Embed Size (px)
Citation preview

Annu. Rev. Phys. Chern. 1993. 44: 145-72 Copyright © 1993 by Annual Reviews Inc. All rights reserved
THE ADIABATIC THEORY
OF H�EAVY-LIGHT-HEAVY
CHEJMICAL REACTIONS*
Rex T. Skodje
Department of Chemistry and Biochemistry, Joint Institute for Laboratory Astrophysics, and the Program in Applied Mathematics, University of Colorado, Boulder, Colorado 80309-0215
KEY WORDS: reaction dynamics, chaos, semiclassical dynamics, resonances
INTRODUCTION Many important chemical reactions consist of a light atom exchange between two heavy atoms or groups of atoms. Using the convention of Polanyi et al (1), we refer to such reactions as Heavy-Light-Heavy (HLH). Usually, but not always, HLH systems are hydrogen-atom transfer reactions. Because HLH reactions are of great practical importance as well as chemically interesting, the dynamics of these systems have been the object of much research (1-48). Some of the issues that have attracted the most attention include: (a) the existence of reactive resonances and, possibly, vibrationally bound states; (b) transition state recrossing effects and oscillating collinear reaction probabilities; (c) the role of light atom tunneling; and (d) the nature of transition state spectroscopy involving electron photodetachment from bound negative ions. We can single out the experimental photo detachment work of Neumark (29, 31, 39) and the theoretical quantum scattering calculations of Schatz (35, 36) for generating new excitement about this class of reactions.
From the standpoint of fundamental dynamical theory, what makes this class of reactions so special are the intrinsic timescales. Under normal circumstances, the light atom will tend to move much faster than the
* In this chapter, matrices are designated by script S, Y.
145 0066-426X/93/1101--o145$05.00
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

146 SKODJE
heavy groups. Thus, we expect that the exchange time, 'exch, which is a characteristic time for the light atom to move across the saddlepoint region, will be significantly shorter than the collision time, 'coli, which is governed by the heavy atom translational motion, i.e.
1.
From the timescale relations, one might infer either great simplicity or great complexity for the reaction dynamics. On the side of simplicity, a separation of times cales may reasonably be expected to result in vibrational adiabaticity. In such a case, the fast degrees of freedom will conserve quantum number (or classical action) and may be eliminated, leaving a lower-dimensional dynamical problem in the slow variables. Such adiabatic separability led Miller (18) to refer to the collinear system I + HI -+
IH + I as the "World's Simplest Model Reaction." On the side of complexity, we might envision that during a slow collision, the light atom may execute complicated and intricate dynamics before the heavy atoms have time to move apart. Indeed, dynamical simulations have revealed that the picture of a single exchange across the barrier is incorrect for many HLH reactions, and instead the light atom may chatter back and forth many times (9). Furthermore, three-dimensional classical trajectory simulations have shown that a HL diatom can actually undergo one or more complete rotations within the collision complex (44).
We take the middle ground between these two points of view. Many HLH reactions can be adiabatically reduced with good accuracy, although the theory is not quite as straightforward as first imagined. However, even the simplified adiabatic dynamics remain deliciously complex, showing multiple exchanges, internal rotations, and such. The challenge to the theoretician is to simplify the reaction dynamics enough to understand them but not so much as to approximate important effects out of existence.
In this article, we review recent efforts to understand the dynamics of HLH reactions (28, 30, 44-50). The goal of this research has been to properly characterize the role of separation of time scales in the reaction dynamics, and thereby formulate a simple and intuitive theoretical picture of thc reaction proccss. Thc methods we have developed have been numerically tested for the I + HI reaction on the Manz-R6melt LEPS A potential surface (7). The first part of the review discusses our treatment of collinear HLH reactions. In terms of this simple restricted geometry case, we expose an essential contradiction in the adiabatic approach. An analogy to a curve crossing shows that there must exist a critical configuration in the course of the reaction where the adiabatic theory fails. To describe the reaction dynamics correctly, we reformulate the adiabatic theory of reactions to resolve the difficulty, and a new picture for the dynamics emerges. We then discuss the three-dimensional (3D) reaction dynamics in terms of an
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

HLH CHEMICAL REACTIONS 147 adiabatic picture that eliminates one fast variable. The reaction process is described as a coherent superposition of two atom-rotor scattering problems. We then present an analysis of the time-independent and timedependent quantum scattering process, the classical reaction dynamics, and the nature of the trapped resonance states. Because 3D reactions generally exhibit much stronger chaotic scattering than do their collinear analogs, the correction factors in the reformulated adiabatic theory can often be eliminated in favor of a simpler statistical treatment.
COLLINEAR REACTION DYNAMICS When reliable methods first emerged to solve the close-coupling equations for collinear reactive scattering, the HLH mass combination was found to be a most problematic case. However, it was soon discovered that the main source of difficulty was the choice of natural collision coordinates (or other closely related coordinate systems), which were inappropriate for HLH reactions because of the extremely high curvature of the reaction path. Several groups (5, 6, 9) proposed the use of hyperspherical coordinates, which proved much more satisfactory. For a collinear A + Be reaction, the hypersphcrical system reduces to the polar coordinates
�A-BC 2 2 P = -- RA-BC +rBC, fiBC 2.
where (RA-BC, rBe) are the a-channel Jacobi coordinates. The Hamiltonian is then wri tlCen as
3.
where fi == IIBC. In this representation, the radius, p, is the scattering (or translational) coordinate while the angle 8 describes the vibrational motion of either the reactant or product diatom. Accurate reaction probabilities for collinear HLH systems could be obtained quite efficiently by using hyperspherical coordinates.
Babamov & Marcus (9) point out that a vibrationally adiabatic treatment of collinear HLH reactions could be formulated in hyperspherical coordinates. The radial motion, which primarily describes heavy atom translation, was taken as slow while the vibrational motion of the angle was fast. In the quantum vibrationally adiabatic picture, the quantum number n of the 8-motion is conserved as the p-coordinate slowly evolves. When classical mechanics is used, the vibrational action is assumed to be a constant of motion. If p is held constant, the potential for the 8-vibration,
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

148 SKODJE
V(p = const, (J), generally takes the form of a double well with the reactant and product channels corresponding to the individual wells. Reaction occurs when there is a net transfer from the reactant well to the product well.
The quantum mechanical vibrationally adiabatic model pennits a separable solution of the two-dimensional Schrodinger equation. Thus, scattering wavefunctions may bc written in the product form
'P± (p, (J) = 'P,;t (p)<Pn«(J; p). 4.
The translational motion is governed by
5.
with the appropriate scattering boundary conditions, and vibrational dynamics solves
6.
If we ignore the nonadiabatic coupling, then the Y-matrix is diagonal in the adiabatic basis and can be represented as Y" = e2i�n where �n represents the phase shifts. For a symmetric HLH reaction, the eigenstates in the double well Schrodinger equation, Equation 6, occur in split pairs of even and odd parity. These eigenstates are de localized between the two wells. To form reactant and product states localized in one of the two wells, we take the linear combinations Xr,p = (<Pn ± <Pn+ 1)/21/2, where n is even. The reactive Y-matrix then becomes an analogous superposition (5),
7.
and the reaction probability becomes simply, PR = sin2 (�n_ �n+I). Since the phase shift difference, �n - (n+ I, tends to increase monotonically with translational energy, the reaction probability is predicted to oscillate strongly as the collision velocities increase. This prediction, and the accuracy of Equation 7, have been borne out by quantum simulations for a number of collinear symmetric HLH reactions.
In terms of classical mechanics, the description of a collinear HLH reaction is elegantly represented as a time-dependent double-well problem (9, 1 8, 30). As illustrated in Figure 1, the reaction begins with the light atom vibrating in the reactant side of the double well. As the heavy atoms approach, the barrier slowly falls but the vibrational action is conserved. The action is computed from the definite integral � Po dfJ/2n, where
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

w
w
w
HLH CHEMICAL REACTIONS 149
00
Figure 1 Time-dependent double-well representation for a HLH reaction and its relation to separatrix crossing. The motion in the double-well potential represents the fast vibration of the light atom, while the slow motion of the heavy atoms gives rise to the time-dependence of the potential. On the left, the reaction proceeds when the trajectory untraps from the entrance chan.nel well as the barrier slowly lowers and then retraps into an exit channel well as the barrier rises again. On the right, the phase space description is shown where the figureeight separatrix slowly shrinks and then expands with time.
Po = ± j2J1(EVib - V(p, 8)), and the instantaneous double-well potential, V(p, 8), is evaluated assuming p is held constant. For a sufficiently energetic collision, the trajectory will eventually pass over the top of the barrier and un trap from the reactant well. During the middle of the collision process, the light atom chatters back and forth between the two heavy atoms. It is not uncommon for the light atom to cross the barrier a dozen or more times. As the heavy atoms move apart during the final phase of the reaction, the barrier rises again and eventually the light atom is retrapped in either the reactant or product side welL The reaction probability is the fraction of trajectories that end up trapped in the product side well.
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

