Embed Size (px)
Citation preview

1. Introduction
2. Development of HCC
3. Receptors and signaling
pathways in HCC: molecular
targets
4. Angiogenesis in HCC:
molecular targets
5. Expert opinion
Review
Targeted therapy ofhepatocellular cancerPiotr Jan WysockiUniversity of Medical Sciences, Department of Chemotherapy, Greater Poland Cancer Center,
ul. Garbary 15, Poznan 61-866, Poland
Importance of the field: Hepatocellular cancer (HCC) is the fifth most common
malignancy worldwide and third leading cause of cancer death. HCC is highly
resistant to conventional systemic therapies, and prognosis for advanced HCC
patients remains poor. However, identification of signaling pathways respon-
sible for HCC growth and progression such as RAS/RAF/MEK/ERK or PI3K/AKT/
mTOR has determined crucial molecular targets and led to development of
novel promising targeted therapies.
Areas covered in this review: This article presents molecular mechanisms
responsible for development and progression of HCC and strategies aimed
to block important molecules involved in signal transduction. It also reviews
the clinical studies evaluating efficacy and safety of novel targeted
approaches for treatment of this malignancy.
What the reader will gain: Inhibition of molecular targets (ligands, membrane
receptors and receptor-associated kinases) represents a promising strategy for
treatment of HCC; in the case of sorafenib, this has already been demon-
strated to significantly improve survival of advanced HCC patients. This article
reviews novel therapeutic approaches that are based on combinations of
different targeted agents with or without classic cytotoxic drugs.
Take home message: Despite significant progress, advanced HCC remains an
incurable disease, and the overall efficacy of recently approved targeted
therapy (sorafenib) remains moderate. It is to be hoped that several ongoing
clinical trials evaluating novel targeted approaches for treatment of HCC will
lead to further improvement in the management of advanced disease.
Keywords: angiogenesis, hepatocellular cancer, mTOR inhibitors,
receptor tyrosine kinase inhibitors, signaling pathways, targeted therapy
Expert Opin. Investig. Drugs (2010) 19(2):265-274
1. Introduction
Hepatocellular cancer (HCC) is the fifth most common malignancy worldwide andthird leading cause of cancer death. In 2000, HCC accounted for 7.5% of cancer inmen and 3.5% in women. There are some certain geographic regions in Asia andAfrica where the incidence of HCC is 40 times higher than in other regions of theworld due to endemic hepatitis B virus (HBV) infection [1-3]. The incidence of HCCis rising globally; however, a much sharper increase has been documented inWesterncountries, mainly because of the high prevalence of hepatitis C virus (HCV)infection [4]. In Western countries, 30 – 40% of HCC cases are diagnosed at anearly stage that is amenable to potentially curative treatments such as surgery(resection or liver transplantation) and locoregional procedures (radiofrequencyablation). Up to 70% of patients diagnosed with early HCC can survive 5 years.However, locally advanced or relapsing (after locoregional treatment) disease has apoor prognosis due to underlying liver disease and lack of effective systemictreatment. Conventional chemotherapy has not been demonstrated to prolongsurvival of patients with locally advanced or metastatic HCC. However, recent
10.1517/13543780903514110 © 2010 Informa UK Ltd ISSN 1354-3784 265All rights reserved: reproduction in whole or in part not permitted
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y N
yu M
edic
al C
ente
r on
05/
13/1
3Fo
r pe
rson
al u
se o
nly.

advances in our understanding of HCC biology and biotech-nology have led to the development of novel molecularlytargeted agents for this malignancy. The aim of this article is toreview the molecular mechanisms responsible for developmentand progression of HCC, and the role of targeted therapies inthe treatment of HCC.
2. Development of HCC
Hepatocarcinogenesis is a multistep process initiated by exter-nal stimuli that lead to genetic changes in hepatocytes or stemcells, resulting in proliferation, apoptosis, dysplasia and neo-plasia [5-7]. Risk factors for HCC development include viralinfections (HBV or HCV), exposure to aflatoxin B, chronicalcohol intake, and cirrhosis. Almost 80% of HCC casesdevelop in cirrhotic liver [5,6]. The majority of HCC casesare related to chronic viral infections. However, the mechan-isms by which HBV or HCV induce malignant transforma-tion seem to be different. HBV is a DNA virus; its DNAintegrates into the host genome, inducing chromosome insta-bility [8,9] and insertional mutations that may activate variousoncogenes such as cyclin A [10,11]. Replication of HBV requiresexpression of several viral proteins, in particular X protein(HBx) [12]. HBx acting as a transactivator upregulates severaloncogenes such as c-myc and c-jun [13,14] and transcriptionalfactors like NF-kB or AP-2 [15,16]. Additionally, HBx activatespromoters of genes encoding IL-8, TNF, TGF-ß andEGFR [17]. HBx that is localized in cytoplasm can stimulateseveral signal transduction pathways: JAK/STAT [18], RAS/RAF/MAPK [19], and Wnt/ß-catenin [20]. Furthermore, HBxmay impair various functions of p53 protein [21] and stimulateproduction of angiogenic factors such as VEGF by HCCcells [22]. Since HCV genome (RNA) does not integrate intothe host genome, the major role in hepatocarcinogenesis issupposed to be played by HCV viral proteins – core, NS3 andNS5A. HCV core protein can promote apoptosis or cellproliferation through interaction with p53 [23] or via upregu-lation of Wnt-1 at the transcriptional level [24]. NS4Aand NS4B proteins mediate translational inhibition anddegradation of various cellular proteins [25].Aflatoxin B, a fungal toxin present in contaminated ground-
nuts, is a very potent mutagen that reacts with guanine inDNA, leading to mutations. Endemic exposure to aflatoxin B
is common in some Asian and African countries and isassociated with a high frequency of HCC-carrying mutationsin p53 at codon 249 [26].
Cirrhosis is present in about 80 – 90% of HCC patientsand constitutes the largest single risk factor. The risk ofdeveloping HCC in cirrhotic liver varies with the underlyingdisease. The highest estimated 5-year cumulative risk isobserved in HCV cirrhosis (17 – 30%), followed by hemo-chromatosis (21%), HBV cirrhosis (10 – 15%), alcoholiccirrhosis (8%), and biliary cirrhosis (4%) [4]. In cirrhotic liver,changes in fat metabolism associated with activation of adi-pocyte-like pathways are supposed to be involved in neoplastictransformation [27,28]. Additionally, steatosis, steatohepatitisand associated oxidative stress are recognized as importantcofactors in HCC development [29].
3. Receptors and signaling pathways in HCC:molecular targets
3.1 EGFREGFR is expressed on the surface of hepatocytes and plays arole in regeneration after liver injury or partial hepatec-tomy [30]. Hepatocarcinogenesis and proliferation of HCCcells depends on stimulation of EGFR by its ligands, TGF-aor EGF [31-33]. Activation of EGFR initiates two signalingpathways, RAS/RAF/MEK/ERK and PI3K/AKT/mTOR,which play a major role in the biology of HCC [7].Thereare two strategies for targeting EGFR: neutralizing monoclo-nal antibodies (cetuximab or panitumumab), and tyrosinekinase inhibitors (TKIs; erlotinib, gefitinib and lapatinib,which also inhibits the HER2 receptor). Monoclonal anti-bodies bind to the extracellular domain of EGFR and blockthe ligand-induced receptor activation; receptor TKIs blockkinase-dependent downstream signaling. Both approachesdemonstrated significant inhibitory activity in vitro againstHCC cell cultures [34,35].
