Embed Size (px)
Citation preview

BIOPOLY MERS VOL. 14, 2159-2179 (1975)
The Conformational Analysis of Adenosine Triphosphate by Classical Potential Energy
Calculations
0. E. MILLNER, JR. and JON A. ANDERSEN, Research and Development Department, Norwich Pharmacal Company, Norwich,
New York 13815
Synopsis
The conformational analysis of adenosine triphosphate was conducted by using classical potential energy calculations. All rotatable bonds were examined, i.e., no dihedral angles were fixed a t predetermined conformations except for the ribofuranose ring, which was held in the C(3’)-endo conformation-the conformation observed for adenosine in the crystal state.
The energy terms included in the total energy expression consist of nonbonded pairwise interaction, electrostatic pairwise interaction, free energy of solvation, and torsional bond potentials.
Two separate approaches were used in the conformational analyses. The first consisted of a sequential fragment approach where four bonds were rotated simultaneously at 30’ increments. Each fragment overlapped the preceding one by a t least one bond. All ro- tors were then simultaneously examined a t their minima and a t f15O. The second ap- proach consisted of a coarse grid search where all rotors were examined simultaneously, but only a t staggered positions. The low-energy conformations thus obtained were then used as starting conformations for a minimization routine based on the method of conju- gate directions. The first approach required about 40 hr of central processing unit (CPU) computer time, while the coarse grid/minimization approach required about 4 hr of CPU time.
Both the sequential fragment approach and the minimization approach yielded lowest- energy conformations which are remarkably similar to the solid-state conformation of C(3’)-endo ATP.
INTRODUCTION
The ubiquity of adenosine triphosphate (ATP) in biological systems has stimulated investigations of the conformation of the ATP molecule by X-ray spectroscopy,1,2 nuclear magnetic resonance s p e c t r o s ~ o p y , ~ ~ molecular orbital calculation^,^ and immunochemical studies.* In addi- tion, specific structural aspects of ATP have been examined by using ul- traviolet spectroscopy? and optical rotatory dispersion.lOJ1
Although several molecular orbital calculation^^^-^^ and classical po- tential energy (CPE) c a l c ~ l a t i o n s ~ ~ J ~ have been performed on nucleo- sides and nucleoside analogs, few calculations on nucleoside triphos- phates have been reported. The authors are aware of one molecular or-
2159
0 1975 by John Wiley & Sons, Inc.

2160 MILLNER AND ANDERSON
bital investigation of ATP7 and of no CPE calculations on nucleoside triphosphates. Furthermore, the molecular orbital investigation of the conformation of ATP by Perahia et al.7 consisted of a reduced treat- ment where only certain rotors were rotated, the remainder being fixed at the crystallographic values. Thus, it seems that a more complete ex- amination of the conformational degrees of freedom for ATP via CPE calculations is warranted. Classical potential energy calculations in the manner of Scheragal* have been used with noticeable success in the con- formational analysis of a variety of molecular type^.'^-^^ While the conformational analysis of large molecules by molecular orbital methods is restricted to the examination of a relatively few conformations, con- formational analysis by CPE calculations allows one to examine a large number of conformations. For example, the conformational analysis of a molecule with four rotors could be performed by allowing all four ro- tors to be rotated simultaneously at 30" increments. While an exami- nation of the total permutations (20,736) by molecular orbital methods would require far too much computer time, the computational time re- quired to evaluate all 20,736 conformations by CPE calculations is feasi- ble.
THEORETICAL METHOD
The theoretical method employed has been described in detail by Scheraga.18 Briefly, the approach taken here entails the determination
TABLE I Variables Used in Calculating dji and eji
Atom rvdw ,a ai x 1024,
A cm3 Npff
H 1.20 0.42b 0.9e 0 (hydroxyl) 1.52 0.59b 7 .Oe 0 (carbonyl) 1.52 0.846 1.0e 0 (ether or phosphate 1.52 0.64b 7.0e
0 (pendant phosphate 1.52 1.ooc 7.0e
C (aliphatic) 1.70 0.93b 5.2e C (aromatic) 1.70 1.22d 5.2e N (primary) 1.55 0.87b 6.le N (aromatic) 1.55 1.03b 6.le
1.80 3.00d 14.3f P
chain oxygen)
0 XY gen )
~ _ _ _ _ _ _______ a Van der Waals'radii taken from Bondi, A. (1964) J. Phys. Chem. 68, 441-456. b Taken from Ketelaar, J. A. A. (1958) Chemical Constitution, Elsevier Pub-
lishing Company, New York, p. 91. c Estimated on the basis of electron density on the pendant phosphate oxygens.
The effect of varying the value from 1.0 to 2.0 was found to be negligible. d From Ref. 27. e From Ref. 18. f From Scott, R. A. & Scheraga, H. A. (1965) J. Chem. Phys. 42,2209-2215.

CONFORMATIONAL ANALYSIS OF ATP 2161
of the molecular conformation which gives a minimum value for Etot in
(1)
E,b represents the nonbonded pairwise interaction energy, U j , which consists of a van der Waals’ repulsive energy contribution and a London attractive energy contribution. These are represented by the first and second terms, respectively, in the Lennard-Jones “6-12” equation (Eq. (2)).
Eq. (1).
Etot = Enb -I- Eel i- Esolv i- Etors
u.. 1J = d.. /r!a- IJ V e../rG. 1J Y (2)
The coefficient eij of the attractive term is obtained from the Slater- Kirkwood equation (Eq. (3))
TABLE I1 Constants for Nonbonded Potential Energy Equationa9b
CaI 370 car 546 874 H 128 202 47 OOH 278 404 89 217 Oc=o 372 552 124 283 369 0.0. 297 443 99 231 300 246 N-NH, 365 553 135 278 356 29 125 Nar 525 640 143 315 414 33 412 456 OPO 422 514 144 321 421 339 418 474 480 P 1150 1410 401 860 1138 917 1134 1291 1303 3567
a Key: C,I = aliphatic carbon;C, = aromatic carbon; OOH = hydroxyl oxygen; Cc=o = carbonyl oxygen; 00- = ether oxygen or phosphate chain oxygen; N-NH, = primary amine nitrogen; Nar = aromatic nitrogen; O p a = pendant phos- phate oxygen.
b The values given are for the interaction of the two atoms found in the column and row corresponding to those values.
where e is the electronic charge; h is Planck’s constant divided by 27r; m is the electronic mass; ai and aj are the atomic polarizabilities of atoms i and j ; and Ni and Nj are the “effective” numbers of valence shell elec- trons of atoms i and j . The coefficient dij of the repulsion term is ob- tained by minimizing U , in Eq. ( 2 ) when rij is equal to the sum of the van der Waals’ radii (rvdw) of atoms i and j . The values used for a,, Ni, and rvdw are given in Table I. The values obtained for eij and dij are given in Tables I1 and 111.

2162 MILLNER AND ANDERSON
TABLE I11 Constants for Nonbonded Potential Energy Equationa?b
dij x kcal a'*/mole
ca1 Car H OOH OC=O 0.0. N-NH, N, 0p.0- P
Ca1 286 car 477 740 H 38 7 1 45 OOH 149 245 17 80 OC=O 209 470 25 108 145 00- 146 224 15 78 100 9 4 N-NH, 203 285 32 110 145 122 27 Nar 352 311 28 117 173 135 166 181 OPQ 225 280 28 125 165 130 173 196 181 P 1015 1280 139 562 750 559 785 900 825 3660
a Key: Gal= aliphatic carbon; Car = aromatic carbon; OOH = hydroxyl oxygen; CC=O = carbonyl oxygen; 0.0. = ether oxygen or phosphate chain oxygen; N-NH = primary amine nitrogen; N, = aromatic nitrogen; 0p.0 = pendant phos- phati oxygen.
b The values given are for the interaction of the two atoms corresponding to the column and row of the respective numerical values.