150 SKODJE
Classical adiabatic theory also predicts that the reaction probability will oscillate versus energy, and provides an intuitive explanation for the effect. In Figure I, we see that a trajectory will be reactive (nonreactive) if it crosses the barrier an odd (even) number of times during the chattering part of the collision ( 17, 18, 30). As the translational energy changes, the net duration of the time-dependent double-well process in Figure I also changes. Thus, the average number of barrier crossings of a typical trajectory increases (or decreases) monotonically with translational energy. The reaction probability oscillates as the mean number of barrier crossings goes back and forth between even and odd values. Thus, the oscillation in P R is properly termed an exchange oscillation, because it is physically related to the process of light-atom exchange.
There are obvious limitations to this simple adiabatic model. Some of the issues left open include: (a) Why does the adiabatic model succeed for some HLH reactions, such as I + HI, but fail for others, such as H + MuH? (b) How light does the exchanged atom have to be for adiabatic approximation to apply? (c) Under what circumstances should one switch to a description based on natural collision coordinates? (d) How can one describe complex formation and vibrational transitions? A not very illuminating resolution to such questions involves implementing a full nOlladiabatic computation. Although such a calculation can in principle be fully converged, from just a basis set calculation it is difficult to obtain physical insight or a simple answer to the above questions. Instead, our approach has been to focus on the classical mechanics and proceed deeply into formal adiabatic theory and nonlinear dynamics to achieve a physical understanding of adiabatic breakdown. We have developed rigorous error criteria that indicate when the adiabatic approach will fail. Furthermore, explicit expressions have been obtained for the vibrational product distributions and for the probability of complex formation.
Adiabatic Theory
Formal adiabatic in variance theory is well established in the mathematics and physics literature (51-56). As in ordinary perturbation theory, one introduces an ordering parameter, A, which is assumed to be small. In adiabatic theory, A reflects the timescale ratio, e.g. A � 'cxch/'coJl' The dynamics is then expanded in an asymptotic series in A. Effectively, one produces a sequence of canonical transformations wherein the generalized action is conserved to successively higher order in A. Thus, the actionangle variables to order n can be written as
n
I" = I J",kh, and 8. k�O
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

HLH CHEMICAL REACTIONS 151
where the tJlme variation of the nearly conserved action is in = O(An+ I) .
The explicit canonical transformations can be obtained by using only a knowledge of the classical Hamiltonian, the generating function for transformation to zero-order action-angle variables, and their derivatives. However, beyond the lowest few orders, the expressions become very complex and require the usc of symbolic manipulators. The usual adiabatic approximation in the sense of Ehrenfest is represented by the zero-order term of this expansion. The breakdown in conservation of the zero-order action, 1== [0, can be estimated by the size of the correction terms, and often, by just the first term, All' One would naturally suspect, therefore, that the accuracy of adiabatic theory for a given HLH reaction could be gauged by the size of such correction terms. However, a fundamental contradiction in adiabatic: theory appears when it is applied to HLH reactions, and the A-expansion of the dynamics fails to exist beyond the first order as a result of unremovable infinities.
Though the timescale ratio for many collinear HLH systems is near 10: 1, the formai adiabatic expansion is ill-defined because separatrix crossings take place when the trajectory traps and untraps in the double well. In the phasl� space diagrams of Figure I, the separatrix is the figure-eight shaped curve that forms a dividing surface between trapped motion in the reactant channel well (region a), trapped motion in the product wcll (rcgion b), and untrapped chattering motion (region c). From a phase-space perspective, the reaction corresponds to a sequence of two separatrix crossings that occur as the separatrix shrinks and expands. The breakdown of adiabatic theory at a separatrix crossings is easy to understand. As the trajectory traps (or un traps) from the channel well, there is an instant, to when the vibrational period goes to infinity. At to a trajectory propagating in the static double-well potential would be caught forever on top of the barrier. This implies that near to no matter how slowly the heavy atoms are moving., the vibration can never be fast compared with the translation motion. In fact, Kruskal's theorem (53) and other foundational theorems of adiabatic theory explicitly exclude cases in which the frequency of the fast variable, OJ = 8Hj8I, passes through zero. Otherwise, the separatrix crossing would lead to divergences, because OJ typically occurs in the denominators of the higher correction terms.
Uniform Adiabatic· Theory
To model HLH reactions usefully, we must fundamentally extend adiabatic theory so that separatrix crossings may be treated. We have developed such a procedure, which allows the dynamics to be modeled through the separatrix crossing region (30, 49, 50, 56). In terms of this uniform adiabatic theory we can compute reaction probabilities,
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.
152 SKODJE
vibrational inelasticities, complex formation probabilities, and the structure of reactivity bands. The details of this method are quite involved and we refer the interested reader to the original papers. Here, I review the motivation and one of the main results.
As discussed by Cary et al (55), a generic slow separatrix crossing may be divided into a sequence of three intervals: before, during, and after. First, substantially before the crossing occurs, the dynamics are adiabatic and the asymptotic expansions are nondivergent. Second, close to the crossing point is a near separatrix region where the adiabatic theory breaks down badly. Finally, well after the separatrix crossing, the dynamics are adiabatic once again. The idea of the method (54-56) is to develop a third (nonadiabatic) asymptotic expansion for the dynamics in the near separatrix region, which is then uniformly matched to the adiabatic expansions known to be valid before and after the crossing. The final result is a map, akin to a classical curve crossing formula, which permits us to follow the dynamics through the separatrix crossing. Thus, the structure of the theory is similar to uniform-WKB theory, where twon-asymptotic expansions of the wavefunction, defined on either side of a classical turning point, are matched together with an Airy function that is an accurate
solution near the turning point. Strong nonadiabatic couplings exist at a separatrix crossing, and the
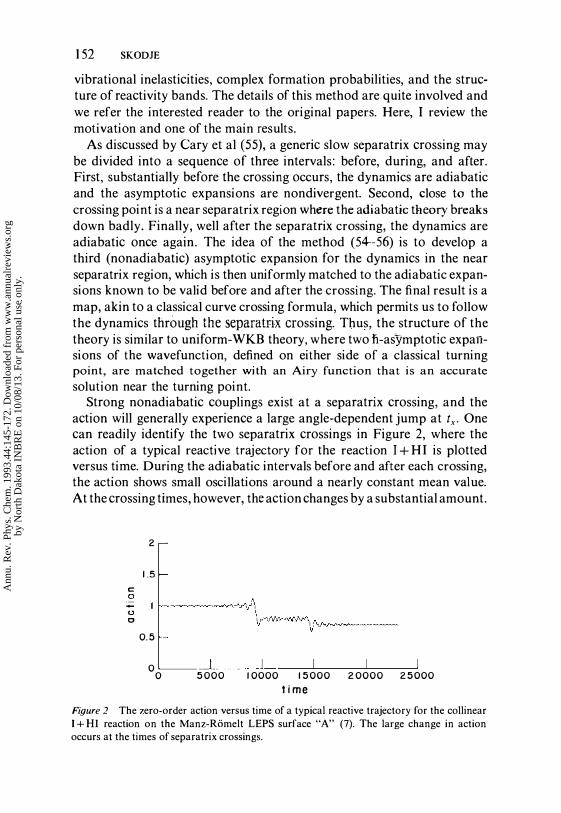
action will generally experience a large angle-dependent jump at tx• One can readily identify the two separatrix crossings in Figure 2, where the action of a typical reactive trajectory for the reaction I + HI is plotted versus time. During the adiabatic intervals before and after each crossing, the action shows small oscillations around a nearly constant mean value. At the crossing times, however, the action changes by a substantial amount.
2
1.5 co 0
+-u 0
0.5
0 0 5000 10000 15000 20000 25000 time
Figure 2 The zero-order action versus time of a typical reactive trajectory for the collinear I + HI reaction on the Manz-Riimelt LEPS surface "A" (7). The large change in action occurs at the times of separatrix crossings.
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

