Embed Size (px)
Citation preview

United Arab Emirates University United Arab Emirates University
Scholarworks@UAEU Scholarworks@UAEU
Medical Education Dissertations Medical Education
11-2018
The Role of Human α7-Nicotinic Acetylcholine Receptors in The Role of Human 7-Nicotinic Acetylcholine Receptors in
Mediating Neuroprotective Action of Curcumin Mediating Neuroprotective Action of Curcumin
Eslam Mohammed Gaber ElNebrisi
Follow this and additional works at: https://scholarworks.uaeu.ac.ae/med_ed_dissertations
Part of the Medicine and Health Sciences Commons
Recommended Citation Recommended Citation Gaber ElNebrisi, Eslam Mohammed, "The Role of Human α7-Nicotinic Acetylcholine Receptors in Mediating Neuroprotective Action of Curcumin" (2018). Medical Education Dissertations. 3. https://scholarworks.uaeu.ac.ae/med_ed_dissertations/3
This Dissertation is brought to you for free and open access by the Medical Education at Scholarworks@UAEU. It has been accepted for inclusion in Medical Education Dissertations by an authorized administrator of Scholarworks@UAEU. For more information, please contact [email protected].

UAEU • rt 1i! )) 0 .l..:l1.a.J I ~ J.SU I L:.IIJ Lo V I Ci.sUJ ~ '\:)' United Arab Emirates University
United Arab E1nirates University
College of Medicine and Health Sciences
THE ROLE OF HUMAN a7-NICOTINIC ACETYLCHOLINE RECEPTORS IN MEDIATING NEUROPROTECTIVE ACTION OF
CURCUMJN
Eslam Mohammed Gaber ElNebrisi
This thesis is submitted in partial fulfilment of the requirements for the degree of
Doctor of Philosophy
Under supervision of Professor Murat Oz
November 2018





vii
Abstract
Curcumin is a polyphenolic compound isolated from the rhizomes of Curcuma
longa. Curcumin has been demonstrated to have antioxidant, anti-inflammatory, and
anticancer properties. Moreover, it has been shown to exhibit beneficial effects in the
treatment of several neurodegenerative diseases including Alzheimer’s and
Parkinson’s diseases. However, the molecular and cellular targets mediating the
pharmacological actions of curcumin remain largely unknown. In this study, the
effects of curcumin application on the functional properties of the nicotinic
acetylcholine receptor, a prototype for ligand gated ion channels were investigated.
Using the two-electrode voltage clamp technique, the results showed that curcumin
co-application caused a significant potentiation of the action of human α7-nicotinic
acetylcholine receptors (α7-nAChR) expressed in Xenopus oocytes. Importantly,
curcumin was found less effective on other nicotinic receptor subunit combinations
and other members of ligand-gated ion channels. Curcumin significantly decreased
desensitization of α7-nAChR suggesting that it acts as a type II PAM. High affinity
binding site for curcumin on α7-nAChR was also verified by molecular docking study.
In the second part of this study, the neuroprotective effects of curcumin in an
animal model of Parkinson’s disease was investigated. Stereotaxic micro-neurosurgery
was successfully established (for the first time in our university) and used to induce
the toxin based (6-hydroxydopamine; 6-OHDA) animal model of Parkinson’s disease.
This was followed by testing the behavior of the animals, tissue collection,
immunohistochemistry, striatal fiber density measurement, and stereological data
analysis. Systemic administration of curcumin alleviated 6-OHDA-induced motor
abnormalities and protected against substantia nigra pars compacta dopaminergic
neuronal loss, through an α7-nAChRs-mediated mechanism. The protective effects of
curcumin were completely reversed by the administration of the α7-nAChR-selective
antagonist methyllycaconitine (MLA).
In summary, the results of this study suggest that α7-nAChR-mediated activation
is an important mechanism for the neuroprotective effects of curcumin in toxin-based
(6-hydroxydopmine; 6-OHDA) animal model of Parkinson’s disease.
Keywords: Curcumin, α7-nAChR, Xenopus oocytes, Parkinson’s disease, 6-OHDA –
neuroprotection

viii
Title and Abstract (in Arabic)
خصائص تفعيل في في مستقبلات الأسيتايل كولين النيكوتينية 7دور وحدات ألفا
الكركم الوقائية للجهاز العصبي
الملخص
تتميز نبتة م. يعتبر الكركم أحد مركبات البولبفينول والمستخلص من جذمور نبات الكرك
لسرطان. وزيادة على ذلك، لمضاد كذلك، و صائص: مضادة للالتهابات، الأكسدةخبعدة الكركم
أمراض ج عدد من الأمراض مثلعرفت هذه النبتة بخصائص دوائية واستخدمت بفاعلية لعلا
ولقد وضعت عدة فرضيات لآلية عمل الكركم التنكس العصبي كداء الزهايمر وداء باركنسون.
وفي هذا السياق ية للنبات.إما عن طريق القنوات الأيونية أو النواقل لكل هذه الخصائص الدوائ
المرتبطة بربيطة أيونية محددة.قمنا بدراسة تأثير مادة الكركم على القنوات الأيونية
وظيفة مستقبلات الأسيتايل كولين النيكوتينية المكونة من لقد بدأنا بدراسة تأثير الكركم على
ثبيت الجهد خدام تقنية ت( و ذلك باستXenopusو المستنسخة في بويضات ضفادع ) 7وحدات ألفا
كهروفسيولوجية استتنتجنا أن مادة التجارب الالكهربائي باستخدام قطبين كهربائيين. من خلال
. 7المكونة من وحدات ألفا مستقبلات الأسيتايل كولين النيكوتينية الكركم لها تأثير محفز على
ية المرتبطة القنوات الأيونموعة وهذا التأثير خاص فقط بهذه المستقبلات دون غيرها من بقية مج
مستقبلات مستقلبات مادة الكركم على ات وثم قمنا بدراسة تأثير مشتق .بربيطة أيونية محددة
لهذه يضا أوقد أعطت تأثير محفز 7المكونة من وحدات ألفا الأسيتايل كولين النيكوتينية
المستقبلات ولكن بدرجة أقل من مادة الكركم.
مادة ل الوقائي تأثيرالفي الجزء الثاني من البحث بدراسة قمنا المعطيات، فإنناوبناءا على هذه
المرض وتطور الكركم على نموذج داء باركنسون. وذلك باستخدام الجرذان وتحريض حدوث
وحقن مادة -للمرة الأولى بجامعة دولة الإمارات العربية المتحدة-من خلال جراحة المخ الدقيقة
قد تبع ذلك اختبار سلوك الحيوانات، جمع الأنسجة، و .هيدروكسيدوبامين-6 الأعصاب السامة
أثبتت النتائج أن الكركم يتمتع . والكيمياء المناعية، وتحليل البيانات المجسمة لجمع النتائج
الأسيتايل كولين النيكوتينية المكونة من عن طريق مستقبلات بخصائص وقائية للجهاز العصبي
( MLAالتأثيرات الوقائية للكركم بإعطاء دواء ميثايلكاكونيتين )تم عكس كما 7وحدات ألفا
.7المضاد الانتقائي لوحدات الألفا

ix
وذات هو آلية مهمة باستخدام الكركم 7 وحدات ألفاتشير نتائج هذه الدراسة إلى أن تنشيط
.لمرض باركنسونفي النموذج الحيواني تأثير وقائي على الجهاز العصبي
المكونة من وحدات ألفا نةمستقبلات الأسيتايل كولين النيكوتي ،كمرالك الرئيسية: البحثم مفاهي
وقاية الجهاز العصبي ،هيدروكسيدوبامين-6 ،داء باركنسون ،7

x
Acknowledgments
First and foremost, I would like to thank my advisors Professor Murat Oz and
Professor Safa Shehab. Professor Oz, you have been encouraging me since I was a
Master student, you were abundantly helpful and offered me invaluable patience,
support and guidance. I would like to thank you for encouraging my research and for
allowing me to grow as a research scientist.
Prof. Shehab, Thank you for the opportunity to work for and learn from you. Thanks for
the patient guidance, encouragement and advice you provided throughout my time as
student in your lab. I consider myself extremely lucky to have a supervisor who cared
so much about my work, and who responded to my questions and queries so promptly.
I must express my deepest gratitude and thanks to Ahmed, my husband, for
supporting me spiritually throughout my study journey. I was continually impressed
by his patience and constant encouragement he provided me with. My lovely kids;
Bara’a, Anas, Rayan, and Yazan, for being patient enough to delay so many activities,
trips, and holidays till I graduate. My special appreciation and thanks to my beloved
family; my father and mother and all my brothers and sisters for their continual prayers
& endless love. I would like to thank my mother and father-in-law, for their daily
prayers every morning.
My thesis committee guided me through all the three years. I would like to convey
my special thanks to the members of the Advisory committee; Dr. Ojha, for his timely
suggestion and valuable contribution at every stage of the research, including both in-
vitro and in-vivo parts. Dr. Bassem Sadek, whom without his knowledge and
assistance this study would not have been successful.

xi
My special thanks are extended to Professor Bassam Ali, the coordinator of the
post-graduate studies for his full support in the past one year. In addition, I must also
appreciate all the guidance and support I have received from Dr. Maryam Al Shamsi,
the former-Assistant Dean of Research of the College of Medicine and Health
Sciences.
I would like to express my sincere gratitude to Professor Eric PK Mensah-Brown
for inspiring me to think bigger, for never-ending support, and for the great effort in
reviewing my thesis.
I would like to extend my thanks to my dearest friends Arwa Al Nahdi, Nermin
Essa, and Shaima Fikri who have given their heart whelming full support all the time.
My thanks also go out of CMHS to AAU, who so kindly participated in this
research by giving generously of their time and collaborating for molecular docking
experiments.
I am grateful to all of those with whom I have had the pleasure to learn during my
study, especial thanks go to Dr. Nassruddin Hammadi for providing me necessary
technical assistance in Animal House and his prompt inspiration. I would like to thank
Dr. Syed Muhammad Nurulain, Mrs. Petrilla Jayaprakash, Dr. Hayate Javid, Mrs.
Anjana Valappil, and Mrs. Sumisha Rehmathulla, for their kind help and co-operation
throughout my work in Prof. Oz and Prof. Shehab laboratories.
Above all, before all and after all, all praise be to Allah for the strength that keeps
me standing and for the hope that keeps me believing that I can do this and still more.

xii
Dedication
To my beloved husband, parents, siblings and
all my supportive family

xiii
Table of Contents
Title ............................................................................................................................... i
Declaration of Original Work ...................................................................................... ii
Copyright .................................................................................................................... iii
Advisory Committee ................................................................................................... iv
Approval of the Doctorate Dissertation ....................................................................... v
Abstract ...................................................................................................................... vii
Title and Abstract (in Arabic) ................................................................................... viii
Acknowledgments ........................................................................................................ x
Dedication .................................................................................................................. xii
Table of Contents ...................................................................................................... xiii
List of Tables........................................................................................................... xviii
List of Figures ........................................................................................................... xix
List of Abbreviations................................................................................................ xxii
Chapter 1: Introduction ................................................................................................ 1
1.1 Curcumin .................................................................................................... 1
1.1.1 Natural Curcumin Analogues and Metabolites .................................... 2
1.1.2 Pharmacokinetics and Pharmacodynamics of Curcumin ...................... 4
1.1.3 Molecular Targets of Curcumin ............................................................ 6
1.1.4 Biological Properties of Curcumin ....................................................... 7
1.1.5 Effects of Curcumin on Different Ion Channels and
Receptors ............................................................................................ 12
1.2 Acetylcholine Receptors .......................................................................... 13
1.2.1 Nicotinic Acetylcholine Receptors ..................................................... 14
1.3 Parkinson’s Disease .................................................................................. 24
1.3.1 Background ......................................................................................... 25
1.3.2 Pathophysiology .................................................................................. 25

xiv
1.3.3 Diagnosis............................................................................................. 27
1.3.4 Treatment ............................................................................................ 31
1.3.5 Prognosis ............................................................................................. 34
1.4 The Neuroprotective Role of Nicotine and Nicotinic Receptors
Against Nigrostriatal Damage .................................................................. 34
1.4.1 Dopaminergic and Cholinergic Systems Correlation and
Dopamine Release .............................................................................. 35
1.4.2 Immune Modulation via Nicotinic Receptors ..................................... 41
1.4.3 Effect of Nicotinic Receptors on L-Dopa Induced
Dyskinesia .......................................................................................... 46
1.4.4 Molecular Neuroprotective Mechanisms of Nicotinic
Acetylcholine Receptors ..................................................................... 52
1.5 Animal Models of Parkinson’s Disease ................................................... 55
1.5.1 The Neurotoxin Model ........................................................................ 55
1.5.2 Genetic Models ................................................................................... 64
1.6 Fundamental Methods of Assessing Structure and Function of
Nigrostriatal Pathway ............................................................................... 67
1.6.1 Dopaminergic Neurons in the Substantia Nigra Pars
Compacta ............................................................................................ 67
1.6.2 Dopaminergic Terminals in the Striatum ............................................ 69
1.6.3 Striatal Dopamine ............................................................................... 69
1.6.4 Lewy Body Aggregates ....................................................................... 69
1.6.5 Behavioral/Motor Assessment ............................................................ 69
Chapter 2: Aims and Objectives ................................................................................ 70
2.1 In-vitro Electrophysiological Study ......................................................... 70
2.2 In-vivo Study ............................................................................................ 70
Chapter 3: Materials and Methods ............................................................................. 72
3.1 Electrophysiological In-vitro Study ......................................................... 72
3.1.1 Female Xenopus Oocytes .................................................................... 72
3.1.2 Chemicals ............................................................................................ 73
3.1.3 Other Materials ................................................................................... 74
3.1.4 Experimental Setup ............................................................................. 75

xv
3.1.5 Preparation of Required Solutions...................................................... 78
3.1.6 Drug Application ................................................................................ 80
3.1.7 Isolation and Maintenance of Oocyte from Xenopus
Laevis ................................................................................................. 81
3.1.8 Oocyte Preparation.............................................................................. 83
3.1.9 Synthesis of cRNA .............................................................................. 85
3.1.10 In-vitro cRNA Synthesis ................................................................... 86
3.1.11 Microinjection of cRNA into Oocytes .............................................. 87
3.1.12 Two Electrode Voltage Clamp.......................................................... 90
3.1.13 Parameters Tested by Electrophysiological Recording .................... 93
3.1.14 Statistical Analysis .......................................................................... 100
3.2 Molecular Docking Experiments............................................................ 101
3.3 In-vivo Study: Animal Model of Parkinson’s Disease ........................... 102
3.3.1 Animals ............................................................................................. 102
3.3.2 Drugs ................................................................................................. 102
3.3.3 Surgical Procedure ............................................................................ 103
3.3.4 Apomorphine-Induced Rotational Behavior ..................................... 107
3.3.5 Histology ........................................................................................... 107
3.3.6 Measurement of Striatal Fiber Density ............................................. 108
3.3.7 Stereological Analysis ...................................................................... 109
3.3.8 Statistical Analysis ............................................................................ 110
Chapter 4: Results .................................................................................................... 111
4.1 Results .................................................................................................... 111
4.1.1 Effects of Curcumin on α7-nicotinic Acetylcholine
Receptors .......................................................................................... 111
4.1.2 Concentration Response Curve ......................................................... 114
4.1.3 Effects of Curcumin on α7-nAChRs are not Mediated by
G-proteins ......................................................................................... 116
4.1.4 Effects of Curcumin on α7-nAChRs are not Mediated by
Protein Kinases ................................................................................. 118
4.1.5 Effects of Curcumin on α7-nAChRs are not Dependent
on Intracellular Ca2+ ......................................................................... 120

