Embed Size (px)
Citation preview

IntroductionEstablishment of the mammalian vasculature is con-trolled by a complex process involving differentiationand assembly of multiple cell lineages. Rapid expansionof blood vessels is necessary for the tissue growthobserved during embryonic development and tumorgrowth. Dysregulation of vascular development canresult in excessive proliferation of vessels, as seen inhemangiomas, retinopathy of prematurity, and venousmalformations (1). Because of the fundamental signif-icance of vascular development to human biology anddisease, there has been much interest in understandingthe molecular mechanisms underlying the develop-ment and proliferation of vessels.
Anatomically, formation of blood vessels can be divid-ed into 2 distinct steps. Vasculogenesis, the initial step,occurs when mesodermally derived angioblasts differ-entiate into vascular endothelial cells and assemble intovascular tubes (2). Subsequently, in a process known asangiogenesis, preexisting vessels sprout new branchesthat become remodeled into mature vessels (3). This steprequires recruitment of vascular mesenchymal cells thatencase endothelial tubes and differentiate into vascularsmooth muscle cells (VSMCs), pericytes, and fibroblaststo promote the assembly and stabilization of the matureblood vessel wall (4). Reciprocal endothelial-mesenchy-mal interactions are essential for angiogenesis.
Recent studies have revealed several signaling pathwaysimportant during vasculogenesis and angiogenesis (1, 4).The secreted ligand angiopoietin-1 (Ang-1) and its recep-tor tyrosine kinase Tie2 (5–8), and ephrinB2 and itsreceptor EphB4 (9, 10), are critical for endothelial-mes-
enchymal interactions during angiogenesis. Within theheart, Ang-1 secretion from the endocardium is also nec-essary for trabeculation of the neighboring myocardium(8). TGF-β and its receptors play a role in vascular stabi-lization and remodeling and function through the Smadfamily of transcription factors (11–14). In contrast tothese molecules that function mainly during angiogen-esis, VEGF, a heparin-binding growth factor with highspecificity for endothelial cells, is a central mediator ofangiogenesis and vasculogenesis (4, 15). There are 5 iso-forms produced from a single VEGF gene by alternativesplicing. VEGF induces endothelial proliferation, pro-motes cell migration, and inhibits apoptosis (3). Inhibi-tion of VEGF activity results in tumor regression, indi-cating that VEGF is necessary for the neovascularizationassociated with tumors (16). VEGF signals through itsreceptor tyrosine kinases, VEGFR-1/Flt-1 and VEGFR-2/KDR/Flk-1, which are expressed specifically on thesurface of vascular endothelial cells and are required forboth vasculogenesis and angiogenesis (17–20). Endothe-lial cells of VEGFR-1 mutant mouse embryos differenti-ate, but do not assemble properly into vessels (18),whereas VEGFR-2 mutants display more severe defectsof vasculogenesis (17). Recently, another VEGF receptor,VEGFR-3, has been shown to be important for theremodeling and maturation of primary vascular net-works into larger blood vessels (21). These data suggestthat VEGF controls unique steps of vascular develop-ment through distinct receptors.
Neuropilin-1 is a specific receptor for the 165-aminoacid form of VEGF (VEGF165) and is a potential regula-tor of VEGF-induced angiogenesis (22). Neuropilin-1
The Journal of Clinical Investigation | February 2000 | Volume 105 | Number 3 261
The basic helix-loop-helix transcription factor, dHAND, is required for vascular development
Hiroyuki Yamagishi,1,2 Eric N. Olson,2 and Deepak Srivastava1,2
1Department of Pediatrics, Division of Cardiology, and2Department of Molecular Biology, University of Texas Southwestern Medical Center, Dallas, Texas 75235, USA
Address correspondence to: Deepak Srivastava, Department of Pediatrics and Molecular Biology, 6000 Harry Hines Boulevard, Dallas, Texas 75235-9148, USA. Phone: (214) 648-1246; Fax: (214) 648-1196; E-mail: [email protected].
Received for publication November 4, 1999, and accepted in revised form December 17, 1999.
Reciprocal interactions between vascular endothelial cells and vascular mesenchymal cells are essen-tial for angiogenesis. Here we show that the basic helix-loop-helix transcription factor,dHAND/Hand2, is expressed in the developing vascular mesenchyme and its derivative, vascularsmooth muscle cells (VSMCs). Targeted deletion of the dHAND gene in mice revealed severe defectsof embryonic and yolk sac vascular development by embryonic day 9.5. Vascular endothelial cellsexpressed most markers of differentiation. Vascular mesenchymal cells migrated appropriately butfailed to make contact with vascular endothelial cells and did not differentiate into VSMCs. In ascreen for genes whose expression was dependent upon dHAND (using subtractive hybridizationcomparing wild-type and dHAND-null hearts), the VEGF165 receptor, neuropilin-1, was found to bedownregulated in dHAND mutants. These results suggest that dHAND is required for vascular devel-opment and regulates angiogenesis, possibly through a VEGF signaling pathway.
J. Clin. Invest. 105:261–270 (2000).

was first identified in the Xenopus tadpole nervous sys-tem (23) and found to play a role in semaphorin-medi-ated axonal chemorepellence (24). Neuropilin-1 isexpressed in the endothelial and mesenchymal cells ofthe murine vasculature (25) and may mediate interac-tions between endothelial and mesenchymal cells.Mouse embryos overexpressing neuropilin-1 exhibit car-diovascular, peripheral nervous system (PNS) and limbdefects (25). Mice deficient in neuropilin-1 have defectsin PNS axonal guidance and die between embryonic day10.5 and 12.5 from cardiovascular defects (26).
Despite recent progress in identification of signalingmolecules and cell-surface receptors that regulateblood vessel formation, little is known currently aboutthe nuclear transcriptional pathways that regulate orare regulated by the vascular signaling cascades. Tissue-specific members of the basic helix-loop-helix (bHLH)family of transcription factors are essential for devel-opment of many cell lineages (27–29), but have notbeen demonstrated to play a role in vascular develop-ment. The bHLH transcription factor dHAND (decidu-um, heart, autonomic nervous system, neural crest-derived)/Hand2 is required for cardiac morphogenesis(30–32). Mice lacking dHAND have a single ventricleand aortic arch defects and die by embryonic day 11.0(E11.0) from heart failure (31). Although the cardiacdefects of dHAND mutants are well characterized ashypoplasia of the right ventricle segment and poor tra-beculation of the remaining ventricle, the role ofdHAND in development of the vasculature has notbeen examined previously. Here we demonstrate thatdHAND is expressed in the developing vasculature andVSMCs and is required for normal vascular develop-ment. The dHAND-null endothelial cells differentiatebut mesenchymal cells fail to differentiate into VSMCs.The vascular defects in dHAND-mutant embryos aresimilar to the angiogenic defects observed in VEGF andFlt1 mutants. In a screen to identify dHAND-depend-ent genes in the heart by suppressive subtractivehybridization, we found that expression of the VEGF165
receptor, neuropilin-1, was dHAND dependent in theheart and portion of the vasculature. These data indi-cate that dHAND plays a role in angiogenesis and mayfunction through a VEGF signaling pathway.