The clinical efficacy of erlotinib in HCC patients has beenevaluated in two Phase II clinical studies. In a study by Philipand colleagues, 38 patients with unresectable or metastaticHCC were receiving 150 mg of erlotinib daily [36]. Expressionof EGFR was detected in 88% of the patients. Objectiveclinical responses were observed in three patients. At 6 monthsof treatment, 32% of patients remained progression-free, andmedian OS (overall survival) was 13 months. In analogicalstudy involving 40 patients, no objective clinical responseswere observed [37]. At 4 months of treatment, 43% of patientsremained progression-free and the median OS was10.75 months. Lapatinib was evaluated in a cohort of40 patients with advanced HCC in a Phase II study. Theobjective response rate was 5%. Median progression-freesurvival (PFS) and OS were 2.3 and 6.2 months, respec-tively [38]. In a similar study in a group of 31 patients, gefitinibinduced 3% of objective responses and 22.6% of SD (stabledisease). Median PFS and OS were 2.8 and 6.8 months,respectively [39].
Article highlights.
. Molecular mechanisms of hepatocellular cancer (HCC)development and progression.
. Receptors and signal transduction molecules astherapeutic targets.
. Molecular targets in HCC-associated angiogenesis.
. Safety and efficacy of novel targeted therapies fortreatment of HCC.
. Promising future strategies for treatment of HCC.
This box summarises key points contained in the article.
Targeted therapy of hepatocellular cancer
266 Expert Opin. Investig. Drugs (2010) 19(2)
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y N
yu M
edic
al C
ente
r on
05/
13/1
3Fo
r pe
rson
al u
se o
nly.

In a Phase II study, 30 patients with advanced or metastaticHCC received cetuximab. No clinical responses were seen,and disease stabilization was observed in 17% of patients.Median PFS and OS were 1.4 and 9.6 months, respec-tively [40]. In a similar study, 27 advanced HCC patientswere administered cetuximab. The disease stabilization ratewas 44.4% (no objective responses) and median PFS,1.8 months [41]. The clinical efficacy of cetuximab combinedwith standard chemotherapy was analyzed in a multicenterPhase II study that included 43 treatment-naive advancedHCC patients. Patients were treated with cetuximab (standardregimen) and chemotherapy, repeated every 2 weeks(gemcitabine + oxaliplatin). The objective response rate was23% and disease stabilization was observed in 65% of patients.Median PFS and OS were not reported [42].
3.2 IGF-R1Aberrant activation of insulin-like growth factor signalingpathway resulting from upregulation and activation ofIGF-1R can be detected in 20% of HCC. Additionally,IGF-II expression is increased in 12 – 44% of HCC sam-ples [43]. In a xenograft HCC model, monoclonal antibodytargeting IGF-1R delayed tumor growth and improvedsurvival of treated animal [44]. In a Phase I clinical study,one patient with heavily pretreated HCC experienced SD of> 9 months following administration of IMC-A12 mono-clonal antibody [45]. A Phase II study evaluating IMC-A12in patients with advanced HCC has been recently initiated.
3.3 PI3K/AKT/mTOR pathwayThe phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR pro-tein cascade is a major signaling pathway associated withreceptor tyrosine kinases (RTKs) that have been identifiedin cancer cells [46]. The second important pathway in HCC isthe RAS/RAF/MAPK cascade [47]. Various RTKs that may beexpressed by cancers cells (e.g., VEGFR-1 [48], PDGFR-a [49],EGFR [50], or c-MET [51]) use the PI3K/Akt/mTOR signalingpathway to shape the phenotype and function of malignantcells. Activation of RTKs leads to activation of PI3K, whichcan be also activated indirectly via RAS. The function of PI3Kis negatively controlled by a phosphatase and tensinhomologue (PTEN). Loss of functional PTEN is frequent(> 55%) in HCC [52]. However, the frequency of PTENmutations is much lower than in other malignancies(range, 0 – 11%) [53]. Activated PI3K upregulates expressionof NF-kB-dependent survival genes such as Bcl-XL [54].PI3K, via a 3-phosphoinositide-dependent protein kinase 1(PDK-1), activates AKT [55]. AKT is a serine/threonine kinasethat phosphorylates and inactivates several pro-apoptoticproteins such as Bad and caspase-9.
One important downstream effector of AKT is mTOR(mammalian target of rapamycin). Activation of the PI3K/AKT/mTOR pathway correlates with poor prognosis inHCC [56]. Phosphorylation of mTOR and its downstreamtarget, S6K1, were detected in 15 and 45% of human HCC
cases, respectively [57]. Through its downstream effectors,activated mTOR regulates numerous cellular processes suchas initiation of mRNA transcription and protein translation.mTOR regulates essential signal transduction pathways and isinvolved in coupling growth stimuli to cell cycle progres-sion [58]. Phosphorylation of mTOR leads to upregulationof hypoxia-inducible factors alpha (HIF-1a and HIF-2a).Transcriptional targets of HIFs are strongly associatedwith metastasis.
The CXCR4, a chemokine receptor that is supposed to beone of the major metastatic mediators, was shown to beupregulated by HIF in HCC [59,60]. Similarly, hypoxiaincreases expression of MMP2 and MMP9 [61]; and HIFupregulates lysyl oxidase, which facilitates metastasis throughalteration of extracellular matrix components such as elastinand collagen [62]. One of the targets of HIF-2a is Oct4, a geneencoding a POU-domain transcription factor that is a keyregulator of stem-cell behavior. It is possible that Oct4 mod-ulates tumor biology through promotion of the growth of‘cancer stem cells’, which seem to be pivotal for maintainingtumor self-renewal and chemotherapy resistance [63]. HIFs areassociated with malignant phenotype of cancer cells, not onlyregulating tumor cell phenotype but also inducing angiogen-esis through expression of VEGF or PDGF [64]. The HIFs arealso responsible for tumor chemoresistance. Platelet-derivedgrowth factor B (PDGF-B) not only stabilizes novel vesselsbut also decreases penetration of anticancer drugs throughincreasing of interstitial hypertension [65]. Another mechanismof chemoresistance mediated by HIFs is upregulation ofMDR1 [66].
The PI3K/AKT/mTOR pathway may be inhibited atvarious levels. PI3K inhibitors such as wortmannin andLY294002 have demonstrated some efficacy in animalHCC models [67]. Another inhibitor, FTY720, was shownto induce apoptosis in HCC cell lines, as well as inhibitinggrowth of HCC xenografts [68]. Activation of AKT can beinhibited by an orally bioavailable alkylphospholipid, perifo-sine, which has been already tested in a few Phase I stud-ies [69,70]. HIFs represent another interesting therapeutictarget. In a murine model, administration of HIF-1aantisense into HCC cells decreased intracellular levels ofHIF-1a and VEGF and increased therapeutic efficacy ofdoxorubicin [71].
