Embed Size (px)
Citation preview


Chapter-2
17
2.1 INTRODUCTION
Non-steroidal anti-inflammatory drugs (NSAIDs) are amongst the
most widely used prescription and over the counter medications for the
treatment of pain and inflammation particularly arthritis. Pain, swelling
and tenderness at the joints are a few symptoms associated with the
arthritis. In the 1970’s, it was demonstrated that aspirin and other
NSAIDs block the formation of prostaglandins (PGs) produced from the
metabolism of arachidonic acid by the enzyme cyclooxygenase (COX),
familiarly known as prostaglandin synthase.32 The COX inhibitors are of
three kinds, namely COX-1, COX-2 and COX-3. Role of COX-3 in
prostaglandin mediated physiological activity is not clear and further
studies are under way. Until very recently, all commercially available
NSAIDs were inhibitors of both COX-1 and COX-2. Selective inhibitors of
COX-2 are now widely recognized as offering promising treatment for
inflammatory conditions without adverse side effects as associated with
consumption of nonselective inhibitors.
3,4-Diaryl substituted isoxazole derivative, valdecoxib 1 (figure 2.1) is
classified as NSAID. Valdecoxib 1 acts as a selective inhibitor of COX-2,
an enzyme that facilitates the formation of prostaglandins, which
mediate the process of inflammation.33 Increased risk of gastrointestinal
ulceration associated with blockade of COX-1 derived prostaglandins led
to the development of selective COX-2 inhibitors.34 Valdecoxib 1 has

Chapter-2
18
been recognized as an effective medication in the treatment of
rheumatoid arthritis, osteoarthritis, and dysmenorrhea.
Figure 2.1: Structure of valdecoxib 1
In view of the growing importance of anti-inflammatory drugs with
selective COX-2 inhibitory activity in day to day life, a systematic study
was taken up to develop an efficient, scalable and commercially viable
new synthetic route that would provide the title compound 1 with high
degree of quality. As this molecule is a drug substance intended for
human use, the quality should be in agreement with regulatory
requirements and ICH guidelines.
2.2 LITERATURE PRECEDENCE
Talley et al. disclosed the process for the preparation of 1 (scheme
2.1).35,36 Deoxybenzoin 2 was treated with hydroxylamine hydrochloride
to result in the oxime intermediate that was in situ deprotonated using
n-butyl lithium to form the oxy-anion that was trapped with ethyl acetate
to result in hydroxyisoxazoline intermediate 3. Compound 3 underwent
dehydration followed by chlorosulfonation upon treatment with
chlorosulfonic acid, which was finally converted to 1 by treating with
ammonium hydroxide.

Chapter-2
19
Scheme 2.1: Synthesis of 1 from 2
Alternatively, 1 was prepared in a stepwise manner (scheme 2.2).
Dehydration of 3 by treatment with sulfuric acid provided isoxazole
derivative 4. Chlorosulfonation with reagents such as chlorosulfonic
acid, SO3-Py complex/phosphorous oxychloride and sulfuric chloride,
provided the chlorosulfonic acid derivative, which in turn was subjected
to amidation with ammonium hydroxide to yield 1.35
Scheme 2.2: Sulfamidation with different reagents
An alternative approach for the synthesis of 1 was described in US
5859257 patent.37 In this process, 2 was first subjected to sulfamidation
by treatment with chlorosulfonic acid followed by ammonium hydroxide
to afford sulfonamide derivative 5. Having installed the required
functionality, build up of the isoxazole ring was carried out by different

Chapter-2
20
methods (scheme 2.3). First method involved the protection of the amine
group in 5 as cyclic disilylamine derivative by treating with 1,2-bis-
(chlorodimethylsilyl)ethane, which upon reaction with hydroxylamine
hydrochloride under basic conditions provided oxime 6. Treatment of 6
with ethyl acetate in presence of lithium diisopropylamide (LDA) followed
by dehydration and deprotection of silyl group in presence of aqueous
TFA afforded 1.
In the second method, masking of the amine functionality in 5 as a
pyrrole ring was obtained by reacting with acetonylacetone, which was
treated with hydroxylamine hydrochloride to yield the oxime 7. By
following the same methodology adopted above, the isoxazole 1 was built
up from 7 (scheme 2.3).
Scheme 2.3: Synthesis of 1 with protection and deprotection approach

Chapter-2
21
In another report, 1 was prepared by the [3+2] cycloaddition between
nitrile oxide and the corresponding alkyne.38 The precursors for the
cycloaddition reaction were prepared as follows.
Compound 8 was treated with ammonium hydroxide to result in
sulfonamide derivative 9. The carbonyl functionality of 9 was converted
to the alkyne group under modified Seyferth-Gilbert homologation
conditions, by treating with LDA, diethyl chlorophosphate followed by
methylation using hexamethylphospharamide (HMPA) and iodomethane
to obtain compound 10. Finally, compound 10 was subjected to [3+2]
cycloaddition with the nitrile oxide (generated in situ from benzaldoxime
11) to produce 1 (scheme 2.4).
Scheme 2.4: Synthesis of 1 utilizing [3+2] cycloaddition
Waldo et al. designed their strategy for the preparation of 1 utilizing
two key reactions namely halo-isoxazolation and Suzuki coupling
reaction.39 Exposure of the ynone 12 to methoxylamine hydrochloride

Chapter-2
22
generated the O-methyl oxime 13, which was subjected to ICl
(chloroiodide) cyclization conditions to yield iodoisoxazole 14. Treatment
of iodoisoxazole 14 under standard Suzuki-Miyaura coupling conditions
with compound 15 afforded 1 (scheme 2.5). It is important to note that
diastereomeric oximes 13 namely E & Z isomers were formed on
treatment of 12 with methoxylamine hydrochloride. These diastereomers
were separable by column chromatography. It had been observed that
only the Z-isomer underwent inter halogen mediated cyclization to
furnish the corresponding iodoisoxazole.
Scheme 2.5: Synthesis of 1 using halo-isoxazolation and Suzuki
coupling reactions
A simpler approach to the synthesis of 1 starting from readily
available raw materials was devised by Nunno and co-workers.40 Lithium
enolate 17 was generated from phenylacetone (16) by treatment with
LDA, which was trapped in situ with nitrile oxide 18 by a [3+2]

Chapter-2
23
cycloaddition to form 3. Dehydration followed by sulfamidation of 3
under known reaction conditions afforded 1 (scheme 2.6).
Scheme 2.6: Synthesis of 1 by [3+2] cycloaddition of 17 and 18
The above reported synthetic routes to 1 indicate the usage of
a) Moisture sensitive and highly flammable reagents like n-butyl
lithium and LDA.
b) Protection and deprotection procedures.
c) Carcinogenic chemicals like diethyl chlorophosphate, hexamethyl-
phospharamide and iodomethane.
d) Purification by column chromatography.
e) Expensive and commercially unavailable raw materials.
With so many hurdles, the above mentioned processes appeared not
to be commercially viable. In view of these drawbacks, a comprehensive
study was undertaken and attempts were made to develop a new, simple,
efficient and scalable synthetic procedure for 1.

Chapter-2
24
2.3 OUR APPROACH
Based on the mechanistic pathway of [3+2] cycloaddition as shown in
figure 2.2 and the reports on the application of enamine as an olefin
equivalent in such type of reactions,41,42 a novel synthetic route involving
an alternative dipolarophile other than those described in the literature
and the known nitrile oxide as 1, 3-dipole was designed to synthesize 1
efficiently.
Figure 2.2: In situ generated nitrile oxide and its [3+2] cycloaddition
with olefin
It was envisioned that the enamine 19 would serve as dipolarophile in
the [3+2] cycloaddition reaction with the nitrile oxide to generate the
pyrrolidinylisoxazoline 21 which could be converted into 1 after few
transformations (scheme 2.7). As the synthetic route is modified, the
formation of new impurities in the sequence can be expected. Hence, the
interest lay in the identification and characterization of the related

Chapter-2
25
compounds. In addition, a detailed study relating to the formation and
synthesis of these related substances is presented in this chapter.
Scheme 2.7: Novel approach to the synthesis of 1
2.4 RESULTS AND DISCUSSION
In our endeavor, the synthesis of 1 was planned from the readily
available starting material 16. The route for the synthesis of 1 was based
on the masking of the ketone functionality as an enamine in order to
localize the formation of the double bond which would be in conjugation
with the benzene ring in the form of a styrene moiety. In this regard, the
ketone group in 16 was transformed into the respective pyrrolidine
derived enamine 19 using a reported protocol.41 The [3+2] cycloaddition42
between 19 and benzonitrile oxide (generated in situ from 20 using

Chapter-2
26
triethylamine in DCM43) afforded 21, which was subjected to
aromatization in the presence of aqueous hydrochloric acid to result in
the isoxazole 4 (scheme 2.7). Spectral data of this compound was found
to be in agreement with the reported data.40 After synthesizing the core
isoxazole entity, the focus was shifted on the decoration of the aromatic
ring with the suitable functionalities. The isoxazole derivative 4 was
subjected to chlorosulfonation in presence of chlorosulfonic acid,
followed by amidation in presence of aqueous ammonia to afford 1 in 50
% yield and 99.0 % purity. Intensive process optimization study was
carried out to improve the yield and quality of final product by various
purification techniques that finally improved the yield to 80 % and
quality to
99.92 %. The synthesized material met the requirement specified by
regulatory authorities, hence qualifying the material for human
consumption. This process has been successfully implemented on Kg
scale in the plant.
Aforementioned synthesis had overcome the disadvantages in prior
art processes.
Having synthesized the target molecule, a systematic study was taken
up for the identification and characterization of the impurities that were
formed during the synthesis of drug substance. Five related substances,
23 (impurity-A), 24 (impurity-B), 25 (impurity-C), 26 (impurity-D) and

Chapter-2
27
27 (impurity-E) were observed by a simple high performance liquid
chromatographic (HPLC) method (figure 2.3). A
U
-0.020
-0.010
0.000
0.010
0.020
0.030
0.040
0.050
0.060
0.070
0.080
0.090
0.100
Minutes
0.00 2.00 4.00 6.00 8.00 10.00 12.00 14.00 16.00 18.00 20.00 22.00 24.00 26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00
IMP
UR
ITY
-C -
6.0
11
IMP
UR
ITY
-D -
6.5
67
IMP
UR
ITY
-A -
9.9
31
Vald
ecoxib
- 1
6.0
15
IMP
UR
ITY
-B -
16.8
11
IMP
UR
ITY
-E -
23.7
05
Figure 2.3: HPLC chromatogram of 1 and its related substances
In this regard LC–MS analysis was performed on the drug substance
to obtain the molecular weights of the related substances that were
present along with the desired product, which would provide insight to
the probable structures. The LC–MS data revealed the molecular weights
as 393, 314, 314, 315 and 611. Based on the LC–MS data and synthetic
pathway, the following tentative structures were predicted (figure 2.4).