xvi
4.1.6 Effects of Curcumin are not Dependent on Changes in
Membrane Potential.......................................................................... 121
4.1.7 Effects of Curcumin at Different Concentrations of
Acetylcholine .................................................................................... 123
4.1.8 Effects of Curcumin on the Desensitization of Nicotinic
Receptors .......................................................................................... 126
4.1.9 Effects of Curcumin on the Specific Binding of [125I]α-
bungarotoxin ..................................................................................... 128
4.1.10 Effects of Curcumin on the Current Mediated by
Different Nicotinic Receptor Subunits and Other
Members of Ligand-Gated Ion Channels ......................................... 130
4.1.11 Effects of Other Curcumin’s Analogues and
Metabolites on the Current Mediated by α7 Nicotinic
Acetylcholine Receptors ................................................................... 132
4.1.12 Docking of Curcumin and Curcumin Derivatives into
the Human α7-nAChR Transmembrane Domain ............................. 134
4.2 In-vivo Results ........................................................................................ 137
4.2.1 Apomorphine-Induced Rotation Test ............................................... 137
4.2.2 Morphological Analysis .................................................................... 140
Chapter 5: Discussion .............................................................................................. 148
5.1 Discussion .............................................................................................. 148
5.1.1 Effects of Curcumin on α7-Nicotinic Acetylcholine
Receptor ............................................................................................ 148
5.1.2 Effects of Curcumin on α7-Nicotinic Receptor are not
Mediated by G-proteins and Protein Kinases, and are
not Dependent on Intracellular Ca2+ Levels, and
Membrane Potential.......................................................................... 149
5.1.3 Effects of Curcumin at Different Concentrations of
Acetylcholine .................................................................................... 151
5.1.4 Effects of Curcumin on the Specific Binding of [125I]α-
bungarotoxin ..................................................................................... 152
5.1.5 Effects of Curcumin on Desensitization of Nicotinic
Receptors .......................................................................................... 153
5.1.6 Docking of Curcumin and Curcumin Derivatives into
the Human α7-nAChR Transmembrane Domain ............................. 154

xvii
5.1.7 Neuroprotective Properties of Curcumin in Parkinson’s
Disease .............................................................................................. 156
Chapter 6: Conclusions ............................................................................................ 170
References ................................................................................................................ 172
List of Publications .................................................................................................. 223
Appendix .................................................................................................................. 224

xviii
List of Tables
Table 1: Curcumin analogues and metabolites ............................................................ 3
Table 2: Motor and non-motor symptoms of Parkinson’s disease ............................. 28
Table 3: UK Parkinson’s disease society Brain Bank clinical
diagnosing criteria ........................................................................................ 30
Table 4: Treatment options of Parkinson’s disease.................................................... 32
Table 5: Chemicals required for the experiments ...................................................... 73
Table 6: Other materials and devices used in the study ............................................. 74
Table 7: Calcium free MBS solution composition ..................................................... 78
Table 8: Antibiotic materials ...................................................................................... 79
Table 9: ND96 solution composition ......................................................................... 79
Table 10: Normal Ringer’s solution composition ...................................................... 80
Table 11: The initial concentration of all subunits .................................................... 88
Table 12: Binding energies of curcumin and curcumin derivatives,
generated from their docking into the human α7-nAChR
transmembrane domain, along with the docking scores of
two known type II PAMs......................................................................... 135
Table 13: nAChRs and PAMs in clinical trials for treatment of PD
(“Home - ClinicalTrials.gov,” n.d.) ......................................................... 166
Table 14: Curcumin in clinical trials for treatment of various
neurodegenerative disorders (“Home - ClinicalTrials.gov,”
n.d.) .......................................................................................................... 168

xix
List of Figures
Figure 1: The source and chemical structure of curcumin ........................................... 1
Figure 2: Molecular targets of curcumin ...................................................................... 7
Figure 3: Therapeutic potential of curcumin ................................................................ 8
Figure 4: Curcumin structural features ...................................................................... 10
Figure 5: The activated forms of the acetylcholine receptor classes ......................... 13
Figure 6: Neuronal nicotinic acetylcholine structure ................................................. 15
Figure 7: Proposed mechanism of activation and desensitization ............................. 17
Figure 8: Types of Allosteric modulators .................................................................. 19
Figure 9: Molecular activation routes of the α7-nAChRs .......................................... 20
Figure 10: Distribution of nicotinic acetylcholine receptors human
brain .......................................................................................................... 22
Figure 11: Proposed mechanism of α7-nAChRs in Parkinson’s
disease ...................................................................................................... 23
Figure 12: Structures of the basal ganglia .................................................................. 24
Figure 13: Lewy body in affected dopaminergic neurons ......................................... 26
Figure 14: Pathophysiology of Parkinson’s disease................................................... 27
Figure 15: Schematic presentation of pathophysiology and treatment
of PD ........................................................................................................ 33
Figure 16: Effect of nicotine treatment on MPTP- lesioned primates ....................... 37
Figure 17: Striatal [3H]dopamine release of wild type and α7-
nicotinic receptor null mutant mice .......................................................... 38
Figure 18: Stimulation of dopamine release in in Drosophila
melanogaster ventral nerve cord (VNC) .................................................. 39
Figure 19: Data of Acetylcholine stimulated dopamine release before
and after bathing with different nicotinic and muscarinic
antagonists ................................................................................................ 40
Figure 20: The role of microglia in health and disease .............................................. 41
Figure 21: Staining of Primary human macrophages with fluorescein
isothiocyanate (FITC)-labelled α-bungarotoxin (α -Bgt,
1.5 mgml21) ............................................................................................. 42
Figure 22: RT–PCR analysis of α7-nAChRs expression on microglia
using N9 and primary cultured microglial cells ....................................... 44
Figure 23: Nicotine inhibits H2O2-induced astrocyte apoptosis
through protection of mitochondrial membrane potential ....................... 45
Figure 24: Nicotine administration reduces L-dopa-induced
dyskinetic-like movements in rats and monkeys ..................................... 48
Figure 25: Effect of TC-8831 and amantadine in combination with L-
DOPA in MPTP-lesioned monkeys ......................................................... 50
Figure 26: Effect of ABT-126 on LID in MPTP treated monkeys ............................ 51

xx
Figure 27 : α7-nicotinic acetylcholine-mediated molecular signaling
mechanism ................................................................................................ 54
Figure 28: Chemical structures of 6-hydroxydopamine (6-OHDA)
and dopamine ........................................................................................... 56
Figure 29: Molecular Mechanisms for different animal models of PD ..................... 58
Figure 30: Chemical structures of MPTP and MPP+ ................................................ 61
Figure 31: Dopaminergic neuronal distribution in striatum and
substantia nigra ......................................................................................... 68
Figure 32: An adult female Xenopus laevis (Professor Murat Oz’s
laboratory) ................................................................................................ 72
Figure 33: Two-electrode voltage-clamp (TEVC) recording set-up
from Xenopus oocytes .............................................................................. 76
Figure 34: The oocyte impaled with two microelectrodes ......................................... 77
Figure 35: Steps of oocyte isolation and preparation ................................................. 82
Figure 36: Frog’s ovarian lobe ................................................................................... 84
Figure 37: Stages of oocyte development .................................................................. 85
Figure 38: Agarose gel analysis of mRNA ................................................................ 86
Figure 39: Microelectrode set-up for cRNA injection ............................................... 89
Figure 40: cRNA injection set-up (Professor Murat Oz’s Laboratory) ..................... 89
Figure 41: Schematic of glass microelectrode assembly ........................................... 91
Figure 42: Schematic illustration of two-electrode voltage clamp
setup using Xenopus oocytes .................................................................... 92
Figure 43: Typical experimental protocol for electrophysiological
recording from oocyte .............................................................................. 94
Figure 44: Radioligand binding assay ........................................................................ 99
Figure 45: Time course of the experiment ............................................................... 104
Figure 46: Stereotaxic surgery to lesion nigrostriatal pathway ................................ 105
Figure 47: The three sites of 6-OHDA intra-striatal injection ................................. 106
Figure 48: The effects of acetylcholine and α-bungarotoxin in
oocytes expressing α7-nAChR ............................................................... 112
Figure 49: Effects of curcumin on α7-nicotinic acetylcholine
receptors ................................................................................................. 113
Figure 50: Effect of curcumin on α7-nicotinic acetylcholine receptors
is time- and concentration-dependent .................................................... 115
Figure 51: Effects of curcumin on α7-nAChR are not mediated by G-
proteins ................................................................................................... 117
Figure 52: Effects of curcumin on α7-nAChR are not mediated by
prtein kinases .......................................................................................... 119
Figure 53: Effects of curcumin on α7-nAChR are not dependent on
intracellular Ca2+ levels .......................................................................... 121
Figure 54: Effects of curcumin are not dependent on changes in
membrane potential ................................................................................ 122

xxi
Figure 55: Effects of curcumin at different concentrations of
acetylcholine .......................................................................................... 124
Figure 56: Acetylcholine concentration response curve .......................................... 125
Figure 57: Effect of curcumin on the desensitization of nicotinic
receptors ................................................................................................. 127
Figure 58: Effects of curcumin on the specific binding of [125I]α-
bungarotoxin .......................................................................................... 129
Figure 59: Effects of curcumin on the current mediated by different
nicotinic receptor subunits and other members of LGICs ..................... 131
Figure 60: Effects of curcumin analogues and metabolites on
Acetylcholine-mediated current ............................................................. 133
Figure 61: The binding mode of curcumin (cyan sticks) obtained
from docking into the human α7-nAChR transmembrane
domain (gray sticks) ............................................................................... 136
Figure 62: Motor performance of the rats was assessed using
apomorphine-induced rotation test (0.25 mg/kg)
expressed as full body turn per minute over 30 min .............................. 138
Figure 63: Apomorphine-induced rotation test in 6-OHDA injected
rats before and after MLA I.P injection ................................................. 139
Figure 64: Photographs of TH immunoreactive fibers ............................................ 141
Figure 65: Striatal TH-immunoreactive fiber density expressed as a
percentage of the fiber density on the lesioned side to the
non-lesioned side .................................................................................... 142
Figure 66: Photomicrographs of coronal sections of SN for TH
immunohistochemistry ........................................................................... 145
Figure 67: Stereological assessment of total numbers of TH-positive
cell bodies in the SN at all three levels; rostral, middle,
and caudal ............................................................................................... 147
Figure 68: Neuroprotective mechanisms of curcumin in PD ................................... 158
Figure 69: Drug-induced rotation test in rats that had 6-OHDA
injection in the right striatum ................................................................. 160
Figure 70: Hypothetical model of Ca2+- dependent cell survival
mechanism .............................................................................................. 165

xxii
List of Abbreviations
ABC Avidin–biotin-complex
AD Alzheimer's disease
ACh Acetylcholine
AP Antro-posterior
AUC Area under the curve
Ba2+ Barium
ANOVA Analysis of variance
BAPTA 1,2-bis(o-aminophenoxy) ethane-N,N,N',N'-tetraacetic acid
BDNF Brain-derived neurotrophic factor
BDMC Bisdemethoxycurcumin
Bcl-2 B-cell lymphoma 2
BDMC Bisdemethoxycurcumin
Ca2+ Calcium
CaCCs Ca2+ activated Cl- channels
CaM Calcium effector protein calmodulin
CD Cyclodextrin
CDK Cycline-dependent kinase
CMC Carboxy methyl cellulose
CNS Central nervous system
COMTi Catechol-O-methyl transferase inhibitor
COX-2 Cycloxygenase-2
CPu Caudate–putamen
CREB cAMP response element-binding
CUR-SL Curcumin-loaded silica liposomes
CUR-FL Curcumin-loaded flexible liposomes
DA Dopamine
DAB Diaminobenzidine
DAT Dopamine transporters
DBS Deep brain stimulation
DhβE Dihydro-β-erythroidine hydrobromide

xxiii
DMC Demethoxycurcumin
DMSO Dimethyl sulfoxide
DV Dorso-ventral
ECD Extracellular domain
ERK/MAPK Extracellular signal-regulated mitogen-activated protein kinase
FGF-2 Fibroblast growth factor-2
GABA G-aminobutyric acid
GDPβS Guanyl-5'-yl thiophosphate; guanosine 5'-(trihydrogen
3-thiodiphosphate 5'-O-(2-thiodiphosphate); 71376-97-1;)
Go-6983 3-[1-[3-(Dimethylamino)propyl]-5-methoxy-1H-indol-3-yl]-4-
(1Hindol-3-yl)-1H-pyrrole-2,5-dione
GPe Globus pallidus external segment
GPi Globus pallidus internal segment
GPx Glutathione peroxidase
HO-1 Hemoxygenase-1
H2O2 Hydrogen Peroxide
HPLC High-performance liquid chromatography
Im Membrane current
ICD Intracellular domain
IGF Insuline-like growth factor
IL Interleukin
iNOS Inducible nitric oxide synthase
IP Intraperitoneal
ISO Isoprenaline
I-V Current-voltage relationships
JAK2 Janus kinase 2
KN-62 l-[N,O-Bis(5-isoquinolinesulfonyl)-N-methyl-∼-tyrosyl]
-4- phenylpiperazine
KT-5720 (9R,10S,12S)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-
methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg,39,29,19-kl]
pyrrolo[3,4- i][1,6]benzodiazocine-10-carboxylic acid, hexyl ester
LB Lewy body
xxiv
L-dopa Levodopa
LEC Liposome-encapsulated curcumin
LID L-dopa-induced dyskinesias
LN Lewy neurites
LPS Lipopolysaccharides
LRRK2 Leucine rich repeat kinase 2
mAChR Muscarinic acetylcholine receptor
MBS Modified barth’s solution
MDMA 3,4-methylenedioxymethamphetamine
MFB Medial forebrain bundle
ML Medio-lateral
MLA Methyllycaconitine
MPP+ 1-methyl-4-phenylpyridinium
MPTP 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
mRNA messenger RNA
NAc Nucleus accumbens
nAChR Nicotinic acetylcholine receptor
NADPH Nicotinamide adenine dinucleotide phosphate hydrogen
NAM Negative allosteric modulator
NEM N-ethylmaleimide
NF-kB Nuclear factor-kappaB
NGF Nerve growth factor
NMS Non-motor symptoms
PAM Positive allosteric modulator
PB Phosphate buffer
PBS Phosphate buffered saline
PCA P-chloroamphetamine
PCR Polymerase chain reaction
PD Parkinson’s disease
PET Positron emission tomography
PINK1 Phosphatase and tensin homolog- induced novel kinase 1
PKA Protein kinase A

xxv
PKC-412 tyrosine kinase inhibitor
PLGA Polylactic-co-glycolic acid
pLGIC Pentameric ligand-gated ion channels
PPAR Peroxisome-proliferator activated receptor
PTEN Phasphatase and tensin homolog
PTX Pertussis toxin
RNA Ribonucleic acid
ROS Reactive oxygen species
SAM Silent allosteric modulator
SN Substantia nigra
SNpc Substantia nigra pars compacta
SNpr Substantia nigra pars reticulata
SOD Superoxide dismutase
STAT Signal transducer and activator of transcription
STN Subthalamic nucleus
TGF Transforming growth factor
TH Tyrosine hydroxylase
THC Tetrahydrocurcumin
TMD Transmembrane domain
TNF- α Tumor necrosis factor- α
Vc Command potential
Vm Membrane potential
VMAT Vesicular monoamine transporter
VTA Ventral tegmental area
5-HT Serotonin or 5-hydroxytryptamine
ΔΨm Mitochondrial membrane potential

