Embed Size (px)
Citation preview

Eur. J . Biochem. 71, 533-537 (1976)
The Dissociation of Flavin Coenzymes from Trypsin-Solubilized NADPH/Cytochrome c (P-450) Reductase of Pig-Liver Microsomes
Gordon E. TROUT
Department of Physiology and Medical Biochemistry, University of Cape Town
Received June S/September 28, 1976)
The change in fluorescence emission at 520 nm after excitation at 365 nm was used to investigate the effect of pH and ionic strength on the dissociation of flavin cofactors from microsomal NADPH/ cytochrome c (P-450) reductase.
In the unmodified enzyme both the FAD and FMN moieties appeared to dissociate at a similar rate and followed first-order kinetics. The rate constant for the dissociation was increased by low pH and high ionic strength, particularly in the range pH 4.4-3.8 (0.02 M acetate buffer) where the rate constants increased 80-fold.
Modification of the enzyme by treatment with p-chloromercuribenzoate enhanced the rate of flavin dissociation and, in the region of pH 4, resulted in a biphasic increase in fluorescence consistent with two simultaneous parallel first-order dissociations. It was concluded that p-chloromercuriben- zoate treatment modified the protein so that the two flavin cofactors dissociated at different rates.
Using the measured rate constants for the dissociations, and the known variation in fluorescence of flavin nucleotides with pH, an analogue computer simulation of the dissociation as well as a manual curve-fitting procedure showed that the biphasic response could be explained as a simultaneous rapid dissociation of FAD and a slower loss of FMN from the protein.
Hepatic microsomal NADPH/cytochromec (P-450) reductase is a flavoprotein component of the cyto- chrome P-450 mixed-function oxidase system and can be cleaved from the microsomal membrane by trypsin digestion [l]. It has been shown that the trypsin- solubilized enzyme has approximately one mole each of FAD and FMN per mole of protein [2]. The activity of the enzyme is dependent to a marked extent on a high ionic strength [ l ] and, with microsomal cyto- chrome h, as substrate, the trypsin-solubilized enzyme is reported to show activity only in the presence of high salt concentrations [3].
The non-covalent binding of flavins to protein is still incompletely understood [4] but a number of amino acid residues including tyrosine, tryptophan and cysteine [5-11] have been shown to be involved in particular proteins. The dissociation of many flavo- proteins may be achieved by treatment with urea, guanidine hydrochloride [12,13] or mercurials [6,9,13] as well as by a number of methods which involve a change in pH [lo] or high concentrations of such salts as ammonium sulphate, potassium bromide or calcium chloride [5,14-171. In the case of microsomal NADPH/cytochrome c reductase, several investiga-
Enzyme. NADPH/cytochrome c (P-450) reductase (EC 1.6.2.4).
tors have indicated that the flavins are dissociable by acid ammonium sulphate [18,19] while photoinacti- vation in the presence of 40% ammonium sulphate at pH 6.8 has been reported to yield an apoprotein which can be reactivated by addition of either FMN or FAD [19]. The high salt concentration necessary for maximum activity of the microsomal NADPH/ cytochrome c (P-450) reductase [I] may simultaneously promote the dissociation of the flavin cofactors re- sulting ultimately in loss of activity as in D-amino acid oxidase [20]. A further feature of the microsomal reductase is that pretreatment with one mole of p-chloromercuribenzoate per mole of protein brings about an enhancement of enzymatic activity whereas larger amounts result in inhibition [18]. As treatment of some flavoproteins with mercurials has resulted in the dissociation of the flavin cofactors, the possibility arises that the modified activity against cytochrome c described above, is associated with a change in flavin binding.
The present investigation was undertaken to clarify the effect of pH and ionic strength on the stability of flavin binding to NADPH/cytochrome c reductase and to assess the role of sulphydryl groups in the protein from a study of the effect of p-chloromercuri- benzoate on the kinetics of dissociation.

