Embed Size (px)
Citation preview

Solid State Blending of Poly(ethylene terephthalate) withPolystyrene: Extent of PET Amorphization andCompositional Effects on Crystallizability
RYAN J. SCHEXNAYDRE,1 BRIAN S. MITCHELL1,2
1Department of Chemical and Biomolecular Engineering, Tulane University, New Orleans, Louisiana 70118
2Tulane Institute for Macromolecular Engineering and Science, Tulane University, New Orleans, Louisiana 70118
Received 29 August 2007; revised 2 March 2008; accepted 31 March 2008DOI: 10.1002/polb.21469Published online in Wiley InterScience (www.interscience.wiley.com).
ABSTRACT: Polystyrene (PS) and poly(ethylene terephthalate) (PET) were blended to-gether in the solid state via cryogenic mechanical attrition (CMA) and in the meltthrough conventional twin-screw extrusion. CMA PS/PET blend morphologies werecharacterized both qualitatively and quantitatively through microscopy and thermalanalysis. Specifically, CMA reduced the dispersed-phase domain size and its distribu-tion relative to simple melt extrusion, although not to the extent attained with addedchemical compatibilizers. CMA also amorphized the PET phase and depressed the PETcold crystallization rate, which was measured by post-CMA nonisothermal MDSC anal-ysis. The PET amorphization efficiency and crystallizability for CMA PS/PET blendswere the highest and lowest, respectively, at the PS/PET phase inversion. These con-comitant phenomena are known to be caused by CMA-induced PET crystal defect for-mation and subsequent entropic stabilization. Such behaviors are linked to theenhanced presence of an uncrystallizable rigid amorphous PET phase, and the weightfraction of this rigid amorphous fraction (RAF PET) was quantified and also maximizednear the PS/PET phase inversion. Moreover, the increased compatibilization andamorphization efficiencies and reduced PET crystallizability were determined to beinterdependent. These studies have verified that CMA of PET with PS is more efficientthan extrusion due to the formation of nonequilibrium, metastable morphologies thatcan be more precisely controlled and better stabilized with an interesting, composition-dependent interplay between PET crystallizability and the extent of PS/PET compatibi-lization. VVC 2008 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 46: 1348–1359, 2008
Keywords: blending; blends; compatibilization; crystallization; immiscible PS/PETblends; polystyrene; processing; rigid amorphous fraction; solid state
INTRODUCTION
Immiscible polystyrene/poly(ethylene terephtha-late), PS/PET, blends can be compatibilized bycryogenic mechanical alloying (CMA), and the
extent of compatibilization was previously deter-mined to be both higher than that of extrudedblends and also dependent upon blend morphol-ogy and composition.1 Because compatibility isdefined as the removal of amorphous PET fromthe bulk to the interphase with PS, a highercompatibilization efficiency means that corre-spondingly higher amounts of amorphous PETare becoming entangled at the interphase withPS. Previous work by Font et al.2,3 have shown
Correspondence to: B. S. Mitchell (E-mail: [email protected])
Journal of Polymer Science: Part B: Polymer Physics, Vol. 46, 1348–1359 (2008)VVC 2008 Wiley Periodicals, Inc.
1348

that PET crystalline content and the crystalliz-ability of the PET amorphous phase are reducedby ball milling, both with and without the pres-ence of poly(butylene terephthalate), PBT. It hasalso been shown that the addition of glassynanosilica to PET introduces significant inter-phase area that directly impacts PET crystalnucleation and growth processes and improvesPET thermal stability and mechanical proper-ties.4
Similarly, the shear strength development forasymmetric, incompatible amorphous PS/PETinterphases is comparable to compatible crystal-line PET/PET interphases, both below and aboveTg.
5 Furthermore, these studies showed that acompatibilized amorphous PS/PS interphaseshows significantly higher shear strength rela-tive to either the incompatible, amorphous PS/PET or the compatible, crystalline PET/PETinterphases. One could then interpolate that acompatibilized, amorphous PS/PET interphasewould have increased strength over any incom-patible heteropolymer analogues. Thus, anyincrease in interphase strength or adhesion isexpected with compatibilization of the PS/PETinterphase. So, it is hypothesized that CMA willcreate stronger PS/PET interphases due to thecompatibilization of the two polymers throughminimization of incompatible PS/PET inter-phases and an increase in compatible, amor-phous PET entanglements with PS.
The effects of CMA on blend morphology andproperties for similar systems, for example,PMMA/PEP and iPP/sPS, illustrate that CMA isnot merely refining a physical mixture—irre-versible structural changes induce correspond-ing alterations in the crystallization behaviorand macroscopic properties.6,7 Moreover, PS/PET studies primarily focus upon PET crystalli-zation changes, for example, qualitative shifts inonset temperature and peak characteristics, formelt-processed blends.8 The explanations of anycompositional dependences of these trends, forexample, cold crystallization peak area, are of-ten brief and do not propose any fundamentalrelationships to the formation or composition de-pendence of compatibilized PS/PET interphases.Although several investigations of chemicallycompatibilized, melt-blended PS/PET systemsprovide semiquantitative estimates of the dis-persed phase size and specific structure-propertyrelationships, these studies are limited to fixedPET-rich compositions, do not specify or quan-tify compatibilization efficiencies, and do not
establish relationships between concomitantphenomena, that is, crystallizability and compa-tibilization.9–11
With regards to these deficiencies, the goalsof this article are to (1) illustrate that CMA pro-duces finer dispersions than extrusion; (2) dem-onstrate that CMA amorphizes the PET phasein PS/PET blends and does so more than millingof pure PET; (3) verify that CMA depresses PETcrystallizability and alters its growth dimension-ality in PS/PET blends; (4) explain any composi-tional dependences of PET amorphization anddepressed crystallizability in CMA PS/PETblends relative to milling of pure PET; (5) relatethese concomitant phenomena to the presence ofuncrystallizable, rigid amorphous PET phase;(6) show how this uncrystallizable PET phase isenhanced in CMA PS/PET blends relative toboth neat and milled PET as well as extrudedblends; and (7) highlight the efficiency of CMAas its processing advantage over extrusion.Although some of these investigations merelypinpoint the CMA technique alone, the lattermajority of this work specifically addresses post-processing stability concerns, that is, thecrystallizability of the affected PET phase istargeted as comparative measure of PS/PETblend demixing that reverses the benefits ofcompatibilization.
EXPERIMENTAL
Materials and Methods
Poly(ethylene terephthalate) (PET), [g] ¼ 0.58,and atactic polystyrene (aPS), Mw ¼ 260,000,were purchased in pellet form from ScientificPolymer Products, Inc. (Ontario, NY). The Mw
for PET is �43,000 g mol�1—an order of magni-tude lower than that of PS. The polymers weremechanically attritted in pure form, that is,cryogenic mechanical milling (CMM), and alsomechanically alloyed via CMA with a SPEX6750 Mixer/Mill. Mixtures of �30, 50, and 70nominal weight percent PET were processed,and the SPEX 6750 was operated at liquid nitro-gen temperature (�196 8C) for 30 min total mill-ing time for each sample within polycarbonatemilling vials. Stainless steel (SS) ends cappedthe vials, preventing sample leakage duringmilling. The mechanism of impact was compres-sive stress transferred to the trapped sample asthe SS impactor collided into the vial ends. Dual
SOLID STATE BLENDING OF PET WITH PS 1349
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

electromagnets provided the force required tooscillate the impactor horizontally at the maxi-mum rate of 30 Hz. Milled powders were imme-diately stored in a dry environment.