HLH CHEMICAL REACTIONS 153
The first order action change, 111, may be analytically computed (mapped) from the initial action-angle variable (Ii, ((Ji) by
9.
where (Px is the initial phase for the singular critical orbit that exactly hits the top of the barrier, and � is the nonadiabaticity parameter defined as � = UOx (54-56). The magnitude of the action change is gauged by �, . which is the ratio of t, the rate of change of the separatrix action at t x, to the frequency of the inverted barrier, Ox' Thus, one achieves a large action jump when the barrier between the wells is broad or when the rate of change of the well depths is large. In terms of the ordering parameter A, � is O(A).
To compute the reaction probability for collinear HLH reactions, we must follow the dynamics through the sequence of two separatrix crossings
occurring at tx and t,," In terms of the phase space regions shown in Figure 1, the trajectories follow the route a ...... c ...... b when reactive and a -+ c -+ a when nonreactive. The action variable experiences a jump at each crossing. The key to determining reactivity in our theory, however, is the behavior of the conjugate angle variable, ((J. Simply put, one can translate the reactivity criterion of an odd number of barrier crossings into a criterion based on the total angle change accrued during the propagation in region c. If the total angle change satisfies the relation 2n � 11((J + ((Ji � n, mod 2n, then the trajectory has experienced an odd number of crossings and is reactive. When � is not equal to zero, the angle change cannot be computed simply by integrating the zero-order frequency, J W(Ii) dt, which is incorrect even to the lowest order. Due to anharmonicity, the action jump occurring at t" modifies the frequency and leads to a corrected angle change
10.
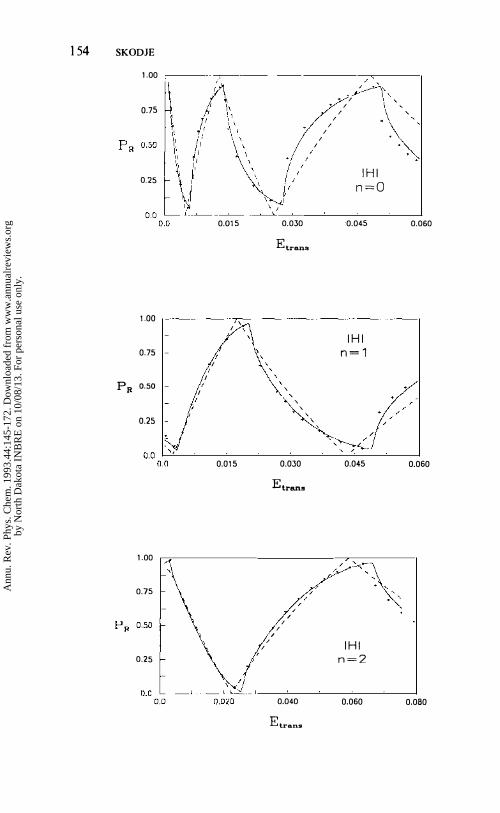
Implementing Equation 10 to order 0(,1,), accurate classical reaction probabilities can be computed without the need for a trajectory calculation. Figure 3 shows the exact quasiclassical reaction probability for I + HI along with the prediction of Equation 10. Good agreement is obtained. The vibrational inelasticity, the complex formation probability, and the structure ofilhe classical reactivity bands have also been computed to high accuracy for this reaction (30, 49, 50).
After a deeper analysis (50), we may define a dimensionless phase mixing parameter,
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.
154 SKODJE
1.00
0.75
0.25
1.00
0.75
PR 0.50
0.25
1.00
0.75
0.25
,
0.Q15
I ,
, I
I I
I I
I I
0.015
0.030
Etrana
,
/ I
\ ,
, ,
, ,
\
0.030
Elrans
/
/ I
/
0.040
/ /
Elrans
I /
\
/ I
+ / /
/ /
IHI n=O
0.045
IHI n=1
0.045
IHI n=2
0.060
',
0.060
0.060
0.080
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

_ it' iJw(IJ 13 - oJ dt�,
Ix I
HLH CHEMICAL REACTIONS 155
11.
which proves invaluable in achieving a physical understanding of the reaction dynamics. For a quasiclassical ensemble in which all the trajectories start with action l;, 13 is interpreted as follows: when the ensemble crosses the separatrix at to the nonadiabatic coupling causes the action to become continuously distributed over an interval of rough· size �, i.e.
<IMI) � Ii;!. During the region c propagation from Ix to lx" the anharmonicity, (lw/al;, causes the ensemble to spread out in phase. 13 measures the extent of this spreading for each energy. 13 is a quantification of the transient chaos associated with separatrix crossing. In the chemical reaction dynamics, if 13 is large (compared to 1) then the ensemble randomizes in region c and the probability of retrapping in either reactant or product well should become roughly equal. Therefore, when 13 is large the regular oscillation of the reaction probability is suppressed, and when 13 is small the oscillation should be large. For the 1+ I:II reaction we have 13 � 0.1, which is consistent with the large observed oscillation. The simple picture is that the oscillation of P R is a by-product of near-integrability in the scattering dynamics and this oscillation can be liquidated by strong chaotic scattering quantified by 13.
Thus, the applicability of adiabatic theory to HLH reactions and the extent of non adiabatic effects can be gauged by the magnitude of three fundamental parameters: A, �, and 13. The separation of timescales is reflected by A. The most usual notion of adiabaticity requires A to be small .enough that the higher perturbation corrections are not important. When separatrix crossing occurs, the correction terms diverge even when A is small, and we need to focus our attention on � and 13. Vibrational inelasticity will be weak when � is small (compared to b) and will be strong when � is large. Finally, the nonadiabatic coupling will significantly affect the phase change and thus the reaction probability when 13 � I, which quantifies the chaotic scattering. Even when � and 13 are large, the uniform adiabatic theory can give quite accurate results.
THREE-DIMENSIONAL REACTIONS The description of HLH reaction dynamics in three dimensions presents a challenging problem. Including rotational degrees offreedom dramatically
Figure 3 The quasiclassical reaction probability for the I + HI (n) reaction versus translational energy, in Hartree. The + 's are the exact results of a simulation, the solid line is the prediction of uniforn1 adiabatic theory, and the dashed line is the primitive adiabatic theory obtained by s.;tting � = O.
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