534 Dissociation of Flavins from Microsomal NADPH/Cytochrome c Reductase
MATERIALS AND METHODS
Trypsin (bovine) was purchased from Miles-Sera- vac, horse heart cytochrome c (grade 111) and NADPH from Sigma Chemicals, and p-chloromercuribenzoate and bovine serum albumin from British Drug Houses. Enzyme assays were performed on a Pye/Unicam SP 1800 double-beam spectrophotometer and other ab- sorption readings on a Beckmann DU/Gilford spectro- photometer. Fluorescence measurements were made with a Perkin Elmer spectrofluorimeter (Model 203) either at timed intervals or, in cases where the fluor- escence change was rapid, on a chart recorder. Acetate buffers were prepared so as to have almost equal ionic strength checked by conductivity measurement with a Radiometer conductivity meter type CDM 2e. NADPH/cytochrome c reductase activity was mea- sured with respect to horse heart cytochrome c using the method of Omura and Takasue [21]. Protein concentrations were determined by the Lowry method [22] using bovine serum albumin as standard protein and molar concentrations of enzyme were calculated assuming a molecular weight for the NADPH/cyto- chrome c reductase of 68800 [23]. These values were used throughout the study for assessing both the protein concentration in the dissociation experiments and for preparing solutions of known protein/mercu- rial ratios. p-Chloromercuribenzoate in pH 7.0 phos- phate buffer was prepared on the day of use and assayed at 232 nm using an absorption coefficient of 16.9 mM-' cm- [24]. Polyacrylamide gel electrophoreses were run on 7.5% gels at pH 8.5 [25] and gels were stained with Coomassie blue. An analogue computer built in the department was used to simulate the dissociation.
Isolation of NADPHICytochrorne c Reductase
Fresh pig liver (1.5 kg) was minced, washed 3 -4 times with cold 0.15% KCl solution to remove excess blood, and the cells disrupted in a Waring blender (3 x 10 s at low speed). Heavy particles were removed by centrifugation at low speed (10 min at 2 000 x g) followed by high-speed (20 min at 10000 x g) to re- move mitochondria. The post-mitochondria1 super- natant was then added to 4 volumes of a calcium/ magnesium solution to aggregate the microsomes [26]. After decanting the bulk of the supernatant, the micro- somes were collected by low-speed centrifugation and washed with calcium/magnesium solution followed by 0.1 M phosphate buffer pH 7.5. The NADPH/cyto- chrome c reductase was freed from the microsome by digestion with trypsin and purified [21] until samples ran as a single band on polyacrylamide gel electrophoresis, showed a specific activity of 12 - 20 pM horse heart cytochrome c reduced min-' mg protein-', and a A,,,/A,,, of 6.9-7.1.
Kinetic Measurements of Fluorescence
The rate of flavin dissociation from the enzyme under various conditions was determined from the increase in fluorescence following dilution of an ali- quot (50 pl) in a suitable buffer (3 ml) and used to determine the first-order rate constant. Fluorescence was excited at 365 nm and the emission measured at 520 nm and a range of protein concentrations (Lowry method) between 0.023 and 0.9 nM were employed. The fluorimeter sensitivity was adjusted for full-scale deflection on a sample of the reductase at the same concentration as in the experiment but in which the flavin cofactors had been released by heat denatura- tion (3 min in boiling water). Care was taken to mini- mise photodecomposition of the sample and all read- ings were made as quickly as possible. The effect of p-chloromercuribenzoate treatment on the fluores- cence of NADPH/cytochrome c reductase was deter- mined on samples pretreated at pH 7.5 for five minutes with one tenth volume of standard p-chloromercuri- benzoate at room temperature so that the ratio of protein to mercurial was either 1 : 1 or 1 : 5. A suitable aliquot was then added rapidly to the buffer and the fluorescence change measured. Kinetic experiments were carried out at room temperature at least in duplicate.