1
Chapter 1: Introduction
1.1 Curcumin
Curcumin is a polyphenolic compound, the main ingredient of turmeric (Curcuma
longa), and a member of the ginger family (Zingiberaceae) (Aggarwal and Sung,
2009). The plant grows largely in India, China and other tropical countries (Aggarwal
et al., 2007). Vogel and Pelletier were the first to report the isolation of a “yellow
coloring-matter” from the rhizomes of Curcuma longa (turmeric) and named it
curcumin in 1815. Vogel found that turmeric is a mixture of many components and
successfully isolated a pure curcumin oil in 1842. In 1910, Milobedeska and Lampe
characterized its structure as diferuloylmethane, or 1,6-heptadiene-3,5-dione-1,7-bis
(4-hydroxy-3-methoxyphenyl)-(1E, 6E) (Figure 1), and three years later they
synthesized curcumin (Gupta et al., 2012). Curcumin exhibits keto–enol tautomerism,
where enol forms predominant in an alkaline medium while keto forms in acidic and
neutral media (Priyadarsini, 2014).
Figure 1: The source and chemical structure of curcumin.
(A) The root of turmeric. (B) Crystallized powder of curcumin. (C) The enol and keto
forms of curcumin [Modified from (Zhang et al., 2013)].
Curcuma Longa
Rhizome
Turmeric
Curcumin (Enol form)
Curcumin (Keto form)
(A) (C) (B)

2
Curcuminoid (the yellow pigmented fraction of turmeric) forms 3-5% of turmeric
and is composed mainly of three derivatives; curcumin (diferuloylmethane, curcumin
I), demethoxycurcumin (DMC, curcumin II), and bisdemethoxycurcumin (BDMC,
curcumin III) (Anand et al., 2008; Goel et al., 2008). Recently, cyclocurcumin has
been isolated as a curcuminoid component of turmeric by Kiuchi et al. (Kiuchi et al.,
1993). Among all, curcumin is the most abundant (~77%) followed by DMC (~17%)
and BDMC (~3%) (Anand et al., 2008; Goel et al., 2008). Curcumin is hydrophobic
in nature and not soluble in neutral solvent e.g: water, but soluble in organic solvents
e.g: dimethylsulfoxide (DMSO), ethanol, and acetone (Priyadarsini, 2009).
1.1.1 Natural Curcumin Analogues and Metabolites
As mentioned earlier, curcumin I, curcumin II, curcumin III, and cyclocurcumin
collectively known as curcuminoid, are considered the natural turmeric analogues
(Table 1.A - D).
There are numerous curcumin metabolites that have been reported and are
available commercially, including tetrahydrocurcumin (THC), dimethyl curcumin,
didemethyl curcumin, Vanillylidenacetone, Di-(tert-Butyl-dimethylsilyl) Curcumin,
O-tert-Butyl-dimethylsilyl Curcumin, and curcumin-d6 (Table 1.E - K).
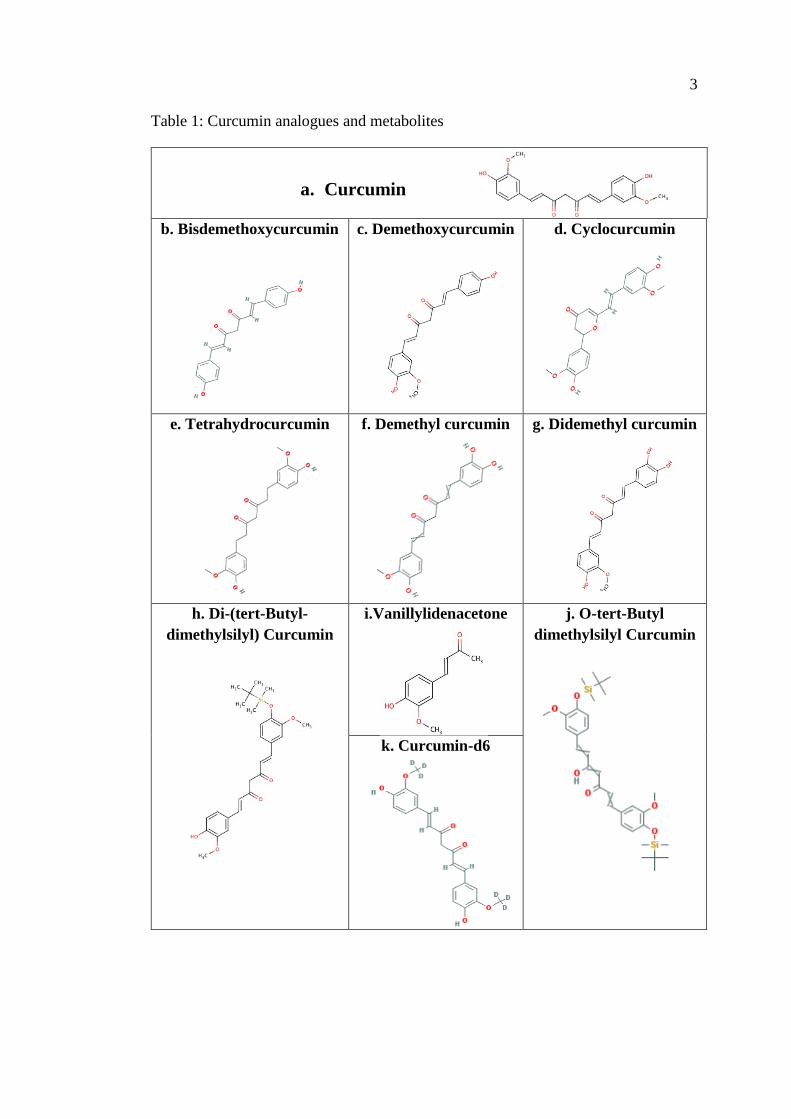
3
Table 1: Curcumin analogues and metabolites
b. Bisdemethoxycurcumin c. Demethoxycurcumin d. Cyclocurcumin
e. Tetrahydrocurcumin f. Demethyl curcumin g. Didemethyl curcumin
h. Di-(tert-Butyl-
dimethylsilyl) Curcumin
i.Vanillylidenacetone j. O-tert-Butyl
dimethylsilyl Curcumin
k. Curcumin-d6
a. Curcumin

4
1.1.2 Pharmacokinetics and Pharmacodynamics of Curcumin
Curcumin has a low bioavailability in plasma and tissue due to its poor absorption,
rapid metabolism, as well as rapid systemic elimination. Various studies have been
conducted on the pharmacokinetics and pharmacodynamics of curcumin, the first of
which was reported by Wahlstrom and Blennow 1978 in Sprague-Dawley rats.
Curcumin had poor absorption from the gut, and nearly 75% of curcumin was excreted
in the feces with negligible amounts being detected in blood plasma (Wahlström and
Blennow, 1978). Holder et al. experimented with deuterium- and tritium-labeled
curcumin given intravenously (I.V) and intra-peritoneally (I.P), and observed that
majority of curcumin was excreted in bile and then in feces (Holder et al., 1978).
Ravindranath in 1980, used three different doses (10, 80, and 400 mg/kg) of tritium-
labeled curcumin, and detected curcumin in tissue of interest after 12 days of
administration. The percent of absorbed curcumin (60-66%) remained constant with
no difference between doses indicating that increasing the dose did not increase
absorption, thus there is a dose-dependent limitation of curcumin’s bioavailability
(Ravindranath and Chandrasekhara, 1981). Pan et al. in 1999, administered 100 mg/kg
of curcumin I.P to mice to investigate the pharmacokinetics of curcumin. After one
hour of administration, curcumin level in spleen, liver, and kidney were 26.1, 26.9,
and 7.5 μg/g, respectively, and a minute trace levels (0.41 mg/g) were detected in the
brain (Pan et al., 1999).
Studies of curcumin’s pharmacokinetics in humans yielded more or less similar
data, a peak plasma level of 0.41–1.75 µM have been obtained after administration of
4 – 8 g of curcumin orally in humans (Cheng et al., 2001). After several experiments,
Perkins, 2002 concluded that humans need a daily dose of 1.6 g curcumin to produce

5
an effect (Perkins et al., 2002). Many groups have shown that the liver is the primary
site of curcumin metabolism, where it undergoes extensive reduction via alcohol
dehydrogenase, followed by conjugation (Garcea et al., 2004; Hoehle et al., 2006;
Wahlström and Blennow, 1978).
Almost all studies verified that unformulated curcumin has poor bioavailability in
animal models and humans. To improve curcumin bioavailability, different
formulations have been developed. For example, a nanocurcumin was developed to
enhance curcumin solubility in aqueous solution. Cheng et al. 2013, prepared a
nanoparticle form of curcumin that yielded a higher plasma concentration and higher
AUC by six times. Moreover, mean residence time in the brain was longer in a mice
model (Cheng et al., 2013).
In another study, Polylactic-co-glycolic acid (PLGA) is one of the forms of
formulated curcumin, was employed and was reported to enhance curcumin
bioavailability by 5.6 folds and also extended curcumin half-life. This was due to
improvement of water solubility of the compound -which is known to be highly
lipophilic as mentioned previously-, more induction of intestinal juices facilitating
ingestion, higher permeability enhancing absorption, and elongating residence time in
the intestine which allowed for more absorption (Xie et al., 2011)
Liposomal curcumin is another formulated curcumin a form of drug carrier which
helps to increase the solubility of the compound. Liposome-encapsulated curcumin
(LEC) increased curcumin bioavailability by facilitating cellular uptake and increasing
its absorption. Different forms have been generated, silica-coated flexible liposomes
loaded with curcumin (CUR-SLs) and curcumin-loaded flexible liposomes (CUR-
FLs), were found to enhance curcumin bioavailability by 7.76- and 2.35 folds higher,
respectively, compared to unformulated curcumin (Li et al., 2012).

6
Cyclodextrin (CD), is a form of cyclic oligosaccharides, which encapsulates and
facilitate its cellular uptake, bioavailability, and elongates its half-life (Prasad et al.,
2014).
CD encapsulated curcumin improves curcumin permeability 1.8 fold across skin
in animal model compared to unformulated curcumin (Rachmawati et al., 2013).
Curcumin bioavailability can be enhanced up to 2000% in humans and to 154% in
rats, by administrating piperine (a component derived from pepper and a known
inhibitor of hepatic and intestinal glucuronidation) along with curcumin. Concomitant
piperine administration with curcumin significantly decreased elimination and half-
life clearance of curcumin (Anand et al., 2007; Shoba et al., 1998).
1.1.3 Molecular Targets of Curcumin
Based on the extensive pieces of evidence from both in-vitro and in-vivo studies,
several molecular targets of curcumin have been identified. Curcumin interacts with
transcription factors, e.g., nuclear factor-kB (NFκB), and signal transducer and
activator of transcription (STAT) proteins (Shishodia et al., 2007), growth factors and
their receptors, e.g. epidermal growth factor receptors and HER2 (Chen et al., 2006;
Soung and Chung, 2011), cytokines, e.g., interleukin 1b (IL-1b), interleukin 6 (IL-6)
(Cho et al., 2007), enzymes, e.g., hemoxygenase-1 (HO-1) (McNally et al., 2007), and
genes regulating cell proliferation and apoptosis (Aoki et al., 2007). This ability of
curcumin to modulate or interact with multiple cell signaling pathways and proteins,
strongly indicates that this polyphenol is an effective multi-targeted compound (Figure
2) (Goel and Aggarwal, 2010; Hasima and Aggarwal, 2012; Rainey et al., 2015;
Ravindran et al., 2009). This conclusion is in line with several recently published
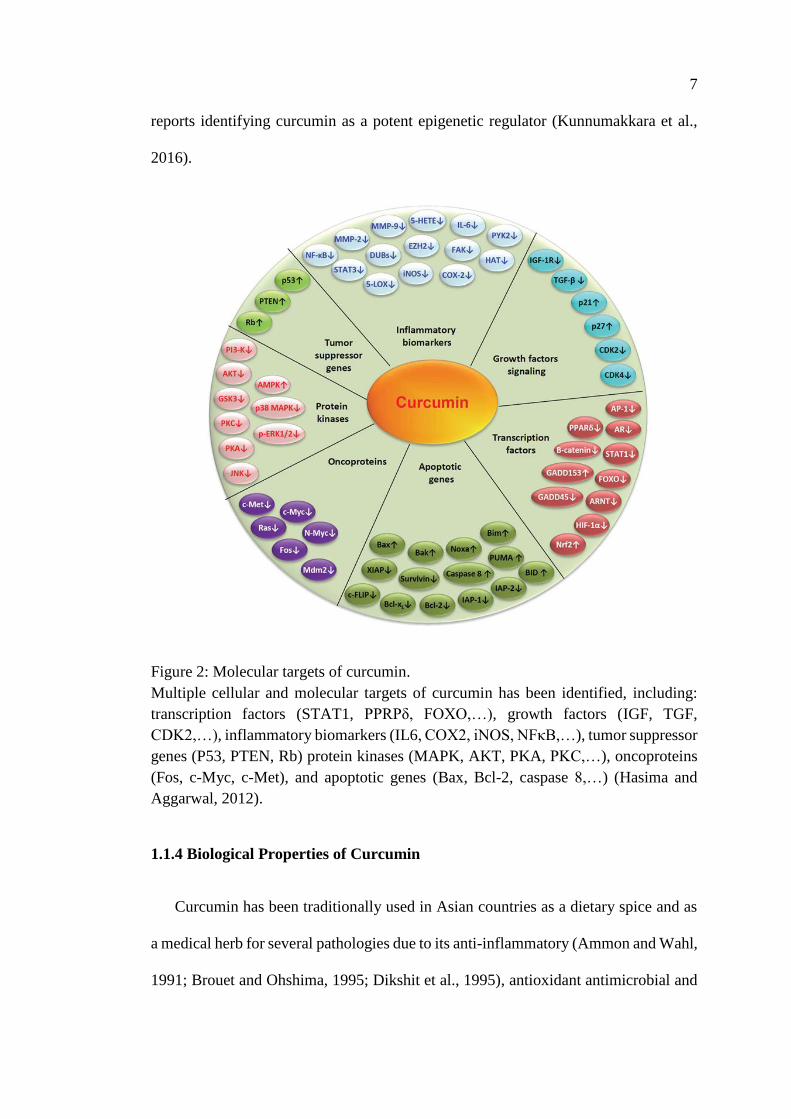
7
reports identifying curcumin as a potent epigenetic regulator (Kunnumakkara et al.,
2016).
Figure 2: Molecular targets of curcumin.
Multiple cellular and molecular targets of curcumin has been identified, including:
transcription factors (STAT1, PPRPδ, FOXO,…), growth factors (IGF, TGF,
CDK2,…), inflammatory biomarkers (IL6, COX2, iNOS, NFκB,…), tumor suppressor
genes (P53, PTEN, Rb) protein kinases (MAPK, AKT, PKA, PKC,…), oncoproteins
(Fos, c-Myc, c-Met), and apoptotic genes (Bax, Bcl-2, caspase 8,…) (Hasima and
Aggarwal, 2012).
1.1.4 Biological Properties of Curcumin
Curcumin has been traditionally used in Asian countries as a dietary spice and as
a medical herb for several pathologies due to its anti-inflammatory (Ammon and Wahl,
1991; Brouet and Ohshima, 1995; Dikshit et al., 1995), antioxidant antimicrobial and

8
anticancer properties (Limtrakul et al., 1997; Rao et al., 1995), anti-arthritic (Deodhar
et al., 1980), hepatoprotective (Kiso et al., 1983), anti-thrombotic (Srivastava et al.,
1985), cardio-protective (Dikshit et al., 1995; Nirmala and Puvanakrishnan, 1996a;
Srivastava et al., 1985), and hypoglycemic properties (Arun and Nalini, 2002; Babu
and Srinivasan, 1995; Srinivasan, 1972). Moreover, it has been shown to exhibit a wide
range of pharmacological activities including the treatment of several
neurodegenerative diseases such as Alzheimer and Parkinson’s diseases (Figure 3). In
the next section, we will be discussing some of the biological properties of curcumin
(Zhou et al., 2011).
Figure 3: Therapeutic potential of curcumin.
Curcumin has been shown to exhibit a wide range of pharmacological activities in
central and peripheral body systems, through its anti-oxidant, anti-inflammatory and
neuroprotective properties (Zhou et al., 2011).