However, the most promising target in the PI3K/AKT/mTOR pathway is represented by mTOR. Inhibitors ofmTOR are currently used as immunosuppressant drugs fol-lowing liver transplantation. The effectiveness of mTORblockade with rapamycin analogues has been tested in pre-clinical HCC models [53]. Rapamycin was shown to inhibitproliferation of HCC cell line in vitro and growth of HCCtumors in animal models [72]. Everolimus (RAD001) effec-tively inhibited growth of HCC in vitro and in vivo, andsignificantly enhanced cytotoxic effect of cisplatin in HCCcell lines [73,74]. Another mTOR inhibitor, sirolimus, wasevaluated in 21 advanced HCC patients [75]. One patient
Wysocki
Expert Opin. Investig. Drugs (2010) 19(2) 267
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y N
yu M
edic
al C
ente
r on
05/
13/1
3Fo
r pe
rson
al u
se o
nly.

experienced partial response, and five remained progression-free at 3 months. Median OS was 6.5 months. A Phase I/IIstudy evaluating the clinical efficacy of the mTOR inhibitoreverolimus (RAD001) in advanced HCC has beenrecently initiated.
3.4 RAS/RAF/MAPK pathwayBeside the PI3K/AKT/mTOR pathway, the MAPK pathwayplays a major role in hepatocarcinogenesis. The MAPKpathway includes a cascade of phosphorylation involvingkinases such as RAS, RAF, mitogen-activated protein extra-cellular kinase (MEK) and extracellular signal-regulated kinase(ERK). The MAPK pathway can be activated by RTKs such asEGFR, HER2, IGF-R1 or c-MET by integrin receptors or bysignaling from ion channels [76]. Activation of the MAPKpathway induces cell proliferation, migration and inhibitionof apoptosis [77]. This pathway is often aberrantly activated inHCC [78,79], and there are several molecular mechanismsresponsible for its activation. HCV core protein can directlyactivate the Raf/MEK/ERK cascade [80]. Loss of RAF kinaseinhibitor protein was demonstrated to stimulate HCC pro-liferation and migration [81]. Overexpression of RAS was alsoobserved in HCC tumors [82]. As in the case of the PI3K/AKT/mTOR pathway, the RAS/RAF/MEK/ERK pathway may beinhibited at various levels. In order to be capable of signaltransduction, RAS must undergo post-translational modifica-tion by incorporation of prenyl moieties (farnesyl andgeranylgeranyl groups).Inhibitors of farnesyl transferase prevent prenylation of
RAS proteins; to date, several inhibitors have been devel-oped. Among them, ABT-100 was shown to prevent thedevelopment of chemically induced HCC in rats [83]. Inhi-bition of MEK was also shown to prevent development ofHCC and increase apoptosis in existing HCC tumors inmice [84]. In another study, administration of MEK inhib-itor resulted in dose-dependent growth inhibition of HCCxenografts [85].Targeting RAF kinase is currently the most promising
targeted approach for treatment of HCC. Sorafenib, amulti-TKI, has demonstrated significant clinical efficacy ina pivotal Phase III trial (SHARP), which led to its approval forHCC treatment by international regulatory agencies [86].Sorafenib tosylate is a bisaryl urea first designed as anin vitro inhibitor of the RAF-1 protein. However, sorafenibwas also shown to inhibit the tyrosine kinases of VEGFR-1,VEGFR-2, VEGFR-3, PDGFR-B, FLT-3 and c-KIT. In thepivotal Phase III study, 602 treatment-naive, advanced HCCpatients were randomly assigned to receive sorafenib 400 mgb.i.d. or placebo [86]. Patients recruited in the study hadnormal liver function (Child–Pugh class A). Sorafenib signif-icantly improved median PFS and OS compared with placebo(5.5 vs 2.8 and 10.7 vs 7.9 months, respectively). The hazardratio (HR) for death in the sorafenib group was significantlyreduced, to 0.69 (95% CI, 0.55 – 0.87; p < 0.001). Theresponse rate was similarly low in both arms: 2% PR (partial
response) and 71% SD in the sorafenib arm and 1% PR and67% SD in the placebo arm. Disease control was significantlyhigher in the sorafenib group (43 vs 32%; p = 0.002). Toxicityof the treatment was acceptable, with diarrhea, weight loss,hand–foot skin reaction and hypophosphatemia being themost frequently reported adverse events.
The efficacy of sorafenib in patients with advanced HCCwas confirmed in another Phase III trial, which involved207 patients from the Asia-Pacific region [87]. Sorafenibsignificantly prolonged median OS compared with placebo(6.5 vs 4.2 months, respectively). The reduction of HR forsurvival in the sorafenib arm (0.68; 95% CI, 0.50 – 0.93;p = 0.014) was comparable to the SHARP trial. Based on thePhase III studies, sorafenib is now approved for the treatmentof patients with advanced HCC with adequate liver function(Child–Pugh class A).
4. Angiogenesis in HCC: molecular targets
As in other human cancers, angiogenesis is pivotal for thedevelopment and progression of HCC. Several studies dem-onstrated that the intensity of angiogenesis in HCC (assessed bymicrovessel density) correlated with the risk of vascular inva-sion, metastasis and patient prognosis [88,89]. VEGF (mainly itsA-isoform, VEGF-A) is one of the most potent angiogenicfactors. The effect of VEGF-A is mediated via two receptors:VEGFR-1 (Flt-1) and VEGFR-2 (KDR). VEGFR-2 mediatesall of the known cellular responses to VEGF, and VEGFR-1 issupposed to modulate VEGFR-2 signaling. In HCC xeno-grafts, tumor growth was demonstrated to be tightly controlledby the level of VEGF expression [90]. The expression of VEGFincreases in parallel to HCC development [91]. A quantitativeanalysis revealed that VEGF was expressed in 64% of encap-sulated HCC and in 78% of non-encapsulated (more aggres-sive) HCC [92]. In another study, expression of VEGF wasdetected in 89% of HCC samples [93].
Expression of VEGF and other angiogenic factors such asbFGF or angiopoietin 2 correlates with vascular density,invasion and metastasis in HCC [94,95]. Moreover, HCCcell lines were demonstrated to express VEGF receptors, whichmay be responsible for VEGF-mediated autocrine stimulationof tumor growth [96]. The VEGF pathway may be targeted bytwo approaches, using either anti-VEGFmonoclonal antibodyor inhibitors of the receptor tyrosine kinase associatedwith VEGFR.
4.1 Targeting VEGFBevacizumab is a humanized monoclonal antibody that neu-tralizes all isoforms of VEGF. This drug is currently approvedby the FDA for treatment of colon, breast and non-small celllung cancers, in combination with chemotherapy. In a HCCxenograft model, bevacizumab significantly decreased vesseldensity and prolonged time to progression of tumor-bearingmice [97]. In a Phase II clinical trial, 46 patients with locallyadvanced HCC were treated with bevacizumab 5 or 10 mg/kg
Targeted therapy of hepatocellular cancer
268 Expert Opin. Investig. Drugs (2010) 19(2)
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y N
yu M
edic
al C
ente
r on
05/
13/1
3Fo
r pe
rson
al u
se o
nly.

i.v. every 2 weeks [98]. Objective clinical responses wereobserved in 13%, and disease stabilization lasting ‡ 6 monthsin 65% of patients. Median PFS was 6.9 months. OS was 53%at 1 year, 28% at 2 years, and 23% at 3 years. In anotherPhase II study, among 38 evaluable patients bevacizumabinduced 16% PR and 47% SD [99].
Treatment based on bevacizumab combined with erlotinibwas evaluated in 40 patients in another Phase II trial. Thepartial response rate was 25%. Median PFS was9 months; OS, 15.65 months. This combination will befurther evaluated in a Phase III clinical study [100].