MethodsSection and whole-mount in situ hybridization. In situhybridization to mouse embryo sections was per-formed with riboprobe labeled with 35S as describedpreviously (30). Antisense riboprobe was synthesizedwith SP6 RNA polymerase (MAXIScript; AmbionInc., Austin, Texas, USA) from dHAND full-lengthcDNA. Hybridization was performed on tissue sec-tions of E9.5 and E10.5 mouse embryos. Afterhybridization, sections were washed and dipped inKodak NTB-2 emulsion. Slides were exposed for 14days and then developed in D19 (Eastman Kodak Co.Scientific Imaging Systems, New Haven, Connecti-cut, USA) for 3 minutes.
Whole-mount RNA in situ hybridizations fordHAND, Ang-1, COUP-TFII, SM22α, and neuropilin-1were performed using digoxigenin-labeled antisenseriboprobes. In situ hybridizations were performed asdescribed previously (31). Briefly, embryos werehybridized with digoxigenin-labeled riboprobes at60°C for 18 hours. After a series of washes, embryoswere incubated with alkaline phosphatase–conjugat-ed anti-digoxigenin antibodies at room temperaturefor 1 hour. After another series of washes, embryoswere incubated in a substrate color-reaction mixture(Roche Molecular Biochemicals, Indianapolis, Indiana,USA) for 10–12 hours in darkness. Color reaction wasterminated by fixing embryos in 4% paraformaldehydeand 0.1% glutaraldehyde.
Breeding and genotyping of mice. Mice heterozygous forthe dHAND mutation were generated as described pre-viously (31). To generate transgenic mice in a dHAND-null background, male mice heterozygous for the Tie2-lacZ transgene (33) and the SM22lacZ transgene (34)were mated to dHAND heterozygous females. Male off-spring that were heterozygous for dHAND and the lacZtransgene were mated with dHAND heterozygousfemales to obtain embryos that expressed the lacZ trans-gene and were heterozygous or homozygous for the tar-geted dHAND allele. Mothers were sacrificed and theiruteri dissected to harvest E8.0–10.5 embryos. GenomicDNA was prepared from tail biopsies or from embry-onic yolk sacs and subjected to PCR and/or Southernblot for genotyping as described previously (31–34).
Immunochemistry. Whole-mount immunochemistrywas performed with mAb MEC13.3 (rat anti-mouseplatelet-derived endothelial adhesion molecule;PharMingen, San Diego, California, USA), againstmouse platelet endothelial cell adhesion molecule-1(PECAM-1) (35). Embryos were fixed in 4% para-formaldehyde/PBS at 4°C overnight and dehydrated inmethanol. Embryos were bleached in 5% hydrogen per-oxide in methanol for 4–5 hours at room temperature,followed by rehydration and blocking in PBSMT (3%skim milk and 0.1% Triton-X-100 in PBS) at room tem-perature twice for 1 hour. Embryos were incubated in10 µg/mL anti-mouse PECAM in PBSMT at 4°C over-night, washed with PBSMT at 4°C and secondaryhorseradish peroxidase–conjugated antibody inPBSMT at 4°C overnight. Embryos were again washedin PBSMT at 4°C and rinsed in PBST (0.1% Triton-X-100, PBS) at room temperature for 20 minutes. For sig-nal detection, embryos were incubated in 0.3 mg/mLdiaminobenzidine (Sigma Chemical Co., St. Louis, Mis-souri, USA) in PBST for 20 minutes, and hydrogen per-oxide was added to a final concentration of 0.03% andincubated overnight. The staining reaction was termi-nated by rinsing the embryos in PBST. Embryos werepostfixed in 4% paraformaldehyde/PBS at 4°C over-night. To better observe the vasculature of the embryos,embryos were dehydrated through graded steps into100% methanol and cleared in a solution of benzyl ben-zoate/benzyl alcohol (2:1).
262 The Journal of Clinical Investigation | February 2000 | Volume 105 | Number 3

β- galactosidase staining. Embryos were dissected in 4°CPBS and fixed in 2% paraformaldehyde/PBS with phe-nol red for 30 minutes on ice. After rinsing with PBS,embryos were incubated at room temperature over-night in 0.1% X-gal, 5 mM potassium ferricyanide, 5mM potassium ferrocyanide, 1 mM magnesium chlo-ride, 0.002% NP-40, 0.01% sodium deoxycholate, PBS,pH 7.0. After staining, the embryos were rinsed in PBSand postfixed at 4°C overnight in 4% paraformalde-hyde and 0.1% glutaraldehyde in PBS. Embryos weredehydrated and cleared in a solution of benzyl ben-zoate/benzyl alcohol (2:1).
Histology. For histologic analysis, embryos were embed-ded in paraffin after fixation. Transverse and sagittal sec-tions were made at 7-µm intervals throughout theembryos. Paraffin was cleared in xylene, and photographsof sections were taken without counterstaining to illus-trate color reaction after either immunochemistry, β-galreaction, or whole-mount in situ hybridization.
Electron microscopy. Embryos were fixed in 2.5% glu-taraldehyde in PBS at 4°C overnight and post-fixedwith 1% osmium tetroxide for 1 hour. After washings,embryos were dehydrated in increasing concentrationsof ethanol and infiltrated and embedded in LK-112medium. Ultrathin transverse sections were cut with adiamond knife, mounted on 200-mesh grids, andstained with uranyl acetate and lead citrate. Sectionswere viewed with and photomicrographs obtainedthrough an electron microscope.
RT-PCR. Total RNA was purified from whole body,hearts, or yolk sacs of wild-type and dHAND-nullembryos at E9.5, and adult mouse heart, aorta, andliver (Trizol; GIBCO BRL, Rockville, Maryland, USA)and A10 cell line (ATCC #CRL-1476; American TypeCulture Collection, Rockville, Maryland, USA). After
RT reaction, PCR analysis of several endothelial mark-ers was performed using oligonucleotides specific forFlk-1, Tie-1, and Tie-2, described previously (17, 36),and numerous other vascular markers (primersequences available upon request). PCR primers specif-ic for dHAND (upper: 5′-TACCAGCTACATCGCCTACCT-3′;lower: 5′-TCACTGCTTGAGCTCCAGGG-3′) and neu-ropilin-1 (upper: 5′- TGAAAAATACCCCAACTG-3′; lower:5′- CCTGAATGATGACACCTCTT-3′) were used under thefollowing conditions: 94°C, 5 minutes; cycles of 94°C,1 minute; 60°C, 1 minute; 72°C, 1 minute; and 72°C,7 minutes. PCR products were analyzed after serialcycles in the linear range of amplification. RNA load-ing was controlled by amplification of the housekeep-ing gene, GAPDH. Negative controls were performedfor each sample using non–reverse transcribed RNA.