Chapter-2
28
Figure 2.4: Chemical structures for the related substances of valdecoxib
To substantiate our claim, efforts were directed for the synthesis of
the respective compounds (figure 2.4). The presence of these related
substances was confirmed by performing the co-injection in HPLC
analysis (figure 2.5) with standards that were synthesized separately.
AU
-0.02
0.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
Minutes
0.00 2.00 4.00 6.00 8.00 10.00 12.00 14.00 16.00 18.00 20.00 22.00 24.00 26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00
IMP
UR
ITY
-C -
6.0
07
IMP
UR
ITY
-D -
6.5
57
IMP
UR
ITY
-A -
9.9
37
Vald
ecoxib
- 1
6.0
22
IMP
UR
ITY
-B -
16.8
40
IMP
UR
ITY
-E -
23.6
96
Figure 2.5: HPLC chromatogram of 1 co-injected with impurities

Chapter-2
29
2.4.1 3-[5-Methyl-4-(4-sulfamoylphenyl)isoxazol-3-yl]benzene
sulfonamide (Impurity-A, 23)
Isoxazole 4 is a pseudo symmetrical molecule containing two
activated aromatic nucleus that are available for the sulfonation reaction.
As the drug substance contains only one sulfonyl group, this means
there exists a need for the controlled monosulfonation of the starting
material. The reaction was optimized in such a manner to maximize the
formation of the main product 1. Even under these circumstances there
is a possibility for the chlorosulfonation at more than one position, which
eventually would be transformed to sulfonamide derivatives by the
subsequent reaction with aqueous ammonia.
Isoxazole derivative 4 was subjected to chlorosulfonation with excess
chlorosulfonic acid followed by reaction with aqueous ammonia to obtain
23 (scheme 2.8, experimental section 2.6.9). The obtained product was
characterized by analytical tools as described.
Scheme 2.8: Synthesis of 23
Mass spectrum of 23 showed protonated molecular ion peak at m/z
394 (figure 2.6) and IR spectrum displayed characteristic absorptions at
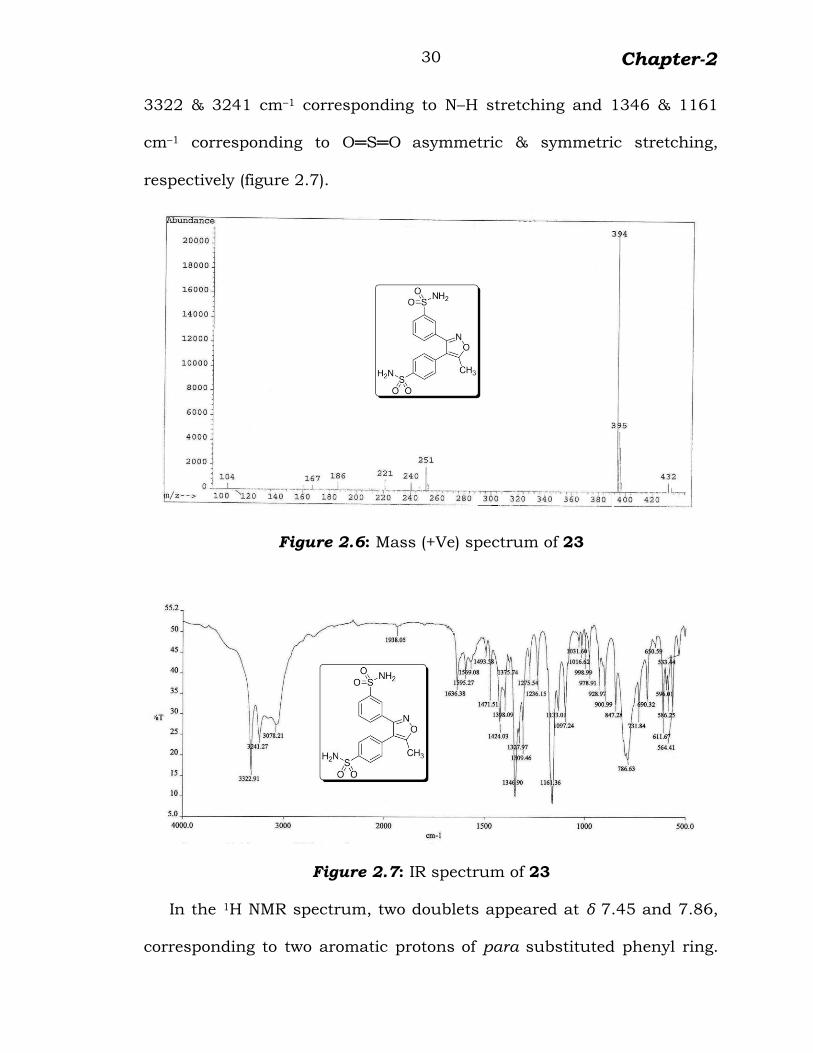
Chapter-2
30
3322 & 3241 cm–1 corresponding to N–H stretching and 1346 & 1161
cm–1 corresponding to O═S═O asymmetric & symmetric stretching,
respectively (figure 2.7).
Figure 2.6: Mass (+Ve) spectrum of 23
Figure 2.7: IR spectrum of 23
In the 1H NMR spectrum, two doublets appeared at δ 7.45 and 7.86,
corresponding to two aromatic protons of para substituted phenyl ring.
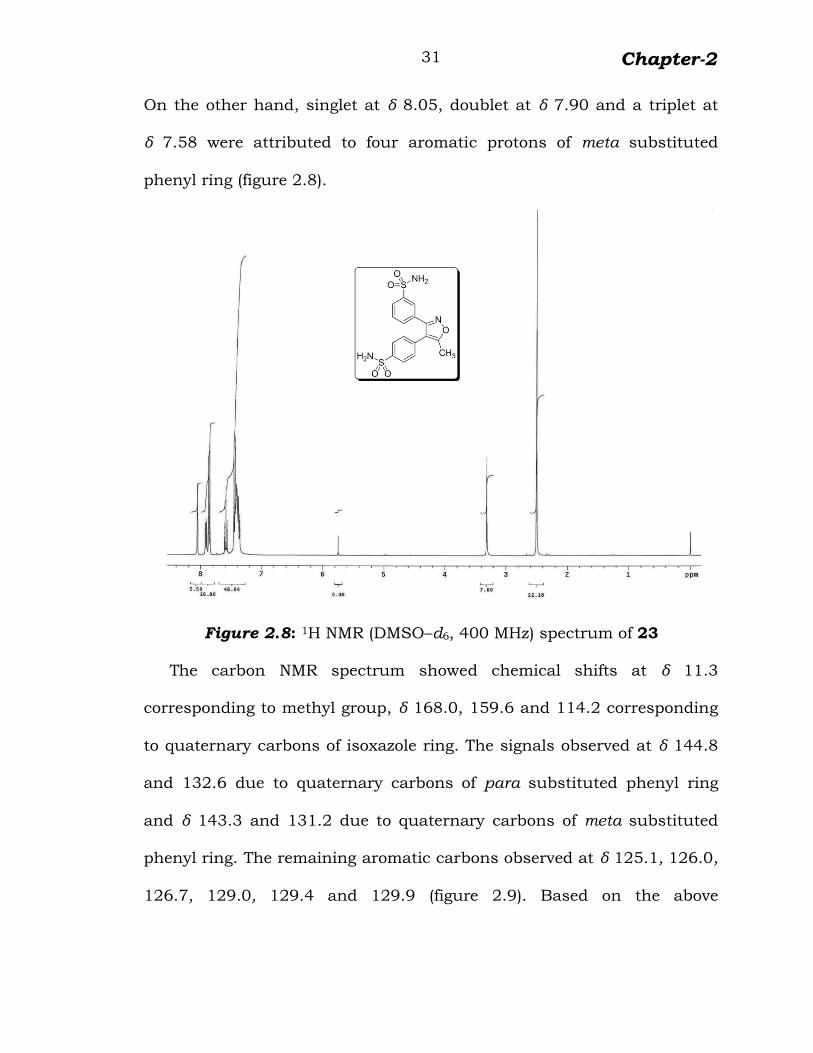
Chapter-2
31
On the other hand, singlet at δ 8.05, doublet at δ 7.90 and a triplet at
δ 7.58 were attributed to four aromatic protons of meta substituted
phenyl ring (figure 2.8).
Figure 2.8: 1H NMR (DMSO–d6, 400 MHz) spectrum of 23
The carbon NMR spectrum showed chemical shifts at δ 11.3
corresponding to methyl group, δ 168.0, 159.6 and 114.2 corresponding
to quaternary carbons of isoxazole ring. The signals observed at δ 144.8
and 132.6 due to quaternary carbons of para substituted phenyl ring
and δ 143.3 and 131.2 due to quaternary carbons of meta substituted
phenyl ring. The remaining aromatic carbons observed at δ 125.1, 126.0,
126.7, 129.0, 129.4 and 129.9 (figure 2.9). Based on the above
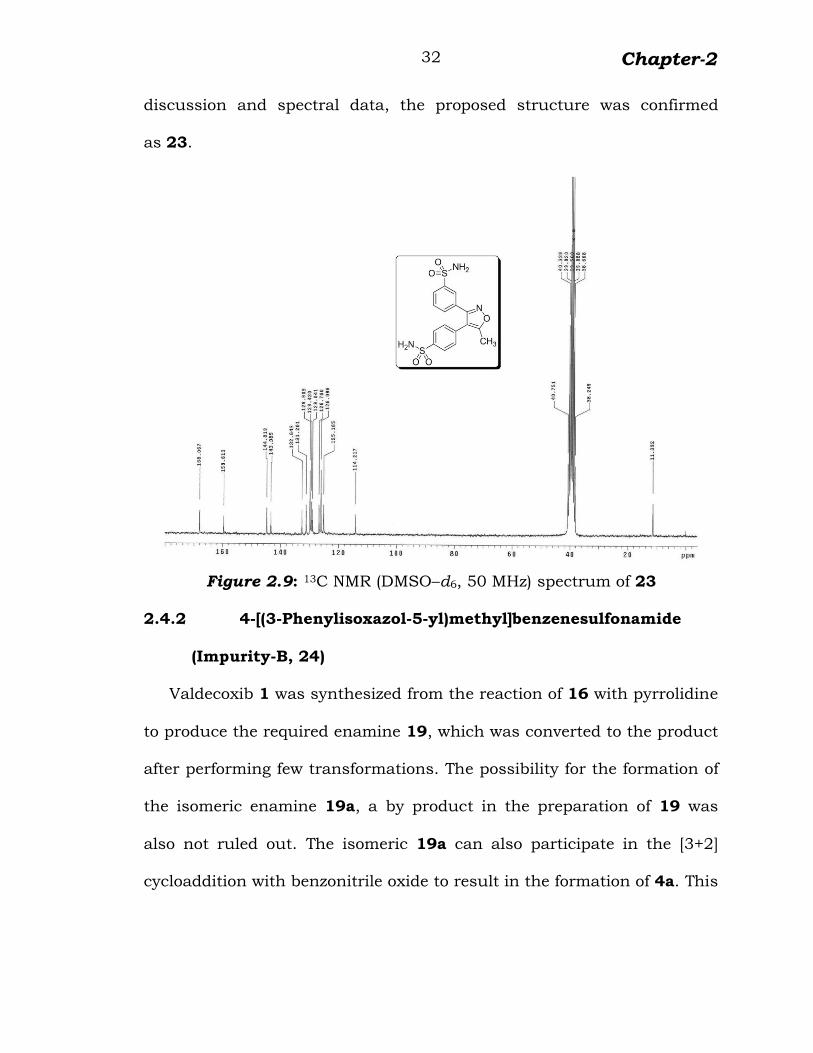
Chapter-2
32
discussion and spectral data, the proposed structure was confirmed
as 23.
Figure 2.9: 13C NMR (DMSO–d6, 50 MHz) spectrum of 23
2.4.2 4-[(3-Phenylisoxazol-5-yl)methyl]benzenesulfonamide
(Impurity-B, 24)
Valdecoxib 1 was synthesized from the reaction of 16 with pyrrolidine
to produce the required enamine 19, which was converted to the product
after performing few transformations. The possibility for the formation of
the isomeric enamine 19a, a by product in the preparation of 19 was
also not ruled out. The isomeric 19a can also participate in the [3+2]
cycloaddition with benzonitrile oxide to result in the formation of 4a. This