A few clinical trials have evaluated various combinations ofbevacizumab with chemotherapy. The efficacy of bevacizumabwith gemcitabine and oxaliplatin in advanced HCC wasevaluated in a Phase II trial [101]. In a group of 30 evaluablepatients, the objective response rate (ORR) was 20%; SD,27%. Median OS and PFS were 9.6 and 5.3 months, respec-tively. Other first-line therapy based on bevacizumab com-bined with capecitabine and oxaliplatin was evaluated in30 patients with advanced HCC [102]. In this trial, 11% ofpatients achieved partial response and 78% disease stabiliza-tion. Median PFS was 5.4 months. Bevacizumab combinedwith capecitabine was evaluated in another Phase II study.Among 45 patients with advanced or metastatic HCC, theobjective response rate (CR + PR) was 16% and SD wasobserved in 44%. Median OS and PFS were 4.1 and10.7 months, respectively [103].
4.2 Targeting VEGF receptorsElucidation of the important role of receptor tyrosine kinasesin angiogenesis has identified novel promising therapeutictargets. The introduction of TKIs such as sunitinib or sor-afenib represents a major breakthrough in the treatment ofrenal cell cancer [104]. In HCC, the first approved multi-kinaseinhibitor, sorafenib, exerts its effect not only againsttumor cells by targeting RAF kinase, but also againstendothelial cells and pericytes by targeting VEGFR-1,VEGFR-2, VEGFR-3 and PDGFR-b, which blocks VEGFand PDGF-dependent angiogenesis.
Sunitinib malate is another orally available multi-kinaseinhibitor that targets receptor tyrosine kinases of VEGFR-1,VEGFR-2, VEGFR-3, PDGFR-a, PDGFR-b, FMS-liketyrosine kinase 3 (FLT-3) and c-KIT. In an European studythat included 45 patients with unresectable HCC, adminis-tration of sunitinib induced 2% objective responses (1 CR)and 40% SD [105]. The median OS was 9.3 months; PFS,2.8 months. In another study conducted in the United States,treatment with sunitinib in 34 patients resulted in 2.9% PRand 50% SD. The median OS and PFS were 9.8 and3.9 months, respectively [106].
Brivanib alaninate is an oral, dual inhibitor of VEGFR andFGFR tyrosine kinases. In a Phase II clinical trial, HCCpatients – either treatment-naive or previously treated withantiangiogenic inhibitor (sorafenib or thalidomide) – receivedbrivanib. Brivanib demonstrated some clinical efficacy in
first- and second-line therapy. The median OS oftreatment-naive patients was 10.0 months [107].
Currently, sunitinib and brivanib are being tested inPhase III studies, and other antiangiogenic multi-kinase inhi-bitors such as pazopanib, vandetanib, or cediranib are beingevaluated in early-phase clinical trials.
5. Expert opinion
Significant progress in cancer molecular biology and biotech-nology resulted in the development of various targetedapproaches for the treatment of HCC (Table 1). Clinical trialsevaluating the efficacy of sorafenib demonstrated that targetedtherapy can significantly improve clinical outcome and pro-long survival of patients with advanced HCC. However, theera of targeted therapies for HCC is just beginning, and manynew drugs are expected to emerge. The cytostatic mechanismof action of targeted agents is reflected by their ability toinduce prolonged disease stabilization rather than cure. There-fore, the development of therapeutic approaches based onmolecularly targeted drugs must take into account that theseagents must have low toxicity to be suitable for the chronictreatment of a malignant disease.
There are still many unanswered questions associated withtargeted approaches in the treatment of HCC. Novel drugsrequire novel biomarkers. Despite the obvious clinical efficacyof sorafenib, many patients turn out to be refractory to thistherapy. Individualization of targeted therapies depends onbiomarkers, which help to predict response to treatment. Inthe case of sorafenib, phosphorylated ERK seems to representsuch a promising biomarker. A Phase II trial evaluating theefficacy of sorafenib revealed that increased levels of phospho-ERK (a downstream target of RAF) in HCC samples pre-dicted a significantly prolonged PFS compared with patientswith low levels of phospho-ERK [108]. In the case of othertargeted drugs currently used for treatment of various malig-nancies (such as bevacizumab, sunitinib or temsirolimus),validated biomarkers are still missing.
The efficacy of sorafenib was demonstrated mostly inpatients with Child–Pugh class A liver function, due toparticular clinical trial inclusion criteria. However, patientswith good liver condition represent only a minority of HCCpatients. Therefore, prospective evaluation of sorafenib inpatients with liver function worse than Child–Pugh class Ais crucial. A recent, retrospective subgroup analysis of theSHARP study (presented at the 44th Annual Meeting of theEuropean Association for the Study of the Liver [EASL])revealed that sorafenib was similarly effective in patients withintermediate and late-stage liver cancer (Barcelona ClinicLiver Cancer stages B and C).
Another aspect that remains to be determined is the efficacyof targeted therapies in the adjuvant setting. Since regrowth ofHCC tumors following surgery or transarterial chemoembo-lization is associated with rapid angiogenesis, antiangiogenicstrategies may prove effective in reducing the relapse rate. The
Wysocki
Expert Opin. Investig. Drugs (2010) 19(2) 269
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y N
yu M
edic
al C
ente
r on
05/
13/1
3Fo
r pe
rson
al u
se o
nly.
rationale for combining local modalities (transcatheterarterial embolization or chemoembolization) with systemictargeted therapies has been recently extensively reviewedby Strebel and Dufour [109]. Studies seeking to determinethe molecular pathways responsible for HCC developmentand progression indicate the existence of crosstalk betweenvarious levels of different signal-transducing cascades.This knowledge is expected to help identify novel mechanismsof resistance to targeted agents and determine moleculartargets in complementary pathways, which must beblocked simultaneously.Sorafenib is the first systemic therapy to demonstrate a
survival benefit for unresectable HCC. To date, sorafenibremains the only approved targeted agent in systemictreatment of HCC; however, it is access to this drug that
remains a major concern. Currently, only the richest countriescan afford to provide novel, approved therapies to all cancerpatients. Unfortunately, the highest incidence of HCC isobserved in poor countries, owing to endemic HBV infection.Therefore, the wide use of novel targeted therapies requirescompromise between governments, healthcare systems andpharmaceutical companies. One has to hope that such acompromise, which may dramatically improve the prog-nosis for HCC patients worldwide, will not remain anunfulfilled dream.
Declaration of interest
The author states no conflict of interest and has received nopayment in preparation of this manuscript.
