Embed Size (px)
Citation preview

Journal of Steroid Biochemistry & Molecular Biology 144 (2014) 348–360
Synthesis of atypical bile acids for use as investigative tools for thegenetic defect of 3b-hydroxy-D5-C27-steroid oxidoreductasedeficiency
Antimo Gioiello a,*, Bruno Cerra a, Wujuan Zhang b, Gian Paolo Vallerini c,Gabriele Costantino c, Francesca De Franco d, Daniela Passeri d, Roberto Pellicciari a,d,Kenneth D.R. Setchell b
aDepartment of Pharmaceutical Sciences, University of Perugia, Via del Liceo 1, I-06122 Perugia, ItalybDivision of Pathology and Laboratory Medicine, Cincinnati Children’s Hospital Medical Center and Department of Pediatrics of the University of CincinnatiCollege of Medicine, Cincinnati, Ohio, USAcDepartment of Farmacy, University of Parma, Viale delle Scienze 27/A, Parma I-43124, Italyd TES Pharma, Via P. Togliatti, 20, Loc Taverne, I-06073 Corciano, Italy
A R T I C L E I N F O
Article history:Received 27 March 2014Received in revised form 28 May 2014Accepted 17 June 2014Available online 19 June 2014
Keywords:Inborn errorsBile acids3b-Hydroxy-D5-C27-steroidoxidoreductaseSynthesisNuclear receptorsDiagnosisGene expressionBiomarkersMass spectrometry
A B S T R A C T
Deficiency of 3b-hydroxy-D5-C27-steroid oxidoreductase (HSD3B7), an enzyme catalyzing the secondstep in the pathway for bile acid synthesis, leads to a complete lack of the primary bile acids, cholic andchenodeoxycholic acids, and the accumulation of 3b,7a-dihydroxy- and 3b,7a,12a-trihydroxy-D5-cholenoic acids. Patients affected by this autosomal recessive genetic defect develop cholestatic liverdisease that is clinically responsive to primary bile acid therapy. Reference standards of these compoundsare needed to facilitate diagnosis and to accurately quantify biochemical responses to therapy.Described are a novel synthesis of atypical bile acids that characterize the HSD3B7 deficiency and theireffect on bile acid-activated nuclear receptors, target genes and cytochromes involved in bile acidhomeostasis and detoxification. The failure of 3b-hydroxy-D5-cholenoic acids to function as FXR, PXRand CAR agonists and to exert hepatoprotective actions explains the mechanism for progressivecholestatic liver disease in patients with HSD3B7 deficiency.
ã 2014 Elsevier Ltd. All rights reserved.
Contents lists available at ScienceDirect
Journal of Steroid Biochemistry & Molecular Biology
journa l homepage: www.e l sev ier .com/ loca te / jsbmb
1. Introduction
In recent years, the interest in the field of bile acids has grownsignificantly as a result of a redefinition of their biological functionin various physiological and pathological conditions [1–3]. Inaddition to their fundamental and classical role of facilitating theabsorption of fats and fat-soluble vitamins, bile acids are now
Abbrevations: HSD3B7, 3b-hydroxy-D5-C27-steroid oxidoreductase; FXR, farne-soid X receptor; PXR, pregnane X receptor; CAR, constitutive androstane receptor;HDCA, hyodeoxycholic acid; FAB, fast atom bombardment; ESI, electrosprayionization; Q-TOF, quadrupole-time-of-flight; MRM, multiple reaction monitoring;BSEP, bile salt exporting pump; SHP, small heterodimer partner; MDR1, multi drugresistance; LDH, lactate dehydrogenase; Q-RT-PCR, quantitative real-time poly-merase chain reaction.* Corresponding author. Tel.: +39 75 5852318; fax: +39 75 5855160.E-mail address: [email protected] (A. Gioiello).
http://dx.doi.org/10.1016/j.jsbmb.2014.06.0080960-0760/ã 2014 Elsevier Ltd. All rights reserved.
considered key players in being ligands for a complex network ofnuclear (‘genomic’ action) and membrane (‘non-genomic’ action)receptors. Thus, bile acids self-regulate their own homeostasis andact as modulators of cholesterol and triglyceride levels, glucosehomeostasis and energy expenditure. These bile acid-mediatedprocesses have been investigated for their relevance in liverdiseases and other facets of the metabolic syndrome, e.g. diabesityand atherosclerosis [4,5], which represent a considerable increas-ing health burden in both developed and developing countries.
Within the enterohepatic circulation, bile acids provideimportantly the primary driving force for the promotion andsecretion of bile, thus protecting the liver from accumulation ofendogenous and exogenous toxins [6]. The failure to synthesizeprimary bile acids, as occurs in well-defined genetic defects in bileacid biosynthesis, leads to progressive cholestatic liver disease.This is exacerbated by the accumulation of atypical hepatotoxicbile acids generated as a result of mutations in genes encoding

A. Gioiello et al. / Journal of Steroid Biochemistry & Molecular Biology 144 (2014) 348–360 349
enzymes critical to bile acid synthesis [7–9]. When undiagnosed,or untreated, many patients will progress to end-stage liver diseaseand eventually require liver transplantation. Oral primary bile acidtherapy is extremely effective at normalizing the biochemicalabnormalities and improving the histology and clinical markers,and patients live an essentially normal life-style except that bileacid therapy is life-long [7,10].
To date, nine inborn errors in bile acid synthesis have beendescribed. The 3b-hydroxy-D5-C27-steroid oxidoreductase(HSD3B7) deficiency is the most common one [11–13]. HSD3B7is an endoplasmic reticulum membrane-bound enzyme that isresponsible for catalysing one of the early steps in bile acidbiosynthesis affecting alterations to the A/B rings of the steroidnucleus (Fig. 1). It is encoded by a gene, HSD3B7 localized onchromosome 16p11.2-12 and numerous mutations have beenreported accounting for the production of an inactive enzyme [14].The failure to catalyse the classical oxido-reduction step insteroidogenesis which includes the oxidation of the C3b-OHand isomerization of the D5 double bond, does not prevent theremaining reactions occurring in the biosynthetic pathway andconsequently individuals with this defect exhibit a distinct bileacid profile that features excessive production of atypical3b-hydroxy-D5-cholenoic acids 1 and 2 (Fig. 1).
As a result, impairment of HSD3B7 leads to the hepaticaccumulation of these abnormal metabolites that cannot be
HO7α-Hydroxy-cholesterol
OH
O OH
7α-Hydroxy-cholest-4-en-3-one
3ß-Hydroxy-C27-Steroid Oxidoreductase
HO
3β,7α-Δ5-cholen-
HO
3β,7α,12Δ5-cholen-
CO2H
HO OH
CO2H
HO OH
OH
Chenodeoxycholic Acid Cholic AcidH H
HO Cholesterol
CYP7A1
Liver
Fig. 1. Atypical bile acids produced in liver and excreted in human urine/serum of pa(HSD3B7) deficiency.
transported across the canalicular membrane [15]. Accordingly,3b-hydroxy-D5-cholenoic acids 1 and 2 are excreted mainly assulfate and glyco-sulfate conjugates 3–6 in human urine (Fig. 1).The presence of these atypical bile acids, concomitant with a lackof primary bile acids in the urine/serum is the basis for diagnosis ofthis genetic defect and for monitoring the biochemical response tooral primary bile acid therapy [7]. In the absence of suitable purereferences compounds, quantification of D5-cholenoic acids hasbeen problematic and has relied on solvolysis/hydrolysis ofconjugate groups followed by gas chromatography with or withoutmass spectrometry [11]. Unless care is taken, solvolysis andhydrolysis leads to formation of by-products derived from thedehydration reaction at the labile allylic 7a-hydroxy group,resulting in inaccurate measurement of concentrations. There istherefore an urgent need for reference compounds to developsensitive and validated analytical assays to better quantify thelevels of these atypical bile acids for diagnosis of this genetic defectand for monitoring the compliance to therapy. This prompted us todevelop a chemical synthesis for the sulfate and glyco-sulfateconjugates of 3b,7a-dihydroxy-D5-cholenoic and 3b,7a,12a-trihydroxy-D5-cholenoic acids.
We describe here the synthesis and bio-chemical characteriza-tion of a series of D5-cholenoic acid analogs, which are thesignature metabolites of the HSD3B7 deficiency in humans (Fig. 1).The availability of these reference compounds now provides the
CO2H
Dihydroxy-24-oic acid (1)
OH
CO2H
α-Trihydroxy-24-oic acid (2)
OH
OH
NaO3SO OH
NaO3SO OH
OHO
R
R=
R
O
HN CO2H
glycosulfate 4
ExcretionR= OH, sulfate 3
Excretion
R= HN CO2H
glycosulfate 6
R= OH, sulfate 5
tients with liver disease caused by a 3b-hydroxy-D5-C27-steroid oxidoreductase

350 A. Gioiello et al. / Journal of Steroid Biochemistry & Molecular Biology 144 (2014) 348–360
means to be able to develop methods for their accuratemeasurement for application in diagnosis and monitoring ofresponse to therapy in patients with HSD3B7 deficiency. Addition-ally, using these synthesized compounds as tools to gain insightsinto the mechanism(s) responsible of the cholestasis and liverdamage in patients with the HSD3B7 deficiency, we report for thefirst time on their cellular hepatocytotoxicity, their affinity towardsa subset of nuclear receptors, namely farnesoid X receptor (FXR),pregnane X receptor (PXR) and constitutive androstane receptor(CAR), and on their effects on genes and cytochromes involved inbile acid homeostasis and detoxification.
2. Materials and methods
2.1. Synthetic chemistry
2.1.1. General method1H NMR spectra were recorded at 200 and 400 MHz, 13C NMR
spectra were recorded at 100.6 and 50.3 MHz using the solventsindicated below. Chemical shifts are reported in ppm. Theabbreviations used are as follows: s, singlet; d, doublet; dd, doubledoublet; t, triplet; dt, double triplet; qt, quartet triplet; m,multiplet; brs, broad singlet; brm, broad multiplet. The finalproducts were purified by flash chromatography on silica gel(0.040–0.063 mm). TLC was performed on aluminium backed silicaplates (silica gel 60 F254). Hyodeoxycholic acid (7) was purchasedfrom Sigma–Aldrich. All the reagents were of analytical grade. Theanalytical HPLC measurements were made on a Shimadzu (Kyoto,Japan) LC-20A Prominence equipped with a CBM-20A communi-cation bus module, two LC-20AD dual piston pumps, a SPD-M20Aphotodiode array detector and a Rheodyne 7725i injector(Rheodyne Inc., Cotati, CA, USA) with a 20 mL stainless steel loop.A Varian 385-LC evaporative light scattering detector (ELSD)(Agilent Technologies, Santa Clara, CA, USA) was utilized for theanalyses. The analog-to-digital conversion of the output signalfrom the ELSD was allowed by a common interface device. Theadopted ELSD conditions for the analysis of all bile salts were: 30 �Cnebulization temperature, 50 �C evaporation temperature, 1.5 Lmin�1 gas flow rate (air) and 2.0 as the gain factor. A GraceSmartRP18 column (Grace, Sedriano, Italy) 250 � 4.6 mm i.d., 5 mm,100 Åwas used as the analytical column. The analyses were carried out ata 1.0 mL min�1
flow rate after previous conditioning by passingthrough the column the selected mobile phase for at least 30 min atthe same eluent velocity. Before being used, all the mobile phaseswere always filtered through a 0.22 mm Millipore filter (Bedford,MA, USA) and then degassed with 20 min sonication. The columntemperature was controlled through a Grace (Sedriano, Italy)heather/chiller (Model 7956 R) thermostat. All the analyses wereconducted at a 25 �C column temperature. Acetonitrile (MeCN) andmethanol (MeOH) were purchased from Sigma–Aldrich (Milano,Italy). Ammonium formate (HCO2NH4), and formic acid (HCO2H)were purchased from Carlo Erba (Milano, Italy). HPLC-grade waterwas obtained from a tandem Milli-Ro/Milli-Q apparatus (Millipore,Bedford, MA, USA). The purity of each final products was estimatedusing two different HPLC methods of analysis: (a) compounds 1and 3. First method: H2O/MeCN (60:40, v/v) + HCO2NH4 100 mM,pH 3.5 (apparent value on the hydro-organic solution); Secondmethod: H2O/MeCN/MeOH (50:40:10, v/v/v) + HCO2NH4 40 mM,pH: 3.5 (apparent value on the hydro-organic solution); (b)compound 4. First method: H2O/MeCN (70:30, v/v) + HCO2NH4
40 mM, pH 3.5 (apparent value on the hydro-organic solution);Second method: H2O/MeOH (40:60, v/v) + HCO2NH4 40 mM, pH 3.5(apparent value on the hydro-organic solution); (c) compound 2.First method: H2O/MeOH (40:60, v/v) + HCO2NH4 40 mM, pH 3.0(apparent value on the hydro-organic solution); Second method:H2O/MeCN (60:40, v/v) + HCO2NH4 40 mM, pH 3.5 (apparent value
on the hydro-organic solution); (d) compounds 5 and 6. Firstmethod: H2O/MeOH (40:60, v/v) + HCO2NH4 40 mM, pH 3.0(apparent value on the hydro-organic solution); Second method:H2O/MeCN (70:30, v/v) + HCO2NH4 40 mM, pH 3.5 (apparent valueon the hydro-organic solution).