Suppressive subtractive hybridization. One microgram oftotal RNA, after DNase treatment, from pooled wild-type or dHAND-null E9.5 hearts was used for generationof cDNA by the SMART cDNA synthesis kit (CLON-TECH Laboratories Inc., Palo Alto, California, USA).PCR-Select (CLONTECH) was used for subtractivehybridization as described by CLONTECH. Briefly, 2pools of RsaI-digested wild-type cDNA were used astester and ligated to unique adapters. RsaI-digesteddHAND-null cDNA was used as a driver and was not lig-ated to any adapter sequences. Hybridization of thetester population with excess driver and subsequentamplification of subtracted species with the 2 uniqueadapters present on the tester cDNA resulted in a poolof PCR fragments theoretically present in wild-type, butnot dHAND-null, cDNA. TA cloning (Invitrogen Corp.,San Diego, California, USA) of the PCR product andsubsequent DNA isolation and sequencing of selectedclones was performed. Forty clones were initially
The Journal of Clinical Investigation | February 2000 | Volume 105 | Number 3 263
Figure 1Expression of dHAND in the developingvasculature. Whole-mount in situhybridization revealed dHAND expressionin the aortic arch arteries (aa) and aorta(ao) of an E10.0 embryo (a). Expression inthe pharyngeal arches (pa), heart (ht), andlimb bud (lb) was also seen. Sagittal sec-tion of an E9.5 embryo (b and c) showedexpression in the aorta and aortic archarteries. Sagittal section of an E10.5embryo (d and e) demonstrated dHANDexpression in the mesenchyme (m) of thethird and fourth aortic arch arteries(arrowheads) and in the pharyngeal arch(pa) mesenchyme. b and d represent phaseimages of c and e, respectively. Expressionof dHAND was also observed in the vascu-lar smooth muscle cell line, A10, and in theadult heart and aorta by RT-PCR (f). RNAintegrity was assayed by amplification ofGAPDH. Yolk sac expression of dHANDwas observed by RT-PCR in f and whole-mount in situ hybridization (arrowheadsindicate vessels) (g). (h) Sense control.

sequenced. Basic local alignment search tool (BLAST)was used for searching sequence homology to DNA andexpressed sequence tag (EST) databases.
ResultsVascular expression of dHAND. The expression of dHANDin the vasculature has not been examined in detail pre-viously. Whole-mount in situ hybridization at E10.0revealed dHAND expression in the aorta and aortic archarteries in addition to the pharyngeal arches and heart(Figure 1a). Expression of dHAND was enhanced in therostral, compared with the caudal, vasculature. Histo-logically, dHAND was expressed in the walls of the dor-sal aorta and aortic arch arteries at E9.5 (Figure 1, b andc) and in the heart (endocardium and myocardium) andpharyngeal arches. The hybridization signal in theblood vessels was wider than the thickness of theendothelium, suggesting that the mesenchymal cellssurrounding the vessels expressed dHAND. At E10.5,dHAND expression was localized to the vascular mes-enchyme between the third and fourth aortic arch arter-ies (Figure 1, d and e), which later gives rise to VSMCsand to the mesenchyme of the pharyngeal arch. The vas-cular expression of dHAND was subsequently down-regulated and became undetectable by in situ hybridiza-tion around E12.5. However, dHAND expression wasdetectable by RT-PCR in the adult mouse aorta (Figure1f). Consistent with aortic expression of dHAND, we
found that dHAND mRNA was present in a vascularcell line (A-10; ATCC #CRL-1476) that is derived fromrat aortic smooth muscle cells (Figure 1f). In additionto expression in the embryo proper, dHAND was alsodetectable in the developing yolk sac. In situ hybridiza-tion detected dHAND expression along the yolk sac ves-sels during the process of remodeling at E9.5–10.0 (Fig-ure 1, g and h). RT-PCR confirmed dHAND expressionin the yolk sac at E9.5 (Figure 1f).
Abnormal vascular development in dHAND mutants.Mouse embryos homozygous-null for dHAND lack aright ventricle and have a poorly trabeculated left ven-tricle, resulting in death by E11.0 from heart failure(31). Although dHAND is also thought to play a rolein the neural crest component of aortic arch develop-ment, we sought to determine its role during vasculo-genesis and/or angiogenesis. Because heart failuremight cause secondary vascular defects, all analyses ofdHAND mutants were performed at E9.5, before anyevidence of cardiac or growth failure. Unlike wild-typeE9.5 embryos that had a grossly visible vascular pat-tern, dHAND mutants did not have an apparent vas-culature, but rather had pooling of blood in extravas-cular regions of the embryo. Whole-mount immuno-chemical staining of E9.5 dHAND-null embryos usinga mAb against the endothelial-specific markerPECAM-1 (Figure 2, a and b) revealed the presence ofdifferentiated endothelial cells. However, the vascula-
264 The Journal of Clinical Investigation | February 2000 | Volume 105 | Number 3
Figure 2Endothelial development in dHAND mutants. Whole-mount immunochemistry revealed that endothelial cells expressed PECAM-1 proteinappropriately in wild-type (a) and mutant (b) E9.5 embryos, but displayed a disorganized pattern in dHAND mutants (b). The rostral por-tion of dHAND-null embryos was more severely affected than the caudal region, where the aorta (ao) and somitic arteries (arrowheads) werevisible. Sagittal section of PECAM-1 antibody–stained wild-type (e) and mutant (f) embryos revealed disorganization of the dorsal aorta ofdHAND-null embryos, where the aortic lumen was evident. Note patency of the aortic arch artery (aa) in the mutant (f). β-galactosidaseactivity in wild type (c) and mutant (d) embryos harboring lacZ under control of the Tie2 promoter revealed disorganization of lacZ expres-sion in dHAND-null embryos compared with wild-type embryos. Transverse sections of wild-type (g) and mutant (h) embryos in the caudalregion demonstrate the dilated nature of caudal vessels of dHAND-null embryos. ht, heart; as, aortic sac; h, head; nt, neural tube; fg, foregut.

ture was grossly disorganized throughout the embryowith the more rostral vessels being more severelyaffected. Histological analysis of E9.5 mutant embryosshowed that the aortic arch arteries were patent butthe rostral aorta was poorly patterned, without anapparent lumen (Figure 2, e and f).
