

3.6 Der 1. Hauptsatz
Ein System, das mit Umgebung Arbeit δW und Wärme δQ austauscht, verändertseine innere Energie U nach
dU = δW + δQ
Vorzeichenkonvention:
• δQ > 0 : dem System zugeführte Wärme
• δQ < 0 : vom System abgegebene Wärme
• δW > 0 : dem System zugeführte Arbeit
• δW < 0 : vom System abgegebene Arbeit
Beachte:
• Energieerhaltung
• Innere Energie U ist eine Zustandsgröße
• dU damit ein vollständiges Differential
• δW und δQ im allgemeinen pfadabhängig, keine vollständigen Differentiale
• Erinnere kalorische Theorie: U ist global erhalten, aber keine Substanz, sonderneine Eigenschaft der Materie
• Interpretation: Es gibt kein Perpetuum mobile (sich ständig Bewegendes)1. Art, d.h. keine Maschine, die nichts anderes macht als Arbeit zu leisten
• Seit 1775: Französische Akademie der Wissenschaften nimmt keine Vorschlägemehr an
Anwendung:Betrachte Zustandsgleichung des idealen Gases
pV = NkT
• Im folgenden N konstant.
• 3 Variablen: p, V und T
Hält man jeweils eine konstant, ergeben sich drei mögliche Prozesse.
24

• 4. Prozess: Adiabatischer Prozess mit δQ=0
Ausgangspunkt: 1. Hauptsatz mit U(V, T )
dU =
(∂U
∂V
)T
dV +
(∂U
∂T
)V
dT
Betrachte ideales Gas
U =3
2NkT
CV =3
2Nk
pV = NkT
• V = V0 = konstant. Isochore Zustandsänderung
Experiment: Erhitzen von Gas in geschlossenem Gefäß von T1 nach T2
(V0, p1, T1)∆Q→ (V0, p2, T2)
In Abhängigkeit von Änderung von T
dU = δQ− p dV︸︷︷︸=0
= δQ = dQ = CV dT
∆U = U2 − U1 =
∫ T2
T1
CV dT = CV (T2 − T1) = ∆Q
In Abhängigkeit von Änderung von p
Mit Ti = piV0Nk
folgt:
∆U =CV V0
Nk(p2 − p1)
Merke :∆U = ∆Q = CV (T2 − T1) (1)
• p = p0 = konstant. Isobare Zustandsänderung
(V1, p0, T1)∆Q,∆W→ (V2, p0, T2)
25

Experiment: Gefäß mit flexibler masseloser Abdeckung unter Luftdruck
dU = δQ+ δW
∆W = −∫ V2
V1
p0 dV = −p0(V2 − V1)
– Beachte: δQ = CV dT nicht anwendbar, da p = const.
– Für ideales Gas: U = 32NkT , unabhängig vom Volumen
– Damit gilt allgemein
dU =
(∂U
∂T
)V
dT = CV dT
Gilt auch für adiabatische Prozesse.
– Damit
∆U =
∫ T2
T1
CV dT = CV (T2 − T1) =CV p0
Nk(V2 − V1)
Damit Wärmeänderung:
∆Q = ∆U −∆W = p0
(CVNk
+ 1
)(V2 − V1)
MitVi =
NkTip0
folgt∆Q = (CV +Nk)(T2 − T1)
Merke:
– ∆Q ist wegen Ausdehnungarbeit im isobaren Fall größer als in isochoren,Gl. (1).
– Für ideales Gas gilt, Beweis folgt:
Cp = CV +Nk =5
2Nk
26

• T = T0 = konstant. Isotherme Zustandsänderung
(V1, p1, T0)∆Q,∆W→ (V2, p2, T0)
Experiment: Expansion in Gefäß im Wärmebad
U =3
2NkT0 → dU = 0
∆W = −∫ V2
V1
p(V )dV = −NkT0
∫ V2
V1
dV
V= −NkT0 log
(V2
V1
)= NkT0 log
(p2
p1
)∆Q = −∆W = NkT0 log
(V2
V1
)• δQ=0. Adiabatischer Prozess, kein Wärmeaustausch
Experiment: Isoliertes Gefäß
(V1, p1, T1)∆W→ (V2, p2, T2)
Mit
dU = δW + δQ︸︷︷︸=0
= δW = dW
dU = CV dT
dW = −pdV = −NkTV
dV
folgt:
CVTdT = −Nk
VdV
CV
∫ T2
T1
dT
T= −Nk
∫ V2
V1
dV
V
CV log
(T2
T1
)= −Nk log
(V2
V1
)T2
T1
=
(V2
V1
)− NkCV
=
(V1
V2
)2/3
Entsprechend, mit Hilfe der Gasgleichung: Die Adiabaten-Gleichungen:
27

(T2
T1
)5/2
=p2
p1
,p1
p2
=
(V2
V1
)5/3
pV -Adiabaten (Isentropen) verlaufen steiler als Isothermen: pV 5/3 = const.
Lessons learned
• Für ideales Gas gilt:
pV = NkT, U =3
2NkT, CV =
3
2Nk
• Wärme ist kein Stoff
• Wärme und Arbeit sind keine exakten Differentiale
• Innere Energie ist exaktes Differential
• Adiabaten verlaufen steiler als Isothermen
Die d-Zoologie1: ∂, d, δ, ∆
• ∂: partielle Ableitung
d
dxf(y(x)) =
∂f
∂y
∂y
∂x
• d: exaktes Differentialdf(x, y) =
∂f
∂xdx+
∂f
∂ydy
mit∂f
∂x∂y=
∂f
∂y∂x
• δ: nicht exaktes Differential
δf(x, y) = g(x, y)dx+ h(x, y)dy
1Auch als Deologie bezeichnet
28

mit∂g
∂y6= ∂h
∂x
• ∆: Endliche Änderung
∆W =
∫ V2
V1
dV....
4 Der 2. Hauptsatz der ThermodynamikExtremalprinzipien in der Physik:
• Klassische Mechanik: Minimierung der Wirkung, Hamilton’sches Prinzip,Euler-Lagrange Gleichungen
• Optik: Fermat’sches Prinzip: Kürzeste optische Weglänge
• Dissipatives mechanisches System: Minimierung der Energie
• Globales ”als ob” vs. lokales ”what’s next”
• Was ist Extremalprinzip für Ausdehnung eines Gases ?
Beachte: Für ideales Gas ändert sich hierbei die innere Energie nicht
• Allgemein: Gibt es ein Extremalprinzip, das für die Einstellung eines Gleich-gewichtes ”verantwortlich” ist ?
29

4.1 Reversible und irreversible Prozesse
• Beobachtung: Im isoliertem System laufen Zustandsänderungen ”von selbst””spontan” ab, bis Gleichgewichtszustand erreicht ist. Diese Art von Änderungensind irreversibel, d.h. kehren sich von selbst nicht um.
Beispiele:
– Aus der Thermodynamik:
∗ Ausdehnung eines Gases von kleinem in grosses Volumen
∗ Temperaturausgleich
– Aus der Mechanik: Pendel mit Reibung
Spontane Umkehrung ist nie beobachtet wurden. Ist aber energetisch (1. Haupt-satz) möglich
• Eigenschaften von irreversiblen thermodynamischen Prozessen:
– Irreversible Prozesse laufen über Nichtgleichgewichtszustände ab
– Zustandsgrößen, z.B. p, haben während des Prozesses keine definiertenWerte. Es gibt räumliche Inhomogenitäten
– Reales Gas: Turbulenz kann entstehen, die am Ende Wärme produziert.
– Klar machen an Ausdehnung von kleinem in grosses Volumen
• Reversible Prozesse laufen über Gleichgewichtszustände
– Idealisierung: Ist System im Gleichgewicht, passiert nix
– Zustandsgrößen haben zu jedem Zeitpunkt definierte Werte.
30
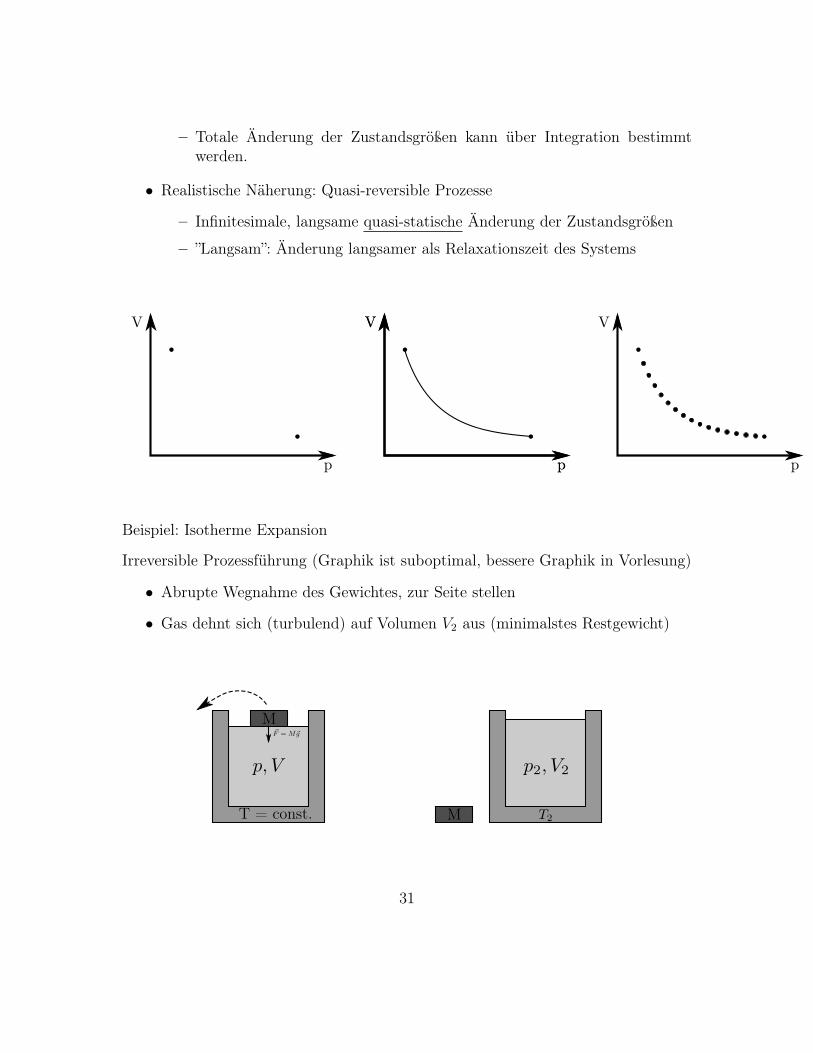
– Totale Änderung der Zustandsgrößen kann über Integration bestimmtwerden.
• Realistische Näherung: Quasi-reversible Prozesse
– Infinitesimale, langsame quasi-statische Änderung der Zustandsgrößen
– ”Langsam”: Änderung langsamer als Relaxationszeit des Systems
Beispiel: Isotherme Expansion
Irreversible Prozessführung (Graphik ist suboptimal, bessere Graphik in Vorlesung)
• Abrupte Wegnahme des Gewichtes, zur Seite stellen
• Gas dehnt sich (turbulend) auf Volumen V2 aus (minimalstes Restgewicht)
T = const. M
M
31

• Es wird keine Arbeit geleistet, δW = 0.
• Prozess irreversibel, da zur Seite gelegtes Gewicht nicht nach oben hüpfen kann
Reversible Prozessführung
• Verkleinere Masse schrittweise um dM und hebe Massestücke an entsprechen-der Höhe auf
• Mache dies so langsam, dass System stets im (Quasi-)Gleichgewicht bleibt
• System leistet Arbeit
∆W = −∫ V2
V1
pdV = −NkT∫ V2
V1
dV
V= −NkT log
(V2
V1
)• Prozess reversibel, da man Massenstücke dM sukzessive wieder auflegen kann.
Der 1. Hauptsatz gilt unabhängig von Prozessführung
• DamitdU = δWrev + δQrev = δWirr + δQirr
• Unter Berücksichtigung des Vorzeichens, Kompression, Expansion
δWirr ≥ δWrev = −pdVδQirr ≤ δQrev = siehe Kap. 4.3
• Beachte: δW = −pdV gilt nur bei reversibler Prozessführung
32

• Bei reversibler Prozessführung wird am wenigsten Arbeit benötigt, bzw. ammeisten verrichtet
• Bei irreversibler Prozessführung wird Teil der Arbeit in (Ab-)wärme verwandelt
• Wieder einmal:
δW und δQ hängen von der Prozessführung ab.
Sie können daher keine Zustandsgrößen sein
Der 1. Hauptsatz reicht zur Beschreibung irreversibler Prozesse nicht aus, daer sie nicht verbietet:Es braucht weitere Zustandsgröße
4.2 Carnot’scher Kreisprozess
Betrachte Kreisprozess
• Arbeitsmedium wird nach Reihe von Zustandsänderungen wieder in Ausgangs-zustand gebracht
• Es gilt ∮dU = 0
Carnot’scher Kreisprozess (1824)
• Beispiel für Wärmekraftmaschine
• Arbeitsmedium: Ideales Gas
• Besteht aus zwei isothermen und zwei adiabatischen Prozessen
33
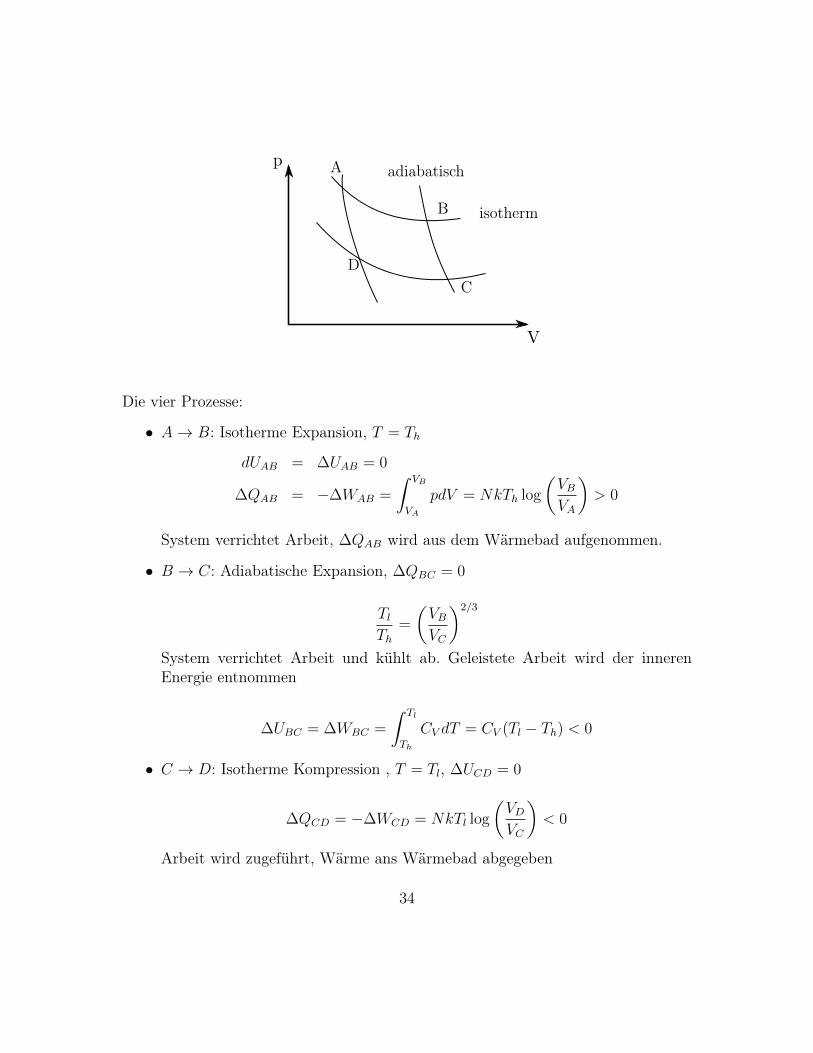
A
B
CD
adiabatisch
isotherm
Die vier Prozesse:
• A→ B: Isotherme Expansion, T = Th
dUAB = ∆UAB = 0
∆QAB = −∆WAB =
∫ VB
VA
pdV = NkTh log
(VBVA
)> 0
System verrichtet Arbeit, ∆QAB wird aus dem Wärmebad aufgenommen.
• B → C: Adiabatische Expansion, ∆QBC = 0
TlTh
=
(VBVC
)2/3
System verrichtet Arbeit und kühlt ab. Geleistete Arbeit wird der innerenEnergie entnommen
∆UBC = ∆WBC =
∫ Tl
Th
CV dT = CV (Tl − Th) < 0
• C → D: Isotherme Kompression , T = Tl, ∆UCD = 0
∆QCD = −∆WCD = NkTl log
(VDVC
)< 0
Arbeit wird zugeführt, Wärme ans Wärmebad abgegeben
34

• D → A: Adiabatische Kompression, ∆QDA = 0
ThTl
=
(VDVA
)2/3
∆UDA = ∆WDA = CV (Th − Tl) > 0
Dem System wird Arbeit zugeführt
Energiebilanz:
∆Utotal =4∑i=1
∆Ui,i+1modulo 4 = CV (Tl − Th) + CV (Th − Tl) = 0
Glück gehabt ! :-)
Wärme und Arbeit:
∆QAB = NkTh log
(VBVA
)> 0 (2)
∆QCD = NkTl log
(VDVC
)< 0 (3)
• Mit (ThTl
)3/2
=VBVC
=VAVD
folgt
∆Qtotal = Nk log
(VBVA
)(Th − Tl) > 0 (4)
• Wärme wird zugeführt
• Und damit wegen ∆U = 0
∆Wtotal = −∆Qtotal < 0
Arbeit wird verrichtet.
• Eine Maschine, die Wärme in Arbeit umwandelt !
35

Aber wie gut ?
• Wirkungsgrad η: Der in Arbeit umgewandelte Teil der aufgenommenen Wärme
η =|∆Wtotal|∆QAB
=∆QAB + ∆QCD
∆QAB
= 1 +∆QCD
∆QAB
= 1− TlTh
< 1 (5)
Wärme kann nie vollständig in Arbeit umgewandelt werden.
• Übung: Man kann zeigen: Im beliebigen Kreisprozess bildet Gl. (5) obereSchranke für η
• Beispiel Wärmekraftwerk:
Th = 800K, Tl = 300K
ηideal = 1− 300K
800K=
5
8≈ 60%
ηreal ≈ 45%
2. week
4.3 Thermodynamische Definition der Entropie
• Gl. (4): Wie zu erwarten: Q im Carnot-Prozess keine Zustandsgröße
• Aus Gl. (2, 3) folgt aber
∆QAB
Th+
∆QCD
Tl= 0
• Zerlege den Prozess in infinitesimal kleine Teilstücke, so folgt:∮δQrev
T= 0
1Tist der integrierende Faktor, der aus dem nicht-exakten Differential δQrev ein
exaktes und damit eine Zustandsgröße macht !
• Man kann zeigen:
– δQT
ist für jeden reversiblen Kreisprozess ein exaktes Differential, Übung ?
36

– Ebenso für jeden reversiblen thermodynamischen Prozess
• Ergo: Es muss eine Zustandsvariable geben, die durch
dS =δQrev
T, SB − SA =
∫ ZustandB
ZustandA
δQrev
T
definiert ist
• Die Größe S heißt Entropie, Clausius (1865)
– Kunstwort aus εν = in und τρoπη = Verwandlung: Entropie
– ”Da die Entropie ebenso wichtig ist, soll sie auch so ähnlich heissen wiedie Energie”
– ”Wandlungspotential”, misst ”Energieentwertung”
– Damit wird auch ”Isentrope” bei adiabatischer Zustandsänderung klar
• Carnot-Maschine im S-T Diagramm
T
S
• Für reversible Prozesse gilt
dS =δQrev
T
• S ist extensiv
37

• Bei irreversibler Prozessführung gilt
δQirr ≤ δQrev = TdS
• In Kapitel 9 ”Entropie revisited” werden wir einen komplett anderen - mi-kroskopischen, statistischen - Zugang zur Entropie kennenlernen und mit demhiesigen - makroskopisch, thermodynamischen - zusammenführen
Entropie des idealen Gases in Abhängigkeit von T und V
• Sei N = const.
• 1. HauptsatzdU = TdS − pdV
mit Zustandsgleichungen
U =3
2NkT, pV = NkT
• Damit 1. Hauptsatz aufgelöst nach dS
dS =3
2Nk
dT
T+Nk
dV
V
Startend von Zustand (T0, V0) mit Entropie S0, integriere auf
S(T, V )− S0(T0, V0) =3
2Nk log
T
T0
+Nk logV
V0
= Nk log
[(T
T0
)3/2(V
V0
)](6)
Ergebnis: Entropie nimmt mit T und V zu
• Beachte: Nicht die komplette N -Abhängigkeit, weil Term µdN in 1. Hauptsatzfehlte
• Betrachte (irreversible) freie Ausdehnung eines idealen Gases von kleinem ingrosses Volumen in geschlossenem System
– Thermisch isoliert: ∆Q = 0
38

– Volumenerhöhung: ∆V > 0
– Es wird keine Arbeit verrichtet: ∆W = 0
– 1. Hauptsatz: ∆U = 0. Ideales Gas: U = U(T ), damit ∆T = 0
– Aber Entropieänderung
∆S = S(T, V1, N)− S(T, V0, N) = Nk logV1
V0
> 0
– Erster Hinweis: Entropie auch jenseits von δQrevT
wichtig
• In Abhängigkeit von (T, p)
S(T, p)− S0(T0, p0) = Nk log
[(T
T0
)5/2(p0
p
)]
• Da Entropie extensiv muss gelten
S(T, p) = Nk
(s0(T0, p0) + log
[(T
T0
)5/2(p0
p
)])(7)
Details in der Statistischen Physik
4.4 Der 2. Hauptsatz
• Clausius (1850)
• Betrachte abgeschlossenes System, es gilt
δQirr ≤ δQrev = TdS
• Im Gleichgewicht:
δQirr = δQrev = 0 =⇒ dS = 0
Ergo: Die Entropie ist in einem abgeschlossenen System im Gleichgewicht ex-tremal.
Minimum oder Maximum ?
39

• Betrachte ein zusammengesetztes, insgesamt isoliertes System
TA 6= TB
t < 0: Subsysteme seien isoliert
t ≥ 0: Subsysteme gehen ins thermische Gleichgewicht
• DaUA + UB = const. =⇒ dUA = −dUB
Mit
dUA = δQ+ δW = dQ = TAdSA
dUB = TBdSB
• Entropie ist extensiv:
dS = dSA + dSB =dUATA
+dUBTB
dS = dUB
(1
TB− 1
TA
)Die Entropie ist nicht erhalten !
– Für TB > TA: Wärme fließt von B nach A → dUB < 0 → dS > 0
– Für TB < TA: Wärme fließt von A nach B → dUB > 0 → dS > 0
– Ist TA 6= TB läuft spontaner Prozess ab und es gilt dS > 0
40

– Ist TA = TB: Thermisches Gleichgewicht und dS = 0
2. Hauptsatz:In einem abgeschlossenen System im thermischen Gleichgewicht gilt:
dS = 0, S = maximal
Für spontane Prozesse gilt:dS > 0
Entropie definiert über ein Extremalprinzip den Gleichgewichtszustand.
Alternative Formulierungen
• Clausius: ”Es gibt keinen thermodynamischen Prozess, der ausschließlich Wär-me von einem kälterem in ein wärmeres Reservoir überführt. Es existiert keineideale Kältemaschine”
Beweis:
– Da Gesamtsystem keine Arbeit leistet noch Wärme austauscht, kann esals abgeschlossen betrachtet werden.
– Sei T1 < T2
41

– Dann:dS1 = −δQ
T1
, dS2 =δQ
T2
DamitdS = dS1 + dS2 = −δQ
T1
+δQ
T2
= δQT1 − T2
T1T2
< 0
Widerspruch
– Beachte: Wärme vom wärmeren zum kälteren Reservoir fliessen lassengeht :-)
• Kelvin: ”Es gibt keinen periodischen2 thermodynamischen Prozess, der aus-schließlich Wärme in Arbeit verwandelt. Es existiert keine ideale Wärmekraft-maschine”
oder
Planck: ”Es gibt kein Perpetuummobile 2. Art: Periodisch arbeitende Maschine,die Wärme zu 100 % in Arbeit umwandelt und speichert”
Beweis:
Wärme Maschine Speicher
• Nach einem Arbeitszyklus
∆UM = ∆Q︸︷︷︸>0
+ ∆W︸︷︷︸<0
= 0
∆Utotal = ∆Q+ ∆W = 0
1. Hauptsatz ist erfüllt
Betrachte 2. Hauptsatz2periodisch ist wichtig, einzelne Prozesse können das. Erinnere isotherme Expansion
42

Nach einem Arbeitszyklus:
∆Stotal = ∆SB + ∆SM + ∆SS ≥ 0
∆SB = −∆Q
T< 0
∆SM = 0
∆SS = 0
=⇒ ∆Stotal < 0
Widerspruch zum 2. Hauptsatz
–• Mit Entropie lautet der 1. Hauptsatz für reversible Zustandsänderungen:
dU = δQrev + δWrev = TdS − pdV + µdN + . . .
Analogie:
Entropie für Austausch vonWärme entspricht Volumen bei Kompressionsarbeitgegen Druck
• ”Wärmewertigkeit”
Für T2 > T1 gilt
dS1 =δQ
T1
>δQ
T2
= dS2
Wärme bei höherer Temperatur ist ”mehr wert” als bei niedriger.
An Beispiel Sonne => Erde => Weltraum klarmachen3. Halb-woche
4.5 Fundamentalrelationen
• Betrachte Kombination von 1. Hauptsatz mit Entropie-Definition für reversibleProzesse
dU = δQrev + δWrev = TdS − pdV + µdN
Innere Energie U in Abhängigkeit der extensiven Zustandsgrößen S, V,N .
43

• S, V,N werden als die natürlichen Variablen der inneren Energie bezeichnet
• Mit
dU =
(∂U
∂S
)V,N
dS +
(∂U
∂V
)S,N
dV +
(∂U
∂N
)V,S
dN
ergeben sich die intensiven Zustandsgrößen
T (S, V,N) =
(∂U
∂S
)V,N
, p(S, V,N) = −(∂U
∂V
)S,N
, µ(S, V,N) =
(∂U
∂N
)V,S
(8)
• Die Kenntins von U(S, V,N) bestimmt das thermodynamische System kom-plett
Daher wird
dU = TdS − pdV + µdN (9)
auch als Fundamentalrelation oder Gibbs’sche Fundamentalform bezeichnet.
Entropie-Darstellung
• Umstellen der Fundamentalrelation nach dS liefert Fundamentalrelation in derEntropie-Darstellung
dS(U, V,N) =1
TdU +
p
TdV − µ
TdN (10)
dS =
(∂S
∂U
)V,N︸ ︷︷ ︸
1/T
dU +
(∂S
∂V
)U,N︸ ︷︷ ︸
p/T
dV +
(∂S
∂N
)U,V︸ ︷︷ ︸
−µ/T
dN
mit
T = T (U, V,N), p = p(U, V,N), µ = µ(U, V,N)
• U, V,N sind die natürlichen Variablen der Entropie
44

Zustandsgleichungen revisited
• Zustandsgleichungen: Relationen, die im Gleichgewicht zwischen den thermo-dynamischen Variablen eines Systems bestehen
• Bestimmungsgleichungen der intensiven Zustandsvariablen Gl. (8) liefernZustandsgleichungen
• Beispiel: Ideales Gas
– Rechnung analog zu Gl. (6, 7) liefert für Entropie in den natürlichen Va-riablen U, V,N :
S(U, V,N) = Nk
(s0(U0, V0, N0) + log
[(U
U0
)3/2(V
V0
)(N0
N
)5/2])
– Damit1
T=
(∂S
∂U
)V,N
=3
2Nk
1
U=⇒ U =
3
2NkT
p
T=
(∂S
∂V
)U,N
=Nk
V=⇒ pV = NkT
Variablentransformation
• Gleichungen (9) und (10) gehen direkt auseinander hervor
• Beachte: T , p, und µ hängen nicht von denselben Variablen ab
• Beispiel Temperatur
T (S, V,N) =: f(S, V,N) Energie-DarstellungT (U, V,N) =: g(U, V,N) Entropie-Darstellung
• Sei N = const.
• Umrechnung T (S, V ) in T (U, V ) per Variablentransformation
45

• Start: T (S, V )
dT (S, V ) =
(∂T
∂S
)V
dS +
(∂T
∂V
)S
dV
Für S → U , betrachte S(U, V )
dS(U, V ) =
(∂S
∂U
)V
dU +
(∂S
∂V
)U
dV
• Eingesetzt:
dT (U, V ) =
(∂T
∂S
)V
[(∂S
∂U
)V
dU +
(∂S
∂V
)U
dV
]+
(∂T
∂V
)S
dV
=
(∂T
∂S
)V
(∂S
∂U
)V
dU +
[(∂T
∂S
)V
(∂S
∂V
)U
+
(∂T
∂V
)S
]dV
• Ergo: f(S, V,N) 6= g(U, V,N)
4.6 Euler-Gleichung und Gibbs-Duhem-Relation
• Extensive Variablen sind homogene Funktionen 1. Grades
Beispiel: Innere Energie
U(λS, λV, λN) = λ1U(S, V,N) (11)
• Intensive Variablen sind homogene Funktionen 0. Grades, z.B. Temperatur
T (λS, λV, λN) = λ0T (S, V,N)
Homogenität hat weitreichende Folgen
• Differentiation von Gl. (11) nach λ ergibt:
d
dλU(λS, λV, λN) =
∂U
∂(λS)
∂(λS)
∂λ︸ ︷︷ ︸=S
+∂U
∂(λV )
∂(λV )
∂λ︸ ︷︷ ︸=V
+∂U
∂(λN)
∂(λN)
∂λ︸ ︷︷ ︸=N
= U(S, V,N)
46

• Gilt ∀ λ, setze λ = 1. Ergibt Euler-Gleichung
U = TS − pV + µN (12)
• Bedeutung: Fundamentalrelation kann trivial integriert werden, obwohl T , pund µ Funktionen von S, V und N sind.
• Betrachte totales Differential der Euler-Gleichung:
dU = TdS+SdT−pdV−V dp+µdN+Ndµ = TdS−pdV+µdN+SdT−V dp+Ndµ
Vergleich mit 1. Hauptsatz:
dU = TdS − pdV + µdN
Es gilt die Gibbs-Duhem-Relation:
SdT − V dp+Ndµ = 0 (13)
• Bedeutung: Die zu den extensiven Variablen Energie-konjugierten intensivenVariablen können nicht unabhängig sein.
• Anschauung:Aus den drei extensiven Zustandsgrößen S, V , und N lassen sich nur zweiintensive S/N und V/N ableiten.
Chemisches Potential des idealen Gases
• Gibbs-Duhem-Relation:
SdT − V dp+Ndµ = 0
oder
dµ(p, T ) = −S(p, T )
NdT +
V (p, T )
Ndp
Mit Gl. (7) und Gasgleichung folgt
dµ(p, T ) = −k
(s0(T0, p0) + log
[(T
T0
)5/2(p
p0
)])dT + kT
dp
p
47

• µ ist Zustandsgröße, Übung ?, hat exaktes Differential, darf über beliebigenWeg integriert werden
• Wähle Weg (i) isobar von T0 nach T und Weg (ii) isotherm von p0 nach p
p
T
µ(p, T ) = µo(p0, T0)−∫ T
T0
(s0k +
5
2k log
(T
T0
))dT + kT
∫ p
p0
dp
p
Mit ∫dx log x = x log x− x
folgt
µ(p, T ) = µo(p0, T0)− s0k(T − T0)− 5
2kT log
(T
T0
)+
5
2k(T − T0) + kT log
p
p0
= µo(p0, T0)− kT log
((T
T0
)5/2p0
p
)+ k(T − T0)
(5
2− s0
)
Chemisches Potential hängt im wesentlichen von kT ab, d.h. der mittlerenkinetischen Energie der Teilchen ab
48