2.1.2. Synthesis of methyl 3a,6a-ditosyloxy-5b-cholan-24-oate (8)To a solution of HDCA (7) (30 g, 76.4 mmol) in MeOH,
p-toluensulphonic acid (p-TSA) (1.5 g, 8.7 mmol) was added, andthe mixture was stirred at room temperature for 18 h. The mixturewas then concentrated under reduced pressure, and the resultingresidue was dissolved in H2O (1 L), extracted with CHCl3(5 � 350 mL), dried over anhydrous Na2SO4, and concentratedunder reduced pressure to give methyl 3a,6a-dihydroxy-5b-cholan-24-oate (31.6 g, 76.4 mmol, quantitative yield) as whitesolid. The solid thus obtained (25 g, 61.5 mmol) was solved infreshly distilled pyridine (70 mL) and then a solution of tosylchloride (TsCl) (35.2 g, 184.5 mmol) in dry pyridine (37.3 mL) wasadded dropwise. The mixture was stirred at room temperature for18 h. Ice chips were gradually added to the mixture, and the solidwas filtered, washed with H2O and dried to give methyl 3a,6a-ditosyloxy-5b-cholan-24-oate (8) (33 g, 46.2 mmol, yield 75%) aswhite solid that was used for the following step without furtherpurification. 1H NMR (CDCl3, 400 MHz): d 0.59 (s, 3H, 18-CH3), 0.80(s, 3H,19-CH3), 0.88 (d, 3H, 21-CH3), 2.47 (s, 6H, 2 � Ar-CH3), 3.66 (s,3H, COOCH3), 4.30 (brm, 1H, 3-CH), 4.78 (brm, 1H, 6-CH), 7.27–7.37and 7.71–7.80 (m, 8H, 2 � C6H4). 13C NMR (CDCl3, 100.6 MHz): d11.9, 18.2, 20.5, 21.6, 21.7, 22.9, 23.9, 26.4, 27.4, 27.9, 30.8, 30.9, 32.1,34.8, 35.2, 36.1, 39.4, 39.5, 42.8, 46.3, 51.5, 55.7, 79.6, 81.7, 127.5,127.6, 129.7, 129.8, 134.5, 144.6, 144.7, 174.6.
2.1.3. Synthesis of methyl 3b-hydroxy-D5-cholen-24-oate (9)To a solution of ditosylate 8 (20.2 g, 28.3 mmol) in DMF (1 L), a
solution of AcOK (2.2 g, 22.6 mmol) in H2O (100 mL) was added,and the mixture was refluxed for 18 h. The solution was cooled atroom temperature, diluted with H2O (150 mL) and extracted withAcOEt (3 � 250 mL). The organic layer was washed with brine(3 � 100 mL), dried over anhydrous Na2SO4, and concentratedunder reduced pressure. The yellow-orange crude oil was dissolvedin MeOH (200 mL) and refluxed for 1 h in the presence of K2CO3
(20 g). After cooling at room temperature, the suspension wastreated with 1 N HCl (200 mL) and extracted with AcOEt(3 � 150 mL). The organic layer was washed with H2O (200 mL)and brine (200 mL), dried over anhydrous Na2SO4, and concentrat-ed under reduced pressure. Recrystallization of the crude oil fromH2O gave a orange solid (16 g) that was purified by flashchromatography (eluent: petroleum ether/AcOEt from 100:0 v/vto 70:30 v/v) to give the desired methyl 3b-hydroxy-D5-cholen-24-oate (9) (6.5 g, 16.9 mmol, yield 60%) as white solid. 1H NMR(CDCl3, 400 MHz): d 0.67 (s, 3H, 18-CH3), 0.92 (d, 3H, 21-CH3), 1.00(s, 3H, 19-CH3), 3.48–3.52 (brm, 1H, 3-CH), 3.66 (s, 3H, COOCH3),5.34 (d, 1H, 6-CH). 13C NMR (CDCl3, 100.6 MHz): d 11.8, 18.3, 19.4,21.0, 24.2, 28.1, 31.0 (2�), 31.6, 31.8, 35.4, 36.5, 37.2, 39.7, 42.3 (2�),50.0, 51.5, 55.7, 56.7, 71.8, 121.6, 140.7, 174.8.
2.1.4. Synthesis of methyl 3b-ter-butyldimethylsilyloxy-D5-cholen-24-oate (10)
To a solution of methyl 3b-hydroxy-D5-cholen-24-oate (9)(4.9 g, 12.6 mmol) in freshly distilled CH2Cl2 under argon atmo-sphere, imidazole (860 mg, 12.9 mmol) and tert-butyldimethylsilylchloride (2 g, 13.3 mmol) were added, and the mixture was stirredat room temperature for 3 h. The mixture was then diluted withH2O (50 mL) and extracted with CHCl3 (3 � 30 mL) and theresulting organic layer was dried over anhydrous Na2SO4, andconcentrated under reduced pressure. The crude (8 g) was filteredthrough a short column of silica gel by elution with CH2Cl2 to give

A. Gioiello et al. / Journal of Steroid Biochemistry & Molecular Biology 144 (2014) 348–360 351
methyl 3b- tert-butyldimethylsilyloxy-D5-cholen-24-oate (10)(5.8 g, 11.5 mmol, yield 91%) as yellowish solid. 1H NMR (CDCl3,400 MHz): d 0.08 (s, 6H, Si-(CH3 )2C(CH3)3), 0.70 (s, 3H, 18-CH3),0.91 (s, 9H, Si-(CH3)2C(CH3)3), 0.96 (d, 3H, 21-CH3), 1.02 (s, 3H,19-CH3), 3.49–3.51 (brm, 1H, 3-CH), 3.69 (s, 3H, COOCH3), 5.34 (d,1H, 6-CH). 13C NMR (CDCl3, 100.6 MHz): d 11.8, 14.3, 18.2, 18.3, 19.4,21.0, 24.2, 28.1, 31.0 (2�), 31.6, 31.8, 35.4, 36.5, 37.2, 39.8, 42.3 42.4,50.2, 51.4, 55.8, 56.8, 72.6, 121.1, 141.6, 174.8.
2.1.5. Synthesis of methyl 3b-ter-butyldimethylsilyloxy-7a-benzoyloxy-D5-cholen-24-oate (11)
To a solution of 3b-tert-butyldimethylsilyloxy-D5-cholen-24-oate (10) (4.9 g, 9.8 mmol) in freshly distilled CH2Cl2 under argonatmosphere, CuBr (2.8 g, 19.6 mmol) was added. The mixture wasrefluxed for 15 min and then a solution of tert-butylperoxybenzoate (7.4 mL, 39.3 mmol) in distilled CH2Cl2 was addeddropwise in 15 min. The mixture was refluxed for 18 h and agreenish color was developed. After cooling at room temperature,the mixture was filtered on Celite washing with CH2Cl2 and themother liquor was evaporated under reduced pressure. Purifica-tion of dark crude oil by flash chromatography (eluent: petroleumether/AcOEt from 100:0 v/v to 95:5 v/v) gave 2.9 g of methyl3b-tert-butyldimethylsilyloxy-7a-benzoyloxy-D5-cholen-24-oate(11) (4.7 mmol, yield 48%). 1H NMR (CDCl3, 400 MHz): d 0.02 (s, 6H,Si-(CH3 )2C(CH3)3), 0.70 (s, 3H, 18-CH3), 0.86 (s, 9H, Si-(CH3)2C(CH3)3), 0.94 (d, 3H, 21-CH3), 1.04 (s, 3H, 19-CH3), 3.48–3.54 (brm,1H, 3-CH ), 3.63 (s, 3H, COOCH3), 5.19 (s, 1H, 7-CH ), 5.67 (d, 1H,6-CH ), 7.46 (t, 2H, OCOC6H 5 meta ), 7.55–7.58 (m, 1H, OCOC6H 5
para), 8.03 (d, 2H, OCOC6H 5 ortho). 13C NMR (CDCl3, 100.6 MHz): d11.4, 14.2, 18.1, 18.2, 18.3, 20.8, 24.0, 25.6, 25.9, 27.9, 31.0, 31.7, 35.3,36.3, 36.9, 37.4, 39.3, 42.4, 43.5, 49.7, 51.4, 55.6, 60.4, 69.3, 71.9,119.4, 128.4, 129.6, 131.1, 132.7, 148.8, 166.1, 174.6.
2.1.6. Synthesis of methyl 3b-hydroxy-7a-benzoyloxy-D5-cholen-24-oate (12)
To a solution of methyl 3b- tert-butyldimethylsilyloxy-7a-benzoyloxy-5-cholen-24-oate (11) (1.41 g, 2.6 mmol) in acetone,a solution of bis-acetonitrile Palladium-(II)-chloride (3 mg,0.11 mmol) in H2O (200 mL, 11.3 mmol) was added, and the mixturewas stirred at room temperature for 7 h. After filtration of thereaction mixture on Celite, eluting with CH2Cl2, the mother liquorwas washed with H2O (100 mL) and brine (100 mL), dried overanhydrous Na2SO4 and concentrated under reduced pressure.Purification of yellow crude oil by flash chromatography (eluent:petroleum ether/AcOEt from 100:0 v/v to 75:25 v/v) gave methyl3b-hydroxy-7a-benzoyloxy-D5-cholen-24-oate (12) (830 mg,1.9 mmol, yield 72%) as yellow solid. 1H NMR (CDCl3, 400 MHz):d 0.71 (s, 3H, 18-CH3), 0.94 (d, 3H, 21-CH3), 1.06 (s, 3H, 19-CH3),3.57–3.59 (brm, 1H, 3-CH), 3.65 (s, 3H, COOCH3), 5.20–5.22 (m, 1H,7-CH), 5.72 (d, 1H, 6-CH), 7.47 (t, 2H, OCOC6H5meta), 7.56–5.78 (m,1H, OCOC6 H 5 para), 8.02 (d, 2H, OCOC6 H 5 para). 13C NMR (CDCl3,100.6 MHz): d 11.4, 18.2, 18.3, 20.8, 24.0, 25.6, 27.9, 31.0 (2�), 31.1,35.3, 36.2, 36.8, 37.3, 39.2, 41.9, 42.3, 43.5, 49.6, 51.5, 55.6, 69.1, 71.1,119.9, 129.5, 131.0, 132.7, 148.0, 166.1, 174.7.