RESULTS AND DISCUSSION The efect o f p H and Ionic Strength on Flavin Dissociution
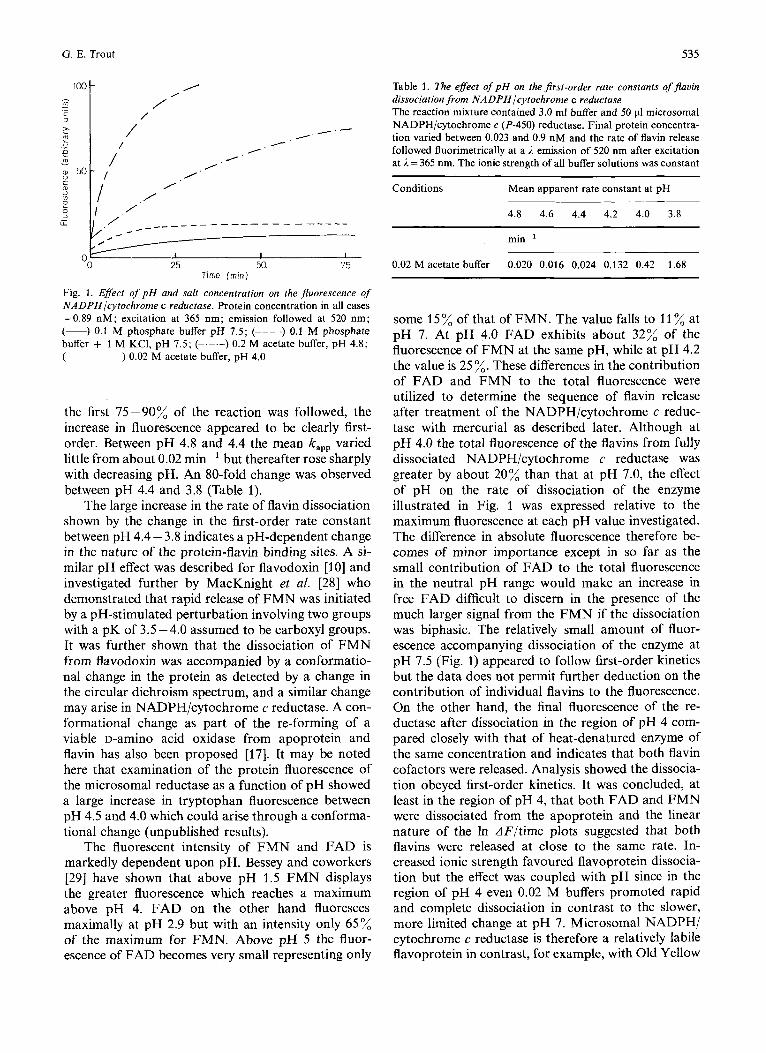
Both ionic strength and pH influenced the dis- sociation of flavin cofactors from NADPH/cyto- chrome c (P-450) reductase (Fig. 1). Within the range pH 7.5 -6.0 (0.1 M phosphate buffer) the dissociation of flavin appeared slow and little influenced by changes in pH. An attempt to estimate the first-order rate constant by the Guggenheim method [17] from the small change in fluorescence observed at pH 7.5 gave a kapp of 0.015 min-'. After about 2 hours the rate of change in fluorescence had become very slow and only some 20% dissociation had taken place. An in- crease in the ionic strength of the buffer by incorpo- ration of 1 M KCI increased the kapp to 0.085 min-' but failed to modify the overall amount of flavin released. It was tentatively concluded that the high ionic strength facilitated the rate of flavin dissociation but had little effect on the dissociation constant. Below pH 6 both the rate and extent of dissociation increased, and in 0.1 M acetate buffer pH 4.0 the rate of dissocia- tion was too great for accurate measurement. On lowering the salt concentration of the acetate buffer (pH 4.0) to 0.02 M, the rate decreased (k,,,=0.42 min-l) and the final fluorescence compared closely with a heat-denatured control. From experiments with 0.02 M acetate buffer at pH 4.8 to 3.8, where
535 G . E. Trout
I loot /
I / /
Time ( rn in )
Fig. 1. Effect of pH and salt concentration on the jluorescence of NADPHlcytochrome c reductase. Protein concentration in all cases =0.89 nM; excitation at 365 nm; emission followed at 520 nm; (-) 0.1 M phosphate buffer pH 7.5; (----) 0.1 M phosphate buffer + 1 M KC1, pH 7.5; (-.-.-) 0.2 M acetate buffer, pH 4.8; (------) 0.02 M acetate buffer, pH 4.0
the first 75-90% of the reaction was followed, the increase in fluorescence appeared to be clearly first- order. Between pH 4.8 and 4.4 the mean kapp varied little from about 0.02 min-' but thereafter rose sharply with decreasing pH. An 80-fold change was observed between pH 4.4 and 3.8 (Table 1).