Chapter-2
33
can further be transformed to 24 under the reaction conditions
employed.
Compound 24 was efficiently synthesized from 16 by reacting with
pyrrolidine to produce the mixture of 19 and 19a (isomer) followed by
[3+2] cycloaddition with benzonitrile oxide (in situ generated from 20) in
dichloromethane and elimination of pyrrolidine ring in presence of conc
hydrochloric acid to yield a mixture of 4 and 4a. Compound 4 was
filtered and the filtrate containing 4a was treated with chlorosulfonic acid
followed by aqueous ammonia to provide 24 (scheme 2.9, experimental
section 2.6.10.1–2.6.10.2).
Scheme 2.9: Synthesis of 24
Chapter-2
34
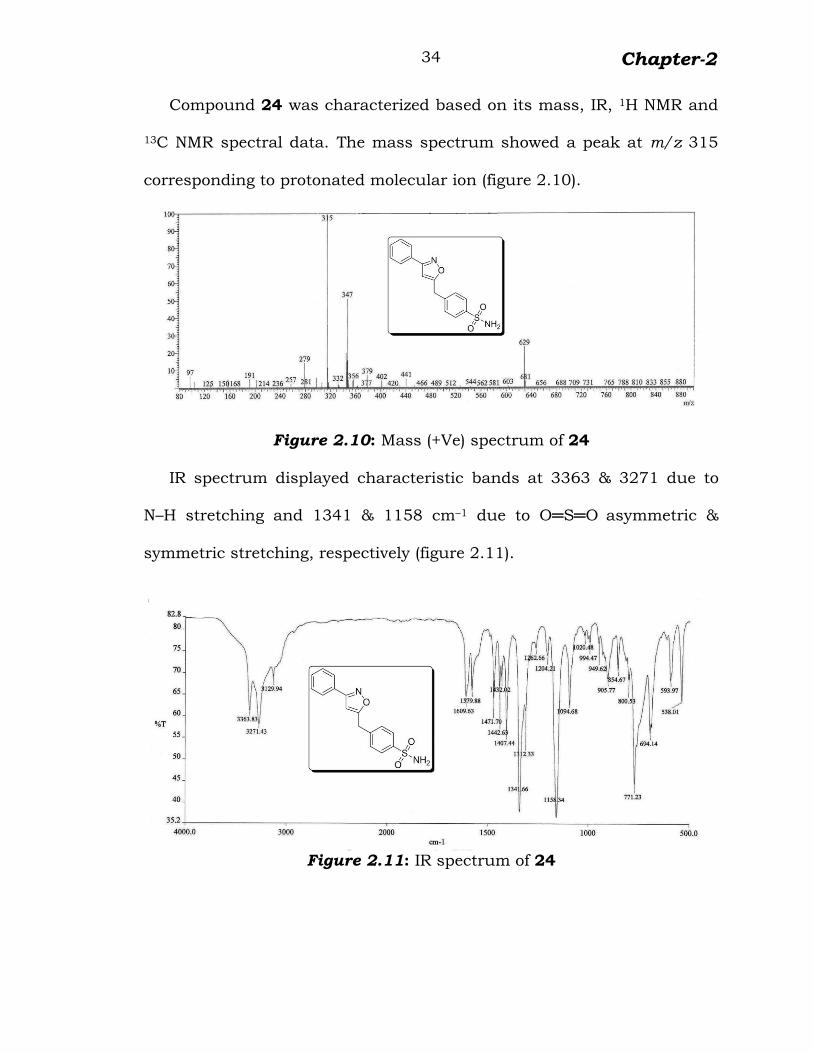
Compound 24 was characterized based on its mass, IR, 1H NMR and
13C NMR spectral data. The mass spectrum showed a peak at m/z 315
corresponding to protonated molecular ion (figure 2.10).
Figure 2.10: Mass (+Ve) spectrum of 24
IR spectrum displayed characteristic bands at 3363 & 3271 due to
N–H stretching and 1341 & 1158 cm–1 due to O═S═O asymmetric &
symmetric stretching, respectively (figure 2.11).
Figure 2.11: IR spectrum of 24
Chapter-2
35
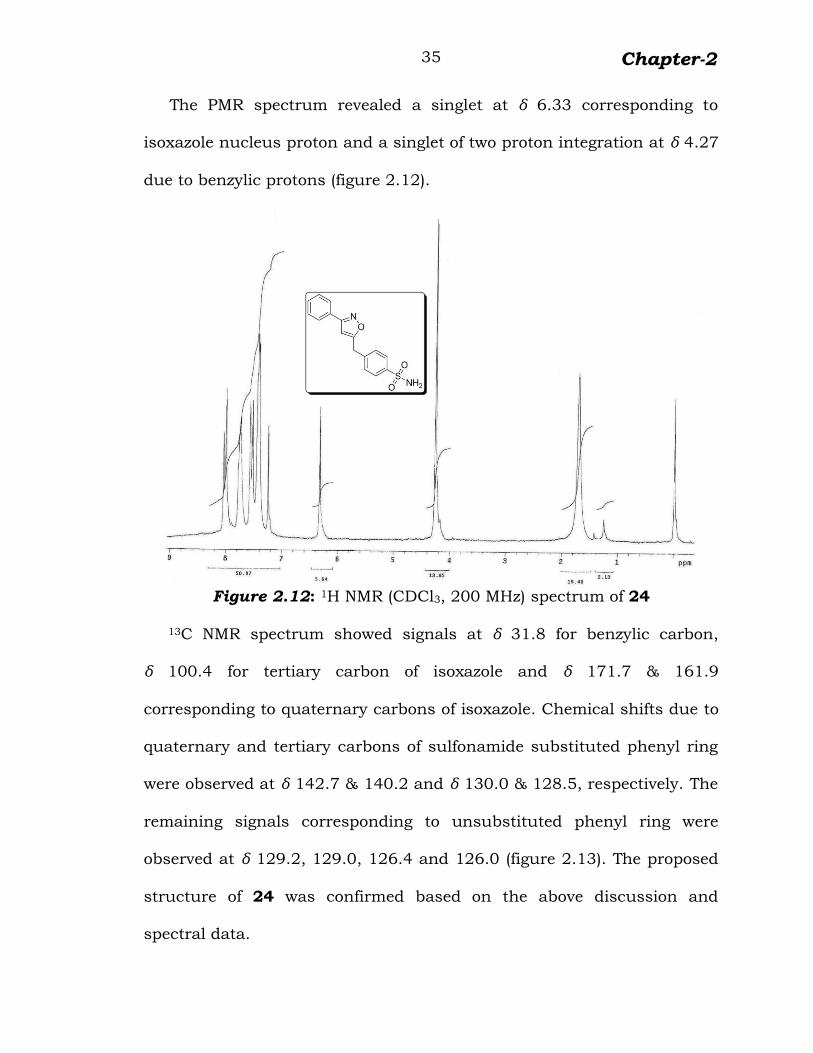
The PMR spectrum revealed a singlet at δ 6.33 corresponding to
isoxazole nucleus proton and a singlet of two proton integration at δ 4.27
due to benzylic protons (figure 2.12).
Figure 2.12: 1H NMR (CDCl3, 200 MHz) spectrum of 24
13C NMR spectrum showed signals at δ 31.8 for benzylic carbon,
δ 100.4 for tertiary carbon of isoxazole and δ 171.7 & 161.9
corresponding to quaternary carbons of isoxazole. Chemical shifts due to
quaternary and tertiary carbons of sulfonamide substituted phenyl ring
were observed at δ 142.7 & 140.2 and δ 130.0 & 128.5, respectively. The
remaining signals corresponding to unsubstituted phenyl ring were
observed at δ 129.2, 129.0, 126.4 and 126.0 (figure 2.13). The proposed
structure of 24 was confirmed based on the above discussion and
spectral data.