9
1.1.4.1 Anti-inflammatory Effects
Inflammation is a physiological process by which our body fight against infections
triggering host immune response. It is a complex interaction that aims at removing the
invading agent or damaged tissue. Over-activation of the immune system and
inflammatory responses may cause further tissue damage (Joe et al., 2004).
Inflammation plays a major role in a number of pathological conditions including:
neurodegenerative, autoimmune, cardiovascular, endocrine, and neoplastic diseases
(Brouet and Ohshima, 1995; Liu and Hong, 2003; Nosalski and Guzik, 2017).
Interaction with and modulation of the effects of various inflammatory mediators by
curcumin demonstrated its anti-inflammatory properties (Aggarwal and Harikumar,
2009; Jurenka, 2009). Curcumin can inhibit inflammatory cytokines, interleukins
(ILs), chemokines, as well as inflammatory enzymes, cycloxygenase-2 (COX-2),
inducible nitric oxide synthase (iNOS) and cyclinD1 (Creţu et al., 2012). Curcumin
has been shown to diminish GFAP level (Yu et al., 2010), suppress NF-kβ activity,
and reduce the levels of tumor necrosis factor- α (TNF-α) (Chen et al., 2014).
1.1.4.2 Anti-oxidant Effects
It is well known that oxidative stress plays a major role in acute, chronic, and
degenerative diseases. Oxidative stress results from imbalance between formation and
neutralization of reactive oxygen species (ROS) in our body, leading to generation of
free radicals and energy failure (Pham-Huy et al., 2008). Curcumin has a strong anti-
oxidant activity compared to vitamin C and E (Toda et al., 1985). The potent activity
of curcumin against pro-oxidants such as superoxide radicals, hydrogen peroxide and
nitric oxide radical, as well as enhancing anti-oxidant enzymes such as catalase,
superoxide dismutase (SOD), glutathione peroxidase (GPx) and heme oxygenase-1

10
(OH-1) results in a decrease in lipid peroxidation and subsequently organ damage
(Jeong et al., 2006; Reddy and Lokesh, 1994, 1992). Curcumin induced heme
oxygenase-1 and protected endothelial cells against oxidative stress (Motterlini et al.,
2000). Curcumin inhibition of free radical formation protected rat myocardial tissue
from isoprenaline (ISO)-induced ischemic injury (Manikandan et al., 2004; Nirmala
and Puvanakrishnan 1996a, 1996b). Curcumin provided anti-oxidant protection
comparable to vitamin E on renal cell lines by its inhibitory effect on lipid
peroxidation, cytolysis, and lipid degradation (Cohly et al., 1998).
The methoxy and phenolic groups on benzene rings and the β-diketone moiety in
the curcumin structure (Figure 4) are thought to be the cause of its anti-oxidant
properties (Sandur et al., 2007; Sreejayan and Rao, 1996).
Figure 4: Curcumin structural features.
Curcumin has three chemical entities in its structure: two aromatic ring systems
containing O-methoxy phenolic groups, connected by a seven carbon linker, consisting
of α, β-unsaturated diketone moiety [Modified from (Bagchi et al., 2015)].
Aromatic
ring
Aromatic
ring

11
1.1.4.3 Neuroprotective Effects
Our previous discussion has shown that both the anti-oxidant and anti-
inflammatory effects of curcumin together form the basis of beneficiary effects of
curcumin in several neurological diseases affecting the central as well as the peripheral
nervous system. Curcumin as a multi-targeted compound can serve as a
neuroprotective agent. Oral administration of (50, 100 and 200 mg/kg) curcumin
protected Swiss albino mice against rotenone-induced dysfunction in the
mitochondrial respiratory chain and conserved the mitochondrial enzyme complex
(Khatri and Juvekar, 2016). Anti-oxidant properties of curcumin improved the levels
of acetylcholine esterase enzyme in mice compared with negative control animals
which was reflected on motor behavioral assessments (Khatri and Juvekar, 2016).
Alzheimer’s disease is a neurodegenerative disorder and the most common cause of
dementia worldwide. The main pathological hallmark is the aggregation of Aβ
amyloid protein plaque formation, which has not been phagocytosed due to microglial
dysfunction. Curcumin has anti-protein aggregation properties (Darvesh et al., 2012),
and could inhibit plaque formation and accumulation, and activated microglial
phagocytic activity (Cole et al., 2007; Ono et al., 2004).
1.1.4.4 Anti-cancer Effects
In 1987, Kuttan and colleagues carried out the first clinical trial to investigate the
anti-cancer properties of curcumin. He included 62 patients having external cancerous
lesions and used an ointment containing ethanol turmeric extract. Patients who
received this treatment reported a significant improvement in their symptoms of pain,
itching, smell, and lesion size (Kuttan et al., 1987). Since this study, several other trials
have been conducted on different types of cancer insuring the dose dependent chemo-

12
preventive effect of curcumin in head and neck, breast, gastrointestinal (colon,
pancreatic, stomach, esophageal and oral carcinogenesis), and cervical cancers (Bayet-
Robert et al., 2010; Cao et al., 2016; Carroll et al., 2011; Cheng et al., 2001; Epelbaum
et al., 2010; Ghalaut et al., 2012; Kim et al., 2011). Curcumin was not tested as a single
anti-cancer agent only, but also as an adjuvant anti-tumor agent and to reduce adverse
effects of other chemotherapeutics (Belcaro et al., 2014; Garcea et al., 2005).
Curcumin can suppress carcinogenesis at different stages of promotion, angiogenesis,
and growth (Conney et al., 1991; Huang et al., 1992; Robinson et al., 2003).
1.1.5 Effects of Curcumin on Different Ion Channels and Receptors
Depending on the above discussion, several types of ligand-gated ion channels and
receptors have been suggested to be involved in mediating pharmacological actions of
curcumin. In this study, we are investigating the effect of curcumin application on the
functional properties of α7-nicotnic acetylcholine receptors mainly and other ligand
gated ion channels.
13
1.2 Acetylcholine Receptors
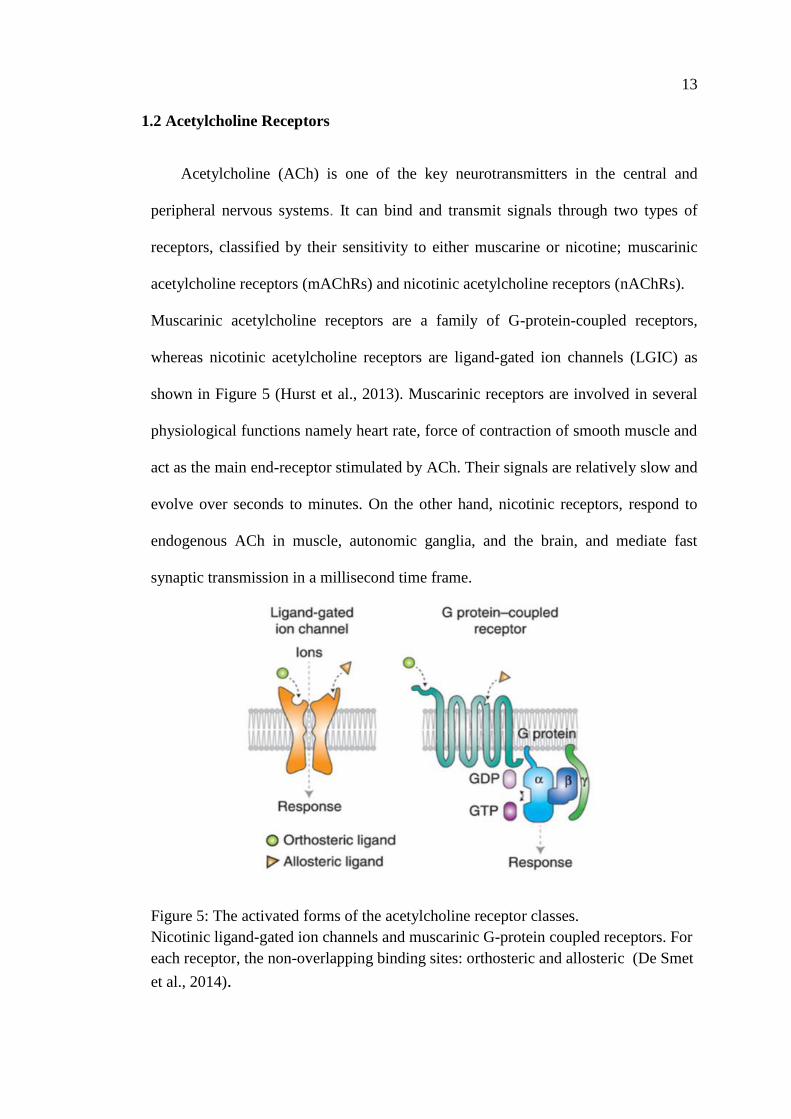
Acetylcholine (ACh) is one of the key neurotransmitters in the central and
peripheral nervous systems. It can bind and transmit signals through two types of
receptors, classified by their sensitivity to either muscarine or nicotine; muscarinic
acetylcholine receptors (mAChRs) and nicotinic acetylcholine receptors (nAChRs).
Muscarinic acetylcholine receptors are a family of G-protein-coupled receptors,
whereas nicotinic acetylcholine receptors are ligand-gated ion channels (LGIC) as
shown in Figure 5 (Hurst et al., 2013). Muscarinic receptors are involved in several
physiological functions namely heart rate, force of contraction of smooth muscle and
act as the main end-receptor stimulated by ACh. Their signals are relatively slow and
evolve over seconds to minutes. On the other hand, nicotinic receptors, respond to
endogenous ACh in muscle, autonomic ganglia, and the brain, and mediate fast
synaptic transmission in a millisecond time frame.
Figure 5: The activated forms of the acetylcholine receptor classes.
Nicotinic ligand-gated ion channels and muscarinic G-protein coupled receptors. For
each receptor, the non-overlapping binding sites: orthosteric and allosteric (De Smet
et al., 2014).

14
The central role of nicotinic receptors in converting chemical stimuli into electrical
signals has involved them broadly in a broad range of physiological functions
including muscle contraction, brain development, cognitive function, learning and
memory, arousal, reward, motor control, analgesia, synaptic plasticity as well as
pathological disorders including Alzheimer's disease, Parkinson's disease, epilepsy
and schizophrenia (Jensen et al., 2005; Lindstrom, 2003; Mineur and Picciotto, 2008;
Posadas et al., 2013). These receptors are the target of pharmacologically administered
nicotine.
1.2.1 Nicotinic Acetylcholine Receptors
Early in the twentieth century, nicotine became a fundamental molecule in the basic
science of pharmacology. In 1905, Langley reported that body muscles contract via a
“receptive substance” in muscles. The identification of the muscle nicotinic
acetylcholine receptors paved the way for the discovery of neurotransmitter receptors
(Langley, 1905). But it was not until 1970s when neuronal nAChRs, were identified
(Changeux et al., 1970; Miledi and Potter, 1971). A decade later, the extended family
of nicotinic receptor has been identified (Patrick et al., 1983).
Nicotinic acetylcholine receptors (nAChRs) were the first of all neurotransmitters
to be identified biochemically and functionally (Lindstrom, 2003). nAChRs are
members of a structurally related family of ligand gated ion channels that also include
receptors for neurotransmitters such as 5-hydroxytryptamine (5-HT), g-aminobutyric
acid (GABA), and glycine (Albuquerque et al., 2009; Hendrickson et al., 2013).
Initially, this class of receptors was named the Cys-loop family as all receptors contain
a conserved two disulfide-bonds cysteines separated by 13 amino acids in their
extracellular amino terminus (Figure 6A). Recent discovery of these receptors in
15
prokaryotic cells but lacking the character of Cys-loop led to the change the in name
from Cys-loop family to pentameric Ligand-gated ion channels (pLGIC) (Tasneem et
al., 2005).
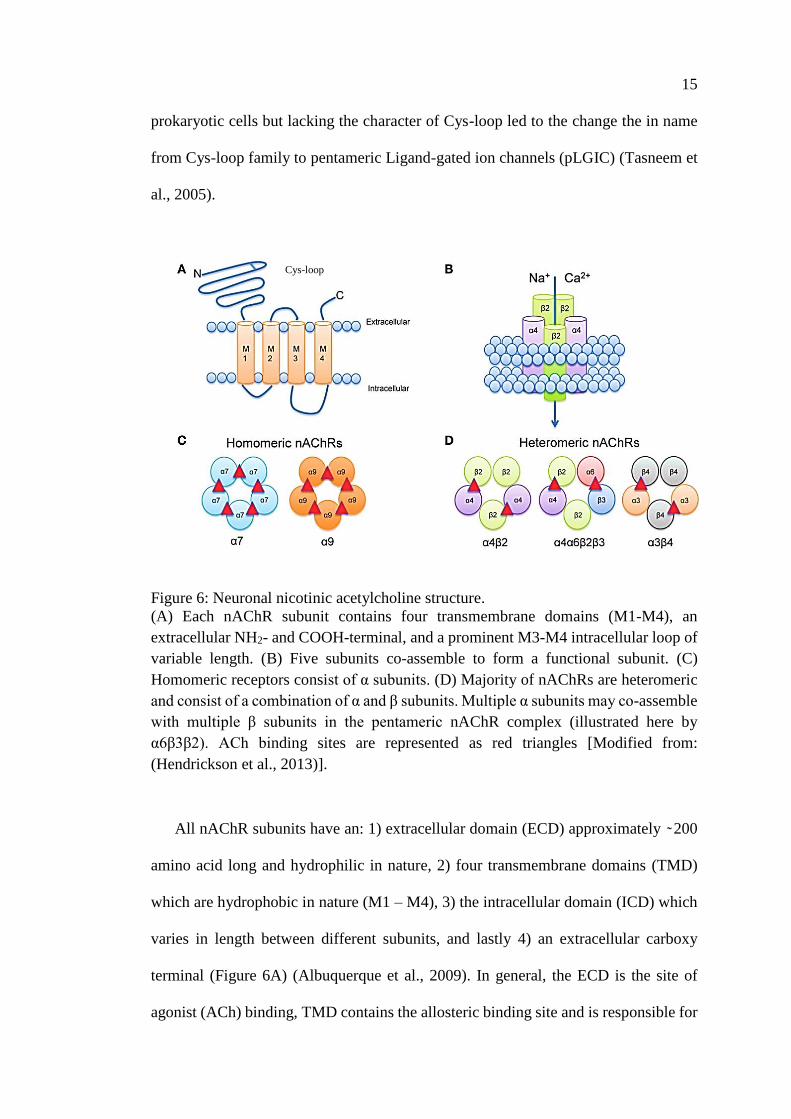
Figure 6: Neuronal nicotinic acetylcholine structure.
(A) Each nAChR subunit contains four transmembrane domains (M1-M4), an
extracellular NH2- and COOH-terminal, and a prominent M3-M4 intracellular loop of
variable length. (B) Five subunits co-assemble to form a functional subunit. (C)
Homomeric receptors consist of α subunits. (D) Majority of nAChRs are heteromeric
and consist of a combination of α and β subunits. Multiple α subunits may co-assemble
with multiple β subunits in the pentameric nAChR complex (illustrated here by
α6β3β2). ACh binding sites are represented as red triangles [Modified from:
(Hendrickson et al., 2013)].
All nAChR subunits have an: 1) extracellular domain (ECD) approximately ̴ 200
amino acid long and hydrophilic in nature, 2) four transmembrane domains (TMD)
which are hydrophobic in nature (M1 – M4), 3) the intracellular domain (ICD) which
varies in length between different subunits, and lastly 4) an extracellular carboxy
terminal (Figure 6A) (Albuquerque et al., 2009). In general, the ECD is the site of
agonist (ACh) binding, TMD contains the allosteric binding site and is responsible for
Cys-loop