Conventional melt extrusion was performedat 220 8C at 100 rpm in a ThermoHaake Rheo-mix counterrotating twin-screw extruder (Wal-tham, MA). The diameter of the extruder is25 mm with L/D ¼ 16 and three heating zones.The temperature of all zones was maintainedconstant for all samples. Pure PS and PET wereextruded first to determine the optimal process-ing parameters such that the PET crystals werefully molten as the blend exited the die. It wasalso ensured that the melt viscosity did notdecrease so much that the extrudate flowed toofast for adequate cooling and pelletizing. BulkPS/PET mixtures that were 30, 50, and 70 nomi-nal weight percent PET were fed into the ex-truder at �120 g per charge. The extruded pel-lets were then dried in an oven at 60 8C for 24 hto remove any water incorporated during extru-date cooling.
MDSC and TGA Characterization and Analysis
Thermal properties of blends and pure compo-nents were studied by modulated differentialscanning calorimetry (MDSC) in a TA Instru-ments DSC 2920 (Newcastle, DE). Prior to datacollection, the instrument was calibrated forbaseline, temperature (indium melting point),and modulation (sapphire specific heat) in drynitrogen at 5 8C min�1 between 60 and 300 8Cwith a modulation amplitude of 0.53 8C and aperiod of 40 s. Dry nitrogen gas at a flow rate of50 mL min�1 was used for the purge gas. Forthe experimental runs, the samples were firstheld isothermally at 60 8C for 5 min to equili-brate. All relevant thermal transitions werethen recorded, for example, the PET crystalliza-tion temperature, Tc, and crystalline meltingpoint, Tm, as well as the corresponding widthsfor these transitions, DTc and DTm. The onsetand final temperatures for both cold crystalliza-tion and melting provided peak widths and lim-its of integration for the evaluation of the heatsof PET crystallization and fusion (DHc and DHm,respectively). These various temperatures andpeak areas were determined accurately with TAInstruments Universal Analysis 2000 software.Blend compositions were accurately determinedfrom a TA Instruments TGA 2950 Thermogravi-tometric Analyzer (Newcastle, DE) calibration
curve that was previously created. AlthoughPET Tg values were of interest for previous PS/PET compatibilization studies,1 they were alsorecorded in this study solely for calculation ofTm � Tg. Three independent samples were runper nominal composition, and appropriate statis-tical methods were used to generate correspond-ing error bars.
Fluorescent Confocal Microscopy
The dispersed-phase domain size distribution inCMA PS/PET blends was determined by laserscanning fluorescent confocal microscopy(LSCFM), that is, a Zeiss LSM 510 confocalmicroscope (Jena, Germany) with a specific laserwith k at or below 468 nm to fluoresce dry-dyedgreen PS microspheres (Duke Scientific Corp.,Palo Alto, CA). These fluorescent tags werenearly monodisperse particles of diameter 6 lm,and control experiments were initially per-formed to verify miscibility in PS and immisci-bility in PET. The optimal blending protocol con-sisted of initially premilling the 5 wt % dye intoPS powders for 15 min and subsequently comil-ling this dye-PS mixture with PET via CMA for30 min. This procedure ensured that the dyespacing in the PS phase was small enough todistinguish it from the PET phase in the confo-cal images yet not incorporating too much dyeso as to create a third dye phase in the PS/PETblends. Although the size of the dye particlescan mask dispersed phase domains smaller than6 lm, it was thought that these smaller domainscan be accounted for and estimated by differen-ces in dye spacing intervals.
The milled PS-dye/PET blends of 30 and 70nominal weight percent PET were then sub-jected to Buehler Specimen Mount Press (LakeBluff, IL) at 8000 psi for an hour at room tem-perature to consolidate the powders into a thinfilm for confocal imaging between two glassmicroscope slides. Since the mechanical pressingoccurred below Tg, the thermal histories of theblends were not compromised. During imaging,the fluorescence intensity was first adjusted toensure maximum contrast and elimination ofother imaging effects. Images were collected asz-stacks with the interval optimized by the Zeisssoftware, which also estimated the domain sizeand its distribution. Three independent regionswithin a sample disk of each nominal composi-tion were analyzed, and appropriate statisticalmethods were used to calculate the average size
1350 SCHEXNAYDRE AND MITCHELL
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

of the dispersed phase and its polydispersity forcomparison to literature blends.
RESULTS AND DISCUSSION
Effects of CMA on PS/PET Blend Morphology
The confocal microscopy image in Figure 1 illus-trates the excellent phase contrast between PSand PET in a CMA 70/30 PS/PET blend. Asstated previously, the fluorescent green dyemicrospheres preferentially ‘‘tag’’ the PS phase.The PET domains in this particular image arenot spherical and are characterized by irregular-ity, that is, angularity and eccentricity. Thesedeviations from sphericity also exist for PSdomains in a CMA 30/70 PS/PET blend andallow for more interphase contact between amor-phous PET and PS phases. PS and PET powdersare increasingly subjected to a combination offracture and cold-welding with milling time,leading to increased defect formation and inter-nal stresses. CMA has been shown to introduceirregularity in dispersed-phase shapes for vari-ous systems, for example, PMMA/PEP and PC/PEEK.7,12 More specifically, CMM of PET wasinvestigated by Zhu et al.13 and was shown to
induce physical interpenetration of chains withconcomitant changes in surface, defect, andstrain energies, that is, the formation of a semi-stable thermodynamic state. Conversely, SEMmicrographs of PS/PET blends processed bymelt mixing and/or extrusion, both with andwithout reactive compatibilizers, clearly showmuch more spheroidal dispersed phases.9–11 Itcan be inferred, therefore, that a metastable,phase-separated state exists at the scale of theimage in Figure 1 because of the nonequilibriumdomain shapes that are characteristic of immis-cible material mixtures.
From such LCSFM images in Figure 1, boththe number-average dispersed-phase domainsize (Dn) and its standard deviation (r) were cal-culated in lm.14 Finer dispersions certainlyresult from increased milling time, for example,a 30/70 PS/PET blend milled for 30 and 300 minhad domain size distributions (Dn 6 r) of 12 63.5 and 8 6 1.5 lm, respectively. However, sev-eral PS/PET blend studies9–11 indicate that alevel of dispersion near 1–2 lm is possible withreactively functionalized copolymers added aschemical compatibilizers in melt extrusion proc-esses. Moreover, even after 10 h of CMM, PETparticle sizes still average only about 10 lm.13
The reduction of the PS/PET domain size distri-bution, therefore, is not the prevalent driver ofPS/PET compatibilization since previous studieshave shown that CMA, particularly at longermilling times, is comparable with conventionalmelt blending—even with the presence of chemi-cal compatibilizers.1 Thus, the increased com-patibility of CMA PS/PET blends must bederived from another quantifiable, milling-in-duced phenomenon that substantially increasesamorphous PET/aPS entanglements at theirinterphase in lieu of intrinsic solid-state domainsize restrictions.