Eel represents the electrostatic component of the total energy and is calculated with the coulombic potential function (Eq. (4)).
Here, qi and qj are the net atomic charges of the respective atoms and rij is their interatomic distance. D is the apparent local dielectric constant and has been assigned the value of 3.5 in this study-a value within the range of dielectric constants examined by Brant and F l ~ r y , ~ ~ who took the experimental dielectric constants of polymers into consideration. The net atomic charges were obtained from Alving and Laki's CNDO/2 results25 on ADP-3 and ribose phosphate and extrapolated to ATPW3. The phosphate atoms, pendant oxygen, and intrachain oxygens were as- signed atomic charges of 0.32, -0.49, and -0.36, respectively. Values of -0.31 and 0.21 were assigned to the oxygen and hydrogen atoms of the y-phosphate hydroxyl. Remaining atomic charge assignments are list- ed in Ref. 24. The numerical factor is included in Eq. (4) to yield ener- gies in units of kcal/mole.
Esolv, representing the contribution to the total energy made by the free energy of solvation, is found by summing the free-energy change for each atom that arises as the result of other approaching atoms (Eq. (5)).
Gi represents the free-energy contribution arising from the removal of solvent from atom i and is calculated according to Eq. (6)

CONFORMATIONAL ANALYSIS OF ATP 2163
where GP represents the free-energy change resulting from the removal of one water molecule from the first hydration shell of atom i. 4(Wi,AJ represents the function shown in Eq. (7)
where Wi is the amount of water removed from atom i by the approach of all other atoms, and Ai is the number of water molecules belonging to the first shell of atom i.
This function was designed to give a value of zero when Wi = 0 and a maximum value of Ai.26 Wi is calculated bywmming the amount of water, q i j removed from atom i by approach of atom j for all approach- ing atoms,
Wi = C qij (8) j # i
and qi; is described by Eq. (9), where Vj is proportional to the volume of the approaching atom j .
(9) g(rij) is a continuous function (Eq. (10)) designed to give qi, a value of zero when the interatomic distance ri, is equal to ro, the sum of the van der Waals’ radii of atoms i and j plus 2.2 A.
4. . i j = V . ,g( r i j )
g(r;j) was designed26 to be a very steep function which would reflect the assumption that when ri; becomes less than ro an amount of water is displaced which is proportional to atom j and that further decreases in rij down to the sum of the van der Waals’ radii should not profoundly affect the solvation. When r > ro, g(rij) is assigned a value of zero.
The values for GP, A;, and V; used in the calculation of the free-ener- gy change due to solvation effects are listed in Table IV.
Etors represents the torsional energy component and is represented by Eq. (11)
(11)
where U$ is the barrier height of rotation about the respective bond, X is the periodicity of the barrier, and 4 is the angular rotational incre- ment. The P-0 bonds were assigned a barrier height of 1.0 kcal/mole while the C(5’)-0 and C(4’)-C(5’) bonds were assigned barrier heights of 3.0 and 3.5 kcal/mole, re~pectively.~~ The barrier height of the glyco- sidic bond was considered to be negligible in accordance with Lakshmi- narayanan and Sasisekharan.16 Each bond was considered to have threefold periodicity.
Etors = (U4/2) (1 + cos X 4)

2164 MILLNER AND ANDERSON
TABLE IV Parameters Employed in the Calculation of Esob
Atom or group
H (amino) H (hydroxyl) 0 (ether, carbonyl,
phosphate ) 0 (hydroxyl) C (aromatic) CH (aromatic) CH (aliphatic) CH, (aliphatic) N (aromatic, amide) P
G: ,a A fa kcal/mole (solvation number)
0.31 0.31 0.94
0.84 0.11 0.11
-0.13 -0.13
0.63
2 2 4
5 2 3 2 3 2 O*
0.102 0.096 0.225c
0.172 0.158 0.269 0.226 0.342 0.110 -
a Taken from Ref. 26. b Calculated by dividing by 30 the volume of the atom or group (in A3) given
C Vi calculated by using an average value for the volume of an oxygen atom from
d The solvation of the phosphorus atoms was assumed to be negligible based
by Bondi (J. Phys. Chem. 68,441-456 (1964)).
those given by Bondi (J . Phys. Chem. 68, 441-456 (1964)).
upon space-filling models.
CONFORMATIONAL ANALYSIS
First Method
The nomenclature of the rotatable bonds is presented in Figure 1. The immensity of the number of possible conformations if one examines all rotors simultaneously at 30' increments of rotation (1211, or almost 9 trillion for ATP-3) makes it apparent that a fragment approach must be taken.
1-Methyltriphosphate. Bond angles and distances required for the coordinates program2* were obtained from refinements by Cruick- shank29 on the X-ray crystallographic data of sodium triphosphate de- termined by Davies and C ~ r b r i d g e . ~ ~ In the conformational analysis of the trivalent anion of l-methyltriphosphate, w3!, w3, and w2t were rotated simultaneously at 30' increments from 0' to 360'. This was followed by simultaneous rotation of 0 2 ' , wg, wl', and ax', 01, 6 respectively. The calculated minimum for each rotor was incorporated in the succeeding calculations. All local secondary minima within 1.5 kcal of the calculat- ed lowest-energy conformation were carried through successive calcula- tions also.
ATP. The geometry of the adenine-ribose moiety (C(3')-endo) was taken from Lai and Marsh.31 The rotors x, $, 4, and w1 were rotated si- multaneously at 30' increments from 0' to 360' starting from eclipsed positions. Rotors w1' through w3' were fixed at their predetermined minima. This was followed by simultaneous rotation of $, 4, w1, and w1'
with x examined at positions found in all conformations within approxi-

CONFORMATIONAL ANALYSIS OF A T P 2165
Fig. 1. Nomenclature of the rotatable bonds examined in the conformational analyses.
mately 2 kcal of the calculated lowest-energy conformation ( O O , 300°, 330O). Next, rotations about a and p were examined simultaneously along with x and II/ to determine the effect of the ribose hydroxyls on the preferred conformations at x and II/.
The results of the fragment approach were further examined by si- multaneous rotations around all rotors, excluding a and p. To do this, each rotor was examined at its predetermined minimum energy confor- mation and at f30°. The results were then refined by repeating the procedure at f 1 5 O increments.
Second Method
A second approach to the conformational analysis of ATP was per- formed in the following manner. Simultaneous rotations were made at all bonds from x through w3’, excluding bonds a and p (Fig. 1). In order to keep the computer time at a practical level, only the energies (Eq. (1)) of the staggered conformations (60°, 180°, 300°, where the starting, eclipsed conformation is Oo) were determined. The results from the grid search were then used in a Powell minimization s u b r o ~ t i n e ~ ~ , ~ ~ based on the method of conjugate directions. Starting conformations for the minimization subroutine were selected from the low-energy con- formations of the grid search in the following manner. Conformations corresponding to various values of x through w3 were considered if, for all three positions of w y , the total conformational energy was within 14 kcal/mole of the grid minimum. This yielded 3 X 36 conformations-36 different permutations of x through w3 and three staggered positions of w3’. From these, only the 36 conformations corresponding to different permutations of x through w3 were used as starting conformations. wgf was given a starting value corresponding to its respective minimum for each of the above 36 conformations. The step sizes were set so that each rotor was incremented by f60° in the first step of the minimiza- tion procedure to ensure that all the conformational space between the eclipsed conformations was examined.
