Embed Size (px)
Citation preview

PRENATAL DIAGNOSISPrenat Diagn 2007; 27: 674–676.Published online 17 May 2007 in Wiley InterScience(www.interscience.wiley.com) DOI: 10.1002/pd.1759
RESEARCH LETTER
The first prenatal diagnosis for veno-occlusive diseaseand immunodeficiency syndrome, an autosomal recessivecondition associated with mutations in SP110
Simon T. Cliffe1,2*, Melanie Wong3, Peter J. Taylor2, Ezia Ruga4, Bridget Wilcken5, Robert Lindeman2,6,Michael F. Buckley1,2 and Tony Roscioli1,2,7
1Centre for Vascular Research, University of New South Wales, Sydney, Australia2Molecular and Cytogenetics Unit, Department of Haematology and Genetics, Prince of Wales Hospital, Sydney, Australia3Department of Allergy, Immunology and Infectious Diseases, The Children’s Hospital at Westmead, Sydney, Australia4Department of Pediatrics, University of Padova, Padova, Italy5Western Sydney Genetics Program, Children’s hospital, Westmead, Australia6School of Medical Sciences, University of New South Wales, Sydney, Australia7Sydney South West Integrated Genetics Service, Royal Prince Alfred hospital, University of Sydney, Australia
Objectives We present the first prenatal diagnosis of familial hepatic veno-occlusive disease with immunod-eficiency (VODI). Homozygous mutations in the gene SP110 are the genetic basis of VODI. The proband inthis report presented at three months of age with hepatomegaly hepatic failure and was found to have hypogam-maglobulinemia. He died one month after hepatic transplant at eight months of age due to hemophagocyticsyndrome. DNA testing detected a homozygous truncating mutation in exon 5; SP110 c.642delC. Prenataltesting was offered to this family in a subsequent pregnancy.
Methods Chorion villus was sampled at 12 weeks’ gestation. DNA was extracted using standard techniques,and sequencing of SP110 exon 5 was performed using flanking primers. Maternal contamination was excludedby examining STR markers in CVS and maternal DNA.
Results A heterozygous SP110 c.642delC mutation was detected in exon 5. This mutation was present inheterozygous form in both parents.
Conclusions The prenatal test result is predictive of a child with a normal immune and hepatic phenotype.This report presents the first prenatal molecular diagnosis for VODI and shows the importance of moleculargenetic research in not only defining the aetiology of syndromes but also in assisting reproductive choicesthrough the collaboration of genetic and feto-maternal services. Copyright 2007 John Wiley & Sons, Ltd.
KEY WORDS: Chorionic villus sample (CVS); veno-occlusive disease with immunodeficiency (VODI)
INTRODUCTION
Familial Veno-Occlusive Disease with Immunodefi-ciency (VODI, OMIM235550) is an autosomal recessivedisorder whose aetiology has been identified recentlyas being due to mutations in the nuclear body pro-tein SP110 (Roscioli et al., 2006). The clinical crite-ria employed to diagnose VODI include: (1) clinicalevidence of immunodeficiency including Pneumocystisjerovici infection, mucocutaneous candidiasis, enterovi-ral or cytomegalovirus infections; (2) hepatomegaly,evidence of hepatic failure or a histological diagnosisof veno-occlusive disease (VOD; also known as sinu-soidal obstruction syndrome) not explained by iatrogenicfactors such as cyclophosphamide treatment or other
*Correspondence to: Simon T. Cliffe, Molecular and CytogeneticsUnit, Department of Haematology and Genetics, Prince of WalesHospital, Barker St Randwick, Sydney, Australia.E-mail: [email protected]
cytoreductive therapy; (3) a pattern of disease consistentwith autosomal recessive inheritance; (4) onset prior tothe age of 12 months. Familial clusters of VOD mayrepresent complications of ingested toxins rather thana genetic aetiology, particularly if they occur in olderage groups and are not reported to be associated withimmunodeficiency (Shah et al., 1987; Al-Hasany andMohamed, 1970). The index of suspicion for VODIshould, however, be high particularly where a combinedimmunodeficiency consistent with this diagnosis is asso-ciated with VOD in a potentially consanguineous couple.