Compositional Dependences for PET ThermalTransitions
The decreases in PET particle size with highermilling times are synonymous with a reductionin the PET crystallite domain size. Generallyspeaking, the amount of amorphization in mate-rials has been shown to be related to the amountof initial crystallinity prior to processing bymeans of a thermodynamic driving force to cre-ate defects.15 So, any amorphization of PETwould create ‘free’ amorphous chains that can beused to entangle with PS chains at their inter-
Figure 1. A laser-scanning confocal fluorescent mi-croscopy (LSCFM) image of a CMA 70/30 PS/PETblend. The green fluorescent tags are miscible in thePS phase, so PET crystalline domains are seen hereas dark regions with no fluorescence. Note the non-spheroidal nature of the PET domains.
SOLID STATE BLENDING OF PET WITH PS 1351
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb
phase. In order for a large quantity of theseamorphous PET chains to access these inter-phase environments, they require sufficient self-diffusion from the PET bulk phase. It is thus im-portant for CMA to simultaneously destroy PETcrystallites and promote fine, stable dispersionsof much smaller and more defective crystallites.
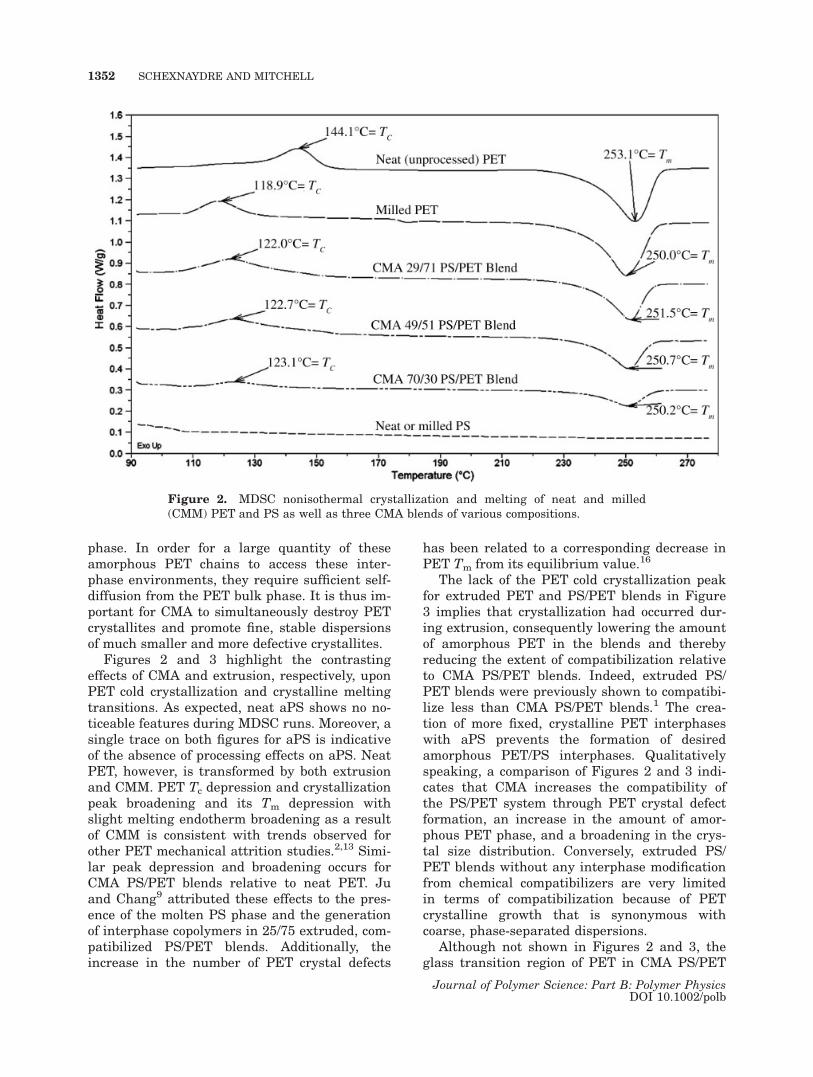
Figures 2 and 3 highlight the contrastingeffects of CMA and extrusion, respectively, uponPET cold crystallization and crystalline meltingtransitions. As expected, neat aPS shows no no-ticeable features during MDSC runs. Moreover, asingle trace on both figures for aPS is indicativeof the absence of processing effects on aPS. NeatPET, however, is transformed by both extrusionand CMM. PET Tc depression and crystallizationpeak broadening and its Tm depression withslight melting endotherm broadening as a resultof CMM is consistent with trends observed forother PET mechanical attrition studies.2,13 Simi-lar peak depression and broadening occurs forCMA PS/PET blends relative to neat PET. Juand Chang9 attributed these effects to the pres-ence of the molten PS phase and the generationof interphase copolymers in 25/75 extruded, com-patibilized PS/PET blends. Additionally, theincrease in the number of PET crystal defects
has been related to a corresponding decrease inPET Tm from its equilibrium value.16
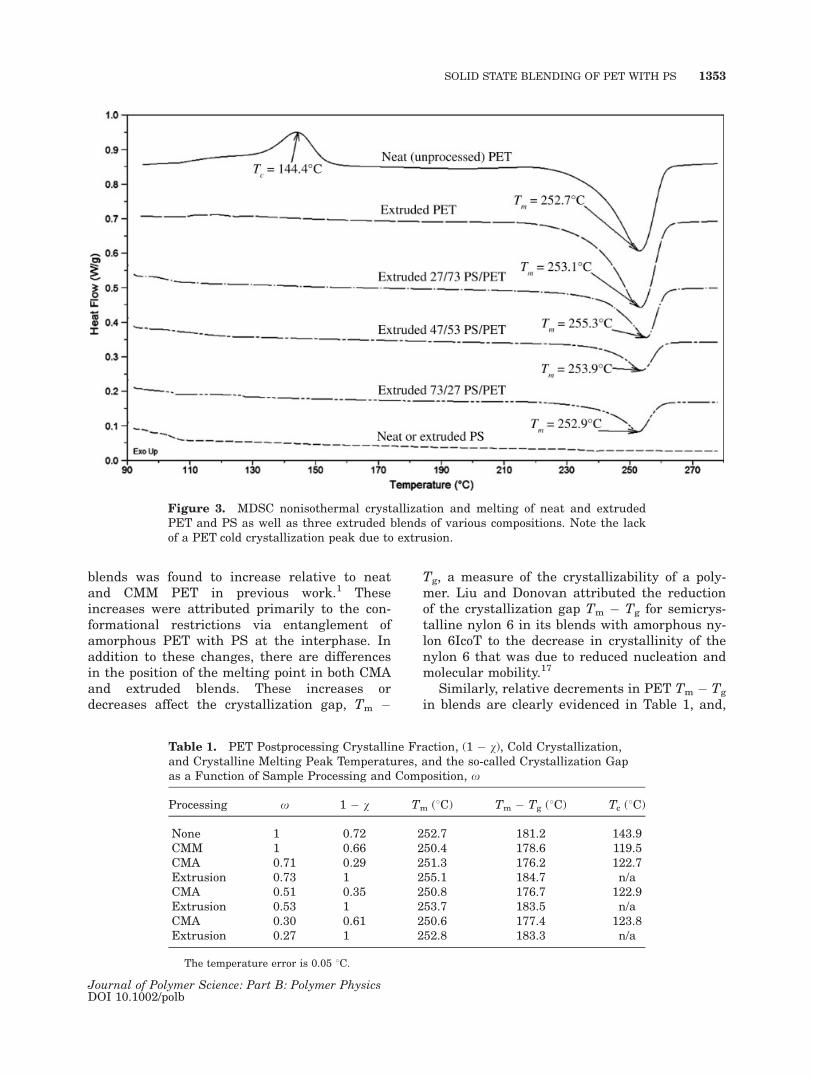
The lack of the PET cold crystallization peakfor extruded PET and PS/PET blends in Figure3 implies that crystallization had occurred dur-ing extrusion, consequently lowering the amountof amorphous PET in the blends and therebyreducing the extent of compatibilization relativeto CMA PS/PET blends. Indeed, extruded PS/PET blends were previously shown to compatibi-lize less than CMA PS/PET blends.1 The crea-tion of more fixed, crystalline PET interphaseswith aPS prevents the formation of desiredamorphous PET/PS interphases. Qualitativelyspeaking, a comparison of Figures 2 and 3 indi-cates that CMA increases the compatibility ofthe PS/PET system through PET crystal defectformation, an increase in the amount of amor-phous PET phase, and a broadening in the crys-tal size distribution. Conversely, extruded PS/PET blends without any interphase modificationfrom chemical compatibilizers are very limitedin terms of compatibilization because of PETcrystalline growth that is synonymous withcoarse, phase-separated dispersions.