• Um neues Teilchen im Gleichgewicht bei Temperatur T und Druck p einemidealen Gas hinzuzufügen, muss unabhängig der Anzahl bereits vorhandenerTeilchen, die Energie µ(p, T ) aufgebracht werden.
Übung: Massenwirkungsgesetz
Lessons learned
• Reversible und irreversible Prozesse
• Carnot’scher Kreisprozess funktioniert nur, weil Arbeit und Wärme keine ex-akten Differentiale sind
• Entropie als exaktes Differential und Zustandsgröße
• 2. Hauptsatz: dS ≥ 0, im Gleichgewicht: S = maximal
• Fundamentalrelationen
• Zustandsgleichungen folgen aus Bestimmungsgleichungen der intensiven Va-riablen
• Euler-Gleichung und Gibbs-Durhem Relation
5 Der 3. Hauptsatz der ThermodynamikAuch als Nernst’sches Theorem (1918) bezeichnet.
• Vor der Hand Existenz eines absoluten Nullpunktes der Temperatur nicht klar.
• Früher Verdacht: Erinnere Gay-Lussac: (1802)
V
T= const. für p = const.
Eingeschränkter Gültigkeitsbereich ?
• Ergibt affine Beziehung, Celsius, Fahrenheit, Kelvin
V = a(T − b)
49
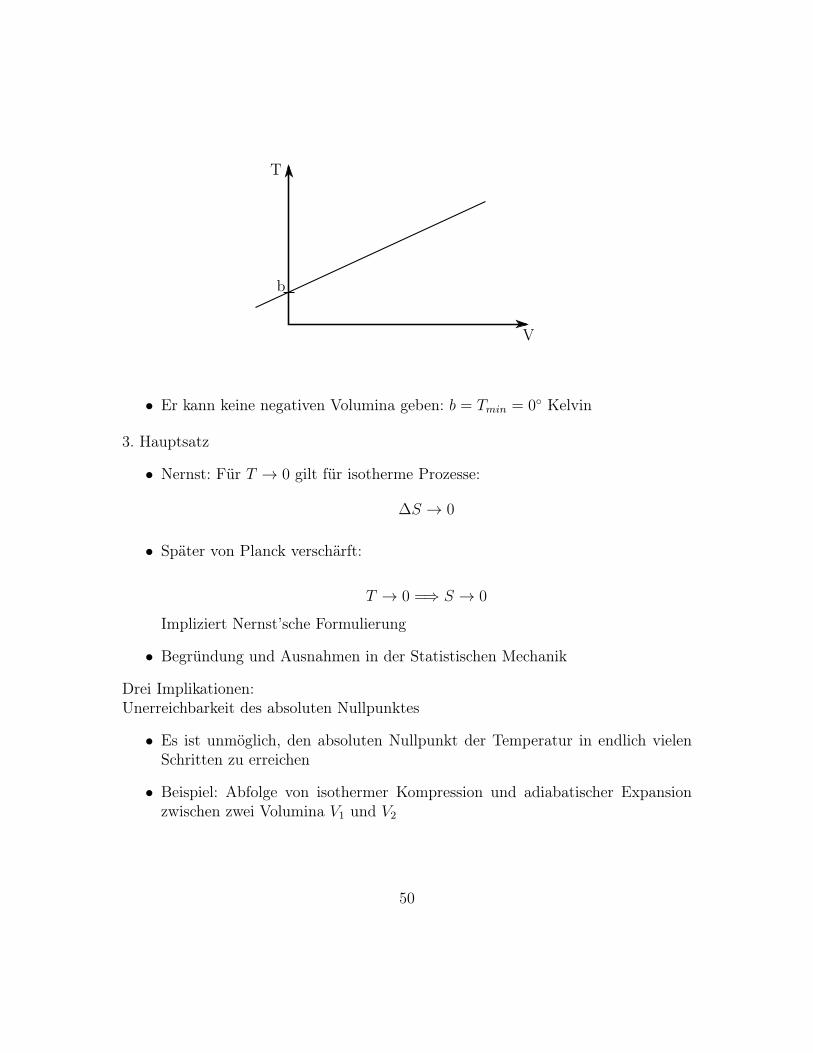
b
T
V
• Er kann keine negativen Volumina geben: b = Tmin = 0 Kelvin
3. Hauptsatz
• Nernst: Für T → 0 gilt für isotherme Prozesse:
∆S → 0
• Später von Planck verschärft:
T → 0 =⇒ S → 0
Impliziert Nernst’sche Formulierung
• Begründung und Ausnahmen in der Statistischen Mechanik
Drei Implikationen:Unerreichbarkeit des absoluten Nullpunktes
• Es ist unmöglich, den absoluten Nullpunkt der Temperatur in endlich vielenSchritten zu erreichen
• Beispiel: Abfolge von isothermer Kompression und adiabatischer Expansionzwischen zwei Volumina V1 und V2
50
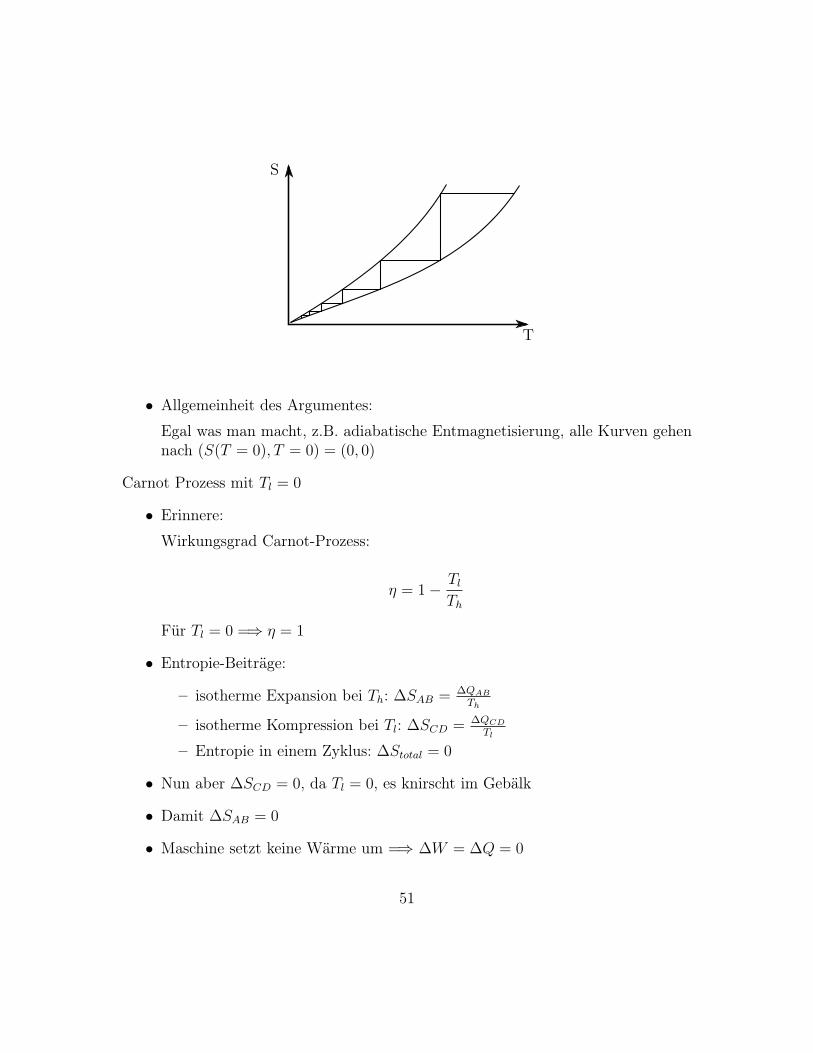
S
T
• Allgemeinheit des Argumentes:
Egal was man macht, z.B. adiabatische Entmagnetisierung, alle Kurven gehennach (S(T = 0), T = 0) = (0, 0)
Carnot Prozess mit Tl = 0
• Erinnere:
Wirkungsgrad Carnot-Prozess:
η = 1− TlTh
Für Tl = 0 =⇒ η = 1
• Entropie-Beiträge:
– isotherme Expansion bei Th: ∆SAB = ∆QABTh
– isotherme Kompression bei Tl: ∆SCD = ∆QCDTl
– Entropie in einem Zyklus: ∆Stotal = 0
• Nun aber ∆SCD = 0, da Tl = 0, es knirscht im Gebälk
• Damit ∆SAB = 0
• Maschine setzt keine Wärme um =⇒ ∆W = ∆Q = 0
51

• Von Wirkungsgrad kann keine Rede sein
• Ergebnis gegen jede (klassische) Intuition
• Irgendwas stimmt da nicht !
Spezifische Wärme
• Erinnere
δQ = CV dT, dS =δQ
T, ideales Gas: CV =
3
2Nk = const.
• DamitdS = CV
dT
T
und
S(T ) = S(T = 0) +
∫ T
0
dT ′CVT ′
3. Haupsatz: S(T = 0) = 0
• Aber Integral
∫ T
0
dT ′CVT ′
= CV log
(T
0
)divergiert !
S(T ) = S(T0) +
∫ T
T0
dT ′CVT ′
sieht für T0 → 0 auch nicht gut aus
• Einzige Chance für Konvergenz: CV ist CV (T ) mit
CV (T )→ 0 für T → 0
und zwar schneller als der Logarithmus für T → 0 divergiert, also schneller alslinear
• Reminder: Das gilt nicht für das ideale Gas.
52

• Noch schlimmer: Gilt nicht für jedes klassische thermodynamische System, sie-he Kap. 11.3 Gleichverteilungssatz
• CV (T )→ 0 für T → 0 löst auch das Problem des Wirkungsgrades des Carnot-Prozesses für T = 0, da er unter der Annahme CV = const. abgeleitet wurde.
• Kann erst in der Quantenmechanik verstanden werden, Kap. 11.4
• Visionäres Ergebnis vor Einführung der Quantenmechanik
Lessons learned
• In der Regel: limT→0 S(T ) = 0
• Unerreichbarkeit des absoluten Nullpunktes
• Wärmekapazität: limT→0C(T ) = 0 in Widerspruch zu klassischer Physik
6 Thermodynamische PotentialeInnere Energie U wird als thermodynamisches Potential bezeichnet, da
• sie eine Energie ist
• ihre Abteilung durch Vergleich der Fundamentalrelation mit den exakten Dif-ferentialen ”Kräfte” liefern á la F = −∂V
∂x
• Beispiel: U in Abhängigkeit von S:
dU(S) =∂U
∂SdS = TdS
Erster Teil Mathematik, zweiter Teil Physik
• Für alle Variablen:
T =
(∂U
∂S
)V,N
, T treibende Kraft für Wärmeaustausch
−p =
(∂U
∂V
)S,N
, p treibende Kraft für Volumenaustausch
µ =
(∂U
∂N
)V,S
, µ treibende Kraft für Teilchenaustausch
53

• Allgemein: Alle Größen, die eine Fundamentalrelation besitzen, werden als ther-modynamisches Potential bezeichnet, z.B. auch die Entropie S = S(U, V,N).
6.1 Prinzip der maximalen Entropie – Prinzip der minimalenEnergie
Klassische Mechanik: Nicht abgeschlossene Systeme streben nach einem Minimumder Energie
• Regentropfen: Potentielle Energie → kinetische Energie → Aufprall → Dissi-pation der Energie in Wärme
• Pendel: Durch Reibung irgendwann in Ruhelage. Wieder Arbeit in Wärmeverwandelt.
• Energie des Teilsystems wird minimiert. Entropie des Gesamtsystems wird ma-ximiert. Gesamtenergie ist erhalten.
Formalisierung:
• Betrachte insgesamt abgeschlossenes System mit zwei (nicht abgeschlossenen)Teilsystemen A (das Pendel) und B (die umgebene Luft), das Bad.
Utotal = UA + UB, Stotal = SA + SB
• Teilsystem A leiste Arbeit δWA < 0 an B, tausche aber keine Wärme mit Baus:
δQA = TdSA = 0, dSA = 0
Pendel kühlt nicht ab.
dUA = δWA < 0
• Teilsystem B, das Bad, nimmt im Allgemeinen
– einen Teil α von dUA als Wärme auf
δQB = −αdUA
54

– den Rest als ArbeitδWB = −(1− α)dUA
Beim Pendel: α = 1
– Damit
dUB = δQB + δWB = −dUA = −δWA > 0
δQB = −αδWA = TdSB > 0
dStotal = dSA + dSB > 0
• Gesamtentropie nimmt zu durch Umwandlung von Arbeit von A in Wärme inB.
• Prozess läuft ab, so lange A Arbeit leisten kann.
• Am Ende hat
– A Zustand minimaler Energie erreicht
– Gesamtsystem Zustand maximaler Entropie erreicht.
T
Sthermisches Gleichgewicht
T
UA
thermisches Gleichgewicht
T => t
55

• Wichtig: Ein Teil der Arbeit von A muss in Wärme für B verwandelt werden,sonst ist Prozess reversibel und läuft nicht von selbst ab.
Bis hier im wesentlichen: System abgeschlossen
• Thermodynamische Potentiale U(S, V,N) & S(T, V,N)
• Gleichgewicht durch Extermalprinzip für Entropie gegeben
Ist das System nicht abgeschlossen, definiere andere thermodynamische Potentiale,für die andere Extremalbedingungen gelten.
• Abhängig von den experimentell zu kontrollierenden Größen
• Entropie in U(S, V,N) schwer experimentell zu kontrollieren
• Relevante Fälle:
– System im Wärmebad: T = const.
– Offenes Reagenzglas: p = const.
• Im folgenden: Die möglichen Kombinationen extern konstant gehaltenen Grö-ßen
• Idee: Konstant gehaltene Größen vereinfachen die thermodynamischen Poten-tiale
4. Wo-che
6.2 Freie Energie
Betrachte geschlossenes System S im Wärmebad B, ersters durch (T, V,N) = const.charakterisiert.
System
dU
BadT, V,N
T
56

• Wärmebad bei z.B. 273 K durch Wasser/Eis-Gemisch
• Energieaustausch zwischen System S und Bad B.
Erster Hauptsatz: dUS + dUB = 0 (14)Zweiter Hauptsatz: dS = dSS + dSB ≥ 0 (15)
Fundamentalrelation: dSB =1
TdUB +
p
TdVB︸︷︷︸=0
−µTdNB︸︷︷︸
=0
=1
TdUB (16)
Aus Gl. (16) und Gl. (14) folgt:
dSB = − 1
TdUS
Eingesetzt in Gl. (15) folgt:
dSS −1
TdUS ≥ 0 → dUS − TdSS ≤ 0
oderd(US − TSS) ≤ 0
• Bedeutung: Im Gleichgewicht wird die Größe US − TSS minimiert.
• Um dies als Extremalprinzip eines thermodynamishen Potentials auszudrücken,definiere (Helmholtz’sche) freie Energie:
F = U − TS
Es giltdF = dU − TdS ≤ 0
Im Gleichgewicht wird:
• innere Energie minimiert
• Entropie maximiert
Die beiden Ziele sind in der Regel gegenläufig
• Balance zwischen energetischem Anteil U und entropischem Anteil −TS, sodass Summe minimal wird
57

• bei niedrigen Temperaturen dominiert energetischer Anteil
• bei hohen Temperaturen dominert entropischer Anteil
• Isotherme Prozesse, die innere Energie vergrößern, also unter Energieaufwandablaufen, sind spontan möglich, wenn Entropiegewinn TdS größer als Energie-aufwand dU .
• Energie kommt aus Wärmebad.
Merke: Freie Energie für isotherme geschlossene Systeme hat analoge Bedeutungwie Entropie für abgeschlossene Systeme
Warum ”freie” Energie ?
• System mit Wärmebad, T = const.
• ErinnereTdS = δQrev ≥ δQirr
Vom Systems S aus gesehen:
dUS − TdSS = δW revS ≤ δW irr
S
oderdFS = d(US − TSS) = δW rev
S ≤ δW irrS
”Freie” Energie, weil die Änderung der freien Energie bei konstanter Temperaturder maximal möglich zu verrichtenden Arbeit entspricht.
Fundamentalrelation der freien Energie
• Erinnere Euler-Gleichung
F = U − TSdF = dU − TdS − SdTdU = TdS − pdV + µdN = dU(S, V,N)
ergibt für dF :
dF = −SdT − pdV + µdN = dF (T, V,N)
58

• Aus den partiellen Ableitungen:
dF =
(∂F
∂T
)V,N︸ ︷︷ ︸
=−S(T,V,N)
dT +
(∂F
∂V
)T,N︸ ︷︷ ︸
=−p(T,V,N)
dV +
(∂F
∂N
)T,V︸ ︷︷ ︸
=µ(T,V,N)
dN
ergeben sich die Zustandsgleichungen für S, V und µ.
6.3 Legendre-Transformation
• Der Übergang von der inneren auf die freie Energie geschieht durch eineLegendre-Transfomation.
• Erinnere Übergang von Lagrange-Funktion L(q, q) auf Hamilton-FunktionH(q, p) in der Klassischen Mechanik.
• Legendre-Transformation ist eine Berührungstransformation
• Allgemein wichtig für Variablen-Transformationen.
• Legendre-Transformation überführt f(x) in Funktion g(u) = L(f(x))
Herleitung der Legendre-Transformation:Formal:
• Gegeben eine Funktion f(x) mit
df =df
dxdx =: u(x)dx, u = f ′(x)
• Gesuchte Funktion g(u) soll
x(u) = −dgdu, dg = −x du, Vorzeichen je nachdem
erfüllen
• Bilde totales Differential von ux
d(ux) = xdu+ udx = −dg + df
Umstellendg = d(f − ux)
59
• Integration:g(u) = f(x(u))− ux(u)
• Zur konkreten Berechnung muss u = f ′(x) invertiert werden:
g(u) = f(f′−1(u))− uf ′−1(u)
• Rücktransformation:
f(x) = g(u(x)) + u(x)x
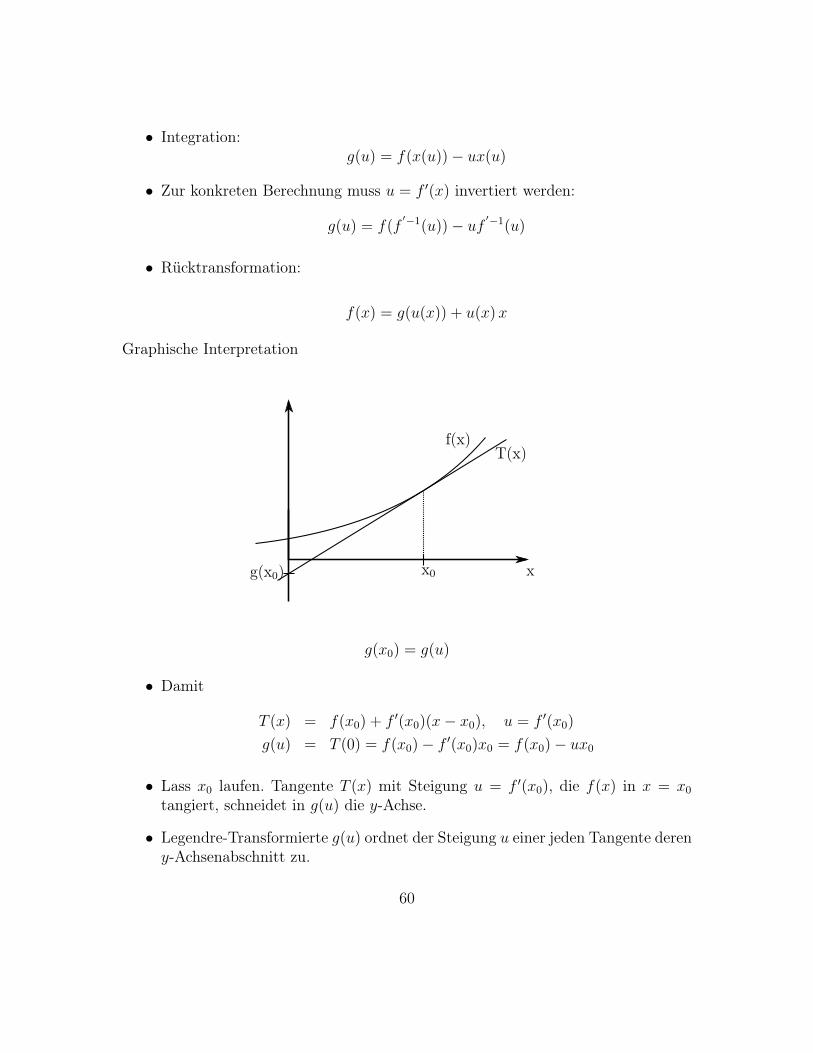
Graphische Interpretation
x
f(x)T(x)
x0g(x0)
g(x0) = g(u)
• Damit
T (x) = f(x0) + f ′(x0)(x− x0), u = f ′(x0)
g(u) = T (0) = f(x0)− f ′(x0)x0 = f(x0)− ux0
• Lass x0 laufen. Tangente T (x) mit Steigung u = f ′(x0), die f(x) in x = x0
tangiert, schneidet in g(u) die y-Achse.
• Legendre-Transformierte g(u) ordnet der Steigung u einer jeden Tangente dereny-Achsenabschnitt zu.
60

• Beschreibung derselben Kurve, nur über andere Variable, u statt x.
Kurzfassung Legendre-Transformation
• Definition:
g(u) = f(x)− ux, u =df
dx
• Wegendg(u) = df(x)− u dx︸︷︷︸
=df(x)
−x du = −x du
hängt g von u, aber nicht von x ab.
Beispiel:
• Betrachtef(x) = x2, x ∈ R+
0 f ′(x) = 2x = u
• Legendre-Transformation:
g(u) = f(x(u))− ux(u) = x2(u)− ux(u)
x = f ′−1(u) : x =u
2
g(u) = f(u/2)− u u2
=
(1
4− 1
2
)u2 = −u
2
4
dg =∂g
∂udu = −u
2du = −xdu
• Rücktransformation:
f(x) = g(u(x)) + u(x)x = −u2(x)
4+ u(x)x
u(x) = 2x
f(x) = −x2 + 2x2 = x2
Legendre-Transformation von der inneren Energie U(S, V,N) auf die freie EnergieF (T, V,N), V und N unterdrückt
dU(S) =∂U
∂SdS = T dS
T= ∂U∂S→ dF (T ) =
∂F
∂TdT = −S dT
Beachte:
61

• S und T Energie-konjugiert
• Erinnere: Lagrange nach Hamilton: q und p Wirkungs-konjugiert
p :=∂L(q, q)
∂q, H(q, p) = q(q, p)p− L(q, q(q, p))
Physikalisch:
• Praktisch einfacher Temperatur als Entropie zu kontrollieren.
• Das heisst geschlossenes statt abgeschlossenes System
• Für diesen Fall ist freie Energie F (T, V,N) das natürliche thermodynamischePotential
6.4 Enthalpie
Wird statt des Volumens der Druck kontrolliert, ergibt sich die Enthalpie H(S, p,N)als natürliches Potential.
• Wichtig für chemische Prozesse, die bei konstantem Atmosphärendruck, offenesReagenzglas, durchführt werden.
• Ausgehend von der inneren Energie U(S, V,N) muss die V -Abhängigkeit durchdie p-Abhängigkeit ersetzt werden
• Von dV zu dp
dU(V ) =∂U
∂VdV = −p dV
−p= ∂U∂V→ dH(p) =
∂H
∂pdp = V dp
• Legendre-Transformation
H = U + pV
dU = TdS − pdV + µdN = dU(S, V,N)
dH = dU + pdV + V dp
ErgibtdH = TdS + V dp+ µN = dH(S, p,N)
62

• Zustandsgleichungen aus
dH =
(∂H
∂S
)P,N︸ ︷︷ ︸
=T (S,p,N)
dS +
(∂H
∂p
)S,N︸ ︷︷ ︸
=V (S,p,N)
dp+
(∂H
∂N
)S,P︸ ︷︷ ︸
=µ(S,p,N)
dN
• Innere und freie Energie praktisch zur Beschreibung isochorer Prozesse wegendV = 0, Enthalpie praktisch wegen dp = 0 für isobare Prozesse.
• System kann gegen konstanten äußern Druck Volumenarbeit leisten
δW revvol = −pdV
1. Hauptsatz (nur Volumenarbeit)
dU = δQ− pdV
Bei konstantem Druck:
dH|p = d(U + pV ) = δQ− pdV + pdV + V dp = δQ+ V dp
Damit
dH|p = δQ
analog zudU |V = δQ
• Betrachte isobaren und adiabatischen Prozess, der aber sonstige Arten vonArbeit mit der Umgebung austauschen kann. Es gilt:
dH|p,adia = δW revsonst ≤ δW irr
sonst
– Enthalpieänderung ist bei isobarer, adiabatischer Zustandsänderung diemaximal vom System leistbare Arbeit
– In einem sich selbst überlassenen isobaren und adiabatischen System lau-fen nur irreversible Prozesse ab. Es gilt
dH ≤ 0
Im Gleichgewicht gilt
dH = 0, H = minimal
63

Enthalpie des idealen Gases:
U =3
2NkT
pV = NkT
H = U + pV =3
2NkT +NkT =
5
2NkT
Spezifische Wärme:• Mit dH|p = δQ
Cp =∂Q
∂T
∣∣∣∣p
=
(∂H
∂T
)p
Cp =5
2Nk
• Erinnere: CV = 32Nk. Cp > CV wegen Volumenarbeit
Praktische Bedeutung der Enthalpie:
• Bei isobaren Prozessen gilt: ∆H = ∆Q
• Enthalpieänderung entspricht dem Wärmeumsatz
• Wichtig bei chemischen Reaktionen
∆H = HProdukte−HEdukte
< 0 → Reaktion läuft spontan ab, exotherm> 0 → Reaktion läuft nicht spontan ab, endotherm
• Aber: Wichtig bei chemischen Reaktionen: Aktivierungsenergie
H
Reaktion
∆H
Aktivierungsenergie
64

• Thermodynamik sagt Enthalpieänderung voraus, aber nicht die Aktivierungs-energie.
• Aktivierungsenergie keine Folge von Gleichgewichtstheorie, da dynamischerProzess
• Enthalpien von vielen Stoffen bekannt
• Da H extensiv und damit additiv, kann Enthalpieänderung leicht vorhergesagtwerden.
Daher hat Enthalpie große Bedeutung in der Chemie.
6.5 Freie Enthalpie
• Die freie Enthalpie G(T, p,N) ergibt sich für Abhängigkeit der experimentellzu kontrollierenden Grössen Temperatur T und Druck p.
• Legendre-Transformation:
G(T, p,N) = U − TS + pV = H − TS = F + pV (17)
• Differentiell:dG = dU − TdS − SdT + V dp+ pdV
• Ergibt Fundamentalrelation:
dG = −SdT + V dp+ µdN
• Zustandsgleichungen folgen aus
−S =
(∂G
∂T
)p,N
, V =
(∂G
∂p
)T,N
, µ =
(∂G
∂N
)T,p
Extremalbedingung der freien Enthalpie
• 1. HauptsatzdU = δQ+ δW = TdS − pdV
2. Haupsatz
dS ≥ δQ
T
65

DamitdU − δW
T=δQ
T≤ dS
oderdU + pdV − TdS ≤ 0
für dT = dp = 0 ist dies grade das Differential der freien Enthalpie
dG = d(U − TS + pV ) = dU − TdS − S dT︸︷︷︸=0
+pdV + V dp︸︷︷︸=0
• Es giltdG ≤ 0, Im Gleichgewicht: G = minimal
• WegenG = F + pV
entspricht die freie Enthalpie G der freien Energie F mit zusätzlicher Volumen-arbeit bei p = const.
Änderung der freien Enthalpie ist maximal leistbare Arbeit bei isothermer undisobarer reversibler Prozessführung
Erinnere Euler-Gleichung:
U = TS − pV + µN
Mit Gl. (17) folgt:
G(T, p,N) = U − TS + pV = µ(T, p)N
Bedeutung: Freie Enthalpie pro Teilchen entspricht chemischem Potential.
Daher freie Enthalpie wichtig bei
• Phasenübergängen, Kap. 14
• Langsamen chemischen Reaktionen, z.B. in Batterien, die stets im thermischenGleichgewicht mit der Umgebung sind, T = const.
66

6.6 Großkanonisches Potential
Die letzte Übung ...
• Bisher stets N = const.
• Jetzt offenes System mit Teilchenaustausch dN 6= 0
• Gesucht: Thermodynamisches Potential mit dµ statt dN
Beachte:
• Die Transformation aller Variablen
Ψ = U − TS + pV − µN
macht keinen Sinn, da wegen Euler-Gleichung (12) gilt
U = TS − pV + µN
• Folge:Ψ ≡ 0
Was ist gute Transformation ?
• Chemisches Potential durch ”Teilchenbad” fixieren.
• Analogie: Austausch von Wärme mit Wärmebad führt zu konstanter Tempera-tur, Austausch von Teilchen mit Teilchenbad führt zu konstantem chemischenPotential
• Teilchenaustausch in der Regel mit Wärmeaustausch verbunden
• Daher liegt Transformation der inneren Energie U(S, V,N) auf PotentialΦ(T, V, µ) nahe
• Φ(T, V, µ) heißt großkanonisches Potential
• Legendre-Transformation:
Φ = U − TS − µN
Fundamentalrelation:
dΦ = −SdT − pdV −Ndµ
67

• Praktisch für isothermische Systeme mit festem chemischen Potential
• Auf Grund der Euler-Gleichung
U = TS − pV + µN
giltΦ = −p(T, µ)V
• In Analogie zu vorigen Potentialen gilt für bei konstanter Temperatur undchemischem Potential:
dΦ ≤ 0
Im GleichgewichtdΦ = 0, Φ = Φminimal
6.7 Maxwell-Relationen
• Auf Grund der Differenzierbarkeit der thermodynamischen Potentiale gilt derSatz von Schwarz, d.h. die 2. Ableitungen vertauschen
• Daraus folgen Beziehungen zwischen thermodynamischen Größen.
• Betrachte die Fundamentalform der inneren Energie:
dU = TdS − pdV + µdN
=
(∂U
∂S
)V,N
dS +
(∂U
∂V
)S,N
dV +
(∂U
∂N
)S,V
dN
Alles hinreichend differenzierbar: Satz von Schwarz:
∂
∂V
((∂U
∂S
)V,N
)S,N
=∂
∂S
((∂U
∂V
)S,N
)V,N
ergibt sich mit
T =
(∂U
∂S
)V,N
p = −(∂U
∂V
)S,N
die Maxwell-Relation (∂T
∂V
)S,N
= −(∂p
∂S
)V,N
Beachte: Ableitungen der nicht-natürlichen Variablen
68

• Analog: (∂T
∂N
)S,V
=
(∂µ
∂S
)V,N
, −(∂p
∂N
)S,V
=
(∂µ
∂V
)S,N
Entsprechend für die anderen thermodynamischen Potentiale:
• Freie Energie: dF = −SdT − pdV + µdN(∂S
∂V
)T,N
=
(∂p
∂T
)V,N
• Enthalpie: dH = TdS + V dp+ µdN(∂T
∂p
)S,N
=
(∂V
∂S
)p,N
• Freie Enthalpie: dG = −SdT + V dp+ µdN
−(∂S
∂p
)T,N
=
(∂V
∂T
)p,N
• Großkanonisches Potential: −SdT − pdV −Ndµ(∂S
∂V
)T,µ
=
(∂p
∂T
)V,µ
• Allgemein: Nützlich zur Berechung unbekannter Größen aus bekannten,resp. nicht messbarer aus messbaren
Guggenheim-Quadrat:
TV
S p
F
H
GU
69