RESULTS AND DISCUSSION
The conformational energy characteristics of adjacent rotors are sum- marized in the energy contour plots shown in Figures 2-9. The con-
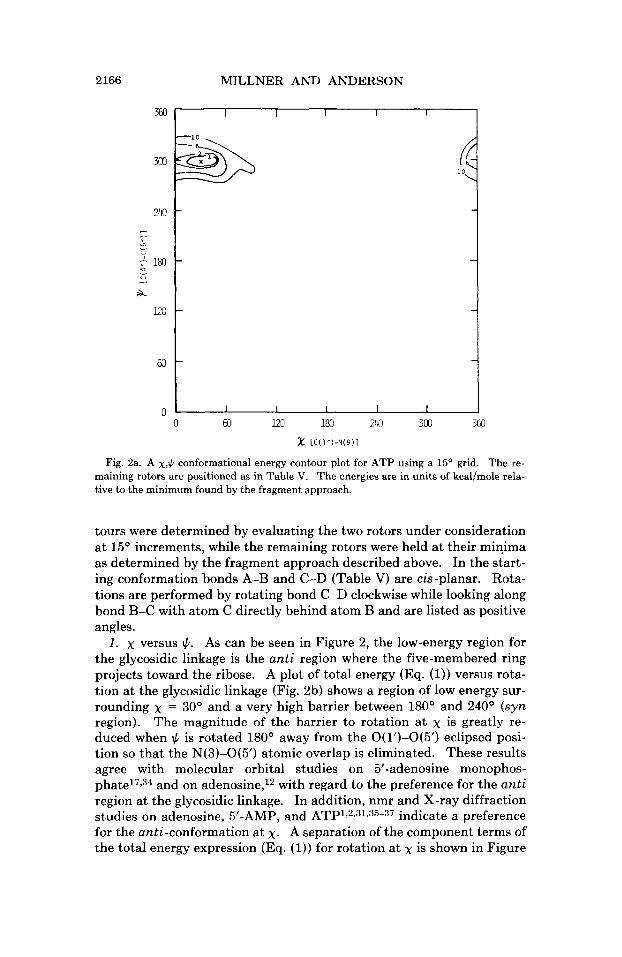
MILLNER AND ANDERSON
0 60 120 180 2110 500 3Go
x CC(I')-N(9)1
Fig. 2a. A x,$ conformational energy contour plot for ATP using a 15' grid. The re- maining rotors are positioned as in Table v. The energies are in units of kcal/mole rela- tive to the minimum found by the fragment approach.
tours were determined by evaluating the two rotors under consideration at 15' increments, while the remaining rotors were held at their minima as determined by the fragment approach described above. In the start- ing conformation bonds A-B and C-D (Table V) are cis-planar. Rota- tions are performed by rotating bond C-D clockwise while looking along bond B-C with atom C directly behind atom B and are listed as positive angles.
1. x versus $. As can be seen in Figure 2, the low-energy region for the glycosidic linkage is the anti region where the five-membered ring projects toward the ribose. A plot of total energy (Eq. (1)) versus rota- tion at the glycosidic linkage (Fig. 2b) shows a region of low energy sur- rounding x = 30' and a very high barrier between 180' and 240' (syn region). The magnitude of the barrier to rotation at x is greatly re- duced when $ is rotated 180' away from the O(1')-O(5') eclipsed posi- tion so that the N(3)-O(5') atomic overlap is eliminated. These results agree with molecular orbital studies on 5'-adenosine monophos-
and on adenosine,12 with regard to the preference for the anti region at the glycosidic linkage. In addition, nmr and X-ray diffraction studies on adenosine, 5'-AMP, and ATP1,2,31,35-37 indicate a preference for the anti-conformation at x. A separation of the component terms of the total energy expression (Eq. (1)) for rotation at x is shown in Figure
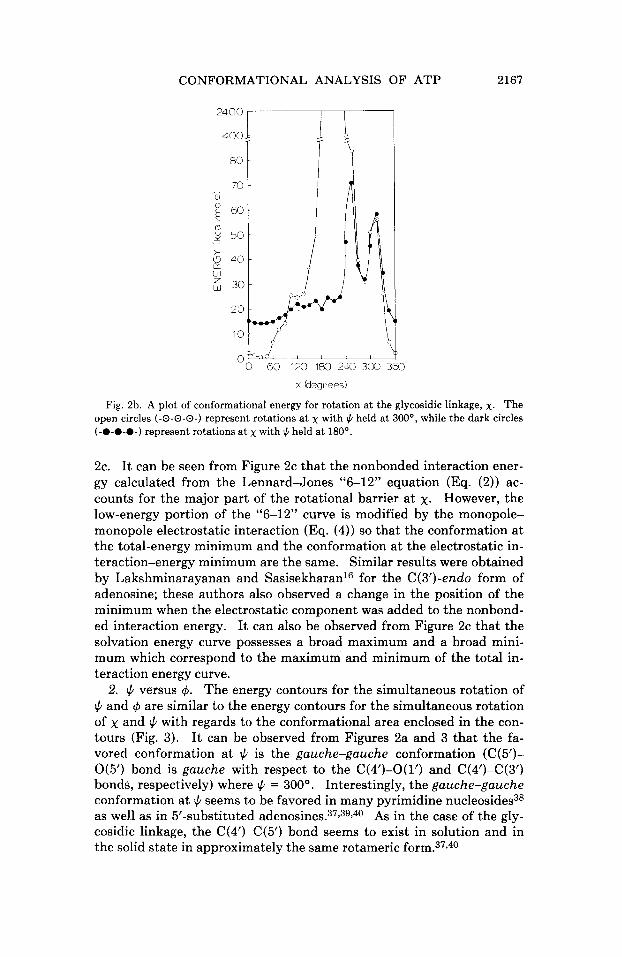
CONFORMATIONAL ANALYSIS OF ATP 2167
o h Go 40 ,&I 24c 3&J 362
x (degrees)
Fig. 2b. A plot of conformational energy for rotation a t the glycosidic linkage, x. The open circles (-@-O-@-) represent rotations a t x with 1c. held a t 300°, while the dark circles (-O-O-O-) represent rotations a t x with J. held a t 180’.
2c. It can be seen from Figure 2c that the nonbonded interaction ener- gy calculated from the Lennard-Jones “6-12” equation (Eq. (2)) ac- counts for the major part of the rotational barrier a t x. However, the low-energy portion of the “6-12” curve is modified by the monopole- monopole electrostatic interaction (Eq. (4)) so that the conformation at the total-energy minimum and the conformation at the electrostatic in- teraction-energy minimum are the same. Similar results were obtained by Lakshminarayanan and Sasisekharanl6 for the C(3’)-endo form of adenosine; these authors also observed a change in the position of the minimum when the electrostatic component was added to the nonbond- ed interaction energy. It can also be observed from Figure 2c that the solvation energy curve possesses a broad maximum and a broad mini- mum which correspond to the maximum and minimum of the total in- teraction energy curve.