The large increase in the rate of flavin dissociation shown by the change in the first-order rate constant between pH 4.4 - 3.8 indicates a pH-dependent change in the nature of the protein-flavin binding sites. A si- milar pH effect was described for flavodoxin [lo] and investigated further by MacKnight et al. [28] who demonstrated that rapid release of FMN was initiated by a pH-stimulated perturbation involving two groups with a pK of 3.5 - 4.0 assumed to be carboxyl groups. It was further shown that the dissociation of FMN from flavodoxin was accompanied by a conformatio- nal change in the protein as detected by a change in the circular dichroism spectrum, and a similar change may arise in NADPH/cytochrome c reductase. A con- formational change as part of the re-forming of a viable D-aminO acid oxidase from apoprotein and flavin has also been proposed [17]. It may be noted here that examination of the protein fluorescence of the microsomal reductase as a function of pH showed a large increase in tryptophan fluorescence between pH 4.5 and 4.0 which could arise through a conforma- tional change (unpublished results).
The fluorescent intensity of FMN and FAD is markedly dependent upon pH. Bessey and coworkers 1291 have shown that above pH 1.5 FMN displays the greater fluorescence which reaches a maximum above pH 4. FAD on the other hand fluoresces maximally at pH 2.9 but with an intensity only 65% of the maximum for FMN. Above pH 5 the fluor- escence of FAD becomes very small representing only
Table 1. The effect of pH on the first-order rate constants of jlavin dissociation from NADPHlcytochrome c reductase The reaction mixture contained 3.0 ml buffer and 50 p1 microsomal NADPH/cytochrome c (P-450) reductase. Final protein concentra- tion varied between 0.023 and 0.9 nM and the rate of flavin release followed fluorimetrically at a 1 emission of 520 nm after excitation at I = 365 nm. The ionic strength of all buffer solutions was constant
Conditions Mean apparent rate constant at pH
4.8 4.6 4.4 4.2 4.0 3.8
min ~ ' 0.02 M acetate buffer 0.020 0.016 0.024 0.132 0.42 1.68
some 15% of that of FMN. The value falls to 11 % at pH 7. At pH 4.0 FAD exhibits about 32% of the fluorescence of FMN at the same pH, while at pH 4.2 the value is 25 %. These differences in the contribution of FAD and FMN to the total fluorescence were utilized to determine the sequence of flavin release after treatment of the NADPH/cytochrome c reduc- tase with mercurial as described later. Although at pH 4.0 the total fluorescence of the flavins from fully dissociated NADPH/cytochrome c reductase was greater by about 20% than that at pH 7.0, the effect of pH on the rate of dissociation of the enzyme illustrated in Fig. 1 was expressed relative to the maximum fluorescence at each pH value investigated. The difference in absolute fluorescence therefore be- comes of minor importance except in so far as the small contribution of FAD to the total fluorescence in the neutral pH range would make an increase in free FAD difficult to discern in the presence of the much larger signal from the FMN if the dissociation was biphasic. The relatively small amount of fluor- escence accompanying dissociation of the enzyme at pH 7.5 (Fig. 1) appeared to follow first-order kinetics but the data does not permit further deduction on the contribution of individual flavins to the fluorescence. On the other hand, the final fluorescence of the re- ductase after dissociation in the region of pH 4 com- pared closely with that of heat-denatured enzyme of the same concentration and indicates that both flavin cofactors were released. Analysis showed the dissocia- tion obeyed first-order kinetics. It was concluded, at least in the region of pH 4, that both FAD and FMN were dissociated from the apoprotein and the linear nature of the In dF/time plots suggested that both flavins were released at close to the same rate. In- creased ionic strength favoured flavoprotein dissocia- tion but the effect was coupled with pH since in the region of pH 4 even 0.02 M buffers promoted rapid and complete dissociation in contrast to the slower, more limited change at pH 7. Microsomal NADPH/ cytochrome c reductase is therefore a relatively labile flavoprotein in contrast, for example, with Old Yellow

536 Dissociation of Flavins from Microsomal NADPH/Cytochrome c Reductase
Enzyme reported to be practically non-dissociable between pH 4 and 11 [ l l ] .