Although not shown in Figures 2 and 3, theglass transition region of PET in CMA PS/PET
Figure 2. MDSC nonisothermal crystallization and melting of neat and milled(CMM) PET and PS as well as three CMA blends of various compositions.
1352 SCHEXNAYDRE AND MITCHELL
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb
blends was found to increase relative to neatand CMM PET in previous work.1 Theseincreases were attributed primarily to the con-formational restrictions via entanglement ofamorphous PET with PS at the interphase. Inaddition to these changes, there are differencesin the position of the melting point in both CMAand extruded blends. These increases ordecreases affect the crystallization gap, Tm �
Tg, a measure of the crystallizability of a poly-mer. Liu and Donovan attributed the reductionof the crystallization gap Tm � Tg for semicrys-talline nylon 6 in its blends with amorphous ny-lon 6IcoT to the decrease in crystallinity of thenylon 6 that was due to reduced nucleation andmolecular mobility.17
Similarly, relative decrements in PET Tm � Tg
in blends are clearly evidenced in Table 1, and,
Figure 3. MDSC nonisothermal crystallization and melting of neat and extrudedPET and PS as well as three extruded blends of various compositions. Note the lackof a PET cold crystallization peak due to extrusion.
Table 1. PET Postprocessing Crystalline Fraction, (1 � v), Cold Crystallization,and Crystalline Melting Peak Temperatures, and the so-called Crystallization Gapas a Function of Sample Processing and Composition, x
Processing x 1 � v Tm (8C) Tm � Tg (8C) Tc (8C)
None 1 0.72 252.7 181.2 143.9CMM 1 0.66 250.4 178.6 119.5CMA 0.71 0.29 251.3 176.2 122.7Extrusion 0.73 1 255.1 184.7 n/aCMA 0.51 0.35 250.8 176.7 122.9Extrusion 0.53 1 253.7 183.5 n/aCMA 0.30 0.61 250.6 177.4 123.8Extrusion 0.27 1 252.8 183.3 n/a
The temperature error is 0.05 8C.
SOLID STATE BLENDING OF PET WITH PS 1353
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb
interestingly enough, the 30/70 PS/PET blendshows the smallest crystallization gap. Thehigher probability of PET crystal destruction dueto a higher quantity of PET, combined with hin-dered PET nucleation related to a correspond-ingly high amount of amorphous PS/PET inter-phase area, are the primary reasons for this mini-mum in the crystallization gap. The increase ofamorphous PET Tg and the decrease of the crys-talline PET Tm cooperatively signify thedepressed crystallizability of the PET phase. ThePET amorphous phase has been shown previouslyto be affected by both CMA and extrusion becausethe position of PET Tg in these processed blendschanges relative to that of pure neat PET.1
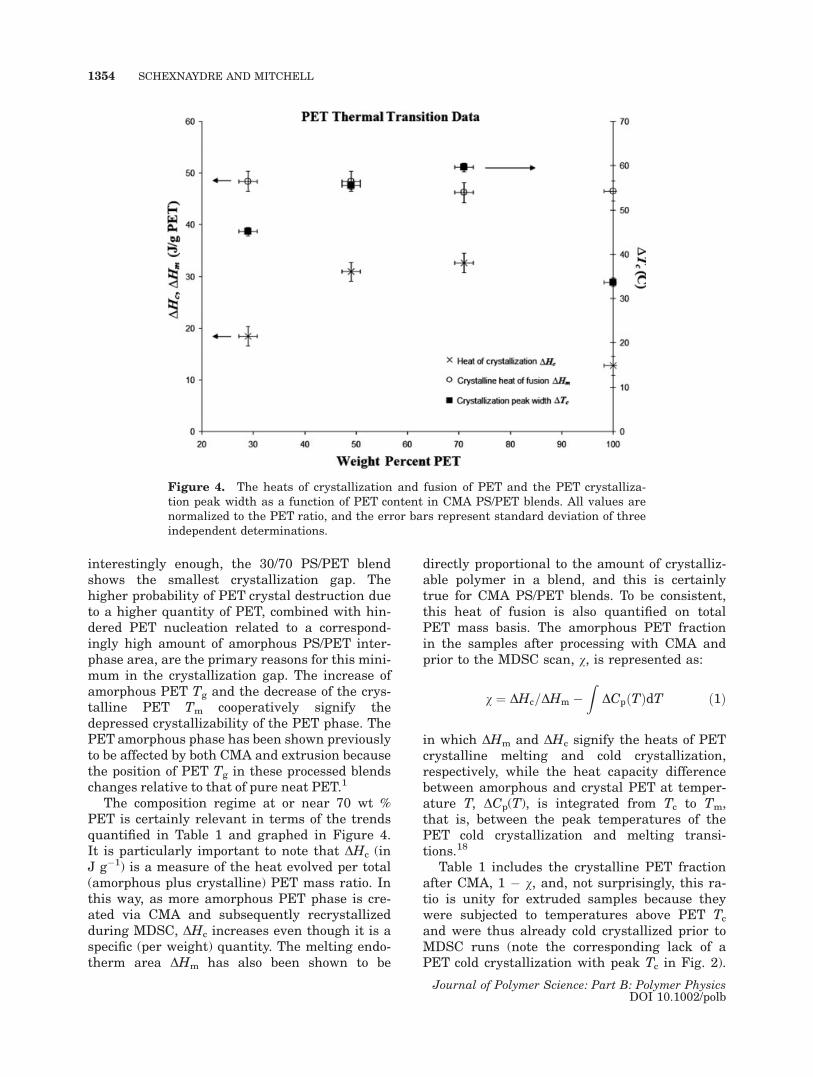
The composition regime at or near 70 wt %PET is certainly relevant in terms of the trendsquantified in Table 1 and graphed in Figure 4.It is particularly important to note that DHc (inJ g�1) is a measure of the heat evolved per total(amorphous plus crystalline) PET mass ratio. Inthis way, as more amorphous PET phase is cre-ated via CMA and subsequently recrystallizedduring MDSC, DHc increases even though it is aspecific (per weight) quantity. The melting endo-therm area DHm has also been shown to be
directly proportional to the amount of crystalliz-able polymer in a blend, and this is certainlytrue for CMA PS/PET blends. To be consistent,this heat of fusion is also quantified on totalPET mass basis. The amorphous PET fractionin the samples after processing with CMA andprior to the MDSC scan, v, is represented as:
v ¼ DHc=DHm �Z
DCpðTÞdT ð1Þ
in which DHm and DHc signify the heats of PETcrystalline melting and cold crystallization,respectively, while the heat capacity differencebetween amorphous and crystal PET at temper-ature T, DCp(T), is integrated from Tc to Tm,that is, between the peak temperatures of thePET cold crystallization and melting transi-tions.18
Table 1 includes the crystalline PET fractionafter CMA, 1 � v, and, not surprisingly, this ra-tio is unity for extruded samples because theywere subjected to temperatures above PET Tc
and were thus already cold crystallized prior toMDSC runs (note the corresponding lack of aPET cold crystallization with peak Tc in Fig. 2).