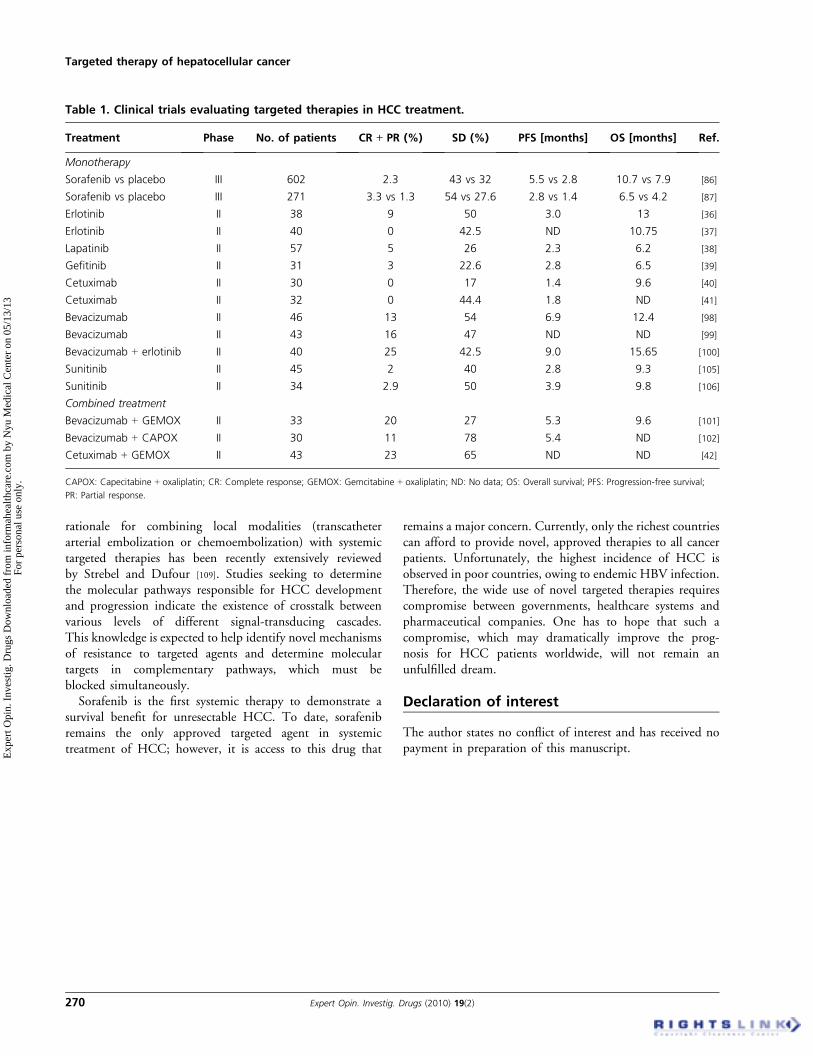
Table 1. Clinical trials evaluating targeted therapies in HCC treatment.
Treatment Phase No. of patients CR ++ PR (%) SD (%) PFS [months] OS [months] Ref.
Monotherapy
Sorafenib vs placebo III 602 2.3 43 vs 32 5.5 vs 2.8 10.7 vs 7.9 [86]
Sorafenib vs placebo III 271 3.3 vs 1.3 54 vs 27.6 2.8 vs 1.4 6.5 vs 4.2 [87]
Erlotinib II 38 9 50 3.0 13 [36]
Erlotinib II 40 0 42.5 ND 10.75 [37]
Lapatinib II 57 5 26 2.3 6.2 [38]
Gefitinib II 31 3 22.6 2.8 6.5 [39]
Cetuximab II 30 0 17 1.4 9.6 [40]
Cetuximab II 32 0 44.4 1.8 ND [41]
Bevacizumab II 46 13 54 6.9 12.4 [98]
Bevacizumab II 43 16 47 ND ND [99]
Bevacizumab + erlotinib II 40 25 42.5 9.0 15.65 [100]
Sunitinib II 45 2 40 2.8 9.3 [105]
Sunitinib II 34 2.9 50 3.9 9.8 [106]
Combined treatment
Bevacizumab + GEMOX II 33 20 27 5.3 9.6 [101]
Bevacizumab + CAPOX II 30 11 78 5.4 ND [102]
Cetuximab + GEMOX II 43 23 65 ND ND [42]
CAPOX: Capecitabine + oxaliplatin; CR: Complete response; GEMOX: Gemcitabine + oxaliplatin; ND: No data; OS: Overall survival; PFS: Progression-free survival;
PR: Partial response.
Targeted therapy of hepatocellular cancer
270 Expert Opin. Investig. Drugs (2010) 19(2)
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y N
yu M
edic
al C
ente
r on
05/
13/1
3Fo
r pe
rson
al u
se o
nly.

Bibliography1. Bosch FX, Ribes J, Diaz M, Cleries R.
Primary liver cancer: worldwide incidence
and trends. Gastroenterology
2004;127(5 Suppl 1):S5-16
2. Parkin DM, Bray F, Ferlay J, Pisani P.
Estimating the world cancer burden:
Globocan 2000. Int J Cancer
2001;94(2):153-6
3. Pisani P, Bray F, Parkin DM. Estimates of
the world-wide prevalence of cancer for 25
sites in the adult population. Int J Cancer
2002;97(1):72-81
4. Fattovich G, Stroffolini T, Zagni I,
Donato F. Hepatocellular carcinoma in
cirrhosis: incidence and risk factors.
Gastroenterology
2004;127(5 Suppl 1):S35-50
5. Farazi PA, DePinho RA. Hepatocellular
carcinoma pathogenesis: from genes to
environment. Nat Rev Cancer
2006;6(9):674-87
6. Thorgeirsson SS, Grisham JW. Molecular
pathogenesis of human hepatocellular
carcinoma. Nat Genet 2002;31(4):339-46
7. Villanueva A, Newell P, Chiang DY, et al.
Genomics and signaling pathways in
hepatocellular carcinoma. Semin Liver Dis
2007;27(1):55-76
8. Brechot C, Pourcel C, Louise A, et al.
Presence of integrated hepatitis B virus
DNA sequences in cellular DNA of human
hepatocellular carcinoma. Nature
1980;286(5772):533-5
9. Murakami Y, Saigo K, Takashima H, et al.
Large scaled analysis of hepatitis B virus
(HBV) DNA integration in HBV related
hepatocellular carcinomas. Gut
2005;54(8):1162-8
10. Minami M, Daimon Y, Mori K, et al.
Hepatitis B virus-related insertional
mutagenesis in chronic hepatitis B patients
as an early drastic genetic change leading to
hepatocarcinogenesis. Oncogene
2005;24(27):4340-8
11. Wang J, Chenivesse X, Henglein B,
Brechot C. Hepatitis B virus integration in
a cyclin A gene in a hepatocellular
carcinoma. Nature 1990;343(6258):555-7
12. Feitelson MA, Duan LX. Hepatitis B
virus X antigen in the pathogenesis of
chronic infections and the development of
hepatocellular carcinoma. Am J Pathol
1997;150(4):1141-57
13. Balsano C, Avantaggiati ML, Natoli G,
et al. Full-length and truncated versions of
the hepatitis B virus (HBV) X protein (pX)
transactivate the cmyc protooncogene at
the transcriptional level. Biochem Biophys
Res Commun 1991;176(3):985-92
14. Twu JS, Lai MY, Chen DS, Robinson WS.
Activation of protooncogene c-jun by
the X protein of hepatitis B virus. Virology
1993;192(1):346-50
15. Chirillo P, Falco M, Puri PL, et al.
Hepatitis B virus pX activates NF-kappa
B-dependent transcription through a
Raf-independent pathway. J Virol
1996;70(1):641-6
16. Seto E, Mitchell PJ, Yen TS.
Transactivation by the hepatitis B virus X
protein depends on AP-2 and other
transcription factors. Nature
1990;344(6261):72-4
17. Andrisani OM, Barnabas S. The
transcriptional function of the hepatitis B
virus X protein and its role in
hepatocarcinogenesis (Review). Int J Oncol
1999;15(2):373-9
18. Lee YH, Yun Y. HBx protein of hepatitis B
virus activates Jak1-STAT signaling.