The Efect of p-Chlovomevcuvibenzoate on Flatin Dissociation
The effect of low pH in promoting flavin dissocia- tion together with the dependence of FAD on a pH below about 5 for significant fluorescence was res- ponsible for the emphasis of the present investigation being conducted in the region of pH 4. Nevertheless some experiments were conducted around neutrality and in all cases studied p-chloromercuribenzoate treat- ment of the enzyme accelerated the dissociation of flavin cofactors. Pretreatment of the reductase with excess p-chloromercuribenzoate at pH 7 increased both the rate and extent of flavin dissociation which appeared virtually complete after 3 h and followed first-order kinetics. A rate constant of 0.02 min-' was determined for an apparent monophasic reaction.
The most significant aspect of p-chloromercuri- benzoate treatment of the reductase was the intro- duction of a marked biphasic increase in fluorescence on dissociation at pH 4.0 - 4.2. Preliminary experi- ments suggested that this biphasic response diminished as the pH increased presumably due, at least in part, to the small contribution from the weak FAD fluor- escence at the higher pH values.
As the enzymatic activity of NADPH/cytochrome c reductase has previously been shown to be markedly affected by reaction withp-chloromercuribenzoate [ 181, two levels of pretreatment were investigated for their effect on flavin dissociation; a 1 : 1 protein/mercurial ratio previously shown to activate the cytochrome c reductase activity of the enzyme, and a 1 : 5 protein/ mercurial ratio which inhibited cytochrome c reductase activity. Spectrophotometric titration of the enzyme [24] showed that five sulphydryl groups readily reacted with p-chloromercuribenzoate at pH 7.5 and 25 "C and as stable absorbance readings were obtained well within the 2 minutes delay between each addition, a preincubation time of five minutes at room tempera- ture and pH 7.5 was therefore adopted as adequate to ensure complete reaction of the available sulphydryl residues and was used throughout the investigation. Both pretreatments accelerated flavin dissociation, and analysis of the data at pH 4.0-4.2 gave biphasic In A F/time plots separable into two parallel first-order reactions [27] from which individual rate constants were estimated. These constants are listed in Table 2 together with the equivalent constants of the mono- phasic reaction at pH 4.2 - 4.0 in the absence of the mercurial compound. The rate constant for the slow stage of the biphasic dissociation after p-chloro- mercuribenzoate treatment at each pH compared favourably with the single constant for the analogous reaction without the mercurial compound. By contrast,
Table 2. The effect of p-chloromercuribenzoate on thejirst-order rate constunts of flavin dissociation from NADPHlcytochrome c re- ductase Freshly prepared p-chloromercuribenzoate photometrically stan- dardised at pH 7.0 ( E ~ ~ ~ = 16900 M-' cm-') was added in calculated amount to the enzyme so the proteinlp-chloromercuribenzoate mo- lar ratio was either 1 : 1 or 1 : 5 and allowed to react for 5 min before a suitable aliquot was rapidly added to 3 ml of the 0.02 M acetate buffer. Fluorescence changes at 520 nm were measured with time after excitation at 365 nm
Conditions Mean apparent rate constants for flavin dissociation in acid solution
min-'
0.02 M acetate buffer alone 0.42 - 0.132 -
0.02 M acetate buffer after treatment with 1 : 1 pro- teinlp-chloromercuri- benzoate 0.37 1.46 0.11 0.90
treatment with 1 : 5 pro- teinlp-chloromercuri- benzoate 0.41 1.78 0.12 1.07
0.02 M acetate buffer after
the rate constant for the rapid phase was elevated by a factor of between 4 and 9 depending upon pH, but appeared to be only slightly affected by increasing the proportion of mercurial to protein ratio from 1 : 1 to 5 : 1. In no case however did the rate constant for the fast dissociation exceed that for the untreated enzyme at the lower pH of 3.8 (Table 1). Hence the effect of the mercurial compound on the dissociation was not greater than can be achieved by a pH change alone. It was concluded that the biphasic characteristic of the dissociation resulted from change in the structure of the protein molecule involving only one sulphydryl group. Modification of this residue with mercurial appeared principally to promote the rapid phase of flavin dissociation.