Figure 4. The heats of crystallization and fusion of PET and the PET crystalliza-tion peak width as a function of PET content in CMA PS/PET blends. All values arenormalized to the PET ratio, and the error bars represent standard deviation of threeindependent determinations.
1354 SCHEXNAYDRE AND MITCHELL
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

As expected, the values for 1 � v in Table 1 areless for CMM PET and CMA PS/PET than forneat PET, thereby giving quantitative proof thatPET crystallinity was destroyed by the millingprocess. Interestingly enough, this postmillingPET crystallinity is less for all CMA PS/PETblends than for CMM PET. This implies that thepresence of PS also promotes amorphization ofthe PET phase. The minimum at 70 wt % PETphysically means that CMA has the highestamorphization efficiency in this PET-rich compo-sition window, that is, for the same amount ofimpact energy, the PET phase in these blends isamorphized the most. According to Jang et al.,the phase inversion for PS/PET blends occurs at60 wt % PET and is known to favor the compati-bility of the blend.11 Thus, this high amorphiza-tion and compatibilization efficiency compositionwindow is near the phase inversion for PS/PET.
In theory, most of the newly-formed amor-phous PET should merely entangle with PS andcontribute solely to the compatibilization be-tween the two polymers. However, there is alsoa thermodynamic driving force for amorphousPET to be recrystallized during heating. If thesenew, free amorphous PET chains are notentangled with PS at the interphase and are nothindered diffusionally, then MDSC heating willsubsequently recrystallize them. This postpro-cessing thermal treatment would then increasethe number of weak crystalline PET/aPS inter-phases and deteriorate mechanical properties.Thus, it is desirable to quantify and comparethe rate of PET cold crystallization for neat andCMM PET to CMA PS/PET blends and is alsorelevant to determine whether the quantifiedamorphous PET phase is stabilized with respectto postmilling recrystallization.
PET Cold Crystallization Kinetic Studies for CMAPS/PET Blends
The rate at which the amorphous PET chainsrecrystallize during cold crystallization providesa quantitative measure of the deviation fromthermodynamic equilibrium as well as the sta-bility of the milled blends. The overall PET crys-tallization kinetics is captured by a modifiednonisothermal Avrami equation:
a¼1�exp��ðktÞn
�¼Aðt;TÞ=Atotal
¼Z
ðdHc=dtÞ�dt=DHc¼Z
dHc=dTÞ�dT=DHc ð2Þ
where a is the relative degree of crystallinity orextent of conversion from the amorphous to crys-talline phase, k is an overall rate constant, and nis the Avrami exponent associated with the crys-tallization mechanism.19,20 The fractional con-version a represents the area fraction of the coldcrystallization peak that has been crystallized attime and temperature t and T, respectively, sincenonisothermal MDSC experiments involve tem-perature-time relationships with a constantheating rate u, that is, t ¼ (T � To)/u where T isthe temperature at time t and To is the tempera-ture at the very beginning (onset) of crystalliza-tion. The integrals dHc /dt and dHc /dT representthe heat evolution at time t or temperature Tduring MDSC runs. Bishara and Shaban21 modi-fied the Avrami equation with a Mandelkern lin-earization, and Wang et al.20 subsequently cor-rected the isothermal crystallization rate con-stant k (also represented as Z) for nonisothermalcrystallization, that is, log Zt ¼ (log Z)/u. Thecrystallization half-time t1/2 is taken as theinverse of the overall rate and also physicallysignifies the time at which 50% of the PET coldcrystallization had been completed.22
The double log Avrami plot in Figure 5 is forthe nonisothermal cold crystallization of PET ina CMA PS/PET 30/70 blend. The abscissa corre-sponds to the logarithm of time in seconds,while the ordinate is related to the logarithm ofthe extent of crystallization. Two differentregions, which correspond to two different typesof cold crystallization, characterize all CMA PS/PET blends. Primary spherulitic crystallizationoccurs at earlier times and shows no deviationfrom linearity in such a double log plot. Second-ary interlamellar crystallization often evolveslater in the overall crystallization process and isidentified with nonlinearities. In studies of PET,it has been well-established that secondary crys-tallization, or growth from defective crystallites,is characterized by more restricted amorphouschains of reduced entropy due the presence ofneighboring lamellar stacks.23 The relative con-tribution of secondary crystallization to the over-all crystallization process was found to bedirectly proportional to the amount of PET inCMA PS/PET blends.