To further examine endothelial development indHAND mutants, transgenic mice containing a lacZmarker under control of the endothelial-specific Tie2promoter (33) were crossed into the dHAND-nullbackground. As observed with PECAM expression,Tie2 expression was intact, but vascular developmentwas disrupted in the dHAND-null embryos at E9.5(Figure 2, c and d). Interestingly, histologic analysisrevealed that the vasculature was formed but dilatedin the caudal portion of the embryo (Figure 2, g andh). The disorganization of the rostral vasculature anddilation of caudal vessels were similar to that seen inVEGF-deficient or MEF2C homozygous-null embryos,although the etiology of this observation in eithermodel is unclear (19, 20, 37).
Because endothelial differentiation appeared rela-tively normal in dHAND mutants, we examined theexpression of several endothelial markers, as well asother factors implicated in vasculogenesis and angio-genesis. By semiquantitative RT-PCR, transcripts ofVEGF and its receptors Flk-1 and Flt-1, and Ang-1 andits receptors, Tie1 and Tie2, were detected at similar lev-els in wild-type and dHAND mutant embryos at E9.5(Figure 3). Similarly, expression of the bHLH tran-scription factor, ARNT (38), and the transcription fac-tor, MEF2C (37), both implicated in vascular develop-ment, were unaffected in dHAND mutants (Figure 3).These results suggest that the vascular defects ofdHAND-null embryos do not result from alteredexpression of the factors examined, although we can-not eliminate the possibility of subtle changes in geneexpression in specific regions of mutant embryos.
Abnormal vascular remodeling in dHAND-null yolk sacs.The vitelline circulation in the embryonic yolk sac rep-resents the earliest circulatory system and is the firstsite of vasculogenesis and angiogenesis in the embryo.Normally, a honeycomb-like vascular plexus is evidentby E8.5; subsequent remodeling of the vasculature(angiogenesis) results in defined vessels by E9.5 (Figure4a). However, dHAND-null yolk sacs failed to form anormal vasculature at E9.5 (Figure 4b). Expression oflacZ under control of the Tie2 promoter revealed thatendothelial cells were present in both wild-type anddHAND-null yolk sacs at E9.5 (Figure 4, c and d). Inwild-type yolk sacs, lacZ expression demarcated the for-mation of large vitelline vessels and a fine network ofsmaller vessels (Figure 4c). In contrast, lacZ expressionin yolk sacs lacking dHAND displayed a honeycomb-like plexus pattern of endothelial cells with no remod-eling into a vascular network (Figure 4d). Histologicanalyses revealed that although capillary-like vesselscontaining blood cells with an endothelial cell lining(Figure 4e) were seen in the wild-type yolk sac, no dis-
tinct blood vessels were evident in the dHAND-mutantyolk sac (Figure 4f). Large cavities containing bloodcells and lined by endothelial cells (Figure 4f) wereobserved in dHAND mutants suggesting that vasculo-genesis proceeds, but angiogenesis is blocked in themutant yolk sac.
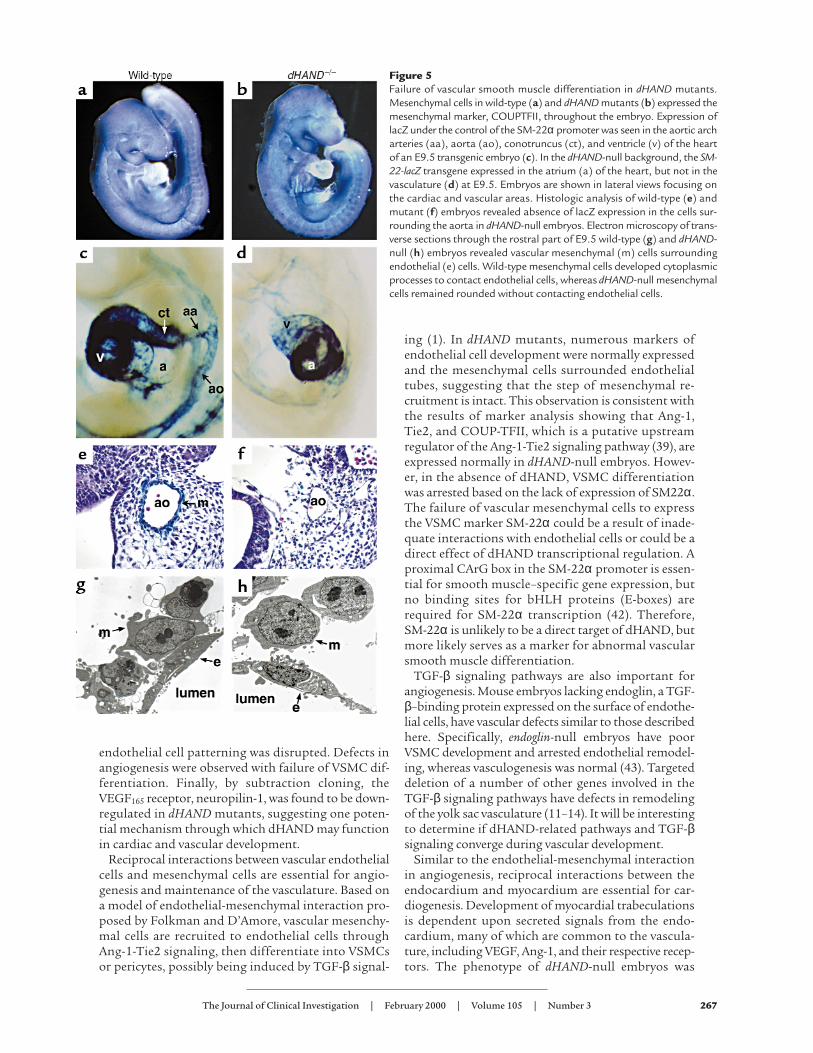
Vascular mesenchyme cells fail to differentiate into VSMCsin dHAND mutants. Mesenchymal cells begin to invadethe vasculature around E9.0 and later differentiateinto VSMCs. Because dHAND was expressed in vas-cular mesenchymal cells and VSMCs, we examined thedevelopment of vascular mesenchymal cells indHAND mutants. Ang-1 and COUP-TFII are expres-sed in mesenchymal cells and play a role in angiogen-esis, possibly in a common molecular pathway (9, 39).Transcripts of these 2 genes were examined indHAND-null embryos by RT-PCR and whole-mountin situ hybridization, and no difference was foundbetween wild-type and dHAND-null embryos (Figure3 and Figure 5, a and b; data not shown).