156 SKODJE
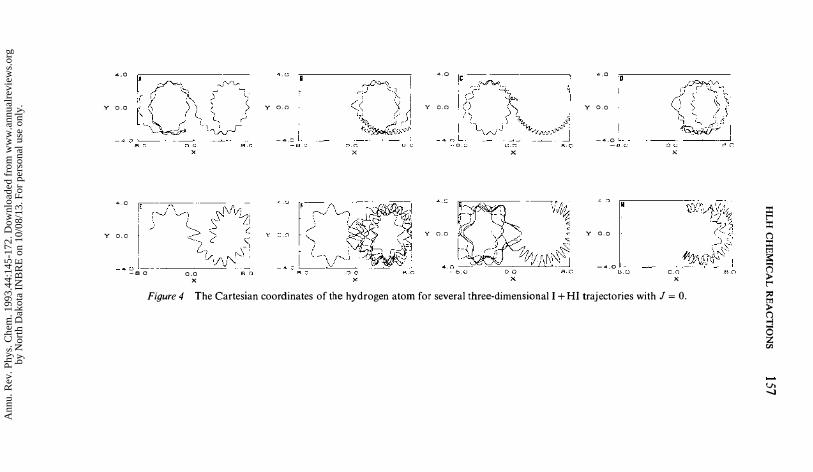
alters the simple collinear dynamics described above. Some of the new effects that emerge in three dimensions are illustrated in Figure 4, where several typical trajectories are shown for our prototype system, I + HI, for J = 0. The Cartesian coordinates (X, Y) of the hydrogen atom are plotted while the iodine atoms (not shown) move slowly along the X-axis. The center of mass is fixed at (0, 0). The IH diatom often undergoes one or more complete rotations within the collision complex, a behavior referred to as rotational chattering. We can also see the influence of effective angular potential barriers, which can reflect back an IH diatom as it attempts to rotate towards a collinear configuration. Such new dynamic effects cause the prominent exchange oscillations in the collinear reaction probability to be absent or greatly suppressed in three dimensions. Figure 5 shows the exact quasiclassical reaction probability for several initial jstates of I + HI that do not show the exchange oscillations.
Our view is that many bimolecular HLH reactions are primarily driven by rotational dynamics. The essential precursor to reaction is the establishment of the proper orientation for hydrogen-atom exchange to occur (generally near collinear), and the subsequent exchange process itself is usually quite rapid. Thus, studying the rotational scattering problem is necessary to learn precisely how the system rotates into the proper orientation. What sets HLH problems apart from other familiar three-atom exchange reactions (such as H + H2) are the intrinsic timescales. The relevant diatomic rotational period tends to be roughly the same as the collision time, while the diatomic vibrational period (and hence the exchange duration) is much shorter. This implies that considerable rotational motion occurs during the collision itself, which leads, for instance, to the rotational chattering seen in Figure 4. Since the reaction coordinate is often largely rotational in character, conventional treatments based on. the collinear reaction dynamics or a tight variational transition state may miss the essential dynamics. Instead, we have constructed a model of the reaction dynamics based on the rotational-scattering problem as the zero-order starting point. This model features the critical orientational effects of the three dimensional reaction.
Our model employs a separation of timescales argument to transform the reaction dynamics into the coherent sum of two atom-rotor scattering problems. Despite the impressive success of the adiabatic model for collinear systems, the extension of adiabatic theory to the three-dimensional reaction dynamics was initially beset with some ambiguity. Were the rotational degrees of freedom to enter as fast or slow variables, and in what coordinate system? Manz et al ( 15) and Kubach (34) suggested that rotational motion should be fast because it is primarily light-atom motion. According to this view, a HLH reaction was similar to a charge-transfer
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.
4.0
Y 0.0
4.0
y 0.0
-4,0 l..1 __ � __ �_::""':�_-'
-8 a 0.0 x
y 0.0
_ 4.::: l..1 .,--_�_-:-__
�
_--:-..J
-S. ::: 0.0 S.C x
4.0
y 0.0
x
y 0.0
4.0
y 0.0 «»1 - 04 -:::. 8l..1 ."'O-�---:
O
'-.
O
�-�--,:-,
x
y 0.0
_ 4.0 l..1 -=-___ -:-�_...::..:'-_-..J
- B . O 0.0 X
Figure 4 The Cartesian coordinates of the hydrogen atom for several three-dimensional I + HI trajectories with J = O.
:I: t"' :I: (") :I: �
� > t"' ::0 til > (") g Z rJ)
VI -...J
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.
158 SKODJE
0.6
0.0
0.0 0.01
Et.re.n.s
n=O j=2,lO
0.02
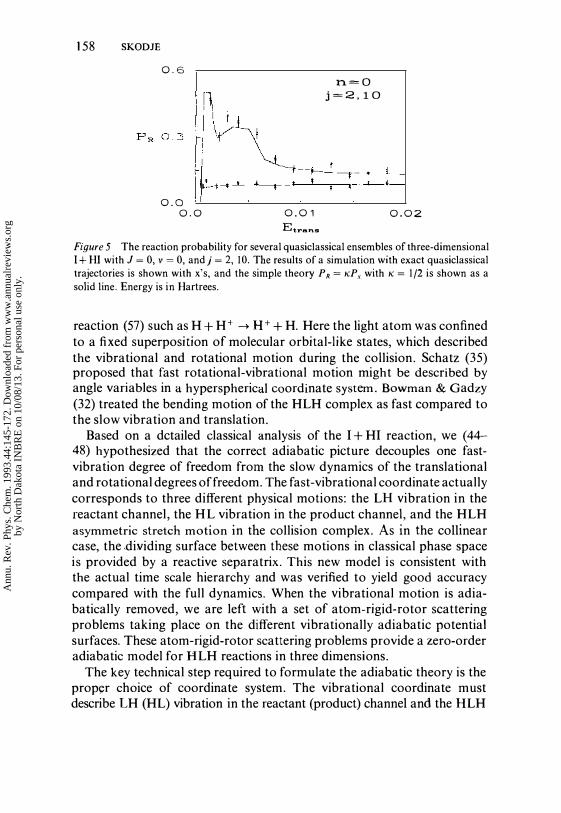
Figure 5 The reaction probability for several quasic1assical ensembles of three-dimensional I + HI with J = 0, v = 0, and j = 2, 10. The results of a simulation with exact quasiclassical trajectories is shown with x's, and the simple theory PR = KP, with K = 1/2 is shown as a
solid line. Energy is in Hartrees.
reaction (57) such as H + H+ � H+ + H. Here the light atom was confined to a fixed superposition of molecular orbital-like states, which described the vibrational and rotational motion during the collision. Schatz (35) proposed that fast rotational-vibrational motion might be described by angle variables in a hyperspherical coordinate system. Bowman & Gadzy (32) treated the bending motion of the HLH complex as fast compared to the slow vibration and translation .
Based on a dctailed classical analysis of the I + HI reaction, we (44-48) hypothesized that the correct adiabatic picture decouples one fastvibration degree of freedom from the slow dynamics of the translational and rotational degrees offreedom. The fast-vibrational coordinate actually corresponds to three different physical motions: the LH vibration in the reactant channel, the HL vibration in the product channel, and the HLH asymmetric stretch motion in the collision complex. As in the collinear case, the .dividing surface between these motions in classical phase space is provided by a reactive separatrix. This new model is consistent with the actual time scale hierarchy and was verified to yield good accuracy compared with the full dynamics. When the vibrational motion is adiabatically removed, we are left with a set of atom-rigid-rotor scattering problems taking place on the different vibration ally adiabatic potential surfaces. These atom-rigid-rotor scattering problems provide a zero-order adiabatic model for HLH reactions in three dimensions.
The key technical step required to formulate the adiabatic theory is the proper choice of coordinate system. The vibrational coordinate must describe LH (HL) vibration in the reactant (product) channel and the HLH
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

HLH CHEMICAL REACTIONS 159
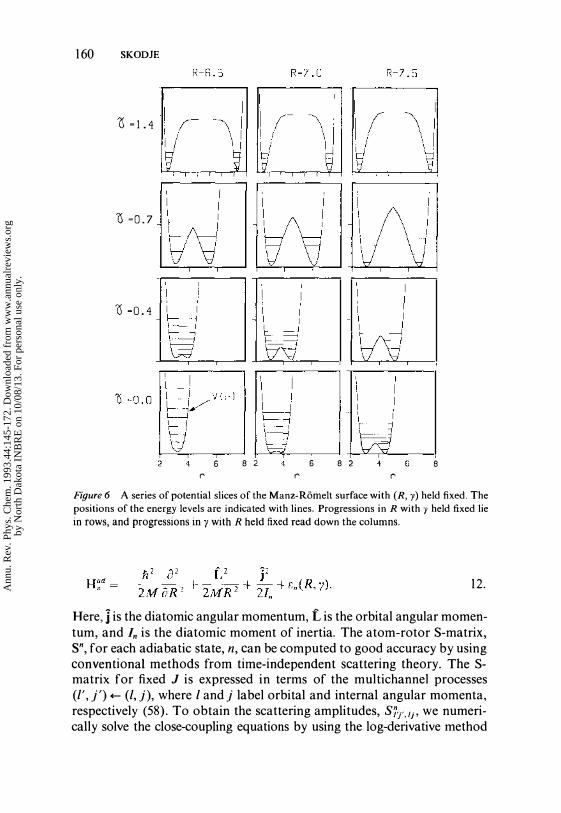
asymmetric stretch for the collision complex. We find that a composite set of usual a- and j3-channel Jacobi coordinates, (R,n ra) with (J equal to a or 13, is ideally suited to the task. For symmetric reactions, we match the coordinate systems when the light atom is midway between the heavy atoms. Thus, when the light atom is closer to the left heavy atom we employ the a-channel coordinates, and when it is closer to the right heavy atom we switch to the j3-channel coordinates. For an asymmetric reaction, the matching should be made along the potential ridge separating reactants from products. The potential is a function of the three scalar coordinates, (Ra, ra, Ya), where Ra = IIRall, ra = Ilrall, and Raora = Raracos Ya . We immediately see that ra correctly describes the fast vibration in all three dynamicallly distinct regions. In the reactant (product) channel, ra = ra (r 13) represe:nts diatomic vibration. In the collision complex, the asymmetric stretch vibration is well modeled by a composite coordinate, ra = r. when rp> r., and ra = rp when r. > rp. (For simplicity in what follows, we shall use the unsubscripted coordinates (R, r, 1'), with the correct subscript implicit.
When the slow coordinates (R, y) are held fixed, the Born-Oppenheimer potential function versus r forms a double well. As in the collinear problem, the reactant and product channels correspond to the right and left wells, respectively. As seen in Figure 6 for I + HI, the exact shape of the double well is a strong function of the coordinates (R, y). The position of the quantum vibrational levels in the static double wells is used to determine the adiabatic potential surfaces, en(R, y), which are crucial in understanding HLH dynamics in three dimensions. Portions of the adiabatic surfaces are shown in Figures 8, 9, II, and 13 (below). These potentials have a strong repulsive core at small R and show pronounced anisotropy. For all n except n = 1, en(R, 1') exhibits a substantial adiabatic well centered at the collinear geometry, y = O. The adiabatic wells prove deep enough to hold many reactive resonances. The wells are surrounded by angular barriers that help to confine the reactive resonances and can repel a diatom attempting to rotate into a collinear configuration. We have explored in detail the adiabatic dynamics for the I + HI reaction by carrying out both classical and quantum computations of the reaction probabilities and computing many of the reactive resonances. We now briefly report some of the main findings.
Quantum Reactive Scattering
When the (ast vibrational degree offreedom is adiabatically decoupled, the reactive collision dynamics reduces to a set of atom-rigid-rotor scattering systems described by the effective Hamiltonians (46-48),
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.
160 SKODJE
O�1.4
o �O.7
'0 �O.4
6 8 2 r
6 8 2 6 8 r r
Figure 6 A series of potential slices of the Manz-Romelt surface with (R, y) held fixed. The positions of the energy levels are indicated with lines. Progressions in R with y held fixed lie in rows, and progressions in y with R held fixed read down the columns.
12.
Here, j is the diatomic angular momentum, t is the orbital angular momentum, and In is the diatomic moment of inertia. The atom-rotor S-matrix, sn, for each adiabatic state, n, can be computed to good accuracy by using conventional methods from time-independent scattering theory. The Smatrix for fixed J is expressed in terms of the multichannel processes (I', }') +- (I, i), where I and} label orbital and internal angular momenta, respectively (58). To obtain the scattering amplitudes, S"j',lj, we numerically solve the close-coupling equations by using the log-derivative method
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