J Biol Chem 1998;273(39):25510-5
19. Benn J, Schneider RJ. Hepatitis B virus
HBx protein activates Ras-GTP complex
formation and establishes a Ras, Raf, MAP
kinase signaling cascade. Proc Natl Acad
Sci USA 1994;91(22):10350-4
20. Cha MY, Kim CM, Park YM, Ryu WS.
Hepatitis B virus X protein is essential for
the activation of Wnt/beta-catenin
signaling in hepatoma cells. Hepatology
2004;39(6):1683-93
21. Ueda H, Ullrich SJ, Gangemi JD, et al.
Functional inactivation but not structural
mutation of p53 causes liver cancer.
Nat Genet 1995;9(1):41-7
22. Lee SW, Lee YM, Bae SK, et al. Human
hepatitis B virus X protein is a possible
mediator of hypoxia-induced angiogenesis
in hepatocarcinogenesis. Biochem Biophys
Res Commun 2000;268(2):456-61
23. Yamanaka T, Kodama T, Doi T.
Subcellular localization of HCV core
protein regulates its ability for p53
activation and p21 suppression.
Biochem Biophys Res Commun
2002;294(3):528-34
24. Fukutomi T, Zhou Y, Kawai S, et al.
Hepatitis C virus core protein stimulates
hepatocyte growth: correlation with
upregulation of wnt-1 expression.
Hepatology 2005;41(5):1096-105
25. Florese RH, Nagano-Fujii M, Iwanaga Y,
et al. Inhibition of protein synthesis by the
nonstructural proteins NS4A and NS4B of
hepatitis C virus. Virus Res
2002;90(1-2):119-31
26. Ozturk M. p53 mutation in hepatocellular
carcinoma after aflatoxin exposure. Lancet
1991;338(8779):1356-9
27. Terasaki S, Kaneko S, Kobayashi K, et al.
Histological features predicting malignant
transformation of nonmalignant
hepatocellular nodules: a prospective
study. Gastroenterology
1998;115(5):1216-22
28. Watanabe S, Horie Y, Kataoka E, et al.
Non-alcoholic steatohepatitis and
hepatocellular carcinoma: lessons from
hepatocyte-specific phosphatase and tensin
homolog (PTEN)-deficient mice.
J Gastroenterol Hepatol
2007;22(Suppl 1):S96-S100
29. Caldwell S, Park SH. The epidemiology of
hepatocellular cancer: from the
perspectives of public health problem to
tumor biology. J Gastroenterol
2009;44(Suppl 19):96-101
30. Taub R. Liver regeneration: from myth to
mechanism. Nat Rev Mol Cell Biol
2004;5(10):836-47
31. Harada K, Shiota G, Kawasaki H.
Transforming growth factor-alpha and
epidermal growth factor receptor in
chronic liver disease and hepatocellular
carcinoma. Liver 1999;19(4):318-25
32. Hisaka T, Yano H, Haramaki M, et al.
Expressions of epidermal growth factor
family and its receptor in hepatocellular
carcinoma cell lines: relationship to cell
proliferation. Int J Oncol
1999;14(3):453-60
33. Ito Y, Takeda T, Higashiyama S, et al.
Expression of heparin binding epidermal
growth factor-like growth factor in
hepatocellular carcinoma: an
immunohistochemical study. Oncol Rep
2001;8(4):903-7
34. Huether A, Hopfner M, Baradari V, et al.
EGFR blockade by cetuximab alone or as
combination therapy for growth control of
hepatocellular cancer. Biochem Pharmacol
2005;70(11):1568-78
35. Hopfner M, Sutter AP, Huether A, et al.
Targeting the epidermal growth factor
Wysocki
Expert Opin. Investig. Drugs (2010) 19(2) 271
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y N
yu M
edic
al C
ente
r on
05/
13/1
3Fo
r pe
rson
al u
se o
nly.

receptor by gefitinib for treatment of
hepatocellular carcinoma. J Hepatol
2004;41(6):1008-16
36. Philip PA, Mahoney MR, Allmer C, et al.
Phase II study of erlotinib (OSI-774) in
patients with advanced hepatocellular
cancer. J Clin Oncol 2005;23(27):6657-63
37. Thomas MB, Chadha R, Glover K, et al.
Phase 2 study of erlotinib in patients with
unresectable hepatocellular carcinoma.
Cancer 2007;110(5):1059-67
38. Ramanathan RK, Belani CP, Singh DA,
et al. A phase II study of lapatinib in
patients with advanced biliary tree and
hepatocellular cancer.
Cancer Chemother Pharmacol
2009;64(4):777-83
39. O’Dwyer PJ, Giantonio BJ, Levy DE, et al.
Gefitinib in advanced unresectable
hepatocellular carcinoma: results from the
Eastern Cooperative Oncology Group’s
Study E1203. J Clin Oncol
(Meeting Abstracts)
2006;24(18 Suppl):4143
40. Zhu AX, Stuart K, Blaszkowsky LS, et al.
Phase 2 study of cetuximab in patients with
advanced hepatocellular carcinoma.
Cancer 2007;110(3):581-9
41. Gruenwald V, Wilkens L, Gebel M, et al.
A phase II open-label study of cetuximab in
unresectable hepatocellular carcinoma:
final results. J Clin Oncol
(Meeting Abstracts)
2007;25(18 Suppl):4598
42. Louafi S, Hebbar M, Rosmorduc O, et al.
Gemcitabine, oxaliplatin (GEMOX) and
cetuximab for treatment of hepatocellular
carcinoma (HCC): results of the phase II
study ERGO. J Clin Oncol
(Meeting Abstracts)
2007;25(18 Suppl):4594
43. Breuhahn K, Longerich T, Schirmacher P.
Dysregulation of growth factor signaling in
human hepatocellular carcinoma.
Oncogene 2006;25(27):3787-800
44. Rowinsky EK, Youssoufian H, Tonra JR,
et al. IMC-A12, a human IgG1
monoclonal antibody to the insulin-like
growth factor I receptor. Clin Cancer Res
2007;13(18 Pt 2):5549s-55s
45. Higano CS, Yu EY, Whiting SH, et al. A
phase I, first in man study of weekly
IMC-A12, a fully human insulin like
growth factor-I receptor IgG1 monoclonal
antibody, in patients with advanced solid
tumors. J Clin Oncol (Meeting Abstracts)
2007;25(18 Suppl):3505
46. Vivanco I, Sawyers CL. The
phosphatidylinositol 3-Kinase AKT
pathway in human cancer. Nat Rev Cancer
2002;2(7):489-501
47. Fang JY, Richardson BC. The MAPK
signalling pathways and colorectal cancer.
Lancet Oncol 2005;6(5):322-7
48. Giannelli G, Sgarra C, Porcelli L, et al.
EGFR and VEGFR as potential target for
biological therapies in HCC cells.