2. $ versus #. The energy contours for the simultaneous rotation of $ and # are similar to the energy contours for the simultaneous rotation of x and $ with regards to the conformational area enclosed in the con- tours (Fig. 3). It can be observed from Figures 2a and 3 that the fa- vored conformation at I) is the gauche-gauche conformation (C(5’)- O(5’) bond is gauche with respect to the C(4’)-O(1’) and C(4’)-C(3’) bonds, respectively) where $ = 300”. Interestingly, the gauche-gauche conformation at $ seems to be favored in many pyrimidine n u c l e o ~ i d e s ~ ~ as well as in 5’-substituted a d e n o s i n e ~ . ~ ~ . ~ ~ , ~ ~ As in the case of the gly- cosidic linkage, the C(4’)-C(5’) bond seems to exist in solution and in the solid state in approximately the same rotameric
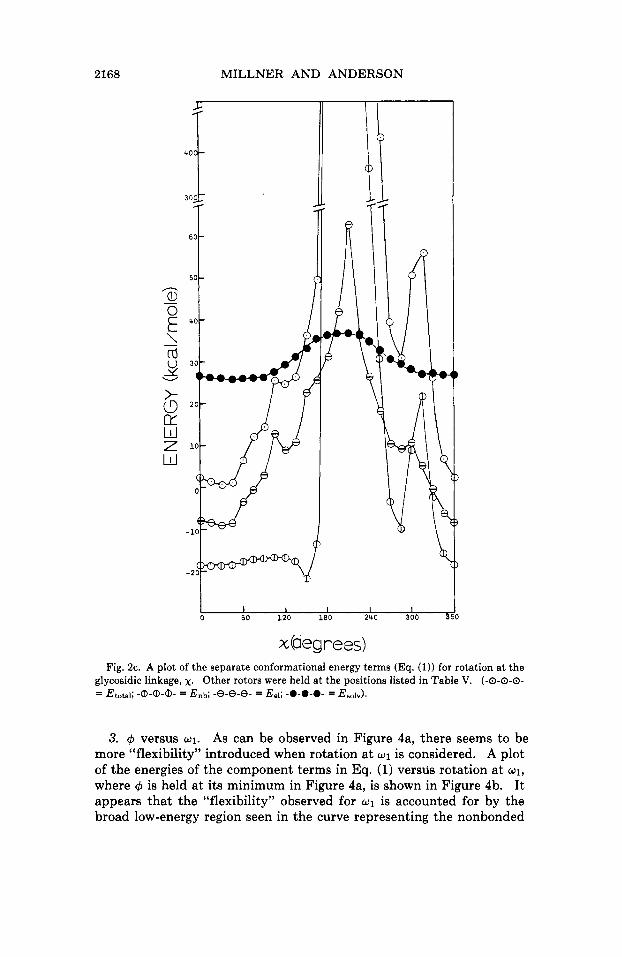
2168 MILLNER AND ANDERSON
50 i
-20 c -v 1 I I I 2QO I 300 I
60 120 180 3
x Oeg reed Fig. 2c. A plot of the separate conformational energy terms (Eq. (1)) for rotation at the
glycosidic linkage, x. Other rotors were held at the positions listed in Table V. (-0-0-0- = E ~ ~ ~ ~ ~ ; am- = E n b ; -8-8-8- = E,~; -0-0-0- = E ~ ~ ~ ~ ) .
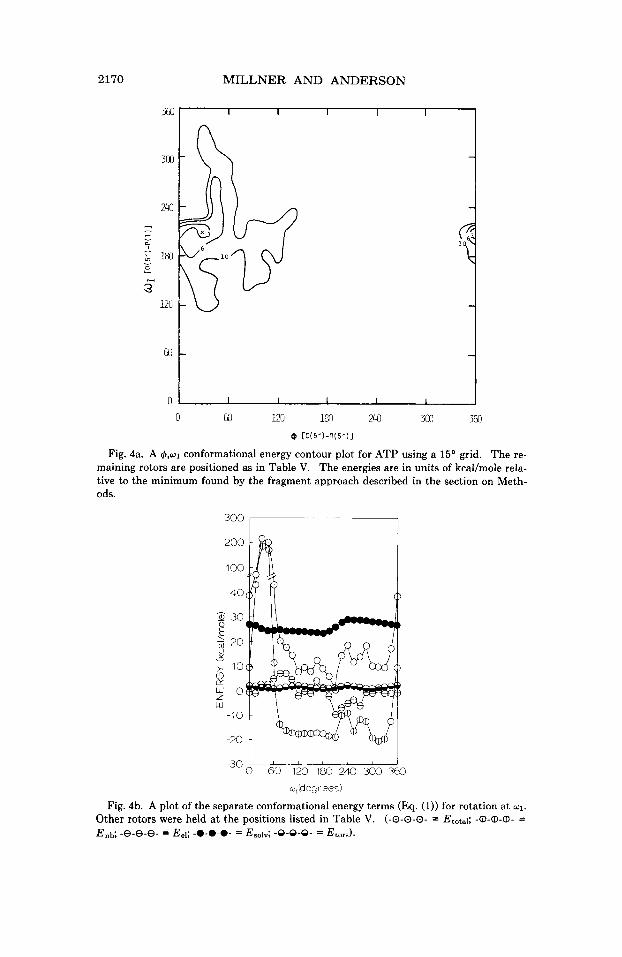
3. rp versus 01. As can be observed in Figure 4a, there seems to be more “flexibility” introduced when rotation at w1 is considered. A plot of the energies of the component terms in Eq. (1) versds rotation at w1,
where rp is held at its minimum in Figure 4a, is shown in Figure 4b. It appears that the “flexibility” observed for w1 is accounted for by the broad low-energy region seen in the curve representing the nonbonded
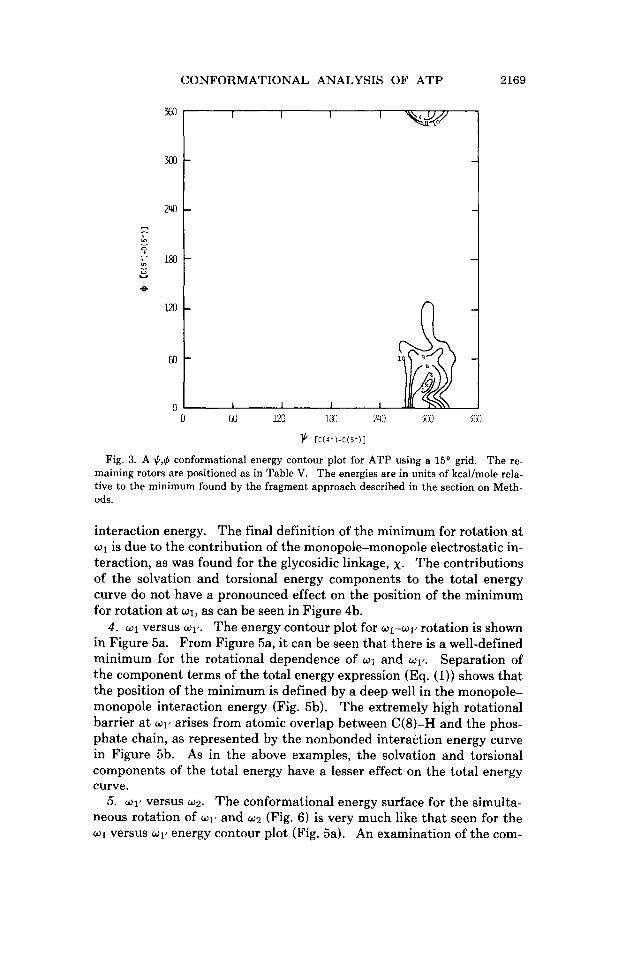
CONFORMATIONAL ANALYSIS OF ATP 2169
1 CC(4’)-C(5’)1 Fig. 3. A $,$ conformational energy contour plot for ATP using a 15’ grid. The re-
maining rotors are positioned as in Table v. The energies are in units of kcal/mole rela- tive to the minimum found by the fragment approach described in the section on Meth- ods.
interaction energy. The final definition of the minimum for rotation at w1 is due to the contribution of the monopole-monopole electrostatic in- teraction, as was found for the glycosidic linkage, x. The contributions of the solvation and torsional energy components to the total energy curve do not have a pronounced effect on the position of the minimum for rotation a t w l , as can be seen in Figure 4b.