• Ecken: die Variablen
• Kanten: die Potentiale der Variablen an den jeweiligen Ecken
• Ableitung eines Potentials nach einer Variablen ist gegenübergelegene Variable
• Pfeile bestimmen Vorzeichen
• Beispiel ∂F∂V
= −p
• Maxwell-Relationen: Ableitung von Variablen längs einer Kante, z.B. ∂V∂S
, beikonstant gehaltener Variablen in der diagonalen Ecken, hier p, ist gleich derAbleitung auf der Kante der gegenüberliegenden Seite, hier ∂T
∂p
∣∣∣S
Lessons learned
• Extremalbedingungen :
– Mechanik: Hamilton’sches Prinzip der minimalen/stationären WirkungS =
∫ t2t1L(x, x)dt → Euler-Lagrange Gleichung
– Optik: Fermat’schen Prinzip des kürzesten optischen Weges
– Thermodynamik: Für abgeschlossenes System gilt: dS ≥ 0. Entropie wirdmaximiert, Energie von Teilsystemen wir minimiert
• Gegeben die unabhängigen Variablen, folgen verschiedene thermodynamischePotentiale mit ihren Extremalbedingungen
• Hin- und herschalten mit Legendre-Transformation
• Idee: Wähle Variablen, die sich konstant halten lassen, macht das Leben ein-facher.
• Maxwell-Relationen koppeln über Satz von Schwarz partielle Ableitungen dernicht-natürlichen Variablen
Kurz-Klausur
70

7 Zusammenfassung Thermodynamik• Gleichgewichtszustände definiert über Zustandsgrößen
– Diese sind exakte Differentiale
• Zustandsgleichungen, Beispiel ideales Gas:
kalorisch : U =3
2NkT
thermisch : pV = NkT
• 1. Hauptsatz: Energieerhaltung
– Wärme ist keine Substanz, sondern Form von Energie(austausch)
– Arbeit und Wärme sind keine exakten Differentiale
• 2. Hauptsatz: Entropie kann in abgeschlossenem System nur erzeugt werden
– Reversible und irreversible Prozesse
– Carnot’scher Kreisprozess, Grenze der Umwandlung von Wärme in Arbeit
– Geht über alle bisherige Physik hinaus.
– Effekt kollektiven Verhaltens vieler Teilchen
• 3. HauptsatzlimT→0
S(T ) = 0
– Unerreichbarkeit des absoluten Temperaturnullpunktes
– Wärmekapazität muss in Widerspruch zur klassischen Theorie tempera-turabhängig sein.
• Thermodynamische Potentiale
– Sag mir Deine konstant gehaltenen Größen, und ich sage Dir Dein be-quemstes thermodynamisches Potential
– Legendre-Transformation
– Erfüllen Extremaleigenschaften.5. Wo-che
71

Teil II
Statistische MechanikAufgabe: Ableitung der makroskopischen Eigenschaften aus den mikroskopischen
Starte mit N -Teilchen Hamiltonian
H(q1, . . . , qN , p1, . . . pN) =N∑i=1
p2i
2m+ V (q1, . . . , qN , p1, . . . pN)
Berechne makroskopische Zustandsgrößen wie Entropie, Temperatur etc.
Inhaltsverzeichnis
Seien Sie verwirrt !
• CV = 32Nk für ideales Gas passt nicht zur Einsicht, dass CV temperaturabhän-
gig sein muss
• ”Entropische Kräfte” bei der freien Energie können nicht verständlich sein
• And, ... there is more to come !
8 Ein bißchen Statistik
8.1 Verteilungen
8.1.1 Zufallsvariablen
Zufallsvariable X:
• Etwas, das eine Wahrscheinlichkeitsdichte oder auch Verteilung pX(x) hat
• Wahrscheinlichkeit, eine Realisierung x in (x, x+dx) zu beobachten, ist pX(x)dx
• pX(x) ≥ 0,∫pX(x) dx = 1
72
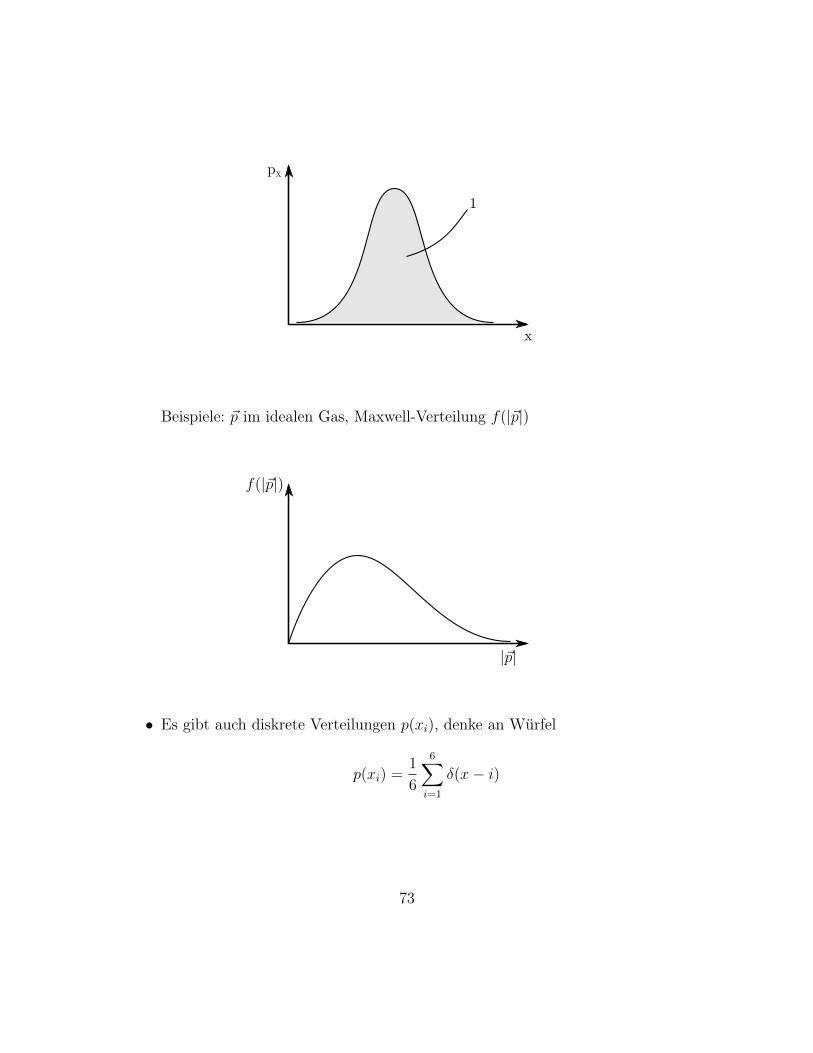
Beispiele: ~p im idealen Gas, Maxwell-Verteilung f(|~p|)
f(|~p|)
|~p|
• Es gibt auch diskrete Verteilungen p(xi), denke an Würfel
p(xi) =1
6
6∑i=1
δ(x− i)
73
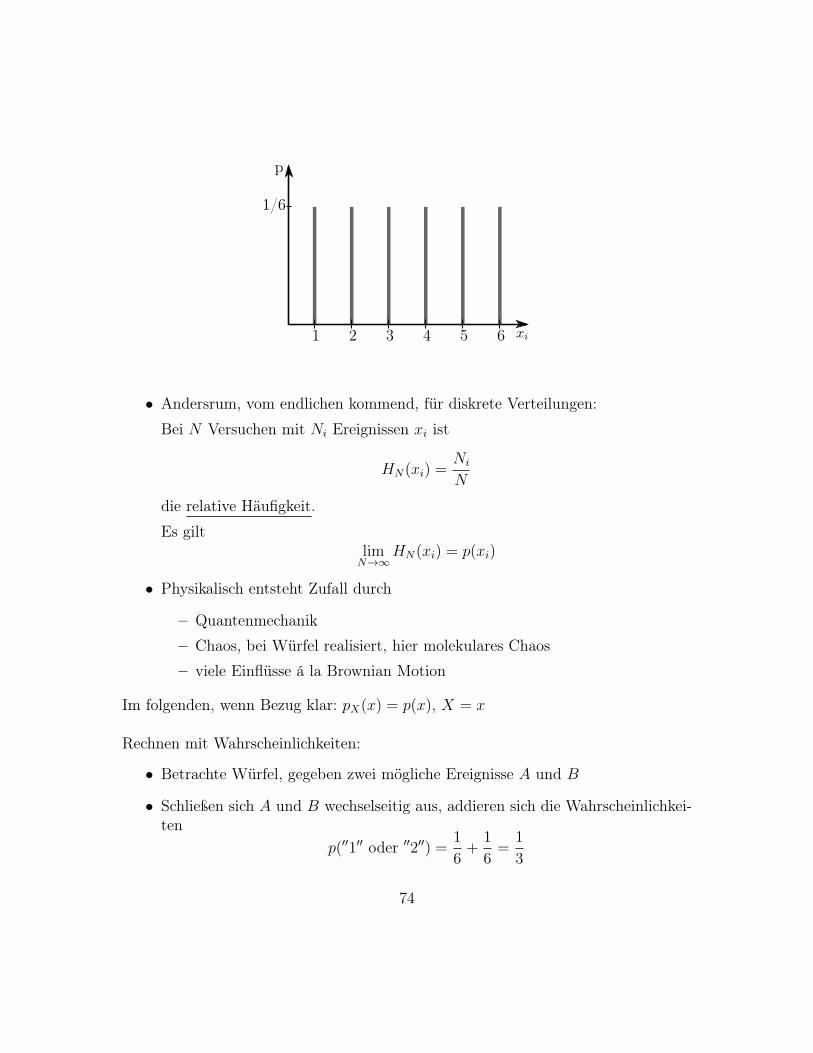
1/6
p
1 2 3 4 5 6
• Andersrum, vom endlichen kommend, für diskrete Verteilungen:
Bei N Versuchen mit Ni Ereignissen xi ist
HN(xi) =Ni
N
die relative Häufigkeit.
Es giltlimN→∞
HN(xi) = p(xi)
• Physikalisch entsteht Zufall durch
– Quantenmechanik
– Chaos, bei Würfel realisiert, hier molekulares Chaos
– viele Einflüsse á la Brownian Motion
Im folgenden, wenn Bezug klar: pX(x) = p(x), X = x
Rechnen mit Wahrscheinlichkeiten:
• Betrachte Würfel, gegeben zwei mögliche Ereignisse A und B
• Schließen sich A und B wechselseitig aus, addieren sich die Wahrscheinlichkei-ten
p(′′1′′ oder ′′2′′) =1
6+
1
6=
1
3
74

• Sind A und B unabhängig, multiplizieren sich die Wahrscheinlichkeiten
p(erst ′′1′′ dann ′′2′′) =1
6· 1
6=
1
36
8.1.2 Momente und Kumulanten
Momente
• Erwartungswert 〈f(x)〉
〈f(x)〉 :=
∫f(x) p(x) dx
Erwartungswert ist eine Zahl
• Momente µk
µk = 〈xk〉 =
∫xk p(x) dx
• 1. Moment: Mittelwert
µ1 = x = µ = 〈x〉 =
∫x p(x) dx
• 2. Momentµ2 = 〈x2〉 =
∫x2 p(x) dx
– Varianz: σ2 = 〈(x− x)2〉 = µ2 − µ21
– Standardabweichung: σ
– Addiert man unabhängige Zufallsvariablen, so sind Varianzen, nicht Stan-dardabweichungen additiv.
• 3. Moment
µ3 =< x3 >=
∫x3p(x) dx
Schiefe: κ = 〈(x− µ)3〉, Maß für Asymmetrie.
75

• 4. MomentKurtosis (Bauchigkeit): γ = 〈(x− µ)4〉/σ4 − 3
“3” erklärt sich weiter unten
Kumulanten
• Charakteristische Funktion oder Erzeugende Funktion
G(k) =< eikx >=
∫dx eikxp(x)
Fouriertransformierte der Dichte p(x)
• Existieren die Momente, i.e. < xn ><∞, Taylor-Entwicklung
G(k) =∞∑n=0
(i k)n
n!< xn >
Ist G(k) bekannt, berechnen sich Momente durch:
dnG(k)
dkn
∣∣∣∣k=0
= in < xn >
• Entwickelt man log(G(k)) nach k, folgt:
log(G(k)) =∞∑n=1
(ik)n
n!κn, G(k) = exp
(ikκ1 −
1
2k2κ2 −
i
6k3κ3 + . . .
)(18)
und man erhält die Kumulanten κn aus:
dn logG(k)
dkn
∣∣∣∣k=0
= inκn
Die ersten 3 lauten
κ1 = µ1
κ2 = µ2 − µ21 = σ2
κ3 = µ3 − 3µ2µ1 + 2µ31
Wichtige Eigenschaften:
76

– Kumulanten sind additiv, daher natürliche GrößenSei
y =N∑i=1
xi
dann folgt
κn(y) =N∑i=1
κn(xi)
Varianzen sind additiv, nicht Standardabweichungen
– Man kann zeigen:
∗ Entweder: Alle Kumulaten außer den ersten beiden verschwinden∗ Oder: Es existieren ∞ viele
8.2 Beispiele für Verteilungen
• Gaußverteilung oder Normalverteilung:
p(x) =1√2πσ
e−(x−µ)2
2σ2
– Bezeichnung: N(µ, σ2)
Standard-Gaußverteilung µ = 0, σ2 = 1: N(0, 1)
In ±1σ liegt 68 % der MasseIn ±1.96σ liegt 95 % der Masse
– Momente von N(0, 1) :
〈xk〉 =
0 für k ungerade
1× 3× · · · × (k − 1) für k gerade
Damit klar, warum bei Kurtosis ”-3”
– Charakteristische Funktion
G(k) = eiµk−12σ2k2
Nur die ersten zwei Kumulanten sind ungleich null.Macht klar, warum Gaußverteilung so besonders
77

Zentraler Grenzwertsatz:
Existeren die ersten beiden Momente, so strebt für N → ∞ die (normierte)Summe von unabhängigen, identisch verteilten (independent, identically distri-buted: iid) Zufallsvariablen gegen eine Gaußverteilung.
– Betrachte N identische Zufallsvariablen xi mit
∗ O.B.d.A.: κ1(xi) =< xi >= 0
∗ κ2(xi) = µ2 − µ21 = σ2
– Bilde:
y =1√N
N∑i=1
xi
Normierung damit σ2y = σ2
xi
– Für y gilt:
p(y) =
∫dx1 . . . dxN p(x1) . . . p(xN) δ
(y −
(x1 + . . .+ xN√
N
))Mit, erinnere QM
δ(z) =
∫dk
2πe−ikz
folgt
p(y) =
∫dk
2πe−iky
∫dx1 . . . dxN p(x1) . . . p(xN) exp
(ik(x1 + . . .+ xN)√
N
)– Mit Erzeugender Funktion∫
dxi p(xi) eikxi√N = G
(k√N
)faktorisiert Integral:
p(y) =
∫dk
2πe−ikyGN
(k√N
)
78

– Mit Gl. (18)
G(k) = exp
ik κ1︸︷︷︸=µ=0
−1
2k2 κ2︸︷︷︸
=σ2
− i6k3κ3 + . . .
(19)
undGN
(k√N
)= exp
(−1
2
k2
NNσ2 − i
6
k3
N3/2Nκ3 + . . .
)(20)
folgt
p(y) =
∫dk
2πe−iky exp
−1
2k2σ2 − i
6
k3
√Nκ3 + . . .︸ ︷︷ ︸
→0 für N→∞
Somit folgt für N →∞
p(y) =1√2πσ
e−y2
2σ2
Wenn 〈xi〉 = µ 6= 0 entsprechend
p(y) =1√2πσ
e−(y−µ)2
2σ2
– Die höheren Kumulanten n > 2 verschwinden mit N .
– Verteilung geht gegen Gauß-VerteilungDarum sind die beiden Größen µ und σ so wichtig.
– Gilt auch wenn xi nicht identisch
– Konvergenzrate, i.e. wie schnell geht es zur Gaußverteilung, hängt i.w. vonder Schiefe ab.
– Bedeutung von Zentralem Grenzwertsatz nicht zu unterschätzen.5. week15Gesetz der großen Zahl
– Mitteln:
79

y =1
N
N∑i=1
xi
Analog zu Gl. (19, 20) mit µ und N statt√N , folgt:
κn(y) =1
Nn−1κn(xi)
Speziell
κ2(y) =σ2x
N
– Ergo:
< y >=< x >, σy =1√Nσx
respektive:σy
< y >=
1√N
σx< x >
Die Schwankung und die relative Schwankung um den Mittelwert ver-schwindet für N →∞ mit 1/
√N
– Oder: Für N → ∞ geht das arithmetische Mittel von identischen, unab-hängigen Zufallsvariablen (auf Gauß’sche Weise) mit Wahrscheinlichkeit1 gegen ihrem Mittelwert.
– Sollte in der Regel für N = 1023 gelten und rechtfertigt die Verwendungdeterministischer Zustandsgrößen
– Ausnahme: Lang-reichweitige Korrelationen bei Phasenübergängen
• Gleichverteilung U(a, b) :
p(x) =
1/(b− a) für a ≤ x ≤ b
0 sonst
• Exponential-Verteilung
p(x) =1
τe−x/τ
80

Es gilt:
µ = τ
σ2 = τ 2
Erwartungswert und Varianz sind keine unabhängigen Parameter.
Ergibt sich bei “konstanter Ausfallrate”
• χ2r Verteilung mit r Freiheitsgraden:
Summe von r quadrierten Gaußverteilungen
”χ2r =
∑ri=1(N(0, 1))2”
y ∼ χ2r, xi ∼ N(0, 1) = pG(xi)
p(y) =
∫dx1 . . . dxr δ(y − (x2
1 + . . .+ x2r))
r∏i=1
pG(xi)
=
∫dx1
1√2πe−x
21/2dx2
1√2πe−x
22/2 . . . dxr
1√2πe−x
2r/2 δ(y − (x2
1 + . . .+ x2r))
=yr/2−1e−y/2
2r/2 Γ(r/2), Γ(z) =
∫ ∞0
tz−1e−t dt
Es gilt:
〈χ2r〉 = r
V ar(χ2r) = 2r ,
d.h. Erwartungswert und Varianz sind keine unabhängigen Parameter.
FOLIE χ2r Verteilungen
χ2 Verteilungen sind additiv:
”χ2r1
+ χ2r2
= χ2r1+r2
”
81

Aus dem zentralen Grenzwertsatz folgt:
limr→∞
χ2r = N(r, 2r)
Beachte :
– χ22 = 1
2e−x/2 ist Exponential-Verteilung mit τ = 2.
• t-Verteilung
t(r, x) =′′N(0, 1)√χ2r/r
′′
=1√r
1
B(1/2, r/2)
(1 +
x2
r
)− 12
(r+1)
, B(a, b) =Γ(a)Γ(b)
Γ(a+ b)
r: Anzahl der Freiheitsgrade
Wichtig beim t-Test: Test auf Gleichheit von Mittelwerten zweier Gaußvertei-lungen.
limr→∞
t(r, x) = N(0, 1), gute Näherung ab N = 30
• F -Verteilung
F (r1, r2, x) =′′χ2r1/r1
χ2r2/r2
′′
= ...
r1, r2: Jeweilige Anzahl der Freiheitsgrade
Wichtig beim F -Test: Test auf Gleichheit von Varianzen zweier Gaußverteilun-gen.
• Cauchy(Lorenz)-Verteilung:
pCauchy(x, a, γ) =1
π
γ
(x− a)2 + γ2
82
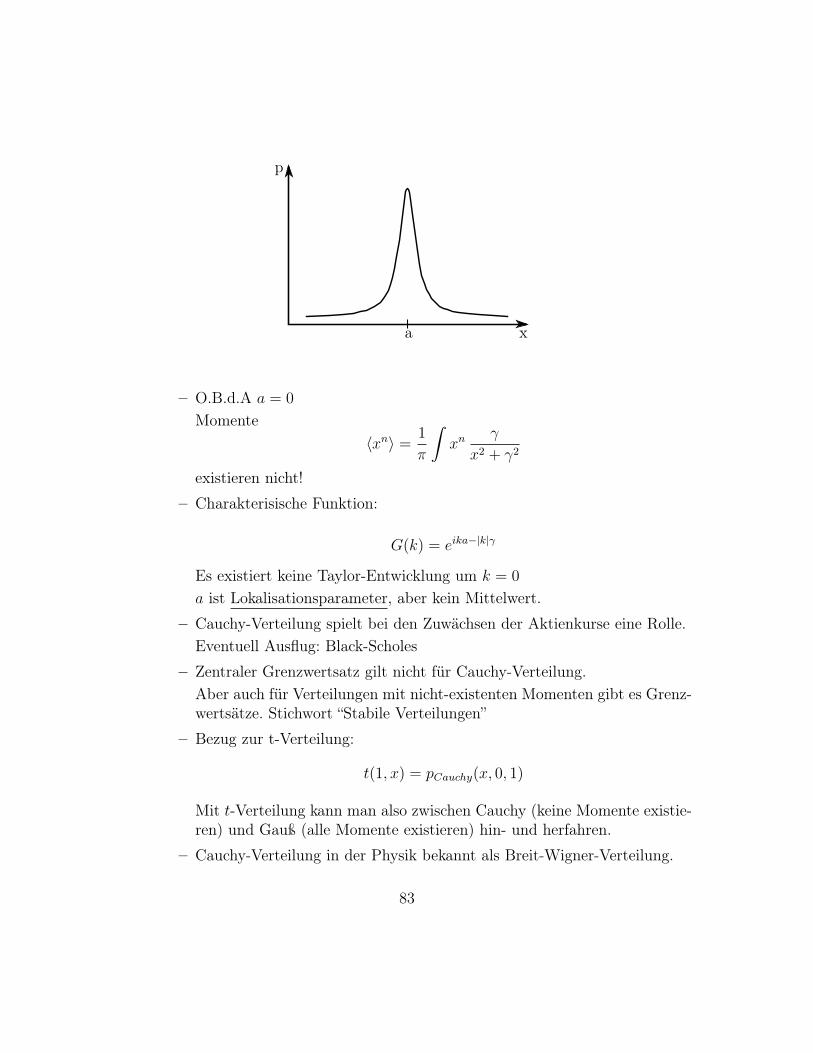
p
xa
– O.B.d.A a = 0
Momente〈xn〉 =
1
π
∫xn
γ
x2 + γ2
existieren nicht!
– Charakterisische Funktion:
G(k) = eika−|k|γ
Es existiert keine Taylor-Entwicklung um k = 0
a ist Lokalisationsparameter, aber kein Mittelwert.
– Cauchy-Verteilung spielt bei den Zuwächsen der Aktienkurse eine Rolle.Eventuell Ausflug: Black-Scholes
– Zentraler Grenzwertsatz gilt nicht für Cauchy-Verteilung.Aber auch für Verteilungen mit nicht-existenten Momenten gibt es Grenz-wertsätze. Stichwort “Stabile Verteilungen”
– Bezug zur t-Verteilung:
t(1, x) = pCauchy(x, 0, 1)
Mit t-Verteilung kann man also zwischen Cauchy (keine Momente existie-ren) und Gauß (alle Momente existieren) hin- und herfahren.
– Cauchy-Verteilung in der Physik bekannt als Breit-Wigner-Verteilung.
83

• Multivariate Gaußverteilung
p(~x) =1
(2π)d/2√|C|
exp
(−1
2(~x− ~µ)TC−1(~x− ~µ)
), d = dim(~x)
mit Kovarianzmatrix C
C = 〈(~x− ~µ)(~x− ~µ)T 〉 =
Cov(X1, X1) · · · Cov(X1, Xn)... . . . ...
Cov(Xn, X1) · · · Cov(Xn, Xn)
Kovarianz, Korrelation allgemein:
Zwei Ererignisse x und y sind statistisch unabhängig, wenn die gemeinsameDichte p(x, y) in die Marginalverteilungen faktorisiert
p(x, y) = p(x) p(y)
Folge: Erwartungswerte faktorisieren
〈xy〉 = 〈x〉〈y〉
Maß für statistische (Un)abhängigkeit
– Kovarianzcov(x, y) = 〈xy〉 − 〈x〉〈y〉
– Normiert auf [−1, 1]: Korrelation
corr(x, y) =cov(x, y)
σxσy
– 1 D Normalverteilung:68 % der Masse in [−σ, σ]99 % der Masse in [−3σ, 3σ]
– 10 D Normalverteilung, C = 1:99 % der Masse außerhalb der [−3σ, 3σ]-Kugel.
– Intuition:
∗ Integration über die Winkel ausführen
84

∗ Verbleibt, d = dim (~x)
Masse innerhalb von Radius r ∼∫ r
0
rd−1 e−r2/2 dr
– Es gibt praktisch nur die längsten Abstände, der Raum ist leer, ”curse ofdimensionality”.
FOLIE χ2r Verteilungen nochmal
6. Wo-che• Binomial Verteilung
B(n, p, k) =
(nk
)pk(1− p)n−k, k = 0, 1, . . . n (21)
Zwei mögliche Ereignisse: x1, x2; p = prob(x1)
Bei n-maliger Durchführung des Experimentes ist B(n, p, k) die Wahrschein-lichkeit, x1 k-mal zu realisieren.
Anwendung: Random Walk
– Die Mutter aller stochastischen Prozesse
– Brown’sche Molekularbewegung
– Diffusion
– Wärmeleitung
– Volltrunkenheit
Betrachte eindimensionalen Random Walk
Physikalische Herleitung:
– N Schritte entlang x-Achse
– m in +x-Richtung, N −m in -x-Richtung
– p+x = p−x = 12
Gesucht: Wahrscheinlichkeit p(m,N) von N Schrittenm in positive x-Richtungzu machen.
– Es gibt N ! Möglichkeiten N Schritte anzuordnen
85

– Schritte +x und −x sind jeweils identisch– Teile durch m! und (N −m)!
– Jeder mögliche Pfad hat Wahrscheinlichkeit 12
N
– Ergibt (Binomial) Verteilung für m Schritte nach rechts
p(m,N) =
(1
2
)NN !
m! (N −m)!
Mathematische Herleitung
– Gl. (21) mit p = 12, n→ N , k → m, et voila !
Verteilung für große N , m, N −m, N m,N −m
– Für große N , m, N −m gilt Stirling’sche Näherung, siehe Übung
log n! = n log n− n
Damit
log p(m,N) = N log1
2+ logN !− logm!− log(n−m)!
= N log1
2+N logN −N −m logm+m− (N −m) log(N −m) + (N −m)
= N log1
2+N logN −m logm− (N −m) log(N −m)
– Entwickle um wahrscheinlichsten Punkt m = m∗, N unterdrückt
log p(m) = log p(m∗)+d log p(m)
dm
∣∣∣∣m∗
(m−m∗)+ 1
2
d2 log p(m)
dm2
∣∣∣∣m∗
(m−m∗)2+. . .
(22)– Bestimmung von m∗:
d log p(m)
dm= −
(logm+
m
m
)+ log(N −m)− N −m
N −m(−1)
= log(N −m)− logm
= logN −mm
!= 0
DamitN −mm
= 1 =⇒ m∗ =N
2, macht Sinn
86

– 2. Ableitung an m∗
d2 log p(m)
dm2
∣∣∣∣m∗
=m∗
N −m∗−m∗ − (N −m∗)
m∗2=
−N(N −m∗)m∗
=−N
(N −N/2)N/2= − 4
N
In Gl. (22) eingesetzt, ergibt, übereinstimmend mit dem zentralen Grenz-wertsatz, eine Gauß-Verteilung
p(m,N) = p(m∗) e−2(m−m∗)2
N
– Vergleich mit Standard-Gauß-Verteilung gibt Varianz
〈(m−m∗)2〉 =N
4
Verteilung für Ort x
– Mitx = m− (N −m) = 2m−N und m∗ =
N
2
gilt
x∗ = 〈x〉 = 0, macht Sinn
Mitm−m∗ =
x+N
2− N
2=x
2
folgt für die Varianz σ2x von x mit
N
4= 〈(m−m∗)2〉 =
⟨(x2
)2⟩
σ2x = N
und damit mit der richtigen Normierung
p(x) =1√
2πNe−
x2
2N
87

– Alternative Formulierung:
x(t) = x(t− 1) + ε(t), ε(t) ∼ N(0, σ2)
Verteilung von ε(t) per Zentralem Grenzwertsatz aus mehreren Schrittenvon oben
Separation der Zeitskalen
Um einen Zeitschritt verschoben
x(t− 1) = x(t− 2) + ε(t− 1)
Damitx(t) = x(t− 2) + ε(t) + ε(t− 1)
Weiter in einander eingesetzt bis t = 0, O.B.d.A.: x(0) = 0
x(t) =t∑i=1
ε(i)
Varianzen sind additivDamit Verteilung der Zufallsvariablen Random Walk
x(t) ∼ N(0, tσ2) =1√2π
1√t σ
exp
(−x(t)2
2 t σ2
)Der Random Walk zeigt Skaleninvarianz
– Definition:Eine Funktion oder eine Zufallsvariable f(z) ist skaleninvariant, wenn siesich bei Reskalierung mit z → az bis auf einen Faktor nicht ändert
f(az) = c(a)f(z) (23)
– Random Walk ist skaleninvariant, da
∗ ε(t) ∼ N(0, σ2)
∗ x ∼ N(0, tσ2)
∗ V ar(x(t, σ2)) = tσ2
88

∗ Multipliziere t mit Faktor a
V ar(x(at, σ2)) = atσ2 = t(aσ2)
∗ Betrachte nicht-quadrierte Größe Standardabweichung SD
SD(x(t, σ2)) =√tσ
SD(x(at, σ2)) =√atσ =
√t(√aσ)
Damit
x(at, σ2) =√ax(t, σ2)
und, unterdrücke σ2, setze f(.) = x(.) und z = t, folgt mit Gl. (23)
c(a) =√a
– Andersrum gelesen: Ein random walk x kann wieder als ε eines anderenrandom walks x′ aufgefasst werden, resp. jedes ε eines random walks xkann wieder als random walk x′′ aufgefasst werden kann.
– Random Walk sieht bei unterschiedlicher Auflösung immer strukturellgleich aus
– Skaleninvarianz wird bei Phasenübergängen, Kap. 14, wichtig
Wieder D = 3
– Random walk in D = 1 und D = 2: Wahrscheinlichkeit, dass er wiederzum Ausgangspunkt zurückkehrt, ist gleich 1
– Random walk in D ≥ 3: Wahrscheinlichkeit, dass er wieder zum Aus-gangspunkt zurückkehrt, ist gleich 0
89

– Ein betrunkener Mann findet immer heim, ein betrunkener Vogel nicht
Diffusion
– Passiver Transportprozess
– Typisches Beispiel für Random Walk
– Fluss J(x, t) gegeben durch
J(x, t) = −D∂p(x, t)∂x
mit D die Diffusionskonstante
– Mit Kontinuitätsgleichung∂p
∂t= −∂J
∂xergibt sich die Diffusionsgleichung oder auch Wärmeleitungsgleichung
∂p
∂t= D
∂2p
∂x2
Übung: Zeige, dass der Random Walk eine Lösung der Diffusionsgleichungist
• Poisson-Verteilung
P (k, λ) =e−λλk
k!, k ∈ N0, λ > 0
– Wahrscheinlichkeit für k Ereignisse in einem Zeitintervall
– λ: Durchschnittliche Anzahl der Ereignisse in Zeitintervall
– Wichtig für Punktprozesse mit konstanter Rate denke an Photonen-Zählprozesse, e-mails pro Stunde
– Für Poisson-Verteilung gilt
µ = σ2 = λ
– Ferner ist sie die Grenzverteilung der Binomialverteilung, siehe Übung:
limn→∞
B(n, k, p) = P (k, λ) wobei limn→∞
np = λ
”Seltene Ereignisse”
90

– Poisson-Verteilung für kleine λ sehr unsymmetrisch. Für große, λ > 30,geht sie gegen Gauß-Verteilung
P (k, λ) =1√2πλ
exp
(−(k − λ)2
2λ
)Kumulative Verteilungen
• Definition:cum(x) =
∫ x
−∞dx′p(x′)
• xα mitcum(xα) = α
nennt man das (100α) % Quantil.
• Definition des Medians:
cum(xMedian) = 0.5
Der mittlere Wert der Verteilung
Schätzer für Median aus N Werten:
– Sortiere N Werte
91

