Embed Size (px)
Citation preview

Paper No. 6
THE EFFECT OF MOLECULAR WEIGHT ON PROPERTIES OF IONICALLY-CURED FLUOROELASTOMERS
By Donald F. Lyons* DuPont Dow Elastomers L.L.C.
Wilmington, DE
Presented at a meeting of the
Rubber Division, American Chemical Society
San Francisco, CA
April 28-30, 2003
*Speaker

ABSTRACT
Three monodisperse vinylidene fluoride/tetrafluoroethylene/hexafluoropropylene terpolymers were prepared by iodine degenerative chain transfer and used to assess the effect of molecular weight on compositions crosslinked with bisphenol AF. MN for the series ranged from 100,000 to 245,000 and polydispersities were ~1.3. Three sets of compounds were prepared from these polymers: unfilled with 3 phr/6 phr Ca(OH)s; filled with 3 phr MgO/6 phr Ca(OH)2; and unfilled with 9 phr MgO as the sole metal oxide. Hardness and tensile properties for the original vulcanizate and after 70 and 336 hours @ 200°C was determined for each compound. This was compared to compression set after similar aging. Compression sets for these compounds were excellent. An analysis of the hardness and compression set data for the unfilled compounds suggests that little if any chain scission is occurring during the first 70 hours of air aging.
2

INTRODUCTION
The use of degenerative chain transfer to prepare fluoroelastomers has been practiced for over 25 years and represents the most successful commercial application of controlled (i.e., “living”) free radical polymerization.1-12 In this process the appropriate fluoroolefins are polymerized in the presence of a perfluoroalkyldiiodide chain transfer agent. Iodine atoms are transferred from the original reagent to the ends of the fluoroelastomer chain. But because the new iodine-carbon bonds have almost exactly the same energy as that of the original perfluoroalkyl reagent, the newly iodo-capped polymer chain will serve as a chain transfer agent itself. In effect, the activity of the original chain transfer agent is never lost. Details of the chemistry are given in references 10-12. One of the great advantages of this process is that the polymer chemist can tailor the molecular weight distribution from narrow to broad by varying details such as diiodide feed rate.
While hundreds of tons of fluoroelastomer are produced by this process yearly, its ability to make monodisperse model polymers for fundamental characterization and cure studies has been underappreciated in the open literature. In this paper, three monodisperse fluoroelastomers were prepared and then cured using the standard bisphenol AF (BpAF) crosslinking chemistry. This allows the effects of molecular weight in fluoroelastomers to be clearly visualized. The fluoroelastomers were standard vinylidene fluoride/tetrafluoroethylene/hexafluoropropylene (VF2/TFE/HFP) terpolymers.
Studies of crosslinked fluoroelastomers are relatively rare and most of them discuss the obsolete diamine cure chemistry and not the currently favored BpAF-based system.13-21 The mechanism of BpAF curing in fluoroelastomers was elucidated by Schmiegel.15-16 The actual cure site is an HFP-VF2-HFP sequence within the backbone. In terpolymers the sequence HFP-VF2-TFE will also serve as a cure site. The protons at such a site are highly acidic and base attack will lead to facile dehydrofluorination. After a rearrangement, a diene structure forms in the backbone and is subsequently attacked by a deprotonated bisphenol. The phenolate displaces fluorine and forms a bond to a backbone carbon. A crosslink is formed after both phenolate moieties of the curative have reacted with (different) polymer chains. Because the specific monomer sequences occur on a statistical basis within a polymer, HFP-containing fluoroelastomers are inherently curable by this chemistry. It is estimated that the concentration of potential cure sites is on the order of 0.6 mol/kg.
The typical compound recipe for a fluoroelastomer BpAF cure includes 3 phr MgO and 6 phr Ca(OH)2. These materials initiate dehydrofluorination and also serve as acid and water acceptors during cure. For best tensile properties and compression set performance, these cure systems require post-cure at temperatures ranging from 200 to 250°C. Recently, compounds that contain 9 phr MgO and no Ca(OH)2 have been explored because they offer the possibility of no or shorter post-cures.22 Calcium hydroxide-free compounds require shorter post-cure times, but longer press-cure times, to achieve the equivalent compression set performance of conventional mixed oxide recipes. It is of interest to compare the effects of these metal oxide packages on well-defined polymers.
3

Therefore three compounds were examined in this work. One is unfilled, with the mixed metal oxide recipe. The second also contains both MgO and Ca(OH)2, but is filled with N990 carbon black. This recipe is the standard reference recipe used to evaluate fluoroelastomers. The third is unfilled with MgO as the only acid acceptor.
4

EXPERIMENTAL
Vinylidene fluoride-tetrafluoroethylene-hexafluoropropylene terpolymers were prepared via emulsion polymerization using standard literature techniques.1-9 The polymerization temperature was 80°C and the initiator used was ammonium persulfate. Isolation was accomplished by coagulation with aluminum sulfate. Different molecular weights were obtained by adding different amounts of perfluoroalkyl diiodide to each recipe. Properties of the three polymers are shown in Table I. These polymers are designated numerically throughout this study, with “1” representing the high molecular weight polymer, “2” the mid, and 3” the low molecular weight polymer.
The magnesium oxide was Elastomag™ 170 (Morton). Calcium hydroxide (HP grade) was supplied by CP Hall. N990 carbon black was supplied by Cancarb. Other curatives were supplied by DuPont Dow Elastomers.
Inherent viscosity was determined in methyl ethyl ketone at 1.0 g/dL concentration. Raw polymer Mooney viscosity information was collected at three different temperatures, 100, 121, and 149°C, according to ASTM D 1646. The lowest molecular weight polymer was too fluid to be measured at any temperature above 100° while the high molecular weight polymer was too stiff to be measured at 100°C. Monomer content was measured by a proprietary FTIR method. Fluorine content is calculated from the compositional data. Glass transition temperatures were determined on a DSC using a 10°C/min heating rate and were based on the inflection point.
Molecular weight data were collected on a Waters GPCV 2000 instrument using dimethyl acetamide containing 0.025M LiCl and 0.002M p-toluenesulfonic acid as solvent at 70°C. The presence of toluenesulfonic acid protonates any residual dimethyl amine and prevents attack on the polymer. Four styrene-divinyl benzene columns were used for separation and the column was calibrated with monodisperse PMMA standards. A refractometer and capillary rheometer, installed in series, were used for sample detection. The polymer samples were also analyzed by a second, noisier, method using similar apparatus except the solvent temperature was 125°C and the standards were polystyrene. The molecular weight data collected with that method agreed with the reported values to within 10%.
Iodine content was measured by neutron activation analysis (General Activation Analysis Inc. Encinitas, CA). ATR analysis was conducted on pressed polymer films on a Nicolet “Protégé” 460 equipped with a SpectraTech “Thunderdome” ATR cell. Residual water was qualitatively detected by increased absorbance in the 3100 – 3700 cm-1 region.
These three polymers were compounded using three different recipes as shown in Table II. Each of the three polymers was compounded with each of the three recipes to form a total of nine compounds. These are named by combining the polymer designation with the compound designation. Thus, F2 represents the medium viscosity polymer compounded with the 30 phr N990 black-containing recipe. Before addition of curatives, the three polymers were “mill-dried”. Polymer was banded on a two roll mill to a thin sheet (~1 mm) and the mill was allowed to warm up from mixing shear. The low molecular weight polymer, which tended to stick to the mill, was heated to ~50°C while the other two polymers were heated to ~90°C. Compounding was carried out on a two-
5

roll mill maintained at a temperature of 40°C. This temperature was dictated by the behavior of the low molecular weight polymer. Curatives were crushed to make a powder before adding to the stock.
Cure characterization was carried out with an Alpha Technologies MDR2000 curemeter. The instrument temperature was set to 177°C during these tests with a frequency of 1.66 Hz and an arc of ±0.5°. Test duration was 12 minutes for all samples.
All test specimens were compression molded at 162°C for thirty minutes. They were then post-cured at 232°C for 16 hours under air. Tensile testing was conducted on dumbbells die-cut out of slabs in accordance with ASTM D412. Samples were aged at 200°C in air for either 70 or 336 hours. All data represents the average of results from three specimens. Testing was conducted at ambient temperature with a strain rate of 50 mm/min.
Hardness (Shore A) was conducted according to ASTM D2240. Compression set on plied disks was determined according to ASTM D395. The cured samples were tested at 200°C in air for either 70 or 336 hours. Samples were removed from the test jigs within 30 seconds of removal from the oven. Measurement was conducted after approximately 45 minutes of cooling at ambient temperature.
6