HLH CHEMICAL REACTIONS 161
in a quasi adiabatic basis (59). Such atom-rigid-rotor calculations are, of course, much less time consuming than full reactive scattering computations.
To compute the reaction probability for a symmetric reaction, recall that the double-well eigenstates for the fast vibration occur in doublets of even and odd parity that are delocalized between the two wells. The reactant and product channel eigenstates, which are localized in one or the other well, are given by linear combinations Xr,p = (cjJ,,± cjJn+ 1)/21/2, where cjJn(r; R, y) are the Qouble-well eigenstates at fixed (R, y), The choice of n is dictated by the initial vibrational state desired. The asymptotic vibrational state LH(v) is correlated to the pair of adiabatic levels, n = 2v and n = 2v + I, Since the even and odd adiabatic scattering states are uncoupled due to symmetry, the reactive S-matrix may be obtained by the corresponding linear combination, SR = (sn - sn+ 1)/2. The total reaction probability from the initial channel (/, j) is
PR = L IYS�j',lj -S�/)JI2, 13, jT
where the sum is over all open final channels. The adiabatic model (46-48) yields accurate reaction probabilities when
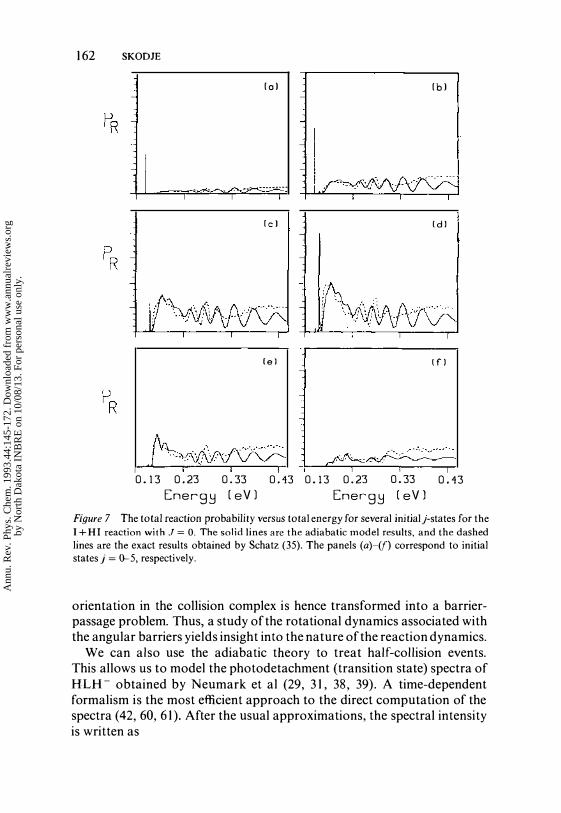
compared to available exact quantum results for I + HI. Figure 7 shows the adiabatic versus exact quantum reaction probabilities of Schatz (35) for a set of initial rotor states with J = O. The agreement is quite good up to about 0,3 eV. At higher energies, Schatz reports loss of unitarity in the full scattering calculation, in which case the comparison is no longer useful. The nonadiabatic effects associated with separatrix crossing are not so great as to spoil the usefulness of the adiabatic model. Though the exact quantum results were available for these comparisons, one should bear in mind that an important advantage of the adiabatic model is that the computations can be (and have been) extended to much higher energy, angular momentum, and vibrational excitation.
The physical picture of quantum HLH reactions is thus based on a coherent superposition of two atom-rigid-rotor scattering problems. These rotational scattering problems take place on the two asymptotically degenerate surfaces, 8n(R, y) and 8n+ I(R, y). The surfaces differ strongly only in the narrow region near the collinear adiabatic well that corresponds to dynamic region c, in which the hydrogen atom can classically exchange back and forth. Hence, if the angular barriers on the adiabatic surfaces should prevent the wavefunction from penetrating region c, we have Sit � sn+ I, and the reaction probability goes to zero. Often the bottleneck for reactivity appears to be the rotation of the rigid diatom into a near collinear configuration to permit exchange. The idea of achieving proper
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.
162 SKODJE
(0)
• • • J"- •• -•••••••••
(e)
(e]
0.13 0.23 0.33 0.43 0.13 0.23 0.33 Ener9� (eVl Ener9� (eVl
(b)
(d)
(f)
0.43
Figure 7 The total reaction probability versus total energy for several initialj·states for the I + HI reaction with J = O. The solid lines are the adiabatic model results, and the dashed lines are the exact results obtained by Schatz (35). The panels (a)-(f) correspond to initial states j = 0-5, respectively.
orientation in the collision complex is hence transformed into a barrierpassage problem. Thus, a study of the rotational dynamics associated with the angular barriers yields insight into the nature of the reaction dynamics.
We can also use the adiabatic theory to treat half-collision events. This allows us to model the photodetachment (transition state) spectra of HLH- obtained by Neumark et al (29, 31, 38, 39). A time-dependent formalism is the most efficient approach to the direct computation of the spectra (42,60,61). After the usual approximations, the spectral intensity is written as
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.
HLH CHEMICAL REACTIONS 163
I(w) � IOOcu <'1'(0) I 'I'(t)eiW1 dt. 14.
The initial wavepacket, '1'(0), is the anion nuclear ground state, which can be inferred by using a normal mode treatment. The time evolution of this packet is carried out by using the adiabatic approximation. Hence, the initial packet is projected onto the adiabatic surfaces by overlapping 'I'(t = 0) with the double well eigenstates, In' i.e.
'1'(0) = :[ Xn fdrx:'I'(O) == I Xn'l'n(O). n n
15.
Each initial atom-rigid-rotor packet, 'l'n(O), is then time-evolved on the appropriate adiabatic surface through an FFT /basis-set propagation scheme (62a).
Classical Reaction Dynamics
Classical mechanics facilitates a physical understanding of the reaction dynamics involved here. When the fast coordinate is adiabatically decoupled, one can easily visualize the dynamics of the slow variables (R, y). Figure 8 shows trajectories for the I + HI system selected from a quasiclassical ensemble with J = O. (The J = 0 case is special in that the classical mechanics are equivalent to the dynamics of a planar collision.) We see very strong rotationally inelastic scattering from the angular barriers, which is clearly responsible for the rotational chattering effect. Since the fast vibrational coordinate has been adiabatically eliminated, the hydro-
(1:::
10.8
9.8
8.8
7.8
6.8
5.8
.......
4.8 +----,----,-----,----,------;,----, 0.00 3.14 6.28 9.42 12.56 15.70 18.84
Figure 8 A quasiclassical ensemble of trajectories in the (R, y) coordinates for the j = 4, v = 0 state of the I + HI system with J = O. The trajectories scatter strongly from the angular barriers of the adiabatic potential.
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

164 SKODJE
gen-exchange process must be viewed in terms of the reactive separatrix. The projection of the reactive separatrix from the full phase space onto configuration space yields a parabolic shaped curve located in the adiabatic well (44). When the trajectory lies outside the separatrix, the hydrogen atom is bound to a single iodine atom. When it is inside the separatrix, the hydrogen atom is undergoing exchange. Thus, a trajectory must first cross the separatrix to be reactive.
The angular barriers form bottlenecks to reaction because they can prevent a trajectory from achieving the proper collinear orientation required for separatrix crossing and reaction. The trajectory flux through the bottleneck may be computed by using exact adiabatic dynamics or, much more simply, by using a transition state-type approximation based on the angular barriers. However, even if a trajectory does surmount the barrier and cross the reactive separatrix, it may still prove to be nonreactive if the hydrogen atom exchanges an even number of times. Thus, the reaction probability is written as PR = KPx, where Px is the separatrix crossing probability. In analogy with transition-state theory, a transmission factor
K is introduced that gives the fraction of separatrix crossing trajectories that are reactive.
Because K could conceal great complexity, this formulation of the reaction may prove inadequate in the absence of further dynamic information. For example, if we apply this approach to collinear HLH dynamics, we find that P x is 8 (Etrans - Ethreshold) and K is exactly P R above Ethreshold. In three dimensions, however, the determination ofK seems to be much easier. From a study of the exact dynamics of I + HI, we conclude that the separatrix crossing trajectories are highly unstable and exhibit the characteristics of chaotic scattering (69). Thus, unlike in collinear dynamics, neighboring trajectories diverge radically by the end of the collision and K may be computed by a statistical theory. A symmetric reaction is then particularly trivial where we have K = 1/2. This obviates the need to implement the uniform adiabatic theory, since the fraction of separatrix crossing trajectories can be computed by using zero-order adiabatic theory. The physical origin of this chaotic behavior appears to lie in the extreme bending instability of the collision complex. When heavy atoms come together with the light atom in the middle, the light atom tends to be expelled from the interaction region with high rotational energy with a nearly random set of phase angles. In Figure 5, a comparison to exact I + HI quasiclassical trajectory simulations shows that this model gives very accurate reactive probabilities. Thus, this simple theory allows an accurate quantitative analysis of the familiar heuristic notions of steric factors and the cone of acceptance for HLH reactions (63-65).
When the reaction is modeled for cases with J # 0, the dynamics are no
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