– Wenn N ungrade:Median = x(N+1)/2
Wenn N grade:
Median =1
2(xN/2 + xN/2+1)
Der Median ist der Maximum-Likelihood Schätzer für θ in der Laplace-Verteilung
p(x) =1
2e−|x−θ|
• Für den Lokationsparameter a der Cauchy-Verteilung auch nicht so schlecht,konsistent, aber nicht optimal
Lessons learned
• Zufallsvariablen sind etwas, das eine Verteilung hat
• Zentraler Grenzwertsatz zeichnet Gaußverteilung aus
• Gesetz der großen Zahl: Für N →∞ werden Verteilungen scharf
• Random Walk als Mutter aller stochastischen Prozesse
9 Entropie revisited• Makroskopisches System ist durch Zustandsgrößen, dem Makrozustand, fest-
gelegt
• Mikrozustände sind aber unbestimmt.
• Mikroskopische Interpretation der Entropie
• Entropie: Maß für die Unkenntnis des Mikrozustandes
92

9.1 Vorspiel
• Betrachte geteiltes Volumen eines abgeschlossenen Systems, alle Teilchen aufeiner Seite.
• Öffne Trennungswand
• Erfahrungstatsache: Die Teilchen verteilen sich gleichmässig links und rechts
• Ausgangszustand wird nie wieder hergestellt
Klassischer Mikrozustand: x1, . . . , xN , p1, . . . , pN
Betrachte System mit drei wechselwirkungsfreien Teilchen auf der linken Seite, diemit gleicher Geschwindigkeit zum Zeitpunkt des Öffnens der Trennungswand in x-Richtung starten
93

• Ausgangszustand wird periodisch wieder hergestellt
• Lasse Teilchen für Zeit T laufen, ändere Impulse zur Zeit T von pT in −pT
• Zum Zeitpunkt 2T sind Teilchen wieder am Ausgangsort
• Zwei Probleme:
– Unschärfe-Relation verbietet exakte Präparation des Ausgangszustandes
– Kleinste Imperfektion der Wände zerstört ”Kohärenz”
• Nichttriviale Ausnahmen hierfür: Spin-Echo Experimente und Tropfen in dieMitte eines Wasserglases
Betrachte 1023 wechselwirkende Teilchen unter den selben Bedingungen
• Chaotische Dynamik bewirkt, dass die Teilchen ihre Anfangsbedingungen ”ver-gessen”
• Das System ist mischend:
Autokorrelationsfunktion 〈x(t)x(t − τ)〉 zerfällt schnell, beim HarmonischenOszillator nie
• Mischungseigenschaft macht die Mikrozustände auf sehr kurzer Zeitskala un-abhängig.
• Oder: Trotz deterministischer Dynamik ergeben sich zeitlich statistisch unab-hängige Mikrozustände
• Oder: Dynamik spielt keine Rolle mehr. Wenn man System nach ∆t anguckt,weiss man nicht, ob man das selbe System vor sich hat oder ein zweites System
94

• Intuition: Alle durch den Makrozustand erlaubten Mikrozustände werden gleichwahrscheinlich
• Anzahl dieser Mikrozustände: Ω
– Für ein Teilchen: Ω(V ) ∝ V
– Für N Teilchen: Ω(V ) ∝ V N
– Für N Teilchen, alle links: Ω(V/2) ∝ (V/2)N
– Für 1023 ist(
12
)1023 ≈ 10−1022
– Argument von oben entspricht exakt einem Mikrozustand, absolut un-wahrscheinlich
6. week15Mathematisierung
• Definition des Makrozustandes: Anzahl der Teilchen, die im Mittel links sind
• Wahrscheinlichkeit, dass ein Teilchen Ti links ist: pi,l = 12
• Wahrscheinlichkeit für k bestimmte Teilchen links:
pk,l =
(1
2
)k• Entsprechend für N − k Teilchen rechts
pN−k,r =
(1
2
)N−k• Gesamtwahrscheinlichkeit:
pk,l pN−k,r =
(1
2
)k (1
2
)N−k=
(1
2
)NGilt ∀ k: Alle Mikrozustände sind gleich wahrscheinlich
Kombinatorik:
• Anzahl der Möglichkeiten, k Teilchen aus N auszuwählen:
N !
k!(N − k)!=
(Nk
)
95

• Wahrscheinlichkeit, dass k beliebige Teilchen links sind:
pk,l =
(Nk
)(1
2
)k• Mittelwert < k > dieser Verteilung
< k >=N∑k=0
kpk,l =N
2
Standardabweichung
σk,l =√< (k− < k >)2 > =
√N/2
Siehe Übung
• Erinnere Gesetz der großen Zahl
Betrachte relative Breite der Verteilung
σk,l〈k〉
=
√2√N
Für N = 1023 ergibt sich sehr scharfe Verteilung
Bedeutung:
• Der Makrozustand mit den meisten ihn realisierenden Mikrozuständen ist derWahrscheinlichste
• Jedes System strebt einem Zustand maximaler mikroskopischer Unordnung an
• ”Maximale Unordnung”, weil ”Teilchen sind irgendwo” unordentlicher ist als”alle Teilchen links”
• Kann man wieder ”als ob” denken á la Hamilton’sches Prinzip der minimalenWirkung
• Extremalprinzip, riecht schon nach Entropie
96

9.2 Formalisierung
Mikrozustand r: Vollständige Beschreibung des mikroskopischen Systems
Beispiele:
• N Würfel sind durch ihre Augenzahlen ai beschrieben
r = (a1, . . . , nN)
• N Spins mit si = ±12durch
r = (s1, . . . , sN)
• Klassisches System mit N Freiheitsgraden im 2N -dimensionalen Phasenraum
r = (q1, . . . , qN , p1, . . . pN)
Grundpostulat der Statistischen Mechanik:In einem abgeschlossenen Systems sind alle zugänglichen Mikrozustände gleich wahr-scheinlich
Kann nicht allgemein bewiesen werden, ist aber plausibel
• N Würfel:
Jedes der 6N möglichen Ergebnisse ist gleich wahrscheinlich
• Quantenmechanik:
Alle Zustände mit E =∑
iEi sind gleich wahrscheinlich
• Klassisches System:
N Teilchen mit Trajektorie (q1(t), . . . , qN(t), p1(t), . . . , pN(t)):
Alle Punkte der Trajektorie gleich wahrscheinlich
Wichtiger Begriff: Die Zustandssumme
Definition:Die Zustandssumme Ω ist die Anzahl aller zugänglichen Mikrozustände
97

• Betrachte abgeschlossenes System mit Energie E = const.
• Realistischer Weise, E ist i.A. quantisiert, (und rechentechnisch einfacher) bisauf Ungenauigkeit ∆E bestimmt
• Damit Mikrokanonische Zustandssumme:
Ω(E) = Anzahl aller zugänglichen Mikrozustände r mit E ≤ Er ≤ E + ∆E
Im Gleichgewicht gilt dann für Wahrscheinlichkeit pr eines Mikrozustandes r
pr =
1Ω(E)
E ≤ Er ≤ E + ∆E
0 sonst
Problem:
• Betrachte Harmonischen Oszillator
H(p, q) =p2
2m+k
2q2 = E
• (Überabzählbar) unendlich viele Phasenraumpunkte
• Frage: Ist die Frage nach der Anzahl von Mikrozuständen überhaupt sinnvoll?Hier dürfen Sie wieder verwirrt sein
98

• System bewegt sich im allgemeinen auf 6N − 1 dimensionaler Energiehyperflä-che mit Flächeninhalt:
σ(E) =
∫d~qd~p δ(E −H(~q, ~p))
Wir nehmen an:
Ω(E) =σ(E)
σ0
σ0 eine Proportionalitätskonstante, siehe unten
• Zur Berechung von σ(E) betrachte gesamtes Phasenraumvolumen ω(E), dasvon E = H(~q, ~p) eingeschlossen wird:
ω(E) =
∫H(~q,~p)≤E
d~qd~p =
∫d~qd~pΘ(E −H(~q, ~p))
Für kleine dE gilt
dω = ω(E + dE)− ω(E) = ω(E) +dω
dEdE − ω(E) =
dω
dEdE
und Satz von Cavalieridω = σ(E)dE
da Volumen = Fläche × Höhe
• Damit
σ(E) =dω
dE
Beispiel: Kugelvolumen ω(R) und ihrer Oberfläche σ(R)
ω(R) =4π
3R3, σ(R) =
dω
dR= 4πR2
SomitΩ(E) =
1
σ0
dω
dE=
1
σ0
d
dE
∫d~qd~pΘ(E −H(~q, (~p))
• Merke: Zustandssumme kann aus (mikroskopischem) Hamiltonian berechnetwerden
99

9.3 Entropie
• Thermodynamik: In einem abgeschlossenen System ist die Entropie im Gleich-gewicht maximal
• Statistische Physik: Alle Mikrozustände sind gleich wahrscheinlich. Folge: Ma-krozustand mit den meisten Mikrozuständen ist der Wahrscheinlichste unddamit der Gleichzugewichtszustand.
Bringe makroskopische Entropie und mikroskopische Zustandssumme zusammen
• Betrachte abgeschlossenes System, das in zwei Teilsysteme unterteilt ist
• Statistische Sichweise:
Wegen Unabhängigkeit der Teilsysteme gilt mit E = E1 + E2, V = V1 + V2,N = N1 +N2:
Ω(E, V,N) = Ω1(E1, V1, N1)Ω2(E2, V2, N2)
mit dem totalen Differential:
dΩ = Ω2dΩ1 + Ω1dΩ2
Division durch ΩdΩ
Ω= d log Ω = d log Ω1 + d log Ω2 (24)
Beachte: log Ω ist extensiv
Im Gleichgewicht gilt:
d log Ω = 0, log Ω = log Ωmax (25)
• Thermodynamische Sichtweise
Entropien sind additiv
S = S(E, V,N) = S1(E1, V1, N1)+S2(E2, V2, N2) → dS = dS1 +dS2 (26)
Gleichgewichtsbedingung
dS = 0, S = Smax (27)
100

• Vergleich von Gl. (24) mit Gl. (26) und Gl. (25) mit Gl. (27) legt nahe, Boltz-mann (1877)
S(E, V,N) = k log Ω(E, V,N)
mit k der Boltzmann-Konstanten
Führt zwei konzeptionell unterschiedliche Definitionen der Entropie zusammen
• Thermodynamische Definition, notwendig um makroskopische Irreversibilitätzu erklären:
∆S =
∫ B
A
δQ
T
• Statistische Definition: Maß für mikroskopische Unordnung
S = k log Ω
7. Wo-che
9.4 Zustandssumme und Entropie des idealen Gases
Vorspiel
• Volumen einer N -dimensionalen Kugel:
VN(R) =
∫dx1 . . . dxN Θ
(R2 −
N∑i=1
x2i
)
mit yi = xi/R
VN(R) = RN
∫dy1 . . . dyN Θ
(1−
N∑i=1
y2i
)= RNCN
CN : Volumen der Einheitskugel
101

• Erinnere: ∫ ∞−∞
dx e−x2
=√π
und damit
I =
∫ ∞−∞
dx1 . . . dxN exp
− (x21 + . . .+ x2
N)︸ ︷︷ ︸=R2
= πN/2
MitdVN(R)
dR= NRN−1CN
Transformation auf Polarkoordinaten
dVN = dx1 . . . dxN = NRN−1CNdR
Winkelanteile automatisch verarbeitet
I = NCN
∫ ∞0
RN−1e−R2
dR
• Mit Variablensubstitution
z = R2,dz
dR= 2R, dR =
dz
2R,
RN−1
2R=
1
2RN−2 =
1
2zN2−1
folgt
I = NCN2
∫ ∞0
dz zN2−1e−z︸ ︷︷ ︸
=Γ(N2
)
= πN/2
und
CN =πN/2
N/2 Γ(N2
)
VN(R) = CNRN =
πN/2
N/2 Γ(N2
)RN
102

• Check: N = 3: Γ(32) = 1
2
√π, C3 = 4π
3und V = 4π
3R3
Betrachte ideales Gas: N Teilchen der Masse m in Volumen V
• Hamiltonian:
H(qi, pi) =N∑i=1
~p2i
2m
• Phasenraumvolumen:
ω(E, V,N) =
∫dq1 . . . dqN
∫dp1 . . . dpN Θ(E −H(qi, pi))
= V N
∫∑i
p2i
2m≤E
dp1 . . . dpN
Impulsintegral entspricht wegen
N∑i=1
p2i ≤ 2mE
dem Volumen einer 3N-dimensionalen Kugel mit Radius√
2mE
Damit:
ω(E, V,N) = V N π3N/2
3N/2 Γ(3N2
)(2mE)3N/2
• Mit
Ω =1
σ0
∂ω
∂Eund S = k log Ω
folgt
HIER IST WAS FALSCH, WIRD NOCH KORRIGIERT
S = k log
V N 1
σ0
π3N/2
Γ(3N2
)(2m)3N/2E
3N2−1︸ ︷︷ ︸
≈E3N2
= k log
((V
σ
)N(2πmE)
3N2
Γ(3N2
)
)
mit σ = σ1/N0
103

• Mit
log Γ(n) = log((n− 1)!)n1≈ (n− 1) log(n− 1)(n− 1) ≈ n log n− n
log1
Γ(3N2
)= −3N
2
(log
3N
2− 1
)folgt
S = Nk
(log
(V
σ(2πmE)
32
)+
3
2− log
((3N
2
) 32
))und schließlich:
S(E, V,N) = Nk
[3
2+ log
(V
σ
(4πmE
3N
) 32
)](28)
• Vergleiche mit Ergebnis der Thermodynamik, Gl. (6) auf Seite 38
• Erinnere:
dU = dE = TdS − pdV N − Abhängigkeit unterdrückt
dS =1
TdE +
p
TdV Physik
dS exaktes Differential
dS =
(∂S
∂E
)V︸ ︷︷ ︸
1/T
dE +
(∂S
∂V
)S︸ ︷︷ ︸
p/T
dV Mathematik
• Thermische und kalorische Zustandsgleichungen des ideales Gases:
1
T=
(∂S
∂E
)V,N
= Nk
(σ
V
)(3N
4πmE
) 32
︸ ︷︷ ︸äußere Ableitung
3
2
V
σ
(4πm
3N
) 32
E12︸ ︷︷ ︸
innere Ableitung
= Nk3
2
1
E
=⇒ E =3
2NkT
p
T=
(∂S
∂V
)E,N
=Nk
V=⇒ pV = NkT
104

Beachte und merke: Ableitung der makroskopischen Gleichungen aus mikroskopi-schen Eigenschaften
Zusammenfassung:
• Betrachte abgeschlossenes System: (E, V,N)
• Zustandssumme Ω(E): Anzahl der zugänglichen Mikrozustände
• Wahrscheinlichkeit für Mikrozustand r: pr = 1Ω(E)
, Grundpostulat
• Berechnung der Anzahl der Mikrozustände:
σ(E) =
∫d~qd~p δ(E −H(~q, ~p))
• Problem: Überabzählbar unendliche viele Mikrozustände
• Annahme:
Ω(E) =σ(E)
σ0
σ0: Proportionalitätskonstante
• Eingeschlossenes Phasenraumvolumen
ω(E) =
∫H(~q,~p)≤E
d~qd~p =
∫d~qd~pΘ(E −H(~q, ~p))
• Satz von Cavalieriσ(E) =
dω
dE
SomitΩ(E) =
1
σ0
dω
dE=
1
σ0
d
dE
∫d~qd~pΘ(E −H(~q, (~p))
• Ideales Gas:
Thermodynamik, Seite 38:
105

– Leite Zustandsgleichungen phänomenologisch her
– Nimm 1. Hauptsatz
– Löse nach dS und integriere
Statistische Physik:
– Hamiltonian:
H(qi, pi) =N∑i=1
~p2i
2m
– Integration über q trivial: V N
– Integration über p: 3N -dimensionale Kugel
– Entropie: S = k log Ω, Gl. (28)
– Ergibt (makroskopische) Zustandsgleichungen aus (mikroskopischem) Ha-miltonian
9.4.1 Gibbs’sches Paradoxon (1887)
Umformulierung von Gl. (28)
S(E, V,N) = Nk
[3
2+ log
(1
σ
V
NN
(4πm
3
(E
N
)) 32
)]Gl. (28) kann nicht richtig sein.
• Die Entropie ist (muss) extensiv (sein)
• Mit E/N der Energie pro Teilchen, V/N dem Volumen pro Teilchen ist Entropiein Gl. (28) nicht extensiv.
• Hier dürfen Sie wieder verwirrt sein. Oder sieht jemand einen Fehler in denobigen Rechnungen ?
Gibbs’sches Paradoxon
• Betrachte abgeschlossenes System mit zwei Teilsystemen A & B, TA = TB = T ,pA = pB
• Druck und Temperatur identisch
• Unterschiedliche Gase in A & B
106

• EntropieSvortotal = SA(T, VA, NA) + SB(T, VB, NB)
• Entferne Trennwand
Snachtotal = SA(T, VA + VB, NA) + SB(T, VA + VB, NB)
• Mit
S = Nk
(3
2+ log
V
σ(2πmkT )2/3
)=
3
2Nk +Nk log V + const.
ist Mischungsentropie
∆Smisch = Snachtotal − Svortotal
= NAk log(VA + VB) +NBk log(VA + VB)−NAk log VA −NBk log VB
∆Smisch = NAk log
(VA + VBVA
)+NBk log
(VA + VBVB
)> 0 (29)
Verwende nun identische Gase
Snachtotal = S(T, VA + VB, NA +NB)
Führt wiederum auf Gl. (29), Zunahme der Entropie
• Das kann aber nicht sein ! Zustände physikalisch nicht unterscheidbar
Makroskopisch findet kein Prozess statt
Herausziehen der Trennwand ist reversibler Prozess: ∆S = 0
Was ist falsch ?
• Klassisch sind Teilchen unterscheidbar, individuell, ”anmalbar” und damitdurchnumerierbar.
Die durchnummerierten Teilchen beider Seiten verteilen sich irreversibel überdas gesamte Volumen, Entropie nimmt zu
Quantenmechanische identische Teilchen sind ununterscheidbar
107

• LösungEs gibt N ! Möglichkeiten N identische Teilchen anzuordnen =⇒ Teile Zu-standssumme durch N !
Ω(E, V,N) =1
N !
σ(E, V,N)
σ0
Ergibt für das ideale Gas:
S(E, V,N) = Nk
[3
2+ log
(V
σ
(4πmE
3N
) 32
)]− k logN !︸ ︷︷ ︸
≈N logN−N
und damit
S(E, V,N) = Nk
[5
2+ log
(V
Nσ
(4πmE
3N
) 32
)](30)
• Unterschiedliche Gase
∆Smisch = NAk log
(VA + VBNA
)+NBk log
(VA + VBNB
)−NAk log
(VANA
)−Nbk log
(VBNB
)= NAk log
(VA + VBNA
NA
VA
)+NBk log
(VA + VBNB
NA
VB
)Gl. (29) bleibt unverändert ∆Smisch > 0
• Identische Gase
∆Smisch = (NA+NB)k log
(VA + VBNA +NB
)−NAk log
(VANA
)−Nbk log
(VBNB
)= 0
daVA + VBNA +NB
=VANA
=VBNB
Druck ändert sich nicht
Merke: Gibbs’scher Korrekturfaktor 1N !
von 1878
• korrigiert Widersprüche einer konsequent klassischen Statistischen Physik
• berücksichtigt die Ununterscheidbarkeit identischer quantenmechanischer Teil-chen
108

9.4.2 Quantenmechanische Berechnung
• Ideales Gas: Keine Wechselwirkung
Schrödinger Gleichung
Hψ(x1, . . . , xn) = Eψ(x1, . . . , xn)
vereinfacht sich zu
H =N∑i=1
hi(xi)
Aus Lösung von 1-Teilchen Schrödinger-Gleichung
hi(xi)φi(xi) = εiφi(xi)
ergibt sich N -Teilchen Wellenfunktion durch Produktansatz3
ψ(x1, . . . , xn) =N∏i=1
φi(xi)
• Dieser löst die N-Teilchen Schrödinger-Gleichung
Hψ =
(∑k
hk
)∏i
φi =
(∑k
εk
)∏i
φi = E∏i
φi = Eψ
mit Gesamtenergie E =∑
i εi
• Beispiel N = 2
(h1 + h2)φ1φ2 = h1φ1︸︷︷︸=ε1φ1
φ2 + φ1 h2φ2︸︷︷︸=ε2φ2
= (ε1 + ε2)φ1φ2 = Eφ1φ2
Ideales Gas:
• Volumen sei dreidimensionaler Kasten mit Kantenlänge L3Kap. 12 Quantengase wird zeigen, dass dies nicht richtig ist
109

• Ein-Teilchen Wellenfunktion
ψnx,ny ,nz = A sin kxx sin kyy sin kzz = A sinnxπx
Lsin
nyπy
Lsin
nzπz
Lnx, ny, nz = 1, 2, . . .
• Impulseigenwerte pro Richtung
pi =~
2Lni
• Ein-Teilchen Energie:
εnx,ny ,nz =(~k)2
2m=
~2
2m(k2x + k2
y + k2z) =
h2
8mL2(n2
x + n2y + n2
z)
• Gesamtenergie:
E =h2
8mL2
3N∑i=1
n2i
Das ist für N 1 wieder eine Kugel, jetzt mit Radius:
R =√
8mEL2/h2
• In analoger Rechnung zu oben ergibt sich:
ω(E, V,N) =
(V
h3
)N(2πmE)3N/2
3N/2 Γ(3N/2)
Beachte:
– Quantenmechanischer Zustand auf klassischen Phasenraum übertragenbraucht ein Volumen h3. Unschärferelation.
– Unterschied zum klassischen Fall: Wir zählen Zustände ab, damit entfälltdas (klassische4) Problem mit σ0, ω(E, V,N) ist dimensionslos
• Zustandssumme
Ω(E, V,N) =
(V
h3
)N(2πmE)3N/2
Γ(3N/2)
Hier braucht man technisch E ≤ Er ≤ E + ∆E
4Wortspielverdacht !
110

• Stirling-Näherung für Γ(3N/2)
• Gibbs-Korrektur 1N !
per Hand, Stirling-Näherung
• Und schließlich
S(E, V,N) = Nk
[5
2+ log
(V
Nh3
(4πmE
3N
) 32
)](31)
• Proportionalitätskontante σ in Gl. (30) ersetzt durch Volumen der Phasen-raumzelle h3
Gibbs-Korrektur immer noch per Hand, wird erst in Kap. 12 Quantengasegelöst
7. Week15Zustandssumme revisited, klassischer Limes
• Erinnereσ(E) =
∫d~qd~p δ(E −H(~q, ~p))
• Damit ZustandssummeΩ(E) =
σ(E)
σ0
Eine Zustandssumme sollte als Anzahl dimensionslos sein.
• Das kriegen wir jetzt geschenkt
Ω(E) =σ(E)
N !h3N
Das bleibt auch im klassischen Limes so
– N ! aus Ununterscheidbarkeit der Teilchen, solange sie ununterscheidbarsind
– h3N aus Phasenraumzelle
• Für sonstige quantenmechanische Effekte gibt es einen klassischen Limes, sieheKap. 11.4
111

• Betrachte thermische de Broglie-Wellenlänge:
λ =h√
2πmkT
Bedeutung: Mittlere Ortsunschärfe, die zur mittleren Energie εkin = 32kT , also
zum mittleren Impuls ¯|~ |p =√
2mεkin gehört
Damit wird Gl. (31) mit Volumen pro Teilchen v = VN
zu
S(E, V,N) = Nk
[5
2+ log
( vλ3
)]• Gültigkeitsbereich der klassischen Näherung
v
λ3 1 Mittleres Volumen pro Teilchen Unschärfevolumen
– geringe Dichten
– hohe Temperaturen
– große Massen
Beispiele:
– Gasförmiges Neon bei 100 K: v/λ3 ∼ 106
– Elektronen im metallischen Leiter bei 300 K: v/λ3 ∼ 10−3
9.5 Shannon-Information
• Shannon (1948) untersuchte die Information, die man in einem verrauschten”Kanal” übermitteln kann. Er führte das ”Bit” ein
• Wieviele Bits sind nötig, um eine bestimmte Information zu übertragen ?
• Zentrale Größe, die Shannon-Information oder Entropie:
I = −N∑i=1
pi log pi
112

Motivation:
• Betrachte Schachbrett, 64 Felder, eine Figur
• Frage: Wo ist die Figur ? pi = 164
• Effizienteste Strategie:
– Halbiere Schachfeld in rechts/links
– Frage: Ist Figur links ? pi = 132
– Je nach Antwort, halbiere rechte oder linke Hälfte
– Frage: Ist Figur oben ? pi = 116
– Und so weiter ...
– Nach 6. Frage: pi = 1
Aus anfänglicher Wahrscheinlichkeit – und damit Unsicherheit – pi wird nachn Fragen Sicherheit, p = 1
Die am Anfang fehlende oder am Ende gewonnene Information ist
2npi = 1
• Definiere InformationIi := n = − log2 pi
Die mittlere Information ist
I =∑i
piIi = −∑i
pi log2 pi
Beispiele:
• Würfel
pi =1
6
damit
113

I = −6∑i=1
1
6log
1
6= log 6
Betrachte manipulierten Würfel, der immer eine 6 liefert
I = 0 wegen 0 log 0 = 0 mit Satz von l’Hospital, 1 log 1 = 0
Bedeutung: Shannon-Information = Maß für Unwissenheit vor der Messungbzw. Neuigkeitswert (Unwissenheitsreduktion) nach der Messung5
• Für mikrokanonisches System gilt für Wahrscheinlichkeit der Mikrozustände r
pr =1
Ω
damit Information
I = −∑i
1
Ωlog
1
Ω= log Ω
Zusammenhang Information I und Entropie S
• Betrachte N Teilchen, die mit Wahrscheinlichkeit pi = ni/N inM unterschied-lichen Zuständen sind
M∑i=1
ni = N
• Ziel: Drücke S = k log Ω direkt durch die pi aus
• Gegeben die Besetzungszahlen ni, ist die mikrokanonische Zustandssumme
Ω(n1, n2, . . . nM) =N !∏i ni!
5Ich hoffe, dass diese Vorlesung nicht zu nieder-entropisch ist :-)
114

• Mit Stirling log n! ≈ n log n− n folgt:
S
k= N logN −N −
∑i
ni log ni +∑i
ni︸ ︷︷ ︸=N
= N logN −N∑i
pi logNpi
= N logN −N logN∑i
pi︸ ︷︷ ︸=1
−N∑i
pi log pi = −N∑i
pi log pi
Damit ist die EntropieS = −kN
∑i
pi log pi (32)
Statistische Mechanik als Anwendung der Informationstheorie
Eigenschaften:
• Es gilt I ≥ 0.
• Extremfall:I = 0, wenn pi = δi i0
Kenntnis über Mikrozustand (und Markrozustand) nimmt bei Messung nichtzu, wenn sich Mikrozusatnd mit Sicherheit in einem bestimmten, bekanntenZustand befindet.
• Die Funktion f(x) = −x log x ist konkav
f
(∑i
µixi
)≥∑i
µif(xi) für 0 ≤ µi ≤ 1,∑i
µi = 1
Ferner:f(0) = f(1) = 0
Sehr seltene und sehr häufige Ereignisse tragen wenig zur mittleren Informationbei
– Sehr seltene, weil sie selten sind
115
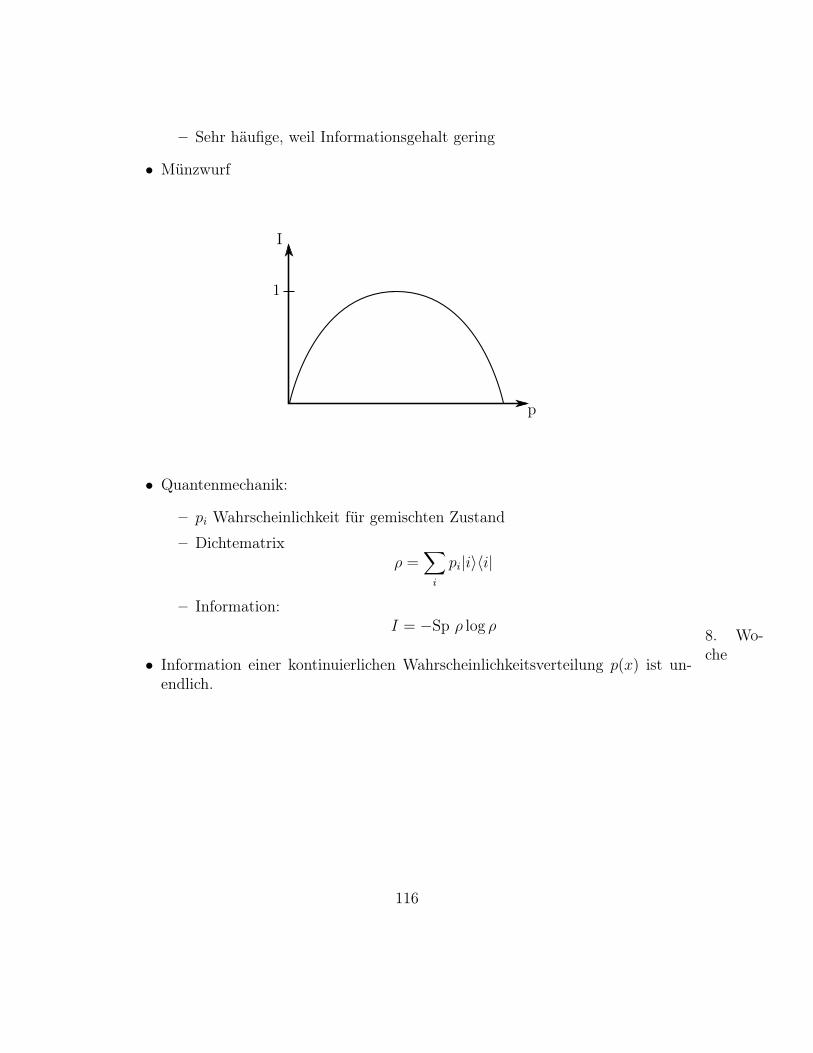
– Sehr häufige, weil Informationsgehalt gering
• Münzwurf
1
I
p
• Quantenmechanik:
– pi Wahrscheinlichkeit für gemischten Zustand
– Dichtematrixρ =
∑i
pi|i〉〈i|
– Information:I = −Sp ρ log ρ 8. Wo-
che• Information einer kontinuierlichen Wahrscheinlichkeitsverteilung p(x) ist un-endlich.
116

Sei pi = p(xi)∆x
I = −∑i
pi log pi = −∑i
p(xi)∆x log (pi(xi)∆x)
= −∑i
p(xi)∆x log p(xi)−∑i
p(xi)∆x log ∆x
∆x → dx→ 0,∑
∆x→∫dx
I = −∫p(x) log p(x)dx− log dx
∫p(x)dx︸ ︷︷ ︸
=1
→∞
Macht Sinn, da jede Realisierung einer kontinuierlichen Wahrscheinlichkeits-verteilung unendlich unwahrscheinlich ist6
Gilt in der klassischen Mechanik. Erst natürliche Beschränkung auf Phasen-raumzelle h3N durch die Quantenmechanik liefert vernünftige Definition
• Für kontinuierliche Verteilungen, betrachte differentielle Entropie
h = −∫p(x) log p(x)
Hat aber keine so schönen Eigenschaften:
– nicht skalierungsinvariant, macht zwar für gegebene skalierbare VerteilungSinn, aber Vergleich verschiedener Verteilungen schwierig
– Kann negativ werden
Landauer-Prinzip (1961)
• Thermodynamische Entropie hatte was mit Wärme zu tun.
• Findet sich das in der statistischen Informations-Definition wieder ?
• Ja ! Landauer-PrinzipDas Löschen eines Bits an Information führt zur Abgabe von Wärme
Q = kT log 2
Experimentelle Bestätigung: 2012, der Drops ist nicht gelutscht6Siehe Douglas Adams ”Per Anhalter durch die Galaxis”: Der unendliche Unwahrscheinlichkeits-
drive.
117