16
the ion pore, permeability and selectivity (especially M2 which is conserved
throughout LGIC), and ICD controls channel conductance (Albuquerque et al., 2009;
Changeux, 2010; Jones et al., 2010; King et al., 2015; Paulo et al., 2009).
Nicotinic acetylcholine receptors have a pentameric structure consisting of five
transmembrane subunits around a central water-filled pore selective for cation (Figure
6B). To date, 16 distinct subunits of nAchRs have been identified in the human
proteome. Subunits are divided into two subgroups, the α and β subunits of which 5
nAChR subunits that are expressed in muscle (α1, β1, γ, δ, and ε) and 11 nAChR
subunits are expressed in nervous tissue (α2-7*, α9, α10, β2-4). This study mainly
focuses on neuronal types of nAChRs. Each nAChR can be either a homomeric,
formed by five identical subunits or heteromeric receptor that result from the
combination of different subunits (Figures 6C & D). The nomenclature for the genes
that encode the nAChR subunits is CHRNxy where CHRN stands for cholinergic
receptor, nicotinic, and xy represents the subunit. For example, CHRNB4 is the gene
for the β4 subunit. The α7 neuronal nicotinic receptor gene, CHRNA7 is located on
the long arm of Chromosome 15, is widely expressed in both the brain (Sinkus et al.,
2015). CHRNA7 was first identified in chicken, α7 subunit immediately attracted
much interest of physiologists and geneticists, since it forms functional homomeric
receptors and has unique features in terms of its 1) genomic structure, 2) localization
and function with high calcium permeability (PCa/PNa≈10), 3) rapid activation and
desensitization by agonist (millisecond scale) (Bertrand et al., 1992; Couturier et al.,
1990), and 4) selective inhibition by α-bungarotoxin (α-Btx) and methyllycaconitine
(MLA) (Couturier et al., 1990; Séguéla et al., 1993; Turek et al., 1995). Because of its
simple organizational structure, the α7 subunit can be used to study structure–function
relationships. For example, mutation of a single amino acid in the channel domain will

17
cause the whole receptor complex to be modified, which provides a better
understanding of receptor function (Hurst et al., 2013). Despite its homomeric
arrangement, α7-nAChR can assemble in a heteromeric form with other subunits; α7β2
heteromeric receptors (Liu et al., 2012, 2009; Moretti et al., 2014; Thomsen et al.,
2015; Zoli et al., 2015).
Depending on the presence, abundance, and timing of ACh binding, nAChRs exist
in different states and undergo spontaneous conformational transitions: closed at rest,
open pore, and desensitized (Figure 7) (Hurst et al., 2013). Prolonged exposure to low
doses of ACh, nicotine, or a nicotinic agonist substantially will lead to desensitization,
stabilizing the receptor in a closed state, unresponsive to further agonist stimulation.
Figure 7: Proposed mechanism of activation and desensitization.
Channel transition between three main conformational states by the binding of agonist:
(A) closed, (B) open, and (C) desensitized, where the channel is having high binding
affinity to agonist, but impermeable to ions [Modified from (Nys et al., 2013)].
A
C B

18
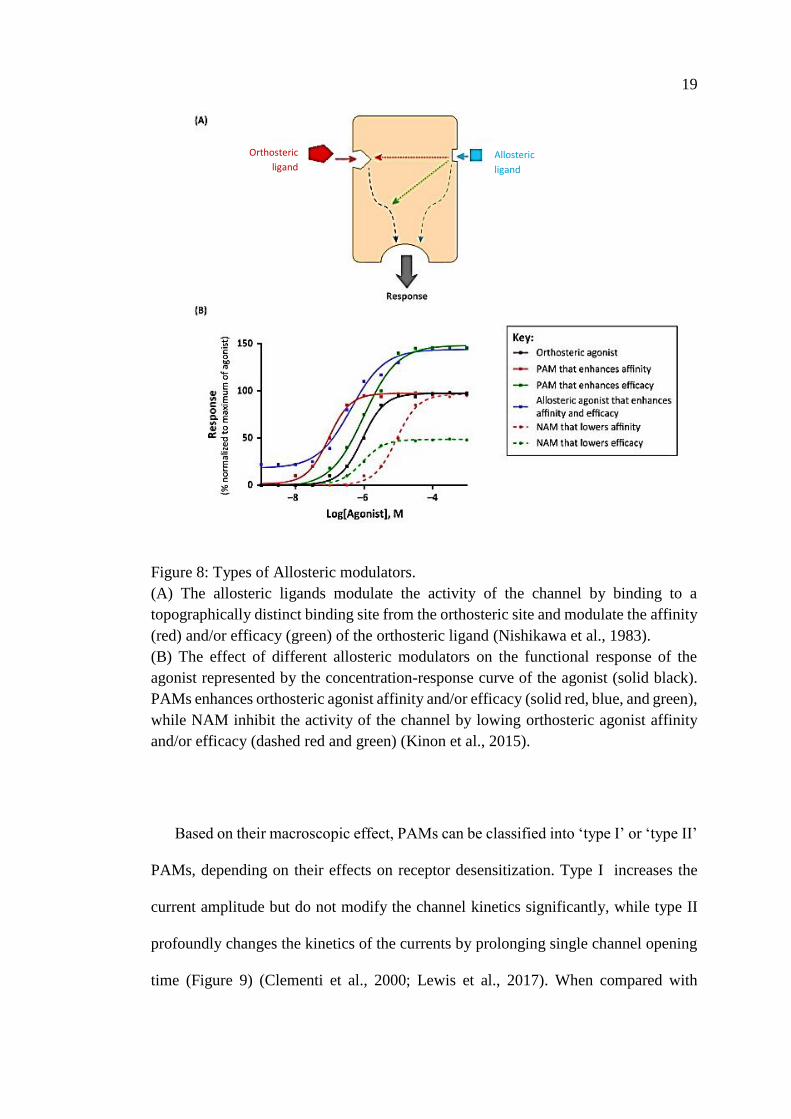
Orthosteric is the term used to describe the binding site for the natural ligand, and
classical agonists are therefore referred to as orthosteric agonists. However, a group of
compounds that lack agonist activity on nAChRs and act via a distinct transmembrane
binding site are described as allosteric modulators, (allo- from the Greek meaning
"other") (Figures 5 & 8) (Flor and Acher, 2012). Allosteric modulators can modulate
the activity of the channel. They are of three types: 1) positive allosteric modulators
(PAMs), that potentiate the activity of the channel but only in the presence of an
agonist with minimal level of desensitization; 2) negative allosteric modulators
(NAMs), inhibiting channel activity upon binding, or acting as open-channel blocker;
3) silent allosteric modulators (SAMs), having no effect on orthosteric activity but
blocking allosteric modulation (Figure 8) (Corradi and Bouzat, 2016).
19
Figure 8: Types of Allosteric modulators.
(A) The allosteric ligands modulate the activity of the channel by binding to a
topographically distinct binding site from the orthosteric site and modulate the affinity
(red) and/or efficacy (green) of the orthosteric ligand (Nishikawa et al., 1983).
(B) The effect of different allosteric modulators on the functional response of the
agonist represented by the concentration-response curve of the agonist (solid black).
PAMs enhances orthosteric agonist affinity and/or efficacy (solid red, blue, and green),
while NAM inhibit the activity of the channel by lowing orthosteric agonist affinity
and/or efficacy (dashed red and green) (Kinon et al., 2015).
Based on their macroscopic effect, PAMs can be classified into ‘type I’ or ‘type II’
PAMs, depending on their effects on receptor desensitization. Type I increases the
current amplitude but do not modify the channel kinetics significantly, while type II
profoundly changes the kinetics of the currents by prolonging single channel opening
time (Figure 9) (Clementi et al., 2000; Lewis et al., 2017). When compared with
Allosteric
ligand
Orthosteric
ligand

20
regular α7-nAChR agonists, PAMs have emerged as an important pharmacological
target because they: 1) have greater structural diversity compared to orthosteric site
which is highly conserved in nAChRs (Yang et al., 2012); 2) allow more flexible
structural form and final effects; 3 have an extra neuroprotective activities, as
activation of α7-nAChR can be inactivated by desensitization, some α7-PAMs has the
ability to desensitize the receptor back to conducting state (Kalappa et al., 2013; Sun
et al., 2013; Uteshev, 2014). Moreover, it has been suggested that neuronal injury
activates cholinergic system, and the presence of PAM will reduce the level of agonist
stimulation required for its neuroprotective effect (Uteshev, 2014).
Figure 9: Molecular activation routes of the α7-nAChRs.
PAM type I and II compounds (grey lines) produce differential enhancement of the
inward currents generated by nicotinic agonists (black line). (A) PAM type I
profoundly increase the current amplitude, (B) PAM type II significantly change the
kinetics of the current by prolonging the single channel opening time.
A B