RESULTS
Polymerization of these materials proceeded uneventfully by standard emulsion polymerization techniques. Iodine contents agreed well with the MN values, indicating that the vast majority of endgroups were iodine terminated. The predicted iodine contents given in Table I were calculated assuming all endgroups are terminated with iodine. As discussed in reference 12, a necessary (but not sufficient) requirement for a monodisperse molecular weight distribution by the degenerative iodine transfer process is that the overwhelming majority of chain ends are iodine.
The compositions of the three polymers are typical of commercially available 68% F fluoroelastomers. Compositions of these polymers are virtually identical, assuring an identical cure site density in all three polymers. As a check on composition, TG (glass transition) values are identical within experimental error, again confirming the similarity of composition between the three samples.
Cure data are given in Table III. The set of MDR curves of the “C” series is shown in Figure 1. For all three sets of compounds the low molecular weight polymers cured fastest, followed by the medium molecular weight and then the high molecular weight polymer. As expected, the M series cured more slowly than the C series. Although ts2 data are similar between the two sets, the M series takes longer to reach its final state of cure. Filled compounds cured faster than unfilled compounds. The final torque was directly dependent on polymer molecular weight.
Physical properties of the cured polymers were analyzed using standard ASTM methods. Although these tests provide less fundamental information than other options, they have the advantage of being easily related to the types of testing typical rubber compounders conduct in their daily business. Since fluoroelastomers are generally used in seals, compression set was of particular interest.
The physical properties of the nine compounds are provided in Table IV. The room-temperature stress-strain properties of the unfilled mixed oxide compounds are shown in Figure 2. Only a modest dependence of tensile strength or elongation on polymer molecular weight was observed, as is expected.
The stress-strain properties of the “M”-series compounds were generally similar to those of the “C” series. They did build less strength at a given elongation, though. This may indicate slightly less curing for this formulation.
The stress-strain properties of the filled compounds show moderate reinforcement. These were filled with 30.0 phr of N990 (“MT”) black. The level and type of filler used in these compounds is typical of a “starting-point formulation” recommended by fluoroelastomer manufacturers. N990 is a large particle size, unstructured black and is used to reduce the overall cost of the formulation without increasing hardness too much. As shown in Figure 3, the stress-strain curve is almost linear. Modulus and ultimate tensile strengths are increased 40-50% over the unfilled values. Maximum elongation decreased only slightly.
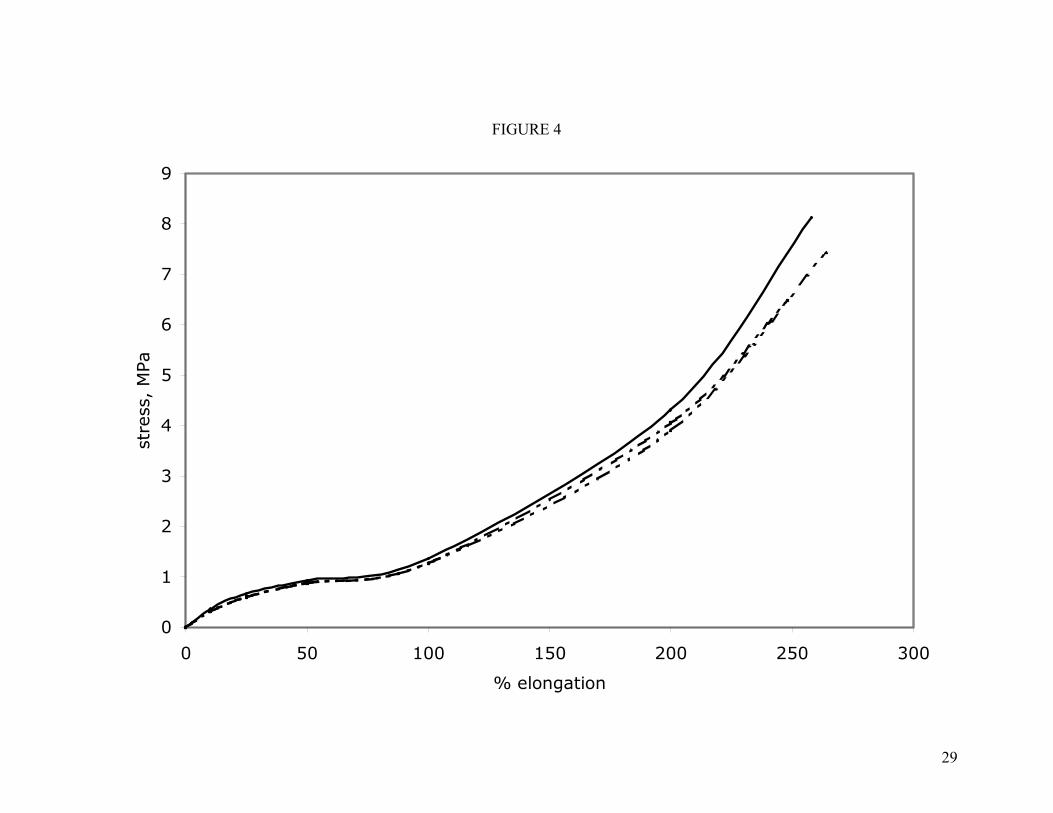
All compounds were aged in air at 200°C for 70 and 336 hours in order to compare their tensile properties under similar conditions to that of the compression set measurement. These properties are also given in Table IV. The effect of aging on stress-
7

strain is shown in Figure 4 for the low molecular weight polymer compounded with mixed oxides. Under these conditions virtually no change is observed in the vulcanizate behavior. Likewise, the tensile properties for vulcanizates from the higher molecular weight polymers were unchanged by these aging conditions. Nor did the set of filled compounds exhibit significant change in their stress-strain properties during aging. 200°C is considered to be well within the usage temperature of BpAF-cured fluoroelastomers so it is gratifying to observe such little change. Typical automotive and aerospace specifications require tensile testing at 250 and 275°C.
In both unfilled compounds, Shore A hardness was related to molecular weight, with the lowest molecular weight producing the lowest hardness and the highest producing the hardest compound. This relationship was clear for both series of unfilled compounds. The relationship between molecular weight and hardness for the filled compound was the opposite. In this series vulcanizates derived from the lowest molecular weight polymer produced high hardness and those derived from the high molecular weight polymer gave low hardness.
Excellent compression set properties were found for this series of polymers (Table V). The unfilled high molecular weight polymers in particular showed compression sets below 5% after 70 hours at 200°C aging. For all three compounds, compression set increased with lower molecular weight. The filled compounds had significantly higher compression sets. Even these compounds, however, exhibited better compression set than expected for a 68% F fluoroelastomer. The fact that at least two of the nine compounds displayed such low compression sets supports the assumption that this terpolymer composition does not exhibit room temperature crystallinity in the absence of strain.
8