• Gibt theoretische Untergrenze für Verlustleistung von Computern
• Gilt nicht für Quantencomputer ...
9.6 Prinzip der maximalen Entropie
Jaynes (1957)
• Generell gilt für eine Wahrscheinlichkeitsverteilung notwendiger Weise nur dieEigenschaft:
N∑i=1
pi = 1
Andere Eigenschaften, z.B. 〈x〉 = µ, V ar(x) = σ2 zeichnen bestimmte Vertei-lungen aus
• Möchte man zusätzliche Eigenschaften haben, sollten auch wirklich nur dieseEigenschaften einbezogen werden.
• Anders formuliert: Jenseits der zusätzlichen Eigenschaften sollte die Verteilungmaximal überraschend, i.e. maximal-entropisch sein
• Führt auf ”Optimierung unter Nebenbedingungen” und damit Lagrange-Multiplikatoren
Beispiel 1:
• Betrachte Würfel. Wir wissen nur∑6
i=1 pi = 1
• Führt auf∂
∂pj
(−∑i
pi log pi − λ∑i
pi
)= 0
Ergebnis:− log pj − 1− λ = 0 =⇒ pj = e−1−λ
• λ ergibt sich aus Normierung∑6
i=1 pi = 1
=⇒ pi =1
6
Beispiel 2:
118

• Betrachte die Ereignisse, bei denen nach N Würfen die Summe der Augenzah-len ai grade einer vorgegebenen Zahl A entspricht
A =∑i
niai = N∑i
piai
bzw. für die mittlere Augenzahl 〈a〉 gilt:
〈a〉 =A
N
• Dies gibt weitere Nebenbedingung∑i
piai = 〈a〉
Lagrange-Multiplikatoren Ansatz:
∂
∂pj
(−∑i
pi log pi − λ1
∑i
pi − λ2
∑i
piai
)= 0
• Ergibt− log pj − 1− λ1 − λ2aj = 0
oderpj = e−1−λ1−λ2aj , pj ∝ e−λ2aj
eine Exponential-Verteilung
pj =e−λ2aj
Z
mit NormierungZ =
∑i
e−λ2ai
”Z” wie Zustandssumme
Bezug zur Statistischen Physik
119

• Sei nun
ai → εi Energieniveaus
A→ E =∑
i niεi = N〈ε〉 = const. Gesamtenergie
N =∑
i ni = const. Teilchenanzahl
• Zentrales Ergebnis der Statistischen Mechanik: Besetzungswahrscheinlichkeitenpi der Energieniveaus εi
pi =e−λ2εi
Z, Z =
∑i
e−λ2εi
• Bestimmung von λ2
Mit Gl. (32)
S = −kN∑i
pi log pi = −kN∑i
pi (−λ2εi − logZ)
= kλ2E + kN∑i
pi logZ = kλ2E + kN logZ
Mit (∂S
∂E
)V,N
=1
T= kλ2
erhalten wirλ2 = β =
1
kTund somit die Boltzmann-Verteilung für die Wahrscheinlichkeit der Besetzungder Energieniveaus:
pi =e−
εikT
Z=e−βεi
Z, Z =
∑i
e−βεi (33)
Merke:
• Im mikrokanonischen System (E, V,N) sind alle Mikrozustände r:(x1, . . . , xN , p1, . . . , pN) mit Energie E gleich wahrscheinlich
pr =1
Ω= const.
120

• Die Energien εi der einzelnen Teilchen i sind aber unterschiedlich.
• Die Besetzungswahrscheinlichkeit pi = ni/N der Energieniveaus ist durch denBoltzmann-Faktor
pi ∝ e−εikT
gegeben
Prinzip der maximalen Entropie hat in vielen Bereichen der Datenanalyse undModellbildung Anwendung, wenn es um ”vorurteilsfreie” maximal-entropischeAnsätze geht
Prinzip der maximalen Entropie für kontinuierliche Verteilungen, Variationsrechnung
• Bei gegebener Varianz maximiert Gaußverteilung die differentielle Entropie füralle p(x) mit Träger (−∞,∞). Sie ist die ”zufälligste” aller Verteilungen.
• Für kompakten Träger ist es die Gleichverteilung
9.7 3. Hauptsatz revisited
Frage: Wie verhalten sich Systeme bei kleinen Werten der inneren Energie ?
• Quantenmechanische Systeme haben üblicherweise genau einen Zustand mitniedrigst möglicher Energie E0, den Grundzustand.
• Im Grenzfall gilt somit
Ω(E) = 1 für E → E0
und damitS(E) = k log Ω(E) = 0 für E → E0 (34)
• Erinnere Zustandssumme des idealen Gases
Ω(E) ∝(E
N
)3N/2
Dieses gilt verallgemeinert für viele Systeme, mit f den Freiheitsgraden desSystems, γ den inneren Freiheitsgraden der Teilchen, z.B. Rotations- oder Vi-brationsfreiheitsgrade von Molekülen, siehe Kap. 11.3
121

Ω(E) ∝(E
f
)γf• Für Anregung von Freiheitsgraden steht die Energie E − E0 zur Verfügung
Ergibt für die Zustandssumme:
Ω(E) ∝ (E − E0)γf
Es folgt:
1
T:=
∂S(E)
∂E= k
∂ log Ω(E)
∂E= kγf
∂ log(E − E0)
∂E∝ γf
E − E0
E→E0→ ∞
Daher bedeutet E → E0 soviel wie T → 0
• Physikalische Bedeutung der Temperatur:
T ∝ E − E0
γf(35)
Temperatur: Verfügbare Energie pro Freiheitsgrad
• Aus Gln. (34, 35) folgt der 3. Hauptsatz:
limT→0
S(T ) = 0
Aber beachte: Frustrierte Zustände
• Betrachte System aus drei Spins mit jeweils paar-weiser negativer Kopplung
Hamiltonian:
H = s1s2 + s2s3 + s3s1
1 23
122

• Alle sechs Zustände sind gleich wahrscheinlich
limT→0
S(T ) = k log 6
9.8 Entropische Kräfte revisited
Erinnere: Freie Energie F = U − TS, isothermisches System im Wärmebad
• Minimierung der Energie gegen Maximierung der Entropie, widerstrebendeKräfte
• Betrachte Aggregationsmodell von zwei ununterscheidbaren Teilchen in Volu-men mit N Gitterzellen
• Dimer sei mit Bindungsenergie UD = −ε energetisch begünstigt
• Vergleiche freie Energie für Dimer mit der für zwei Monomere
• Pure Energieminimierung würde Dimer bevorzugen.
• Aber: Monomerzustand hat mehr mögliche Positionen =⇒ entropisch bevor-zugt
• Energie zur ”Überstimmung” von Bindungsenergie ε kommt aus Wärmebad
123

Betrachte Dimer
• Es gibt N − 1 Positionen für den Dimer, Bindungsenergie UD = −ε
• Damit freie Energie:
FD = UD − TSD = −ε− kT log(N − 1) ≈ −ε− kT logN
Betrachte Monomer
• Anzahl der Mikrozustände:
ΩM = Ωtotal − ΩD =N !
2! (N − 2)!− (N − 1) =
N(N − 1)
2− (N − 1)
• MitN(N − 1)
2− (N − 1) =
(N
2− 1
)(N − 1)
Freie Energie:
FM = UM︸︷︷︸=0
−TSM = −kT log
[(N
2− 1
)(N − 1)
]≈ −kT log
N2
2
• Stabilerer Zustand: Der mit der kleineren Freien Energie
124

• Bei niedriger Temperatur Dimer, bei hoher Monomer, bei T0 mit FD = FMgleich stabil.
−ε− kT0 log(N − 1) = −kT0 log
[(N
2− 1
)(N − 1)
]ε = kT0 log
(N
2− 1
)T0 ≈
ε
k log(N/2)
Lessons learned
• Statistische Entropie drückt Unwissenheit über Mikrozustand aus
• Verbindung von statistischer und thermodynamischer Entropie
• Entropie des ideales Gases in drei Schritten
– Klassisch
– Gibbs-Korrektur N ! aus Mischungsentropie
– Quantenmechanisch: Richtiger Normierungsfaktor h3N
Gibbs-Korrektur immer noch per Hand, Kap. 12
– Unzulänglichkeit der rein klassischen Statistischen Physik
• Verbindung zur Shannon’schen Informationstheorie
• Prinzip der maximalen Entropie, Boltzmann-Verteilung der Besetzungszahlen
• Verständnis des 3. Hauptsatzes
• Freie Energie und entropische Kräfte
8. Week15
10 GesamtheitenAuch als Ensemble bezeichnet.
125

Big picture: In der Thermodynamik hatten wir in Kap. 6 Thermodynamische Poten-tiale basierend auf makroskopischen Zustandsvariablen eingeführt
• Abgeschlossene Systeme:
Innere Energie: U(S, V,N)
Entropie: S(T, V,N)
• Geschlossene Systeme im Wärmebad
Freie Energie: F (T, V,N)
• Offene Systeme im Wärme- und Teuilchenbad
Großkanonisches Potential Φ(T, V, µ)
Jetzt: Blick darauf aus mikroskopischer Sicht.
Ausgangspunkt:
• Grundpostulat der Statistischen Mechanik: Im abgeschlossenen System sindalle Mikrozustände mit der Energie E gleich wahrscheinlich.
=⇒ Zustandssumme ergab sich durch einfaches Abzählen der Mikrozustände
• In nicht abgeschlossenen Systemen treten Mikrozustände mit unterschiedlicherEnergie auf. Mikrozustände sind nicht gleich wahrscheinlich.
=⇒ Zustandssumme muss dies durch eine Gewichtsfunktion ρ(q, p) berücksich-tigen.
10.1 Ergodenhypothese
• Will man den Erwartungswert 〈f〉 einer Größe f eines thermodynamischenSystems bestimmen, so ist der natürliche Ansatz das Zeit-Mittel :
〈f〉Z = limT→∞
1
T
∫ T
0
dtf(q(t), p(t)) (36)
Auf Grund der Komplexität der Dynamik nicht realistisch auszurechnen
126

• Aber: Die Dynamik des Systems induziert eine Dichte ρ(q, p) im Phasenraum
Damit sollte sich der Erwartungswert 〈f〉 auch durch das Schar-Mittel oderEnsemble-Mittel mit Phasenraumdichte ρ(q, p) bestimmen lassen:
〈f〉S =
∫dqdp
h3Nρ(q, p)f(q, p) (37)
• Intuition:
Schar-Mittel mittelt über viele ”Kopien” des makroskopischen Systems, die je-weils in einem bestimmten Mikrozustand gemäß der Zeitentwicklung sind.
• Ergodenhypothese besagt
〈f〉S = 〈f〉Z
Anschauung:
• N -maliges würfeln mit einem Würfel, Zeitmittel, sollte statistisch das selbeergeben wie einmaliges Würfeln mit N Würfeln, Scharmittel
Wann gilt Ergodenhypothese ?
• Boltzmann (1887): Jede Trajektorie geht durch jeden erlaubten Punkt des Pha-senraums
• Erwies sich als zu streng.
• Quasi-Ergodenhypothese: Jede Trajektorie, abgesehen von einer Menge vomMaß Null, kommt jedem erlaubten Punkt des Phasenraumes beliebig nahe
Anschaulich machen am Torus, kommensurabel, inkommensurabel, generisch
Die zwei typischen Ausnahmen:
• Abhängigkeit von den Anfangsbedingungen im Doppelmulden-Potential
127
q
W
W=E, E einzeichnen
Wird bei Phasenübergängen Kap. 14 relevant
• Algebraisch langsame Dynamik
– Typische Dynamik des Mischens:
〈x(t)x(t− τ)〉 ∼ e−λτ
Exponentiell: Immer schnell– Langsam: Algebraisches Mischen, speziell in Nicht-Gleichgewichts Syste-
men:〈x(t)x(t− τ)〉 ∼ τ−α
Beispiele: Gläser, komplizierte Energie-Landschaft
128

Zurück zur Statistischen Mechanik
• Zeitmittel viel zu kompliziert zum expliziten Ausrechnen
• ρ in Scharmittel i.a. auch zu kompliziert zum explizierten Ausrechnen
• Aber, Ergodenhypothese sagt, dass es die Dichte ρ gibt
• Es wird sich zeigen, dass die Zustandssumme die zentrale Größe ist
Das Programm im folgenden
• Betrachte drei verschiedene physikalische Situationen
• Leite Zustandssummen her
• Einmal physikalisch
• Einmal unter Anwendung der Methode der maximalen Entropie
• Rechne alles aus fürs ideale Gas
10.2 Mikrokanonische Gesamtheit
• Betrachte abgeschlossenes System
• Volumen und Teilchenzahl sind fest, Energie auf kleines Intervall [E,E + ∆]beschränkt
Physikalische Herleitung
• Grundpostulat: Alle Mikrozustände mit gleicher Energie sind gleich wahr-scheinlich
• Zustandssumme:
Ω(E, V,N) = Anzahl aller zugänglichen Mikrozustände r mit E ≤ Er ≤ E+∆
• Boltzmann-Faktor e−βεi gibt Wahrscheinlichkeit für Vorliegen eines Teilchensmit Energie εi, siehe Gl. (33)
• Entropie:S(E, V,N) = k log Ω(E, V,N)
129

• Alles weitere für ideales Gas folgt wie oben
Prinzip der maximalen Entropie:
• Einzige Nebenbedingung an pi:∑
i pi = 1
∂
∂pj
(−∑i
pi log pi − λ∑i
pi
)= 0
ergibt
− log pi − 1− λ = 0 → pi = e−1−λ =1
Ω9. Wo-che
10.3 Kanonische Gesamtheit
• Mikrokanonische Gesamtheit war über vorgegebene Energie in abgeschlossenemSystem definiert
• Jedes beliebige Sytem kann unter Aufnahme einer hinreichend großen Umge-bung in ein mikrokanonisches verwandelt werden
• Aber: Rechnungen in der mikrokanonischen Gesamtheit sind oft schwierig
• Ferner: Umgebung interessiert in der Regel nicht.
• In Praxis oft Temperatur vorgegeben.
• Betrachte sehr großes Wärmebad B, in das System S eingebettet ist. ErlaubtEnergieaustausch
• Erinnere freie Energie F mit natürlichen Variablen (T, V,N)
Berechnung der Wahrscheinlichkeitsverteilung der Mikrozustände r
• Im Gesamtsystem giltES + EB = E = const.
Bad groß gegen System:ESE
= 1− EBE 1
Da ES, EB 6= const., kann System S Mikrozustände r mit unterschiedlichenEnergien Er annehmen.
130

• Frage: Wahrscheinlichkeit pr für einen Mikrozustand r mit Energie Er ?
pr ∝ # Mikrozustände in Gesamtsystem S & B, für die S in Mikrozustand rmit ErDa Mikrozustand r in S ein bestimmter ist:
Wahrscheinlichkeit pr für Mikrozustand mit Er im System S:
pr ∝ ΩB(E − Er)
Da Er E ≈ EB und log ΩB extensiv, Taylorentwicklung von log ΩB
log ΩB(E − Er) ≈ log ΩB(E)− ∂ log ΩB(E)
∂E︸ ︷︷ ︸=β
Er + . . .
mitβ =
1
kT=
1
k
∂S
∂E=∂ log Ω(E)
∂E≈ ∂ log ΩB(E)
∂E
• Damitpr ∝ ΩB(E − Er) ≈ ΩB(E) e−βEr
Ergibt
pr =1
Ze−βEr Boltzmann-Faktor
Z =∑r
e−βEr Kanonische Zustandssumme
• Merke: Ableitung der Entropie des Wärmebades bringt dessen Temperatur β =1kT
ins Spiel.
• Beachte:
– Kanonische Gesamtheit: Boltzmann-Faktor e−βEr gibt Wahrscheinlichkeitfür Mikrozustand r mit Energie Er
– Für feste Energie Er des Systems S sind alle zugänglichen Mikrozuständer wieder gleich wahrscheinlich. Mikrokanonische Situation.
131

– Mikrokanonisches Gesamtheit: Alle Mikrozustände r gleich wahrschein-lich. Boltzmann-Faktor e−βεi gibt Wahrscheinlichkeit für Vorliegen einesTeilchens mit Energie εi, siehe Gl. (33)
– Ab jetzt r → i
• Thermodynamische Größen f ergeben sich in kanonischer Gesamtheit als Mit-telwerte:
〈f〉 =∑i
e−βEi
Zf(Ei)
Ganz im Sinne des Schar-Mittels
Ableitung über das Prinzip der maximalen Entropie
• Volumen und Teilchenzahl sind fest
• Temperatur ist vorgegeben, Temperatur bestimmt Erwartungswert der Energie〈E〉
• Nebenbedingungen pi:∑
i pi = 1, E:∑
i piEi = 〈E〉
∂
∂pj
(−∑i
pi log pi − λ∑i
pi − β∑i
piEi
)= 0
ergibt
− log pi − 1− λ− βEi = 0 → pi =1
Ze−βEi
So einfach kann das gehen :-) Klausur
Die kanonische Zustandssumme
Z =∑i
e−βEi
mathematisch nur ein Normierungsfaktor, ist zentral für die Statistische Mechanik
Beispiele:
132

• Innere Energie
U ist Mittelwert der Mikrozustandsenergien des Systems
U = 〈E〉 =∑i
piEi =1
Z
∑i
e−βEiEi
Mit∂Z
∂β= −
∑i
e−βEiEi
und∂ logZ
∂β= − 1
Z
∑i
e−βEiEi
ist
U = − 1
Z
∂Z
∂β= −∂ logZ
∂β= kT 2 ∂ logZ
∂T
• Entropie
Erinnere Gl. (32), allerdings ohne FaktorN , da hierN -Teilchen Mikrozustände,nicht Einteilchenzustände
S = −k∑i
pi log pi
= − kZ
∑i
e−βEi loge−βEi
Z
= kβ1
Z
∑i
Eie−βEi
︸ ︷︷ ︸=〈E〉=U
+k
ZlogZ
∑i
e−βEi︸ ︷︷ ︸=Z
=U
T+ k logZ = kT
∂ logZ
∂T+ k logZ
• Freie Energie
F = U − TS = U − T(U
T+ k logZ
)ergibt
133

F = −kT logZ
Bringt mikroskopische Zustandssumme mit makroskopischem thermodynamischemPotential zusammen
Analogie:
– Freie Energie F (T, V,N) ist thermodynamisches Potential zur kanonischenZustandssumme Z(T, V,N)
– Entropie S(E, V,N) ist thermodynamisches Potential zur mikrokanoni-schen Zustandssumme Ω(E, V,N)
Kanonische Zustandssumme: Die wichtigste Größe in der Statistischen Mechanik
Merke: Alles folgt aus einer Normierungsgröße !
Zusammenhang kanonische und mikrokanonische Zustandssumme:
• Kanonische Zustandssumme
Z =∑i
e−βEi
Index i läuft über alle Mikrozustände mit Energie Ei, inklusive Mehrfachzäh-lungen
• Fasse Mikrozustände gleicher Energie zusammen, so folgt
Z =∑k
Ω(Ek) e−βEk (38)
mit Ω(Ek) der Anzahl der Mikrozustände mit Energie Ek, Index k läuft überEnergiewerte. Keine Mehrfachzählungen mehr.
• Ω(Ek) ist mikrokanonische Zustandssumme für Energie Ek
• Merke: Kanonische Zustandssumme ist thermische Mittlung über die mikroka-nonische Zustandssummen
134

• Gl. (38) mit Argumenten, V,N unterdrückt
Z(β) =∑k
Ω(Ek) e−βEk
stellt eine diskretisierte Laplace-Transformation oder Z-Transformation dar:
Laplace-Transformation:
f(s) =
∫ ∞0
dx f(x)e−sx, s ∈ C
– Wird oft zum Lösen von Differentialgleichungen verwendet
– Mits = iω, ω ∈ R
ergibt sich Fourier-Transformation
Ideales Gas
• Kontinuumslimit
i → (q1, . . . , qN , p1, . . . , pN)∑i
→ 1
h3N
∫dqdp
Z =1
h3N
∫dqdp e−βH(q,p)
• N -Teilchen Zustandssumme aus 1-Teilchen Zustandssumme
Z(β, V,N) =1
N !Z(β, V, 1)N
Gibbs-Korrektur nach wie vor per Hand.
• 1-Teilchen Zustandssumme
Z(β, V, 1) = V1
h3
∫dp e−βp
2/(2m) = V
(2πm
βh2
) 32
=V
λ3
Erinnere: λ: Thermische de Broglie Wellenlänge
135

• N -Teilchen Zustandssumme
Z(β, V,N) =1
N !
V N
λ3N
Erinnere, wie schmerzhaft die Rechnung in der mikrokanonischen Gesamtheitwar, Kap. 9.4
• Freie Energie
F = −kT logZ = −NkT
(1 + log
(V
N
(2πmkT
h2
)3/2))
damit folgt:
p = −(∂F
∂V
)T,N
=NkT
V
S = −(∂F
∂T
)V,N
= Nk
(5
2+ log
(V
N
(2πmkT
h2
)3/2))
Identische Ergebnisse wie in der mikrokanonischen Gesamtheit9. Week1510. Halb-woche
10.4 Großkanonische Gesamtheit
Betrachte offenes System, bei dem sowohl Energie als auch Teilchen ausgetauschtwerden können.
Physikalische Ableitung der Zustandssumme
• In Analogie zur Ableitung der kanonischen Gesamtheit, S: System, B: Bad
ES + EB = E, NS +NB = N
Wahrscheinlichkeit pi,Ni für Mikrozustand mit Energie Ei und Teilchenzahl Ni
pi,Ni ∝ ΩB(E − Ei, N −Ni)
136

• Wieder Taylor-Entwicklung:
log ΩB(E − Ei, N −Ni) = log ΩB(E,N)− ∂ log ΩB
∂EEi −
∂ log ΩB
∂NNi + . . .
• Mit
∂ log ΩB
∂E=
1
k
∂S
∂E=
1
kT= β
∂ log ΩB
∂N=
1
k
∂S
∂N=−µkT
= −βµ
Damit folgt:
pi,Ni =1
Ye−β(Ei−µNi) Wahrscheinlichkeitsverteilung
Y (T, V, µ) =∑i
e−β(Ei−µNi) Großkanonische Zustandssumme
Ableitung mit der Maximum-Entropie Methode:
• Die Erwartungswerte von Energie E und Teilchenzahl N sind vorgegeben
• Nebenbedingungen:
∑i
pi = 1,∑i
piEi = 〈E〉,∑i
piNi = 〈N〉
Damit
∂
∂pj
(−∑i
pi log pi + λ∑i
pi − β∑i
piEi + βµ∑i
piNi
)= 0
Ergibt:
− log pi − 1 + λ− βEi + βµNi = 0 → pi =1
Ye−β(Ei−µNi)
Oft benutzt: Fugazität z = eβµ
137

• Großkanonische Zustandssumme:
Y (T, V, µ) =∑i
e−β(Ei−µNi) =∞∑N=0
eβµN∑i
e−βEi
Erinnere kanonische Zustandssumme:
Z(T, V,N) =∑i
e−βEi
ergibt wieder diskretisierte Laplace-Transformation:
Y (T, V, µ) =∞∑N=0
Z(T, V,N) eβµN
• Großkanonische Zustandssumme: Mittelung über kanonische Zustandssummenmit unterschiedlichen Teilchenanzahlen
Analog zu: Kanonische Zustandssumme: Thermische Mittlung über mikroka-nonische Zustandssummen
Aus großkanonischer Zustandssumme folgen thermodynamische Größen:
• Innere Energie
∂ log Y
∂β=
1
Y
∑i
e−β(Ei−µNi)(−Ei + µNi)
= −〈Ei〉+ µ〈Ni〉 = −U + µN
Damit
U = −∂ log Y
∂β+ µN
• Teilchenzahl
∂ log Y
∂µ=
1
Y
∑i
e−β(Ei−µNi)βNi = β〈Ni〉 = βN
N =1
β
∂ log Y
∂µ= z
∂ log Y
∂z
138

• Entropie
S = −k∑i
pi log pi = −k∑i
pi(−β(Ei − µNi)− log Y )
= kβ∑i
piEi︸ ︷︷ ︸〈Ei〉=U
−kβµ∑i
piNi︸ ︷︷ ︸〈Ni〉=N
+k log Y∑i
pi︸ ︷︷ ︸=1
DamitS =
1
T(U − µN) + k log Y
Beziehung zum Großkanonisches Potential Φ, Kap. 6.6
• Erinnere Legendre-Transformation
Φ = U − TS − µN
Φ = U − T(
1
T(U − µN) + k log Y
)− µN
Φ = −kT log Y
in Analogie zu F = −kT logZ
• Euler-Gleichung
Φ = U − TS − µN = −pV =⇒ Y = exp
(pV
kT
)Betrachte nicht-wechselwirkende ununterscheidbare Teilchen
• Kanonische Zustandssumme
Z(T, V,N) =1
N !ZN(T, V, 1)
Großkanonische Zustandssumme
Y (T, V, µ) =∑N
1
N !(zZ(T, V, 1))N = exp (zZ(T, V, 1))
• Großkanische Zustandssumme folgt direkt aus kanonischer Ein-Teilchen Zu-standssumme
139

• Beispiel ideales Gas:
Z(T, V, 1) =V
λ3, λ =
h√2πmkT
Φ(T, V, µ) = −kT log Y = −kTeβµV(
2πmkT
h2
) 32
10.5 Äquivalenz der Gesamtheiten
Im thermodynamischen Limes wird alles gleich
Gesamtschau ideales Gas• Mikrokanonische Gesamtheit
S = k log Ω = kN
(5
2+ log
((V
N
)(4πmE
Nh2
)3/2))
1
T=
∂S
∂E= Nk
3
2
1
E=⇒ E =
3
2NkT
p
T=
∂S
∂V=Nk
V=⇒ pV = NkT
• Kanonische Gesamtheit
Z(T, V,N) =1
N !
(V
λ3
)NE = − ∂
∂βlogZ = kT 2 ∂
∂TlogZ =
3
2NkT
p
kT=
∂
∂VlogZ =
N
V=⇒ pV = NkT
• Großkanonische Gesamtheit
Y (T, V, z) =∑N
zNZ(T, V,N) = expzV
λ3
N = z∂
∂zlog Y =
zV
λ3= log Y
E = − ∂
∂βlog Y + µN =
3
2kT
zV
λ3=
3
2NkT
pV
kT= log Y =
zV
λ3= N =⇒ pV = NkT
140
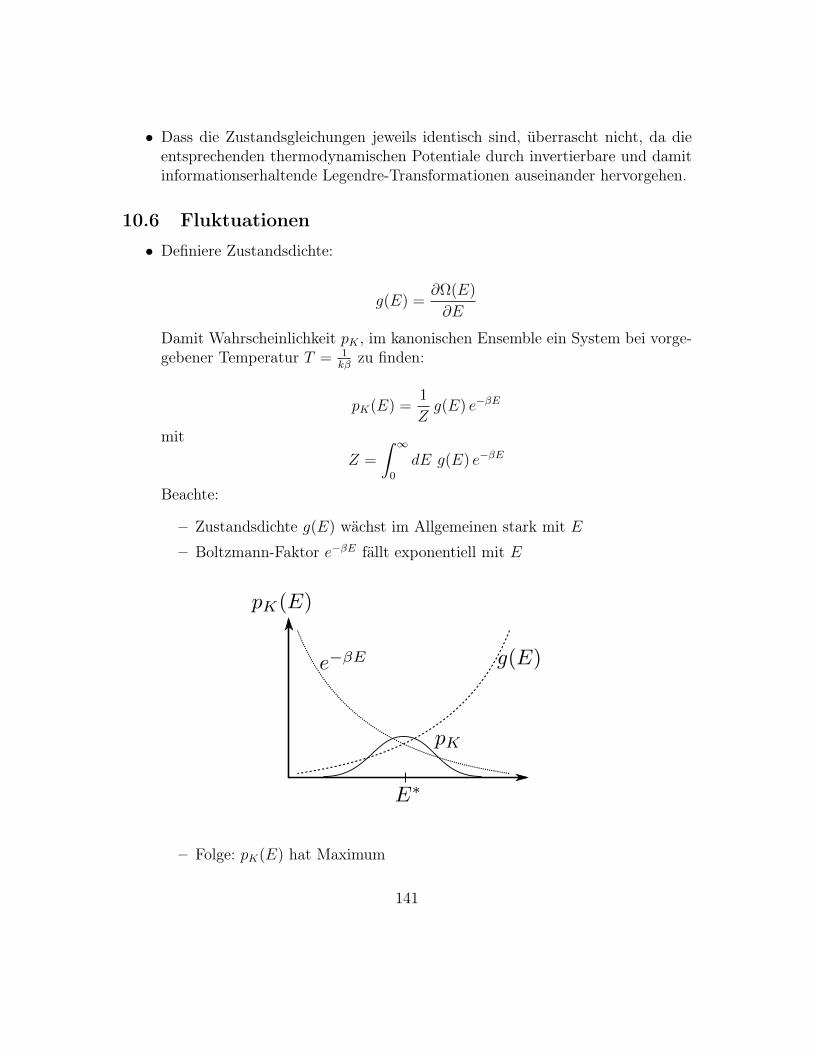
• Dass die Zustandsgleichungen jeweils identisch sind, überrascht nicht, da dieentsprechenden thermodynamischen Potentiale durch invertierbare und damitinformationserhaltende Legendre-Transformationen auseinander hervorgehen.
10.6 Fluktuationen
• Definiere Zustandsdichte:
g(E) =∂Ω(E)
∂E
Damit Wahrscheinlichkeit pK , im kanonischen Ensemble ein System bei vorge-gebener Temperatur T = 1
kβzu finden:
pK(E) =1
Zg(E) e−βE
mitZ =
∫ ∞0
dE g(E) e−βE
Beachte:
– Zustandsdichte g(E) wächst im Allgemeinen stark mit E
– Boltzmann-Faktor e−βE fällt exponentiell mit E
– Folge: pK(E) hat Maximum
141

• Maximumsbedingung
∂pK(E)
∂E=
1
Z
(∂g
∂E− gβ
)e−βE
!= 0
oder1
g
∂g
∂E
∣∣∣∣E=E∗
=1
kT
Mit ∂Ω(E) = g(E)∂E
∂ log Ω
∂E
∣∣∣∣E=E∗
=1
kToder
∂S
∂E
∣∣∣∣E=E∗
=1
T
Das macht Sinn, da es ja grade die Vorschrift war, in der mikrokanonischenGesamtheit die Temperatur aus der Entropie zu berechnen
• Wahrscheinlichste Energie entspricht Mittelwert im kanonischen Ensemble
E∗ =1
Z
∫ ∞0
dE g(E) e−βEE = − 1
Z
∂Z
∂β= − ∂
∂βlogZ = U
Differenziere die Gleichung nach β, beachte Z = Z(β)
∂U
∂β= − 1
Z
∫ ∞0
dE g(E) e−βE E2 +1
Z2
(∫ ∞0
dE g(E) e−βEE
)2
= −(〈E2〉 − 〈E〉2)
Erinnere Varianzσ2 = 〈x2〉 − 〈x〉2
• Damitσ2E = −∂U
∂β= kT 2∂U
∂T= kT 2CV (39)
Fluktuationen hängen von Wärmekapazität ab.
• Relative SchwankungenσE〈E〉
=
√kT 2CVU
Wärmekapazität und innere Energie sind extensiv, i.e. ∝ N
142