SM22α is an early marker for differentiated VSMCsand marks the developing vasculature only after vas-cular mesenchymal cells differentiate into VSMCsaround E9.0 (40, 41). Unlike Ang-1 and COUP-TFII,SM22α was not detected in the dHAND-null vascula-ture by in situ hybridization (data not shown). Togenetically confirm the absence of SM22α expression,transgenic mice containing a lacZ marker under con-trol of a SM22α arterial VSMC-specific promoter (34)were crossed into the dHAND-null background. No
The Journal of Clinical Investigation | February 2000 | Volume 105 | Number 3 265
Figure 3Normal endothelial cell differentiation in dHAND-null embryos.Semiquantitative RT-PCR analysis of wild-type (+/+) and dHAND-null(–/–) embryos using primers specific to the endothelial cell markersFlk-1, Tie1, Tie2, Flt-1, and VEGF revealed equal levels of expressionsuggesting normal differentiation and quantity of endothelial cells indHAND mutants. Similarly, the mesenchymal marker, Ang-1, wasunaffected in dHAND mutants, as were the transcription factorsMEF2C and ARNT. GAPDH expression indicates equal RNA loading.

expression of lacZ was detected in the vasculature ofmice lacking dHAND at E9.5 or E10.5 (Figure 5, c andd). Ventricular expression of SM22α was not detectedin dHAND mutants, although atrial expression waspresent (Figure 5, c and d). Histologic analysis con-firmed absence of SM22α-expressing cells around thevasculature of mutants (Figure 5, e and f). These resultssuggest that mesenchymal cells were present in theabsence of dHAND, but fail to differentiate intoSM22α-expressing VSMCs.
To more accurately determine if vascular mesenchy-mal cells had migrated to the developing vasculatureand to assess the relationship of vascular endothelialcells with mesenchymal cells, we performed electronmicroscopy of wild-type and dHAND-null E9.5 embryos.Transverse sections revealed that mesenchymal cells hadmigrated around the aorta of dHAND mutants, similarto wild-type (Figure 5, g and h). However, dHAND-nullmesenchymal cells remained rounded without cyto-plasmic processes that normally make contact withendothelial cells. Endothelial cells also began to appearabnormal at this stage, possibly secondary to insuffi-cient interactions with the mesenchyme. Together, theseobservations suggest a role for dHAND in promotingvascular mesenchyme development and possiblyendothelial-mesenchymal interactions.
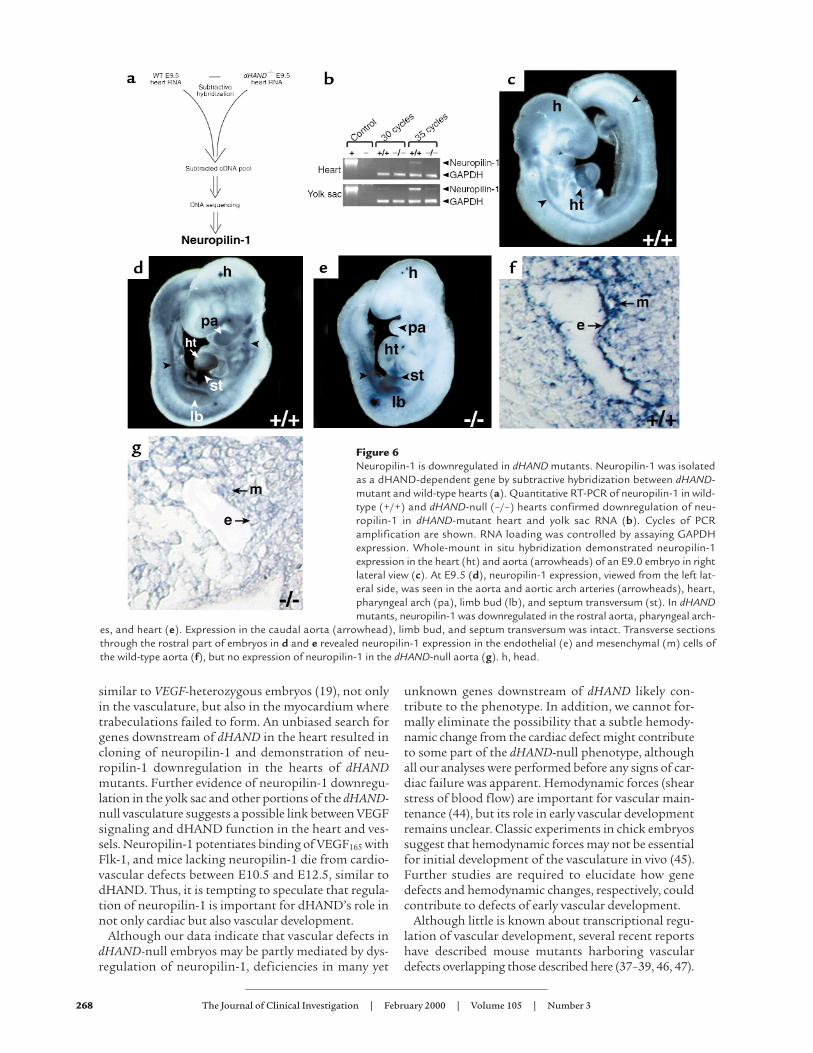
Neuropilin-1 functions downstream of dHAND. To gaininsight into potential mechanisms through whichdHAND might function, we sought to identify dHAND-dependent genes by subtraction cloning. Subtraction
cloning was performed by suppressive-subtrac-tive hybridization in which E9.5 dHAND-nullheart cDNA was subtracted from wild-type E9.5heart cDNA, resulting in isolation of genes thatwere expressed in wild-type, but not dHAND-mutant, hearts (Figure 6a). Whereas numerousgenes were identified and will be described else-where, one of the dHAND-dependent factorsrepresented neuropilin-1, the VEGF165 receptor(22). Downregulation of neuropilin-1 indHAND-mutant hearts was confirmed by semi-quantitative RT-PCR (Figure 6b).
The expression pattern of neuropilin-1 inearly embryonic development has not beendescribed previously. In addition to its expres-sion in the heart, neuropilin-1 mRNA wasfound to be most prominent in the aorta, aor-
tic arch arteries, pharyngeal arches, and limb bud ofwild-type embryos by whole-mount in situ hybridiza-tion (Figure 6, c and d). The expression of neuropilin-1 described previously in the PNS and placenta andthat described here is very similar to that of dHANDexpression. Because neuropilin-1 was downregulatedin the hearts of dHAND mutants, we examined thespatial regulation of neuropilin-1 expression indHAND-null embryos. Neuropilin-1 was downregu-lated in the ventricle of the heart, as expected (Figure6e). Interestingly, neuropilin-1 mRNA was severelydownregulated in the rostral aorta and pharyngealarches but was still detectable in the caudal aorta, cor-relating with the expression pattern of dHAND.Expression in the limb buds and septum transversumwas normal in dHAND mutants (Figure 6e), providinginternal controls for RNA integrity within the embryo.Histologic analysis also demonstrated downregulationof neuropilin-1 expression in the rostral dHAND-nullvasculature (Figure 6, f and g). Neuropilin-1 expres-sion was also seen in the yolk sac of E9.5 wild-typeembryos (Figure 6b). Transcripts of neuropilin-1 weredownregulated in the dHAND-null yolk sac by semi-quantitative RT-PCR (Figure 6b).