DISCUSSION Three VF2-TFE-HFP terpolymer elastomers were prepared with MN ranging from
100,000 to 245,000. Polydispersities of these polymers were typical of that possible via degenerative transfer. Narrower molecular weight distributions are difficult to produce due to termination reactions in which one or both polymer chains is iodine-free. Chain transfer to polymer has also been implicated as a source of widening polydispersity.12 MN of the high molecular weight polymer is similar to that reported for a high molecular weight VF2-HFP dipolymer.13,18
The Mooney viscosities of this series spanned that commercially available in 68% F fluoroelastomers. Mooney viscosity is usually considered to depend on MW. Because this series of polymers is so monodisperse it is impossible to determine which moment of the molecular weight distribution has influenced it more.
The influence of cure rate on molecular weight was unexpected. One explanation for this behavior is that the low and mid-molecular weight polymers contained more water than the high molecular weight polymer. Trace amounts of water accelerate the BpAF cure and indeed are necessary for efficient curing. Excess water may explain why the low molecular weight polymer cured faster. ATR-IR analysis of the polymer revealed a higher retained moisture content than the other two samples due to inefficient drying after coagulation. All of the polymer samples were therefore mill dried before compounding as a precaution, but it is possible that some water may have been trapped nonetheless. The observation that the mid-molecular weight polymer cured faster than the high sample is more difficult to explain by this hypothesis. This polymer was easily dried after preparation and had a similar moisture content to the high molecular weight polymer.
If excess moisture is ruled out, then the cure rate dependence indicates that the rate limiting step in this BpAF cure depends on polymer diffusion, not small molecule (accelerator or BpAF) diffusion. This suggests that, although the accelerator is necessary for efficient dehydrofluorination, individual chains must still diffuse close to the metal oxide particles in order for it to occur. Once dehydrofluorination has occurred, attack by BpAF occurs quickly. Venkateswarlu, et al, followed the disappearance of BpAF during fluoroelastomer cure and found evidence for rapid monofunctionalization of BpAF before difunctionalization (which generates crosslinks) occurred.19 In that study, the concentration of free BpAF significantly declined during the cure induction period while the compound viscosity remained unchanged. The authors concluded that the majority of BpAF monofunctionally attached to the polymer backbone before these pendant BpAF moieties attacked another polymer chain. Thus, a simplified cure mechanism is
[R4P+], η177 Polymer + base -----------------------> Dehydrofluorinated polymer (1)
Dehydrofluorinated polymer + BpAF ---------------> pendant BpAF (2)
9

Pendant BpAF + dehydrofluorinated polymer ----------------> crosslink (3)
where [R4P+] represents the accelerator and η177 is the viscosity of the compound at 177°C, the curemeter temperature. The relative rates of these reactions are ordered (2) > (3) > (1). The dependence of scorch time on molecular weight may be especially marked in this series of polymers because they essentially lack ionically charged initiator residues normally present in fluoroelastomers. It is estimated that over 95% of the chain ends in these polymers are non-ionic in nature. Ionically charged endgroups would increase the solubility of accelerator (BTPPC) in the rubber medium as well as confounding the effect of molecular weight since transient ionomeric networks can be formed.
Calcium hydroxide-free recipes have recently been publicized because they require shorter post-cure to achieve desired compression sets. The penalty for this beneficial behavior is longer press cure time. In the compounds studied here, ts2 takes about 7% longer for the “M” series vs the “C” series. Full cure, as indicated by tc90 and tc95, takes 30% longer in the “M” series. Therefore the major cure retardation is occurring when most or all the BpAF is attached to a chain and mobile base, necessary for further dehydrofluorination, is becoming scarce. (Note that early in the cure cycle, deprotonated BpAF can itself serve as base and initiate dehydrofluorination.) Later in the cure cycle the speed of dehydrofluorination will be more directly related to the basicity of the metal oxide and Ca(OH)2 is a stronger base than MgO.
The effect of carbon black on the kinetics of fluoroelastomer curing is not well studied. Perhaps it is better to say that since almost all published formulations contain 30 phr N990 black, the behavior of unfilled compounds is what is really unfamiliar. N990 black is characterized by a low level of surface acidity and aqueous slurries prepared from it are even slightly basic.23 Because the vast majority of its surface oxygen is present as phenolic or hydroquinone moieties, it is not surprising that the presence of N990 black would accelerate the cure since these structures can also participate in nucleophilic attack on the polymer.
Tensile properties of these materials were unremarkable and it is clear that molecular weight distribution did not affect stress-strain behavior in these samples. The values found for the filled stock are normal for VF2/TFE/HFP fluoroelastomers cured with these levels of curatives. The stress-strain curves of the unfilled vulcanizates turned markedly upward after about 100% elongation. This is an elongation much shorter than that typically associated with limited extensibility of chains. Rather, this phenomenon is consistent with the strain-induced crystallization previously seen in VF2-TFE-HFP terpolymer elastomers.24 In that work, strain-induced crystallization was also observed at about 100% elongation. Both 50 and 100% moduli of the unfilled compounds are inversely proportional to the inverse of MN, as predicted by Flory,25 but the curve is not linear. With only three data points it is impossible to assess the effect of experimental error on this correlation.
While the tensile properties of these polymers are similar to standard commercial products, the compression sets are significantly better. Following the treatment of Baldwin26, changes in crosslink density upon compression set were investigated on the C series of compounds to determine the fraction, f, of crosslinks originally present that will
10

be fissionable either mechanically or chemically during aging. The original network chain density is νt
0. During aging, some of the original crosslinks will fail while others remain intact. The stable chains are given by the equation
νs0 = (1-f) νt
0 (4)
where νs0 represents the density of stable (unchanged) chains. Now the sample is
compressed and heated. During this operation, a new network is superimposed on the original and the total network chain density after heat aging is designated as νt
a. The change in total network chain density after aging is given by
∆νt = νta – νt
0 (5)
The ratio between stable chains formed in the compressed state, νsa, and present in
the original vulcanizate is given by
νsa/ νs
0 = (∆νt + fνt0)/[ νt
0(1-f)] (6)
For a standard ASTM compression set test protocol, Baldwin developed the following equation to predict the ratio of stable chains formed during aging (νs
*) to those present originally (νs
0).
νs* = 0.563 *[1-(1-C.S./400)3] (7)
νs0 [(1-C.S./400)3 – 0.753]
where C.S. is the compression set with 25% compression. Thus f can be determined with compression set data and knowledge of original and aged network chain density.
Network chain density was calculated as follows. Young’s modulus, E, was determined on these unfilled compounds by correlation with their Shore A hardnesses.27 It was assumed that the metal oxides did not significantly reinforce the cured rubber. Then, according to the statistical theory of rubber elasticity28, the molecular weight of the average network strand, Mc, is given by
E = 3ρRT/Mc (8)
11

where ρ is the density of the elastomer, R is the gas constant, and T is absolute temperature. For these samples and by using hardness data, Mc is determined by total (entanglement + chemical) crosslink density. For all three polymers, hardness increased during aging, indicating a net increase in crosslink density during aging. Mc values are calculated in Table VI. With an average mer weight of 85, the number of monomer units in a network strand ranges from 40 to 58. Use of equation 8 does assume a Gaussian distribution of crosslinked chain lengths within the sample, which may not be valid for the complex fluoroelastomer cure chemistry, relying as it does on phase transfer agents and heterogeneous curatives. These Mc values, however, are in good agreement with those calculated on unfilled vulcanizates of Viton® A-Hv that were also presumably cured with BpAF.18
In Table VII, network chain density values for the original sample (νt0) and after
aging at 70 hours and 336 hours at 200°C (νt70 and νt
336, respectively) derived from Mc, νs
70/ νs0, νs
336/ νs0, f 70, and f336 are given for the three samples. These results imply that
crosslink disassociation during the first 70 hours aging at 200°C is practically negligible in all three compounds. All compression set incurred during the first 70 hours of aging is due to additional crosslinks formed within the sample. Some crosslink degradation is observed during the 336 hour aging period – minimal in the high molecular weight sample, but increasingly important in the mid and low molecular weight polymer. Note that the standard ASTM method was followed for handling the pellet after releasing the jig. More relaxation would be expected if these pellets were warmed in an unstressed state after the test; this would lead to lower values of f.
As expected, compression set values decreased with increasing molecular weight. More quantitative analysis requires knowledge of sol-gel fractions of the vulcanizate, which was not obtained in this study.
It is of interest to compare the effective chain length determined by the Young’s modulus with that based on the concentration of BpAF crosslinking agent alone. Assuming a BpAF concentration of 1.1 x 10-4 mol/cc, and using Flory’s correction for dangling chain ends,25 Mc is predicted to be about 15,000 for these vulcanizates. Of course, entanglements will reduce Mc by providing physical crosslinks. But in comparison to other systems in which the contributions of entanglements are calculated, the effect on fluoroelastomer seems extreme.29,30
At least three factors may be responsible for the anomalously high crosslink density. First, fluoroelastomers are known to exhibit strong dipole-dipole interactions between polymer chains which could cause errors in the hardness measurement due to slow relaxation of chains.13 Secondly, bisphenol AF is a rigid molecule and this rigidity may be so great that it introduces a larger than normal amount of entanglements into the network. But an additional factor is that crosslinks are generated in the presence of phosphonium accelerator and metal oxides without the presence of BpAF.15,16 Polymer #1 was milled with 1.8 phr BTPPC, 3.0 phr MgO, and 6.0 phr Ca(OH)2 only. The compound turned dark tan and about 11% of it would not dissolve in DMAC. This same compound was heated in a 177°C oven for 10 minutes which rendered about 17% insoluble in DMAC. It is irrelevant to this discussion whether the crosslinking occurred before or after exposure to solvent since the 16 hour post-cure that all of the fully
12