21
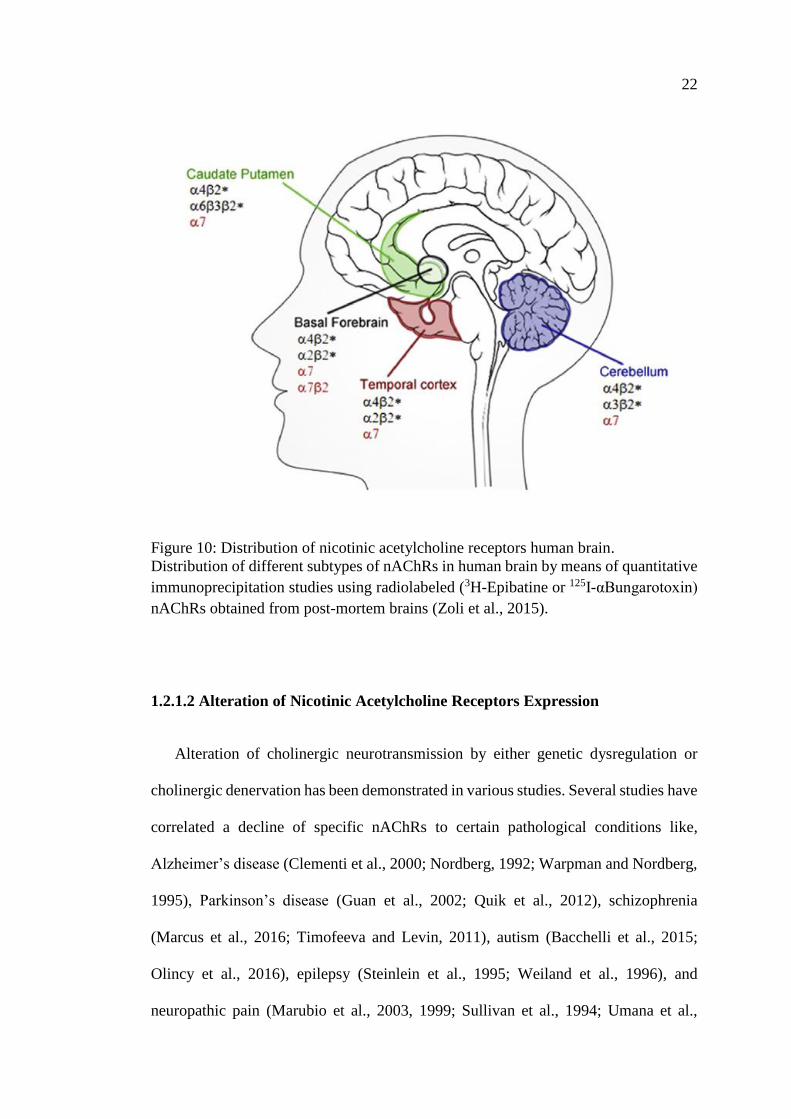
1.2.1.1 Distribution of Nicotinic Acetylcholine Receptors
The current existing data regarding the distribution on nAChRs suggest that it is
relatively conservative in all vertebrate species. The distribution of nAChRs is not
restricted to well-defined brain cholinergic pathways. The structure and localization of
the different nAChR subtypes have been investigated using a number of
complementary techniques, including in situ hybridization and PCR for specific
subunit RNAs, immune-precipitation for protein subunits, imaging by
autoradiography, PET and SPECT, and functional electrophysiological or
neurotransmitter release assays (Mineur and Picciotto, 2008). However, despite the
incomplete nature of the studies performed to date, it is possible to draw a map of
nAChR distribution in the human brain as shown in Figure 10, with the α7 and β2*
being the most widely distributed throughout the mammalian brain (the asterisk denote
the possibility of different subunits) (Millar and Gotti, 2009). The regional distribution
of nAChRs subtypes has been described in the temporal cortex, cerebellum, striatum
and basal forebrain and have been localized in a variety of brain structures, in particular
the thalamus, cortex and the striatum (Zoli et al., 2015).
Grady et al. examined the mouse brain using in situ hybridization to characterize
the mRNA expression pattern of nAChRs (Grady et al., 2007). Grady’s laboratory
demonstrated that the ventral tegmental area (VTA) and substantia nigra (SN)
expressed variable concentrations of α 3-7, β2, and β3. Also, they reported the presence
of heteromeric nAChRs subunits; α4α6β2β3, α6β2β3, α6β2, α4β2, and α4α5β2, on
dopaminergic terminals of mouse striatum (Grady et al., 2007; Le Novere et al., 1996).
Several studies have demonstrated a decline of specific nAChRs in Parkinson’s disease
(Guan et al., 2002).
22
Figure 10: Distribution of nicotinic acetylcholine receptors human brain.
Distribution of different subtypes of nAChRs in human brain by means of quantitative
immunoprecipitation studies using radiolabeled (3H-Epibatine or 125I-αBungarotoxin)
nAChRs obtained from post-mortem brains (Zoli et al., 2015).
1.2.1.2 Alteration of Nicotinic Acetylcholine Receptors Expression
Alteration of cholinergic neurotransmission by either genetic dysregulation or
cholinergic denervation has been demonstrated in various studies. Several studies have
correlated a decline of specific nAChRs to certain pathological conditions like,
Alzheimer’s disease (Clementi et al., 2000; Nordberg, 1992; Warpman and Nordberg,
1995), Parkinson’s disease (Guan et al., 2002; Quik et al., 2012), schizophrenia
(Marcus et al., 2016; Timofeeva and Levin, 2011), autism (Bacchelli et al., 2015;
Olincy et al., 2016), epilepsy (Steinlein et al., 1995; Weiland et al., 1996), and
neuropathic pain (Marubio et al., 2003, 1999; Sullivan et al., 1994; Umana et al.,
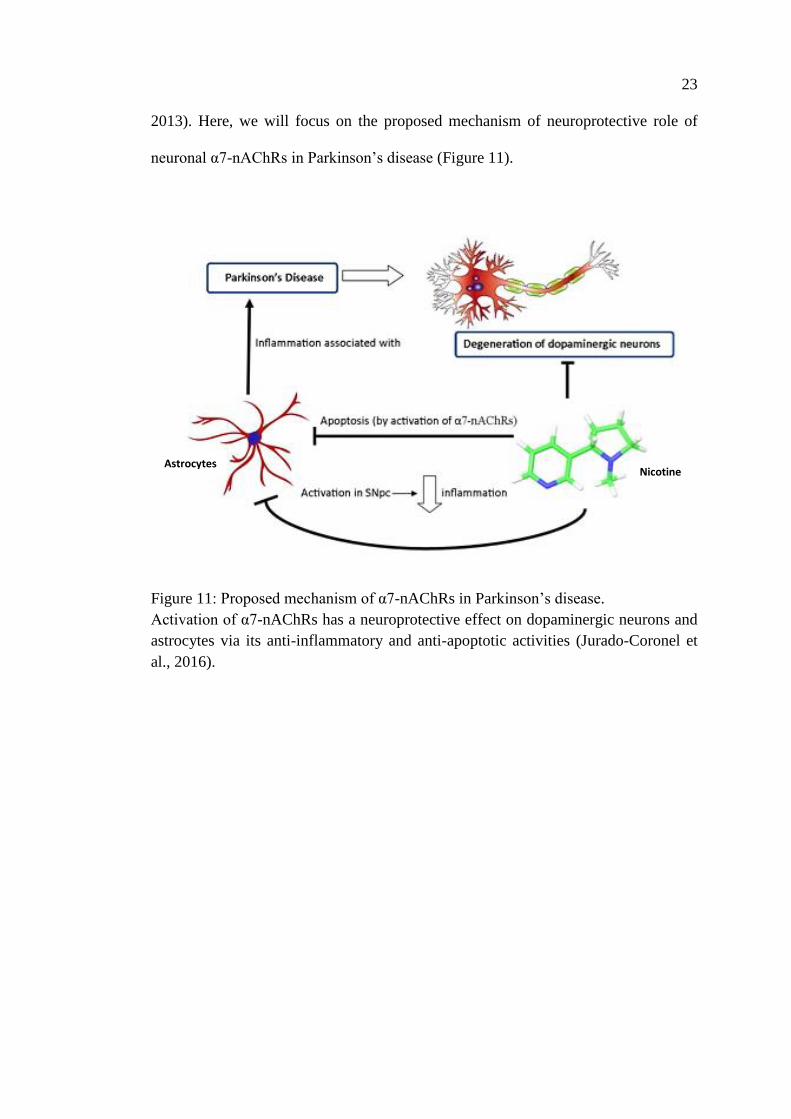
23
2013). Here, we will focus on the proposed mechanism of neuroprotective role of
neuronal α7-nAChRs in Parkinson’s disease (Figure 11).
Figure 11: Proposed mechanism of α7-nAChRs in Parkinson’s disease.
Activation of α7-nAChRs has a neuroprotective effect on dopaminergic neurons and
astrocytes via its anti-inflammatory and anti-apoptotic activities (Jurado-Coronel et
al., 2016).
Astrocytes Nicotine
24
1.3 Parkinson’s Disease
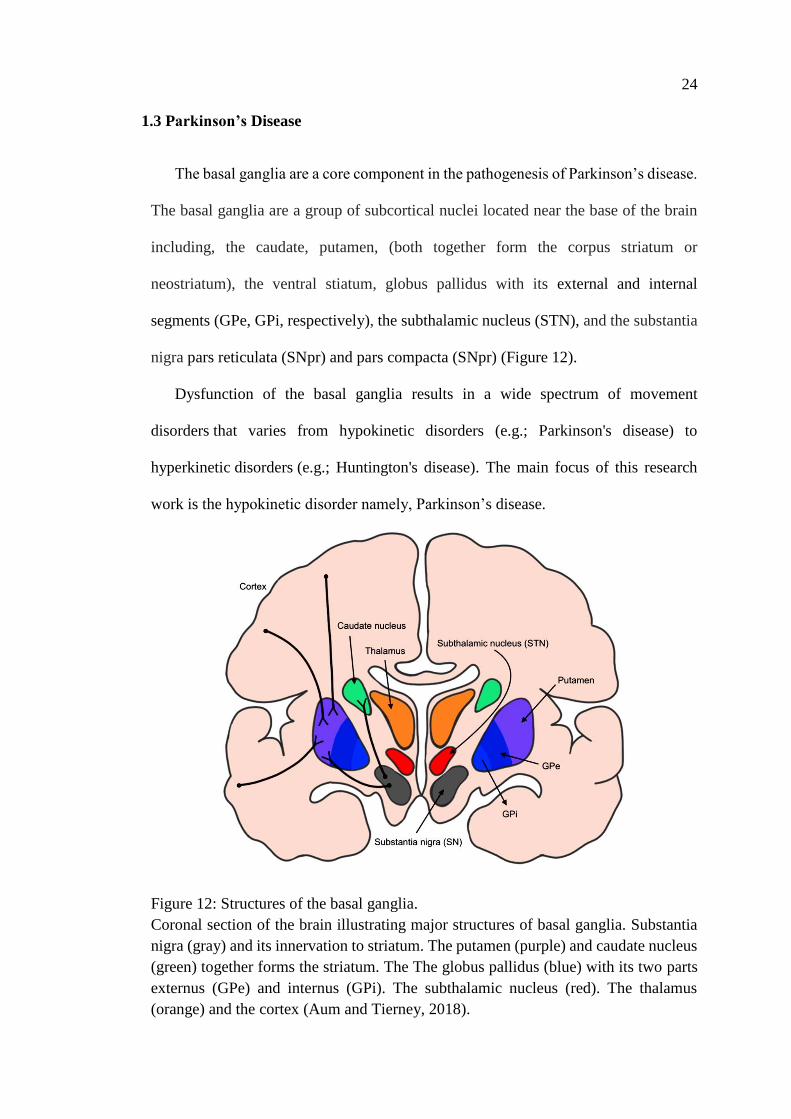
The basal ganglia are a core component in the pathogenesis of Parkinson’s disease.
The basal ganglia are a group of subcortical nuclei located near the base of the brain
including, the caudate, putamen, (both together form the corpus striatum or
neostriatum), the ventral stiatum, globus pallidus with its external and internal
segments (GPe, GPi, respectively), the subthalamic nucleus (STN), and the substantia
nigra pars reticulata (SNpr) and pars compacta (SNpr) (Figure 12).
Dysfunction of the basal ganglia results in a wide spectrum of movement
disorders that varies from hypokinetic disorders (e.g.; Parkinson's disease) to
hyperkinetic disorders (e.g.; Huntington's disease). The main focus of this research
work is the hypokinetic disorder namely, Parkinson’s disease.
Figure 12: Structures of the basal ganglia.
Coronal section of the brain illustrating major structures of basal ganglia. Substantia
nigra (gray) and its innervation to striatum. The putamen (purple) and caudate nucleus
(green) together forms the striatum. The The globus pallidus (blue) with its two parts
externus (GPe) and internus (GPi). The subthalamic nucleus (red). The thalamus
(orange) and the cortex (Aum and Tierney, 2018).

25
1.3.1 Background
Parkinson’s disease (PD) is the second most common neurodegenerative disease
after Alzheimer's disease (AD). Parkinson’s disease was first described by an English
physician and surgeon, James Parkinson in his Essay on the Shaking Palsy in 1817,
which was called later Parkinson’s disease (PD) by Jean-Marie Charcot (Parkinson,
2002). PD is an age-related disorder. The prevalence of the disease increases with
advancing age. The prevalence is around 1% over the age of 60 and 0.3% of all ages
in industrialized countries (de Lau and Breteler, 2006).
1.3.2 Pathophysiology
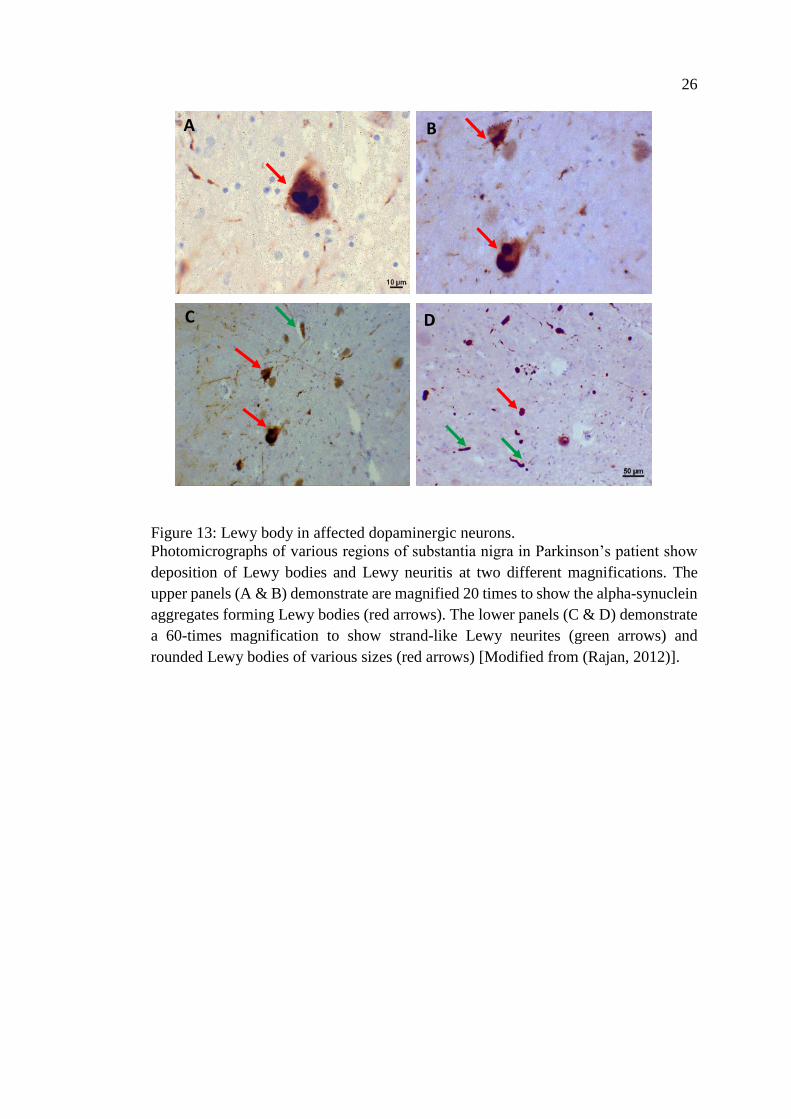
Parkinson is a slowly progressive multisystem disorder rather than just a disease
involving massive neuropathological alterations in the brain. Pathologically, the
hallmark of the disease is the phosphorylation of alpha synuclein protein and formation
of proteinaceous inclusions, Lewy bodies (LB) in neurons (Figure 13) and Lewy
neurites (LN) in axons and dendrites as well as degeneration of dopaminergic
nigrostriatal neurons (Braak et al., 1994; Del Tredici and Braak, 2016).
Other central nervous system (CNS) neurotransmitter systems are also affected to
varying degrees including cholinergic, GABA-ergic, glutamatergic, tryptaminergic,
noradrenergic and adrenergic nerve cells that may show similar damage in their
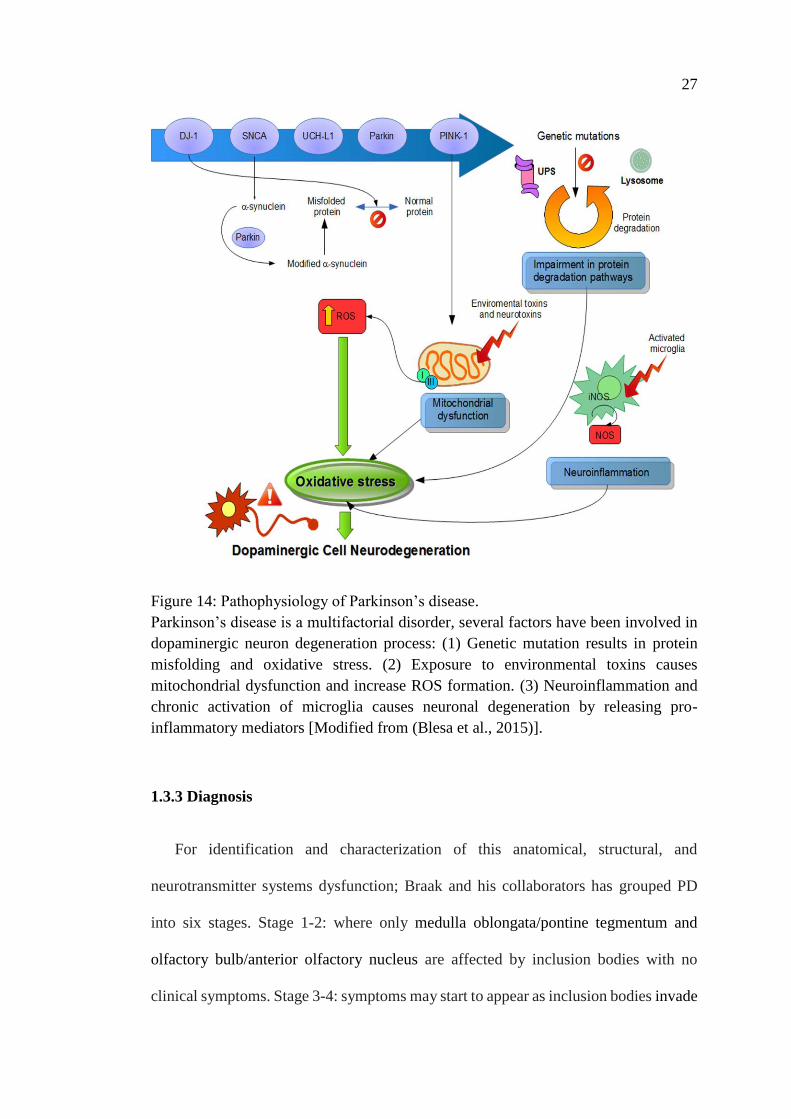
cytoskeletons (Braak and Braak, 2000). Mechanistically, some environmental insults
and/or gene mutations contribute to the degenerative changes observed in Parkinson’s
disease, causing mitochondrial dysfunction, oxidative stress, modifications in protein
handling, adaptations in immune-modulators, as well as alterations in other molecular
and cellular functions (Figure 14) (Franco-Iborra et al., 2016; Olanow and McNaught,
2011; Schapira and Jenner, 2011).
26
Figure 13: Lewy body in affected dopaminergic neurons.
Photomicrographs of various regions of substantia nigra in Parkinson’s patient show
deposition of Lewy bodies and Lewy neuritis at two different magnifications. The
upper panels (A & B) demonstrate are magnified 20 times to show the alpha-synuclein
aggregates forming Lewy bodies (red arrows). The lower panels (C & D) demonstrate
a 60-times magnification to show strand-like Lewy neurites (green arrows) and
rounded Lewy bodies of various sizes (red arrows) [Modified from (Rajan, 2012)].
D C
A B
27
Figure 14: Pathophysiology of Parkinson’s disease.
Parkinson’s disease is a multifactorial disorder, several factors have been involved in
dopaminergic neuron degeneration process: (1) Genetic mutation results in protein
misfolding and oxidative stress. (2) Exposure to environmental toxins causes
mitochondrial dysfunction and increase ROS formation. (3) Neuroinflammation and
chronic activation of microglia causes neuronal degeneration by releasing pro-
inflammatory mediators [Modified from (Blesa et al., 2015)].
1.3.3 Diagnosis
For identification and characterization of this anatomical, structural, and
neurotransmitter systems dysfunction; Braak and his collaborators has grouped PD
into six stages. Stage 1-2: where only medulla oblongata/pontine tegmentum and
olfactory bulb/anterior olfactory nucleus are affected by inclusion bodies with no
clinical symptoms. Stage 3-4: symptoms may start to appear as inclusion bodies invade
28
the substantia nigra and other nuclei of the midbrain and forebrain. Stage 5-6: end
stage of the disease as the neocortex is affected with a wide range of clinical
manifestation (Braak et al., 2004, 2003). The clinical symptoms of PD can be
categorized into motor and non-motor symptoms as shown in Table 2.
Table 2: Motor and non-motor symptoms of Parkinson’s disease
Motor symptoms Reference
Limb rigidity
Cogwheel phenomenon
Shuffling gait lack
Arm swing while walking.
Expressionless face (hypomimia)
Micrographia
Limb tremor
Resting pill-rolling
Loss of balance and falls
Freezing of movements
Postural instability
Speech disturbances
Swallowing problems
Dribbling of saliva
Dystonia
Postural deformities
(Jankovic, 2008)
(Virmani et al., 2015)
(Virmani et al., 2015)
(Williams et al., 2006)
(Perez-Lloret et al., 2012)
(Kalf et al., 2011)
(Kalf et al., 2012)
(Tolosa and Compta, 2006)
(Doherty et al., 2011)
Non-Motor symptoms Reference
Orthostatic hypotension
Constipation
Excessive sweating
Urinary control disturbance
Sleep disturbances
Visual hallucinations and illusions
Cognitive impairment and
Dementia
Anhedonia
Depression and anxiety
Loss of smell and taste
Limb pain
(Lahrmann et al., 2006)
(Jost, 2003)
(Hirayama, 2006)
(Jost, 2003)
(Monderer and Thorpy, 2009)
(Onofrj et al., 2007)
(Williams-Gray et al., 2006)
(Pont-Sunyer et al., 2015)
(Reijnders et al., 2008)
(Doty et al., 1988)
(Williams and Lees, 2009)