An attempt was made to identify which of the flavin coenzymes was associated with the first and second phases of the dissociation respectively from a qualitative model simulated on an analogue com- puter. Of the two possibilities uiz. a slow change in FAD+a fast change in FMN, or a fast change in FAD + a slow change in FMN, only the curves ob- tained for the latter sequence appeared to resemble the experimentally observed change. This was con- firmed by calculating two hypothetical curves from the known rate constants, the overall fluorescence change, and an estimate of the contribution of FAD to the total fluorescence at pH 4.2 [29] which could be compared with the experimental curve (Fig. 2). It was concluded that the sequence of dissociation

G. E. Trout 5 3 1
100 t and diminish the binding forces of the flavin nucleo- tides for the protein. It seems more likely, however, that the mercurial compound binds to a critical sul- phydryl group in the protein and brings about a conformational change accounting, not only for the increased activity noted [19] but also facilitates flavin dissociation, more particularly FAD, especially at low pH.
Council is gratefully acknowledged. The financial support of the South African Medical Research
I I
1 2 3 4 5 6 Time (rnin)
Fig. 2. Diflerentiul dissociation of' flaoins from p-chloromercuri- benzoate-modified NADPH/cytvchrome c reductase. Comparison of observed increase in fluorescence of NADPH/cytochrome c re- ductase at pH 4.2 (0.02 M acetate buffer) after treatment with p-chloromercuribenzoate (1 : 1 protein : p-chloromercuribenzoate) with calculated curves in which the rapid phase in the dissociation is either FAD (-----) or FMN (-.-.-). Observed dissociation (-), k , =0.84 min-', k,=0.08 min-'. Protein concentration (final) = 0.88 nM
was a rapid loss of FAD simultaneously with a slower loss of FMN. If so, this sequence differs from that in the NADPH/cytochrome c reductase subunit of Escherichiu Cnli sulfite reductase in which p-chloro- mercuriphenylsulphonate treatment stimulates the release of FMN but leaves FAD firmly bound to the protein [9].
The mechanism of the induced by the mercurial compound is not known though it is clear that a sul- phydryl group is important in maintaining the inte- grity of certain flavoproteins [6,9]. Apart from the role of sulphydryl groups as active participants in the oxidation/reduction process of some flavoproteins as postulated for glutathione reductase [8] and thio- redoxin reductase [12], chemical and kinetic evidence is available which suggests that, in some flavoproteins at least. a sulphydryl group is directly involved in flavin/protein binding [6,9]. However in the case of flavodoxin where modification of the protein with sulphydryl specific reagents markedly affects the bind- ing of FMN, crystallographic studies show that no direct contact exists between the FMN and cysteine, and that the nearest sulphydryl group is directed away from the flavin [30]. It is established therefore that modification of a cysteine residue can influence flavin binding even though the cysteine is not directly in- volved in providing a sulphur-flavin bond. In addition to these effects on sulphydryl residues, mercuric ions and methyl mercury are also known to bind to the purine/pyrimidine base of nucleotides [31] and p-chlo- romercuribenzoate may act in an analogous manner
REFERENCES 1
2 3
4. 5 . 6 . 7. 8.