2.1.7. Synthesis of 3b,7a-dihydroxy-D5-cholen-24-oic acid (1)A solution of methyl 3b-hydroxy-7a-benzoyloxy-D5-cholen-
24-oate (12) (395 mg, 0.78 mmol) in a solution of NaOH in MeOH(10% w/v) (8 mL) was refluxed for 24 h. The mixture wasconcentrated under reduced pressure and the residue wasdissolved in H2O (15 mL) and extracted with Et2O (3 � 20 mL).The aqueous phase was treated at 0 �C with 0.1 N HCl until pH 4 andextracted with CHCl3/MeOH 90:10 v/v (8 � 30 mL). The purificationof crude by automatic chromatography (Column: SNAP KP-C18-HS12 g; eluent: H2O/MeOH from 50:50 v/v to 30:70 v/v) gave 3b,7a-dihydroxy-D5-cholen-24-oic acid (1) (225 mg, 0.59 mmol, yield
75%) as light yellow amorphous solid. mp: 212–213 �C. 1H NMR(CD3OD + 2 gtt D2O, 400 MHz): d 0.72 (s, 3H, 18-CH3), 0.97 (d, 3H,21-CH3), 1.00 (s, 3H, 19-CH3), 3.49–3.52 (brm, 1H, 3-CH), 3.77 (s, 1H,7-CH), 5.55 (d, 1H, 6-CH). 13C NMR (CD3OD + 2 gtt D2O, 100.6 MHz):d 12.1, 18.7, 18.8, 21.8, 29.2, 32.3, 36.7, 38.0, 38.4, 38.9 (2�), 40.5,42.7, 43.2, 43.3, 48.4, 49.7, 50.6, 57.0, 65.8, 72.0, 124.7, 146.7, 178.7.IR (nmax in cm�1): 3510, 3250 (OH), 2600, 1710 (CO2H). Purity fromHPLC: 1� method: 100%; 2� method: 99.7%.
2.1.8. Synthesis of 3b-sulfooxy-7a-hydroxy-D5-cholen-24-oic acid,disodium salt (3)
To a solution of 3b,7a-dihydroxy-D5-cholen-24-oic acid (1)(186 mg, 0.48 mmol) in freshly distilled pyridine (5 mL), at 0 �C andunder argon atmosphere, a solution of chlorosulphonic acid(180 mL, 2.8 mmol) in freshly distilled pyridine (5 mL) was added.After 30 min, the mixture was allowed to warm to roomtemperature and stirred for additional 2 h. Then, 3 N aqueoussolution of NaOH (10 mL) was added and the mixture was stirred atroom temperature for 30 min. The alkaline aqueous phase wasextracted with cool Et2O (3 � 30 mL), concentrated under reducedpressure and filtered on RP-8 cartridge, eluting first with H2O andthen with H2O/MeOH 50:50 v/v. The purification of crude by MPLC(Column: Lobar 310-25 LiChroprep RP-18, 40–62 mm; eluent: H2O/MeOH 75:25 v/v) gave 3b-sulfooxy-7a-hydroxy-D5-cholen-24-oicacid as disodium salt (3) (237 mg, 0.46 mmol, yield 96%) as lightbrown amorphous solid. mp: >215 �C. 1H NMR (d6-DMSO,400 MHz): d 0.64 (s, 3H, 18-CH3), 0.88 (d, 3H, 21-CH3), 0.96 (s,3H, 19-CH3), 3.59 (s, 1H, 7-CH), 3.95–4.00 (brm, 1H, 3-CH), 5.43 (d,1H, 6-CH). 13C NMR (d6-DMSO, 100.6 MHz): d 12.0, 18.2, 18.9, 20.7,24.1, 28.2, 29.1, 33.2, 35.6, 35.8, 36.9, 37.2, 37.7, 38.8, 39.3, 41.7, 42.2,49.4, 56.4, 63.7, 75.3, 125.6, 143.5, 178.1. IR (nmax in cm�1): 3554(OH), 2554, 1610 (CO2
�). Purity from HPLC: 1� method: 98.9%; 2�
method: 95.5%.
2.1.9. Synthesis of glyco-conjugate of 3b-sulfooxy-7a-hydroxy-D5-cholen-24-oic acid, disodium salt (4)
To a solution of 3b-sulfooxy-7a-hydroxy-D5-cholen-24-oicacid, disodium salt (3) (190 mg, 0.37 mmol) in DMF (5 mL), DMT-MM (306 mg, 1.11 mmol) and Et3N (0.26 mL, 1.85 mmol) wereadded and the resulting suspension was stirred for 15 min at roomtemperature. Then, glycine ethyl ester hydrochloride (309 mg,2.22 mmol) was added and the mixture was stirred at roomtemperature for additional 3 h. 1 N aqueous solution of NaOH(20 mL, 2.3 mmol) was added and the mixture was stirred for 2 h at40 �C. The mixture was cooled at room temperature and poured incool acetone. The precipitate was filtered, washed with cool Et2Oand purified by automatic chromatography (Column: SNAP KP-C18-HS 12 g; eluent: H2O/MeOH from 100:0 v/v to 95:5 v/v) to givethe desired glyco conjugate of 3b-sulfooxy-7a-hydroxy-D5-cholen-24-oic acid, disodium salt (4) (125 mg, 0,24 mmol, yield65%) as light yellow amorphous solid. mp: >215 �C. 1H NMR (D2O,400 MHz): d 0.60 (s, 3H, 18-CH3), 0.88 (d, 3H, 21-CH3), 0.91 (s, 3H,19-CH3), 3.78 (s, 1H, 7-CH), 3.59–3.64 (brs, 1H, NH-CH 2-CO2), 4.10–4.16 (brm, 1H, 3-CH), 5.56 (d, 1H, 6-CH). 13C NMR (D2O, 100.6 MHz):d 11.4,17.9,18.1, 20.6, 23.8, 27.7, 28.2, 31.6, 32.4, 35.2, 36.6, 37.0, 37.1,38.6, 38.9, 41.7, 41.9, 43.3, 49.0, 54.7, 64.9, 79.8, 124.0, 145.2, 176.8,177.1. IR (nmax in cm�1): 3303 (OH), 2937, 1657 (CO2), 1592 (CO),1221, 1072, 1050, 965. Purity from HPLC: 1� method: 95.4%; 2�
method: 95.6%.
2.1.10. Synthesis of methyl 3a,12a-dihydroxy-7-oxo-5b-cholan-24-oate (14)
To a solution of 7-ketodeoxycholic acid (13), (5.0 g, 12.3 mmol)in MeOH (100 mL), p-TSA monohydrate (234 mg, 1.23 mmol) wasadded and the mixture was sonicated for 2 h. MeOH was removedby evaporation under reduced pressure, the residue solved in H2O

352 A. Gioiello et al. / Journal of Steroid Biochemistry & Molecular Biology 144 (2014) 348–360
(100 mL) and extracted with AcOEt (3 � 80 mL). The combinedorganic phases were washed with aqueous saturated solution ofNaHCO3 and brine, dried over anhydrous Na2SO4 and concentratedunder reduced pressure, affording methyl 3a,12a-dihydroxy-7-oxo-5b-cholan-24-oate (14) (5.17 g, 12.3 mmol, quantitative yield)as a white solid, that was used for the following step withoutfurther purification. 1H NMR (CDCl3, 400 MHz): d 0.67 (s, 3H, 18-CH3), 0.96 (d, 3H, 21-CH3), 1.16 (s, 3H, 19-CH3), 2.81–2.85 (m, 1H, 6-CH), 3.56 (m,1H, 3-CH), 3.65 (s, 3H, CO2CH3), 3.99 (s,1H,12-CH). 13CNMR (CDCl3, 100.6 MHz): d 12.8, 17.4, 22.8, 24.3, 27.6, 29.2, 29.7,30.9, 31.1, 34.1, 34.7, 35.0, 36.0, 37.3, 40.6, 45.3, 46.0, 46.4, 46.5, 49.5,51.5, 70.8, 72.1, 174.7, 211.8.
2.1.11. Synthesis of methyl 3b,12a-dihydroxy-7-oxo-5b-cholan-24-oate (15)
DEAD (2.5 mL, 15.9 mmol) was added to a solution of PPh3
(4.18 g, 15.9 mmol) in freshly distilled THF (240 mL), cooled to 0 �C,under argon atmosphere. The mixture was stirred for 30 min at thistemperature, then a solution of 14 (4.47 g, 10.6 mmol) and p -NBA(2.13 g, 12.8 mmol) in freshly distilled THF (120 mL) was added. Thereaction mixture was allowed to warm to room temperature andstirred for additional 3.5 h. The reaction mixture was treated withaqueous saturated solution of NaHCO3 and extracted with AcOEt(3 � 70 mL). The organic phase was washed with brine, dried overanhydrous Na2SO4 and concentrated under reduced pressure. Theresidue was dissolved in a 10% w/v solution of NaOH in MeOH(25 mL) and the reaction mixture was stirred at room temperaturefor 6 h. MeOH was removed by evaporation under reducedpressure and the residue was solved in water. The mixture waswashed with Et2O, in order to remove residual PPh3. The aqueousphase was acidified (pH 1) with 3 N aqueous HCl and extractedwith AcOEt (3 � 50 mL). The organic phase was dried overanhydrous Na2SO4 and concentrated under reduced pressure.The residue was solved in MeOH (80 mL), then p -TSA monohydrate(172 mg, 0.90 mmol) was added. The mixture was sonicated for 2 h.The reaction mixture was concentrated under reduced pressureand the residue was solved in water and extracted with AcOEt(3 � 50 mL). The organic phase was washed with aqueoussaturated solution of NaHCO3 and brine, dried over anhydrousNa2SO4, filtered and concentrated under reduced pressure. Thecrude was purified by flash chromatography (eluent: CHCl3/MeOHfrom 100:0 v/v to 95:5 v/v) to give methyl 3b,12a-dihydroxy-7-oxo-5b-cholan-24-oate (15) (3.8 g, 9.04 mmol, yield 85%), as awhite solid. 1H NMR (CDCl3, 200 MHz): d 0.69 (s, 3H, 18-CH3), 0.96(d, 3H, 21-CH3), 1.22 (s, 3H, 19-CH3), 2.82–2.92 (m, 1H, 6-CH), 3.67(s, 3H, CO2CH3), 4.01 (s, 1H, 12-CH). 4.08 (s, 1H, 3-CH). 13C NMR(CDCl3, 50.3 MHz): d 12.8, 17.3, 23.2, 24.1, 27.2, 27.5, 28.8, 29.3, 30.7,30.9, 34.4, 34.8, 35.1, 35.3, 40.6, 41.0, 44.9, 46.5 (2�), 49.4, 51.4, 66.1,72.0, 174.5, 212.1.
2.1.12. Synthesis of methyl 3b,12a-dihydroxy-7-oxo-D5-cholen-24-oate (16)
A stirred solution of freshly distilled diisopropylamine (10.8 mL,76.7 mmol) in freshly distilled THF (75 mL) was prepared underargon atmosphere and cooled to -78 �C. Butyllithium (2.5 M inhexanes, 28.6 mL, 71.6 mmol), was added dropwise, via an argonconditioned dropping funnel. After stirring at �78 �C for 30 min,TMSCl (13.0 mL, 102 mmol) was added dropwise. After stirring foradditional 30 min, a solution of 15 (4.30 g, 10.2 mmol) in freshlydistilled THF (30 mL) was added dropwise taking care thetemperature not to raise over �70 �C during the addition. Thereaction mixture was kept under stirring at -78 �C for 1 h, thentriethylamine (30 mL) was added dropwise. The mixture wasallowed to warm to room temperature then aqueous saturatedsolution of NaHCO3 (25 mL) was added. The organic phase wasseparated and washed several times with H2O and brine, dried over
anhydrous Na2SO4 and concentrated under reduced pressure. To asolution of trimethylsilylenolether thus obtained, in freshlydistilled CH3CN (150 mL), DTBMP (3.18 g, 15.5 mmol) and DDQ(7.03 g, 30.9 mmol) were added and the reaction mixture wasstirred at room temperature overnight, under argon atmosphere.The reaction mixture was cooled to 0 �C, then 1 N HCl was added(pH 5) and the mixture was stirred at 0 �C for 1 h. The mixture wasdiluted with brine and extracted with Et2O (3 � 100 mL). Theorganic phase was washed with aqueous saturated solution ofNaHCO3 and brine, dried over anhydrous Na2SO4, and concentratedunder reduced pressure. The crude was purified by flashchromatography (eluent: Et2O/AcOEt, from 70:30 v/v to 50:50v/v), affording methyl 3b,12a-dihydroxy-7-oxo-D5-cholen-24-oate (16) (1.95 g, 4.66 mmol, yield 60%) as a white solid. 1H NMR(CDCl3, 400 MHz): d 0.70 (s, 3H, 18-CH3), 0.97 (d, 3H, 21-CH3), 1.17(s, 3H, 19-CH3), 3.64 (m, 1H, 3-CH), 3.65 (s, 3H, CO2CH3), 4.04 (s, 1H,12-CH), 5.69 (d, 1H, 6-CH). 13C NMR (CDCl3, 100.6 MHz): d 12.8, 17.1,17.6, 25.6, 27.7, 28.5, 30.9, 31.1 (2�), 34.9, 36.2, 37.8, 41.4, 41.8, 43.4,45.2, 46.3, 47.0, 51.5, 70.4, 72.0, 126.1, 165.4, 174.6, 201.7.