The data in Table 2 correspond to least-square fits of linear primary crystallization tothe double logarithm of eq 2. The changes in theAvrami exponents with the addition of PS corre-spond to changes in the nucleation process forcrystallization, and the increased t1/2s for CMA
SOLID STATE BLENDING OF PET WITH PS 1355
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb
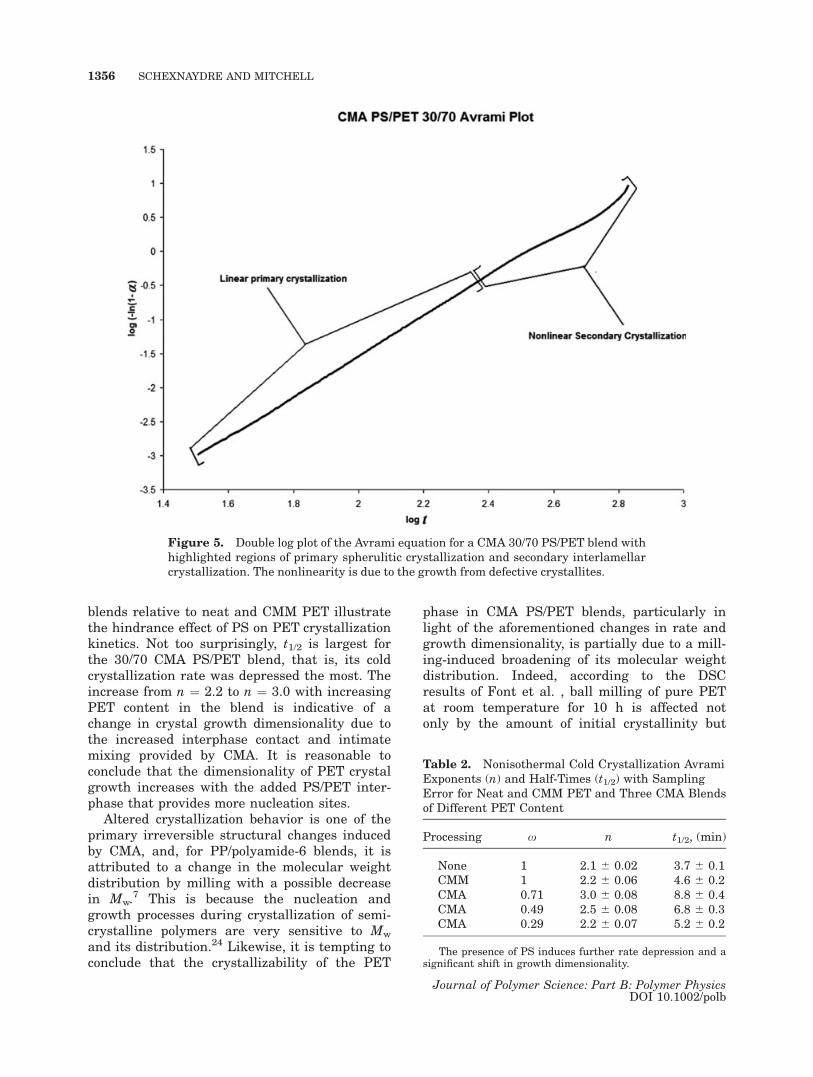
blends relative to neat and CMM PET illustratethe hindrance effect of PS on PET crystallizationkinetics. Not too surprisingly, t1/2 is largest forthe 30/70 CMA PS/PET blend, that is, its coldcrystallization rate was depressed the most. Theincrease from n ¼ 2.2 to n ¼ 3.0 with increasingPET content in the blend is indicative of achange in crystal growth dimensionality due tothe increased interphase contact and intimatemixing provided by CMA. It is reasonable toconclude that the dimensionality of PET crystalgrowth increases with the added PS/PET inter-phase that provides more nucleation sites.
Altered crystallization behavior is one of theprimary irreversible structural changes inducedby CMA, and, for PP/polyamide-6 blends, it isattributed to a change in the molecular weightdistribution by milling with a possible decreasein Mw.
7 This is because the nucleation andgrowth processes during crystallization of semi-crystalline polymers are very sensitive to Mw
and its distribution.24 Likewise, it is tempting toconclude that the crystallizability of the PET
phase in CMA PS/PET blends, particularly inlight of the aforementioned changes in rate andgrowth dimensionality, is partially due to a mill-ing-induced broadening of its molecular weightdistribution. Indeed, according to the DSCresults of Font et al. , ball milling of pure PETat room temperature for 10 h is affected notonly by the amount of initial crystallinity but
Table 2. Nonisothermal Cold Crystallization AvramiExponents (n) and Half-Times (t1/2) with SamplingError for Neat and CMM PET and Three CMA Blendsof Different PET Content
Processing x n t1/2, (min)
None 1 2.1 6 0.02 3.7 6 0.1CMM 1 2.2 6 0.06 4.6 6 0.2CMA 0.71 3.0 6 0.08 8.8 6 0.4CMA 0.49 2.5 6 0.08 6.8 6 0.3CMA 0.29 2.2 6 0.07 5.2 6 0.2
The presence of PS induces further rate depression and asignificant shift in growth dimensionality.
Figure 5. Double log plot of the Avrami equation for a CMA 30/70 PS/PET blend withhighlighted regions of primary spherulitic crystallization and secondary interlamellarcrystallization. The nonlinearity is due to the growth from defective crystallites.
1356 SCHEXNAYDRE AND MITCHELL
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

also by changes in the molecular weight distri-bution.2 However, gel permeation chromatogra-phy results of semicrystalline polyethylene oxide(PEO), amorphous polyvinylpyrrolidone (PVP),and their blends do not show significant molecu-lar weight degradation during cryogenic millingfor 30 min or less.1 This is primarily because oftheir inability to form stable mechanoradicalswhich exist long enough to avoid recombination.Neither PS nor PET can degrade into stableproducts at cryogenic temperatures due to sig-nificant structure-related energy barriers. It isreasonable to assume that very little, if any, mo-lecular weight degradation occurs for the PS/PET system at cryogenic temperatures and rela-tively short milling times.
Thus, the irreversible changes in the PETphase structure for CMA PS/PET blends must bedue to the milling-induced enhancement and sta-bilization of an uncrystallizable PET phase. Thisis confirmed by the increasing melting pointwith increasing PET content in CMA blends inTable 1 because thinner PET lamellae, charac-teristic of lower crystallinity, are stabilized bytheir reduced entropy, that is, Tm ¼ DHm/DSm.This reduced entropy is due to the enhancementof the so-called rigid amorphous phase, or frac-tion, (RAF) of PET, a conformationally restrictedamorphous PET phase that is directly related tothe degree PET crystal imperfection.25 The re-stricted conformations in the RAF PET phaseare those trans PET chains that came from thedestruction of the crystalline lattice by CMAbecause it has been shown by Tzavalas et al.that crystalline and RAF PET consist fully oftrans conformers.26 The decreased trans/gaucheconformation ratio with lower crystallinity isthus due to the increase of gauche conformers inthe fully amorphous phase as well as the re-moval of trans conformers from the fully crystal-line phase. RAF PET, therefore, retards thegrowth of crystalline PET by essentially forminga barrier to the fully (mobile) amorphous phase.