σE〈E〉
= O(
1√N
)Erinnere Gesetz der großen Zahl
• Damit
limN→∞
pK(E) = pMK(E) = δ(E − 〈E〉)
• Analoge Rechnung gilt für Beziehung kanonischer und großkanonischer Ge-samtheit.
Lessons learned
• Ergodenhypothese als Grundlage der Theorie der Gesamtheiten
• Normierungsgröße Zustandssumme von zentraler Bedeutung
• Mikrokanonische Gesamtheit ⇐⇒ Abgeschlossenes System
• Kanonische Gesamtheit ⇐⇒ Austausch von Wärme, isothermes System
• Großkanonische Gesamtheit ⇐⇒ Austausch von Wärme und Teilchen
• Verteilungen folgen (auch) aus Maximum-Entropie Prinzip
• Im thermodynamischen Limes werden die Gesamtheiten identisch
11 Anwendungen
11.1 Maxwell’sche Geschwindigkeitsverteilung
• Hatten wir bisher für ideales Gas, 2. Übung
• Gegeben Gas mit Hamilton-Funktion
H(q, p) =N∑i=1
~pi2m
+ V (~q1, . . . , ~qN)
143

Wie ist Wahrscheinlichkeit, ein Teilchen im Impuls-Intervall p < |~pi| < p + dpzu finden ?
• Kanonische Wahrscheinlichkeitsdichte
ρ(q, p) =1
Ze−βH(q,p)
• Integriere über alle Ortskoordinaten und N − 1 Impulskoordinaten, ergibtMarginal-Verteilung für Impuls eines einzelnen Teilchen:
p(~p) =1
(2πmkT )3/2e−
~p2
2mkT
Für Wahrscheinlichkeit, dass Impulsbetrag in [p, p + dp] liegt, integriere überKugelschale
p(p) =4πp2
(2πmkT )3/2e−
p2
2mkT
• Beachte: Gilt klassisch nicht nur für ideale Gase, sondern auch für beliebigeinterne und äußere Potentiale.
• Quantenmechanisch: Abhängigkeit der Impulsverteilung vom Potential
11.2 Barometrische Höhenformel
• Betrachte Gas im Schwerefeld der Erde:
H(q, p) =N∑i=1
p2i
2m−mgqi +
1
2
∑i 6=j
Wij(qi − qj)
• Integriert man in
ρ(q, p) =1
Ze−βH(q,p)
über alle Impuls- und N − 1 Ortskoordinaten, so folgt barometrische Höhen-formel:
p(~q) = const e−m~g~q/kT
144

• Beachte: Für Anwendung Temperaturabhängigkeit zu berücksichtigen
Temperatur nimmt in guter Näherung linear mit der Höhe ab
∆h = const(
1 +1
273(T1 − T2)
)(log p1 − log p2),
11.3 Gleichverteilungs- und Virialsatz
• Sei x = (q1 . . . , q3N , p1, . . . , p3N) der klassische Mikrozustand, H(x) dieHamilton-Funktion und f(x) eine beliebige Observable.
• Dann gilt ⟨f(x)
∂H(x)
∂xi
⟩= kT
⟨∂f(x)
∂xi
⟩• Beweis für die kanonische Gesamtheit:
Das Integral einer Ableitung verschwindet, wenn die abgeleitete Größe für großeArgumente verschwindet, Beweis per partieller Integration
1
Z
∫d6Nx
∂
∂xif(x) e−
H(x)kT =
⟨∂f(x)
∂xi
⟩− 1
kT
⟨f(x)
∂H(x)
∂xi
⟩= 0
• Betrachte f(x) = xj, so folgt der Gleichverteilungssatz⟨xj∂H(x)
∂xi
⟩= δijkT
• Betrachte H(x) = Hkin(p1, . . . , p3N) + V (q1, . . . , q3N) mit
Hkin =3N∑i=1
p2i
2m
Mit xj = pj gilt ⟨pi∂H(x)
∂pi
⟩=
⟨p2i
m
⟩= kT
〈Ekin〉 =
⟨p2i
2m
⟩=
1
2kT
145

Jeder Impulsfreiheitsgrad liefert einen Beitrag von 12kT zur kinetischen Energie.
Insgesamt:
3N∑i=1
⟨pi∂H(x)
∂pi
⟩= 2 〈Ekin〉 = 3NkT
〈Ekin〉 =3
2NkT
Erinnere ideales Gas, innere Energie = kinetische Energie
U =3
2NkT
• Hängt das Potential quadratisch von q ab
V (q1, . . . , q3N) =3N∑i=1
αiq2i =
3N∑i=1
Vi(qi)
folgt mit xj = qj ⟨qi∂H(x)
∂qi
⟩= 2 〈Vi〉 = kT
Somit analog zu oben
〈V 〉 =3
2NkT
• Merke: Jeder quadratische Term in der Hamilton-Funktion trägt 12kT zur Ener-
gie bei
Allgemeine homogene Potentiale
• Ist das Potential homogen vom Grade n
V (λq) = λnV (q), V (qi) = αqni
so folgt:
146

⟨qi∂V (q)
∂qi
⟩= nα 〈qni 〉 = kT
und〈V 〉 =
3
nNkT
Bei quadratischer q-Abhängigkeit erhält man:
E =1
2fNkT
und somit:
CV =∂E
∂T=
1
2fNk
Intuition:
• Temperatur entspricht mittlerer kinetischer Energie pro Teilchen
• Fügt man System Energie zu, so trägt ein Teil zur Erhöhung der kinetischenEnergie - und damit der Temperatur - bei
• Der andere dient der Erhöhung der potentiellen Energie
• Beispiel harmonischer Oszillator
– Virialtheorem:〈E〉 = 2〈Ekin〉 = 2〈Epot〉
– Nur die Hälfte der Energie geht in Temperaturerhöhung
– Im Vergleich zum idealen Gas muss das Doppelte an Energie zugeführtwerden, um die gleiche Temperaturerhöhung zu erreichen.
f : Thermodynamische Freiheitsgrade
• Festkörper, in dem Atome durch lineare elastische Kräfte gebunden ist: f = 6
H =~p2
2m+k
2~q2
147

• Zweiatomiges Gas: f = 7, 3 für Schwerpunkt, 2 für Rotation, 2 für Vibration
H =1
2m
(p2x + p2
y + p2z
)+
1
2I
(p2φ
sin2 θ+ p2
θ
)+
(p2ξ
2mr
+mrω
2ξ2
2
)• Freiheitsgrade pro Teilchen in der Klassischen Mechanik: 6
• Freiheitsgrade in der Statistischen Physik: Angeregte Freiheitsgrade, im Unter-schied zur Klassischen Mechanik. Z.B. Ortsfreiheitsgrade im idealen Gas nichtanregbar
Problem:
• Jeder thermodynamische Freiheitsgrade trägt gleich bei, unabhängig von z.B.Massen und Steifheit elastischer Bindungen
• Betrachte Gas aus viel-atomigen Molekulen
– Sehr viele ”Verformungs”-Freiheitsgrade → CV sollte sehr groß
– Experimentell nicht bestätigt
– Ad hoc Lösung: ”Friere” sehr starre Freiheitsgrade bei niedrigen Tempe-raturen ”ein”
– Erst die Quantenmechanik kann dieses Dilemma lösen
11.4 Thermodynamische Freiheitsgrade in Quantensystemen
Betrachte eindimensionalen quantenmechanischen Oszillator
• Kanonische Zustandssumme
Z =∞∑n=0
e−βEn , En =
(n+
1
2
)~ω
Damit
Z = e−β~ω2
∞∑n=0
(e−β~ω
)n=
e−β~ω2
1− e−β~ω
Historischer Moment: Unsere erste nicht triviale Zustandssumme !
148

• Nutze den Formalismus aus Kap. 10.3:
E = − ∂
∂βlogZ
= − ∂
∂β
(−β~ω
2− log
(1− e−β~ω
))=
~ω2
+~ω e−β~ω
1− e−β~ω
=~ω2
+~ω
eβ~ω − 1
• Damit folgt für:
T → 0 E → 12~ω CV → 0 : Einfrieren von Freiheitsgraden
T →∞ E → 12~ω + ~ω
1+β~ω−1= 1
2~ω + kT CV → k : klassisches Ergebnis für f = 2
• Beachte: Zweistufige klassische Näherung
1. CV von 0 nach CV = k
2. Teilchen unterscheidbar
Zustandssumme molekularer idealer Gase
• Zusätzlich zur Molekülbewegung auch innere Freiheitsgrade anregbar
– Anregung der Elektronen
– Schwingungen
– Rotationen
• Wenn Freiheitsgrade separieren, ist Gesamt-Hamiltonian additiv
H = Htrans +Hel +Hvib +Hrot
149

– Translation und innere Anregung separieren exakt durch Abkopplung derSchwerpunktsbewegung
– Born-Opppenheimer-Näherung zur Abkopplung von Atomkern und Elek-tronen gut auf Grund des großen Massenunterschiedes
– Schwingungen und Rotationen separieren für kleine Schwingungsauslen-kungen, sonst beeinflussen sie das Trägheitsmoment der Rotation
• Gilt Separation, so ergibt sich Zustandssumme als Produkt der einzelnen Zu-standssummen
Z(T, V, 1) = ZtransZelZvibZrot
– Ztrans = V/λ3, die Zustandssumme des idealen Gases
– Zel = e−βε0 . Nur Grundzustand besetzt, da 300K ∼ 25 meV, Anregungs-energien liegen im Bereich von eV
– Zvib: Betrachte zweiatomiges Molekül mit kleinen Schwingungsauslenkun-gen in harmonischer Näherung: En = (n+ 1
2)~ω.
Zvib =∞∑n=0
e−β(n+1/2)~ω =e−β~ω/2
1− e−β~ω
– Zrot hängt von Geometrie des Moleküls ab, im allgemeinen drei Trägheits-achsenBetrachte zweiatomiges Molekül
∗ Trägheitsdmoment I∗ Energieeigenwerte
Erot =~2
2IJ(J + 1), J = 0, 1, . . . , (2J + 1)− fache Entartung
∗ Zustandssumme
Zrot =∞∑J=0
(2J + 1) exp
(−β ~
2
2IJ(J + 1)
)• Gesamtzustandssumme
150
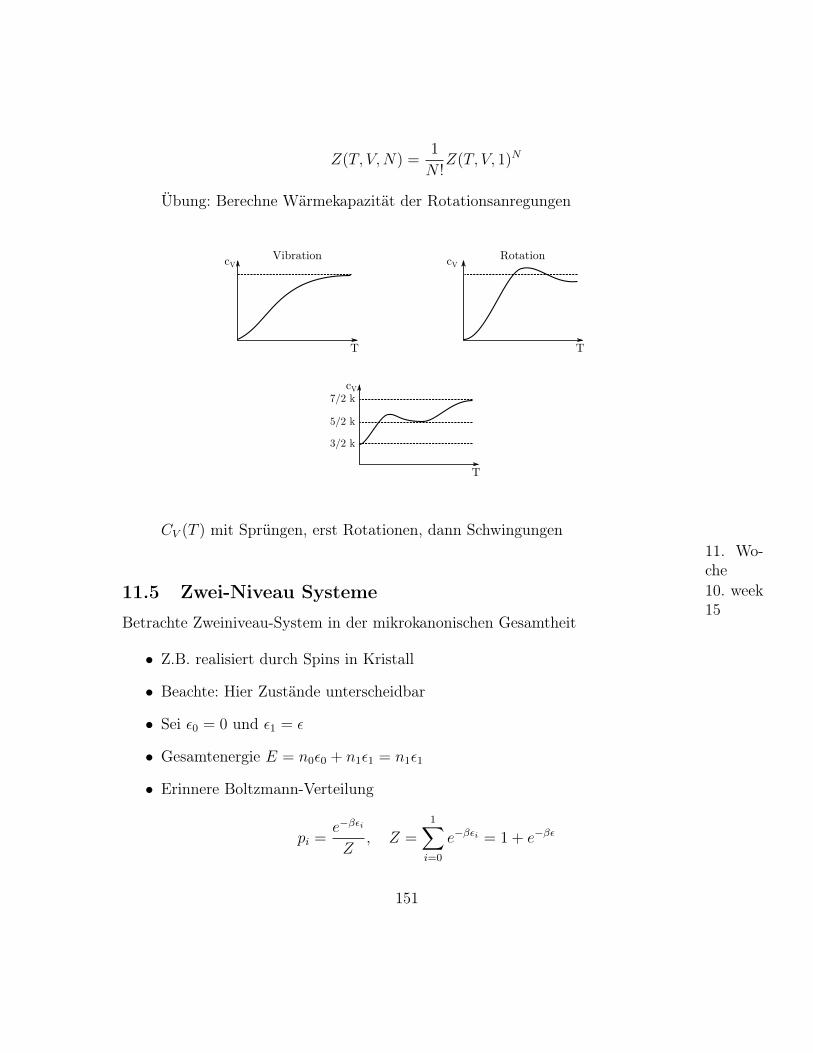
Z(T, V,N) =1
N !Z(T, V, 1)N
Übung: Berechne Wärmekapazität der Rotationsanregungen
CV (T ) mit Sprüngen, erst Rotationen, dann Schwingungen11. Wo-che10. week15
11.5 Zwei-Niveau Systeme
Betrachte Zweiniveau-System in der mikrokanonischen Gesamtheit
• Z.B. realisiert durch Spins in Kristall
• Beachte: Hier Zustände unterscheidbar
• Sei ε0 = 0 und ε1 = ε
• Gesamtenergie E = n0ε0 + n1ε1 = n1ε1
• Erinnere Boltzmann-Verteilung
pi =e−βεi
Z, Z =
1∑i=0
e−βεi = 1 + e−βε
151
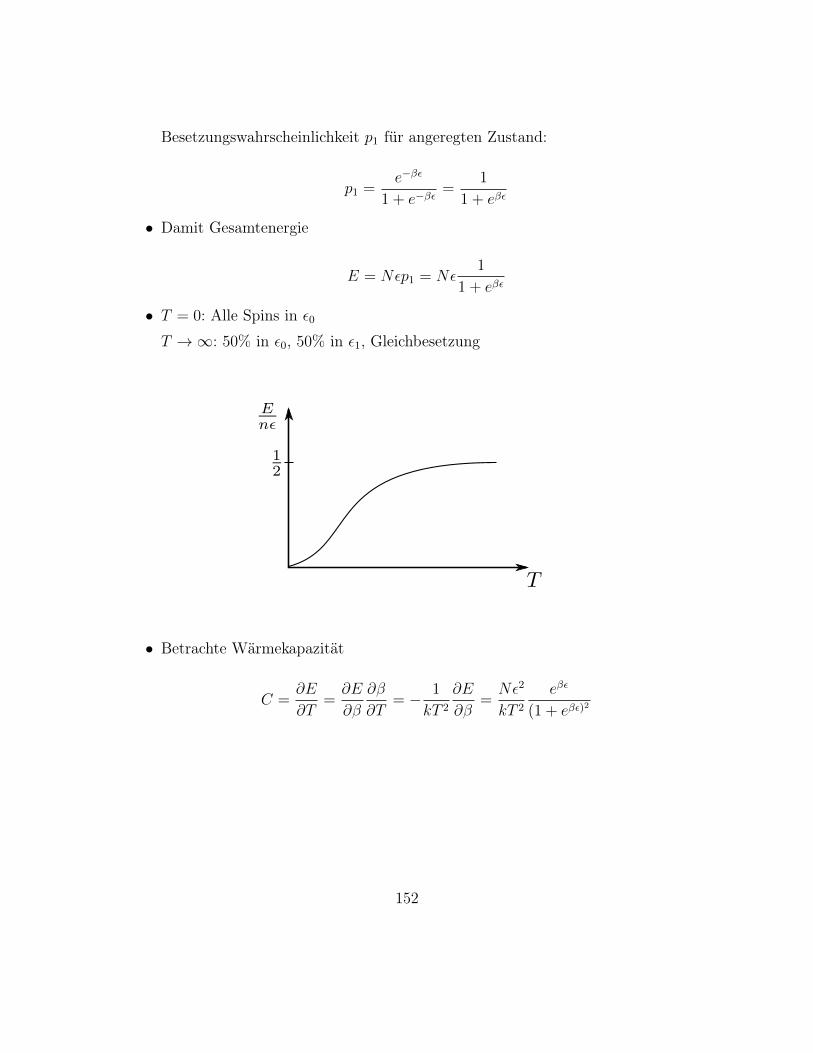
Besetzungswahrscheinlichkeit p1 für angeregten Zustand:
p1 =e−βε
1 + e−βε=
1
1 + eβε
• Damit Gesamtenergie
E = Nεp1 = Nε1
1 + eβε
• T = 0: Alle Spins in ε0T →∞: 50% in ε0, 50% in ε1, Gleichbesetzung
• Betrachte Wärmekapazität
C =∂E
∂T=∂E
∂β
∂β
∂T= − 1
kT 2
∂E
∂β=Nε2
kT 2
eβε
(1 + eβε)2
152
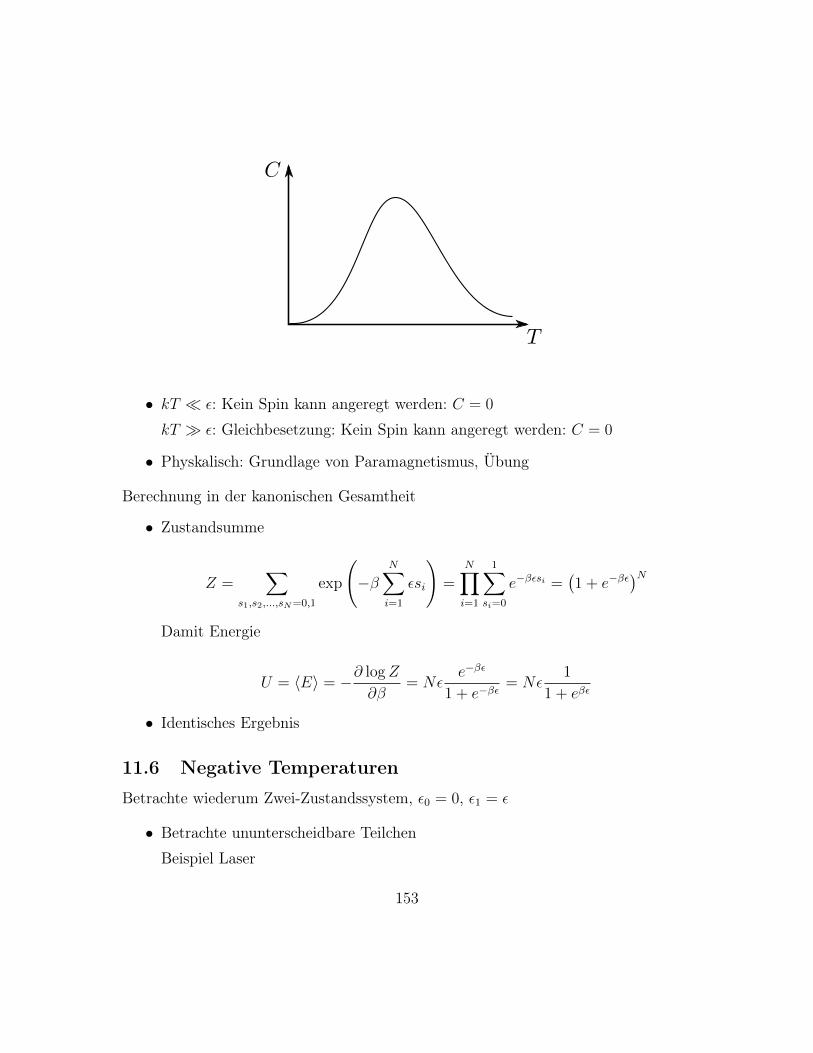
• kT ε: Kein Spin kann angeregt werden: C = 0
kT ε: Gleichbesetzung: Kein Spin kann angeregt werden: C = 0
• Physkalisch: Grundlage von Paramagnetismus, Übung
Berechnung in der kanonischen Gesamtheit
• Zustandsumme
Z =∑
s1,s2,...,sN=0,1
exp
(−β
N∑i=1
εsi
)=
N∏i=1
1∑si=0
e−βεsi =(1 + e−βε
)NDamit Energie
U = 〈E〉 = −∂ logZ
∂β= Nε
e−βε
1 + e−βε= Nε
1
1 + eβε
• Identisches Ergebnis
11.6 Negative Temperaturen
Betrachte wiederum Zwei-Zustandssystem, ε0 = 0, ε1 = ε
• Betrachte ununterscheidbare Teilchen
Beispiel Laser
153

• Erinnere1
T=∂S
∂U
• Energie des Systems, n Anzahl in ε1:
U = nε, n =U
ε
• Anzahl der Mikrozustände
Ω =N !
n!(N − n)!
Betrachte Entropie, verwende Stirling’sche Formel:
S
k= log Ω = −n log
n
N− (N − n) log
N − nN
1
T= k
∂ log Ω
∂U= k
∂ log Ω
∂n
∂n
∂U
• Da ∂n∂U
= 1/ε, folgt
1
T=
k
ε
(− log
n
N− 1 + log
N − nN
+ 1
)= −k
εlog
n/N
1− n/N= −k
εlog
U/(Nε)
1− U/(Nε)
=k
εlog
AGAa
mit AG Anteil der Teilchen im Grundzustand, Aa Anteil der Teilchen im ange-regten Zustand.
154

• Betrachte positive Temperaturen
– Bei kleiner Energie sind die meisten Teilchen im Grundzustand
AGAa
> 1, logAGAa
> 0
Temperatur ist positiv– Bei Anwesenheit eines Bades wird das System Energie aufnehmen, um
Entropie zu vergrößern.– Merke: Systeme mit positiver Temperatur absorbieren Energie um Entro-
pie zu erhöhen
• Betrachte T =∞
– Es giltAGAa
= 1, logAGAa
= 0
– Entropie kann durch Energieaufnahme nicht zunehmen
• Betrachte negative Temperaturen
– Es giltAGAa
< 1, logAGAa
< 0
Temperatur ist negativ
155

– Aufnahme von Energie würde Entropie verringern– 2. Hauptsatz: Entropie-Maximierung– Systeme mit negativer Temperatur geben Energie ab, um Entropie zu
erhöhen– Systeme mit negativer Temperatur sind ”heißer” als Systeme mit positiver
Temperatur– ”Heissheit”: Bestreben Energie abzugeben.
• Ist das alles physikalisch ?
– Negative Temperaturen gibt es nur in Energie-sättigenden Systemen.– Die meisten natürlichen Systeme sind nicht Energie-sättigend.– Energie-Aufnahme führt in der Regel zu höherer Entropie
Realisierungen negativer Temperaturen:
• Geht nicht durch normales Wärmebad
• Braucht Nicht-Gleichgewichtsprozess, um sogenannte Inversion herzustellen
(i) Richte Spins durch Magnetfeld aus, drehe dann Magnetfeld um
Während Relaxationszeit negative Temperatur
(ii) ”Pumpen” im Laser
• Da es Nicht-Gleichgewichtszustand braucht, wundert es nicht, dass eineTemperatur-Definition aus dem Gleichgewicht zu komischen Folgerungenkommt.
11.7 Relativistisches ideales Gas
Betrachte relativistisches ideales Gas
• Hamiltonian
H =N∑i=1
(√(mc2)2 + (c~pi)2 −mc2
)=
N∑i=1
mc2
(1 +
(~pimc
)2)1/2
− 1
Ruhemasse abgezogen, so dass nur kinetische Energie bleibt
156

• Zustandssumme faktorisiert
Z(T, V,N) =1
N !(Z(T, V, 1)N
Ein-Teilchen Zustandssumme
Z(T, V, 1) =1
h3
∫d3q
∫d3p exp (−βH)
• Koordinatenintegral liefert Volumen
Für Impulsintegral: Kugelkoordinaten, Winkelanteile ausintegriert
Z(T, V, 1) =4πV
h3exp(βmc2)
∫ ∞0
dp p2 exp
(−βmc2
(1 +
( p
mc
)2)1/2
)
Variablensubstitution, häufig nützlich bei relativistischen Problemen
p
mc= sinhx, dp = mc coshxdx,
(1 +
( p
mc
)2)1/2
= coshx
Ferner
u = βmc2 Verhältnis Ruheenergie zu thermischer Energieu → ∞ nicht-relativistischer Fallu → 0 ultra-relativistischer Fall
Damit
Z(T, V, 1) =4πV
h3(mc)3eu
∫ ∞0
dx coshx sinh2 x exp(−u coshx)
• Mitcoshx sinhx =
1
2sinh(2x)
wird Integral zu
1
2
∫ ∞0
dx sinh(2x) sinhx e−u coshx
157

Schlage nach: ∫ ∞0
dx sinh(γx) sinhxe−u coshx =γ
uKγ(u)
mit Kγ(u) Zylinderfunktion oder modifizierte Besselfunktion
• DamitZ(T, V, 1) =
4πV
h3(mc)3eu
K2(u)
u
• Reihenentwicklung von K2(u) bekannt, kompliziert, nicht einsichtsreich
• Grenzverhalten von K2(u)
– u→∞: Thermische Energie klein gegen Ruheenergie
K2(u) ≈√π/(2u)e−u
Damit
Z(T, V, 1) ≈ 4πV
h3(mc)3
(1
βmc2
)3/2√π/2 = V
(2πmkT
h2
)3/2
das bekannte nicht-relativistische Ergebnis– u→ 0: Ultra-relativistischer Fall, siehe Übung
K2(u) ≈ 2
u2, ferner eu ≈ 1
Zustandssumme des ultra-relativistischen idealen Gases:
Z(T, V, 1) ≈ 8πV
h3(mc)3
(1
βmc2
)3
= 8πV
(kT
hc
)3
Z(T, V,N) =1
N !
(8πV
(kT
hc
)3)N
• Mit Hilfe der Stirling’schen Formel, ergibt sich die freie Energie für ultra-relativistisches Gas
F (T, V,N) = −kT logZ(T, V,N) = −NkT
(1 + log
(8πV
N
(kT
hc
)3))
158
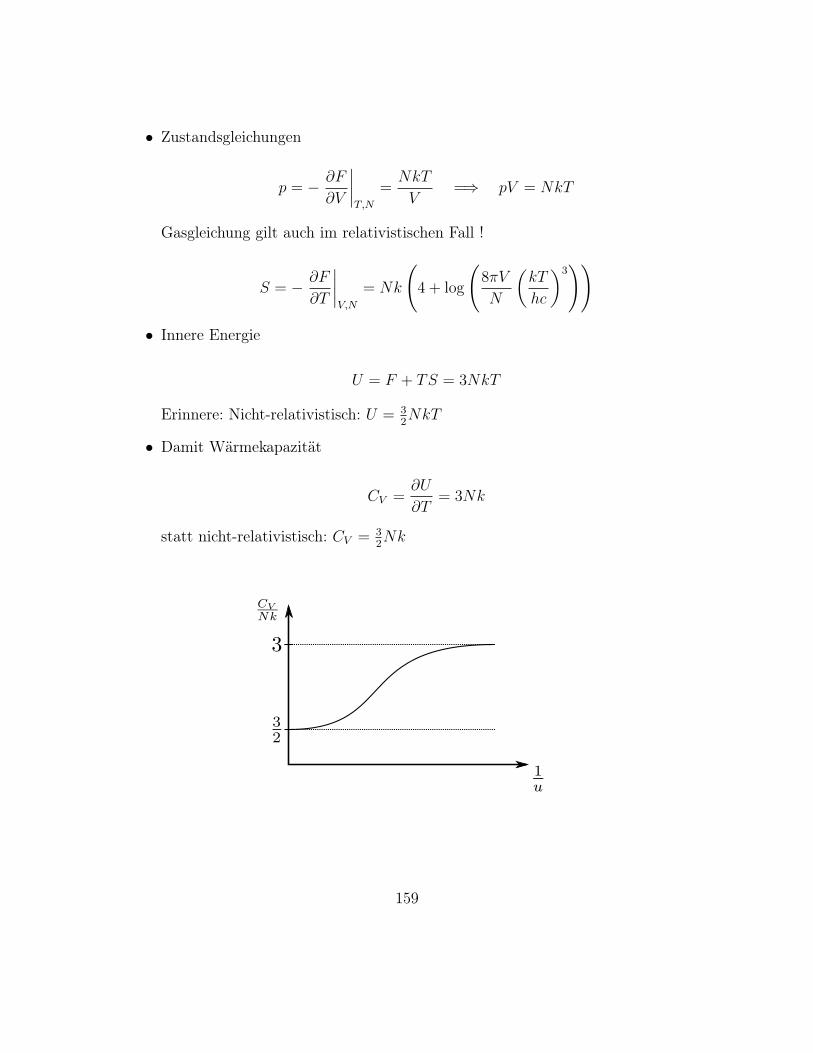
• Zustandsgleichungen
p = − ∂F
∂V
∣∣∣∣T,N
=NkT
V=⇒ pV = NkT
Gasgleichung gilt auch im relativistischen Fall !
S = − ∂F
∂T
∣∣∣∣V,N
= Nk
(4 + log
(8πV
N
(kT
hc
)3))
• Innere Energie
U = F + TS = 3NkT
Erinnere: Nicht-relativistisch: U = 32NkT
• Damit Wärmekapazität
CV =∂U
∂T= 3Nk
statt nicht-relativistisch: CV = 32Nk
159

Lessons learned
• Gleichverteilungssatz: Jeder quadratisch in der Hamilton-Funktion auftretendeTerm trägt mit 1
2kT zur mittleren Energie bei
• Fundamentale Limitation einer rein klassischen Statistischen Physik
• Einfrieren von Freiheitsgraden in der Quantenmechanik
• Negative Temperaturen in Energie-sättigenden Systemen
• Relavistische Thermodynamik: Ideales Gas: U = 3NkT , CV = 3Nk
12 Quantengase
12.1 Symmetrie der Vielteilchenwellenfunktionen
• Betrachte N nicht wechselwirkende Teilchen mit Hamiltonian
H =∑i
hi(xi, pi)
Eigenwertgleichung für Einteilchen Hamiltonian hi
hi|ki〉 = εki |ki〉
mit Eigenzuständen |ki〉, ki Quantenzahl
• Kanonische Zustandssumme
Z(T, V,N) = Sp e−βH =∑r
e−βEr =∑ki
exp(−β∑
εki
)• Betrachte zunächst unterscheidbare Teilchen
Dann Gesamtwellenfunktion Produkt der Ein-Teilchenzustände:
|k1, k2, . . . , kN〉 = |k1〉|k2〉 . . . |kN〉
160

Eigenwertgleichung für System:
H|k1, . . . , kN〉 =∑i
hi|k1〉 . . . |kN〉 =∑i
|k1〉 . . . hi|ki〉 . . . |kN〉
=∑i
εki |k1, . . . , kN〉
Zustandssumme:
ZU(T, V,N) = Sp e−βH =∑k1
. . .∑kN
〈k1| . . . 〈kN |e−βH |k1〉 . . . |kN〉
=
(∑k1
〈k1|e−βh1|k1〉
)· · ·
(∑kN
〈kN |e−βhN |kN〉
)= Z(T, V, 1)N
Klassische (Maxwell-)Boltzmann Statistik führt mit Gibbs-Korrektur zu Zu-standssumme
ZMB(T, V,N) =1
N !Z(T, V, 1)N
Teilchen komplett unabhängig
In Quantenmechanik wird Ununterscheidbarkeit fundamental
• Betrachte zwei Teilchen mit Gesamtwellenfunktion Ψ(r1, r2) und Permutions-operator P :
P Ψ(r1, r2) = Ψ(r2, r1)
Nochmalige Anwendung:
P Ψ(r2, r1) = Ψ(r1, r2)
damit∀Ψ : P 2 Ψ(r1, r2) = Ψ(r1, r2) =⇒ P 2 = 1
Folglich hat P Eigenwerte λ = ±1.
Zudem gilt [P,H] = 0
161