2.1.13. Synthesis of 3b,12a-dihydroxy-7-oxo-D5-cholen-24-oic acid(17)
A solution of 16 (760 mg, 1.82 mmol) in a solution of NaOH inMeOH (10% w/v) (20 mL) was stirred at room temperatureovernight. The reaction mixture was concentrated under reducedpressure and the residue was dissolved in water and washed withEt2O (3 � 30 mL). The aqueous phase was cooled to �5 �C andcarefully acidified (pH 4.5) with 1 N HCl. A white precipitate wasformed and filtered affording 3b,12a-dihydroxy-7-oxo-D5-cholen-24-oic acid (17) (720 mg, 1.78 mmol, yield 98%) as a whitesolid, that was used for the following step without furtherpurification. 1H NMR (d 6-DMSO, 200 MHz): d 0.61 (s, 3H, 18-CH3),0.92 (d, 3H, 21-CH3), 1.11 (s, 3H, 19-CH3), 3.84 (s, 1H, 3-CH), 4.32 (s,1H, 12-CH), 4.93 (brs, 2H, 3-O H + 12-OH), 5.58 (d, 1H, 6-CH), 11.98(bs, 1H, CO2 H). 13C NMR (d 6-DMSO, 100.6 MHz): d 12.7, 17.0, 17.3,25.8, 27.7 (2�), 28.6, 30.9, 31.0, 35.0, 36.0, 37.9, 41.0, 41.7, 43.3, 45.1,45.4, 46.6, 69.4, 125.0, 70.3, 167.7, 175.7, 202.4.
2.1.14. Synthesis of 3b,7a,12a-trihydroxy-D5-cholen-24-oic acid (2)L-Selectride1 (1.0 M in THF, 2.47 mL, 2.47 mmol) was added
dropwise to a fine suspension of compound 17 (200 mg,0.49 mmol) in freshly distilled THF (7 mL), cooled to �78 �C, underargon atmosphere. The reaction mixture was stirred at thistemperature overnight. The mixture was allowed to warm to�15 �C, slowly quenched with H2O, carefully acidified with 1 N HCl(pH 4.5) and extracted with AcOEt (3 � 30 mL). The organic phasewas dried over anhydrous Na2SO4 and concentrated under reducedpressure. Purification by MPLC (Column: Lobar 310-25 LiChroprepRP-18 , 40–62 mm; eluent: H2O/MeOH from 90:10 to 80:20 v/v)afforded 3b,7a,12a-trihydroxy-D5-cholen-24-oic acid (2) (70 mg,0.16 mmol, yield 33%), as colorless crystals. mp: 245–248 �C. 1HNMR (d 6-DMSO, 400 MHz): d 0.61 (s, 3H, 18-CH3), 0.87 (s, 3H, 19-CH3), 0.90 (d, 3H, 21-CH3), 3.31 (m, 1H, 3-CH), 3.58 (s, 1H, 7-CH),3.82 (s, 1H, 12-CH), 5.39 (d, 1H, 6-CH). 13C NMR (d 6-DMSO,100.6 MHz): d 12.8, 17.8, 18.2, 23.8, 27.8, 28.7, 31.7, 33.3, 36.2, 36.8,36.9, 38.0, 40.1, 40.3, 40.5, 42.6, 45.8, 46.8, 63.8, 70.2, 71.4, 125.1,143.9, 177.6. IR (nmax in cm�1): 2876 and 2949 (OH), 1627 (CO2
�).Purity from HPLC: 1� method: 99.4%; 2� method: 98.9%.
2.1.15. Synthesis of 3b-sulfooxy-7a,12a-dihydroxy-D5-cholen-24-oicacid disodium salt (5)
SO3/pyridine complex was prepared adding dropwise, at 0 �C,chlorosulfonic acid (20 mL, 0.30 mmol) to freshly distilled pyridine(2.5 mL) under argon atmosphere. The white solid obtained washeated with a heat gun until it melted. The pale yellow liquid(0.5 mL) was added dropwise to a solution of 2 (120 mg,

A. Gioiello et al. / Journal of Steroid Biochemistry & Molecular Biology 144 (2014) 348–360 353
0.30 mmol) in freshly distilled pyridine (2 mL), cooled to -10 �C. Thereaction mixture was allowed to warm to room temperature andstirred overnight. The mixture was diluted with water and washedwith Et2O, in order to remove most of pyridine. 3 N aqueoussolution of NaOH (1.72 mL, 5.16 mmol) was added and the reactionmixture was stirred for 30 min. The mixture was concentratedunder reduced pressure and the crude was filtered on RP-8cartridge, eluting first with H2O and then with H2O/MeOH from90:10 v/v to 50:50 v/v. Purification by MPLC (Column: Lobar 310-25LiChroprep RP-18, 40–62 mm; eluent: H2O/MeOH from 100:0 v/v to75:25 v/v) afforded 3b-sulfooxy-7a,12a-dihydroxy-D5-cholen-24-oic acid disodium salt (5) (100 mg, 0.19 mmol, yield 91%) as awhite solid. mp: 251–255 �C. 1H NMR (d 6-DMSO, 400 MHz): d 0.62(s, 3H 18-CH3), 0.89 (s, 3H,19-CH3), 0.92 (d, 3H, 21-CH3), 3.59 (s, 1H,7-CH), 3.84 (s, 1H, 12-CH), 3.91 (m, 1H, 3-CH), 5.43 (d, 1H, 6-CH). 13CNMR (d6-DMSO, 100.6 MHz): d 12.9, 17.8, 18.2, 23.8, 27.9, 28.8 (2�),29.2, 33.3, 36.0 (2�), 36.3, 36.8, 36.9, 38.1, 39.3, 45.8, 46.9, 63.9,71.4, 75.4, 125.8, 143.5, 178.4. IR (nmax in cm�1): 2967 and 2869(OH), 1560 (CO2
�). Purity from HPLC: 1� method: 98.9%; 2�
method: 99.8%.
2.1.16. Synthesis of glyco-conjugate of 3b-sulfooxy-7a,12a-dihydroxy-D5-cholen-24-oic acid, disodium salt (6)
To a stirred suspension of 5 (100 mg, 0.19 mmol) in DMF (5 mL),DMT-MM (159 mg, 0.57 mmol) and Et3N (0.14 mL, 0.95 mmol) wereadded and the resulting suspension was stirred for 15 min at roomtemperature. Then, glycine ethyl ester hydrochloride (159 mg,1.14 mmol) was added and the mixture was stirred at roomtemperature for additional 3 h. 1 N aqueous solution of NaOH(17 mL, 17.0 mmol) was added and the reaction mixture was stirredfor 2 h at 40 �C. After cooling to room temperature, the mixture wasfiltered on RP-8 cartridge, eluting first with H2O and then withH2O/MeOH from 90:10 v/v to 50:50 v/v. Purification by MPLC(Column: Lobar 310-25 LiChroprep RP-18 , 40–62 mm; eluent:H2O/MeOH from 100:0 v/v to 90:10 v/v) afforded glyco-conjugateof 3b-sulfooxy-7a,12a-dihydroxy-D5-cholen-24-oic acid, diso-dium salt (6) (66 mg, 0.11 mmol, yield 60%) as a pale yellow solid.mp: >260 �C. 1H NMR (d6-DMSO, 400 MHz): d 0.60 (s, 3H 18-CH3),0.87 (s, 3H,19-CH3), 0.92 (d, 3H, 21-CH3), 3.25 (d, 2H, NH-CH2-CO2),3.31 (s, 1H, 7-CH), 3.82 (s, 1H, 12-CH ), 3.88 (m, 1H, 3-CH), 5.41 (d,1H, 6-CH), 7.01 (brs, 1H, NH). 13C NMR (d 6-DMSO, 100.6 MHz): d12.7, 17.5, 18.1, 23.7, 27.7, 28.7, 29.1, 32.2, 33.1, 35.6, 36.2, 36.8 (2�),38.0, 44.5, 45.8, 46.5, 63.8, 71.3, 75.3, 125.7, 143.5, 171.3, 171.9. IR(nmax in cm�1): 3303 (OH), 2937,1657 (CO2
�),1592 (CO),1221,1072,1050, 965. Purity from HPLC: 1� method: 95.8%; 2� method: 95.5%.
2.2. ESI-MS/MS analysis
Accurate mass measurement was performed on Waters XevoG2-S Q-TOF interfaced with Aquity UPLC system (Milford, MA). MSanalysis for 3b-sulfate-D5-cholen-24-oic acid was performed innegative mode with full scan at m/z 50–1200 Da range. Nitrogenwas used as both cone gas (50 L/h) and desolvation gas (1000 L/h).Source temperature and desolvation temperature were set at120 �C and 500 �C, respectively. The capillary voltage and conevoltage were set at 1500 and 40 V, respectively.
Table 1Inhibition of hCYP450 (CYP) enzymes.
Cmpd CYP1A2 CYP2C19
Ref. Cmpd Ref. Cmpd
Furafylline: IC50 = 0.9 mM Miconazole: IC50 = 0.031 mM
1 >10 mM 0.041 � 0.003 mM
2 >10 mM 0.027 � 0.006 mM
2.3. Biology
2.3.1. AlphaScreen assayActivation of the nuclear receptor was determined by using a
recruitment coactivator assay, namely AlphaScreen technology,according to a previously reported protocol. Briefly, assays wereconducted in white, low-volume, 384-well OptiPlate using a finalvolume of 25 mL containing 10 nM glutathione transferase-taggedNR-LBD protein and 30 nM biotinylated coactivator peptide. Thestimulation was carried out with 1 mL of bile acids solubilized in100% DMSO, for 30 min at room temperature. Luminescence wasread in EnVision microplate analyzer after incubation withdetection mix (acceptor and donor beads) for 4 h at roomtemperature in the dark. Dose response curves were performedin triplicate, and the results were the mean of at least twoexperiments [31].
2.3.2. Cytotoxicity assaysCell viability was evaluated by measuring ATP levels using
CellTiter-Glo (Promega), according to the manufacturer’s instruc-tions. LCA was used as bile acid comparator for cell cytotoxicity,whereas tamoxifen (Sigma) was used as a control of the assay. Cellnecrosis was evaluated by measuring the release of lactatedehydrogenase (LDH) from the necrotic cells using CytoTox-ONE,a homogeneous membrane integrity assay (Promega), according tomanufacturer’s instructions. For analyses of cell viability (ATPlevels), apoptosis and necrosis (LDH release), 2 � 104 HepG2 cellswere stimulated with test compounds at concentrations rangingfrom 100 nM to 300 mM in a white 96-well microplate for 4 h at37 �C [31].