HLH CHEMICAL REACTIONS 165
longer equivalent to a planar collision and require an additional angular coordinate in the description. A body-fixed coordinate system where the Jacobi vector R is fixed along the Z-axis is most useful. When the vibration is adiabatilcally removed, the dynamics are expressed in terms of the three scalar coordinates (R, y, ¢), where R = II R II and (y, ¢) are the spherical polar coordinates of the IH-diatom in the body-fixed frame. Figure 9 shows a typical reactive trajectory for J = 3 along with a contour of Bo(R, y, ¢} = Bo(R, y). The adiabatic well forms a crater at the north pole of the adiabatic potential surface, and the angular barriers form the rim about the crater. The trajectory spirals inward and, to be reactive, must surmount the angular barrier. The Coriolis coupling is responsible for the trajectory"s deviations from planarity.
Semiclassical Dynamics
Although the classical description of three-dimensional HLH reactions can afford great insight into the dynamics, some effects do require a quantum mechanical interpretation. Consider the quasi classical and quantal determinations of P R for I + HI, shown in Figures 5 and 7, respectively.
Figure 9 A representative trajectory for j = 4, v = 0, and J = 3 in the I + HI system. The trajectory is plotted in a body-fixed coordinate system where the vector R,,1Il is fixed in the z-direction. The adiabatic surface is roughly spherical and the angular barriers surround the adiabatic well, which forms a crater near the collinear configuration.
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.
166 SKODJE
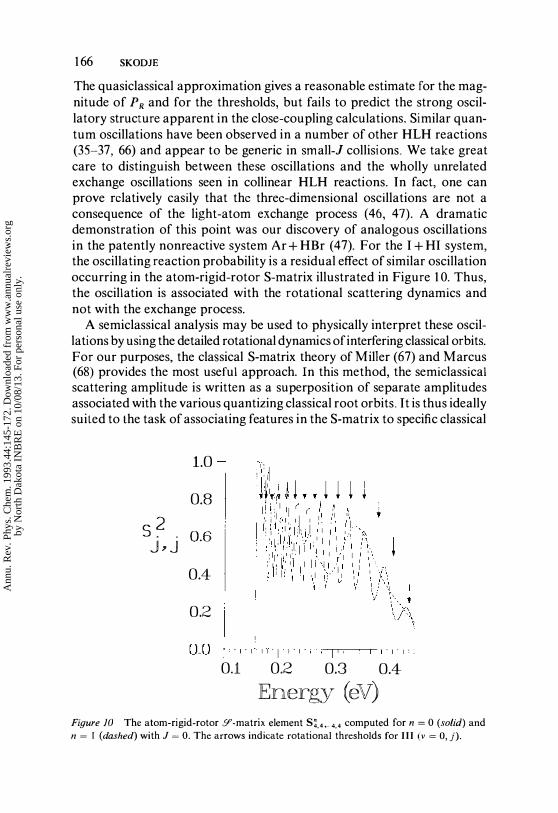
The quasiclassical approximation gives a reasonable estimate for the magnitude of PR and for the thresholds, but fails to predict the strong oscillatory structure apparent in the close-coupling calculations. Similar quantum oscillations have been observed in a number of other HLH reactions (35-37, 66) and appear to be generic in small-l collisions. We take great care to distinguish between these oscillations and the wholly unrelated exchange oscillations seen in collinear HLH reactions. In fact, one can prove relatively casily that the threc-dimensional oscillations are not a consequence of the light-atom exchange process (46, 47). A dramatic demonstration of this point was our discovery of analogous oscillations in the patently nonreactive system Ar + HBr (47). For the I + HI system, the oscillating reaction probability is a residual effect of similar oscillation occurring in the atom-rigid-rotor S-matrix illustrated in Figure 10. Thus, the oscillation is associated with the rotational scattering dynamics and not with the exchange process.
A semiclassical analysis may be used to physically interpret these oscillations by using the detailed rotational dynamics of interfering classical orbits. F or our purposes, the classical S-matrix theory of Miller (67) and Marcus (68) provides the most useful approach. In this method, the semiclassical scattering amplitude is written as a superposition of separate amplitudes associated with the various quantizing classical root orbits. It is thus ideally suited to the task of associating features in the S-matrix to specific classical
1.0
0.8 2 S. " 06
J .. J . 0.4
0.2
0.1 0.2 0.3 0.4 Energy (eV)
Figure 10 The atom-rigid-rotor 9'-matrix element S�,4�4,4 computed for n = 0 (solid) and II = I (dashed) with J = O. The arrows indicate rotational thresholds for III (v = 0, j).
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.
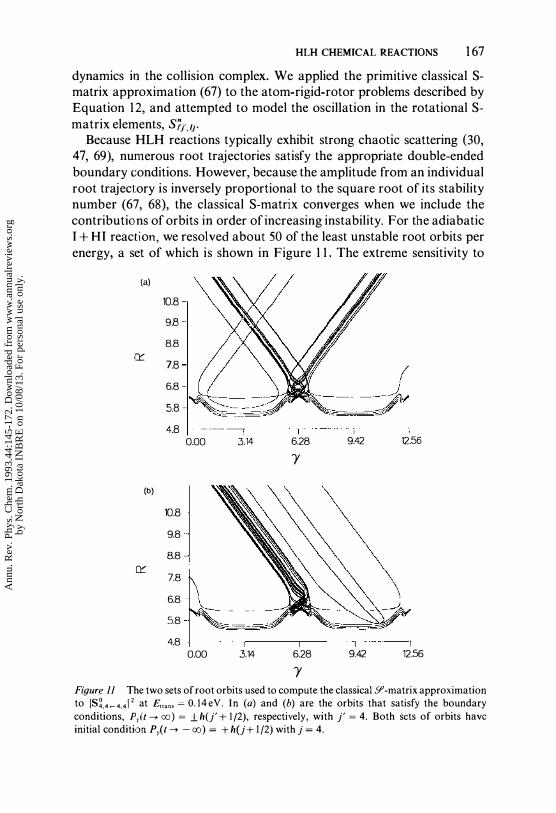
HLH CHEMICAL REACTIONS 167
dynamics in the collision complex. We applied the primitive classical Smatrix approximation (67) to the atom-rigid-rotor problems described by Equation 12, and attempted to model the oscillation in the rotational Smatrix elements, Sh,lj'
Because HLH reactions typically exhibit strong chaotic scattering (30, 47, 69), numerous root trajectories satisfy the appropriate double-ended boundary conditions. However, because the amplitude from an individual root trajectory is inversely proportional to the square root of its stability number (67, 68), the classical S-matrix converges when we include the contributions of orbits in order of increasing instability. For the adiabatic I + HI reaction, we resolved about 50 of the least unstable root orbits per energy, a set of which is shown in Figure 1 1. The extreme sensitivity to
(a)
10.8
9.8
8.8 0:::
7.8
6.8
5.8
4.8 0.00 3.14 6.28 9.42 12.56
Y
(b)
10.8
9.8
8.8 0:::
7.8
6.8
5.8
4.8 0.00 3.14 6,28 9.42 12.56
Y Figure 11 The two sets of root orbits used to compute the classical51' -matrix approximation to IS�,4_4,412 at E",n, = O,14eV. In (a) and (b) are the orbits that satisfy the boundary conditions, Pr(t --'00) = ±h(j' + 1/2), respectively, with j' = 4, Both scts of orbits have initial condition Pr(t --. - 00) = +h(j+ 1/2) with j = 4,
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.
168 SKODJE
initial conditions implicit in chaotic scattering (69) might lead one to expect that the scattering amplitudes associated with various root orbits will add with almost random phases (70). To our great amazement, however, we found that the orbits exhibit a phase coherence for J + HI and do not randomly cancel. As shown in Figure 12, the classical S-matrix method gives a qualitatively reasonable representation of the oscillating transition probability.
Although the quantitative errors in the semiclassical approximation are sufficiently great to disqualify it as a computational method, it nevertheless yields important insight into the scattering dynamics. Careful examination of the individual contributions to the classical S-matrix discloses that the oscillation results from interference between specific families of orbits. Two competing mechanisms for the oscillation were discovered. The first is interference between direct collisions and chattering collisions in which the IH -diatom rotates completely within the collision complex. The second mechanism involves interference between orbits that scatter from the top and bottom of the collinear barrier, respectively. This second mechanism leads to a Bragg-like condition for the oscillations, with a "lattice spacing" related to the barrier height.
Reactive Resonances
Because they provide very sensitive probes into the potential and dynamics in the transition-state region, reactive resonances are of great interest to dynamicists. HLH reactions seem to be particularly fertile ground for the
2 . 0 0
P 1 . 0 0
0 . 0 0 . 0 0 . 1 0 0 . 2 0 0 . 3 0
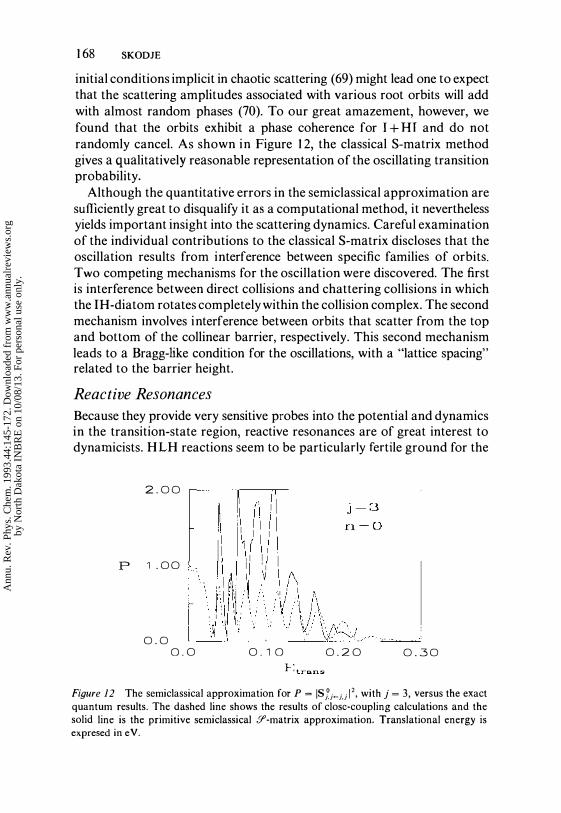
Figure 12 The semiclassical approximation for P = ISJj�}.j12, with j = 3, versus the exact quantum results. The dashed line shows the results of close-coupling calculations and the solid line is the primitive semiclassical Y-matrix approximation. Translational energy is expresed in eV.
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