Chapter-2
36
Figure 2.13: 13C NMR (DMSO–d6, 50 MHz) spectrum of 24
2.4.3 3-(5-Methyl-3-phenylisoxazol-4-yl)benzenesulfonamide
(Impurity-C, 25)
During the chlorosulfonation of 4, probability for the formation of
meta isomer 22a also exists along with desired product 22. Compound
22a upon further reaction with aqueous ammonia would lead to the
formation of 25.
Compound 25 was independently prepared by performing the
chlorosulfonation followed by amidation on compound 4.
Chlorosulfonation on compound 4 resulted in a mixture of 22 and 22a.
Compound 22 was removed from the mixture by filtration. Thus,
obtained filtrate containing meta isomer 22a and minimum amount of 22

Chapter-2
37
was treated with aqueous ammonia resulting in the precipitation of solid
material 1 which was filtered. The filtrate was subjected to the isolation
of 25 (scheme 2.10, experimental section 2.6.11).
Scheme 2.10: Synthesis of 25
The meta sulfonamide 25 was characterized by mass and IR spectra,
in which a peak at m/z 315 corresponding to protonated molecular ion
was observed in the mass spectrum (figure 2.14) and IR spectrum
displayed characteristic absorptions at 3337 & 3247 cm–1 corresponding
to N–H stretching and 1334 & 1165 cm–1 corresponding to O═S═O
asymmetric & symmetric stretching respectively (figure 2.15).
Figure 2.14: Mass (+Ve) spectrum of 25
Chapter-2
38
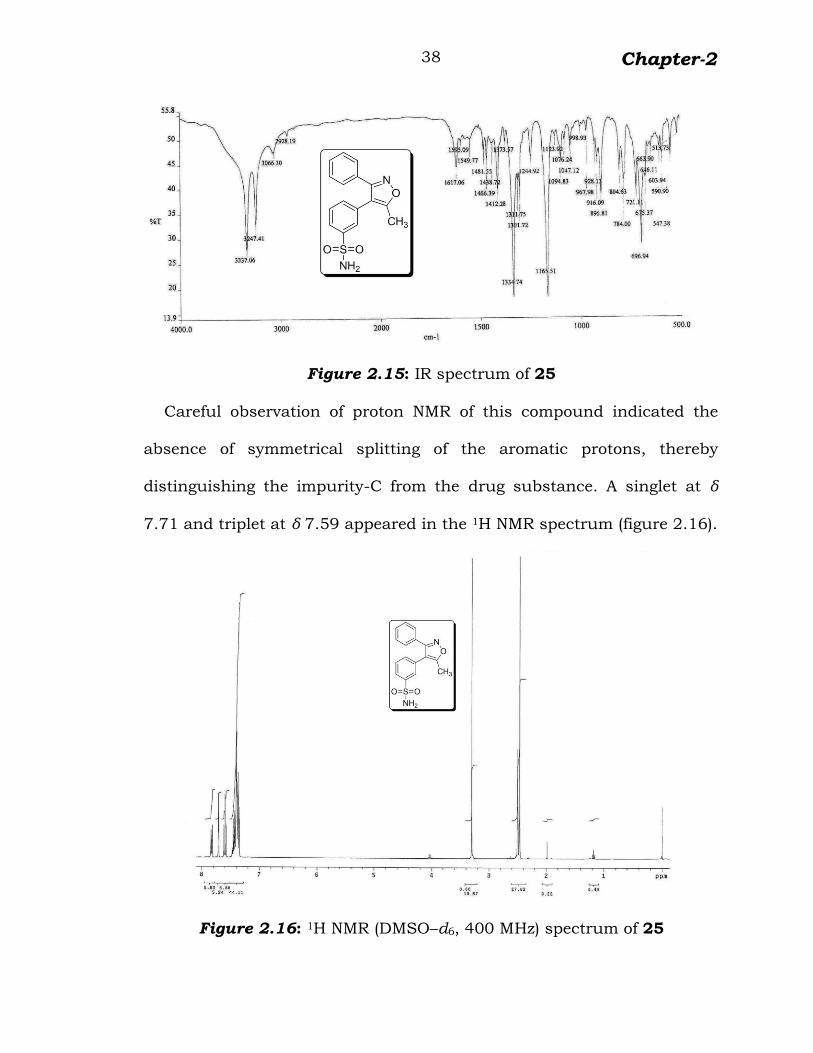
Figure 2.15: IR spectrum of 25
Careful observation of proton NMR of this compound indicated the
absence of symmetrical splitting of the aromatic protons, thereby
distinguishing the impurity-C from the drug substance. A singlet at δ
7.71 and triplet at δ 7.59 appeared in the 1H NMR spectrum (figure 2.16).
Figure 2.16: 1H NMR (DMSO–d6, 400 MHz) spectrum of 25