2.3.3. Quantitative real-time PCRRNA was extracted using RNeasy Plus Kit (QIAGEN, Valencia,
CA) and quantified with Quant-iT RiboGreen (Invitrogen), andcDNA synthesis was carried out using 1 mg of RNA, 250 ng ofrandom primers, RNaseOUT, and SuperScriptIII (all from Invitro-gen), according to manufacturer’s protocol. Fifty nanograms oftemplate was used in 20 mL of SYBR Green PCR Master MIX finalvolume containing 0.3 M concentrations of each primer. Allreactions were performed in triplicate, and the thermal cyclingconditions were as follows: 3 min at 95 �C, followed by 45 cycles of95 �C for 10 s, and 60 �C for 30 s in iCycler iQ5 instrument (Bio-RadLaboratories, Hercules, CA). The relative gene expression wasexpressed as 2CT. The primers used were designed with BeaconDesigner software (Premier Biosoft, Palo Alto, CA) and areindicated below:
hSHP. Fw: ATCCTCTTCAACCCCGATGTG; Rw: ACTTCACACAG-CACCCAGTG
hCYP7A1. Fw: GTGCCAATCCTCTTGAGTTCC; Rw:ATGCTTCTGTGCCCAAATGC
hBSEP. Fw: GATGGCGTTAGAGTTGGCTTC; Rw: GCACA-CAAACTTCCCACAAAC
hCYP3A4. Fw: CATTGCTGTCTCCAACCTTCAC; Rw: CCCGCCTCA-GATTTCTCACC
hCYP2C19. Fw: AGCAATGGAAAGAGATGGAAGGAG; Rw: GAG-CACAGCCCAGGATGAAAG
CYP3A4 CYP2C9
Ref. Cmpd Ref. Cmpd
Ketoconazole: IC50 = 0.04 mM Sulfaphenazole: IC50 = 0.2 mM
> 10 mM > 10 mM> 10 mM > 10 mM

354 A. Gioiello et al. / Journal of Steroid Biochemistry & Molecular Biology 144 (2014) 348–360
hMDR1. Fw: TTCGTTTCCTTTAGGTCTTTCCAC; Rw:CTTCTTCTTTGCTCCTCCATTGC
2.3.4. CYP450 inhibition assayRecombinant CYP450 proteins (Baculosomes; Invitrogen) and
fluorogenic substrates (Vivid; Invitrogen) were used in a fluores-cent homogenous assay. CYP450 enzymes and reference inhibitorsare indicated in Table 1. The concentration of substrates used wasbelow their Michaelis–Menten constant (Km) value in a reactionwith P450 isozymes, insuring detection of even weak CYP450inhibitors. The assay was performed in triplicate in a black, low-volume 384-well plate, in 25 ml of reaction mixture. Therobustness and reproducibility of the assay has been validatedby the Z0 factor.
3. Results
3.1. Synthesis of D5-cholen-24-oic acid derivatives
For the synthesis of derivatives 1, 3–4, we adopted the syntheticstrategy shown in Scheme 1. Previously reported methods forobtaining 3b,7a-dihydroxy-D5-cholenoic acid (1) were based onthe extraction and isolation of the compound from the urine andbile of patients with elevated levels of these atypical bile acids [11]or from animal bile [16] or by the chemical synthesis from 3-keto-lithocholic, where the overall yield was only about 7% [17]. Sinceboth approaches suffer from limitations including low yields,tedious work-ups and purifications, and stereoselectivity issues,we developed an alternative, novel synthetic route for the rapidpreparation of D5-cholenoic acid 1, 3–4. In our case, the synthesisinvolved the use of 3a,6a-dihydroxy-5b-cholan-24-oic acid, alsoknown as hyodeoxycholic acid (HDCA, 7) as the starting material,
Scheme 1. Synthesis of D5-cholen-24-oic acids derivatives 1,3–4. Reagents and conditTBDMSiCl, imidazole, CH2Cl2, r.t.; (e) CuBr, t-BuO2COPh, CH2Cl2, reflux; (f) Pd(CH3CN)2triethylamine, DMT-MM, glycine ethyl ester hydrochloride, DMF, r.t.; (l) 1 N aqueous N
which was treated with a catalytic amount of p-toluenesulfonicacid (p-TSA) in MeOH to give the corresponding methyl ester, inquantitative yield (Scheme 1). Methyl hyodeoxycholate was thenreacted with tosyl chloride (TsCl) in freshly distilled pyridine atroom temperature to generate the corresponding 3a,6a-ditosylate8, in 75% yield. When methyl 3a,6a-ditosyloxy-5b-cholan-24-oate (8) was refluxed in a solution of DMF/H2O (v/v) in the presenceof 0.8 equiv. of potassium acetate (AcOK), the simultaneousinversion of the hydroxyl group at the C3 position and theelimination at C6 position took place. The improvement of theexperimental protocol for the Walden inversion [18] affordedmethyl 3b-hydroxy-D5-cholen-24-oate (9) in 60% yield afterchromatographic purification on silica gel (Scheme 1). This D5-cholenoate product was then protected at the C3-hydroxy groupwith tert-butyldimethylsilyl choride (TBDMSiCl) in freshly distilledCH2Cl2 (91% yield). The regio- and stereoselective allylic oxidationat the C7a position was successfully accomplished by using tert-butylperoxy benzoate in the presence of CuBr as catalyst in CH2Cl2[19–21], to yield the methyl 3b-tert -butyldimethylsilyloxy-7a-benzoyloxy-D5-cholen-24-oate (11) in 48% yield, after silica gelchromatography. Reaction of 11 with Pd(CH3CN)2Cl2 followed byalkali hydrolysis (NaOH/MeOH) and medium flash chromatogra-phy afforded 3b,7a-dihydroxy-D5-cholen-24-oic acid (1) in 75%yield. The corresponding C3-sulfated analogue (3) was achieved byregioselective sulfation reaction (Scheme 1) [22]. A solution ofchlorosulfonic acid in dry pyridine was added to a solution of 1 infreshly distilled pyridine at 0 �C under N2 atmosphere. Reversephase chromatography of the crude reaction mixture gave thedesired 3b-sulfoxy-7a-hydroxy-D5-cholen-24-oate disodium salt(3), in nearly quantitative yield from 1. Finally, the N -acylamidation of 3 with glycine ethyl ester was carried out by using4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium
ions : (a) p-TSA, MeOH, r.t.; (b) TsCl, pyridine, r.t.; (c) AcOK, DMF/H2O, reflux; (d)Cl2, acetone/H2O, r.t.; (g) NaOH, MeOH, reflux; (h) ClSO3H, pyridine, 0 �C ! r.t.; (i)aOH, 40 �C.

Scheme 2. Synthesis of D5-cholen-24-oic acids derivatives 2, 5–6 . Reagents and conditions : (a) MeOH, p-TSA, ultrasounds; (b) PPh3, DEAD, p-NBA, THF; (c) NaOH, MeOH; (d)MeOH, p-TSA, ultrasounds; (e) LDA, TMSCl, THF, �78 �C; (f) DTBMP, DDQ, CH3CN; (g) 1 N aqueous HCl, pH 5, 0 �C to r.t.; (h) NaOH, MeOH; (i) 1 N aqueous HCl, pH 4.5, �5 �C to r.t.; (j) L-Selectride1, THF, �78 �C; (k) 1 N aqueous HCl, pH 4.5, 0 �C to r.t.; (l) ClSO3H, pyridine, �10 �C to r.t.; (m) triethylamine, DMT-MM, glycine ethyl ester hydrochloride,DMF, r.t.; (n) 1 M aqueous NaOH, 40 �C.
A. Gioiello et al. / Journal of Steroid Biochemistry & Molecular Biology 144 (2014) 348–360 355
chloride (DMT-MM) as a coupling agent in DMF [23,24]. Hydrolysisof the terminal ester group with 1 N aqueous NaOH followed byreverse phase medium pressure chromatography (RP-MPLC)afforded the glyco-conjugate disodium salt (4), in 65% isolatedyield from 2 (Scheme 1).
The preparation of the corresponding 7a,12a-dihydroxyderivatives 2, 5–6 required a different synthetic approach due tolack of a commercial available source of 3a,6a,12a-trihydroxy-5b-cholan-24-oic acid. To the best of our knowledge, syntheticmethods for these compounds have to date never been reported.As illustrated in Scheme 2, we sought to use the 7-oxo-deoxycholicacid (7-KDCA) (13) as the starting material and it was treated withcatalytic amounts of p-TSA in MeOH to give the methyl ester 14 inquantitative yield. The inversion of the stereochemistry at C3position was obtained by a Mitsunobu reaction using p -nitro-benzoic acid (p-NBA). Alkaline hydrolysis of resulting C-7 benzoateintermediate with methanolic NaOH followed by esterificationreaction of the C-24 carboxylic moiety in the usual manner,furnished methyl 3b,12a-dihydroxy-7-oxo-5b-cholan-24-oate(15) in 85% yield from 14, after flash chromatographic purification.Treatment of 15 with LDA in THF at �78 �C followed by the reactionof the enolate with trimethylchlorosilane afforded thecorresponding silyl enol ether which was treated with2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and 2,6-di-tert-butyl-4-methylpyridine (DTBMP) in freshly distilled CH3CNat room temperature [25], and subsequently submitted to acidictreatment (1 N aqueous HCl, pH 5), to furnish methyl 3b,12a-dihydroxy-7-oxo-D5-cholen-24-oate (16), in 60% yield afterpurification. 3b,12a-Dihydroxy-7-oxo-D5-cholen-24-oic acid(17) was obtained from methyl ester 16 by alkaline hydrolysis(NaOH/MeOH). Chemo- and stereo-selective reduction of carbonylgroup at the C-7 position was carried out using L-selectride asreducing agent in freshly distilled THF at room temperature [26].Thus, after purification by medium pressure reverse phasechromatography, 3b,7a,12a-trihydroxy-D5-cholen-24-oic acid(2) was isolated in 33% of yield from 17 (Scheme 2). Bothcompounds 5 and 6 were prepared according to the sameprocedures previously reported for 3 and 4 (Scheme 2). Thus,3b-sulfooxy-7a,12a-dihydroxy-D5-cholen-24-oate disodium salt
(5) and the glyco-conjugate of 3b-sulfooxy-7a,12a-dihydroxy-D5-cholen-24-oate disodium salt (6) were obtained in 90% and 60%yield, respectively from 2.