29
1.3.3.1 Motor Symptoms
Dopamine (DA) is neurotransmitter involved in coordination of movement. When
sixty to eighty percent of dopamine producing cells in the substantia nigra are
defunctionalized the extra pyramidal system loses the ability to effectively promote
movement and the motor symptoms of Parkinson's disease begin to appear (Chung et
al., 2001). UK PD Brain Bank has established specific criteria for diagnosis of
Parkinson’s disease and presented it in three steps (Table 2). Step one which is based
mainly on the patient’s motor symptoms include bradykinesia, defined as delay in
initiation of voluntary movement associated with progressive reduction in patient’s
speed and amplitude of repetitive actions (Antonini et al., 2013; Berardelli et al., 2001;
Goetz et al., 2007). It is one of the main and core symptoms that if found, in association
with one of the following additional symptoms i.e., resting tremor, muscular rigidity,
or postural instability- are a criterion for the diagnosis of PD (Hughes et al., 1992).
Step two; is to exclude any other cause for the motor symptoms such as parkinsonian
syndrome. Step three; establishes three supportive measures. Namely, unilateral onset
of symptoms, persistent asymmetry of clinical symptoms, induction of dyskinesia by
the dopaminergic treatment, and good response to levodopa treatment.
1.3.3.2 Non-Motor Symptoms
Clinical heterogeneity of Parkinson’s disease reflects the multisystem involvement
in the pathophysiology of the disease. Therefore, the International Movement
Disorders Society included a range of Non-motor symptoms (NMS) to the diagnostic
criteria of Parkinson’s disease (Postuma et al., 2015). It has been reported that NMS
starts to appear as early as ten years before the diagnosis of PD, outlining a prodromal
or an asymptomatic stage of the disease. There is a wide range of symptoms (Table 3),
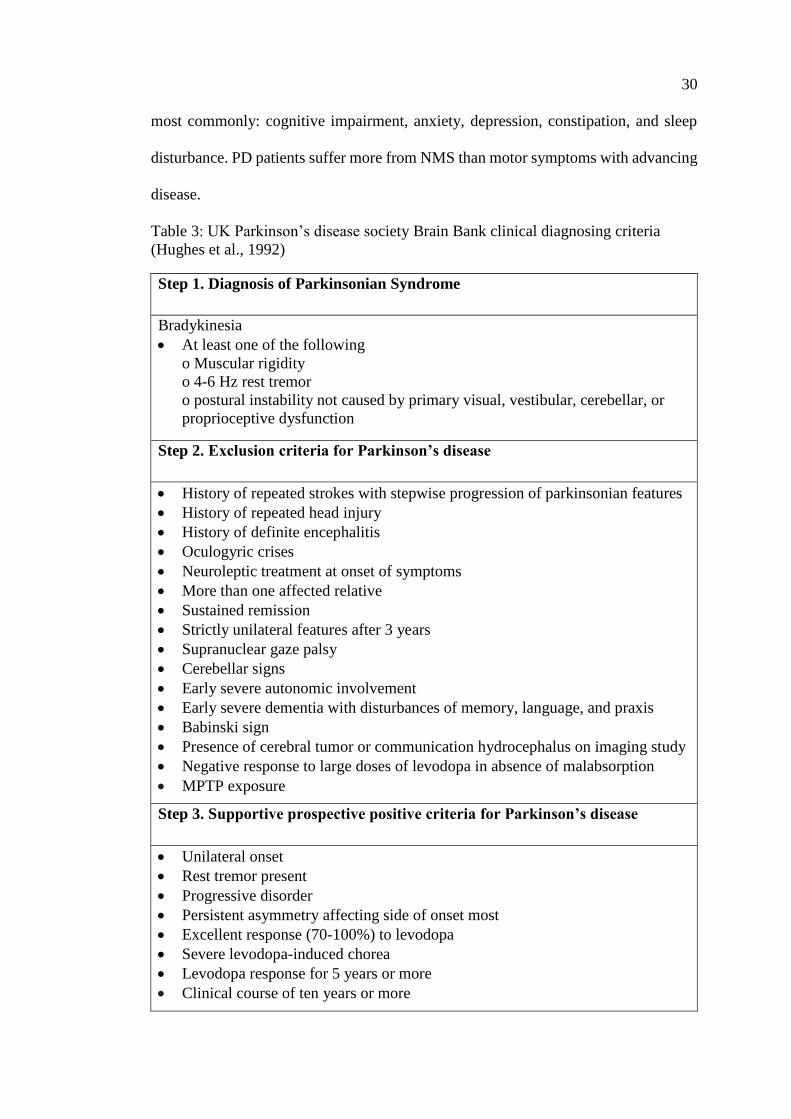
30
most commonly: cognitive impairment, anxiety, depression, constipation, and sleep
disturbance. PD patients suffer more from NMS than motor symptoms with advancing
disease.
Table 3: UK Parkinson’s disease society Brain Bank clinical diagnosing criteria
(Hughes et al., 1992)
Step 1. Diagnosis of Parkinsonian Syndrome
Bradykinesia
• At least one of the following
o Muscular rigidity
o 4-6 Hz rest tremor
o postural instability not caused by primary visual, vestibular, cerebellar, or
proprioceptive dysfunction
Step 2. Exclusion criteria for Parkinson’s disease
• History of repeated strokes with stepwise progression of parkinsonian features
• History of repeated head injury
• History of definite encephalitis
• Oculogyric crises
• Neuroleptic treatment at onset of symptoms
• More than one affected relative
• Sustained remission
• Strictly unilateral features after 3 years
• Supranuclear gaze palsy
• Cerebellar signs
• Early severe autonomic involvement
• Early severe dementia with disturbances of memory, language, and praxis
• Babinski sign
• Presence of cerebral tumor or communication hydrocephalus on imaging study
• Negative response to large doses of levodopa in absence of malabsorption
• MPTP exposure
Step 3. Supportive prospective positive criteria for Parkinson’s disease
• Unilateral onset
• Rest tremor present
• Progressive disorder
• Persistent asymmetry affecting side of onset most
• Excellent response (70-100%) to levodopa
• Severe levodopa-induced chorea
• Levodopa response for 5 years or more
• Clinical course of ten years or more

31
1.3.4 Treatment
The etiology of PD is multifactorial. To date, there is no drug that cures or stops
disease progression. Being mainly a dysfunction in dopaminergic system in the brain,
Levodopa or L-dopa (L-3,4-dihydroxyphenylalanine) was introduced in 1960s as a
prodrug of dopamine enhancing intracerebral dopamine concentration. Since its
approval by the FDA in 1970, L-dopa has been the gold standard treatment for
Parkinson’s disease. Though, after several months to years of treatment with L-dopa,
patients develop the adverse effects of dyskinesias (Brotchie and Jenner, 2011; Huot
et al., 2013; Iravani et al., 2012). This is known as L-dopa-induced dyskinesias (LIDs)
and it will be discussed in greater details later. With the limitation of L-dopa use, other
strategies have been implemented to enhance dopamine release, such as; dopamine
agonist, monoamine oxidase type B inhibitors (MAO), catechol-O-methyl transferase
inhibitors (COMTIs), anticholinergic, beta-blocker, antipsychotic, and antiviral (Table
3 & Figure 15) (Connolly and Lang, 2014; Gazewood et al., 2013a). Surgical
intervention became an option with deep brain stimulation (DBS) in selected PD
patients (Benabid et al., 2009; Morgante et al., 2007; Rizzone et al., 2014).
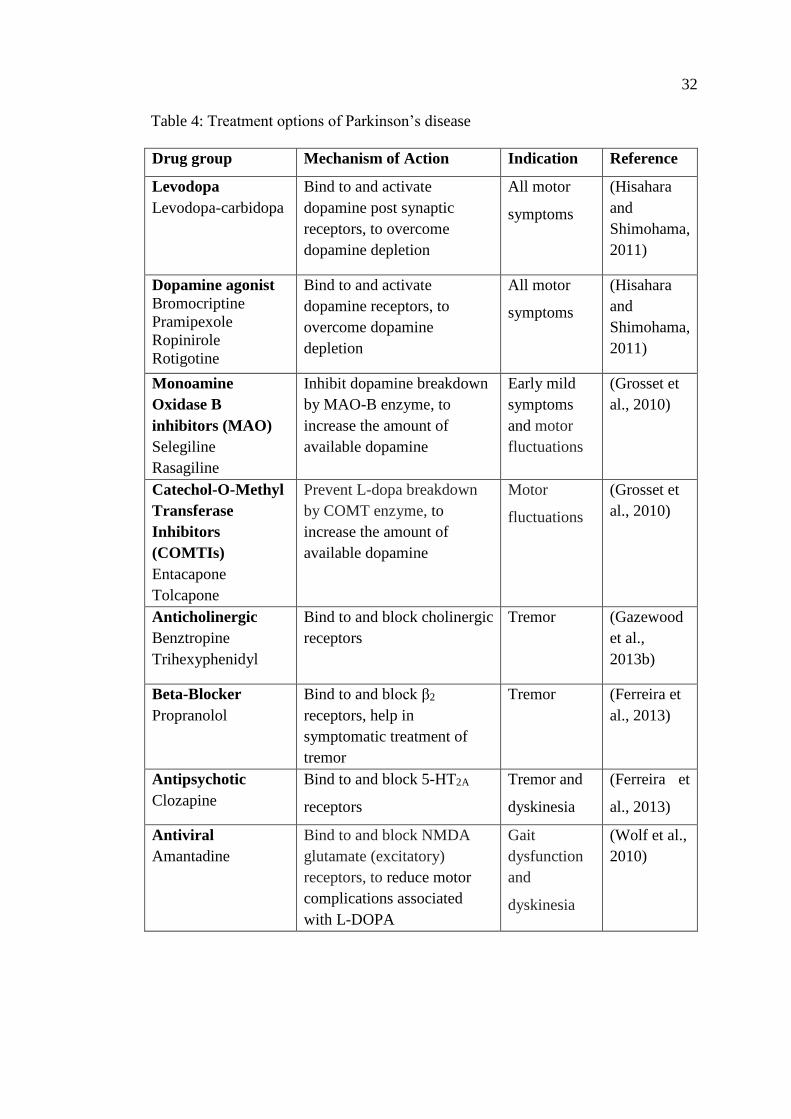
32
Table 4: Treatment options of Parkinson’s disease
Drug group Mechanism of Action Indication Reference
Levodopa
Levodopa-carbidopa
Bind to and activate
dopamine post synaptic
receptors, to overcome
dopamine depletion
All motor
symptoms
(Hisahara
and
Shimohama,
2011)
Dopamine agonist
Bromocriptine
Pramipexole
Ropinirole
Rotigotine
Bind to and activate
dopamine receptors, to
overcome dopamine
depletion
All motor
symptoms
(Hisahara
and
Shimohama,
2011)
Monoamine
Oxidase B
inhibitors (MAO)
Selegiline
Rasagiline
Inhibit dopamine breakdown
by MAO-B enzyme, to
increase the amount of
available dopamine
Early mild
symptoms
and motor
fluctuations
(Grosset et
al., 2010)
Catechol-O-Methyl
Transferase
Inhibitors
(COMTIs)
Entacapone
Tolcapone
Prevent L-dopa breakdown
by COMT enzyme, to
increase the amount of
available dopamine
Motor
fluctuations
(Grosset et
al., 2010)
Anticholinergic
Benztropine
Trihexyphenidyl
Bind to and block cholinergic
receptors
Tremor (Gazewood
et al.,
2013b)
Beta-Blocker
Propranolol
Bind to and block β2
receptors, help in
symptomatic treatment of
tremor
Tremor (Ferreira et
al., 2013)
Antipsychotic
Clozapine
Bind to and block 5-HT2A
receptors
Tremor and
dyskinesia
(Ferreira et
al., 2013)
Antiviral
Amantadine
Bind to and block NMDA
glutamate (excitatory)
receptors, to reduce motor
complications associated
with L-DOPA
Gait
dysfunction
and
dyskinesia
(Wolf et al.,
2010)
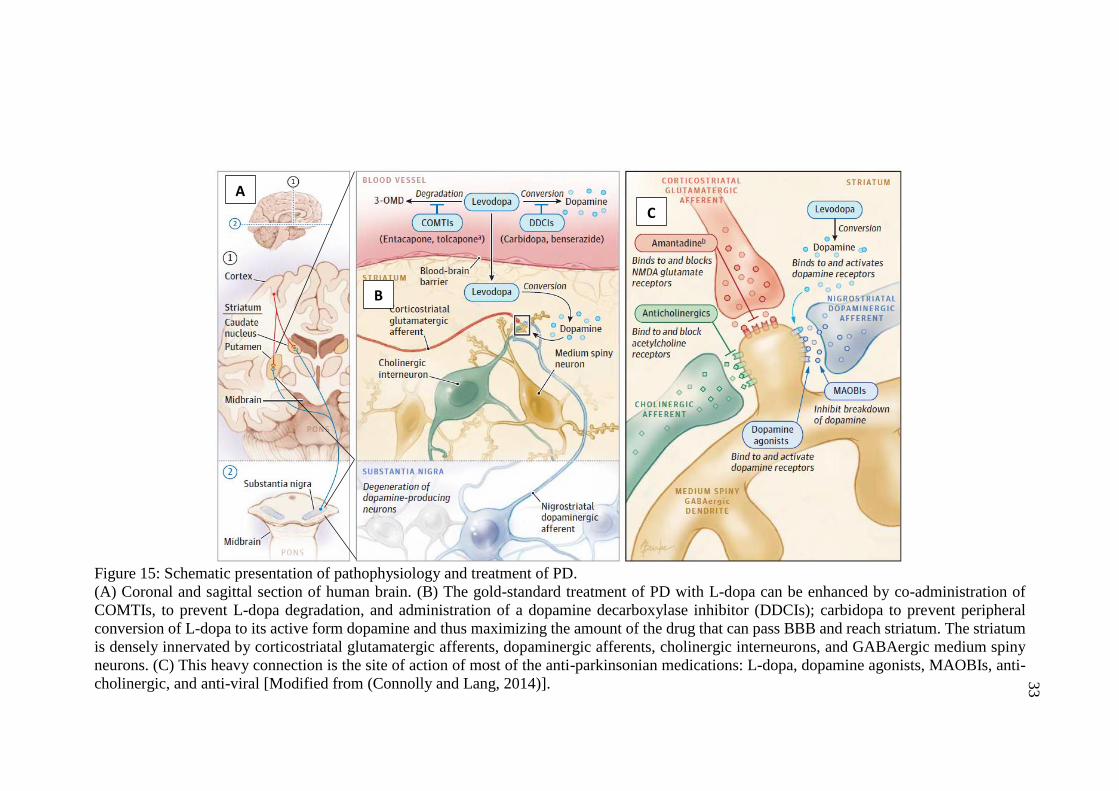
33
Figure 15: Schematic presentation of pathophysiology and treatment of PD.
(A) Coronal and sagittal section of human brain. (B) The gold-standard treatment of PD with L-dopa can be enhanced by co-administration of
COMTIs, to prevent L-dopa degradation, and administration of a dopamine decarboxylase inhibitor (DDCIs); carbidopa to prevent peripheral
conversion of L-dopa to its active form dopamine and thus maximizing the amount of the drug that can pass BBB and reach striatum. The striatum
is densely innervated by corticostriatal glutamatergic afferents, dopaminergic afferents, cholinergic interneurons, and GABAergic medium spiny
neurons. (C) This heavy connection is the site of action of most of the anti-parkinsonian medications: L-dopa, dopamine agonists, MAOBIs, anti-
cholinergic, and anti-viral [Modified from (Connolly and Lang, 2014)].
A
C
B