• Gemeinsame Eigenzustände zu H und P definieren offensichtlich zwei Artenvon Teilchen
• Spin-Statistik Theorem, Pauli, 1940
– Bosonen, Spin ganz-zahlig, S = 0, 1, . . ., besitzen eine symmetrische Viel-teilchenwellenfunktion: ΨS = P ΨS
Besetzungszahlen der Zustände können alle Werte 0, 1, . . .∞ annehmen,siehe unten
– Fermionen, Spin halb-zahlig, S = 12, 3
2, . . ., besitzen eine anti-symmetrische
Vielteilchenwellenfunktion: ΨA = −P ΨA
Besetzungszahlen können die Werte 0, 1 annehmen, siehe unten
Zusammenfassend:PΨ(r1, r2) = (−1)2SΨ(r2, r1) (40)
Folge: Reiner Produktansatz mit Ein-Teilchen Wellenfunktion φi(ri)
Ψ(r1, r2) = φa(r1)φb(r2)
geht für ununterscheidbare Teilchen nicht durch.
• (Anti)symmetrisierung:
ΨS(r1, r2) =1√2
(φa(r1)φb(r2) + φa(r2)φb(r1))
ΨA(r1, r2) =1√2
(φa(r1)φb(r2)− φa(r2)φb(r1))
Folge: Pauli-Prinzip: Für identische Einteilchen-Wellenfunktionen verschwindetdie antisymmetrische Gesamtwellenfunktion
φa = φb =⇒ ΨA = 0
Bedeutung: Es können nicht zwei Fermionen in dem selben Zustand sein.
Gl. (40) hat Auswirkung auf Zustandssumme.
• Beispiel zwei Teilchen in zwei Zuständen, ε0 = 0, ε1 = ε.
Fermionen S = 12mit Spin up, Bosonen S = 0
162

– Abbildung ergänzen– Fermionen: ZF = e−βε
– Bosonen: ZB = 1 + e−βε + e−2βε
– Maxwell-Boltzmann: ZMB = 1+2e−βε+e−2βε
2
2! im Nenner: Gibbs’scher Korrekturfaktor
Betrachte N Teilchen
• Permutationsoperator:
PijΨ(r1, . . . , ri, . . . , rj, . . . , rN) = Ψ(r1, . . . , rj, . . . , ri, . . . , rN)
Damit
ΨS(r1, . . . , rN) = A∑P
P Ψ(r1, . . . , rN)
ΨA(r1, . . . , rN) =1√N !
∑P
(−1)PP Ψ(r1, . . . , rN)
mit(−1)P =
+1 für gerade Anzahl von Vertauschungen−1 für ungerade Anzahl von Vertauschungen
Normierungsfaktor A abhängig davon, wie viele der Quantenzahlen gleich sind,aber im wesentlichen auch 1√
N !
• Da Ψ in alle Erwartungswerte quadratisch eingeht, ergibt sich die Gibbs-Korrektur 1
N !aus der Quantenmechanik
• Geht man vom Produktansatz von Einteilchen-Wellenfunktionen aus, folgt fürden antisymmetrischen Fall:
ΨA(r1, . . . , rN) =1√N !
∑P
(−1)PP φa(r1) . . . , φl(rN)
Dieses läßt sich als Determinante schreiben:
ΨA(r1, . . . , rN) =1√N !
det
φa(r1) . . . φa(rN)φb(r1) . . . φb(rN)
......
φl(r1) . . . φl(rN)
163

die Slater-Determinante
Diese verschwindet in Übereinstimmung mit dem Pauli-Prinzip, wenn zwei Zei-len gleich sind.
12.2 Besetzungszahl-Darstellung
• Für ununterscheidbare Teilchen ist Mikrozustand durch Besetzungszahlen nkder Einteilchenzustände |k〉 eindeutig gegeben.
Es gilt, Summe läuft über Zustände:
E =∞∑k=1
nkεk, N =∞∑k=1
nk (41)
• Aus dem Zusammenhang zwischen mikrokanonischer und kanonischer Gesamt-heit folgt:
Z =∑n1
′∑n2
′. . .Ω(n1, n2, . . .) exp(−β(n1ε1 + n2ε2 + . . .))
=∑nk
′Ω(nk) exp
(−β∑k
nkεk
)
Strich an Summe bedeutet, dass nur über Besetzungszahlen summiert wird, diedie Nebenbedingung N =
∑∞k=1 nk aus Gl. (41) erfüllen.
• Eindeutigkeit des Mikrozustands heißt Ω(nk) = 1
• Im Gegensatz zu klassischer Maxwell-Boltzmann Statistik. Unterscheidbarkeitführt zu
Ω(nk) =N !
n1!n2! . . .
Mit Gibbs’scher Korrektur und längerer Rechnung folgt
ZMB =1
N !
∑nk
′ N !
n1!n2! . . .exp
(−β∑k
nkεk
)
=1
N !
(∞∑k=1
e−βεk
)N
=1
N !Z(T, V, 1)N
164

Teilchen sind unabhängig
• Obwohl Teilchen im (hier betrachteten Falle) quantenmechanischen Falle nichtim eigentlichen Sinne miteinander wechselwirken, sind sie nicht unabhängig,da die allgemeine Nebenbedingung N =
∑∞k=1 nk und für Fermionen noch
nk = 0, 1 berücksichigt werden muss.
• Da in Quantenmechanik der Mikrozustand durch die Besetzungszahlen nkfestgelegt ist, gilt
Ω(nk) = 1
und damit
ZQM =∑nk
′exp
(−β∑k
nkεk
)
• Wegen Nebenbedingung N =∑∞
k=1 nk ist kanonische Zustandssumme schwerzu berechnen.
Das ist der Preis, den man für die Besetzungszahl-Darstellung zahlen muss.
• Erinnere: Mikrokanonische Zustandssumme war wegen Nebenbedingung δ(E−E0) oder E0 < E < E + ∆ schwer zu berechnen. Darum damals lieber kanoni-sche Zustandssumme.
• Entsprechend hier: Übergang zur großkanonischen Zustandssumme
Y (T, V, µ) =∑nk
exp
(−β∑k
nk(εk − µ)
)
und Wahl des chemischen Potentials so, dass N =∑
k nk
• Summe im Exponenten: Zustandssumme faktorisiert
Y =∑n1
∑n2
· · ·
(∏k
e−βnk(εk−µ)
)
=
(∑n1
e−βn1(ε1−µ)
)(∑n2
e−βn2(ε2−µ)
)· · ·
=∏k
(∑nk
e−βnk(εk−µ)
)=∏k
f(εk)
165

• Um die Summation in der Funktion f(εk) ausführen zu können, muß man wissenwelche Besetzungszahlen quantenmechanisch erlaubt sind
– Fermi(-Dirac) Statistik für Fermionen, fF (εk)
– Bose(-Einstein) Statistik für Bosonen, fB(εk) 12. Wo-che• Mittlere Besetzungzahl für gegebenes Energieniveau εi:
〈ni〉 = − 1
β
∂
∂εilog Y
12.3 Ideales Fermi-Gas
• Für Fermionen gilt ni = 0, 1
Zustandssumme
Y =1∑
n1=0
1∑n2=0
. . . e−β(ε1−µ)n1e−β(ε2−µ)n2 . . . =∏i
1∑ni=0
e−β(εi−µ)ni
und
fF (εi) =1∑
ni=0
e−β(εi−µ)ni = 1 + e−β(εi−µ)
DamitY (T, V, µ) =
∏i
(1 + e−β(εi−µ)
)• Mittlere Besetzungzahl 〈nj〉, Fermi-Dirac-Verteilung
〈nj〉 = − 1
β
∂
∂εjlog Y =
e−β(εj−µ)
1 + e−β(εj−µ)=
1
eβ(εj−µ) + 1
• Um die Teilchenzahl richtig zu machen, wähle chemisches Potential so, dassgilt
N(T, V, µ) =
∫ ∞0
dε g(ε)1
eβ(ε−µ) + 1
mit g(ε) der Energiezustandsdichte 11. week15
166

Physikalische Interpretation:
• Pauli-Prinzip: Keine zwei Fermionen im selben Zustand.
• Im Grenzfall T → 0, β →∞ ergibt sich eine Stufenfunktion:
〈n〉 = limβ→∞
1
eβ(ε−µ) + 1=
1 ε < µ0 ε > µ
= Θ(µ− ε)
• Für T = 0: Zustände werden energetisch von unten aufgefüllt, bis zurFermikante: εF = µ, µ ≥ εi. Es kostet Energie, neue Teilchen ins System zubringen
• Für Elektronen: Jedes Energieniveau mit zwei Elektronen, Spin −1/2 und Spin+1/2, besetzt. g(ε) = 2
• Fermi-Kante ist im Limes β →∞, T → 0, scharf.
• Bei endlichen Temperaturen T > 0: ”Aufweichung der Fermi-Kante”. Elektro-nen im Bereich kT unterhalb der Fermikante können angeregt werden.
Beispiel
• Natrium, jedes Atom gebe ein Elektron an Elektronengas ab
• µ = 3.1eV , Fermi-Temperatur TF = µ/k = 36.000K
167

• Energien der bevölkerten Niveaus thermische Energie kT der Umgebungs-temperatur und typischen coulomb’schen Wechselwirkungsenergien zwi-schen Elektronen
=⇒ Valenzelektronen eines Metalls sind ideales Elektronengas
Merke: Obwohl keine klassische Wechselwirkung, gibt es auf Grund des Pauli-Prinzips eine effektive Abstossung
12.4 Ideales Bose-Gas
• Für Bosonen gilt ni = 0, 1, 2, . . .
• Zustandssumme
Y =∞∑
n1=0
∞∑n2=0
. . . e−β(ε1−µ)n1e−β(ε2−µ)n2 . . . =∏i
∞∑ni=0
e−β(εi−µ)ni
Mit geometrischer Reihe
fB(εi) =∞∑ni=0
e−β(εi−µ)ni =1
1− e−β(εi−µ)
DamitY (T, V, µ) =
∏i
1
1− e−β(εi−µ)
• Mittlere Besetzungszahl 〈nj〉, Bose-Einstein Verteilung
〈nj〉 = − 1
β
∂
∂εjlog Y =
e−β(εj−µ)
1− e−β(εj−µ)=
1
eβ(εj−µ) − 1
Zeigt für β(εj − µ)→ 0 eine Divergenz
• −1 im Unterschied zur +1 bei Fermi-Dirac hat Folgen:
Aus
0 ≤ 〈ni〉
168

folgt
eβ(εi−µ) > 1
und damit: εi > µ.
Mit ε0 = 0 gilt µ ≤ 0. Teilchen wollen ins System. µ = 0, wenn keine Teilchen-zahlerhaltung gilt
Merke: Obwohl keine klassische Wechselwirkung, gibt es eine effektive Anziehung
Bose-Einstein Kondensation:
• Vorhersage: Bose & Einstein 1924
Experimentelle Bestätigung (für 10 Sekunden :-): 1995 durch Cornell, Ketterle& Wiemann, 2001 Nobelpreis
• Für T → 0 gehen alle Bosonen in den Grundzustand, T = 200nK
• Kondensation im Impuls-, resp. Energieraum, nicht im Ortsraum
• Makroskopisch bevölkerter Quantenzustand mit p = E = 0
• Phasenübergang, siehe Kap. 14 Phasenübergänge
169
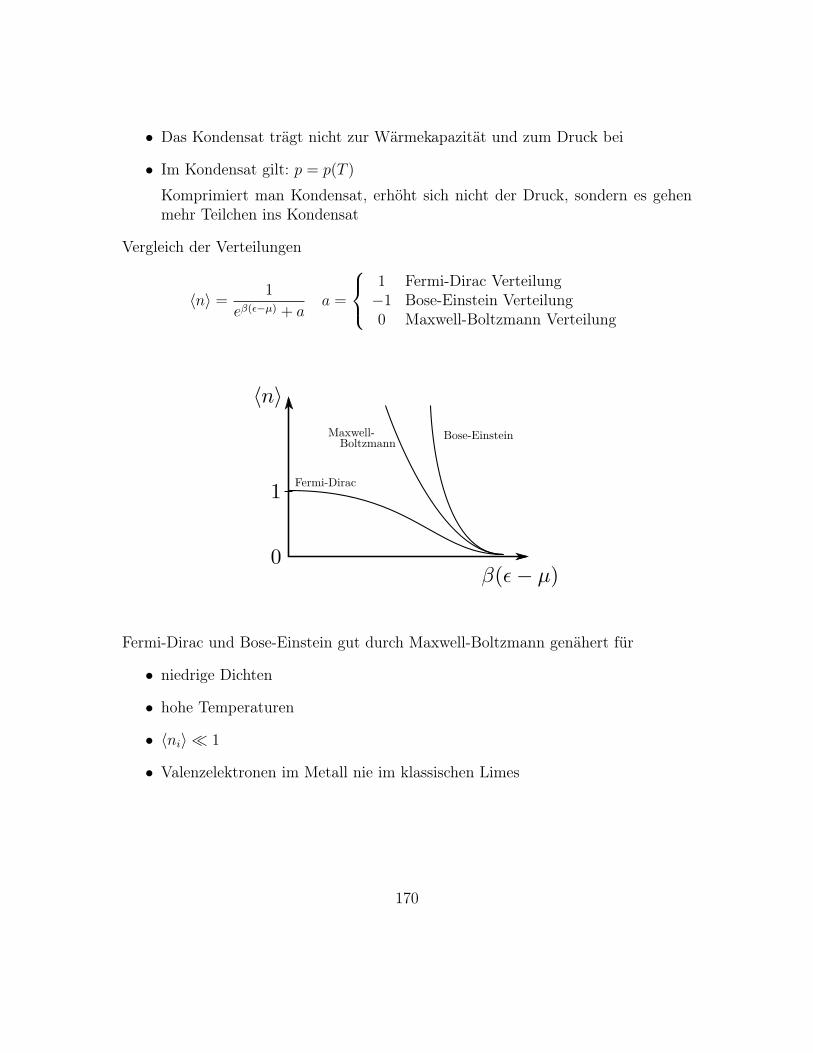
• Das Kondensat trägt nicht zur Wärmekapazität und zum Druck bei
• Im Kondensat gilt: p = p(T )
Komprimiert man Kondensat, erhöht sich nicht der Druck, sondern es gehenmehr Teilchen ins Kondensat
Vergleich der Verteilungen
〈n〉 =1
eβ(ε−µ) + aa =
1 Fermi-Dirac Verteilung−1 Bose-Einstein Verteilung0 Maxwell-Boltzmann Verteilung
Fermi-Dirac und Bose-Einstein gut durch Maxwell-Boltzmann genähert für
• niedrige Dichten
• hohe Temperaturen
• 〈ni〉 1
• Valenzelektronen im Metall nie im klassischen Limes
170
12.5 Das Planck’sche Strahlungsgesetz
Betrachte Schwarzen Strahler, Kirchhoff 1860:
• Erhitze evakuierten Hohlraum
• Messe spektrale Energie-Verteilung durch kleines Loch
Hier reicht eine Kurve :-)
• Gilt für alle Systeme, bei denen abgegebene Strahlung auf thermischen Effektenberuht, z.B. Sonne und kosmische Hintergrundstrahlung, nicht aber Laser, LED
Elektromagnetisches Feld:
• System mit unendlich vielen Freiheitsgraden
• Klassisch: Wellenfelder, lassen sich in ebene Wellen e−i(ωt−kx) zerlegen
• Quantenmechanik: Photonen, Impuls: p = ~k, Energie E = ~ω, keine Ruhe-masse =⇒ v = c
• Kein Erhaltungssatz für Photonen =⇒ µ = 0
• Photonen: p 6= 0 =⇒ keine Bose-Einstein Kondensation
Klassische Behandlung:
171

• Wellengleichung
∆E =1
c2
∂2
∂t2E
Betrachte Kubus, Kantenlänge L: Lösung wegen Randbedingung E = 0
E = E0 sinn1πx
Lsin
n2πy
Lsin
n3πz
Lsinωt
In Wellengleichung eingesetzt
n21 + n2
2 + n23 =
L2
π2ω2
• Kugelnäherung: Anzahl N der Moden
N =π
3
(n2
1 + n22 + n2
3
)3/2 ∝ ω3
Die 3 in den Exponenten kommt von der Raumdimension D
Kumulative Größe
• Modendichte g(ω) durch Ableiten
g(ω) =dN
dω∝ ω2
• Energiedichte g(E), Gleichverteilungssatz Kap. 11.3
g(E) =dE
dω∝ kT ω2
Multiplikation mit kT und Ableiten kommutiert
• Geometrie der Abstrahlung und Vorfaktoren richtig machen
Ergibt Strahlungsdichte u(T, ω)
u(T, ω) =kT
π2c3ω2 Rayleigh-Jeans Gesetz
172
• Divergiert mit ω: Ultraviolettkatastrophe
• Integrierte Strahlungsdichte
u(T ) =
∫ ∞0
dω u(T, ω) =∞
• Physikalischer Grund: Gleichverteilungssatz für unendlich viele Freiheitsgrade
∞ · kT =∞
Planck’sches Strahlungsgesetz, 14.12.1900, Geburtsstunde der Quantenmechanik
u(T, ω) =~π2c3
ω3
e~ωkT − 1
Entstehungsgeschichte
• Schwarzkörperstrahlung war sehr gut vermessen
173
• Ultraviolettkatastrophe war bekannt
• Planck: Anfangs, 1899, ”geratene” Interpolation7
– ”Ich dachte mir nicht viel dabei”
– ”Akt der Verzweiflung”
– Tiefe Bedeutung wurde ihm erst 1900 klar
• Planck betrachtete unterscheidbare Oszillatoren mit Maxwell-Boltzmann Sta-tistik, einen für jede Frequenz ω und visionär/gezwungen E = ~ω. Führt aufdas selbe Ergebnis wie quantenmechanische Ableitung
Quantenmechanische Ableitung:
• Zahl der Photonen dN im Frequenzintervall dω
dN(ω) = 〈nω〉 g(ω) dω
mit Zustandsdichte g(ω)
• Zustandsdichte, analog zu oben
g(ω) ∝ ω2
7Keine Ahnnug, wie er das ohne Computer gemacht hat.
174

Bose-Einstein Verteilung
〈nε〉 =1
eβε − 1
• Mit ε = ~ω, Photonendichte n = N/V und alle Vorfaktoren richtig:
dn(ω)
dω=
1
π2c3
ω2
eβ~ω − 1
EnergiedichtedE(ω)
dω= ~ω
dn(ω)
dω
• Damit final Strahlungsdichte:
u(T, ω) =~π2c3
ω3
e~ωkT − 1
Planck’sche Strahlungsformel
Vergleich mit Klassik:
• Modendichte identisch: ∝ ω2:
• Wahrscheinlichkeit, Mode zu bevölkern
– Klassisch: Gleich für alle Moden
– Quantenmechanisch:1
e~ωkT − 1
• Energie pro Mode:
– Klassisch: kT
– Quantenmechanisch: ~ω
Zwei Lesarten :
• QM: Ununterscheidbare Teilchen mit Bose-Einstein Statistik
175

• Planck: Unterscheidbare Oszillatoren mit Maxwell-Boltzmann Statisitk mit dis-kreten Anregungsstufen
Für das Maximum der Verteilung gilt:
~ωmax = 2.82 kT Wien’sches Verschiebungsgesetz
Für die integrierte Strahlungsdichte gilt:
u(T ) =π2k4
60~c2T 4 Stefan-Boltzmann Gesetz
Grenzfälle
• ~ω kT
u(T, ω) =~ω3
π2c3e−
~ωkT Wien’sches Gesetz
Abbildung ist Quatsch, x-Achse ~ω/kT
• ~ω kT
u(T, ω) =~π2c3
ω3
e~ωkT − 1
≈ ~π2c3
ω3
1 + ~ωkT− 1
u(T, ω) =ω2
π2c3kT Rayleigh-Jeans
176

~ ist verschwunden
Übergang Quantenmechanik → Klassik
Merke:
• Klassisches Rayleigh-Jeans Gesetz ergibt unendlichen Wert für Gesamtenergiedes Strahlungsfeldes
• Wie bei spezifischer Wärme, Kap. 11.4: Anregbarkeit von Zuständen ist in derKlassischen Physik falsch
• Nächster Beweis: Es gibt keine konsistente klassische Statistische Physik
Anwendungen:
• Bestimmung der Temperatur der Sonnenoberfläche: 5800 K. Maximum imsichtbaren Licht
• Schlechter Wirkungsgrad von Glühlampen. T muss kleiner sein als Schmelz-temperatur von Wolfram: 3683 K. Maximum im Infraroten
Kann man alles auch mit: ”Sage mir Deine Zustandssumme und ich sage Dir, wasDu tust” machen
12.6 Phononen und spezifische Wärme von Festkörpern
Phononen: Quasi-Teilchen: Anregung eines elastischen Feldes
Quantenmechanisch: Zweite Quantisierung
• Speziell Festkörperphysik: Anregung von Gitterschwingungen
• In linearer Näherung: Harmonische Schwingungen der Atome um Gleichge-wichtslage
• Normalkoordinaten: Ergibt 3N unabhängige Normalschwingungen
• 3N harmonische Oszillatoren mit Schwingungsfrequenzen ωi
• Phononen sind Bosonen
177

• Im Quantenfall kanonische Zustandssumme, erinnere Kap. 11.4
Eindimensionaler quantenmechanischer Oszillator
Zustandssumme
Z =∞∑n=0
e−βEn , En =
(n+
1
2
)~ω
Z = e−β~ω2
∞∑n=0
(e−β~ω
)n=
e−β~ω2
1− e−β~ω
• Betrachte Logarithmus
logZ = −3N∑i=1
(β~ωi
2+ log
(1− e−β~ωi
))
Führe Verteilungsfunktion g(ω) für die Frequenzen ein, kontinuierliche Nähe-rung
logZ = −∫ ∞
0
dω g(ω)
(β~ω
2+ log
(1− e−β~ω
))• Ergibt für Energie
E = −∂ logZ
∂β=
∫ ∞0
dω g(ω)
(~ω2
+~ω
eβ~ω − 1
)(42)
und für spezifische Wärme
CV =∂E
∂T= kβ2
∫ ∞0
dω g(ω)(~ω)2eβ~ω
(eβ~ω − 1)2
Ansätze fur g(ω):
• Einstein’scher Ansatz
• Debye’scher Ansatz
Einstein’scher Ansatz
• Es gibt nur eine mittlere Frequenz ωE
178
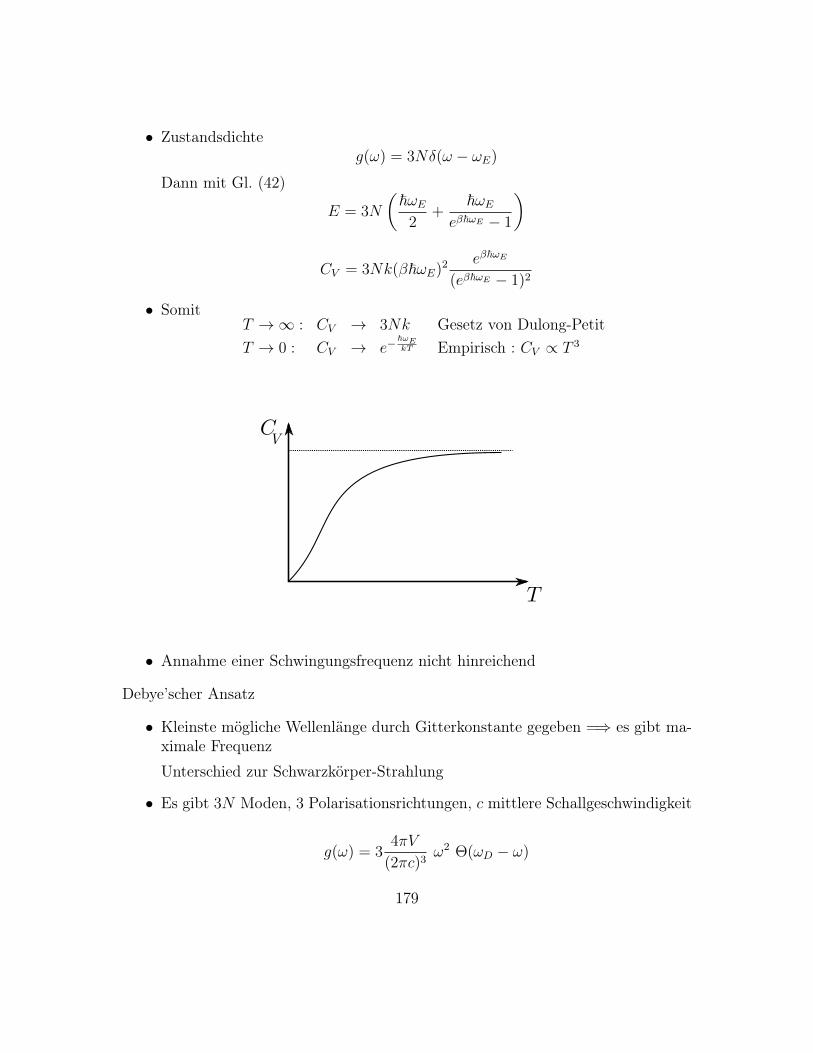
• Zustandsdichteg(ω) = 3Nδ(ω − ωE)
Dann mit Gl. (42)
E = 3N
(~ωE
2+
~ωEeβ~ωE − 1
)
CV = 3Nk(β~ωE)2 eβ~ωE
(eβ~ωE − 1)2
• SomitT →∞ : CV → 3Nk Gesetz von Dulong-PetitT → 0 : CV → e−
~ωEkT Empirisch : CV ∝ T 3
• Annahme einer Schwingungsfrequenz nicht hinreichend
Debye’scher Ansatz
• Kleinste mögliche Wellenlänge durch Gitterkonstante gegeben =⇒ es gibt ma-ximale Frequenz
Unterschied zur Schwarzkörper-Strahlung
• Es gibt 3N Moden, 3 Polarisationsrichtungen, c mittlere Schallgeschwindigkeit
g(ω) = 34πV
(2πc)3ω2 Θ(ωD − ω)
179

ω2 analog zur Zustandsdichte bei Rayleigh-Jeans, Planck Gesetz
• Abschneidefrequenz ωD aus
3N =
∫ ∞0
dω g(ω) =4πV
(2πc)3ω3D
Damit
g(ω) =9N
ω3D
ω2 Θ(ωD − ω)
• Es folgt mit E0 Nullpunktsenergie und Gl. (42)
E = E0 +9N
ω3D
∫ ωD
0
dω~ω3
eβ~ω − 1
x = β~ω
E = E0 +9N(kT )4
(~ωD)3
∫ β~ωD
0
dxx3
ex − 1= E0 + 3NkT D (β~ωD)
mitD(x) =
3
x3
∫ x
0
dx′x′3
ex′ − 1
• Grenzfall kT ~ωD, d.h. x 1:
D(x) =π4
5
1
x3
E = E0 +3
5π4N
(kT )4
(~ωD)3
CV =12
5π4N
(kT )3
(~ωD)3
Empirischer Verlauf wird richtig wiedergegeben
• Grenzfall kT ~ωD, d.h. x 1:
D(x) =3
x3
∫ x
0
dx′x′3
1 + x′ − 1=
3
x3
∫ x
0
dx′x′2 = 1
E = E0 + 3NkT
CV = 3Nk
180

Lessons learned
• Quantenmechanische Vielteilchen-Wellenfunktion müssen symmetrisch oderantisymmetrisch sein. Bosonen, ni = 0, 1, 2, . . .∞, Fermionen, ni = 0, 1
• Dadurch Abweichungen von klassischer Maxwell-Boltzmann Statistik mitGibbs-Korrektur
• Fermionen: Fermi-Dirac Verteilung: Effektive Abstossung
• Bosonen: Bose-Einstein Verteilung: Effektive Anziehung
• Klassische Ableitung der Schwarz-Körper Strahlung zeigt Versagen einer reinklassischen Statistischen Physik
• Spezifische Wärme von Festkörpern: Verschiedene Zustandsdichten =⇒ ver-schiedene Zustandssummen =⇒ verschiedene Physik
13. Wo-che
13 NäherungsverfahrenBisher: Nur freie Teilchen betrachet. Schalte jetzt Wechselwirkungen ein.
Zoo der Näherungsverfahren:
• Störungsrechnung: H = H0 + λW , Entwicklung nach λ
• Quasiklassische Entwicklung: Entwicklungsparameter ~, WKB-Näherung
• Hoch-Temperatur Entwicklung: T0/T , siehe letztes Kapitel
• Tief-Temperatur Entwicklung: T/T0, dito
• Entwicklung um kritischen Punkt: T−TcTc
, Kap. 14
• ε-Entwicklung: ε = D − 3, D Raumdimensionen
• Virialentwicklung: n = N/V , Kap. 13.1
• Molekularfeldnäherung, kein Entwicklungsparameter, Selbstkonsistenz-Methode, Kap. 13.2
181

• Renormierungsgruppen-Theorie, Kap. 13.3
13.1 Virialentwicklung
Entwicklung nach Potenzen der Teilchenzahldichte n = N/V
• Erinnere ideales Gas, mit z = eβµ
Z(1) =V
λ3, Z(N) =
Z(1)N
N !, Y (µ) =
∞∑N=0
zNZ(N) = ezV/λ3
, log Y = N =pV
kT
Allgemein gilt:
Z(1) =V
λ3log Y =
pV
kT, N = z
∂
∂zlog Y
Z(2) beschreibt Zweiteilchen-WechselwirkungZ(3) beschreibt Dreiteilchen-Wechselwirkung, die sich nicht auf Zweiteilchen-wechselwirkungen zurückführen lassen
• Kanonische Zustandssumme mit 2-Teilchen Wechselwirkung
H =∑i
p2i
2m+∑i 6=j
w(|qi − qj|)
Z(2) =1
2!h6
∫d3q1d
3q2d3p1d
3p2 exp
(−β(p2
1
2m+
p22
2m+ w(|q1 − q2|)
))=
V
2λ6
∫d3q e−βw(q)
• Idee: Bei geringer Dichte geht es gegen das ideale Gas, entwickle nach Teilchen-zahldichte n = N/V
pV
kT= log Y = N(1 + nB1(T, V ) + n2B2(T, V ) . . .)
• Taylor-Entwicklungen
Y = 1 + zZ(1) + z2Z(2) + . . .
pV
kT= log Y = zZ(1) + z2
(Z(2)− 1
2Z(1)2
)+ . . . (43)
N = z∂
∂zlog Y = zZ(1) + 2z2
(Z(2)− 1
2Z(1)2
)+ . . . (44)
182

Eliminiere z aus Gln. (43, 44), ergibt:
pV
kT= N −
(N
Z(1)
)21
V
(Z(2)− 1
2Z(1)2
)+ . . .
• Mit1
2Z(1)2 =
1
2
V 2
λ6=
1
2
V
λ6
∫d3q
folgtpV
kT= N − N2
2V
∫d3q(e−βw(q) − 1
)+ . . .
oder
pV
kT= N(1 + nB1(T, V ) + . . .), B1(T, V ) = −1
2
∫d3q(e−βw(q) − 1
)B1(T, V ): 1. Virialkoeffizient
Höhere Virialkoeffizienten durch höhere Ordnungen Taylorentwicklung inGln. (43, 44)
• BeachteZ(1) =
V
λ3gilt klassisch wie quantenmechanisch
Abgeleitete Gleichung für B1 gilt nur klassisch
• Höhere Ordnungen der Entwicklung führen auf dieMayer’sche Cluster-Entwicklung
13.1.1 Van der Waals Gas
Zustandsgleichung für ein reales Gas.Annahmen:
• Bei kleinen Abständen stoßen sich Teilchen ab. Näherung: Harte Kugeln:
w(r) =∞ für r < r0
183