3.2. Electrospray ionization (ESI)-tandem mass spectrometricdetermination of D5-cholen-24-oic sulfates
Patients with a HSD3B7 deficiency synthesize high levels of3b,7a-dihydroxy- and 3b,7a,12a-trihydroxy-D5-cholenoic acids(1 and 2) which are efficiently sulfated at the C-3 position andglycine-conjugated in the side-chain and then excreted in urine(Fig. 1) [11]. The routine screening for HSD3B7 deficiency isbased on the identification of these sulfated bile acids (3–6) byfast atom bombardment ionization-mass spectrometry (FAB–MS)[7]. Electrospray ionization (ESI) mass spectrometry may also beused for identification of these atypical bile acid conjugates [27].The authentic standards of 3b-hydroxy-D5-cholenoic acidconjugates were obtained for the first time by multistep,regio/stereochemistry-controlled synthesis (Schemes 1 and 2).These were analyzed by ESI–MS with direct infusion on a Q-TOFmass spectrometer, which confirmed their molecular weightsconsistent with the synthesized structures and enabled estab-lishment of optimal ionization parameters for development of aclinical assay. The 3b-sulfoxy-7a-hydroxy-D5-cholen-24-oic acid(3) and 3b-sulfoxy-7a,12a-dihydroxy-D5-cholen-24-oic acid (5)exhibited predominantly a singly charged negative ion [M-H]�
and in a Q-TOF instrument gave accurate mass measurements ofm/z 469.2245 and 485.2207, respectively (Fig. 2), consistent withelemental compositions of C24H37O7S and C24H37O8S, respective-ly. The glycine-conjugates 4 and 6 showed singly chargeddeprotonated ions at m/z 526.2551 and 542.2429, correspondingto elemental compositions of C26H40NO8S and C26H40NO9S, butdoubly charged ions at m/z 262.6208 and 270.6175, respectively,were also evident. A high cone energy setting (60 eV) favouredsingly charged ions, while lower cone setting promotedformation of doubly charged ions. Thus, the parent ion scans(data not shown) of these conjugated bile acids yield singledeprotonated molecular ions with negligible fragmentation,which confirms source stability of the molecule under ESI
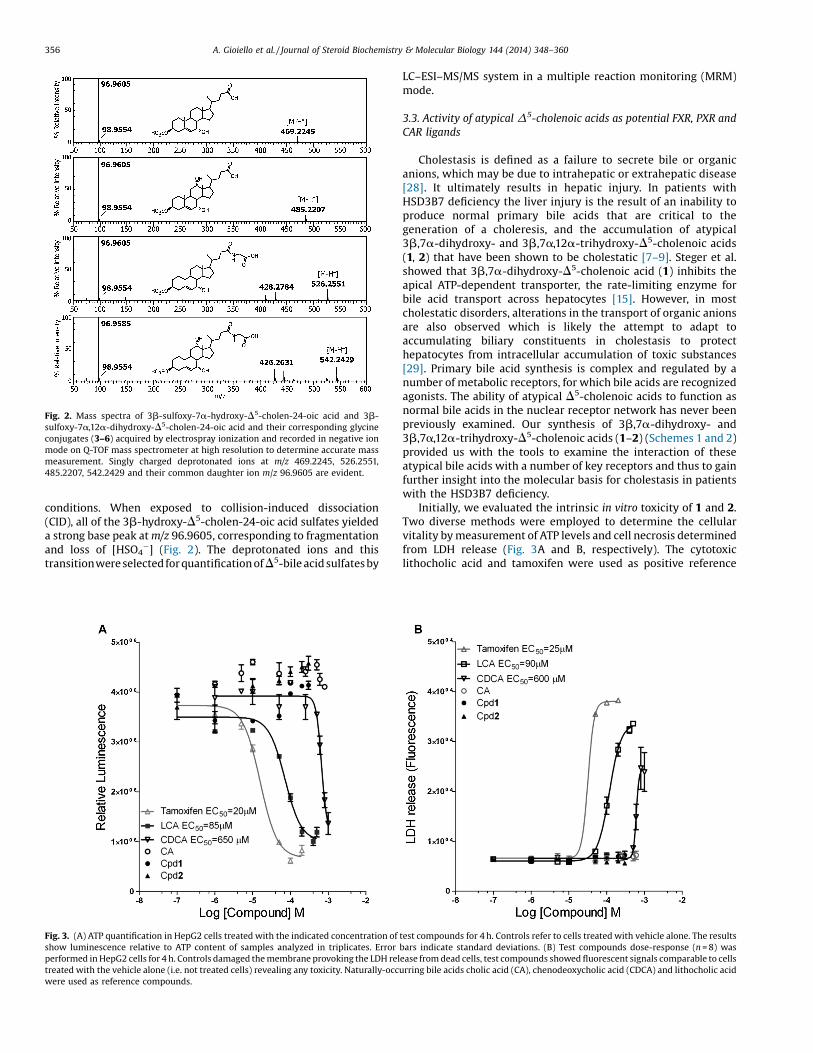
Fig. 2. Mass spectra of 3b-sulfoxy-7a-hydroxy-D5-cholen-24-oic acid and 3b-sulfoxy-7a,12a-dihydroxy-D5-cholen-24-oic acid and their corresponding glycineconjugates (3–6) acquired by electrospray ionization and recorded in negative ionmode on Q-TOF mass spectrometer at high resolution to determine accurate massmeasurement. Singly charged deprotonated ions at m/z 469.2245, 526.2551,485.2207, 542.2429 and their common daughter ion m/z 96.9605 are evident.
356 A. Gioiello et al. / Journal of Steroid Biochemistry & Molecular Biology 144 (2014) 348–360
conditions. When exposed to collision-induced dissociation(CID), all of the 3b-hydroxy-D5-cholen-24-oic acid sulfates yieldeda strong base peak at m/z 96.9605, corresponding to fragmentationand loss of [HSO4
�] (Fig. 2). The deprotonated ions and thistransitionwere selected for quantification ofD5-bile acid sulfates by
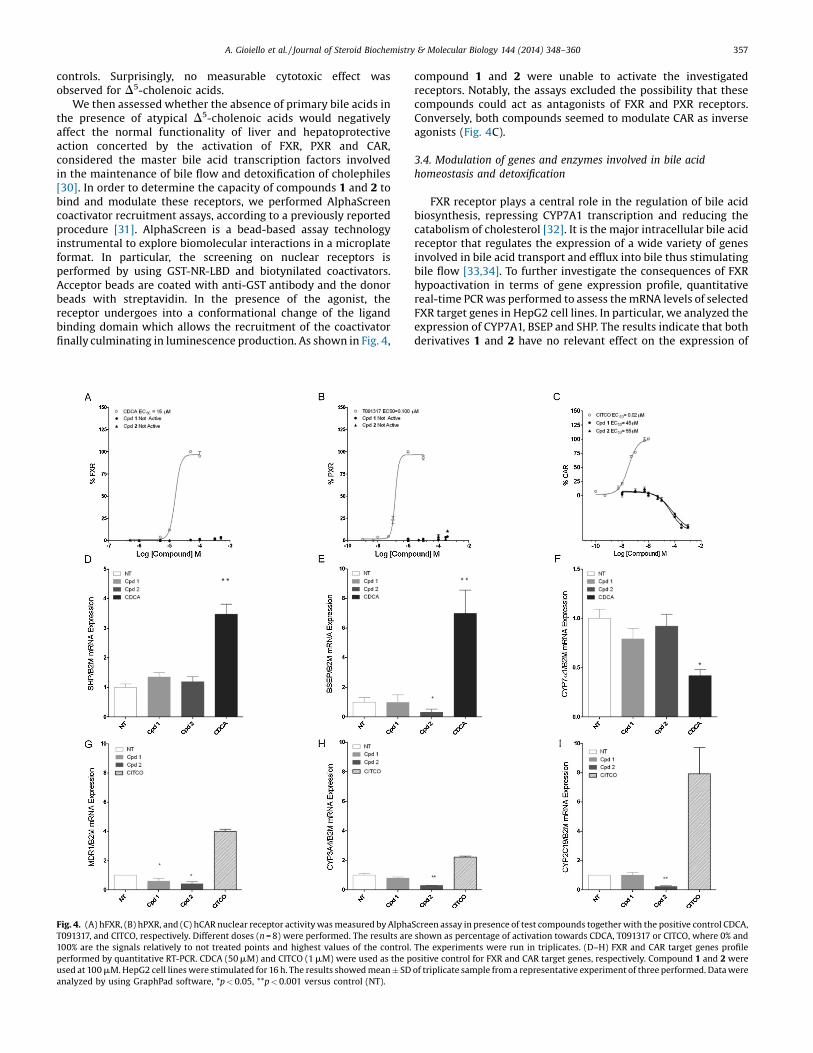
Fig. 3. (A) ATP quantification in HepG2 cells treated with the indicated concentration of
show luminescence relative to ATP content of samples analyzed in triplicates. Error
performed in HepG2 cells for 4 h. Controls damaged the membrane provoking the LDH reltreated with the vehicle alone (i.e. not treated cells) revealing any toxicity. Naturally-occuwere used as reference compounds.
LC–ESI–MS/MS system in a multiple reaction monitoring (MRM)mode.
3.3. Activity of atypical D5-cholenoic acids as potential FXR, PXR andCAR ligands
Cholestasis is defined as a failure to secrete bile or organicanions, which may be due to intrahepatic or extrahepatic disease[28]. It ultimately results in hepatic injury. In patients withHSD3B7 deficiency the liver injury is the result of an inability toproduce normal primary bile acids that are critical to thegeneration of a choleresis, and the accumulation of atypical3b,7a-dihydroxy- and 3b,7a,12a-trihydroxy-D5-cholenoic acids(1, 2) that have been shown to be cholestatic [7–9]. Steger et al.showed that 3b,7a-dihydroxy-D5-cholenoic acid (1) inhibits theapical ATP-dependent transporter, the rate-limiting enzyme forbile acid transport across hepatocytes [15]. However, in mostcholestatic disorders, alterations in the transport of organic anionsare also observed which is likely the attempt to adapt toaccumulating biliary constituents in cholestasis to protecthepatocytes from intracellular accumulation of toxic substances[29]. Primary bile acid synthesis is complex and regulated by anumber of metabolic receptors, for which bile acids are recognizedagonists. The ability of atypical D5-cholenoic acids to function asnormal bile acids in the nuclear receptor network has never beenpreviously examined. Our synthesis of 3b,7a-dihydroxy- and3b,7a,12a-trihydroxy-D5-cholenoic acids (1–2) (Schemes 1 and 2)provided us with the tools to examine the interaction of theseatypical bile acids with a number of key receptors and thus to gainfurther insight into the molecular basis for cholestasis in patientswith the HSD3B7 deficiency.
Initially, we evaluated the intrinsic in vitro toxicity of 1 and 2.Two diverse methods were employed to determine the cellularvitality by measurement of ATP levels and cell necrosis determinedfrom LDH release (Fig. 3A and B, respectively). The cytotoxiclithocholic acid and tamoxifen were used as positive reference
test compounds for 4 h. Controls refer to cells treated with vehicle alone. The resultsbars indicate standard deviations. (B) Test compounds dose-response (n = 8) wasease from dead cells, test compounds showed fluorescent signals comparable to cellsrring bile acids cholic acid (CA), chenodeoxycholic acid (CDCA) and lithocholic acid
A. Gioiello et al. / Journal of Steroid Biochemistry & Molecular Biology 144 (2014) 348–360 357
controls. Surprisingly, no measurable cytotoxic effect wasobserved for D5-cholenoic acids.
We then assessed whether the absence of primary bile acids inthe presence of atypical D5-cholenoic acids would negativelyaffect the normal functionality of liver and hepatoprotectiveaction concerted by the activation of FXR, PXR and CAR,considered the master bile acid transcription factors involvedin the maintenance of bile flow and detoxification of cholephiles[30]. In order to determine the capacity of compounds 1 and 2 tobind and modulate these receptors, we performed AlphaScreencoactivator recruitment assays, according to a previously reportedprocedure [31]. AlphaScreen is a bead-based assay technologyinstrumental to explore biomolecular interactions in a microplateformat. In particular, the screening on nuclear receptors isperformed by using GST-NR-LBD and biotynilated coactivators.Acceptor beads are coated with anti-GST antibody and the donorbeads with streptavidin. In the presence of the agonist, thereceptor undergoes into a conformational change of the ligandbinding domain which allows the recruitment of the coactivatorfinally culminating in luminescence production. As shown in Fig. 4,
Fig. 4. (A) hFXR, (B) hPXR, and (C) hCAR nuclear receptor activity was measured by AlphaT091317, and CITCO, respectively. Different doses (n = 8) were performed. The results are100% are the signals relatively to not treated points and highest values of the control.performed by quantitative RT-PCR. CDCA (50 mM) and CITCO (1 mM) were used as the pused at 100 mM. HepG2 cell lines were stimulated for 16 h. The results showed mean � SDanalyzed by using GraphPad software, *p < 0.05, **p < 0.001 versus control (NT).
compound 1 and 2 were unable to activate the investigatedreceptors. Notably, the assays excluded the possibility that thesecompounds could act as antagonists of FXR and PXR receptors.Conversely, both compounds seemed to modulate CAR as inverseagonists (Fig. 4C).