Cancer Lett 2008;262(2):257-64
49. Oseini AM, Roberts LR. PDGFRalpha: a
new therapeutic target in the treatment of
hepatocellular carcinoma? Expert Opin
Ther Targets 2009;13(4):443-54
50. Franovic A, Gunaratnam L, Smith K, et al.
Translational up-regulation of the EGFR
by tumor hypoxia provides a
nonmutational explanation for its
overexpression in human cancer. Proc Natl
Acad Sci USA 2007;104(32):13092-7
51. Xiao GH, Jeffers M, Bellacosa A, et al.
Anti-apoptotic signaling by hepatocyte
growth factor/Met via the
phosphatidylinositol 3-kinase/Akt and
mitogen-activated protein kinase
pathways. Proc Natl Acad Sci USA
2001;98(1):247-52
52. Wang L, Wang WL, Zhang Y, et al.
Epigenetic and genetic alterations of
PTEN in hepatocellular carcinoma.
Hepatol Res 2007;37(5):389-96
53. Villanueva A, Chiang DY, Newell P, et al.
Pivotal role of mTOR signaling in
hepatocellular carcinoma.
Gastroenterology
2008;135(6):1972-83, 83 e1-11
54. Rogers R, Ouellet G, Brown C, et al.
Cross-talk between the Akt and
NF-kappaB signaling pathways inhibits
MEHP-induced germ cell apoptosis.
Toxicol Sci 2008;106(2):497-508
55. Bellacosa A, Chan TO, Ahmed NN, et al.
Akt activation by growth factors is a
multiple-step process: the role of the PH
domain. Oncogene 1998;17(3):313-25
56. Schmitz KJ, Wohlschlaeger J, Lang H,
et al. Activation of the ERK and AKT
signalling pathway predicts poor prognosis
in hepatocellular carcinoma and ERK
activation in cancer tissue is associated with
hepatitis C virus infection. J Hepatol
2008;48(1):83-90
57. Sahin F, Kannangai R, Adegbola O, et al.
mTOR and P70 S6 kinase expression in
primary liver neoplasms. Clin Cancer Res
2004;10(24):8421-5
58. Faivre S, Kroemer G, Raymond E. Current
development of mTOR inhibitors as
anticancer agents. Nat Rev Drug Discov
2006;5(8):671-88
59. Liu H, Pan Z, Li A, et al. Roles of
chemokine receptor 4 (CXCR4) and
chemokine ligand 12 (CXCL12) in
metastasis of hepatocellular carcinoma
cells. Cell Mol Immunol 2008;5(5):373-8
60. Xiang ZL, Zeng ZC, Tang ZY, et al.
Chemokine receptor CXCR4 expression in
hepatocellular carcinoma patients increases
the risk of bone metastases and poor
survival. BMC Cancer 2009;9:176
61. Leufgen H, Bihl MP, Rudiger JJ, et al.
Collagenase expression and activity is
modulated by the interaction of collagen
types, hypoxia, and nutrition in human
lung cells. J Cell Physiol
2005;204(1):146-54
62. Erler JT, Bennewith KL, Nicolau M, et al.
Lysyl oxidase is essential for
hypoxia-induced metastasis. Nature
2006;440(7088):1222-6
63. Covello KL, Kehler J, Yu H, et al.
HIF-2alpha regulates Oct-4: effects of
hypoxia on stem cell function, embryonic
development, and tumor growth.
Genes Dev 2006;20(5):557-70
64. Gordan JD, Simon MC.
Hypoxia-inducible factors: central
regulators of the tumor phenotype.
Curr Opin Genet Dev 2007;17(1):71-7
65. Levitzki A. PDGF receptor kinase
inhibitors for the treatment of PDGF
driven diseases. Cytokine Growth
Factor Rev 2004;15(4):229-35
66. Semenza GL. Targeting HIF-1 for cancer
therapy. Nat Rev Cancer
2003;3(10):721-32
67. Amaravadi R, Thompson CB. The survival
kinases Akt and Pim as potential
pharmacological targets. J Clin Invest
2005;115(10):2618-24
68. Lee TK, Man K, Ho JW, et al. FTY720
induces apoptosis of human hepatoma cell
lines through PI3-K-mediated Akt
dephosphorylation. Carcinogenesis
2004;25(12):2397-405
69. Crul M, Rosing H, de Klerk GJ, et al.
Phase I and pharmacological study of daily
oral administration of perifosine
(D-21266) in patients with advanced solid
Targeted therapy of hepatocellular cancer
272 Expert Opin. Investig. Drugs (2010) 19(2)
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y N
yu M
edic
al C
ente
r on
05/
13/1
3Fo
r pe
rson
al u
se o
nly.

tumours. Eur J Cancer
2002;38(12):1615-21
70. Van Ummersen L, Binger K, Volkman J,
et al. A phase I trial of perifosine (NSC
639966) on a loading dose/maintenance
dose schedule in patients with advanced
cancer. Clin Cancer Res
2004;10(22):7450-6
71. Liu F, Wang P, Jiang X, et al. Antisense
hypoxia-inducible factor 1alpha gene
therapy enhances the therapeutic efficacy of
doxorubicin to combat hepatocellular
carcinoma. Cancer Sci
2008;99(10):2055-61
72. Semela D, Piguet AC, Kolev M, et al.
Vascular remodeling and antitumoral
effects of mTOR inhibition in a rat model
of hepatocellular carcinoma. J Hepatol
2007;46(5):840-8
73. Huynh H, Chow KH, Soo KC, et al.
RAD001 (everolimus) inhibits tumour
growth in xenograft models of human
hepatocellular carcinoma. J Cell Mol Med
2009;13(7):1371-80
74. Pang RW, Poon RT. From molecular
biology to targeted therapies for
hepatocellular carcinoma: the future is
now. Oncology 2007;72(Suppl 1):30-44
75. Rizell M, Andersson M, Cahlin C, et al.
Effects of the mTOR inhibitor sirolimus in
patients with hepatocellular and
cholangiocellular cancer. Int J Clin Oncol
2008;13(1):66-70
76. Lev S, Moreno H, Martinez R, et al.
Protein tyrosine kinase PYK2 involved in
Ca(2+)-induced regulation of ion channel
and MAP kinase functions. Nature
1995;376(6543):737-45
77. Dhillon AS, Hagan S, Rath O, Kolch W.
MAP kinase signalling pathways in cancer.
Oncogene 2007;26(22):3279-90
78. McKillop IH, Schmidt CM, Cahill PA,
Sitzmann JV. Altered expression of
mitogen-activated protein kinases in a rat
model of experimental hepatocellular
carcinoma. Hepatology
1997;26(6):1484-91
79. Schmidt CM, McKillop IH, Cahill PA,
Sitzmann JV. Increased MAPK expression
and activity in primary human
hepatocellular carcinoma.
Biochem Biophys Res Commun
1997;236(1):54-8
80. Giambartolomei S, Covone F, Levrero M,
Balsano C. Sustained activation of the Raf/
MEK/Erk pathway in response to EGF in
stable cell lines expressing the Hepatitis C
Virus (HCV) core protein. Oncogene
2001;20(20):2606-10
81. Lee HC, Tian B, Sedivy JM, et al. Loss of
Raf kinase inhibitor protein promotes cell
proliferation and migration of human
hepatoma cells. Gastroenterology
2006;131(4):1208-17
82. Coleman WB. Mechanisms of human
hepatocarcinogenesis. Curr Mol Med
2003;3(6):573-88
83. Carloni V, Vizzutti F, Pantaleo P.
Farnesyltransferase inhibitor, ABT-100, is
a potent liver cancer chemopreventive
agent. Clin Cancer Res
2005;11(11):4266-74
84. Wentz SC, Wu H, Yip-Schneider MT,
et al. Targeting MEK is effective
chemoprevention of hepatocellular
carcinoma in TGF-alpha-transgenic mice.