DiscussionThe results in this study demonstrate that dHAND isexpressed in the developing vasculature and VSMCsand plays a role in vascular development. Whereasendothelial cell differentiation and vasculogenesiswere relatively unaffected in dHAND mutants,
266 The Journal of Clinical Investigation | February 2000 | Volume 105 | Number 3
Figure 4Yolk sac vascular defects in dHAND-null embryos. Unlikewild-type yolk sacs at E9.5 (a), dHAND-null yolk sacs atE9.5 display absence of visible vessels (b). Expression of lacZunder control of the Tie2 promoter in wild-type (c) andmutant (d) yolk sacs revealed a honeycomb-like vascularplexus in dHAND-mutants rather than the remodeled ves-sels present in the wild-type (e) and (f) represent histologicsections of c and d, respectively. Arrowheads indicate ves-sels; arrows indicate endothelial cells.
endothelial cell patterning was disrupted. Defects inangiogenesis were observed with failure of VSMC dif-ferentiation. Finally, by subtraction cloning, theVEGF165 receptor, neuropilin-1, was found to be down-regulated in dHAND mutants, suggesting one poten-tial mechanism through which dHAND may functionin cardiac and vascular development.
Reciprocal interactions between vascular endothelialcells and mesenchymal cells are essential for angio-genesis and maintenance of the vasculature. Based ona model of endothelial-mesenchymal interaction pro-posed by Folkman and D’Amore, vascular mesenchy-mal cells are recruited to endothelial cells throughAng-1-Tie2 signaling, then differentiate into VSMCsor pericytes, possibly being induced by TGF-β signal-
ing (1). In dHAND mutants, numerous markers ofendothelial cell development were normally expressedand the mesenchymal cells surrounded endothelialtubes, suggesting that the step of mesenchymal re-cruitment is intact. This observation is consistent withthe results of marker analysis showing that Ang-1,Tie2, and COUP-TFII, which is a putative upstreamregulator of the Ang-1-Tie2 signaling pathway (39), areexpressed normally in dHAND-null embryos. Howev-er, in the absence of dHAND, VSMC differentiationwas arrested based on the lack of expression of SM22α.The failure of vascular mesenchymal cells to expressthe VSMC marker SM-22α could be a result of inade-quate interactions with endothelial cells or could be adirect effect of dHAND transcriptional regulation. Aproximal CArG box in the SM-22α promoter is essen-tial for smooth muscle–specific gene expression, butno binding sites for bHLH proteins (E-boxes) arerequired for SM-22α transcription (42). Therefore,SM-22α is unlikely to be a direct target of dHAND, butmore likely serves as a marker for abnormal vascularsmooth muscle differentiation.
TGF-β signaling pathways are also important forangiogenesis. Mouse embryos lacking endoglin, a TGF-β–binding protein expressed on the surface of endothe-lial cells, have vascular defects similar to those describedhere. Specifically, endoglin-null embryos have poorVSMC development and arrested endothelial remodel-ing, whereas vasculogenesis was normal (43). Targeteddeletion of a number of other genes involved in theTGF-β signaling pathways have defects in remodelingof the yolk sac vasculature (11–14). It will be interestingto determine if dHAND-related pathways and TGF-βsignaling converge during vascular development.
Similar to the endothelial-mesenchymal interactionin angiogenesis, reciprocal interactions between theendocardium and myocardium are essential for car-diogenesis. Development of myocardial trabeculationsis dependent upon secreted signals from the endo-cardium, many of which are common to the vascula-ture, including VEGF, Ang-1, and their respective recep-tors. The phenotype of dHAND-null embryos was
The Journal of Clinical Investigation | February 2000 | Volume 105 | Number 3 267
Figure 5Failure of vascular smooth muscle differentiation in dHAND mutants.Mesenchymal cells in wild-type (a) and dHAND mutants (b) expressed themesenchymal marker, COUPTFII, throughout the embryo. Expression oflacZ under the control of the SM-22α promoter was seen in the aortic archarteries (aa), aorta (ao), conotruncus (ct), and ventricle (v) of the heartof an E9.5 transgenic embryo (c). In the dHAND-null background, the SM-22-lacZ transgene expressed in the atrium (a) of the heart, but not in thevasculature (d) at E9.5. Embryos are shown in lateral views focusing onthe cardiac and vascular areas. Histologic analysis of wild-type (e) andmutant (f) embryos revealed absence of lacZ expression in the cells sur-rounding the aorta in dHAND-null embryos. Electron microscopy of trans-verse sections through the rostral part of E9.5 wild-type (g) and dHAND-null (h) embryos revealed vascular mesenchymal (m) cells surroundingendothelial (e) cells. Wild-type mesenchymal cells developed cytoplasmicprocesses to contact endothelial cells, whereas dHAND-null mesenchymalcells remained rounded without contacting endothelial cells.
similar to VEGF-heterozygous embryos (19), not onlyin the vasculature, but also in the myocardium wheretrabeculations failed to form. An unbiased search forgenes downstream of dHAND in the heart resulted incloning of neuropilin-1 and demonstration of neu-ropilin-1 downregulation in the hearts of dHANDmutants. Further evidence of neuropilin-1 downregu-lation in the yolk sac and other portions of the dHAND-null vasculature suggests a possible link between VEGFsignaling and dHAND function in the heart and ves-sels. Neuropilin-1 potentiates binding of VEGF165 withFlk-1, and mice lacking neuropilin-1 die from cardio-vascular defects between E10.5 and E12.5, similar todHAND. Thus, it is tempting to speculate that regula-tion of neuropilin-1 is important for dHAND’s role innot only cardiac but also vascular development.
Although our data indicate that vascular defects indHAND-null embryos may be partly mediated by dys-regulation of neuropilin-1, deficiencies in many yet
unknown genes downstream of dHAND likely con-tribute to the phenotype. In addition, we cannot for-mally eliminate the possibility that a subtle hemody-namic change from the cardiac defect might contributeto some part of the dHAND-null phenotype, althoughall our analyses were performed before any signs of car-diac failure was apparent. Hemodynamic forces (shearstress of blood flow) are important for vascular main-tenance (44), but its role in early vascular developmentremains unclear. Classic experiments in chick embryossuggest that hemodynamic forces may not be essentialfor initial development of the vasculature in vivo (45).Further studies are required to elucidate how genedefects and hemodynamic changes, respectively, couldcontribute to defects of early vascular development.
Although little is known about transcriptional regu-lation of vascular development, several recent reportshave described mouse mutants harboring vasculardefects overlapping those described here (37–39, 46, 47).