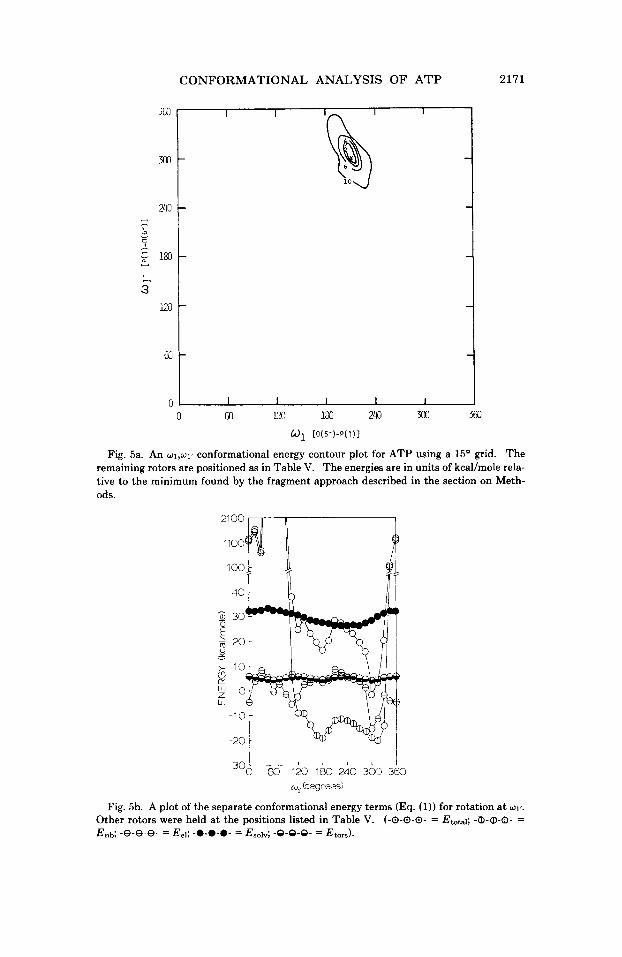
4. w1 versus WIT. The energy contour plot for w1-w1’ rotation is shown in Figure 5a. From Figure 5a, it can be seen that there is a well-defined minimum for the rotational dependence of w1 and w1‘. Separation of the component terms of the total energy expression (Eq. (1)) shows that the position of the minimum is defined by a deep well in the monopole- monopole interaction energy (Fig. 5b). The extremely high rotational barrier a t w1’ arises from atomic overlap between C(8)-H and the phos- phate chain, as represented by the nonbonded interaction energy curve in Figure 5b. As in the above examples, the solvation and torsional components of the total energy have a lesser effect on the total energy curve.
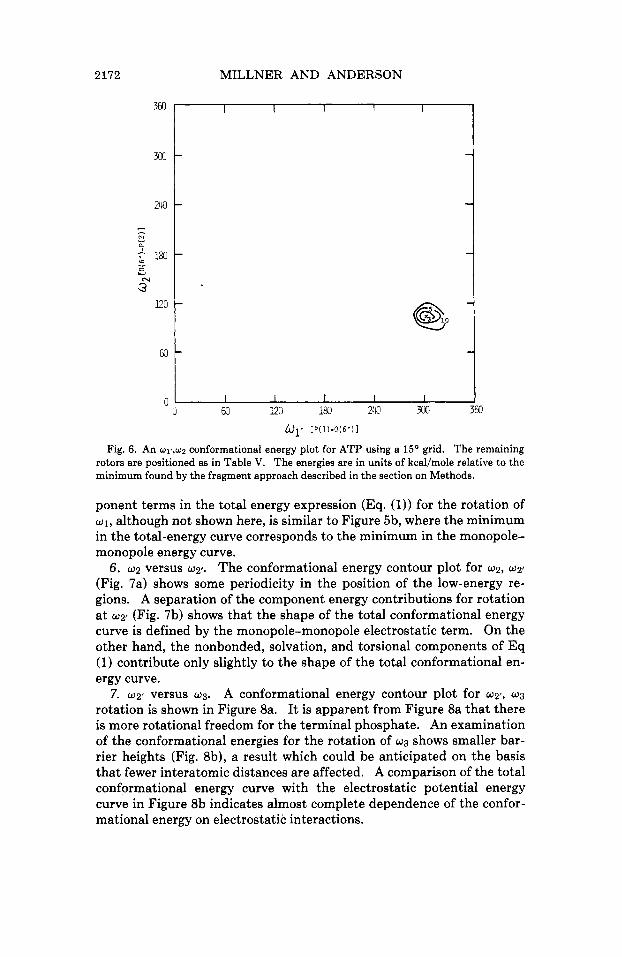
5. 01’ versus w2. The conformational energy surface for the simulta- neous rotation of 01’ and w2 (Fig. 6) is very much like that seen for the w1 versus w1’ energy contour plot (Fig. 5a). An examination of the com-
2170 MILLNER AND ANDERSON
7co I I I I I
-
-
-
6 0 - -
200
100
40
I 0 63 120 1 8 0 240 3oc) 3fX
- 3 0 ' ' ' ' ' '
o,(degrees)
Fig. 4b. A plot of the separate conformational energy terms (Eq. (1)) for rotation at w1.
Other rotors were held at the positions listed in Table V. (-0-0-0- = Etotsl; -@@-a- = En& -8-8-8- = Eel; -0-0-0- = EsolV; -9-9-9- = EtorJ.
CONFORMATIONAL ANALYSIS OF ATP 2171
0 0 wl 120 120 2'10 300
w1 "5-)-P(1)1
Fig. 5a. An w1,w1' conformational energy contour plot for ATP using a 15' grid. The remaining rotors are positioned as in Table V. The energies are in units of kcal/mole rela- tive to the minimum found by the fragment approach described in the section on Meth- ods.
'C - 30 ? 2 20
$? g o
Y t 10 -
W
-10
-20
w,.(degre?sj
Fig. 5b. A plot of the separate conformational energy terms (Eq. (1)) for rotation at w1'.
Other rotors were held at the positions listed in Table V. (-0-0-0- = Etohl; -0-CD-0- = En& -8-8-8- = Eel; -0-0-0- = Esolv; -9-9-9- = Et,,,).
2172 MILLNER AND ANDERSON
360 I I 1 I I I
300
F- - N f - f la0
3" - 0 Y
1 Fig. 6. An w1,,w2 conformational energy plot for ATP using a 1 5 O grid. The remaining
rotors are positioned as in Table V. The energies are in units of kcal/mole relative to the minimum found by the fragment approach described in the section on Methods.
ponent terms in the total energy expression (Eq. (1)) for the rotation of w1, although not shown here, is similar to Figure 5b, where the minimum in the total-energy curve corresponds to the minimum in the monopole- monopole energy curve.