9.
10. 11. 12. 13.
14. 15.
16.
17. 18.
19.
20.
21.
22.
23.
24. 25. 26.
27.
28.
29.
30.
31.
Phillips, A. H. & Langdon, R. J. (1962) J. Biol. Chem. 237,
Iyanagi, T. & Mason, H. S. (1973) Biochemistry, /2,2297 -2308. Bilimorid, M. H. & Kamin, H. (1973) Ann. N . Y . Acud. Sci.
Shifrin, S. & Kaplan, N. 0. (1960) Adc. Enzymol. 22. 337-415. Strittmatter, P. (1961) J . Bid. Chem. 236, 2329-2335. Mayhew, S. G. (1971) Biochim. Biophys. A m , 235, 289-302. McCormick, D. B. (1970) Experientiu (Busel) 26, 243 -244. Colman, R. F. & Black, S. (1965) J . Biol. Chern. 240, 1796-
Siegel, L. M., Davis, P. S. & Kamin, H. (1974) J . B i d Chem.
Hinkson, .I. W. (1968) Biochemi.rtry, 7, 2666-2672. Theorell, H. (1955) Disc. Furaduy Soc. 20, 224 - 231. Thelander, L. (1968) Eztr. J . Biochem. 4, 407 -422. Knight, E., Jr & Hardy, R. W. F. (1967) J . Biol. Chm7. 42.
Warburg, 0. & Christian, W. (1938) Biochern. Z . 298, 368-377. Kanda, M., Brady, F. O., Rajagopalan, K. V. & Handler, P.
Komai, H., Massey, V. & Palmer, G. (1969) J . Biol. Ckem. 244.
Massey, V. & Curti, B. (1966) J . Biol. Chem. 241, 3417-3423. Masters, B. S. S., Kamin, H., Gibson, Q. H. &Williams, C. H.
Baggott, J . P. & Langdon, R. G. (1970) J . Biol. Chem. 245.
Dixon, M. & Kleppe, K. (1965) Biochim. Biophys. Acra, 96,
Omura, T. & Takasue, S. (1970) J . Biochem. (Jupun) 67, 249-
Lowry, 0. H., Rosebrough, N. J., Farr, A. L. & Randall. R. J.
Masters, B. S. S. & Ziegler, D. M. (1971) Arch. Biochem. Bio-
Boyer, P. D. (1954) J . Am. Chem. Soc. 76, 4331. Davis, B. J. (1964) Ann. N . Y . Acud. Sci. 121, 404-427. Kamath, S. A., Kummerow, F. A. & Narayan, K. A. (1971)
Frost, A. A. & Pearson, R. G. (1961) Kinetics and Mechanism,
MacKnight, M. L., Gillard, .I. M. & Tollin, G. (1973) Bio-
Bessey, 0. A., Lowry, 0. H. & Love. R. H. (1949) J. Biol. Chem.
Burnett, R. M., Darling, G. D., Kendall, D. S., Le Quesne, M. E., Mayhew. S. G. & Ludwig, M. L. (1974) J . B i d . Chem.
2652-2660.
212,423 -448.
1803.
249, 1572 - 1586.
1370-1374.
(1972) J. Biol. Chem. 247, 765-770.
1692 - 1700.
Jr (1965) J . Biol. Chem. 240, 921-931.
5888 - 5896.
357 - 367.
257.
(1951) J . Biol. Chem. 193, 265-275.
phys. 145, 358 - 364.
Febs Lett. 17, 90-92.
2nd edn., pp. 49, 162- 164, Willey, New York.
chemistry, 12, 4200 - 4206.
180,755 - 769.
249,4383-4392. Simpson, R. B. (1964) J. Am. Chem. Soc. 86, 2059-2065.
G. E. Trout, Department of Physiology and Medical Biochemistry, University of Cape Town Medical School, Observatory, Cape Town, South Africa