compounded materials received in this study would eliminate any kinetic barriers to reaction. Therefore the number of chemical crosslinks is greater than that expected on the basis of BpAF content alone.
Fogiel found the chemical crosslink density of Viton® A-HV cured with a bisphenol to be much higher than that theoretically possible if the bisphenol is simply acting as a tetrafunctional crosslink site.13 It is not clear whether he considered the effect of BpAF-free crosslinks in his analysis.
Compression set performance of the Ca(OH)2-free series is worse than that of the mixed oxide recipes and much worse than expected based on previous work with similar cure recipes.22 A second set of compounds with the same recipe was prepared from these polymers and similar compression set values were found. At present we have no explanation for this behavior unless the absence of calcium hydroxide makes these compounds more sensitive to residual moisture in the gum. As discussed above, residual moisture was present in the low molecular weight polymer and additional efforts were required to remove it. Conventional mixed oxide fluoroelastomer compounds are sensitive to ambient humidity (the “summer/winter” effect), but we do not have enough experience with Ca(OH)2-free recipes to know whether they will behave differently.
The carbon black-filled compounds exhibited poorer compression sets than the unfilled compounds. Because of flaws in the network, slower stress relaxation, and oxidation of the black itself during aging, this behavior is not unexpected. What is notable about these compounds is that the compression set values are not only much lower than typical terpolymer values, but that there is a significant difference between the behavior of the unfilled and filled compounds; for commercial VF2/TFE/HFP fluoroelastomers, at least, little effect on compression set performance has been found as carbon black content is changed.31
13

CONCLUSION
The use of degenerative iodine chain transfer has allowed the preparation of three well defined VF2/TFE/HFP terpolymer fluoroelastomers. The molecular weights of these three polymers spanned the Mooney viscosity range of commercially available materials. The distinct differences in molecular weight allowed identification of a molecular weight effect in cure chemistry. Stress-strain properties of the three polymers were similar to those of commercial grades, indicating that molecular weight distribution does not play a major role in determining their tensile behavior. Compression set, on the other hand, were excellent for both unfilled and filled compounds. An unexplained phenomenon, though, was the poor compression sets observed with a calcium hydroxide-free formulation. The effect of residual moisture on the performance of these compounds is worth further study.
14

ACKNOWLEDGEMENTS
I wish to thank R. Stevens for his help in arranging evaluation of these polymers, Y. Brun (DuPont) for providing the GPC method, and W. Schmiegel for helpful discussions.
Viton® is a registered trademark of DuPont Dow Elastomers.
The information set forth herein is furnished free of charge and is based on technical data that DuPont Dow Elastomers believes to be reliable. It is intended for use by persons having technical skill, at their own discretion and risk. Handling precaution information is given with the understanding that those using it will satisfy themselves that their particular conditions of use present no health or safety hazards. Because conditions of product use and disposal are outside our control, we make no warranties, express or implied, and assume no liability in connection with any use of this information. As with any material, evaluation of any compound under end-use conditions prior to specification is essential. Nothing herein is to be taken as a license to operate or a recommendation to infringe on any patents. While the information presented here is accurate at the time of publication, specifications can change. Please check www.dupont-dow.com for the most up-to-date information. CAUTION: Do not use in medical applications involving permanent implantation in the human body. For other medical applications, discuss with a DuPont Dow Elastomers customer service representative and read Medical Caution Statement H-69237.
15

REFERENCES
1. M. Tatemoto and T. Nakagawa (to Daikin), U. S. Patent 4,158,678, Jun. 19, 1979.
2. M. Tatemoto, T. Suzuki, M. Tomoda, Y. Furusaka, and Y. Ueta (to Daikin), U.S. Patent 4,243,770, Jan. 6, 1981.
3. D. P. Carlson (to DuPont), U. S. Patent 5,037,921, Aug. 6, 1991.
4. D. P. Carlson (to DuPont), U. S. Patent 5,102,965, Apr. 7, 1992.
5. W. W. Schmiegel (to DuPont Dow Elastomers), U. S. Patent 6,429,271, Aug. 6, 2002.
6. V. Arcella, G. Brinati, M. Albano, and V. Tortelli (to Ausimont), U.S. Patent 5,585,449, Dec. 17, 1996.
7. V. Arcella, G. Brinati, M. Albano, and V. Tortelli (to Ausimont), U. S. Patent 5,625,019, Apr. 29, 1997.
8. T. Enokida, H. Akimoto, and H. Tatsu (to Nippon Mektron), U. S. Patent 5,969,066, Oct. 19, 1999.
9. S. Saito and H. Tatsu (to Nippon Mektron) U. S. Patent 6,011,129, Jan. 4, 2000.
10. M. Oka and M. Tatemoto, Contemporary Topics in Polymer Science, Plenum Press, New York, 1984, Vol. 4, p 763.
11. B. Améduri and B. Boutevin, J. Fluorine Chem., 100, 97, (1999).
12. M. Apostolo, V. Arcella, G. Storti, and M. Morbidelli, Macromolecules, 35, 6154 (2002).
13. T. L. Smith and W. H. Chu, J. Polym. Sci, A-2, 10, 133 (1972).
14. A. W. Fogiel, J. Polym. Sci., Symp., 53, 333 (1975) and references therein.
15. W. W. Schmiegel, Kaut. Gummi Kunst., 31, 137, (1978).
16. W. W. Schmiegel, Angew. Macromol. Chem., 76/77, 39 (1979).
17. A. Neppel, M. v. Kuzenko, and J. Guttenberger, Rubber Chem. Technol., 56, 12 (1983).
18. D. J. Plazek, I.-C. Choy, F. N. Kelley, E. von Meerwall, and L.-J. Su, Rubber Chem. Technol., 56, 866 (1983).
19. P. Venkateswarlu, R. E. Kolb, and R. A. Guenthner, Polym. Preprints, Am. Chem. Soc., 31(1), 360 (1990).
20. A. N. Theodore and R. O. Carter III, J. Appl. Polym. Sci., 49, 1071 (1993).
21. A. N. Theodore, M. Zinbo, and R. O. Carter, III, J. Appl. Polym. Sci., 61, 2065 (1996).
22. P. Mayor-Lopez, “Meeting Needs for Low, No-post-cure Viton®” technical data sheet, DuPont Dow Elastomers, L. L. C., 2002.
23. D. Rivin, Rubber Chem. Technol., 36, 729 (1963).
16