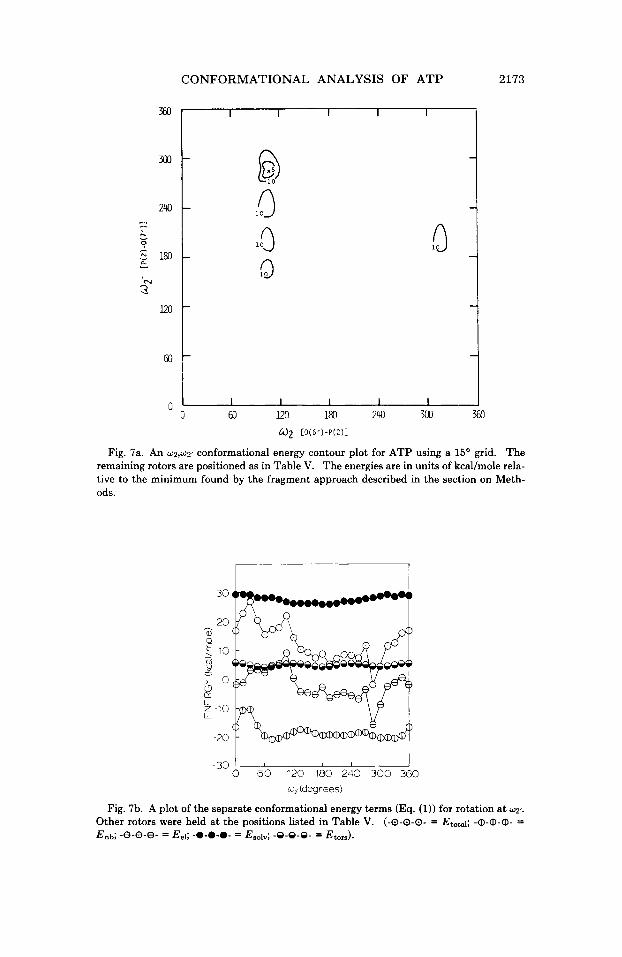
6. w2 versus w2'. The conformational energy contour plot for w2, w2'
(Fig. 7a) shows some periodicity in the position of the low-energy re- gions. A separation of the component energy contributions for rotation at w2' (Fig. 7b) shows that the shape of the total conformational energy curve is defined by the monopole-monopole electrostatic term. On the other hand, the nonbonded, solvation, and torsional components of Eq (1) contribute only slightly to the shape of the total conformational en- ergy curve.
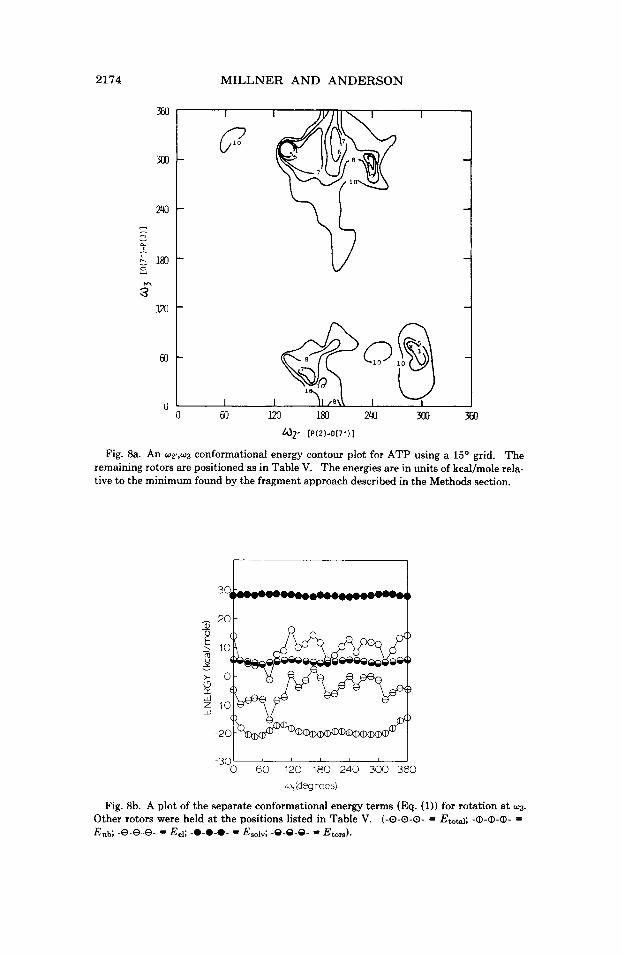
7. w21 versus 0 3 . A conformational energy contour plot for W T , w3
rotation is shown in Figure 8a. It is apparent from Figure 8a that there is more rotational freedom for the terminal phosphate. An examination of the conformational energies for the rotation of w3 shows smaller bar- rier heights (Fig. 8b), a result which could be anticipated on the basis that fewer interatomic distances are affected. A comparison of the total conformational energy curve with the electrostatic potential energy curve in Figure 8b indicates almost complete dependence of the confor- mational energy on electrostatik interactions.
CONFORMATIONAL ANALYSIS OF ATP 2173
0 0 60 I20 180 2110 300 360
W2 [O(G*)-P(Z)I
Fig. 7a. An w2,wz’ conformational energy contour plot for ATP using a 15’ grid. The remaining rotors are positioned as in Table V. The energies are in units of kcal/mole rela- tive to the minimum found by the fragment approach described in the section on Meth- ods.
33 0 60 120 180 240 300 360
o, (degrees)
Fig. 7b. A plot of the separate conformational energy terms (Eq. (1)) for rotation a t WZ,.
Other rotors were held a t the positions listed in Table V. (-0-0-0- = E b d ; -a-a)-a)- = Enb; -8-8-8- = Eel; -0-0-0- = EsOiv; -9-9-9- = Et,,,).
MILLNER AND ANDERSON
0
w2- [P(2)-0(7')1
Fig. 8a. An wy,w3 conformational energy contour plot for ATP using a 15O grid. The remaining rotors are positioned as in Table V. The energies are in units of kcal/mole rela- tive to the minimum found by the fragment approach described in the Methods section.
LLI
-2
-30 ' I
w,(degrees)
0 GO 120 180 240 300 360
Fig. 8h. A plot of the separate conformational energy terms (Eq. (1)) for rotation a t w3.
Other rotors were held at the positions listed in Table V. (-0-0-0- = Etotal; -Q-Q-Q- = Enb; -8-8-8- = Eel; -0-0-0- = Esolv; -0-9-8- = Eb,).
CONFORMATIONAL ANALYSIS OF ATP 2175
360
m
24c - - 0 m 0
a - - 2 la Y
3" I20
Go
0
W j I N ~ - ) - P ( ~ ) I
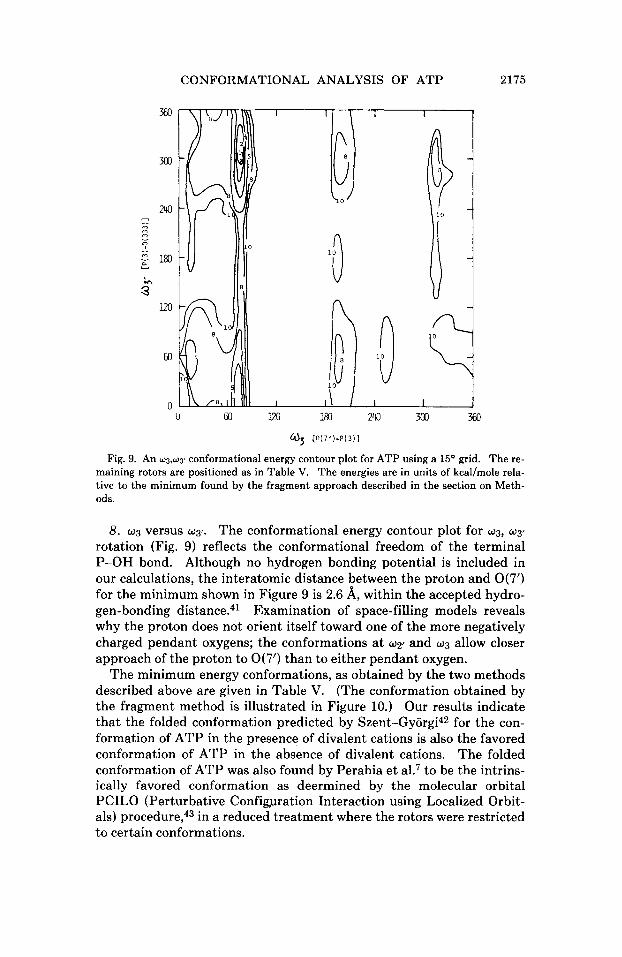
Fig. 9. An w3,wy conformational energy contour plot for ATP using a 15O grid. The re- maining rotors are positioned as in Table v. The energies are in units of kcal/mole rela- tive to the minimum found by the fragment approach described in the section on Meth- ods.