3.4. Modulation of genes and enzymes involved in bile acidhomeostasis and detoxification
FXR receptor plays a central role in the regulation of bile acidbiosynthesis, repressing CYP7A1 transcription and reducing thecatabolism of cholesterol [32]. It is the major intracellular bile acidreceptor that regulates the expression of a wide variety of genesinvolved in bile acid transport and efflux into bile thus stimulatingbile flow [33,34]. To further investigate the consequences of FXRhypoactivation in terms of gene expression profile, quantitativereal-time PCR was performed to assess the mRNA levels of selectedFXR target genes in HepG2 cell lines. In particular, we analyzed theexpression of CYP7A1, BSEP and SHP. The results indicate that bothderivatives 1 and 2 have no relevant effect on the expression of
Screen assay in presence of test compounds together with the positive control CDCA, shown as percentage of activation towards CDCA, T091317 or CITCO, where 0% and
The experiments were run in triplicates. (D–H) FXR and CAR target genes profileositive control for FXR and CAR target genes, respectively. Compound 1 and 2 were
of triplicate sample from a representative experiment of three performed. Data were

358 A. Gioiello et al. / Journal of Steroid Biochemistry & Molecular Biology 144 (2014) 348–360
these genes (Fig. 4D–F), further supporting the notion that they areunable to behave as normal bile acids in hepatic function. The onlysignificant alterations was observed for BSEP mRNA expressionwhich was negatively regulated by compound 2 (Fig. 4E).
Finding evidence of the behavior of the compounds 1 and 2 asinverse agonists of CAR, we further investigated their effects ontarget genes modulated by CAR, including MDR1, CYP3A4 andCYP2C19 (Fig. 4G–I) [35]. We determined the mRNA expression ofthese genes in HepG2 cells by stimulation with 3b-hydroxy-D5-cholenoic acids at concentration of 100 mM. The results indicatedthat both 1 and 2 repressed the expression of MDR1 in hepatic celllines, while only trihydroxy analogue 2 was found to besignificantly effective in down-regulating CYP2C19 and CYP3A4gene expression (Fig. 4G–I). Furthermore, since cytochrome P450proteins are differentially altered in severe chronic liver diseases[36], we examined the alteration of CYP450 profile uponadministration of 3b,7a-dihydroxy- and 3b,7a,12a-trihydroxy-D5-cholenoic acids (1 and 2). Previous in vitro studies indicatedthat total P450 levels and P450-supported catalytic activities tendto be unaltered in patients with mild to moderate hepatic disease,but are generally repressed in those with liver failure or severecirrhosis [37]. When tested by a fluorescent homogenous assay, 1and 2 had no effect on hCYP1A2 and hCYP3A4, hCYP2C9, but theystrongly inhibited hCYP2C19 (Table 1). Noteworthy, the activity ofthe mentioned CYPs was not modified by administration of theprimary bile acids, chenodeoxycholic acid and cholic acid (data notshown).
4. Discussion
We have described for the first time a novel synthetic route toachieve 3b-hydroxy-D5-cholenoic analogs 1–6 from the commer-cially available bile acids, hyodeoxycholic acid and 7-oxo-deoxy-cholic acid (Schemes 1 and 2). The desired products were achievedin good overall yield with a high degree of regio- and
Fig. 5. D5-Cholen-24-oic acids are not agonists of FXR, PXR and CAR receptor. In contrasinjury by adaptive mechanisms including the repression of bile acid biosynthesis via SHPbile acid detoxification pathways. This results in the stimulation of abnormal bile acid
stereoselectivity. Previously reported methods for obtaining3b,7a-dihydroxy-D5-cholenoic acid (1) were based on the labori-ous extraction and isolation techniques [11,16] or by the synthesisfrom 3-keto-lithocholic acid [17]. Conversely, the synthesis of3b,7a,12a-dihydroxy derivatives (2, 5–6) have to date never beenreported. Our synthetic approach to 3b-hydroxy-D5-cholenoicanalogs (1–6) provided chemical tools for the development of avalidate mass clinical assay for diagnosis and monitoring ofresponses to bile acid therapy in patients with liver disease causedby HSD3B7 [11]. In such patients, accurate measurement of theirconcentrations has been hampered by the previous lack of purestandards, a limitation that we have now overcome. Since theseatypical bile acids are not present in the urine and plasma of healthyinfants, these reference compounds can be appropriately added tothese biological matrices without concerns of interferences.
Moreover, the availability of 3b-hydroxy-D5-cholenoic acids(1–2) has also enabled investigations into the interaction of theseatypical bile acids with a number of key nuclear receptors, targetgenes and cytochrome P450s involved in bile acid homeostasis anddetoxification. We have shown in vitro that compound 1 and 2 arenot toxic to HepG2 cell lines (Fig. 3), and that their detrimentaleffects on hepatic function is more likely due to a lack ofhepatoprotective action exerted by the nuclear receptors FXR, PXRand CAR (Fig. 5). In line with previous reports [38–41], these3b-hydroxy-D5-cholenoic acids fail to function as agonists forthese nuclear receptors. Indeed, the effective binding of biliaryderivatives on both PXR and CAR receptor is still elusive, it wasdemonstrated that epimerization of the hydroxyl group at theC3-position had a detrimental effect on the activity of the resultingcompounds towards FXR [38,39]. Conversely, a ‘flat’ configurationof the A/B ring of the steroidal body as occurs in 5a-(allo-BAs) andD5-BAs, may match the FXR activity of their natural 5b counterpart[40,41].
This lack of agonist activity of 1 and 2 is an importantobservation because activation of FXR by primary bile acids
t to normal bile acids, atypical D5-cholen-24-oic acids are unable to counteract liver activation and CYP7A1 repression, alteration of export pumps as BSEP, induction of
production and accumulation leading to a vicious circle causing liver diseases.

A. Gioiello et al. / Journal of Steroid Biochemistry & Molecular Biology 144 (2014) 348–360 359
induces their conjugation and secretion from hepatocytes into thebile to promote bile flow and to protect against the accumulation ofhepatotoxic bile acids in cholestatic liver diseases. Moreover,stimulation of PXR and CAR mediates bile acid transport andmetabolism in the hepatocyte mitigating the harmful effects ofcholephiles, in particular of bile acids that are cytotoxic at highconcentrations, by activation of hepatic detoxification pathways[42]. Noteworthy, the failure to produce adequate amounts ofprimary bile acids combined with the production of atypical bileacid metabolites that were reported to inhibit the apical,ATP-dependent transport system in a competitive manner(Km= 16 mM), indicates that this class of steroids are cholestaticagents by inhibiting the canalicular excretory system [15]. In linewith these findings, the failed activation of FXR in patients withinborn errors in bile acid biosynthesis would reduce bile flow at thebile duct, causing the intrahepatic accumulation of cytotoxicmetabolites as D5-cholenoic acids. This consideration is supportedby the failure to upregulate BSEP (Fig. 4E), which is known tomediate bile acid excretion into the biliary tract [43]. Remarkably,the trihydroxy derivative 2 represses BSEP expression, in contrastto the protective mechanisms afforded by bile acid signallingduring cholestasis, where BSEP mRNA levels are upregulated toprotect hepatocytes against the accumulation of toxic bile acids[44] (Fig. 5). Furthermore, our results strongly support the notionthat a failure to activate FXR and to repress CYP7A1 expression(Fig. 4F) accounts for the sustained production of very highconcentrations of atypical bile acids leading to a vicious cycle ofprogressive hepatic injury. Remarkably, while endogenous bileacids activate detoxification pathways mainly mediated by PXRand CAR in hepatocytes [45,46], compounds 1 and 2 were foundunable to activate PXR while they behaved as inverse agonists ofCAR receptor. Notably, a marked inhibition of CAR target genesincluding MDR1, CYP2C19 and CYP3A4 mRNA was observed in thepresence of 3b,7a,12a-trihydroxy-D5-cholen-24-oic acid (2)(Fig. 4G–I) thus ruling out the absence of a defense mechanismto toxic metabolites from hepatocytes. In particular, inhibition ofCYP3A4 and CYP2C19 can decrease the metabolic rate of specificprescribed drugs metabolized by these CYPs, resulting inbioaccumulation and toxicity with severe consequences for humanhealth. In line with previous observations on severe chronicdiseases, compounds 1 and 2 were found to have diverse ability tointeract with CYP450s activity. In particular, it was shown that theyhad no effect on the majority of CYP450 enzymes investigated withthe exception of inhibiting CYP2C19. Collectively, these findingsconfirm the crucial role of bile acids as key sensors of liver functionwhile having clinical implications for the use of drugs in patientswith liver diseases. Our studies also emphasize the need to betterunderstand the metabolic fate of currently used therapeuticagents.
5. Conclusions
Since the discovery of first inborn error in bile acid biosynthesis[47], there have been total of nine genetic defects in bile acidsynthesis pathway ranging from abnormality in alteration of eithernuclear or side chain portion of cholesterol [9]. Among the variousinherited disorders, HSD3B7 deficiency is the most commonlyoccurring defect. This presents as progressive cholestatic liverdisease causing morbidity or mortality in affected patients[11–13,48]. A unique bile acid pattern in HSD3B7 deficient patienturine revealed by fast atom bombardment ionization massspectrometry (FAB–MS)techniquedefined four signature molecules,namely 3b-sulfate-D5-cholen-24-oic acid conjugates 3–6. To date,quantification of these atypical bile acids has been problematicbecause of the highly labile allylic hydroxyl group that extensivelyundergoes dehydration during conventional methods for bile acid
analysis. To circumvent this problem, bile acid sulfates 3–6 wereprepared and employed as tools for developing a sensitive, preciseand robust analytical assay for the diagnosis of HSD3B7 defect,monitor and assessment of patient's therapeutic response. More-over, the availability of these metabolites, namely 3b-hydroxy-D5-cholenoic acids which are present in high concentration in the liverof these patients, has permitted insights into the molecular basis fortheir cholestatic effects. Thus, we demonstrate for the first timetheir inability to function as FXR, PXR and CAR agonists and to exerthepatoprotective actions, which in normal physiological conditionsare efficiently coordinated by endogenous bile acids. Accordingly,atypical D5-bile acids are found to sustain their own synthesis andaccumulation by the maintenance of CYP7A1 activity and by theinhibition of their canalicular transport and detoxification. Insummary, our results not only contribute to a better understandingof cholestatic conditions in patients with HSD3B7 deficiency, butalso provide fundamental tools for the accurate clinical identifica-tion of this genetic defect, which if not diagnosed early in life oruntreated, may be fatal.
Acknowlegments
KDRS is a founder and Scientific Director of Asklepion Pharma-ceuticals, LLC., Baltimore, Maryland. The synthesis work was fundedby an unrestricted grant from Asklepion Pharmaceuticals, LLC.
References
[1] R. Sharma, A. Long, J.F. Gilmer, Advances in bile acid chemistry, Curr. Med.Chem. (2011) 4029–4052 18.
[2] C. Thomas, R. Pellicciari, M. Pruzanski, J. Auwerx, K. Schoonjans, Targeting bile-acid signalling for metabolic diseases, Nat. Rev. Drug Disc. 7 (2008) 678–693.
[3] A.F. Hofmann, L.R. Hagey, Bile acids: chemistry, pathochemistry, biology,pathobiology, and therapeutics, Cell. Mol. Life Sci. 65 (2008) 2461–2483.
[4] C. Thomas, A. Gioiello, L. Noriega, A. Strehle, J. Oury, G. Rizzo, A. Macchiarulo, H.Yamamoto, C. Mataki, M. Pruzanski, R. Pellicciari, J. Auwerx, K. Schoonjans,TGR5-mediated bile acid sensing controls glucose homeostasis, Cell Metab. 10(2009) 167–177.
[5] T.W.H. Pols, M. Nomura, T. Harach, G. Lo Sasso, M.H. Oosterveer, C. Thomas, G.Rizzo, A. Gioiello, L. Adorini, R. Pellicciari, J. Auwerx, K. Schoonjans, TGR5activation inhibits atherosclerosis by reducing macrophage inflammation andlipid loading, Cell Metab. 14 (2011) 747–757.
[6] S. Modica, R.M. Gadaleta, A. Moschetta, Deciphering the nuclear bile acidreceptor FXR paradigm, Nucl. Recept. Signal. 8 (2010) e005.