Twenty children from nine Australian Lebanese fam-ilies have been diagnosed with VODI over a periodof 30 years. These patients with VODI presented at3–7 months with either a combined T and B cell immun-odeficiency or VOD or with both. All known VODIpatients in Australia have had hypogammaglobuline-mia at presentation, with absent germinal centres andplasma cells but normal T cell numbers suggestive of aB-cell maturation defect. VODI has a high mortality in
Copyright 2007 John Wiley & Sons, Ltd. Received: 1 January 2007Revised: 26 March 2007
Accepted: 27 March 2007Published online: 17 May 2007
FIRST PRENATAL DIAGNOSIS FOR CONDITIONS ASSOCIATED WITH MUTATIONS IN SP110 675
childhood, but if the child survives the acute present-ing illness, the immunodeficiency responds favourably toregular intravenous immunoglobulin (IVIG) and pneu-mocystis prophylaxis (Roscioli et al., 2006).
VODI was originally described in Australians ofLebanese origin by (Mellis and Bale, 1976). The major-ity of children who have been ascertained subsequent tothis description have also been of Lebanese ethnic ori-gin with a prevalence of VODI in the Sydney Lebanesepopulation estimated to be 1 in 2500 (Roscioli et al.,2006). Two previous case reports of familial disordersresembling VODI have appeared in the literature. Thefirst is a case report of two siblings with VOD in theLebanese literature, but without mention of immunode-ficiency (Firzli, 1978). The significance of this omissionis, however, difficult to interpret, given the scarcity ofdiagnostic immunopathology investigations at that time.These families have not been available for molecularcharacterization. A further case report from Lebanon oftwo siblings of non-consanguineous Lebanese parentshas been published, one of whom survived VODI withdemonstrated humoral and cellular immune dysfunction(Etzioni et al., 1987). There are two sporadic cases ofVODI known, one as a case report in the Spanish liter-ature (Manzanares Lopez-Manzanares et al., 1992) andthe second from Padua in northern Italy (Ruga, 2006).The prevalence of VODI in children of non-Lebaneseorigin has yet to be defined, but these sporadic casesindicate that the VODI phenotype is not specific to theLebanese population.
In previous work done by our group, the VODIcandidate region was identified through homozygositymapping and the genetic basis of the disorder was shownto be due to truncating mutations in the SP110 gene(Roscioli et al., 2006). SP110 (Speckled 110kDa), aninterferon gamma inducible gene, was first recognizedin 1993 (Kadereit et al., 1993). It encodes a full-length713 amino acid protein containing a SAND-, planthomeobox- and Bromo-domain, which are commonfeatures in modular proteins involved in chromatin-mediated gene transcription (Bloch et al., 1996). Thefunction of SP110 has not been fully characterized,however it is known to a nuclear protein with a role inthe transcriptional coactivation of retinoic acid receptor-mediated pathways (Bloch et al., 2000, Watashi et al.,2003). SP110 is also one of a number of proteinsthat interact with the Promyelocytic Leukaemia Nuclearbody, which has probable roles in apoptosis, cancerprotection, and in viral response (Wang et al., 1998a,b).