HLH CHEMICAL REACTIONS 169
search for resonance states because of the generic occurrence of deep adiabatic potential wells. Adiabatic models predict the existence of reactive resonances when bound or quasi bound states are trapped in the wells of the adiabatic potential surfaces (71 , 72a).
The recent experimental work of Neumark et al (29, 3 1 , 38, 39) is partially responsible for many recent theoretical studies of three-dimensional resonances in HLH systems. The I + HI system, in particular, has been analyzed by using a wide variety of techniques. Clary & Connor (16) have obtained the lowest states with the quantum-stabilization method. Schatz (35) has identified six resonances in reactive-scattering calculations. Manz et al ( 1 5), Bowman & Glazdy (32), and Kubach (33) have applied various adiabatic models that assume two fast variables and one slow (internal) variable. We have used the one-fast, two-slow adiabatic model presented above to treat the I + HI system. We now provide a few of the details of our calculations.
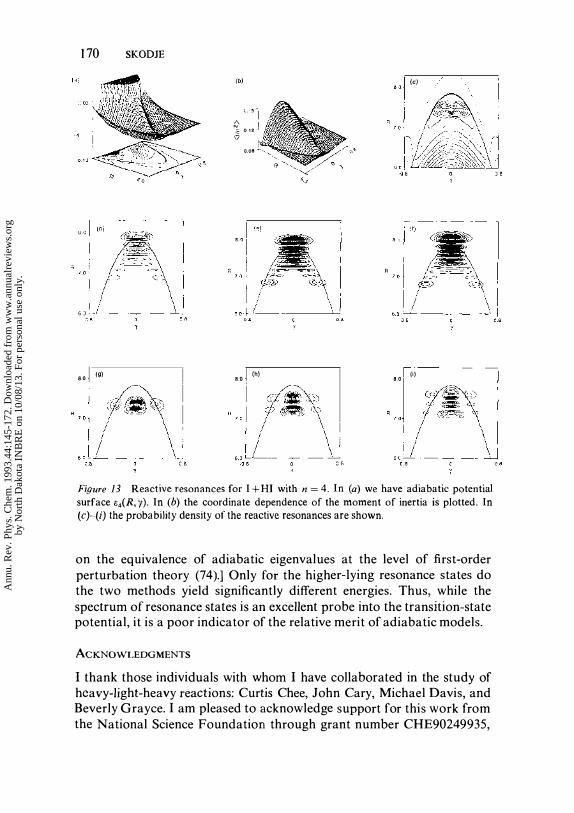
We (45) applied the quantum stabilization method (73) to determine the J = ° resonances for each adiabatic Hamiltonian, H�d, for n between ° and 6. Nonadiabatic coupling was not included. Products of harmonic oscillator states in R and Legendre polynomials in cos (y) comprised the basis set. The range parameter of the harmonic oscillator states served as the stabilization parameter. We have identified 65 stabilized resonance states for the I + HI system; specifically, on the n = 0--6 adiabatic surfaces we found 3 , 0, 10, 3, 1 7, 10, and 22 resonances, respectively. All the resonance states can be labeled with the three quantum numbers, (VR' vY ' vr), which correspond to the R, y, and r degrees of freedom, respectively. Since r represents the asymmetric stretch motion in the chattering region, this designation is similar to the usual scheme for linear triatomics. Many new states were predicted in this study, including several progressions of bend-excited resonances. The adiabatic method achieved nearly perfe:ct agreement with the exact quantum resonance energies for the states that were available. Figure 1 3 shows the probability densities obtained for several highly excited resonance states. It is seen that the extreme instability of the collinear dynamics leads to a bifurcation of the probability density at small values of R.
One might suspect that a comparison of available exact quantum resonance energies with those obtained with the various adiabatic theories would provide a clear test as to which adiabatic method is best. Unfortunately, for those states for which we have nearly exact results, the method of Kubach (34) and our method (45) give nearly identical data. Remarkably, the energies of the low-lying states are insensitive to whether the bend is regarded as fast or slow. [A similar observation for stretchbend Hamiltonians was rationalized by appeal to an argument based
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.
170 SKODJE
8.0 (9)
(D)
(;-..;: 0 12 V
0.06
(h) 8.0
(e)
8.0 (i)
Figure 13 Reactive resonances for I + HI with n = 4. In (a) we have adiabatic potential
surface !;.cR, y). In (b) the coordinate dependence of the moment of inertia is plotted. In (c)�(i) the probability density of the reactive resonances are shown.
on the equivalence of adiabatic eigenvalues at the level of first-order perturbation theory (74).] Only for the higher-lying resonance states do the two methods yield significantly different energies. Thus, while the spectrum of resonance states is an excellent probe into the transition-state potential, it is a poor indicator of the relative merit of adiabatic models.
ACKNOWLEDGMENTS
I thank those individuals with whom I have collaborated in the study of heavy-light-heavy reactions: Curtis Chee, John Cary, Michael Davis, and Beverly Grayce. I am pleased to acknowledge support for this work from the National Science Foundation through grant number CHE90249935,
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