[7] K.D.R. Setchell, J.E. Heubi, Defects in bile acid biosynthesis-diagnosis andtreatment, J. Pediatr. Gastroenterol. Nutr. 43 (2006) S17–S22.
[8] K.D.R. Setchell, K. Bove, J.E. Heubi, Disorders of bile acid synthesis, fifth ed., in:W.A. Walker, O. Goulet, R.E. Kleinman, P.M. Sherman, B.L. Shneider, I.R.Sanderson (Eds.), Pediatric Gastrointestinal Disease, vol. 1, B.C. Decker Inc,Hamilton, Ontario, Canada, 2009, pp. 1069–1094.
[9] K.D.R. Setchell, Disorders of bile acid synthesis and metabolism - a metabolicbasis for liver disease, in: F.G. Suchy, W.F. Balistreri (Eds.), Liver Disease inChildren, vol. 4, Lippincott Williams & Wilkins, 2013, pp. 736–766.
[10] E. Gonzales, M.F. Gerhardt, M. Fabre, K.D.R. Setchell, A. Davit-Spraul, I. Vincent,J.E. Heubi, O. Bernard, E. Jacquemin, Oral cholic acid for hereditary defects ofprimary bile acid synthesis: a safe and effective long-term therapy,Gastroenterology 137 (2009) 1310–1320.
[11] P.T. Clayton, J.V. Leonard, A.M. Lawson, K.D.R. Setchell, S. Andersson, B. Egestad,J. Sjovall, Familial giant cell hepatitis associated with synthesis of 3 beta, 7alpha-dihydroxy-and 3 beta,7 alpha, 12 alpha-trihydroxy-5-cholenoic acids, J.Clin. Invest. 79 (1987) 1031–1038.
[12] P. Subramaniam, P.T. Clayton, B.C. Portmann, G. Mieli-Vergani, N. Hadzi�c,Variable clinical spectrum of the most common inborn error of bile acidmetabolism-3beta-hydroxy-delta 5-C27-steroid dehydrogenase deficiency, J.Pediatr. Gastroenterol. Nutr. 50 (2010) 61–66.
[13] E. Jacquemin, K.D.R. Setchell, N.C. O'Connell, O. Bernard, A new cause ofprogressive intrahepatic cholestasis: 3 beta-hydroxy-C27-steroid dehydroge-nase/isomerase deficiency, J. Pediatr. 125 (1994) 379–384.
[14] J.B. Cheng, E. Jacquemin, M. Gerhardt, R. Nazer, D. Cresteil, J.E. Heubi, K.D.R.Setchell, D.W. Russel, Molecular genetics of 3beta-hydroxy-Delta5-C27-steroid oxidoreductase deficiency in 16 patients with loss of bile acidsynthesis and liver disease, J. Clin. Endocrinol. Metab. 88 (2003) 1833–1841.
[15] B. Stieger, J. Zhang, B. O'Neill, J. Sjövall, P.J. Meier, Transport of taurine conjugatesof 7alpha-hydroxy-3-oxo-4-cholenoic acid and 3beta,7alpha-dihydroxy-5-cholenoic acid in rat liver plasma membrane vesicles, in: G.P. Van Berge-Henegouwen, B. Van Hock, J. De Groote (Eds.), Cholestatic Liver Diseases, KluwerAcademic Press, Dordrecht, The Netherlands, 1994, pp. 82–87.

360 A. Gioiello et al. / Journal of Steroid Biochemistry & Molecular Biology 144 (2014) 348–360
[16] K. Yamasaki, Y. Ayaki, H. Yamasaki, Isolation of 3b,7a-dihydroxychol-5-en-24-oic acid, an intermediate of chenodeoxycholic acid biogenesis, from fistula bileof the rat and hen, J. Biochem. 70 (1971) 715–718.
[17] M. Thoma, R. Mahara, H. Takeshita, T. Kurosawa, Convenient synthesis of3b,12a-3b,7a-, and 3b,7b-dihydroxy-5-cholen-24-oic acids: unusual bileacids in human biological fluids, Steroids 48 (1986) 331–338.
[18] G. Kakiyama, A. Muto, M. Shimada, N. Mano, J. Goto, A.F. Hofmann, T. Iida,Chemical synthesis of 3b-sulfooxy-7b-hydroxy-24-nor-5-cholenoic acid: aninternal standard for mass spectrometric analysis of the abnormal D5-bileacids occurring in Niemann-Pick disease, Steroids 74 (2009) 766–772.
[19] M.S. Kharash, G. Sosnovsky, N.C. Yang, Reaction of t-butyl peresters. I. Thereaction of peresters with olefins, J. Am. Chem. Soc. 81 (1959) 5819–5824.
[20] J.M. Kochi, The copper salt catalyzed peroxide reactions, J. Am. Chem. Soc. 83(1961) 3162–3163.
[21] M.B. Adrus, X. Chen, Catalytic enantioselective allylic oxidation of olefins withcopper(I) catalysts and new perester oxidants, Tetrahedron 53 (1997)16229–16240.
[22] R.H. Palmer, M.G. Bolt, Bile acid sulfates. Synthesis of lithocholic acid sulfatesand their identification in human bile, J. Lipid Res. 12 (1971) 671–679.
[23] M. Kunishima, C. Kawachi, F. Iwasaki, K. Terao, S. Tani, Synthesis andcharacterization of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholi-nium chloride, Tetrahedron Lett. 40 (1999) 5327–5330.
[24] M. Kunishima, C. Kawachi, K. Hioki, K. Terao, S. Tani, Formation ofcarboxamides by direct condensation of carboxylic acids and amines inalcohols using a new alcohol- and water-soluble condensing agent: DMT-MM,Tetrahedron 57 (2001) 1551–1558.
[25] H. Zhang, M.S. Reddy, S. Phoenix, P. Deslongchamps, Total Synthesis ofouabagenin and ouabain, Angew. Chem. Int. Ed. 47 (2008) 1272–1274.
[26] J.J. Poza, C. Jiménez, J. Rodrìguez, J-based analysis and DFT–NMR assignmentsof natural complex molecules: application to 3b,7-dihydroxy-5,6-epoxycho-lestanes, Eur. J. Org. Chem. 23 (2008) 3960–3969.
[27] P.T. Clayton, Applications of mass spectrometry in the study of inborn errors ofmetabolism, J. Inherit. Metab. Dis. 24 (2001) 139–150.
[28] M, Trauner, P.J. Meier, J.L. Boyer, Molecular pathogenesis of cholestasis, N. Engl.J. Med. 339 (1998) 1217–1227.
[29] G. Zollner, M. Trauner, Nuclear receptors as therapeutic targets in cholestaticliver diseases, Br. J. Pharm. 156 (2009) 7–27.
[30] M. Vacca, C. Degirolamo, V. Massafra, L. Polimeno, R. Mariani-Costantini,G. Palasciano, A. Moschetta, Nuclear receptors in regenerating liver andhepatocellular carcinoma, Mol. Cell. Endocrinol. 368 (2013) 108–119.
[31] G. Rizzo, D. Passeri, F. De Franco, G. Ciaccioli, L. Donadio, G. Rizzo, S. Orlandi,B. Sadeghpour, X.X. Wang, T. Jiang, M. Levi, M. Pruzanski, L. Adorini, Functionalcharacterization of the semisynthetic bile acid derivative INT-767, a dualfarnesoid X receptor and TGR5 agonist, Mol. Pharmacol. 78 (2010) 617–630.
[32] J.Y.L. Chiang, R. Kimmel, C. Weinberger, D. Stroup, Farnesoid X receptorresponds to bile acids and represses cholesterol 7alpha-hydroxylase gene(CYP7A1) transcription, J. Biol. Chem. 275 (2000) 10918–10924.
[33] T. Matsubara, F. Li, F.J. Gonzalez, FXR signaling in the enterohepatic system,Mol. Cell. Endocrinol. 368 (2013) 17–29.
[34] T. Li, J.Y.L. Chiang, Nuclear receptors in bile acid metabolism, Drug Metab. Rev.45 (2013) 145–155.
[35] Y. Chen, S.S. Ferguson, M. Negishi, G.A. Goldstein, Identification of constitutiveandrostane receptor and glucocorticoid receptor binding sites in the CYP2C19promoter, Mol. Pharmacol. 64 (2003) 316–324.
[36] J. George, M. Murray, K. Byth, G.C. Farrell, Differential alterations ofcytochrome P450 proteins in livers from patients with severe chronic liverdisease, Hepatology 21 (1995) 120–128.
[37] G.C. Farrell, L. Zaluzny, Portal vein ligation selectively lowers hepaticcytochrome P450 levels in rats, Gastroenterology 85 (1983) 275–282.
[38] T. Fujino, M. Une, T. Imanaka, K. Inoue, T. Nishimaki-Mogami, Structure-activity relationship of bile acids and bile acids analogs in regard to FXRactivation, J. Lipid Res. 45 (2004) 132–138.
[39] L.Z. Mi, S. Devarakonda, J.M. Harp, Q. Han, R. Pellicciari, T.M. Willson,S. Khorasanizadeh, F. Rastinejad, Structural basis for bile acid binding andactivation of the nuclear receptor FXR, Mol. Cell. 11 (2003) 1093–1100.
[40] Y. Iguchi, K. Kihira, T. Nishimaki-Mogami, M. Une, Structure-activityrelationship of bile alcohols as human farnesoid X receptor agonist, Steroids75 (2010) 95–100.
[41] J. Vaquero, M.J. Monte, M. Dominguez, J. Muntanè, Different activation ofthe human farnesoid X receptor depends on the pattern of expressedisoforms and the bile acid pool composition, Biochem. Pharmacol. 86(2013) 926–939.
[42] J. Chen, K. Raymond, Nuclear receptors, bile-acid detoxification, andcholestasis, Lancet 367 (2006) 454–456.
[43] B. Stieger, Role of the bile salt export pump, BSEP, in acquired forms ofcholestasis, Drug Metabol. Rev. 42 (2010) 437–445.
[44] P.J. Meier, B. Stieger, Bile salt transporters, Annu. Rev. Physiol. 64 (2002)635–661.
[45] C. Gnerre, S. Blattler, M.R. Kaufmann, R. Looser, U.A. Meyer, Regulation ofCYP3A4 by the bile acid receptor FXR: evidence for functional binding sites inthe CYP3A4 gene, Pharmacogenetics 14 (2004) 635–645.
[46] G. Bertilsson, J. Heidrich, K. Svensson, M. Asman, L. Jendeberg, M. Sydow-Backman, R. Ohlsson, H. Postlind, P. Blomquist, A. Berkenstam, Identification ofa human nuclear receptor defines a new signaling pathway for CYP3Ainduction, Proc. Natl. Acad. Sci. U.S.A. 95 (1998) 12208–12213.
[47] T. Setoguchi, G. Salen, G.S. Tint, E.H. Mosbach, A biochemical abnormality incerebrotendinous xanthomatosis. Impairment of bile acid biosynthesisassociated with incomplete degradation of the cholesterol side chain, J. Clin.Invest. 53 (1974) 1393–1401.
[48] K.D.R. Setchell, F.J. Suchy, M.B. Welsh, L. Zimmer-Nechemias, J.E. Heubi, W.F.Balistreri, Delta 4-3-oxosteroid 5 beta-reductase deficiency described inidentical twins with neonatal hepatitis. A new inborn error in bile acidsynthesis, J. Clin. Invest. 82 (1988) 2148–2157.


![Pyrethrin Biosynthesis: The Cytochrome P450 Oxidoreductase ...Pyrethrin Biosynthesis: The Cytochrome P450 Oxidoreductase CYP82Q3 Converts Jasmolone To Pyrethrolone1[OPEN] Wei Li,a](https://img.pdfslide.net/doc/110x75/5e2d08c0200c602a86070292/pyrethrin-biosynthesis-the-cytochrome-p450-oxidoreductase-pyrethrin-biosynthesis.jpg)

