Figure 1—Pedigree showing proband (II.1) and patient (II.2)
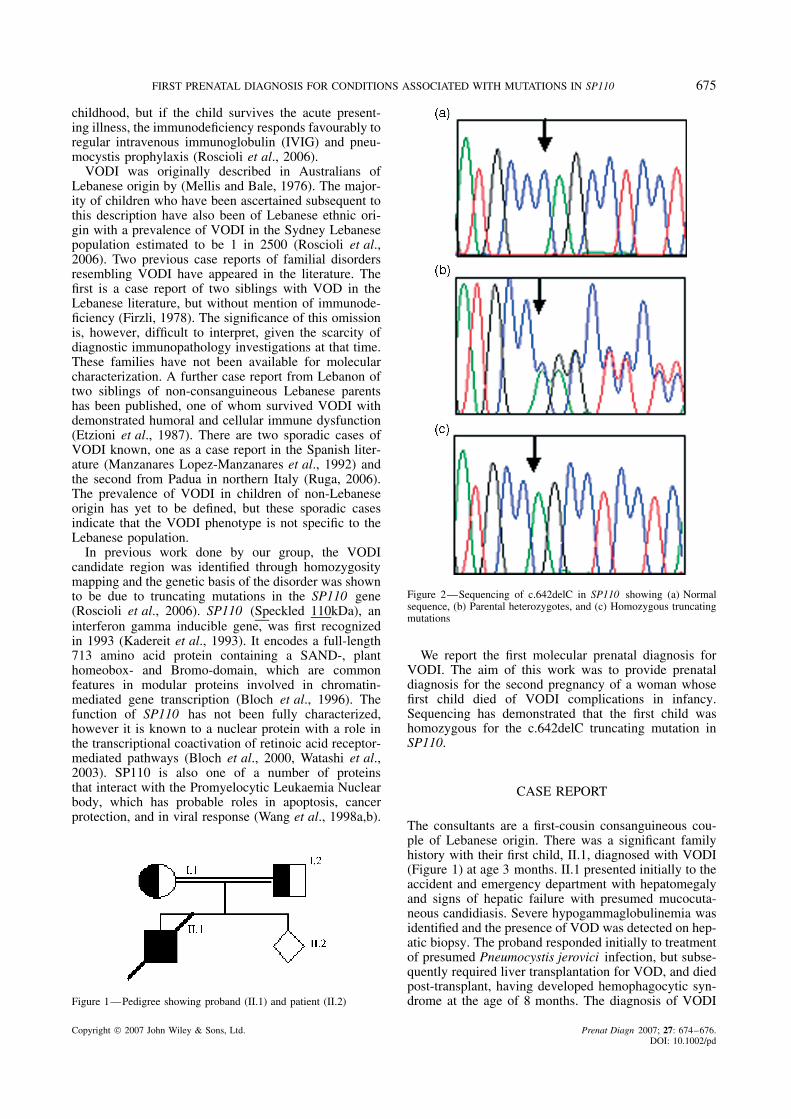
Figure 2—Sequencing of c.642delC in SP110 showing (a) Normalsequence, (b) Parental heterozygotes, and (c) Homozygous truncatingmutations
We report the first molecular prenatal diagnosis forVODI. The aim of this work was to provide prenataldiagnosis for the second pregnancy of a woman whosefirst child died of VODI complications in infancy.Sequencing has demonstrated that the first child washomozygous for the c.642delC truncating mutation inSP110.
CASE REPORT
The consultants are a first-cousin consanguineous cou-ple of Lebanese origin. There was a significant familyhistory with their first child, II.1, diagnosed with VODI(Figure 1) at age 3 months. II.1 presented initially to theaccident and emergency department with hepatomegalyand signs of hepatic failure with presumed mucocuta-neous candidiasis. Severe hypogammaglobulinemia wasidentified and the presence of VOD was detected on hep-atic biopsy. The proband responded initially to treatmentof presumed Pneumocystis jerovici infection, but subse-quently required liver transplantation for VOD, and diedpost-transplant, having developed hemophagocytic syn-drome at the age of 8 months. The diagnosis of VODI
Copyright 2007 John Wiley & Sons, Ltd. Prenat Diagn 2007; 27: 674–676.DOI: 10.1002/pd

676 S. T. CLIFFE ET AL.
was confirmed in the proband by the detection of ahomozygous truncating c.642delC mutation in exon 5of SP110. This mutation was detected in heterozygousform in both unaffected parents (Figure 2).
The proband’s parents requested a genetic consul-tation to discuss the possibility of a prenatal diagno-sis for VODI in the first trimester of their subsequentpregnancy, II.2. The couple chose to pursue prena-tal testing after fully informed consent was obtained.Chorionic villi were sampled at 12 weeks’ gestation,and fetal DNA was extracted using a standard tech-nique. PCR oligonucleotide primers were designed thatflanked exon 5 of SP110, based on Genbank refer-ence Sequence NM 080424 (primer sequences and PCRconditions available from the authors on request) andbidirectional sequencing of SP110 was performed. Theprenatal test result detected a heterozygous c.642delCmutation in exon 5 of SP110, predictive of a child witha normal immune and hepatic phenotype. STR markersin the CVS-extracted DNA were compared to maternalalleles at 8 unlinked loci to exclude maternal contami-nation. The karyotype was normal. Postnatal testing ofimmune function was normal and the child has remainedwell without any features of VODI.