24. Y.-H. Hsu and J. E. Mark, Polym. Eng. Sci., 27, 1203 (1987).
25. P. J. Flory, Ind. Eng. Chem., 38, 417 (1946).
26. F. P. Baldwin, Rubber Chem. Technol., 43, 1040 (1970).
27. B. J. Briscoe and K. S. Sebastian, Rubber Chem. Technol., 66, 827 (1993).
28. L. R. G. Treloar, “Physics of Rubber Elasticity”, 2nd ed., Oxford University Press, London, 1958.
29. L. Mullins, J. Appl. Polym. Sci., 2, 1 (1959).
30. S. Tamura and K. Murakami, J. Appl. Polym. Sci., 16, 1149 (1972).
31. “Viton® B-601C data sheet”, DuPont Dow Elastomers, L. L. C., 1998.
17
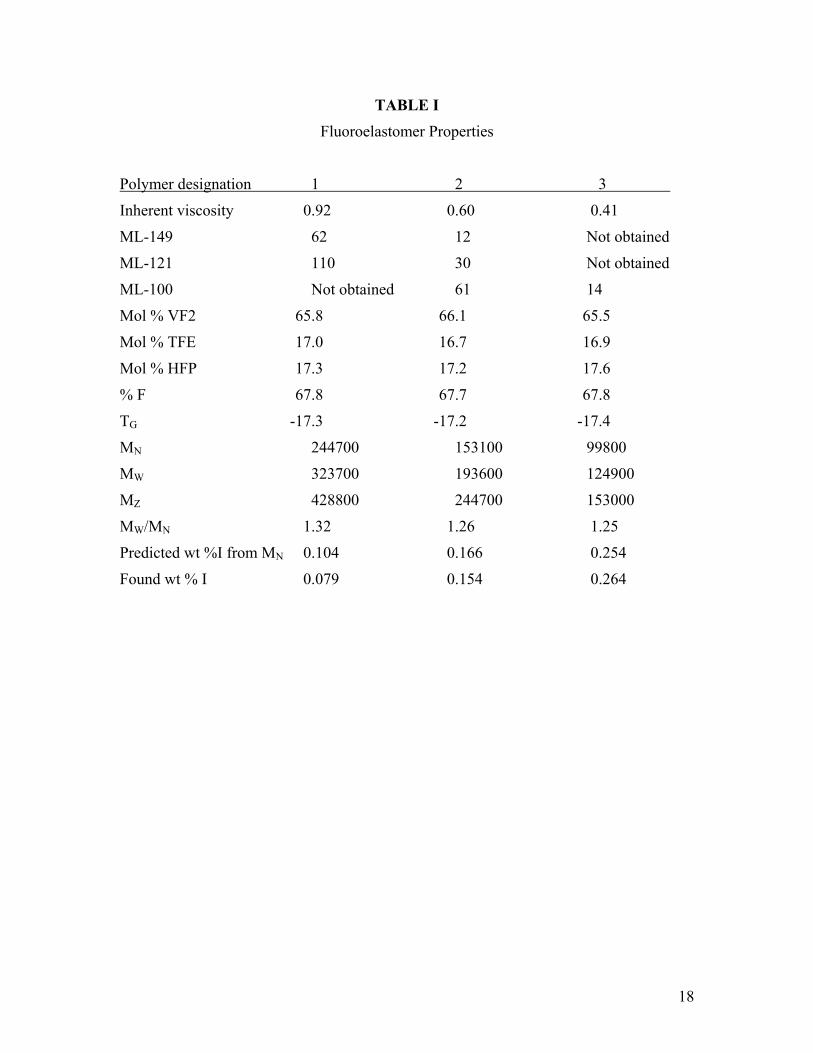
TABLE I
Fluoroelastomer Properties
Polymer designation 1 2 3
Inherent viscosity 0.92 0.60 0.41
ML-149 62 12 Not obtained
ML-121 110 30 Not obtained
ML-100 Not obtained 61 14
Mol % VF2 65.8 66.1 65.5
Mol % TFE 17.0 16.7 16.9
Mol % HFP 17.3 17.2 17.6
% F 67.8 67.7 67.8
TG -17.3 -17.2 -17.4
MN 244700 153100 99800
MW 323700 193600 124900
MZ 428800 244700 153000
MW/MN 1.32 1.26 1.25
Predicted wt %I from MN 0.104 0.166 0.254
Found wt % I 0.079 0.154 0.264
18
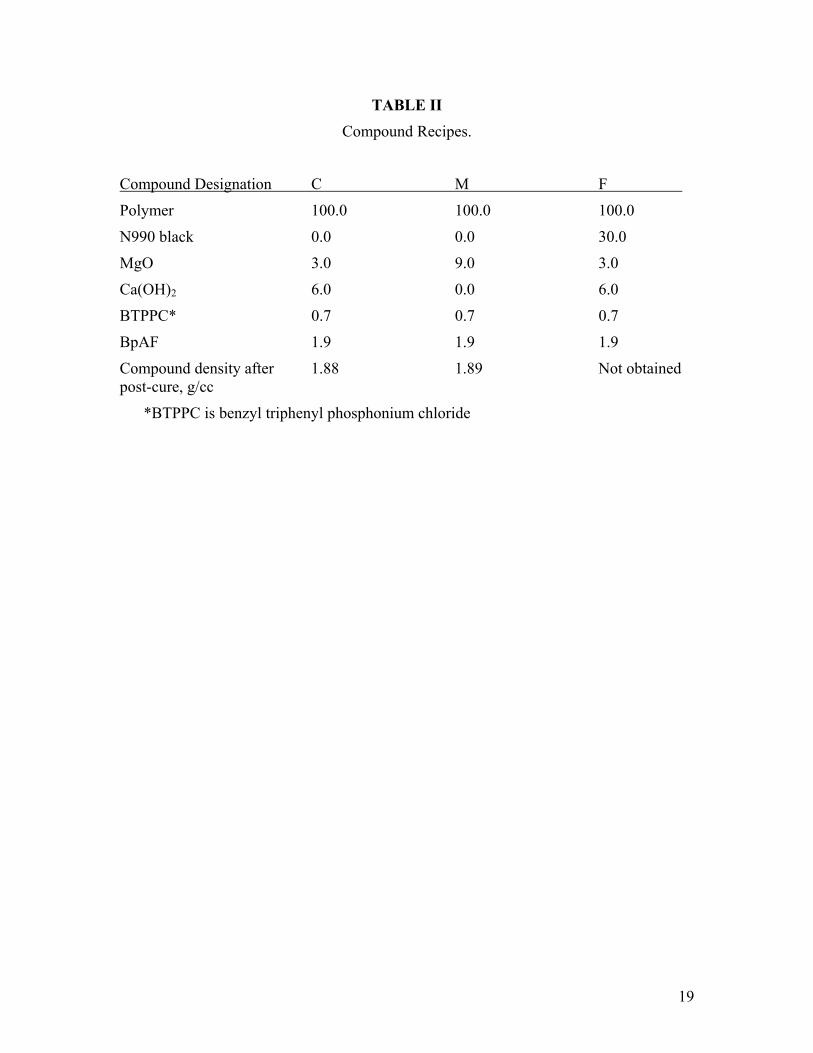
TABLE II
Compound Recipes.