• Bei größeren Abständen durch Polarisation schwache Anziehung. Van der WaalsKraft:
w(r) = −w0
(r0
r
)6
für r ≥ r0
mit w0 kT
B1(T ) = −2π
∫ ∞0
dr r2(e−βw(r) − 1)
= 2π
∫ r0
0
dr r2 − 2π
∫ ∞r0
dr r2(e−βw(r) − 1
)≈ 2π
∫ r0
0
dr r2 + 2πβw0
∫ ∞r0
dr r2(r0
r
)6
=2π
3r3
0
(1− w0
kT
)= b− a
kT
• Qualitativ ist Virialkoeffizient unabhängig von Details der Wechselwirkungdurch 2 Parameter, a und b charakterisiert:
– a: Anziehende Wechselwirkung, mit kT gewichtet, mindert Druck
– b: Ausgeschlossenes Volumen, b ∝ r30
184
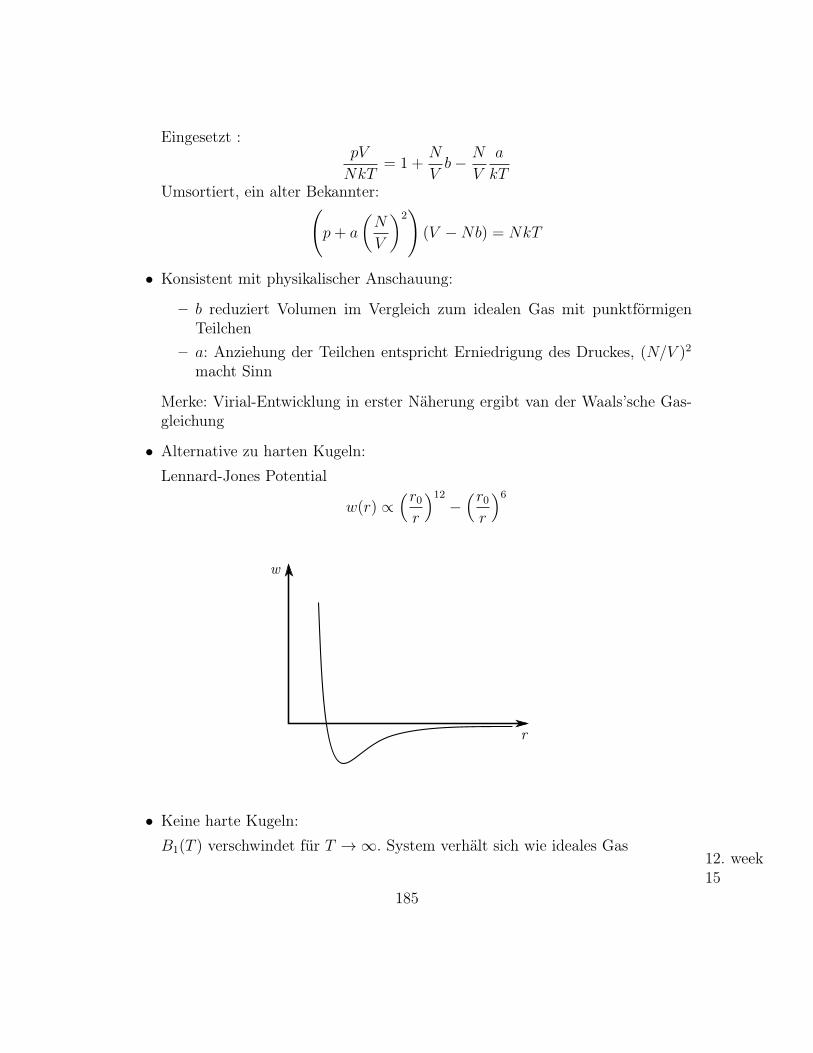
Eingesetzt :pV
NkT= 1 +
N
Vb− N
V
a
kTUmsortiert, ein alter Bekannter:(
p+ a
(N
V
)2)
(V −Nb) = NkT
• Konsistent mit physikalischer Anschauung:
– b reduziert Volumen im Vergleich zum idealen Gas mit punktförmigenTeilchen
– a: Anziehung der Teilchen entspricht Erniedrigung des Druckes, (N/V )2
macht Sinn
Merke: Virial-Entwicklung in erster Näherung ergibt van der Waals’sche Gas-gleichung
• Alternative zu harten Kugeln:Lennard-Jones Potential
w(r) ∝(r0
r
)12
−(r0
r
)6
• Keine harte Kugeln:B1(T ) verschwindet für T →∞. System verhält sich wie ideales Gas
12. week15
185

13.2 Molekularfeldnäherung
Mean-Field Approximation
Drei Themen in diesem Kapitel
• Molekularfeldnäherung
• Ising-Modell, 1925
• Phasenübergang
Betrachte Ising-Modell in einer Dimension
• Ein-dimensionaler Ring von N Spins σi mit Nächste-Nachbarn-Wechselwirkungin äußerem Magnetfeld
H(σ1, σ2, . . . , σN) = −I∑n.n.
σiσk − µBN∑i=1
σi, σN+1 = σ1, σi ∈ −1, 1
µ: magnetisches Moment der Spins
I: Spin-Spin Austauschwechselwirkung
B: Externes magnetisches Feld
• Lässt sich in 1D (Ising) und 2D (Onsager, 1944) analytisch lösen:Transfermatrix-Methode
• 1D: kein Phasenübergang
≥ 2D: Phasenübergang: Für T < TC spontane Magnetisierung
• In 3D bis heute nur simulativ lösbar
• Ursprünglich: Modell für Ferro-Magnetismus, aber auch gut für
– Ordnungs-Unordnungs-Übergang in binären Legierungen
– Spingläser, amorphe Strukturen mit S(T = 0) 6= 0, siehe Kap. 9.7
– Gehirnmodelle, Neuronen statt Spins
Idee der Molekularfeldnäherung:
• Spins wechselwirkungen nicht individuell mit ihren nächsten q Nachbarn
186

• Sondern mit einem mittlerem Feld, das sich aus der mittleren Stellung der qNachbar-Spins ergibt
• Das heißt−I∑n.n.
σiσk → −Iq〈σ〉∑i
σi,
In Worten: Ersetze σk durch seinen thermodynamischen Mittelwert σk → 〈σ〉
Im Detail :
• Betrachte Identität
σiσk = σi〈σk〉+ 〈σi〉σk − 〈σi〉〈σk〉+ (σi − 〈σi〉)(σk − 〈σk〉)
Mittelwerte 〈σi〉 und 〈σk〉 hängen wegen Translationsinvarianz nicht von i, resp.k ab
• Damit Wechselwirkungsterm
HI(σ1, . . . , σN) = −I∑n.n.
σi〈σ〉+ 〈σ〉σk − 〈σ〉2 + (σi − 〈σ〉)(σk − 〈σ〉)
Jeder Zentralspin σi mit Nachbar σk ist auch Nachbarspin zu Zentralspin σk:Die ersten beiden Summen sind identisch.
• Es gibt qN/2 verschiedene Paare. Somit gehören zu jedem Spin σi q/2 verschie-dene Paare
Damit
HI(σ1, . . . , σN) = −Iq〈σ〉∑i
σi + Iq
2N〈σ〉2 − I
∑n.n.
(σi − 〈σ〉)(σk − 〈σ〉)
Immer noch exakt
– 1. Term hat die Form, die wir suchen
– 2. Term ist konstant
– 3. Term: Die Fluktuationen der Spins um mittleren Wert
187

• Mean-field Näherung: Vernachlässige den 3. Term
HmfI (σ1, . . . , σN) = −Iq〈σ〉
∑i
σi + Iq
2N〈σ〉2
Bilde Erwartungswert
U = 〈HmfI 〉 = −IqN〈σ〉2 + I
q
2N〈σ〉2 = −I q
2N〈σ〉2
Für 〈σ〉 = 1 folgt U = −I q2N , macht Sinn, da jedes verschiedene Paar −I
beiträgt
• HmfI enthält Erwartungswert 〈σ〉, der am Ende berechnet werden will.
Führt auf ein Selbstkonsistenz-Problem zur Bestimmung von 〈σ〉Entspricht Hartree-Fock Näherung in der Quantenmechanik
• Nehme äußeres Magnetfeld wieder mit und formuliere durch die Spins verur-sachtes mittleres Magnetfeld als
Bmf =qI〈σ〉µ
Ergibt:Hmf (σi, . . . , σN) = −I q
2N〈σ〉2 − µ(Bmf +B)
∑i
σi
Beachte: Summe von Ein-Teilchen Hamilton-Funktionen, aberSelbstkonsistenz-Problem für 〈σ〉
Berechnung des magnetischen Dipolmomentes D, der Magnetisierung m
• Dipolmoment D:
D = µ
⟨N∑i
σi
⟩= Nµ〈σ〉 (45)
Ableitung aus der freien Energie F
D = − ∂
∂BF (N,B, T, 〈σ〉)|N,T,〈σ〉 (46)
Beide Gleichnungen zusammen geben implizite Gleichung für 〈σ〉
188

• Zustandssumme
Z(N, T,B, 〈σ〉) =∑σ1=±1
. . .∑σn=±1
exp
(−1
2βqNI〈σ〉2
)exp
(βµ(Bmf +B)
N∑i=1
σi
)
= exp
(−1
2βqNI〈σ〉2
)(∑σ=±1
exp(βµ(Bmf +B)σ
))N
= exp
(−1
2βqNI〈σ〉2
)(2 cosh
(βµ(Bmf +B)
))NFreie Energie:
F (N,B, T, 〈σ〉) = −kT logZ =1
2qNI〈σ〉2 −NkT log
(2 cosh
(βµ(Bmf +B)
))• Bestimmungsgleichung für 〈σ〉 nach Gln. (45, 46)
〈σ〉 = tanh
(βµ
(qI
µ〈σ〉+B
))Substituiere
x = βqI〈σ〉+ βµB
Ergibt1
βqI(x− βµB) = tanh x
Bestimmung von x und damit 〈σ〉 aus Schnittpunkt einer Grade ax + b mittanhx
• Betrachte B = 0, setze
Tc =qI
k,
T
Tc=
1
βqI
Ergibt:
T
Tcx = tanhx
189

Achsenbeschriftung Quatsch
– T > Tc: Nur triviale Lösung bei x = 〈σ〉 = 0
– T < Tc: Neben x = 0 auch nicht-triviale Lösungen ±x0 = ±βqI〈σ〉 6= 0
– Spontane Magnetisierung, ein Phasenübergang !
• Analytische Lösung: Dies stimmt nicht :-)
– Liegt an Vernachlässigung der Fluktuationen, die in einer Dimension be-sonders schlimm ist, da wenig Nachbarn.
– ab D = 2: qualitativ richtig
– ab D = 4: quantitativ richtig
• Entwicklung um Tc
Entwickle tanhx für kleine x, Abbruch nach 3. Ordnung
T
Tcx = x− 1
3x3 + . . .
x0 =
(3
(1− T
Tc
))1/2
, 〈σ〉 =T
Tcx0
• 〈σ〉 heißt Ordnungsparameter des Phasenübergangs, siehe Kap. 14.1
Exponent 1/2 ist der kritische Exponent β, siehe Kap. 14.2
190

• Am kritischen Punkt gilt für B 6= 0
D3 =3
kTB
Magnetisierung reagiert nicht-linear auf angelegtes Magnetfeld
Molekularfeldnäherung abstrakt oder hand-waving :-)
• Betrachte:
– Wechselwirkungspotential: W (x− x′)– Mikroskopische Ein-Teilchendichte: ρ(x) =
∑Ni=1 δ(x− xi)
– Makroskopische Ein-Teilchendichte: n1(x) = 〈ρ(x)〉
• Bei Berechungen tauchen Terme folgender Form auf⟨exp
(−β∫d3x′ W (x− x′)ρ(x′)
)⟩In der Molekularfeldnäherung wird das genähert durch
exp
(−β∫d3x′ W (x− x′)〈ρ(x′)〉
)≈ exp
(−β∫d3x′ W (x− x′)n1(x′)
)Statt Erwartungswert der Exponentialfunktion nun Exponentialfunktion desErwartungswertes
Gute Näherung wenn∫d3x′W (x− x′)ρ(x′) =
∑i
W (x− xi) ≈∫d3x′W (x− x′)n1(x′)
Gültigkeitsbereich der Molekularfeldnäherung:
• Mittelung über hinreichend viele Wechselwirkungspartner, mittlerer Teilchen-abstand klein gegen mittlere Reichweite der Wechselwirkung
– hohe Dichten
– langreichweitige Wechselwirkungen
191

• kleine Korrelationen
Allgemein
• Es gibt keinen Entwicklungsparameter
• Komplimentär zur Virialentwicklung
13.3 Renormierungsgruppentheorie
Mächtiges Werkzeug, Wilson 1971, Nobelpreis 1987
• Statistische Physik: speziell zur Bestimmung kritischer Exponenten, Kap. 14.2
• Quantenfeldtheorie
Betrachte ein-dimensionales Ising-Modell mit Nächste-Nachbar Wechselwirkung
• HamiltonianH = −I
∑i
σiσi+1
• Zustandssumme:
Z =∑σi
exp[K(σ1σ2 + σ2σ3 + σ3σ4 + . . .)] (47)
mitK =
I
kT, σi = ±1
Grundidee:
• Drücke die Spinkette von N Spins durch eine mit N/2 Spins aus. Dazu mussdie Kopplung renormiert werden.
• Iteriere dies: Sukzessive teilweises Ausführung der Zustandssumme, quanten-mechisch: Partielle Spurbildung
Im Detail
192

• Fasse alle geraden Terme für i zusammen und führe die Summation über σimit i gerade aus
Z =∑σi
eK(σ1σ2+σ2σ3) · eK(σ3σ4+σ4σ5) · . . .
=∑
σi,i=ungrade
[eK(σ1+σ3) + e−K(σ1+σ3)
]·[eK(σ3+σ5) + e−K(σ3+σ5)
]· . . .
• Zentral: Suche neue Kopplung K ′ und Funktion f(K), so dass
eK(σ1+σ3) + e−K(σ1+σ3) = f(K) eK′σ1σ3
”Renormieren”
• Zur Bestimmung von K ′ und f(K), betrachte mögliche Kombinationen von σ1
und σ3
(σ1, σ3) = (1, 1) e2K + e−2K = 2 cosh(2K) = f(K)eK′
(σ1, σ3) = (−1,−1) e2K + e−2K = 2 cosh(2K) = f(K)eK′
(σ1, σ3) = (1,−1) 2 = f(K)e−K′
(σ1, σ3) = (−1, 1) 2 = f(K)e−K′
Multipiziere obere und untere Gleichungen
4 cosh(2K) = f 2(K)
f(K) = 2√
cosh(2K)
Teile obere durch untere Gleichungen
cosh(2K) = e2K′
K ′ =1
2log(cosh(2K))
• Als Iteration geschrieben, ergibt die Renormierungsgruppengleichung
K(i) =1
2log(cosh(2K(i−1)))
193

oder auchtanhK(i) = (tanhK(i−1))2
13. weekhalbeWoche15
Fixpunkt K∗ gegeben durch
K∗ =1
2log(cosh(2K∗))
odertanhK∗ = (tanhK∗)2
Zwei Lösungen
tanhK = 0 =⇒ K∗1 = 0 entspricht T =∞ oder I = 0
tanhK = 1 =⇒ K∗2 =∞ entspricht T = 0
Zentrale Frage:
Für welche Anfangsbedingungen wird welcher Fixpunkt durch Iteration er-reicht?
• Mathematische Lösung:
Untersuche Stabilität des Fixpunktes∣∣∣∣ ddKK(i)
∣∣∣∣K∗
=
< 1 stabil> 1 instabil
Interpretation: Stabil: Auslenkung aus Fixpunkt wird weggedämpft
• Hier:
– K∗1 ist stabil
– K∗2 ist instabil
– Nur, wenn man auf K∗2 , i.e. I 6= 0, T = 0 startet, bleibt man da
– Für alle anderen K(0) läuft es auf wechselwirkungsfreies Sytem zu =⇒ Esgibt keinen Phasenübergang bei endlicher Temperatur
194
Graphische Lösung
Ising-Modell in 2 Dimensionen
•• Kompliziertere Geometrie
Ein-Teilchen Beitrag zur Zustandssumme:∑σ
eK(σ1+σ2+σ3+σ4)σ = f(K)eK′(σ1σ2+σ3σ4)+L′(σ1σ3+σ2σ4)+M ′(σ1σ2σ3σ4)
195
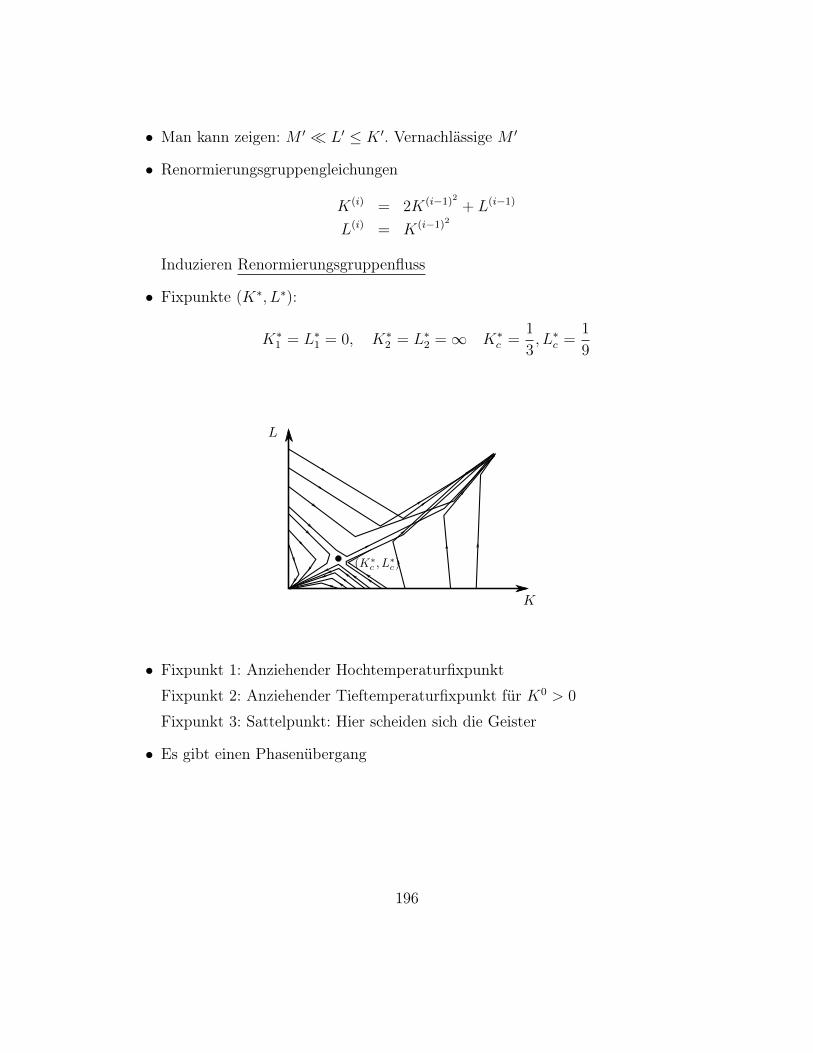
• Man kann zeigen: M ′ L′ ≤ K ′. Vernachlässige M ′
• Renormierungsgruppengleichungen
K(i) = 2K(i−1)2 + L(i−1)
L(i) = K(i−1)2
Induzieren Renormierungsgruppenfluss
• Fixpunkte (K∗, L∗):
K∗1 = L∗1 = 0, K∗2 = L∗2 =∞ K∗c =1
3, L∗c =
1
9
• Fixpunkt 1: Anziehender Hochtemperaturfixpunkt
Fixpunkt 2: Anziehender Tieftemperaturfixpunkt für K0 > 0
Fixpunkt 3: Sattelpunkt: Hier scheiden sich die Geister
• Es gibt einen Phasenübergang
196

Lessons learned
• Virial-Entwicklung entwickelt nach Teilchenzahldichte
• 1. Ordnung ergibt van der Waals’sche Gasgleichung
• Molekularfeldnäherung ist Selbstkonsistenzbedingung, keine Entwicklung nacheinem Parameter. Benötigt hohe Dichten relativ zur Reichweite der Wechsel-wirkung & Korrelationslänge
• Ising Modell als Modell für Paramagnetismus8
• Virial-Entwicklung und Molekularfeldnäherung haben komplementäre Gültig-keitsbereiche
• Renormierungsgruppen-Theorie: Ändere Skala, renormiere Wechselwirkung.Form bleibt identisch. Iteriere, untersuche Fixpunkte & deren Einzugsbereich
14. Wo-che
14 Phasenübergänge
14.1 Klassifikation von Phasenübergängen
Ordnungsparameter ψ
• Makroskopische Größe mit
ψ = 0 für T > Tcψ > 0 für T < Tc
• Flüssigkeit/Gas: Dichtedifferenz ∆ρ = ρGas − ρflüssig
• 2D Ising-Modell: Magnetisierung m
• Bose-Einstein Kondensation: Anzahl N0 Teilchen im Grundzustand
Thermodynamische Potentiale sind am Phasenübergang stetig, aber nicht analytisch
• Phasenübergang 1. Ordnung
197
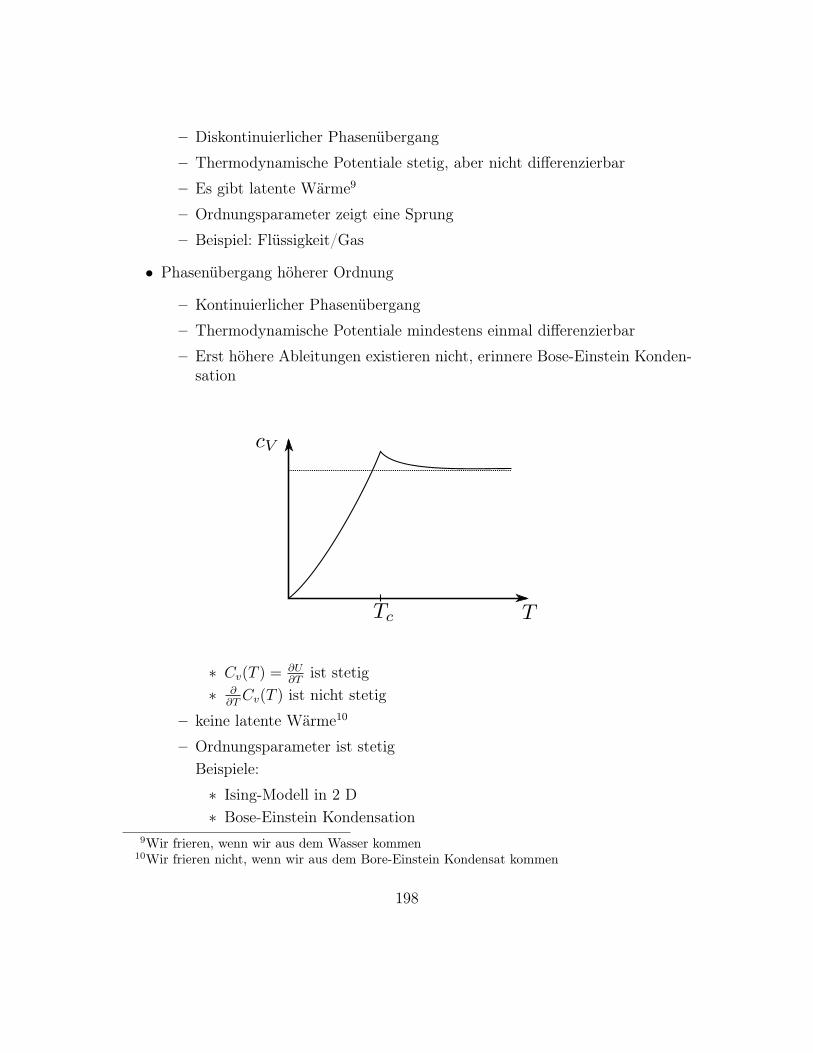
– Diskontinuierlicher Phasenübergang
– Thermodynamische Potentiale stetig, aber nicht differenzierbar
– Es gibt latente Wärme9
– Ordnungsparameter zeigt eine Sprung
– Beispiel: Flüssigkeit/Gas
• Phasenübergang höherer Ordnung
– Kontinuierlicher Phasenübergang
– Thermodynamische Potentiale mindestens einmal differenzierbar
– Erst höhere Ableitungen existieren nicht, erinnere Bose-Einstein Konden-sation
∗ Cv(T ) = ∂U∂T
ist stetig∗ ∂
∂TCv(T ) ist nicht stetig
– keine latente Wärme10
– Ordnungsparameter ist stetigBeispiele:
∗ Ising-Modell in 2 D∗ Bose-Einstein Kondensation
9Wir frieren, wenn wir aus dem Wasser kommen10Wir frieren nicht, wenn wir aus dem Bore-Einstein Kondensat kommen
198

14.2 Skalierungen
Betrachte kontinuierliche Phasenübergänge, Beispiel Ising-Modell in 2D
• Charakteristische Größen zeigen am Phasenübergang Potenzverhalten mitkritischen Exponenten: (
T − TcTc
)a= ta
• Ordnungsparameter, hier Magnetisierung m.
Für T < Tc:
m|B=0 ∝ |t|β Mean-Field Näherung: β =
1
2, exakt: β =
1
8
• Analog zu Gl. (39), Kap. 10.6 Fluktuationen
∂U
∂β= . . . = −(〈E2〉 − 〈E〉2) = −σ2
E
folgt für Suszeptibilität
χ =∂m
∂B
∣∣∣∣B=0
=1
kT(〈m2〉 − 〈m〉2) ∝ σ2
m
und damit Fluktuationen des Ordnungsparameters :
χ ∝ |t|−γ Mean-Field: γ = 1, exakt: γ =7
4
• Kovarianzfunktion G(|~ri − ~rj|) = G(r), hier:
G(r) = 〈σiσj〉 − 〈σi〉〈σj〉
Nicht am kritischen Punkt:
G(r) ∝ e−r/ξ, ξ : Korrelationslänge
Am kritischen Punkt divergiert ξ:
199

ξ|B=0 ∝ |t|−ν Mean-Field: ν =
1
2, exakt: ν = 1
und es gilt
G(r) ∝ 1
rD−2+η
• Für Genießer:
– Dies bedeuted Skaleninvarianz
G(r) = e−r/ξ, G(ar) = e−ar/ξ, aber G(r) =1
rb, G(ar) =
1
ab1
rb
– Deswegen funktioniert Renormierungsgruppentheorie
• Kritische Exponenten hängen nur ab von
– Dimension des Ordnungsparameters
– Symmetrien des Hamiltonians
– Dimension D des Raumes
Ironie der Geschichte: Erinnere ursprüngliches Ziel der Statistischen Physik :-)
• Systeme mit gleichen kritischen Exponenten sind in der selbenUniversalitätsklasse
• Kritische Exponenten sind nicht unabhängig
2 = α + 2β + γ
γ = ν(2− η)
νD = 2− α
14.3 Landau-Theorie
Basiert auf
• Ordnungsparameter ψ
• Symmetrien des Systems
• keiner mikroskopischen Information
200
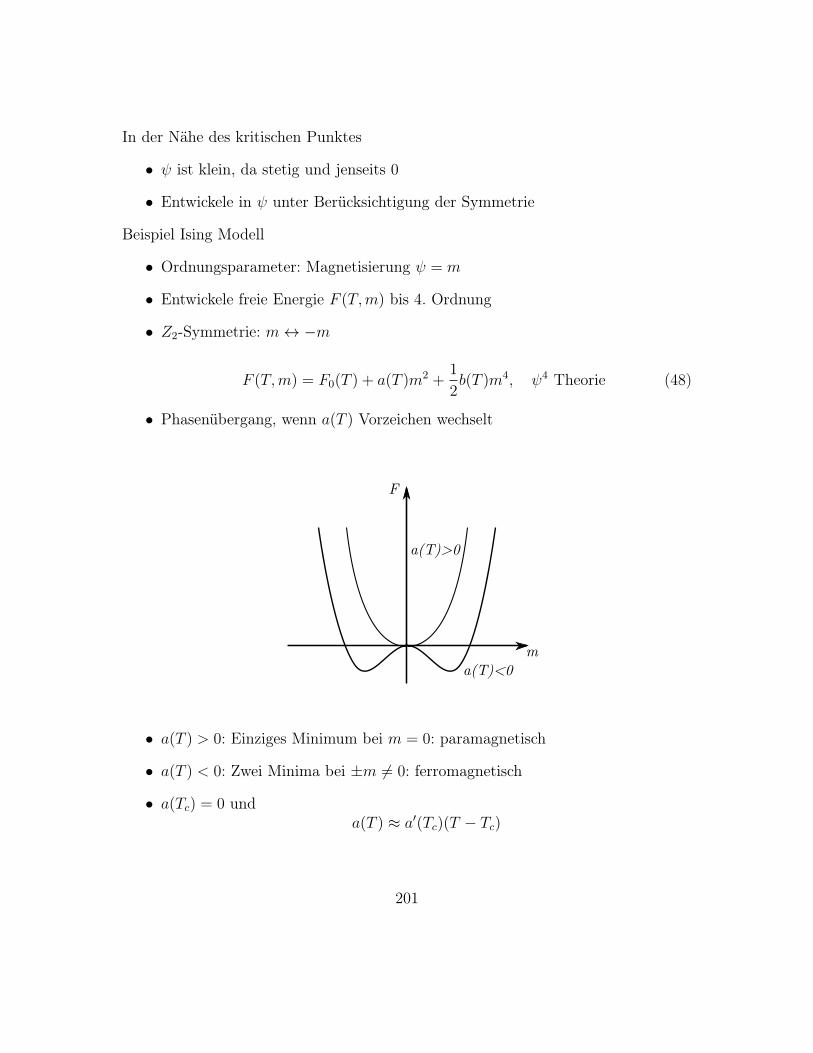
In der Nähe des kritischen Punktes
• ψ ist klein, da stetig und jenseits 0
• Entwickele in ψ unter Berücksichtigung der Symmetrie
Beispiel Ising Modell
• Ordnungsparameter: Magnetisierung ψ = m
• Entwickele freie Energie F (T,m) bis 4. Ordnung
• Z2-Symmetrie: m↔ −m
F (T,m) = F0(T ) + a(T )m2 +1
2b(T )m4, ψ4 Theorie (48)
• Phasenübergang, wenn a(T ) Vorzeichen wechselt
• a(T ) > 0: Einziges Minimum bei m = 0: paramagnetisch
• a(T ) < 0: Zwei Minima bei ±m 6= 0: ferromagnetisch
• a(Tc) = 0 unda(T ) ≈ a′(Tc)(T − Tc)
201

• b(T ) > 0
Um TC : b(T ) ≈ b(Tc) = b
Bestimmung der Magnetisierung:
• Differenziere Gl. (48) und setze Ableitung gleich 0:
0!= 2a(T )m+ 2bm3
a(T ) = −2bm2
Damit
m(T ) =
0 a(T ) > 0
±(|a(T )|b
)1/2
a(T ) < 0
• Da a(T ) ∝ T−TcTC
= t
m(T ) ∝ ±|t|β, β =1
2
wie in der Mean-Field Approximation, gilt allgemein
• Ohne jede mikroskopische Information !
Spontane Symmetriebrechung :
• Symmetrie im Grundzustand ist kleiner als Symmetrie des Hamiltonian
• Tiefer Zusammenhang zwischen Statistischer Physik und Quantenfeldtheorie
• Von hier ist es nicht mehr weit zum Higgs-Boson
Lessons learned
• Phasenübergänge unterscheiden sich nach Analytizität
• Charakteristische Größen zeigen am kritischen Punkt Skalierungsverhalten
• Landau’s ψ4 Theorie: Skalierung nur auf Grund von Symmetrien und Dimen-sion des Ordnungsparameters, keine mikroskopischen Details
14. weekhalbeWoche15
202

15 DanksagungDieses Skript beruht in wesentlichen Teilen auf dem Buch ”Thermodynamik undStatistische Physik” von W. Greiner, L. Neise und H. Stöcker. Viele Anregungenentstammen dem Vorlesungsskript zur gleichnamigen Vorlesung von G. Stock undden Büchern ”Stastistische Mechanik” von H. Römer und T. Filk und dem Buch”Statistical Physics” meines akademischen Lehrers J. Honerkamp. Last, not leastdanke ich D. Bachmann für das gründliche Korrektur lesen des Skriptes.
203





