268 The Journal of Clinical Investigation | February 2000 | Volume 105 | Number 3
Figure 6Neuropilin-1 is downregulated in dHAND mutants. Neuropilin-1 was isolatedas a dHAND-dependent gene by subtractive hybridization between dHAND-mutant and wild-type hearts (a). Quantitative RT-PCR of neuropilin-1 in wild-type (+/+) and dHAND-null (–/–) hearts confirmed downregulation of neu-ropilin-1 in dHAND-mutant heart and yolk sac RNA (b). Cycles of PCRamplification are shown. RNA loading was controlled by assaying GAPDHexpression. Whole-mount in situ hybridization demonstrated neuropilin-1expression in the heart (ht) and aorta (arrowheads) of an E9.0 embryo in rightlateral view (c). At E9.5 (d), neuropilin-1 expression, viewed from the left lat-eral side, was seen in the aorta and aortic arch arteries (arrowheads), heart,pharyngeal arch (pa), limb bud (lb), and septum transversum (st). In dHANDmutants, neuropilin-1 was downregulated in the rostral aorta, pharyngeal arch-
es, and heart (e). Expression in the caudal aorta (arrowhead), limb bud, and septum transversum was intact. Transverse sectionsthrough the rostral part of embryos in d and e revealed neuropilin-1 expression in the endothelial (e) and mesenchymal (m) cells ofthe wild-type aorta (f), but no expression of neuropilin-1 in the dHAND-null aorta (g). h, head.

A member of the myocyte enhancer factor-2 family oftranscription factors, MEF2C, is required for VSMC dif-ferentiation and vascular morphogenesis (37). Embryoslacking MEF2C or dHAND have overlapping pheno-types in the heart (48) and vasculature (37). The MyoDfamily of bHLH proteins and the MEF2 family of tran-scription factors interact with one another and functionsynergistically during skeletal myogenesis (49). BecausedHAND, rather than MyoD family members, isexpressed in smooth and cardiac muscle, it is possiblethat dHAND functions together with MEF2 factorsduring cardiovascular development. The phenotypesafter genetic alteration of dHAND and MEF2C in miceis consistent with such a model warranting furtherstudies of direct interaction among these proteins.
The studies here showed that an unbiased screen forpotential mediators of dHAND function in the cardio-vascular system may have provided potential insight intothe mechanism through which dHAND might supportcardiovascular development. Subtraction of dHAND-nullheart from wild-type heart cDNA provided a tractableapproach to screening for many genes in molecular path-ways regulated by dHAND (50). This type of screenselects genes that may be one or several molecular stepsdownstream. Taking advantage of this approach, wefound neuropilin-1 as one of many genes downstream ofdHAND. This could be a powerful method for function-al genomics and can be used for any genes that are tar-geted in mice. Whether neuropilin-1 is a direct or indirecttarget for dHAND transcriptional activation remains tobe determined and awaits identification and analysis ofthe neuropilin-1 promoter. Given the important role ofVEGF signaling pathways in development and disease, itwill be interesting to determine how neuropilin-1 andother dHAND-dependent genes are involved in vascularmaintenance during embryogenesis, tumorigenesis, andother pathologic conditions.
After submission of this manuscript, a detailed analy-sis of the vascular defects in neuropilin-1 mutant embryoswas reported (Kawasaki, T., et al. 1999. A requirement ofneuropilin-1 in embryonic vessel formation. Development.126:4895–4902). The vascular abnormalities overlappedthose described here and those observed in endothelin-1mutants where dHAND is downregulated.
AcknowledgementsThe authors thank T. Sato for use of Tie2-lacZ trans-genic mice and helpful discussions, X. Chao for tech-nical assistance, S. Tsai for the COUPTFII probe,Michael Bennett for electron microscopy, and S. John-son for manuscript preparation and graphics. Thiswork was supported by grants from the NIH/NHLBI(R01 HL5781-01 and R01 HLDE62591-01) and Marchof Dimes to D. Srivastava.
1. Folkman, J., and D’Amore, P.A. 1996. Blood vessel formation: what is itsmolecular basis? Cell. 87:1153–1155.
2. Risau, W., and Flamme, I. 1995. Vasculogenesis. Annu. Rev. Cell Dev. Biol.11:73–91.
3. Risau, W. 1997. Mechanisms of angiogenesis. Nature. 386:671–674.4. Hanahan, D. 1997. Signaling vascular morphogenesis and maintenance.
Science. 277:48–50.5. Dumont, D.J., et al. 1994. Dominant-negative and targeted null muta-
tions in the endothelial receptor tyrosine kinase, tek, reveal a critical invasculogenesis of the embryo. Genes Dev. 8:1897–1909.
6. Sato, T.N., et al. 1995. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 376:70–74.
7. Davis, S., et al. 1996. Isolation of angiopoietien-1, a ligand for the TIE2receptor, by secretion-trap expression cloning. Cell. 87:1161–1169.
8. Suri, C., et al. 1996. Requisite role of angiopoietin-1, a ligand for the Tie2receptor, during embryonic angiogenesis. Cell. 87:1171–1180.
9. Wang, H.U., Chen, Z., and Anderson, D.J. 1998. Molecular distinctionand angiogenic interaction between embryonic arteries and veinsrevealed by ephrin-B2 and its receptor Eph-B4. Cell. 93:741–753.
10. Adams, R.H., et al. 1999. Roles of ephrinB ligands and EphB receptors incardiovascular development: demarcation of arterial/venous domains,vascular morphogenesis, and sprouting angiogenesis. Genes Dev.13:295–306.
11. Dickson, M.C., et al. 1995. Defective haematopoiesis and vasculogenesisin transforming growth factor-beta 1 knock out mice. Development.121:1845–1854.
12. Oshima, M., Oshima, H., and Taketo, M.M. 1996. TGF-beta receptortype II deficiency results in defects of yolk sac hematopoiesis and vascu-logenesis. Dev. Biol. 179:297–302.
13. Chang, H., et al. 1999. Smad5 knockout mice die at mid-gestation dueto multiple embryonic and extraembryonic defects. Development.126:1631–1642.
14. Yang, X., et al. 1999. Angiogenesis defects and mesenchymal apoptosisin mice lacking SMAD5. Development. 126:1571–1580.
15. Neufeld, G., Cohen, T., Gengrinovitch, S., Poltorak, Z. 1999. Vascularendothelial growth factor (VEGF) and its receptors. FASEB J. 13:9–22.
16. Kim, K.J., et al. 1993. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature.362:841–844.
17. Shalaby, F., et al. 1995. Failure of blood-island formation and vasculo-genesis in Flk-1-deficient mice. Nature. 376:62–66.
18. Fong, G.H., Rossant, J., Gertsenstein, M., and Breitman, M.L. 1995. Roleof the Flt-1 receptor tyrosine kinase in regulating the assembly of vas-cular endothelium. Nature. 376:66–70.