Compound Designation C M F
Polymer 100.0 100.0 100.0
N990 black 0.0 0.0 30.0
MgO 3.0 9.0 3.0
Ca(OH)2 6.0 0.0 6.0
BTPPC* 0.7 0.7 0.7
BpAF 1.9 1.9 1.9
Compound density after 1.88 1.89 Not obtained post-cure, g/cc
*BTPPC is benzyl triphenyl phosphonium chloride
19
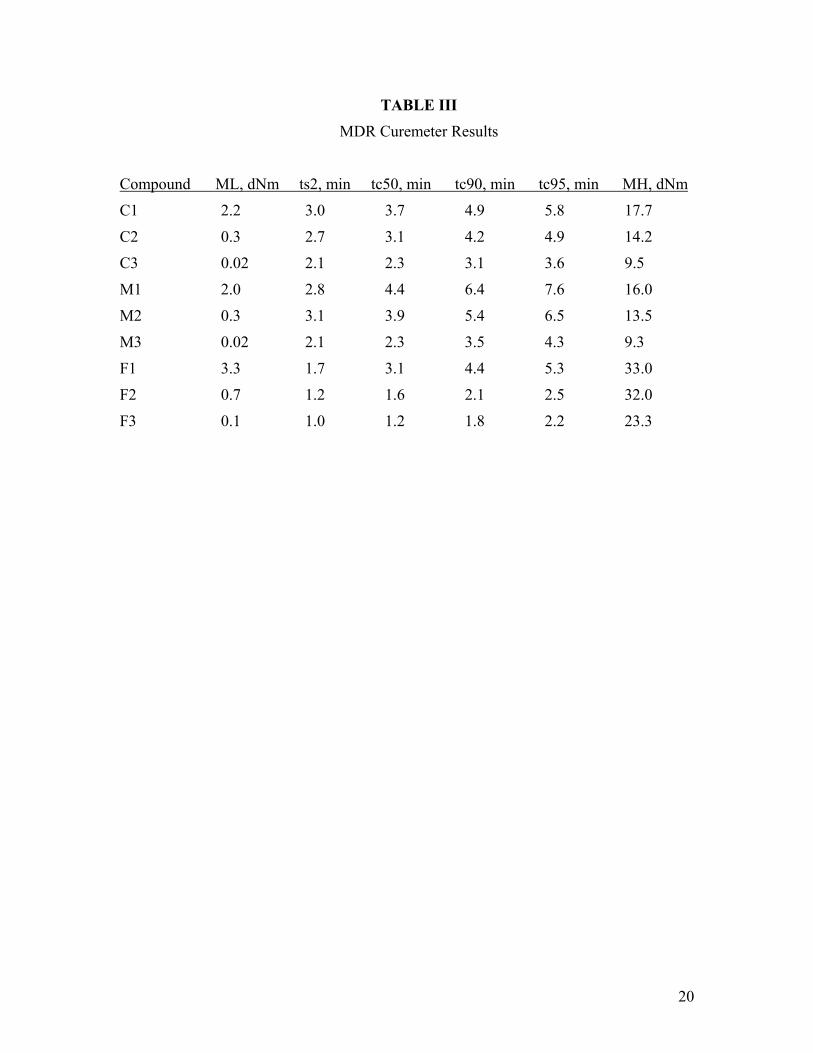
TABLE III
MDR Curemeter Results
Compound ML, dNm ts2, min tc50, min tc90, min tc95, min MH, dNm
C1 2.2 3.0 3.7 4.9 5.8 17.7
C2 0.3 2.7 3.1 4.2 4.9 14.2
C3 0.02 2.1 2.3 3.1 3.6 9.5
M1 2.0 2.8 4.4 6.4 7.6 16.0
M2 0.3 3.1 3.9 5.4 6.5 13.5
M3 0.02 2.1 2.3 3.5 4.3 9.3
F1 3.3 1.7 3.1 4.4 5.3 33.0
F2 0.7 1.2 1.6 2.1 2.5 32.0
F3 0.1 1.0 1.2 1.8 2.2 23.3
20
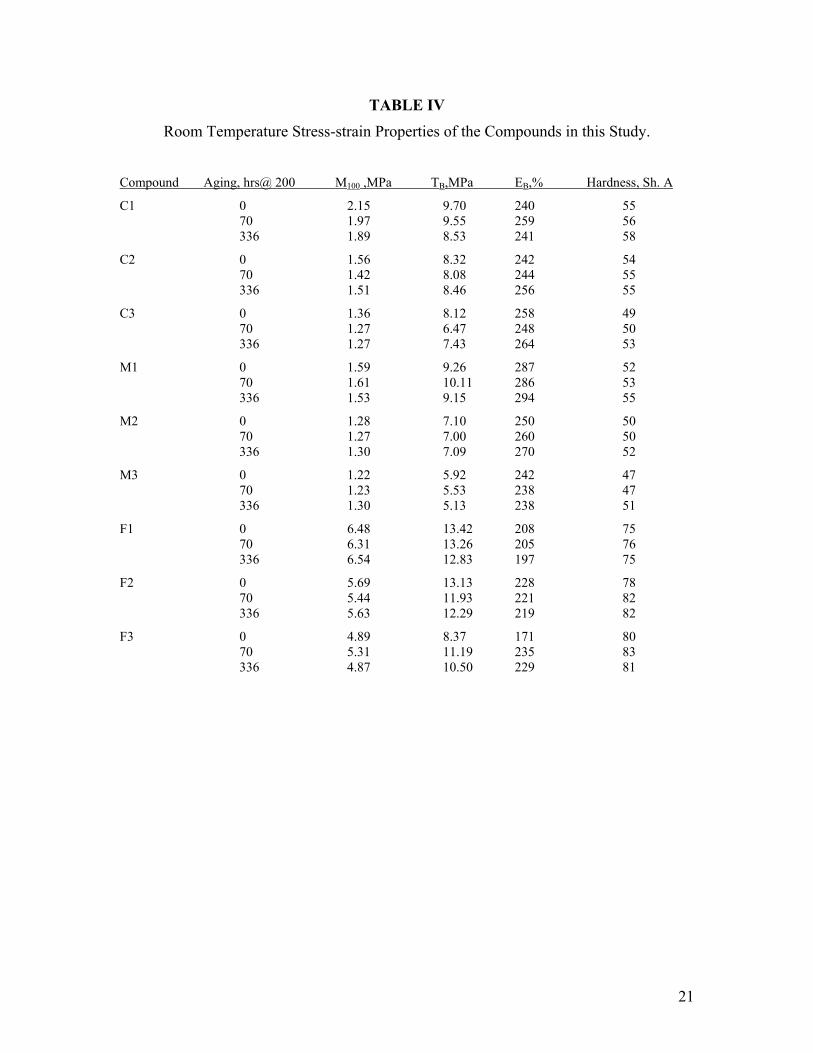
TABLE IV
Room Temperature Stress-strain Properties of the Compounds in this Study.
Compound Aging, hrs@ 200 M100 ,MPa TB,MPa EB,% Hardness, Sh. A
C1 0 2.15 9.70 240 55 70 1.97 9.55 259 56 336 1.89 8.53 241 58
C2 0 1.56 8.32 242 54 70 1.42 8.08 244 55 336 1.51 8.46 256 55
C3 0 1.36 8.12 258 49 70 1.27 6.47 248 50 336 1.27 7.43 264 53
M1 0 1.59 9.26 287 52 70 1.61 10.11 286 53 336 1.53 9.15 294 55