HLH CHEMICAL REACTIONS 171
the Council on Research and Creative Work at the University of Colorado, the Sloan Foundation, and the National Center for Supercomputer Applications.
Literature Cited
1 . Cowley, L. T., Home, D. S., Polanyi, J. C 1 97 1 . Chern. Phys. Lett. 12 : 1 44-49
la . Parr, C A., Polanyi, J. C, Wong, W. H. 1 973. J. Chern. Phys. 5!1: 5-20
2. White, J. M., Thompson, D. L. 1 974. J. Chern. Phys. 6 1 : 7 1 9-32
3. Levy, M. R. 1 979. Prog. React. Kinet. 1 0: 1-252
4. Tamagake, K., Setser, D. W., Sung, J. P. 1 980 . .T. Chern. Phys. 73: 2203-1 7
4a. Langford, A. 0., Bierbaum, V. M., Leone, S. R. 1 985. J. Chern. Phys. 83: 39 1 3- 1 8
5. Hauke, G., Manz, J . , Romeit, J. 1 980. J. Chern. Phys. 73: 5040-44
6. Kaye, J. A., Kuppermann, A. 1 98 1 . Chern. Phys. Lett. 77: 573-79
7. Manz, J., Romeit, J. 1 980. Chern. Phys. Lett. 76: 337--40; 1 980. Chern. Phys. Lett. !I I : 179-84
8. Jonathan, N. B. H., Sellers, P. V., Stace, A. J. 1 98 1 . Mol. Phys. 43: 2 1 5-28
9. Babamov, V. K., Marcus, R. A. 1 98 1 . J. Chern. Phys. 74: 1 790-98
10. Aqui1anti, V. , Grossi, G., Lagana, A. 1 982. Chern. Phys. Lett. 93: 1 79-83
1 I . Kaye, J. A., Kuppermann, A. 1 982. Chern. Phys. Lett. 92: 574-79
12 . Pollak, E., Wyatt, R. E. 1982. J. Chern. Phys. 77: 2689-91
1 3 . Bondi, D. K., Connor, J. N. L., Manz, J., Romelt, J . 1 983. Mol. Phys. 50: 467-88
1 4. Garrett, B. C, Truh1ar, D. G., Wagner, A. F. 1 98 3. J. Chern. Phys. 78: 4400-1 3
1 5. Manz, J., Mayer, R., Romelt, J . 1 983. Chern. Pllys. Lett. 96: 607- 1 2
16 . Clary, D. C . , Connor, J. N. L. 1 983. Chern. Pllys. Lett. 94: 8 1-84
16a. Clary, D. C, Connor, J. N. L. 1 984. J. Phys. Ch.·rn. 88: 275R-64
17. Pollak, E. 1 983. J. Chern. Phys. 78: 1 228-36
1 8 . Hiller, C, Manz, J., Miller, W. H., Romelt, J . 1 983 . 1. Chern. Phys. 78: 3850-56
19. Persky, A., Broida, M. 1 984. J. Chern. Phys. 8 1 : 4352-62
20. Nakamura, H. 1 984. J. Phys. Chern. 88: 48 1 2-23
2 1 . Manz, L. Schor, H. H. R. 1 984. Chern. Phys. Leu. 1 07: 549-54 22. Lopez, V., Babamov, V. K. , Marcus, R.
A. 1984 . .l. Chern. Phys. 8 1 : 3962-66
23. Coveney, P. V., Child, M. S. , Romeit, 1 . 1 985. Chern. Phys. Lett. 120: 349-55
24. Loesch, H. 1 987. Chern. Phys. 1 1 2: 85-93
25. Rakestraw, D. J., McKendrick, K. G., Zare, R. N. 1 987. J. Chern. Phys. 87: 7341-42
26. Last, I., Baer, M. 1 987. J. Chern. Phys. 86: 5534-39
27. Amacc, B., Connor, J. N. L., Whitehead, J. C, Jakubetz, W., Schatz, G. C 1 987. Faraday Discuss. Chern. Soc. 84: 387-403
28. Skodje, R. T., Davis, M. 1. 1 988 . J. Chern. Phys. 88: 2429-56 29. Weaver, A. , Metz, R. B., Bradforth, S.
E., Neumark, D. M. 1988. J. Phys. Chern. 92: 5558-60
30. Skodje, R. T. 1 989 . .l. Chern. Phys. 90: 6 193-62 1 2
3 1 . Kitsopoulos, T . N . , Waller, 1 . M., Loeser, J . G., Neumark, D. M. 1 989. Chern. Phys. Lell. 1 59: 300-6
32. Bowman, J. M., Gazdy, B. 1 989. J. Phys. Chern. 93: 5 129-35
33. Gazdy, B., Bowman, J. M. 1 989. J. Chern. Phys. 9 1 : 46 1 5 24
34. Kubach, C 1 989. Chern. Phys. Lett. 1 64: 475-79
34a. Kubach, C, Nguyen Vien, G., Richard-Viard, M. 199 1 . J. Chern. Phys. 94: 1 929-3)\
35. Schatz, G. C 1 989 . .l. Chern. Phys. 90: 3582-89, 4847-54
35a. Schatz, G. C 1 988. Chern. Phys. Lett. 1 50 : 92-98
36. Schatz, G. C 1 990 . .l. Phys. Chern. 94: 61 57-64
37. Schatz, G. C 1 990 . .l. Chern. Soc. Faraday Trans. I !l6: 1 729-35
38. Metz, R. B., Weaver, A., Bradforth, S. E., Kitsopoulos, T. N., Neumark, D. M . 1 990 . .l. Phys. Chern. 94: 1 377-88
39. Waller, 1. M., Kitsopoulo�, T. N., Neumark, D. M. 1 990. J. Phys. Chern. 94: 2240-41
40. Koizumi, H., Schatz, G. C. 1 99 1 . Advances in Molecular Vibrations and Collision Dynarnics 1 A: 1 39-64. Greenwich, CT: JAI Press
4 1 . Schatz, G. C, Sokolovski, D., Connor, J. N. L. 1 99 1 . Faraday Discuss. Chern. Soc. 9 1 : 1 7-3 \
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.

1 72 SKODJE
42. Metz, R. B., Neumark, D. M. 1992. J. Chern. Phys. 97: 962-72
43. Metz, R. 8., Bradforth, S. E., Neumark, D. M . 1992. Adv. Chern. Phys. 8 1 : 1-6 1
44. Skodje, R. T. 199 1 . J. Chern. Phys. 95: 7234-48
45. Grayce, B. B., Skodje, R. T. 199 1 . J. Chern. Phys. 95: 7249-62
46. Grayce, 8. 8., Skodje, R. T. 1992. J. Phys. Chern. 96: 4 1 34-37
47. Grayce, B. B., Skodje, R. T., Hutson, J. M. 1993. J. Chern. Phys. To be published
48. Grayce, B. 8., Skodje, R. T. J. Chern. Phys. 1993. 98: 3929-44
49. Cary, J. R., Skodje, R. T. 1988. Phys. Rev. Lett. 6 1 : 1795-98
50. Cary, J. R., Skodje, R. T. 1989. Physica D 36: 287-3 1 6
5 1 . Lichtenberg, A. J., Lieberman, M . A. 1983. In Regular and Stochastic Motion. New York: Springer
52. Lenard, A. 1959. Ann. Phys. NY 6: 261 � 76
53. Kruskal, M. 1962. J. Math. Phys. 3: 806-28
54. Timofeev, A. V. 1978. Zh. Eksp. Teat. Fiz. 75: 1303-8 [Sov. Phys. JETP 48: 656-59]
55. Cary, J. R., Escande, D. F., Tennyson, J. L. 1986. Phys. Rev. A 34: 4256-75
56. Skodje, R. T., Cary, J. R. 1 988. Comput. Phys. Rep. 8: 221-9 1
57. Massey, H . S . W., Smith, R . A . 1933. Proc. Roy. Soc. London Ser. A 142: 142-48. Also see: Mott, N. F., Massey, H . S. W. 1965. Theory oj Atomic Collisions, p. 430. Oxford: Oxford Univ. Press. 3rd ed.
58. Arthurs, A. M . , Da1garno, A. 1 960. Proc. R. Soc. London A256: 540-48
59. Manolopoulos, D. E. 1988. PhD thesis. Cambridge Univ.
60. Heller, E. J. 1 978. J. Chern. Phys. 68: 3891-96
6 1 . Lorquet, A. J., Lorquet, J. c., Delwiche, J., Hubin-Franskin, M. J. 1982. J. Chern. Phys. 76: 4692-99
62. Kouri, D. J., Mowrey, R. C. 1987. J. Chern. Phys. 86: 2087-94
62a. Gray, S. K., Wozny, C. J. 1989. J. Chern. Phys. 9 1 : 767 1-84
63. Blackwell, B. A., Polanyi, J. c., Sloan, J. J. 1979. J. Chern. Phys. 70: 299-306
64. Levine, R. D., Bernstein, R. 8. 1984. Chem. Phys. Lett. 105: 467-7 1
65. Loesch, H. 1986. Chern. Phys. 104: 2 13-27
66. Schatz, G. C. 1989. Isr. J. Chem. 29: 361-67
67. Miller, W. H. 1 974. Adv. Chem. Phys. 25: 69-177
68. Marcus, R. A. 1973. J. Chern. Phys. 59: 5 135--44
69. Eckhardt, B., Jung, C. 1986. J. Phys. A Gen. Phys. 19: L829-33
70. Rankin, C. c., Miller, W. H. 197 1 . J. Chem. Phys. 55: 3 1 50-56
7 1 . Kuppermann, A. 198 1 . In Potential Energy SurJaces and Dynamics Calculations, ed. D. G. Truhlar, pp. 375-404. New York: Plenum
72. Garrett, B. C., Schwenke, D. W., Skodje, R. T., Thirumalai, D., Thompson, T. C., Truhlar, D. G. 1 984. Am. Chem. Soc. Symp. Ser. 263: 375-400
72a. Rome1t, J., Pollak, E. 1984. Am. Chem. Soc. Symp. Ser. 263: 353-74
73. Hazi, A., Taylor, H. S. 197 1 . Phys. Rev. A I : 1 109-20
74. Johnson, B. R., Skodje, R. T., Reinhardt, W. P. 1984. Chem. Phys. Lett. 1 12: 396-402
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
45-1
72. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by N
orth
Dak
ota
INB
RE
on
10/0
8/13
. For
per
sona
l use
onl
y.





