Chapter-2
39
CMR spectrum displayed characteristic δ values at 167.0, 160.4 and
115.0 for isoxazole ring and chemical shifts corresponding to
sulfonamide substituted phenyl ring at δ 148.7, 147.4, 129.1, 128.0,
126.4 and 126.0. The remaining signals corresponding to unsubstituted
phenyl ring were observed at δ 129.8, 128.9, 128.2 and 127.9 (figure
2.17). Based on the above spectral data observations, the proposed
structure of 25 was confirmed.
Figure 2.17: 13C NMR (DMSO–d6, 100 MHz) spectrum of 25
2.4.4 4-(5-Methyl-3-phenylisoxazol-4-yl)benzenesulfonic acid
(Impurity-D, 26)
The final step in the synthesis of 1 involves amidation of 22 with
aqueous ammonia. During this reaction, 22 can undergo hydrolysis in
the presence of water to form the sulfonic acid derivative 26. This
Chapter-2
40
compound was independently prepared by subjecting the
sulfonylchloride derivative 22 to hydrolysis in a mixture of
tetrahydrofuran and water under reflux conditions (scheme 2.11,
experimental section 2.6.12).
Scheme 2.11: Synthesis of sulfonic acid derivative 26
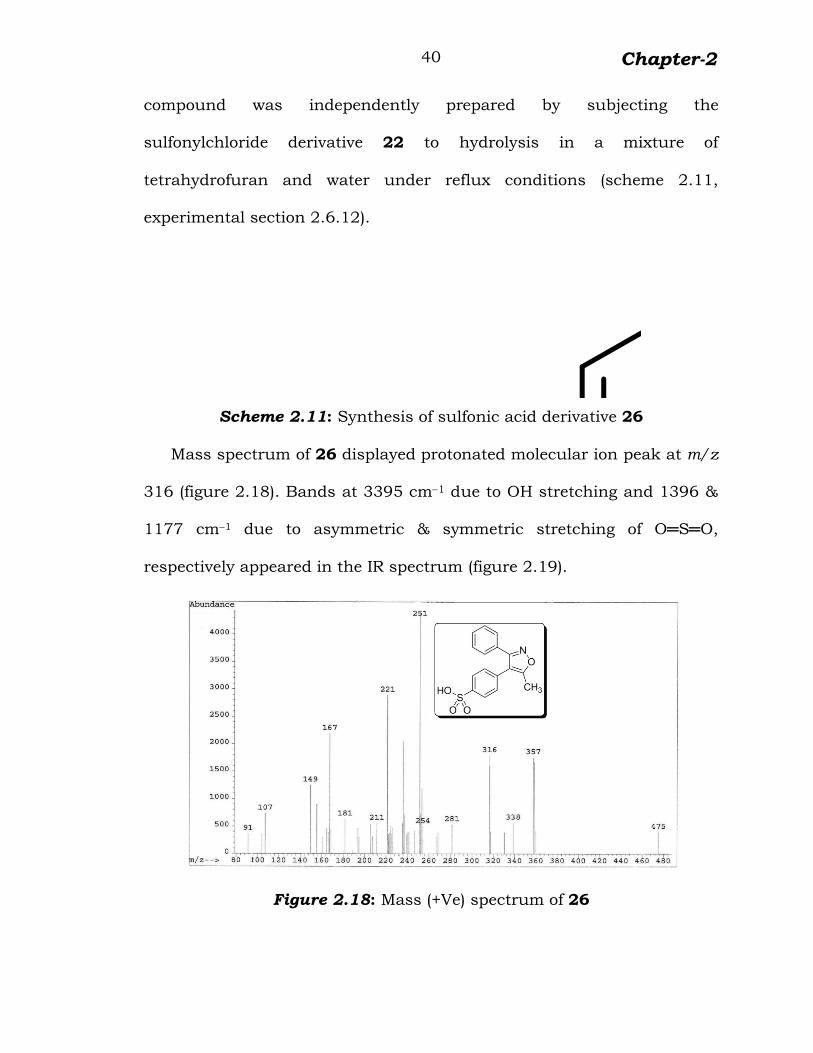
Mass spectrum of 26 displayed protonated molecular ion peak at m/z
316 (figure 2.18). Bands at 3395 cm–1 due to OH stretching and 1396 &
1177 cm–1 due to asymmetric & symmetric stretching of O═S═O,
respectively appeared in the IR spectrum (figure 2.19).
Figure 2.18: Mass (+Ve) spectrum of 26
Chapter-2
41
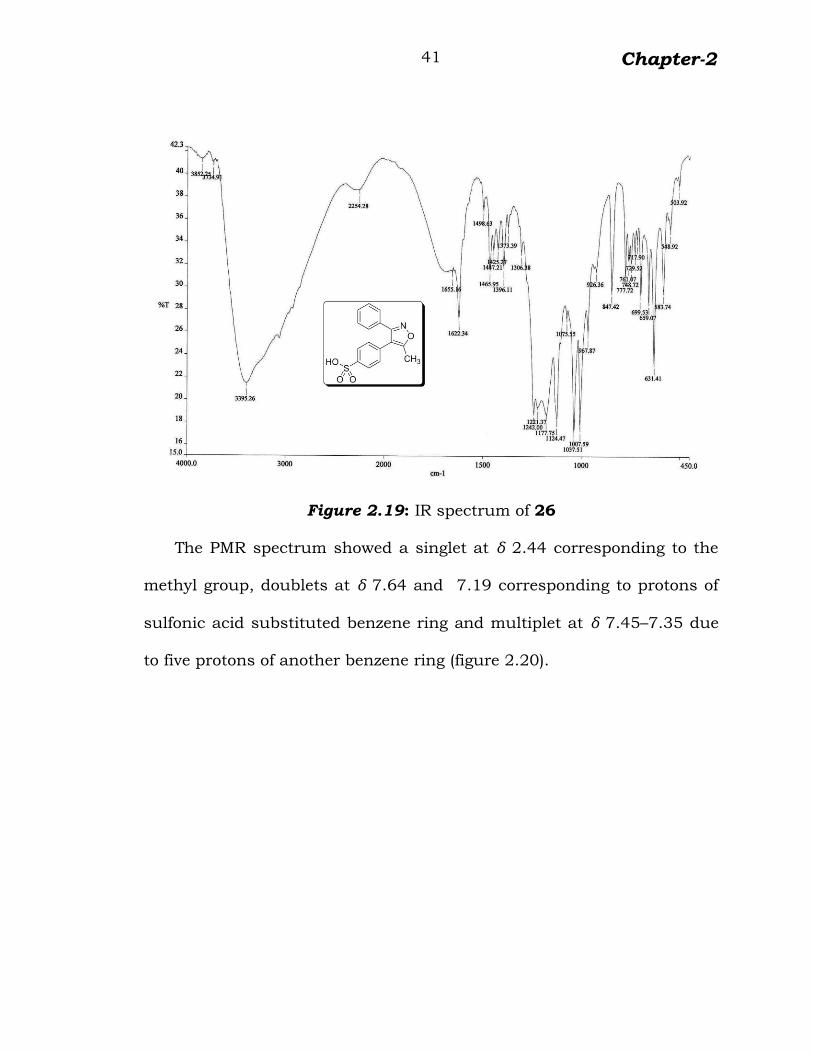
Figure 2.19: IR spectrum of 26
The PMR spectrum showed a singlet at δ 2.44 corresponding to the
methyl group, doublets at δ 7.64 and 7.19 corresponding to protons of
sulfonic acid substituted benzene ring and multiplet at δ 7.45–7.35 due
to five protons of another benzene ring (figure 2.20).

Chapter-2
42
Figure 2.20: 1H NMR (DMSO–d6, 400 MHz) spectrum of 26
In the 13C NMR spectrum, methyl carbon at δ 11.3 and quaternary
carbons of isoxazole at δ 167.0, 160.5 and 114.8 were observed. Signals
at δ 147.4, 129.8, 128.6 and 126.0 represent the sulfonic acid
substituted phenyl ring carbons. Signals corresponding to carbons of
unsubstituted phenyl ring were appeared at δ 129.6, 128.9, 128.7 and
128.0 (figure 2.21). Above discussion and spectral data confirmed the
proposed structure of 26.