M2 0 1.28 7.10 250 50 70 1.27 7.00 260 50 336 1.30 7.09 270 52
M3 0 1.22 5.92 242 47 70 1.23 5.53 238 47 336 1.30 5.13 238 51
F1 0 6.48 13.42 208 75 70 6.31 13.26 205 76 336 6.54 12.83 197 75
F2 0 5.69 13.13 228 78 70 5.44 11.93 221 82 336 5.63 12.29 219 82
F3 0 4.89 8.37 171 80 70 5.31 11.19 235 83 336 4.87 10.50 229 81
21
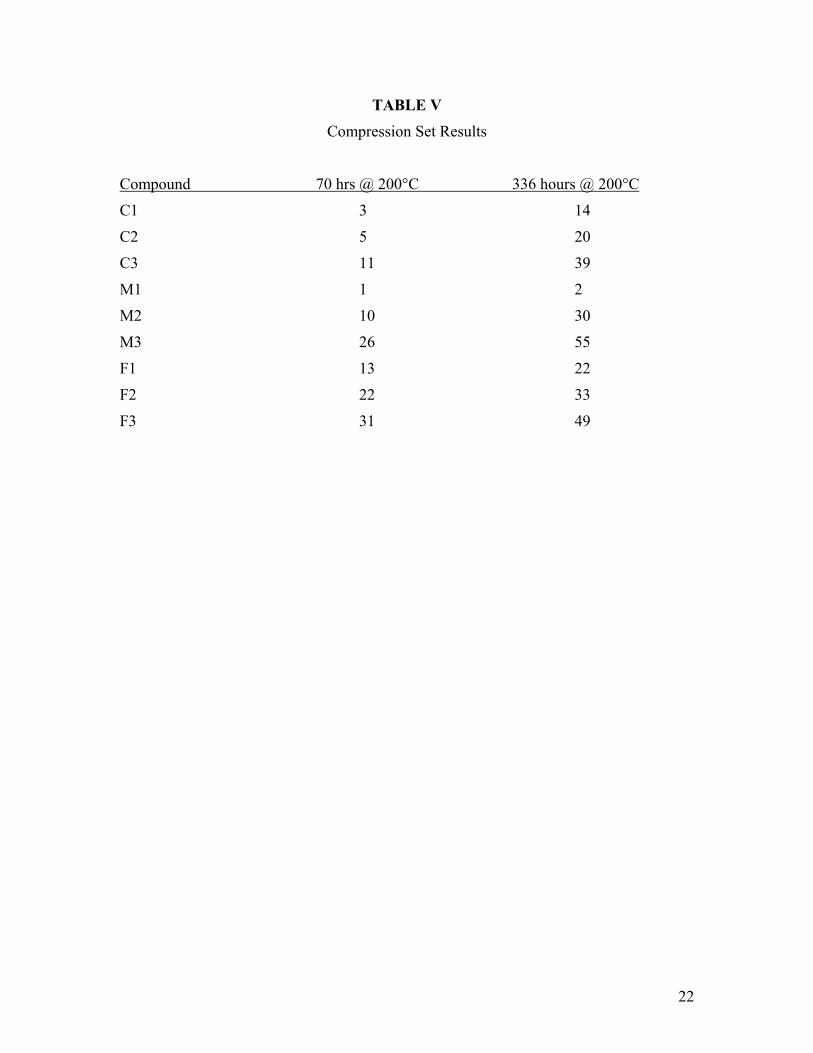
TABLE V
Compression Set Results
Compound 70 hrs @ 200°C 336 hours @ 200°C
C1 3 14
C2 5 20
C3 11 39
M1 1 2
M2 10 30
M3 26 55
F1 13 22
F2 22 33
F3 31 49
22
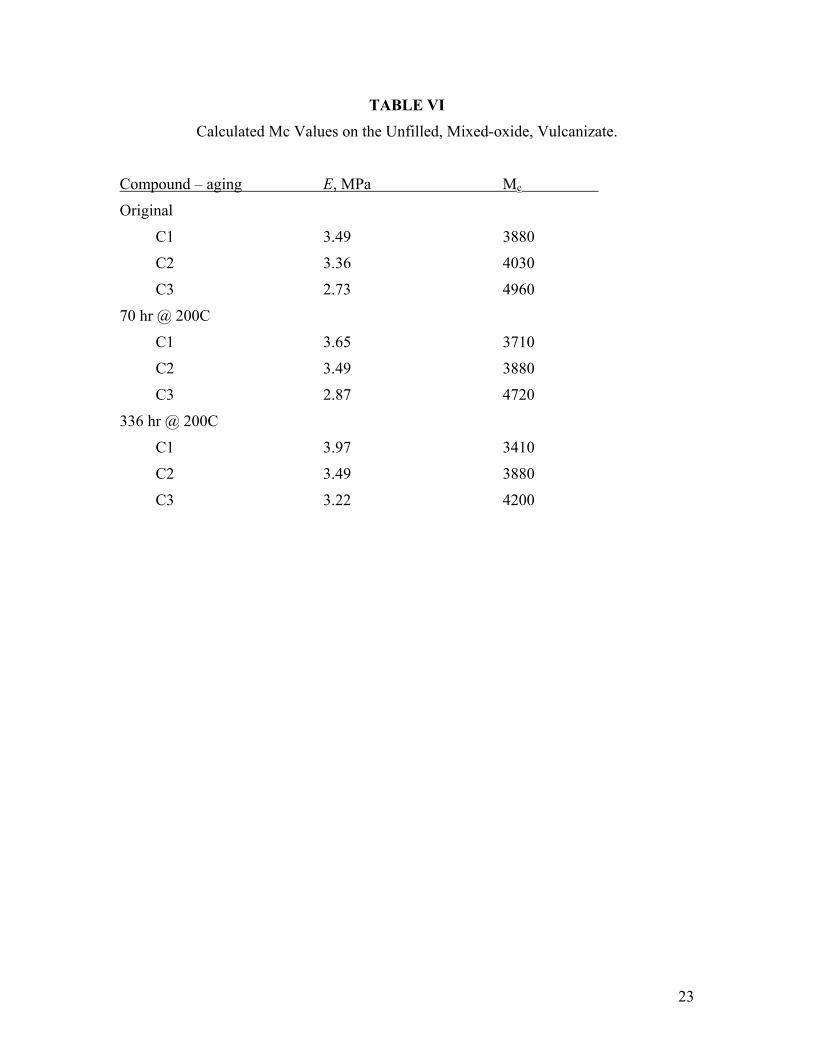
TABLE VI
Calculated Mc Values on the Unfilled, Mixed-oxide, Vulcanizate.
Compound – aging E, MPa Mc
Original
C1 3.49 3880
C2 3.36 4030
C3 2.73 4960
70 hr @ 200C
C1 3.65 3710
C2 3.49 3880
C3 2.87 4720
336 hr @ 200C
C1 3.97 3410
C2 3.49 3880
C3 3.22 4200
23
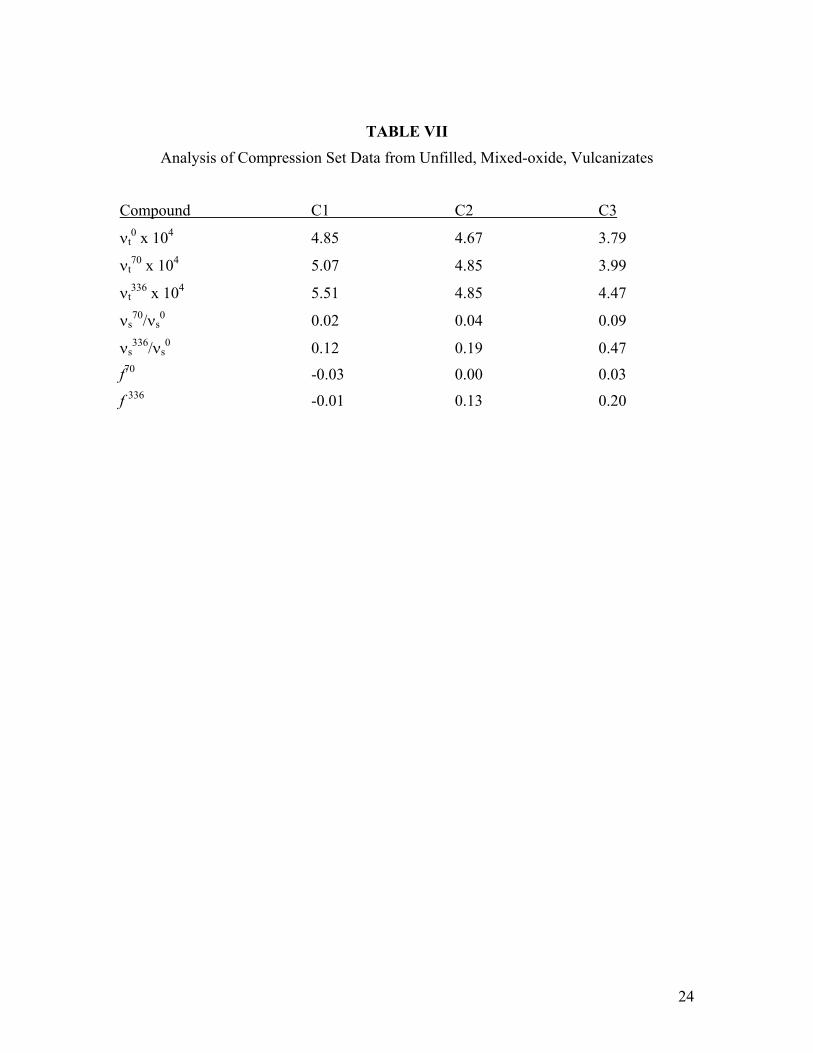
TABLE VII Analysis of Compression Set Data from Unfilled, Mixed-oxide, Vulcanizates
Compound C1 C2 C3
νt0 x 104 4.85 4.67 3.79
νt70 x 104 5.07 4.85 3.99
νt336 x 104 5.51 4.85 4.47
νs70/νs
0 0.02 0.04 0.09
νs336/νs
0 0.12 0.19 0.47
f70 -0.03 0.00 0.03
f 336 -0.01 0.13 0.20
24