Quantitative Estimates of RAF PET in CMA PS/PETBlends
This RAF PET affects not only the vitrificationand devitrification of the amorphous PET struc-ture and the local melting behavior of crystalsbut also macroscopic mechanical behavior. Thus,it is necessary to quantify the amount and dis-tribution of RAF PET in neat and CMM PET aswell as CMA and extruded blends and funda-
mentally relate this information to the extent ofcompatibilization. Androsch et al. define theRAF as the ratio of uncrystallizable PET to thetotal amount of amorphous PET and have shownthat this ratio increases with respect to crystal-linity, reaching a maximum near 20 wt % crys-tallinity.25 The total amount of rigid amorphousfraction in the blend should also include anyRAF PS that would form via processing. How-ever, as mentioned previously, the PS phase hasvery little crystalline content, and there is alsono similar elevation of PS Tg relative to that ofneat PS as was evidenced for the PET phase,that is, the amount of PS entangled at the inter-phase with amorphous PET is small.1
As such, the amount of RAF in the PETphase alone is most relevant and provides anappropriate comparator of processing efficiencybecause its formation accounts for compatibi-lized amorphous PET/PS interphases and inter-lamellar PET nanophase. It is calculated as:
vPETRAF ¼ 1� vc � va¼ 1� ðDHm � DHcÞ=DH�
m � DCp=DCop ð3Þ
in which vPETRAF is the weight fraction of RAFPET remaining after considering both the crys-talline PET weight fraction vc and the fullyamorphous PET weight fraction va. DHc andDHm are the PET heats of crystallization andfusion, respectively, as previously defined. Thisway of defining vc is consistent with the litera-ture.27 The parameter DH�
m is the heat of fusionfor 100% crystalline PET and is taken as 129 J/g.15 The ratio DCp/DCo
p represents the ratio ofthe relaxation strengths of the amorphous PETphases in the blend to that of a completelyamorphous PET phase.28,29 The heat capacitychange at PET Tg in the CMA blend, DCp, waspreviously investigated and obtained duringMDSC runs calibrated for heat capacity meas-urements.1 The value of DCo
p of 77 J K�1 mol�1
(or 0.405 J K�1 g�1) is typically used for thefully amorphous PET sample and is based uponbreaking up the PET repeating unit into foursmall beads of 11 J K�1 mol�1 each and a largebead with a heat capacity increase triple that ofa small bead.25,30
Table 3 contains the bulk crystalline, bulkamorphous, and RAF PET weight fractions forneat and CMM PET as well as extruded andCMA PS/PET blends of various compositions.Not surprisingly, RAF PET exists in neat andCMM PET. However, the small decrease in crys-
SOLID STATE BLENDING OF PET WITH PS 1357
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb
tallinity due to CMM does not generate muchmore RAF PET, that is, the weight fractionincreases negligibly from 0.08 to 0.09. Extrudedblend samples are relatively more crystallinethan CMA samples and are characterized by rel-atively low amounts of RAF PET. Conversely,CMA blends contain more total amorphous PET,that is, the sum of fully amorphous and RAFPET. Not surprisingly, the amount of RAF PETin CMA blends is related to their composition.The PET phase in the 70/30 PS/PET blend doesnot change much relative to neat or CMM PET,so the small increase in RAF PET fraction from0.09 to 0.11 is due to the correspondingly smallamount of PET/PS interphase entanglements.As PET content increases towards the phaseinversion, significantly more RAF PET is gener-ated as the crystalline PET phase is increasinglydestroyed.
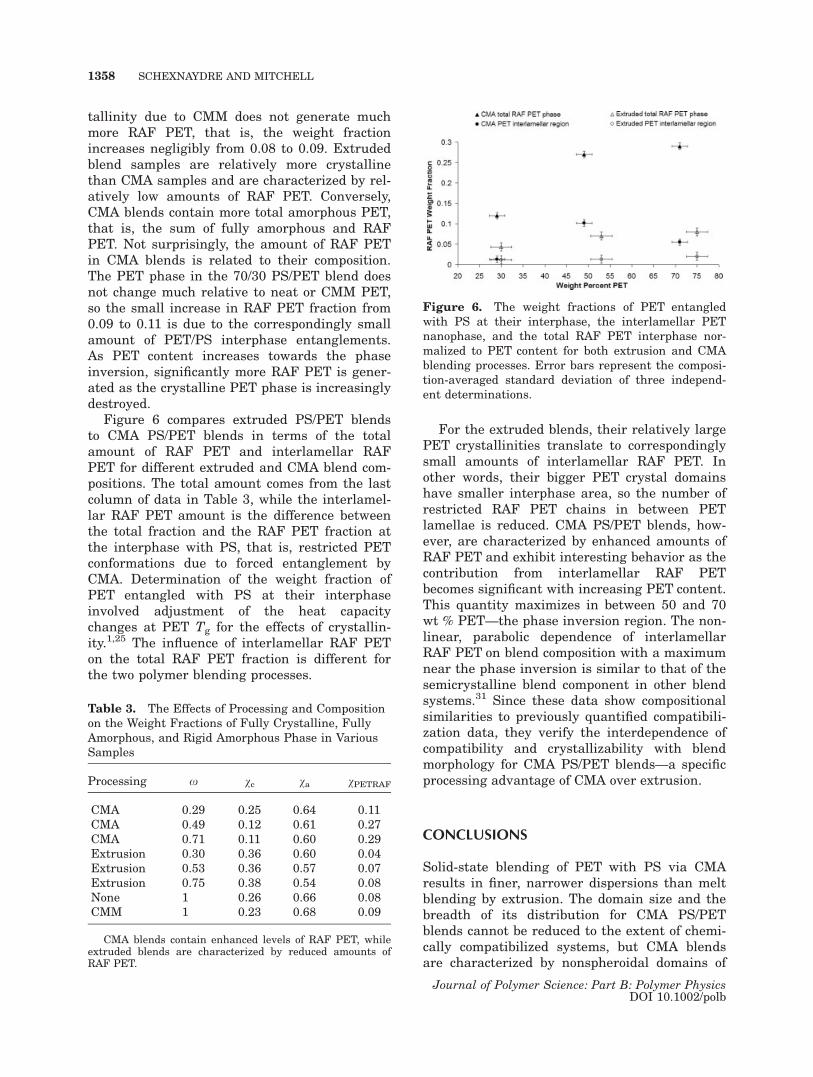
Figure 6 compares extruded PS/PET blendsto CMA PS/PET blends in terms of the totalamount of RAF PET and interlamellar RAFPET for different extruded and CMA blend com-positions. The total amount comes from the lastcolumn of data in Table 3, while the interlamel-lar RAF PET amount is the difference betweenthe total fraction and the RAF PET fraction atthe interphase with PS, that is, restricted PETconformations due to forced entanglement byCMA. Determination of the weight fraction ofPET entangled with PS at their interphaseinvolved adjustment of the heat capacitychanges at PET Tg for the effects of crystallin-ity.1,25 The influence of interlamellar RAF PETon the total RAF PET fraction is different forthe two polymer blending processes.
For the extruded blends, their relatively largePET crystallinities translate to correspondinglysmall amounts of interlamellar RAF PET. Inother words, their bigger PET crystal domainshave smaller interphase area, so the number ofrestricted RAF PET chains in between PETlamellae is reduced. CMA PS/PET blends, how-ever, are characterized by enhanced amounts ofRAF PET and exhibit interesting behavior as thecontribution from interlamellar RAF PETbecomes significant with increasing PET content.This quantity maximizes in between 50 and 70wt % PET—the phase inversion region. The non-linear, parabolic dependence of interlamellarRAF PET on blend composition with a maximumnear the phase inversion is similar to that of thesemicrystalline blend component in other blendsystems.31 Since these data show compositionalsimilarities to previously quantified compatibili-zation data, they verify the interdependence ofcompatibility and crystallizability with blendmorphology for CMA PS/PET blends—a specificprocessing advantage of CMA over extrusion.
CONCLUSIONS
Solid-state blending of PET with PS via CMAresults in finer, narrower dispersions than meltblending by extrusion. The domain size and thebreadth of its distribution for CMA PS/PETblends cannot be reduced to the extent of chemi-cally compatibilized systems, but CMA blendsare characterized by nonspheroidal domains of
Table 3. The Effects of Processing and Compositionon the Weight Fractions of Fully Crystalline, FullyAmorphous, and Rigid Amorphous Phase in VariousSamples
Processing x vc va vPETRAF
CMA 0.29 0.25 0.64 0.11CMA 0.49 0.12 0.61 0.27CMA 0.71 0.11 0.60 0.29Extrusion 0.30 0.36 0.60 0.04Extrusion 0.53 0.36 0.57 0.07Extrusion 0.75 0.38 0.54 0.08None 1 0.26 0.66 0.08CMM 1 0.23 0.68 0.09
CMA blends contain enhanced levels of RAF PET, whileextruded blends are characterized by reduced amounts ofRAF PET.