J Gastrointest Surg 2008;12(1):30-7
85. Huynh H, Soo KC, Chow PK, Tran E.
Targeted inhibition of the extracellular
signal-regulated kinase kinase pathway
with AZD6244 (ARRY-142886) in the
treatment of hepatocellular carcinoma.
Mol Cancer Ther 2007;6(1):138-46
86. Llovet JM, Ricci S, Mazzaferro V, et al.
Sorafenib in advanced hepatocellular
carcinoma. N Engl J Med
2008;359(4):378-90
87. Cheng AL, Kang YK, Chen Z, et al.
Efficacy and safety of sorafenib in patients
in the Asia-Pacific region with advanced
hepatocellular carcinoma: a phase III
randomised, double-blind,
placebo-controlled trial. Lancet Oncol
2009;10(1):25-34
88. Poon RT, Ng IO, Lau C, et al. Tumor
microvessel density as a predictor of
recurrence after resection of hepatocellular
carcinoma: a prospective study.
J Clin Oncol 2002;20(7):1775-85
89. Yao DF, Wu XH, Zhu Y, et al.
Quantitative analysis of vascular
endothelial growth factor, microvascular
density and their clinicopathologic features
in human hepatocellular carcinoma.
Hepatobiliary Pancreat Dis Int
2005;4(2):220-6
90. Yoshiji H, Kuriyama S, Yoshii J, et al.
Vascular endothelial growth factor tightly
regulates in vivo development of murine
hepatocellular carcinoma cells. Hepatology
1998;28(6):1489-96
91. Park YN, Kim YB, Yang KM, Park C.
Increased expression of vascular endothelial
growth factor and angiogenesis in the early
stage of multistep hepatocarcinogenesis.
Arch Pathol Lab Med 2000;124(7):1061-5
92. Zhao J, Hu J, Cai J, et al. Vascular
endothelial growth factor expression in
serum of patients with hepatocellular
carcinoma. Chin Med J (Engl)
2003;116(5):772-6
93. Huang GW, Yang LY, Lu WQ. Expression
of hypoxia-inducible factor 1alpha and
vascular endothelial growth factor in
hepatocellular carcinoma: impact on
neovascularization and survival.
World J Gastroenterol
2005;11(11):1705-8
94. Imura S, Miyake H, Izumi K, et al.
Correlation of vascular endothelial cell
proliferation with microvessel density and
expression of vascular endothelial growth
factor and basic fibroblast growth factor in
hepatocellular carcinoma. J Med Invest
2004;51(3-4):202-9
95. Moon WS, Rhyu KH, Kang MJ, et al.
Overexpression of VEGF and angiopoietin
2: a key to high vascularity of
hepatocellular carcinoma? Mod Pathol
2003;16(6):552-7
96. Liu Y, Poon RT, Li Q, et al. Both
antiangiogenesis- and
angiogenesis-independent effects are
responsible for hepatocellular carcinoma
growth arrest by tyrosine kinase inhibitor
PTK787/ZK222584. Cancer Res
2005;65(9):3691-9
97. Finn RS, Bentley G, Britten CD, et al.
Targeting vascular endothelial growth
factor with the monoclonal antibody
bevacizumab inhibits human
hepatocellular carcinoma cells growing in
an orthotopic mouse model. Liver Int
2009;29(2):284-90
98. Siegel AB, Cohen EI, Ocean A, et al.
Phase II trial evaluating the clinical and
biologic effects of bevacizumab in
unresectable hepatocellular carcinoma.
J Clin Oncol 2008;26(18):2992-8
99. Boige V, Baey C, Dromain C, et al.
Circulating endothelial cells (CEC) and
angiogenic proteins monitoring in patients
(pts) with advanced hepatocellular
carcinoma (HCC) treated with
bevacizumab. J Clin Oncol
(Meeting Abstracts) 2009;27(15S):4597
100. Thomas MB, Morris JS, Chadha R, et al.
Phase II trial of the combination of
Wysocki
Expert Opin. Investig. Drugs (2010) 19(2) 273
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y N
yu M
edic
al C
ente
r on
05/
13/1
3Fo
r pe
rson
al u
se o
nly.

bevacizumab and erlotinib in patients who
have advanced hepatocellular carcinoma.
J Clin Oncol 2009;27(6):843-50
101. Zhu AX, Blaszkowsky LS, Ryan DP, et al.
Phase II study of gemcitabine and
oxaliplatin in combination with
bevacizumab in patients with advanced
hepatocellular carcinoma. J Clin Oncol
2006;24(12):1898-903
102. Sun W, Haller DG, Mykulowycz K, et al.
Combination of capecitabine, oxaliplatin
with bevacizumab in treatment of
advanced hepatocellular carcinoma
(HCC): a phase II study. J Clin Oncol
(Meeting Abstracts)
2007;25(18 Suppl):4574
103. Hsu C, Yang T, Toh H, et al.
Modified-dose capecitabine + bevacizumab
for the treatment of advanced/metastatic
hepatocellular carcinoma (HCC): a
phase II, single-arm study. J Clin Oncol
(Meeting Abstracts)
2007;25(18 Suppl):15190
104. Wysocki PJ, Zolnierek J, Szczylik C,
Mackiewicz A. Targeted therapy of renal
cell cancer. Curr Opin Investig Drugs
2008;9(6):570-5
105. Koeberle D, Montemurro M, Samaras P,
et al. Continuous sunitinib treatment in
patients with unresectable hepatocellular
carcinoma (HCC): a multicenter phase II
trial (SAKK 77/06 and SASL 23). J Clin
Oncol (Meeting Abstracts)
2009;27(15S):4591
106. Zhu AX, Sahani DV, Duda DG, et al.
Efficacy, safety, and potential biomarkers
of sunitinib monotherapy in advanced
hepatocellular carcinoma: a phase II study.
J Clin Oncol 2009;27(18):3027-35
107. Raoul JL, Finn RS, Kang YK, et al. An
open-label phase II study of first- and
second-line treatment with brivanib in
patients with hepatocellular carcinoma
(HCC). J Clin Oncol (Meeting Abstracts)
2009;27(15S):4577
108. Abou-Alfa GK, Schwartz L, Ricci S, et al.
Phase II study of sorafenib in patients with
advanced hepatocellular carcinoma.
J Clin Oncol 2006;24(26):4293-300
109. Strebel BM, Dufour JF. Combined
approach to hepatocellular carcinoma: a
new treatment concept for nonresectable
disease. Expert Rev Anticancer Ther
2008;8(11):1743-9
AffiliationPiotr Jan Wysocki MD PhD
Chair of Medical Biotechnology,
University of Medical Sciences,
Department of Chemotherapy,
Greater Poland Cancer Center,
ul. Garbary 15, Poznan 61-866, Poland
Tel: +48 61 885 0620; Fax: +48 61 885 0694;
E-mail: [email protected]
Targeted therapy of hepatocellular cancer
274 Expert Opin. Investig. Drugs (2010) 19(2)
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y N
yu M
edic
al C
ente
r on
05/
13/1
3Fo
r pe
rson
al u
se o
nly.





