LIST OF FIGURES
Figure 1. MDR curves of the unfilled, mixed-oxide, compounds. Compound designations on figure.
Figure 2. Room-temperature stress-strain properties of unfilled compounds cured with the mixed oxide system. Key: ( ) C1; ( ) C2; ( ) C3.
Figure 3. Room-temperature stress-strain properties of filled compounds cured with the mixed oxide system. Key: ( ) F1; ( ) F2; ( ) F3.
Figure 4. Effect of 200°C aging on the stress-strain properties of the unfilled low molecular weight polymer (C3) cured with the mixed-oxide recipe. Curves obtained at room temperature. Key: ( ) original; ( ) aged 70 hours at 200°C; ( ) aged 336 hours at 200°C.
25
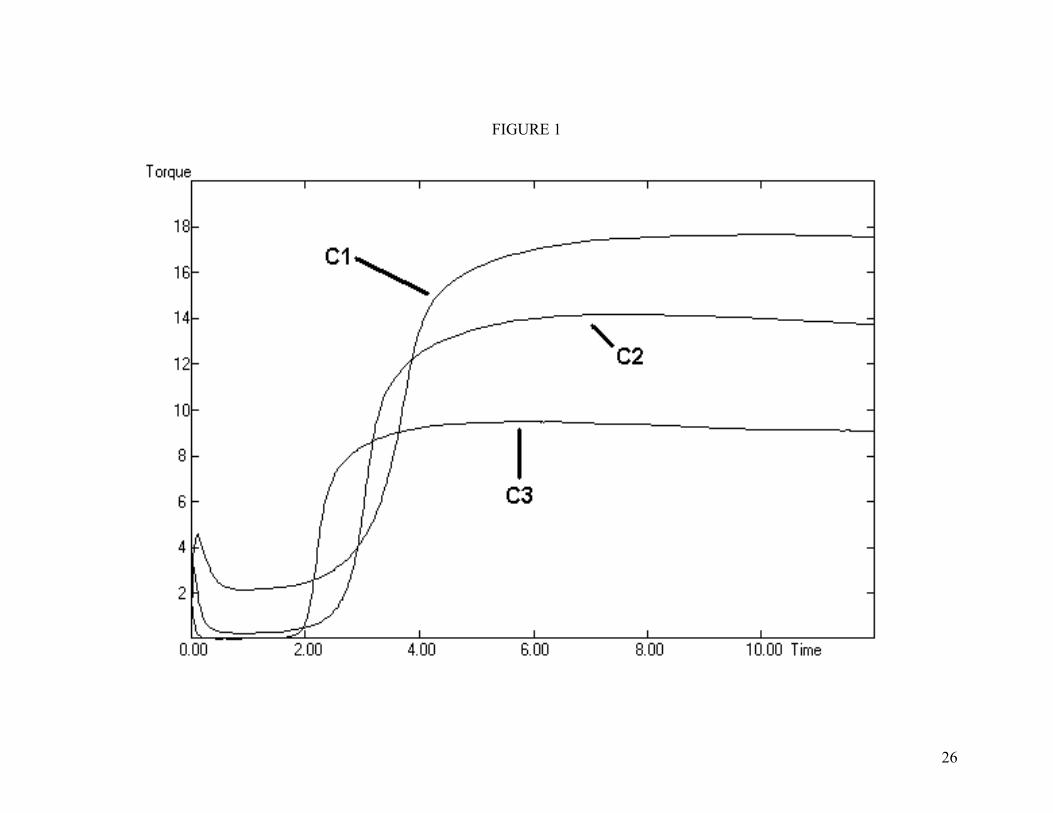
FIGURE 1
26
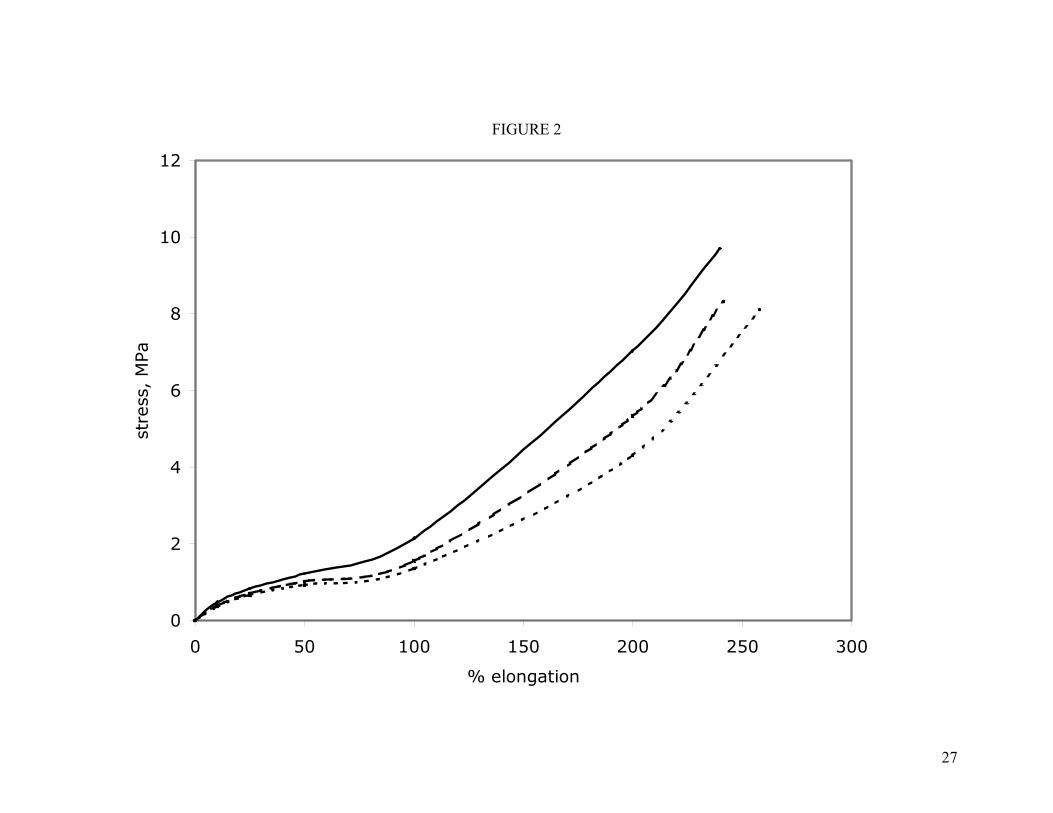
FIGURE 2
0
2
4
6
8
10
12
0 50 100 150 200 250 300
% elongation
stre
ss,
MPa
27
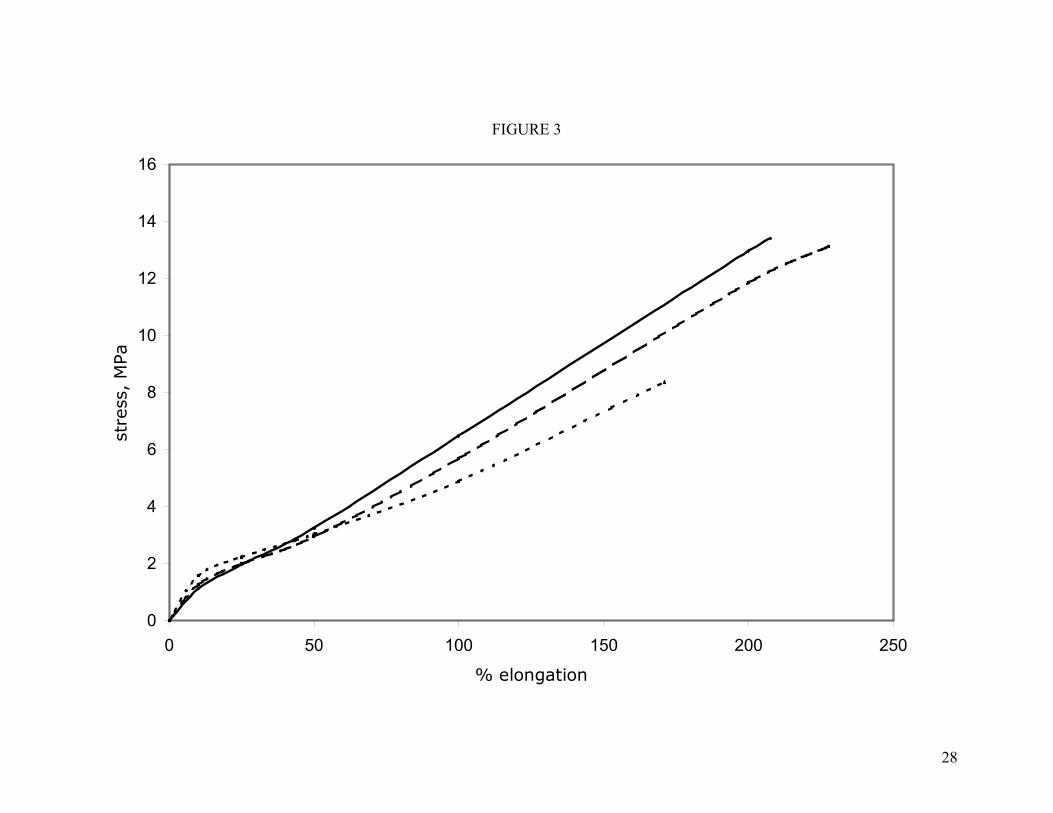
FIGURE 3
0
2
4
6
8
10
12
14
16
0 50 100 150 200 250
% elongation
stre
ss,
MPa
28
29
0
1
2
3
4
5
6
7
8
9
0 50 100 150 200 250 300
% elongation
stre
ss,
MPa
FIGURE 4





