Figure 6. The weight fractions of PET entangledwith PS at their interphase, the interlamellar PETnanophase, and the total RAF PET interphase nor-malized to PET content for both extrusion and CMAblending processes. Error bars represent the composi-tion-averaged standard deviation of three independ-ent determinations.
1358 SCHEXNAYDRE AND MITCHELL
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

increased interphase area. CMA amorphizes thePET phase in PS/PET blends more so than CMMPET, and the amorphization efficiency increaseswith the PET content of the blends. Conversely,extrusion crystallizes PET and severely limits theformation of amorphous PET/aPS entanglements,thereby creating unstable, phase-separated dis-persions. PET cold crystallization rate studieswith nonisothermal MDSC reheating reveal thatthe PET phase within CMA PS/PET blends arecharacterized by lower recrystallization rates andsignificant changes in growth dimensionality rel-ative to CMM PET. Additionally, the recrystalli-zation rate decreased with increasing PET con-tent in CMA PS/PET blends. Both the PETamorphization and post-CMA crystallizabilitytrends correlate well with the extent of PS/PETcompatibilization, that is, the optimum composi-tion is at or near the phase inversion.
CMA concomitantly facilitates the formationof two types of amorphous PET phase fromdestruction of crystallites—the fully amorphousPET chains in the bulk and a rigid amorphousnanophase that suppresses PET recrystalliza-tion. The latter, known as RAF PET, caused devi-ations from linearity in Avrami plots due to sec-ondary crystallization from defective, smallercrystallites. Although present in neat and CMMPET, this RAF PET is more pronounced in CMAPS/PET blends, particularly near the phaseinversion. Extruded blends, however, are charac-terized by diminished levels of RAF PET—partic-ularly negligible amounts of interlamellar RAFPET. Thus, the primary processing advantages ofCMA over extrusion—the ability to direct theinterplay between compatibility and crystalliz-ability through precise compositional control andthe creation of nonspheroidal blend morphologiesof enhanced, intimate contact between PS andPET—were certainly quantified in this study.
The authors thank Dr. Hongxia Zhou for her helpwith thermal analysis data acquisition. Financial sup-port for this project was provided by NASA grantNNC06AA18A.
REFERENCES AND NOTES
1. Schexnaydre, R. J.; Mitchell, B. S. Polym Eng Sci2008, 48, 649–655.
2. Font, J.; Muntasell, J.; Cesari, E. ThermochimActa 1999, 333, 169–172.
3. Font, J.; Muntasell, J.; Cesari, E. Mater Res Bull1999, 34, 157–165.
4. Chae, D. W.; Kim, B. C. J Mater Sci 2007, 42,1238–1244.
5. Boiko, Y.; Guerin, G.; Marikhin, V. A.; Prud’-homme, R. E. Polymer 2001, 42, 8695–8702.
6. Stranz, M.; Koster, U. J Mater Sci 2004, 39,5275–5277.
7. Smith, A. P.; Ade, H.; Balik, C. M.; Koch, C. C.;Smith, S. D.; Spontak, R. J. Macromolecules2000, 33, 2595–2604.
8. Tankhiwale, S.; Gupta, M. C.; Viswanath, S. G.J Polym Plast Technol Eng 2002, 41, 171–181.
9. Ju, M. Y.; Chang, F. C. Polymer 41 2000, 41,1719–1730.
10. Lee, J.; Park, K.; Yoo, D.; Suh, K. J Polym SciPart B: Polym Phys 2000, 38, 1396–1404.
11. Jang, L. W.; Lee, K. H.; Lee, D. C.; Yoon, J. S.;Chin, I. J.; Choi, H. J.; Lee, K. H. J Polym SciPart B: Polym Phys 2000, 78, 1998–2007.
12. Martin, J. P.; McCartney, S. R.; Kander, R. G.J Mater Sci 2003, 38, 195–200.
13. Zhu, Y. G.; Li, Z. Q.; Zhang, D.; Tanimoto, T.J Appl Polym Sci 2006, 99, 2868–2873.
14. Tol, R. T.; Mathot, V. B. F.; Groeninckx, G. Poly-mer, 2005, 46, 383–396.
15. Zhang, D. L. Prog Mater Sci 2004, 49, 537–560.16. Tankhiwale, S.; Gupta, M. C.; Viswanath, S. G.
J Polym Plast Technol Eng 2000, 39, 543–552.17. Liu, Y.; Donovan, J. Polymer 1995, 36, 4797–4803.18. Desprez, S.; Descamps, M. J Noncrystalline Solids
2006, 352, 4480–4485.19. Wellen, R. M.; Rabello, M. S. J Mater Sci 2005,
40, 6099–6104.20. Wang, Y.; Shen, C.; Li, H.; Li, Q.; Chen, J. J Appl
Polym Sci 2004, 91, 309–314.21. Bishara, A.; Shaban, H. I. J Appl Polym Sci 2006,
101, 3565–3571.22. Bian, J.; Ye, S. R.; Feng, L. X. J Polym Sci Part
B: Polym Phys 2003, 41, 2135–2144.23. Wang, Z. G.; Hsiao, B. S.; Sauer, B. B.; Kampert,
W. G.; Polymer 1999, 40, 4615–4627.24. de Carvalho, B.; Bretas, R. E. S. J Appl Polym Sci
1998, 68, 1159–1176.25. Androsch, R.; Wunderlich, B. Polymer 2005, 46,
12556–12566.26. Tzavalas, S.; Drakonakis, V.; Mouzakis, D. E.; Fi-
scher, D.; Gregoriou, V. G. Macromolecules 2006,39, 9150–9156.
27. Vedula, J.; Tonelli, A. E. J Polym Sci Part B:Polym Phys 2007, 45, 735–746.
28. Hadac, J.; Slobodian, P.; Saha, P. J Mater Sci2007, 42, 3724–3731.
29. Rastogi, R.; Vellinga, W. P.; Rastogi, S.; Schick,C.; Meijer, H. E. H. J Polym Sci Part B: PolymPhys 2000, 42, 2092–2106.
30. Pyda M. ATHAS Data Bank. Available at: http://web.utk.edu/�athas/databank/, 1994.
31. El-Taweel, S. H.; Hohne, G. W. H.; Mansour, A.A.; Stoll, B.; Seliger H. Polymer 2004, 45, 983–992.
SOLID STATE BLENDING OF PET WITH PS 1359
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb







![Co poly (ethylene terephthalate-p-oxybenzoate ......A series of co [ poly (ethylene terephthalate-p-oxybenzoate) ] thermotropic copolyesters with various compositions were prepared](https://img.pdfslide.net/doc/110x75/613821260ad5d206764911ff/co-poly-ethylene-terephthalate-p-oxybenzoate-a-series-of-co-poly-ethylene.jpg)





