DISCUSSION
Although the aetiology of VODI has only been describedrecently, the full clinical spectrum of the VODI pheno-type segregates perfectly with homozygous truncatingmutations of SP110. The disorder is associated withsignificant morbidity and a 90% mortality even withtreatment with intravenous immunoglobulin and pneu-mocystis prophylaxis. As the long-term efficacy of thetreatment is as yet unknown, and this family had alreadyexperienced the death of one affected child, they soughtprenatal diagnosis. This represents the first time molec-ular prenatal testing has been provided for VODI viaanalysis of SP110 mutations.
The identification of the genetic basis of VODI hasmade molecular prenatal diagnosis in an additionalinherited immunodeficiency possible. Further investiga-tions may extend the phenotype of immunodeficiencieswhere prenatal molecular diagnosis is able to be offeredand the clinical spectrum of VODI may be clarified whenadditional cases with clinical variability of disease areascertained. This report describes a simple and effectivemethod for diagnostic testing in a prenatal setting.
ACKNOWLEDGEMENTS
The authors wish to thank the family for participatingin this study and SEALS molecular and cytogeneticslaboratory for providing infrastructure. The work wassupported by the Australian NHMRC and an NHMRCpostgraduate research scholarship.
REFERENCES
Al-Hasany M, Mohamed AS. 1970. Veno-occlusive disease of theliver in Iraq. Nine cases occurring in three bedouin families. ArchDis Child 45: 722–724.
Bloch DB, Delamonte SM, Guigaouri P, Filippov A, Bloch KD.1996. Identification and characterization of a leukocyte-specificcomponent of the nuclear body. J Biol Chem 271: 29198–29204.
Bloch DB, Nakajima A, Gulick T, et al. 2000. Sp110 localizes to thePML-Sp100 nuclear body and may function as a nuclear hormonereceptor transcriptional coactivator. Mol Cell Biol 20: 6138–6146.
Etzioni A, Benderly A, Rosenthal E, et al. 1987. Defective humoraland cellular immune functions associated with venoocclusivedisease of the liver. J Pediatr 110: 549–554.
Firzli SS. 1978. Veno-occlusive familial hepatic disease. J Pediatr 92:862.
Kadereit S, Gewert DR, Galabru J, Hovanessian AG, Meurs EF.1993. Molecular-cloning of 2 new interferon-induced, highly relatednuclear phosphoproteins. J Biol Chem 268: 24432–24441.
Manzanares Lopez-Manzanares J, Moreno Villares JM, MedinaMonzon C, et al. 1992. Veno-occlusive disease of the liverassociated with humoral and cellular immunodeficiency. An EspPediatr 36: 314–316.
Mellis C, Bale PM. 1976. Familial hepatic venoocclusive disease withprobable immune deficiency. J Pediatr 88: 236–242.
Roscioli T, Cliffe ST, Bloch DB, et al. 2006. Mutations in the geneencoding the PML nuclear body protein Sp110 are associated withimmunodeficiency and hepatic veno-occlusive disease. Nat Genet38: 620–622.
Ruga EM, Guariso G, D’Antiga L, et al. 2006. Hepatic veno-occlusive disease with immunodeficiency syndrome: case report.In 12th Meeting of the European Society for Immunodeficiencies(ESID), Budapest.
Shah PK, Chittora MD, Deo IN, Khangarot D, Vyas MM, Tak RS.1987. Veno-occlusive disease of liver—a familial episode. J AssocPhys India 35: 240–242.
Wang ZG, Delva L, Gaboli M, et al. 1998a. Role of PML in cellgrowth and the retinoic acid pathway. Science 279: 1547–1551.
Wang ZG, Ruggero D, Ronchetti S, et al. 1998b. Pml is essential formultiple apoptotic pathways. Nat Genet 20: 266–272.
Watashi K, Hijikata M, Tagawa A, Doi T, Marusawa H, Shimo-tohno K. 2003. Modulation of retinoid signaling by a cytoplasmicviral protein via sequestration of Sp110b, a potent transcriptionalcorepressor of retinoic acid receptor, from the nucleus. Mol CellBiol 23: 7498–7509.
Copyright 2007 John Wiley & Sons, Ltd. Prenat Diagn 2007; 27: 674–676.DOI: 10.1002/pd





