Chapter-2
43
Figure 2.21: 13C NMR (DMSO–d6, 100 MHz) spectrum of 26
2.4.5 4-(5-Methyl-3-phenylisoxazol-4-yl)-N-[4-(5-methyl-3-
phenylisoxazol-4-yl)phenylsulfonyl]benzenesulfonamide
(Impurity-E, 27)
The formation of 27 was explained on the basis that a reaction of 1
with staring material 22 could occur. To substantiate our claims on its
formation, a reaction was performed between 1 and compound 22 in
pyridine, which provided 27 (scheme 2.12, experimental section 2.6.13).
Compound 27 was characterized as described below.
Scheme 2.12: Synthesis of 27
Chapter-2
44
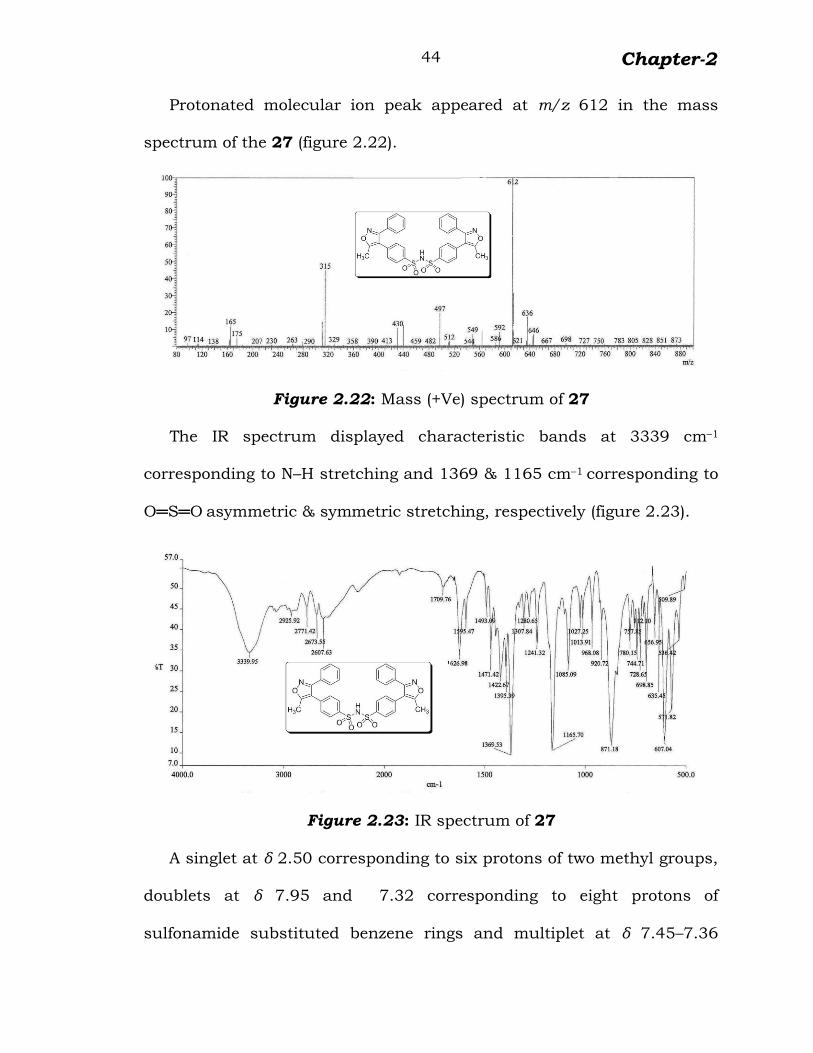
Protonated molecular ion peak appeared at m/z 612 in the mass
spectrum of the 27 (figure 2.22).
Figure 2.22: Mass (+Ve) spectrum of 27
The IR spectrum displayed characteristic bands at 3339 cm–1
corresponding to N–H stretching and 1369 & 1165 cm–1 corresponding to
O═S═O asymmetric & symmetric stretching, respectively (figure 2.23).
Figure 2.23: IR spectrum of 27
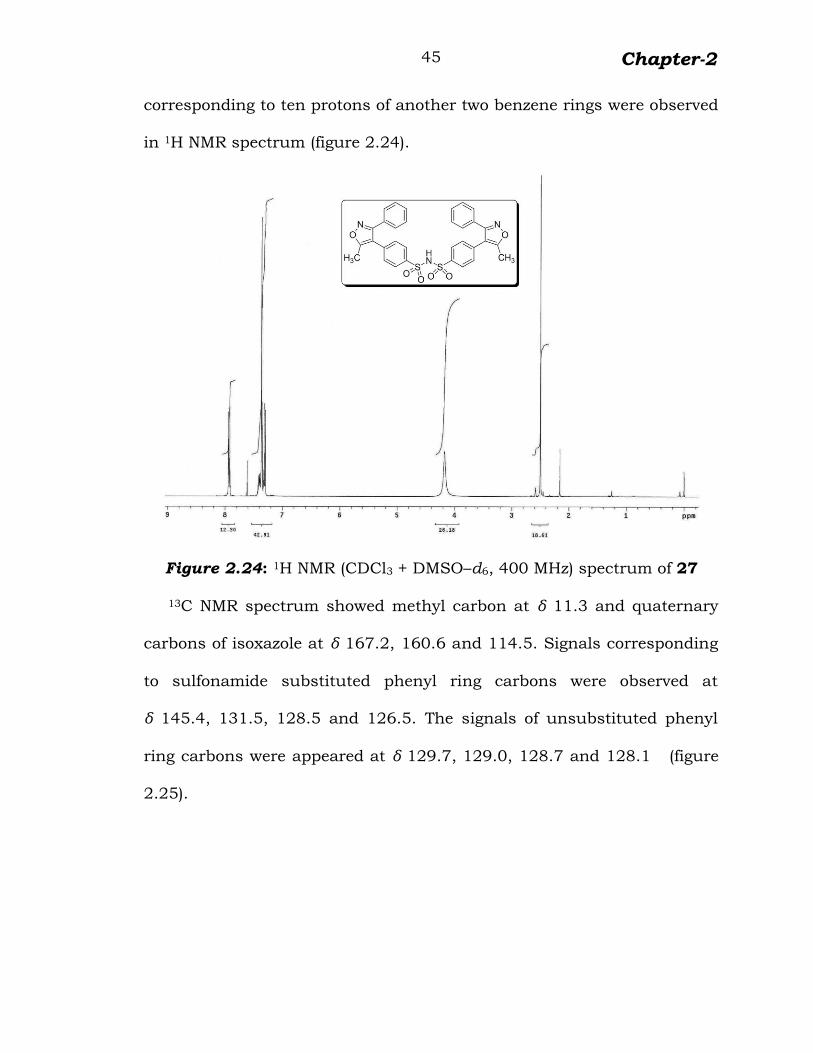
A singlet at δ 2.50 corresponding to six protons of two methyl groups,
doublets at δ 7.95 and 7.32 corresponding to eight protons of
sulfonamide substituted benzene rings and multiplet at δ 7.45–7.36
Chapter-2
45
corresponding to ten protons of another two benzene rings were observed
in 1H NMR spectrum (figure 2.24).
Figure 2.24: 1H NMR (CDCl3 + DMSO–d6, 400 MHz) spectrum of 27
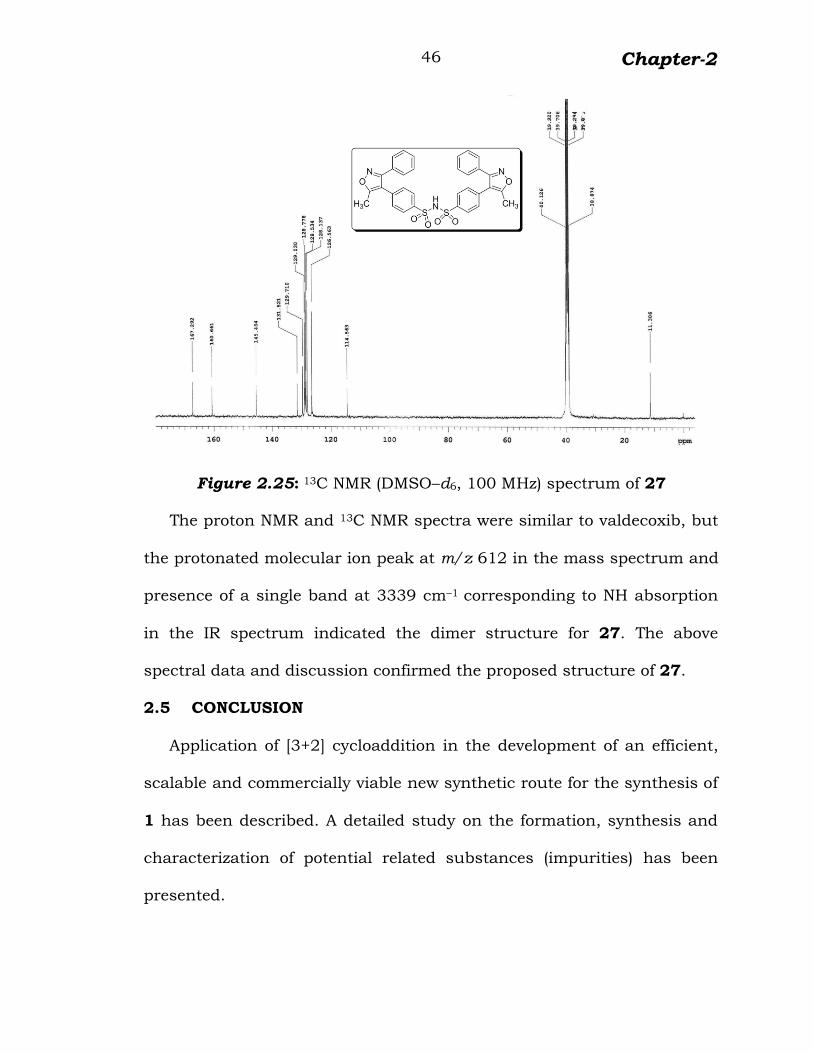
13C NMR spectrum showed methyl carbon at δ 11.3 and quaternary
carbons of isoxazole at δ 167.2, 160.6 and 114.5. Signals corresponding
to sulfonamide substituted phenyl ring carbons were observed at
δ 145.4, 131.5, 128.5 and 126.5. The signals of unsubstituted phenyl
ring carbons were appeared at δ 129.7, 129.0, 128.7 and 128.1 (figure
2.25).
Chapter-2
46
Figure 2.25: 13C NMR (DMSO–d6, 100 MHz) spectrum of 27
The proton NMR and 13C NMR spectra were similar to valdecoxib, but
the protonated molecular ion peak at m/z 612 in the mass spectrum and
presence of a single band at 3339 cm–1 corresponding to NH absorption
in the IR spectrum indicated the dimer structure for 27. The above
spectral data and discussion confirmed the proposed structure of 27.
2.5 CONCLUSION
Application of [3+2] cycloaddition in the development of an efficient,
scalable and commercially viable new synthetic route for the synthesis of
1 has been described. A detailed study on the formation, synthesis and
characterization of potential related substances (impurities) has been
presented.

Chapter-2
47
2.6 EXPERIMENTAL SECTION
The 1H NMR and 13C NMR spectra were measured in CDCl3,
DMSO–d6, CDCl3 + DMSO–d6 and CDCl3 + CD3CN using 200 MHz on a
Gemini–2000 (200 MHz) and 400 MHz on mercury plus (Varian 400 MHz)
FT–NMR spectrometer. The FT–IR spectra were recorded in the solid state
as KBr dispersion using Perkin–Elmer 1650 FT–IR spectrophotometer.
The mass spectrum (70 eV) was recorded on HP–5989A LC–MS
spectrometer. The solvents and reagents were used without further
purification.
2.6.1 High Performance Liquid Chromatography (HPLC)
An in-house liquid chromatographic gradient method was developed
for the separation of all possible related substances of valdecoxib. An
agilent 1100 series HPLC (auto sampler) equipped with binary pump,
static mixer and UV-VIS detector (Agilent Technologies, USA) was used.
Mobile phase-A is buffer (Dissolved 1.36 g of KH2PO4 and 0.22 g of 1-
octane sulfonic acid sodium salt in 1000 mL milli Q water and adjusted
the pH to 3.3 with dil.H3PO4) and mobile phase-B is acetonitrile and
water in 7:3 ratio (v/v). Zorbax CN 150 mm x 4.6 mm x 3.5 µm column
(Agilent Technologies, USA) was used. A timed gradient program of T
(min)/%B: 0/35, 5/35, 20/60, 25/95, 30/95, 35/35, 40/35 with 1.0
mL/min of flow rate, 27 °C of column oven temperature was used.
Column eluent was monitored by UV at 240 nm.

Chapter-2
48
2.6.2 Benzaldoxime 11
A solution of hydroxylamine hydrochloride (32.5 g, 0.467 mol) in
water (90.0 mL) was added portion wise to the mixture of sodium
hydroxide (46 g, 1.150 mol), water (68 mL) and benzaldehyde (45 g,
0.424 mol) at 25–35 °C. The resultant reaction mixture was stirred at
25–35 °C for 30–45 min. Thereafter the pH of reaction mixture was
adjusted to 6.5–7.5 with hydrochloric acid (~11 mL) and extracted with
dichloromethane (3 x 75 mL). The combined organic layers were
concentrated below 50 °C to afford 49 g (95 %) of 11 as a brown color
residue.
Purity by HPLC: 99.0 %.
IR (KBr, cm–1): 3334, 3064, 2923, 1634, 1578, 1494, 755, 691.
1H NMR (200 MHz, CDCl3): δ 9.22 (bs, 1H), 8.18 (s, 1H), 7.70–7.22 (m,
5H).
MS (m/z): 122 (M+ + H).
2.6.3 1-[(1-Methyl-2-phenyl)ethenyl]pyrrolidine (19)