19. Carmeliet, P., et al. 1996. Abnormal blood vessel development and lethal-ity in embryos lacking a single VEGF allele. Nature. 380:435–439.
20. Ferrara, N., et al. 1996. Heterozygous embryonic lethality induced by tar-geted inactivation of the VEGF gene. Nature. 380:439–442.
21. Dumont, D.J., et al. 1998. Cardiovascular failure in mouse embryos defi-cient in VEGF receptor-3. Science. 282:946–949.
22. Soker, S., Takashima, S., Miao, H.Q., Neufeld, G., and Klagsburn, M.1998. Neuropilin-1 is expressed by endothelial and tumor cells as an iso-form-specific receptor for vascular endothelial growth factor. Cell.92:735–745.
23. Takagi, S., Tsuj, T., Amagai, T., Takamatsu, T., and Fujisawa, H. 1987.Specific cell surface labels in the visual centers of xenopus laevis tadpoleidentified using monoclonal antibodies. Dev. Biol. 122:90–100.
24. He, Z., and Tessier-Lavigne, M. 1997. Neuropilin is a receptor for theaxonal chemorepellent semaphorin III. Cell. 90:739–751.
25. Kitsukawa, T., Shimono, A., Kawakami, A., Kondoh, H., and Fujisawa,H. 1995. Overexpression of a membrane protein, neuropilin, in chimericmice causes anomalies in the cardiovascular system, nervous system andlimbs. Development. 121:4309–4318.
26. Kitsukawa, T., et al. 1997. Neuropilin-semaphorin III/D-mediatedchemorepulsive signals play a crucial role in peripheral nerve projectionin mice. Neuron. 19:995–1005.
27. Jan, Y.N., and Jan, L.Y. 1993. HLH proteins, fly neurogenesis, and verte-brate myogenesis. Cell. 75:827–830.
28. Olson, E.N., and Klein, W.H. 1994. bHLH factors in muscle develop-ment: dead lines and commitments, what to leave in and what to leaveout. Genes Dev. 8:1–8.
29. Shivdasani, R.A., Mayer, E.L., Orkin, S.H. 1995. Absence of blood for-mation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCL.Nature. 373:432–434.
30. Srivastava, D., Cserjesi, P., and Olson, E.N. 1995. A subclass of bHLHproteins required for cardiogenesis. Science. 70:1995–1999.
31. Srivastava, D., et al. 1997. Regulation of cardiac mesoderm and neuralcrest by the bHLH protein, dHAND. Nat. Genet. 16:154–160.
32. Thomas, T., Yamagishi, H., Overbeek, P.A., Olson, E.N., and Srivastava,D. 1998. The bHLH factors, dHAND and eHAND, specify pulmonaryand systemic cardiac ventricles independent of left-right sidedness. Dev.Biol. 196:228–236.
33. Schlaeger, T.M., Qin, Y., Fujiwara, Y., Magram, J., and Sato, T.N. 1995.Vascular endothelial cell lineage-specific promoter in transgenic mice.Development. 121:1089–1098.
34. Li, L., Miano, J.M., Mercer, B., and Olson, E.N. 1996. Expression of theSM22 promoter in transgenic mice provides evidence for distinct tran-scriptional regulatory programs in vascular and visceral smooth muscle
The Journal of Clinical Investigation | February 2000 | Volume 105 | Number 3 269

cells. J. Cell Biol. 132:1–11.35. Baldwin, H.S., et al. 1994. Platelet endothelial cell adhesion molecule-1
(PECAM-1/CD31): alternatively spliced, functionally distinct isoformsexpressed during mammalian cardiovascular development. Development.120:2539–2553.
36. Iwama, A., et al. 1993. Molecular cloning and characterization of mouseTIE and TEK receptor tyrosine kinase genes and their expression inhematopoietic stem cells. Biochem. Biophys. Res. Commun. 195:301–309.
37. Lin, Q., et al. 1998. Requirement of the MADS-box transcription factorMEF2C for vascular development. Development. 125:4565–4574.
38. Maltepe, E., Schmidt, J.V., Baunoch, D., Bradfield, C.A., and Simon, C.M.1997. Abnormal angiogenesis and response to glucose and oxygen dep-rivation in mice lacking the protein ARNT. Nature. 386:403–407.
39. Pereira, F.A., Qiu, Y., Zhou, G., Tsai, M.J., and Tsai, S.Y. 1999. The orphannuclear receptor COUP-TFII is required for angiogenesis and heartdevelopment. Genes Dev. 13:1037–1049.
40. Solway, J., et al. 1995. Structure and expression of a smooth muscle cell-specific gene, SM22 alpha. J. Biol. Chem. 270:13460–13469.
41. Li, L., Miano, J.M., Cserjesi, P., and Olson, E.N. 1996. SM22 alpha, amarker of adult smooth muscle, is expressed in multiple myogenic line-ages during embryogenesis. Circ. Res. 78:188–195.
42. Li, L., Liu, Z.-C., Mercer, M., Overbeek, P., and Olson, E.N. 1997. Distinctserum response factor-mediated regulatory networks governing SM22
transcription in smooth, skeletal and cardiac muscle cells. Dev. Biol.187:311–321.
43. Li, D.Y., et al. 1999. Defective angiogenesis in mice lacking endoglin. Sci-ence. 284:1534–1537.
44. Papadaki, M., Eskin, S.G. 1997. Effects of fluid shear stress on gene reg-ulation of vascular cells. Biotechnol. Prog. 13:209–221.
45. Chapman, W.B. 1918. The effect of the heart-beat upon the developmentof the vascular system in the chick. Am. J. Anat. 23:175–203.
46. Kuo, C.T., et al. 1997. The LKLF transcription factor is required for nor-mal tunica media formation and blood vessel stabilization duringmurine embryogenesis. Genes Dev. 11:2996–3006.
47. Tanaka, M., Chen, Z., Bartunkova, S., Yamasaki, N., and Izumo, S. 1999.The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of mul-tiple genes essential for heart development. Development. 126:1269–1280.
48. Lin, Q., Schwarz, J., Bucana, C., and Olson, E.N. 1997. Control of mousecardiac morphogenesis and myogenesis by transcription factor MEF2C.Science. 276:1404–1407.
49. Molkentin, J.D., Black, B.L., Martin, J.F., and Olson, E.N. 1995. Cooper-ative activation of muscle gene expression by MEF2 and myogenic bHLHproteins. Cell. 83:1125–1136.
50. Yamagishi, H., Garg, V., Matsuoka, R., Thomas, T., and Srivastava, D.1999. A molecular pathway revealing a genetic basis for human cardiacand craniofacial defects. Science. 283:1158–1161.
270 The Journal of Clinical Investigation | February 2000 | Volume 105 | Number 3





