8. wg versus wg~. The conformational energy contour plot for wg, wg'
rotation (Fig. 9) reflects the conformational freedom of the terminal P-OH bond. Although no hydrogen bonding potential is included in our calculations, the interatomic distance between the proton and O(7') for the minimum shown in Figure 9 is 2.6 8, within the accepted hydro- gen-bonding distance.41 Examination of space-filling models reveals why the proton does not orient itself toward one of the more negatively charged pendant oxygens; the conformations at up' and wg allow closer approach of the proton to O(7') than to either pendant oxygen.
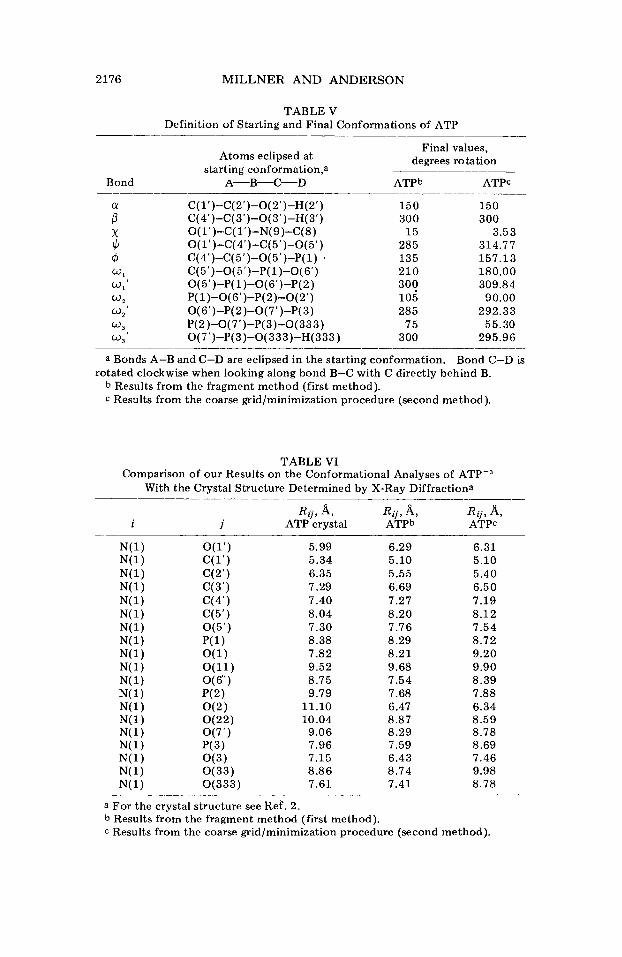
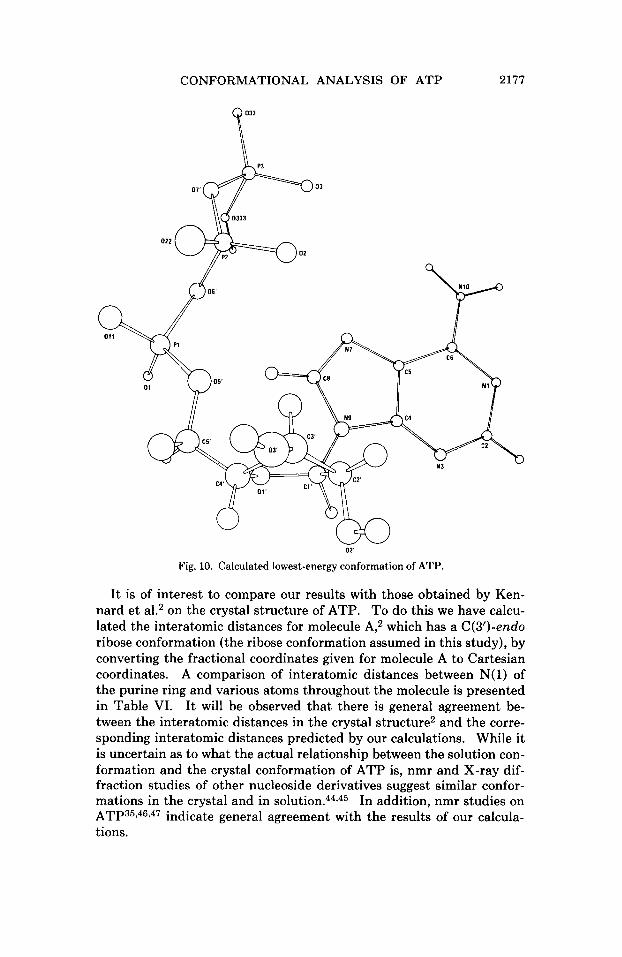
The minimum energy conformations, as obtained by the two methods described above are given in Table V. (The conformation obtained by the fragment method is illustrated in Figure 10.) Our results indicate that the folded conformation predicted by S~ent-GyOrgi~~ for the con- formation of ATP in the presence of divalent cations is also the favored conformation of ATP in the absence of divalent cations. The folded conformation of ATP was also found by Perahia et al.7 to be the intrins- ically favored conformation as deermined by the molecular orbital PCILO (Perturbative Configpration Interaction using Localized Orbit- als) p r o ~ e d u r e , ~ ~ in a reduced treatment where the rotors were restricted to certain conformations.
2176 MILLNER AND ANDERSON
TABLE V Definition of Starting and Final Conformations of ATP
Bond
Atoms eclipsed at starting conformation,a
A-B-C-D _________________
C( 1‘)-C( 2’)-O( 2’)-H(2‘) C( 4’)-C(3’)-0(3’)-H( 3‘) O(l‘)-C( 1’)-N(9)-C(8) O( 1‘ )-C( 4’)-C( 5’)-O( 5’)
C(5’)-O( 5‘)-P( 1 )-O( 6’) O( 5’)-P( 1)-0(6’)-P(2) P( 1)-O( 6’)-P( 2)-O(2‘) O(6‘)-P( 2)-0(7’)-P(3) P( 2 )-O( 7 ‘ )-P( 3)-O( 33 3 ) O( 7’)-P( 3)-O( 333)-H( 333 )
C(4‘)-C(5’)-0(5’)-P( 1) ’
Final values, degrees rotation
ATPb ATPc
250 300
15 285 135 210 30q 105 285
75 300
150 300
3.53 314.77 157.13 180.00 309.84 90.00
292.33 55.30
295.96
a Bonds A-B and C-D are eclipsed in the starting conformation. rotated clockwise when looking along bond B-C with C directly behind B.
b Results from the fragment method (first method). c Results from the coarse grid/minimization procedure (second method).
Bond C-D is
TABLE VI Comparison of our Results on the Conformational Analyses of ATP-3
With the Crystal Structure Determined by X-Ray Diffractiona
2
5.99 5.34 6.35 7.29 7.40 8.04 7.30 8.38 7.82 9.52 8.75 9.79
11.10 10.04 9.06 7.96 7.15 8.86 7.61
6.29 5.10 5.55 6.69 7.27 8.20 7.76 8.29 8.21 9.68 7.54 7.68 6.47 8.87 8.29 7.59 6.43 8.74 7.41
6.31 5.10 5.40 6.50 7.19 8.12 7.54 8.72 9.20 9.90 8.39 7.88 6.34 8.59 8.78 8.69 7.46 9.98 8.78
a For the crystal structure see Ref. 2. b Results from the fragment method (first method). c Results from the coarse grid/minimization procedure (second method).
CONFORMATIONAL ANALYSIS OF A T P 2177
Fig. 10. Calculated lowest-energy conformation of ATP.