Chapter-2
49
A solution of phenylacetone (16, 45 g, 0.133 mol), pyrrolidine (42.5 g,
0.597 mol) and cyclohexane (370 mL) was maintained under azeotropic
reflux (70–80 °C) conditions for 8 h. Thereafter, cyclohexane and excess
pyrrolidine were removed from the reaction mixture under reduced
pressure below 50 °C to provide the 19 [residue was directly used in next
step without further isolation (63.1 g; crude yield was considered as
100 %)].
IR (KBr, cm–1): 3016, 2965, 2871, 1610, 1566, 1436, 1353, 775, 741.
1H NMR (200 MHz, CDCl3): δ 7.40–6.91 (m, 5H), 3.69 (s, 1H), 3.27 (t, J
═ 6.6 Hz, 2H), 2.87 (t, J ═ 6.6 Hz, 2H), 2.13 (s, 3H), 1.92 (t, J ═ 6.6 Hz,
2H), 1.69 (t, J ═ 6.6 Hz, 2H).
MS (m/z): 187 (M+ + H).
2.6.4 Chlorobenzaldoxime 20
To a solution of N-chlorosuccinimide (63.4 g, 0.475 mol) in
N, N-dimethylformamide (240 mL) was added a solution of 11 (48 g,
0.396 mol) in N, N-dimethylformamide (48 mL) over 60 min at 25–35 °C.
The resulted reaction mixture was stirred for 1–1.5 h and poured into
water (720 mL) at 25–35 °C. Thereafter, reaction mixture was stirred for
30–45 min and extracted with dichloromethane (3 x 100 mL). The
combined organic layers were washed with a solution of sodium

Chapter-2
50
hydrosulphite (hydrose) (4.8 g) and water (95 mL) followed by water (2 x
190 mL). The organic layer was concentrated under atmospheric
pressure below 50 °C followed by under reduced pressure below 50 °C to
afford 20 [residue was directly used in next step without further
isolation (61.6 g; crude yield was considered as 100 %)].
IR (KBr, cm–1): 3273, 3063, 2900, 1629, 1492, 1450, 995, 936, 766,
691.
1H NMR (200 MHz, CDCl3): δ 8.53 (s, 1H), 7.83 (d, J ═ 7.0 Hz, 2H),
7.50–7.35 (m, 3H).
MS (m/z): 156 (M+ + H).
2.6.5 3,4-Diphenyl-5-methyl-5-pyrrolidinylisoxazoline (21)
To a stirred solution of 19 (63 g, 0.336 mol) and triethylamine (53 g,
0.523 mol) in dichloromethane (575 mL) was added a solution of 20
(61 g, 0.392 mol) in dichloromethane (95 mL) at 5–10 °C. The
temperature of reaction mixture was raised to 25–35 °C and stirred for
2–3 h. Subsequently, water (360 mL) was added to the reaction mixture
and stirred for 30 min. The organic layer was separated and
concentrated below 55 °C under atmospheric pressure to provide 21

Chapter-2
51
[residue was directly used in next step without further isolation (121.3 g;
crude yield was considered as 100 %)].
IR (KBr, cm–1): 2915, 1600, 1495, 1455, 1391, 1350, 1113, 1076, 756,
709.
1H NMR (200 MHz, CDCl3): δ 7.70–7.20 (m, 10H), 5.50 (s, 1H), 3.80–
2.90 (m, 4H), 2.40–1.60 (m, 4H), 1.45 (s, 3H).
MS (m/z): 307 (M+ + H).
2.6.6 3,4-Diphenyl-5-methylisoxazole (4)
To a stirred mixture of 21 (121 g, 0.395 mol) and water (500 mL) was
added conc hydrochloric acid (36 %, 225 mL, 2.250 mol) at 40–50 °C.
The resulted reaction mixture was heated to 98–102 °C and maintained
for 1.5–2.5 h at the same temperature. Thereafter, reaction mixture was
cooled to 25–35 °C and the product was extracted with dichloromethane
(2 x 200 mL). The combined organic layers were washed with water
(150 mL) and concentrated under atmospheric pressure below 50 °C
followed by under reduced pressure below 50 °C. Subsequently,
isopropyl alcohol (75 mL) was added and maintained for 15–30 min at
45–50 °C. The resulted reaction mixture was cooled to 0–5 °C and stirred
for 45–60 min. The precipitated solid was filtered and washed with

Chapter-2
52
chilled isopropyl alcohol (5 mL). The filtered compound was recrystallised
from isopropyl alcohol to obtain 24 g (26 %) of 4.
Purity by HPLC: 99.8 %.
IR (KBr, cm–1): 3050, 2928, 1618, 1597, 1463, 1433, 1415, 1377, 1304,
1240, 1074, 916, 768, 697.
1H NMR (200 MHz, CDCl3 + CD3CN): δ 7.45–7.11 (m, 10H), 2.44 (s, 3H).
MS (m/z): 236 (M+ + H).
2.6.7 4-(5-Methyl-3-phenyl-4-isoxazolyl)benzenesulfonyl
chloride (22)
To a stirred solution of chlorosulfonic acid (98 g, 0.841 mol) in
dichloromethane (75 mL) was added a solution of 4 (25 g, 0.106 mol) in
dichloromethane (50 mL) at 0–10 °C. The reaction mixture was heated to
reflux and stirred for 9–11 h, then cooled to 25–35 °C and quenched into
chilled water (175 mL) below 10 °C. Thereafter, temperature of the
reaction mixture was raised to 25–35 °C and organic layer was
separated. The aqueous layer was extracted with dichloromethane
(2 x 60 mL). The combined organic layers were washed with water
(3 x 100 mL) and concentrated under atmospheric pressure below 60 °C.
Cyclohexane (250 mL) was added to reaction mixture and heated to

Chapter-2
53
reflux for 15–30 min. Water (75 mL) was added to the reaction mixture
and maintained for 15–30 min under reflux. The organic layer was
separated, cooled to 25–35 °C and stirred for 45 min. The precipitated
solid was filtered and washed with cyclohexane (22 mL). Recrystallization
from cyclohexane was repeated twice and dried under vacuum at 50–55
°C to obtain 21.3 g (60 %) of 22.
Purity by HPLC: 98.7 %.
IR (KBr, cm–1): 3090, 3063, 1625, 1590, 1491, 1464, 1396, 1383, 1191,
782, 754.
1H NMR (200 MHz, CDCl3 + CD3CN): δ 8.02 (d, J ═ 8.6 Hz, 2H), 7.50–
7.28 (m, 7H), 2.53 (s, 3H).
13C NMR (50 MHz, DMSO–d6): 167.1, 160.6, 147.2, 129.9, 129.6, 129.0,
128.7, 128.6, 128.1, 126.0, 114.8, 11.3.
MS (m/z): 334 (M+ + H).
2.6.8 4-(5-Methyl-3-phenyl-4-isoxazolyl)benzenesulfonamide (1)
To a stirred solution of 22 (20 g, 0.060 mol) in dichloromethane (120
mL) was added charcoal (1 g) and stirred for 30–45 min. Thereafter,
charcoal was filtered and washed with dichloromethane (40 mL). To the
combined filtrate was added aqueous ammonia (15 %, 90 mL, 0.794 mol)

Chapter-2
54
at 20–30 °C for 1–1.5 h. Subsequently, dichloromethane was distilled off
from the reaction mixture below 45 °C under atmospheric pressure, then
cooled to 5–10 °C and stirred for 30–45 min. The precipitated solid was
filtered and washed with water (20 mL). The wet compound was charged
into water (100 mL), stirred for 45–60 min and filtered the solid. Washed
with water (20 mL) and dried under vacuum at 80–85 °C to afford 15 g
(80 %) of 1.
Purity by HPLC: 99.92 %.
IR (KBr, cm–1): 3378, 3250, 2926, 1622, 1595, 1563, 1465, 1392, 1333,
1151, 784.
1H NMR (400 MHz, DMSO–d6): δ 7.84 (d, J ═ 8.2 Hz, 2H), 7.50–7.32 (m,
9H), 2.47 (s, 3H).
13C NMR (100 MHz, DMSO–d6): 167.5, 160.6, 143.3, 133.3, 130.0,
129.7, 128.7, 128.4, 128.1, 126.1, 114.2, 11.3.
MS (m/z): 315 (M+ + H).
Anal. for C16H14N2O3S: calcd: C, 61.13; H, 4.49; N, 8.92; S, 10.19.
Found: C, 60.89; H, 4.35; N, 8.98; S, 10.21.

Chapter-2
55
2.6.9 3-[5-Methyl-4-(4-sulfamoylphenyl)isoxazol-3-yl]-benzene
sulfonamide (Impurity-A, 23)
To a stirred solution of chlorosulfonic acid (347 g, 2.978 mol) in
dichloromethane (400 mL) was added a solution of 4 (20 g, 0.085 mol) in
dichloromethane (40 mL) at 0–10 °C. The resulted reaction mixture was
refluxed for 7–8 h followed by cooling to below 10 °C and quenched with
water (900 mL). The organic layer was separated and the aqueous layer
was extracted with dichloromethane (2 x 100 mL). Aqueous ammonia
(15 %, 260 mL, 2.294 mol) was added to the combined organic layer at
25–35 °C and stirred for 1 h. The precipitated solid was filtered and dried
at 80–85 °C to provide 23.5 g (70 %) of 23.
Purity by HPLC: 97.0 %.
IR (KBr, cm–1): 3322, 3241, 1636, 1424, 1346, 1161, 786.
1H NMR (400 MHz, DMSO–d6): δ 8.05 (s, 1H), 7.90 (d, J ═ 8.0 Hz, 1H),
7.86 (d, J ═ 8.4 Hz, 2H), 7.58 (t, J ═ 8.0 Hz, 1H), 7.45 (d, J ═ 8.4 Hz, 2H),
7.42–7.35 (m, 5H), 2.48 (s, 3H).
13C NMR (50 MHz, DMSO–d6): 168.0, 159.6, 144.8, 143.3, 132.6, 131.2,
129.9, 129.4, 129.0, 126.7, 126.0, 125.1, 114.2, 11.3.