It is of interest to compare our results with those obtained by Ken- nard et a1.2 on the crystal structure of ATP. To do this we have calcu- lated the interatomic distances for molecule A,2 which has a C(3’)-endo ribose conformation (the ribose conformation assumed in this study), by converting the fractional coordinates given for molecule A to Cartesian coordinates. A comparison of interatomic distances between N( 1) of the purine ring and various atoms throughout the molecule is presented in Table VI. It will be observed that there is general agreement be- tween the interatomic distances in the crystal structure2 and the corre- sponding interatomic distances predicted by our calculations. While it is uncertain as to what the actual relationship between the solution con- formation and the crystal conformation of ATP is, nmr and X-ray dif- fraction studies of other nucleoside derivatives suggest similar confor- mations in the crystal and in s o l ~ t i o n . ~ ~ , ~ ~ In addition, nmr studies on ATP35,46,47 indicate general agreement with the results of our calcula- tions.

2178 MILLNER AND ANDERSON
The authors gratefully acknowledge the assistance of Dr. Rodney P. Basson, Dr. Victor Day, and Mr. Guy Pierce in computerizing the methods used in this study and Dr. Lester Shipman for his helpful suggestions regarding the Powell minimization procedure. The authors also express their appreciation to Ms. Loretta Downey, Ms. Betty Palmer, and Ms. Carol Titus for their technical assistance in the preparation of computer input.
References
1. Kennard, O., Isaacs, N. W., Coppola, J. C., Kirby, A. J., Warren, S., Motherwell, W. D. S., Watson, D. G., Wampler, W. L., Chenery, D. H., Larson, A. C., Kerr, K. A. & di San- severino, L. R. (1970) Nature 225,333-336.
2. Kennard, O., Isaacs, N. W., Motherwell, W. D. S., Coppola, J. C., Wampler, D. L., Larson, A. C. & Watson, D. G. (1971) Proc. Royal SOC. (London), Ser. A 325,401-436.
3. Cohn, M. & Hughes, T. R. (1960) J. Biol. Chem. 235,3250-3253. 4. Cohn, M. & Hughes, T. R. (1962) J. Biol. Chem. 237,176-181. 5. Happe, J. A. & Morales, M. (1966) J. Amer. Chem. Soc. 88,2077-2088. 6. Glassman, T. A., Cooper, C., Harrison, L. W. & Swift, T. J. (1971) Biochemistry, 10,
7. Perahia, D., Pullman, B. & Saran, A. (1972) Biochem. Biophys. Res. Comm. 47,
8. Estrada-Parra, S. & Garcia-Ortigoza, E. (1972) Zmmunochem. 9,799-807. 9. Brintzinger, H. (1963) Biochim. Biophys. Acta 77,343-345.
843-851.
1284-1289.
10. Lin, C. Y., Urry, D. W. & Eyring, H. (1964) Biochem. Biophys. Res. Comm. 17,
11. McCormick, W. G. & Levedahl, B. H. (1959) Biochim. Biophys. Acta 34,303-308. 12. Berthod, H. & Pullman, B. (1971) Biochim. Biophys. Acta 232,595-606. 13. Jordan, F. & Pullman, B. (1968) Theor. Chim. Acta (Berl.) 9,242-252. 14. Jordan, F. (1973) Biopolymers 12,243-255. 15. Saran, A., Perahia, D. & Pullman, B. (1973) Theor. Chim. Acta (Berl.) 30,31-44. 16. Lakshminarayanan, A. V. & Sasisekharan, V. (1969) Biopolymers 8,475-488. 17. Jordan, F. (1973) J. Theor. Biol. 41,375-395. 18. Scheraga, H. A. (1968) Adu. Phys. Org. Chem. 6,103-184. 19. Ponnuswamy, P. K., Warme, P. K. & Scheraga, H. A. (1973) Proc. Natl. Acad. Sci.
( U S ) 70,830-833. 20. Ponnuswamy, P. K., McGuire, R. F. & Scheraga, H. A. (1973) Znt. J. Peptide Pro-
tein Res. 5,73-84. 21. Platzer, K. E. B., Momany, F. A. & Scheraga, H. A. (1972) Znt. J. Peptide Protein
Res. 4,187-200. 22. Scheraga, H. A. (1973) Jerusalem Symposia on Quantum Chemistry and Biochem-
istry, Bergmann, E. D. & Pullman, B., Eds., Vol. 5, Academic Press, New York, pp. 51-68. 23. Sasisekharan, V. (1973) in Jerusalem Symposia on Quantum Chemistry and Bio-
chemistry, Bergmann, E. D. & Pullman, B., Eds., Vol. 5, Academic Press, New York, pp. 247-258.
642-647.
24. Brant, D. A. & Flory, P. J. (1965) J. Amer. Chem. SOC. 87,2791-2800. 25. Alving, R. E. & Laki, K. (1972) J. Theor. Biol. 34,199-214. 26. Gibson, K. D. & Scheraga, H. A. (1967) Proc. Natl. Acad. Sci. (U.S.) 58,420427. 27. Olson, W. K. & Flory, P. J. (1972) Biopolymers 11,25-36. 28. Kuznesof, P. M. (1966) Program No. 94, Quantum Chemistry Program Exchange,
29. Cruickshank, D. W. J. (1964) Acta Cryst. 17,674-675. 30. Davies, D. R. & Corbridge, D. E. G. (1958) Acta Cryst. 11,315-319. 31. Lai, T. F. & Marsh, R. E. (1972) Acta Cryst. B28,1982-1989. 32. Harwell Subroutine Library (1973) Subroutine VA04, Theoretical Physics Division,
Atomic Energy Research Establishment, Harwell, Berkshire, England. 33. Powell, M. J. D.41964) Computer J. 7,155-162.
Indiana University.

CONFORMATIONAL ANALYSIS OF ATP 2179
34. Vasilescu, D., Lespinasse, J. N., Camous, F. & Cornillon, R. (1972) FEBS Lett . 27,
35. Evans, F. E. & Sarma, R. H. (1974) FEBS Lett. 41,253-255. 36. Son, T.-D. & Chachaty, C. (1973) Biochim. Biophys. Acta 335,l-13. 37. Kraut, J. & Jensen, L. H. (1963) Acta Cryst. 16,79-88. 38. Hruska, F. E., Smith, A. A. & Dalton, J. G. (1971) J. Amer. Chem. SOC. 93, 4334-
39. Yathindra, N. & Sundaralingam, M. (1973) Biopolymers 12,297-314. 40. Follman, H. & Gremels, G. (1974) Eur. J. Biochem. 47,187-197. 41. Hamilton, W. C. & Ibers, J. A. (1968) Hydrogen Bonding in Solids, Benjamin, New
42. Szent-Gyorgi, A. (1960) Introduction to a Submolecular Biology, Academic Press,
43. Diner, S., Malrieu, J. P., Jordan, F. & Gilbert, M. (1969) Theoret. Chim. Acta 15,
44. Blandin, M., Son, T.-D., Catlin, J. C. & Guschlbauer, W. (1974) Biochim. Biophys.
45. Suck, D., Saenger, W., Main, P., Germain, G. & Declercq, J.-P. (1974) Biochim. Bio-
46. Nauman, C. F., Prijs, B. & Sigel, H. (1974) Eur. J. Biochem. 41,209-216. 47. Lam, Y.-F., Kuntz, G. P. P. & Kotowycz, G. (1974) J .Amer. Chem. SOC. 96, 1834-
335-337.
4336.
York.
New York.
100-110.
Acta 361,249-256.
phys. Acta 361,257-265.
1839.
Received April 2, 1975 Accepted May 19,1975





