Chapter-2
56
MS (m/z): 394 (M+ + H).
2.6.10.1 5-Benzyl-3-phenylisoxazole (4a)
A solution of 16 (100 g, 0.745 mol) and pyrrolidine (88 g, 1.237 mol)
in cyclohexane (740 mL) was maintained under azeotropic reflux
conditions until water collection stops. The resulted reaction mass was
concentrated under reduced pressure below 70 °C followed by addition
dichloromethane (1000 mL) and triethylamine (116 g, 1.146 mol) at
25–35 °C. To the resulted reaction mixture was added a solution of 20
(125 g, 0.803 mol) in dichloromethane (200 mL) and stirred at 25–35 °C
for 3 h. Subsequently, the reaction mixture was quenched with water
(750 mL) and organic layer was separated. The aqueous layer was
extracted with dichloromethane (200 mL) and the combined organic
layers were concentrated under reduced pressure below 55 °C.
Thereafter, conc hydrochloric acid (350 mL) and water (700 mL) were
charged to the reaction mixture. The resulted reaction mixture was
refluxed for 3 h at 100 °C, cooled to 25–35 °C and charged
dichloromethane (450 mL). Organic layer was separated and aqueous
layer was extracted with dichloromethane (200 mL). The combined
organic layers were concentrated under reduced pressure below 50 °C.

Chapter-2
57
After cooling the reaction mixture to 25–35 °C, isopropyl alcohol (100 mL)
was charged and filtered the solid (4). The obtained mother liquor was
concentrated below 60 °C under reduced pressure, charged acetone (200
mL) at 25–35 °C and stirred for 1h. Filtered the solid and dried at
40–45 °C to obtain 26 g (15 %) of 4a.
Purity by HPLC: 98.5 %.
IR (KBr, cm–1): 2918, 1597, 1578, 1408, 775.
1H NMR (200 MHz, CDCl3): δ 7.82–7.70 (m, 2H), 7.53–7.22 (m, 8H), 6.33
(s, 1H), 4.13 (s, 2H).
MS (m/z): 236 (M+ + H).
2.6.10.2 4-[(3-Phenylisoxazol-5-yl)methyl]benzenesulfonamide
(Impurity-B, 24)
To a solution of chlorosulfonic acid (79.2 g, 0.680 mol) in
dichloromethane (50 mL) was added a solution of 4a (20 g, 0.085 mol) in
dichloromethane (50 mL) at 0–5 °C. The reaction mixture was stirred at
40 °C for 3–5 h and quenched with water (140 mL) below 10 °C. The
layers were separated and the product was extracted with
dichloromethane (2 x 50 mL) from aqueous layer. Thereafter, aqueous

Chapter-2
58
ammonia solution (15 %, 130 mL 1.147 mol) was added to the combined
organic layer at 25–35 °C and stirred for 1–2 h. The precipitated solid
was filtered and washed with water (40 mL) and dichloromethane (20 mL)
successively. Wet solid was dried at 80–85 °C to afford 16 g (60 %) of 24.
Purity by HPLC: 99.5 %.
IR (KBr, cm–1): 3363, 3271, 1609, 1341, 1158, 771.
1H NMR (200 MHz, CDCl3): δ 8.03 (d, J ═ 8.4 Hz, 2H), 7.91–7.65 (m,
2H), 7.56 (d, J ═ 8.4 Hz, 2H), 7.44–7.35 (m, 3H), 6.33 (s, 1H), 4.27 (s,
2H).
13C NMR (100 MHz, DMSO–d6): 171.7, 161.9, 142.7, 140.2, 130.0,
129.2, 129.0, 128.5, 126.4, 126.0, 100.4, 31.8.
MS (m/z): 315 (M+ + H).
2.6.11 3-(5-Methyl-3-phenylisoxazol-4-yl)benzenesulfonamide
(Impurity-C, 25)
To a solution of chlorosulfonic acid (40 g, 0.343 mol) in
dichloromethane (40 mL) was added a solution of 4 (10 g, 0.042 mol) in
dichloromethane (10 mL) at 0–10 °C. The reaction mixture was refluxed
for 7–8 h. The resulted reaction mixture was quenched with water

Chapter-2
59
(100 mL) below 10 °C. The organic layer was separated and the aqueous
layer was extracted with dichloromethane (2 x 50 mL). The combined
organic layers were concentrated below 45 °C under reduced pressure.
Thereafter, cyclohexane (100 mL) was added to the residue at 25–35 °C
and stirred for complete solid separation. The precipitated solid (22) was
filtered and filtrate was concentrated below 60 °C under reduced
pressure to obtain 22a (not isolated) along with some quantity of 22 (not
quantified). To a solution of 22a (crude) and 22 in dichloromethane
(100 mL) was added aqueous ammonia (15 %, 65 mL, 0.573 mol) at
25–35 °C. The resulted reaction mixture was stirred for 1 h at 25–35 °C.
The organic layer was separated and the aqueous layer was extracted
with dichloromethane (2 x 20 mL). The combined organic layers were
concentrated under reduced pressure below 45 °C, dichloromethane
(10 mL) was added at 25–35 °C and cooled to 10–15 °C. After stirring for
1 h, the precipitated solid (1) was filtered. The filtrate was concentrated
and repeated the crystallization twice using dichloromethane to remove 1
completely. The filtrate (free from 1) was concentrated below 45 °C under
reduced pressure. Ethyl acetate was added to the resulted residue and
refluxed for 30 min followed by cooling to 0–5 °C and stirred for 1 h. The
precipitated solid was filtered and dried at 80–85 °C to provide 1.3 g
(10 %) of 25.
Purity by HPLC: 96.0 %.
IR (KBr, cm–1): 3337, 3247, 1334, 1165.

Chapter-2
60
1H NMR (400 MHz, DMSO–d6): δ 7.83 (d, J ═ 7.8 Hz, 1H), 7.71 (s, 1H),
7.59 (t, J ═ 7.8 Hz, 1H), 7.49–7.36 (m, 8H), 2.45 (s, 3H).
13C NMR (100 MHz, DMSO–d6): 167.0, 160.4, 148.7, 147.4, 129.8,
129.6, 129.1, 128.9, 128.7, 128.2, 128.0, 127.9, 126.4, 126.0, 115.0,
11.1.
MS (m/z): 315 (M+ + H).
2.6.12 4-(5-Methyl-3-phenylisoxazol-4-yl)benzenesulfonic acid
(Impurity-D, 26)
A mixture of 22 (20 g, 0.060 mol), water (65 mL) and tetrahydrofuran
(280 mL) was heated to reflux and maintained for 24 h. Solvent was
distilled completely from the reaction mixture below 80 °C under reduced
pressure. Reaction mixture was cooled to 25–35 °C, toluene (200 mL) was
added and stirred for 1 h. The solid was filtered and dried at 50–55 °C to
obtain 18.5 g (98.4 %) of 26.
Purity by HPLC: 99.7 %.
IR (KBr, cm–1): 3395, 1622, 1396, 1177.
1H NMR (400 MHz, DMSO–d6): δ 7.64 (d, J ═ 8.0 Hz, 2H), 7.45–7.35 (m,
5H), 7.19 (d, J ═ 8.0 Hz, 2H), 2.44 (s, 3H).
13C NMR (100 MHz, DMSO–d6): 167.0, 160.5, 147.4, 129.8, 129.6,

Chapter-2
61
128.9, 128.7, 128.6, 128.0, 126.0, 114.8, 11.2.
MS (m/z): 316 (M+ + H).
2.6.13 4-(5-Methyl-3-phenylisoxazol-4-yl)-N-[4-(5-methyl-3-
phenylisoxazol-4-yl)phenylsulfonyl]benzenesulfonamide
(Impurity-E, 27)
A mixture of 1 (10 g, 0.032 mol), 22 (16 g, 0.048 mol) in pyridine (50
mL) was heated to 110–115 °C and stirred for 9 h at the same
temperature. The reaction mixture was concentrated under reduced
pressure at 95 °C. The resulted solid was recrystallized from isopropyl
alcohol (300 mL) and the wet compound was again recrystallized from
the mixture of water (100 mL) and acetone (100 mL). The wet solid was
dried at 80–85 °C to afford 12.5 g (64 %) of 27.
Purity by HPLC: 98.0 %.
IR (KBr, cm–1): 3339, 1626, 1369, 1165, 871.
1H NMR (400 MHz, CDCl3 + DMSO–d6): δ 7.95 (d, J ═ 8.2 Hz, 4H), 7.45–
7.36 (m, 10H), 7.32 (d, J ═ 8.2 Hz, 4H), 2.50 (s, 6H).
13C NMR (100 MHz, DMSO–d6): 167.2, 160.6, 145.4, 131.5, 129.7,
129.0, 128.7, 128.5, 128.1, 126.5, 114.5, 11.3.
MS (m/z): 612 (M+ + H).







![Synthetic Routes towards 2-thia-7,8-diaza-cyclopenta[l ...277519/FULLTEXT01.pdf · 1-thia-7,8-diaza-cyclopenta[l]phenanthrene for Molecular Electronics Applications ... Kemin som](https://img.pdfslide.net/doc/110x75/5d1b94de88c993dc468d14cb/synthetic-routes-towards-2-thia-78-diaza-cyclopental-277519fulltext01pdf.jpg)







![Modular Synthesis of pH-Sensitive Fluorescent Diaza[4]helicenes · 2014-10-10 · FULL PAPER E. Vauthey, J. Lacour et al. atoms.[10a] First, compound 6 was prepared by using N,N-](https://img.pdfslide.net/doc/110x75/5e6aa5e3fc371e442638f61a/modular-synthesis-of-ph-sensitive-fluorescent-diaza4-2014-10-10-full-paper-e.jpg)