34
1.3.5 Prognosis
Parkinson’s disease is a complex disorder, and with advancing age it is expected
that disease progression will vary between patients (Poewe, 2006). NMS precedes the
motor symptoms and is usually mild and unilateral. Later, it progresses to the
contralateral side but still remains very responsive to treatment, in what is called the
honeymoon period. However, with time treatment efficacy starts to decline and
patients’ symptoms and disability worsen, affecting their quality of life with the need
of home care and frequent hospital admissions (Low et al., 2015; Parashos et al., 2002).
Based on several studies, life expectancy of PD patients ranges from 6.9 to 14.3 years
(Macleod et al., 2014). Thus, the need for development of strategies to stop, slow
down, or preferably reverse the neurodegenerative process is mandatory. So, the next
question is how an interaction at nAChRs level may lead to an overall functional effect
such as protection against nigrostriatal damage. Three mechanisms suggested in recent
studies will be discussed in following sections.
1.4 The Neuroprotective Role of Nicotine and Nicotinic Receptors Against
Nigrostriatal Damage
Cigarette smoking is a well-known health hazard and a risk factor of serious
chronic disorders, including cardiovascular disorders, lung disease, and cancers.
However, unexpectedly, cigarette use appears to confer beneficial effects in
Parkinson’s disease. The initial evidence that nicotine which acts on nAChRs, may be
useful as a therapy for Parkinson’s disease stemmed from the results of various
epidemiological findings in the early 1960s (Allam et al., 2004; Elbaz and Moisan,
2008; Gorell et al., 1999; Noyce et al., 2012; Tanner, 2010; Wirdefeldt et al., 2011).
Nicotine is a highly lipophilic compound and the active addictive ingredient of

35
tobacco. After smoking, chewing, or sniffing, nicotine rapidly crosses the blood-brain
barrier and binds to nicotinic receptors. Nicotine is likely to mediate neuroprotective
effects, as many studies have shown a reduced risk of PD in current and former
smokers (Fahn, 2010; Nicoletti et al., 2010; Tanner, 2010). This apparent
neuroprotection against Parkinson’s disease is correlated with smoking duration,
intensity, and recentness, and is reduced with smoking cessation. Moreover, the
incidence of Parkinson’s disease within twin pairs was less in the twin that smoked
compared to the nonsmoker (Tanner et al., 2002), suggesting that the neuroprotective
effects of tobacco are independent of genetic background. Furthermore, clinical trials
with nicotine patches showed an attenuation of PD symptoms in PD patients
(Fagerström et al., 1994). In addition, recent studies showed that nicotine
administration reduces dyskinesia which is a major side effect of L-dopa used as the
primary treatment for Parkinson’s disease (Bordia et al., 2008; Huot et al., 2013;
Iravani et al., 2012; Quik et al., 2007). All these observations raised the possibility that
nAChR stimulation may be useful in Parkinson’s disease management (Bordia et al.,
2015; Huot et al., 2013; Iravani et al., 2012).
1.4.1 Dopaminergic and Cholinergic Systems Correlation and Dopamine Release
Dopamine inputs to the striatum arise from midbrain dopamine neurons located in
the ventral tegmental area (VTA) and substantia nigra pars compacta (SNpc), which
innervates the ventral striatum (nucleus accumbens (NAc)) and dorsal striatum
(caudate–putamen (CPu)) (Gerfen et al., 1987; Voorn et al., 2004). The striatum is
innervated by a high density of axonal varicosities of dopaminergic nerves forming
dopaminergic synapses (Descarries and Mechawar, 2000; Pickel et al., 1981).

36
The cholinergic striatal interneurons are the primary source of cholinergic input to
the striatum, containing both muscarinic and nicotinic receptors (Zhou et al., 2002,
2001). Nicotinic acetylcholine receptors are principal modulators of neuronal
excitability throughout the central nervous system. Presynaptic nAChRs influences the
release of neurotransmitters, while the postsynaptic nAChRs participate in fast
postsynaptic neurotransmission as an excitatory input in the hippocampus, and
subcortical areas including the ventral tegmental area (Dani, 2001; Mameli-Engvall et
al., 2006; Mansvelder et al., 2002; Pyakurel et al., 2018).
Functional evidence for the specificity of the cholinergic innervation to the VTA
and the SN came from studies of Blaha and Winn (Blaha et al., 1996; Blaha and Winn,
1993). Blaha and Winn measured dopamine efflux following nicotinic agonist
administration before and after performing a lesion in the VTA. In both cases, a
potentiation of DA efflux from the VTA or SN was noticed as a functional evidence
of cholinergic innervation to this area. Using electrical stimulation of the VTA, Forster
and Blaha demonstrated that DA efflux in the striatum is mediated through nicotinic
and glutamatergic receptors in the SN (Forster and Blaha, 2003). Later in 2006, Quik
and colleagues demonstrated that the levels of striatal tyrosine hydroxylase (TH),
dopamine transporter, vesicular monoamine transporter, dopamine and nicotinic
receptors were greater in nicotine-treated MPTP-lesioned parkinsonian animals than
in lesioned animals not receiving nicotine (Figure 16) (Quik et al., 2006).
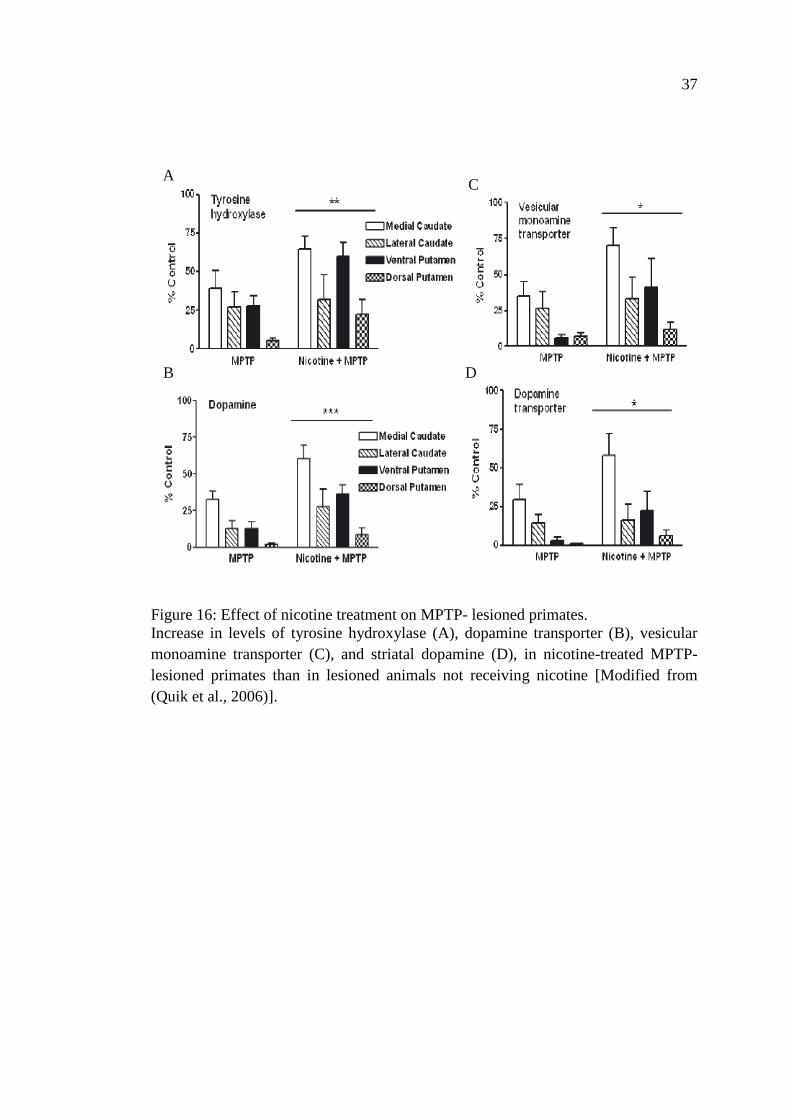
37
Figure 16: Effect of nicotine treatment on MPTP- lesioned primates.
Increase in levels of tyrosine hydroxylase (A), dopamine transporter (B), vesicular
monoamine transporter (C), and striatal dopamine (D), in nicotine-treated MPTP-
lesioned primates than in lesioned animals not receiving nicotine [Modified from
(Quik et al., 2006)].
A
B
C
D
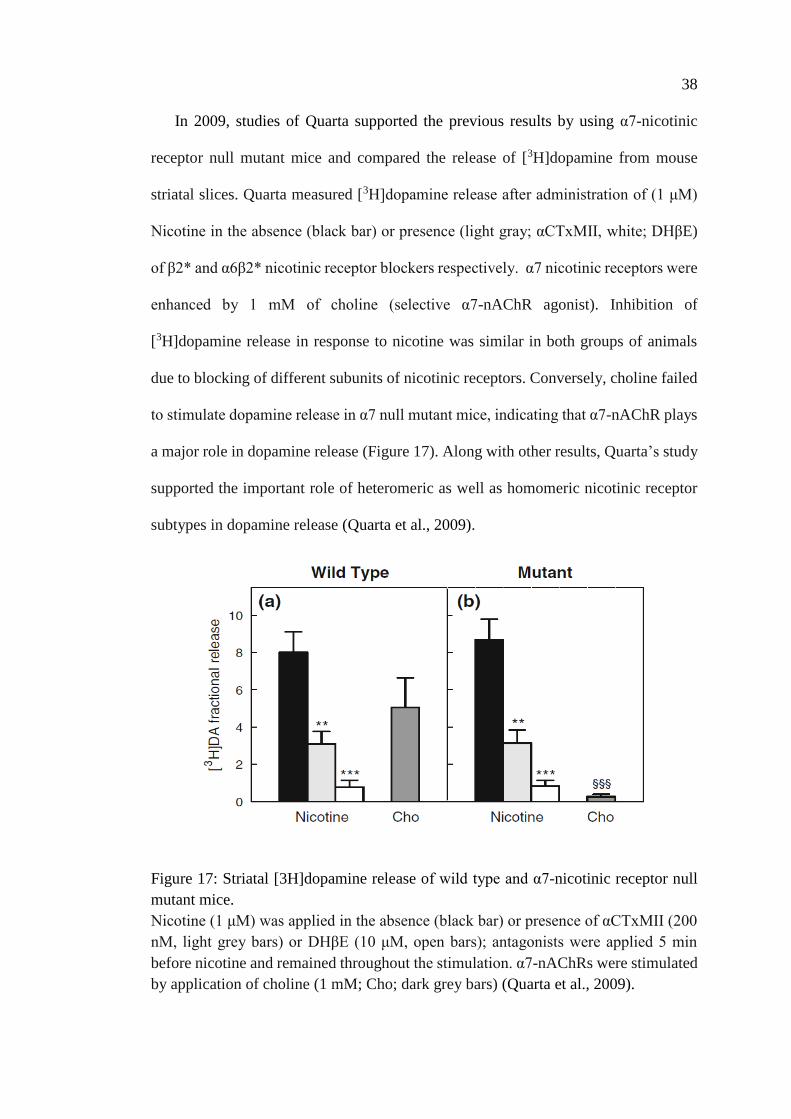
38
In 2009, studies of Quarta supported the previous results by using α7-nicotinic
receptor null mutant mice and compared the release of [3H]dopamine from mouse
striatal slices. Quarta measured [3H]dopamine release after administration of (1 μM)
Nicotine in the absence (black bar) or presence (light gray; αCTxMII, white; DHβE)
of β2* and α6β2* nicotinic receptor blockers respectively. α7 nicotinic receptors were
enhanced by 1 mM of choline (selective α7-nAChR agonist). Inhibition of
[3H]dopamine release in response to nicotine was similar in both groups of animals
due to blocking of different subunits of nicotinic receptors. Conversely, choline failed
to stimulate dopamine release in α7 null mutant mice, indicating that α7-nAChR plays
a major role in dopamine release (Figure 17). Along with other results, Quarta’s study
supported the important role of heteromeric as well as homomeric nicotinic receptor
subtypes in dopamine release (Quarta et al., 2009).
Figure 17: Striatal [3H]dopamine release of wild type and α7-nicotinic receptor null
mutant mice.
Nicotine (1 μM) was applied in the absence (black bar) or presence of αCTxMII (200
nM, light grey bars) or DHβE (10 μM, open bars); antagonists were applied 5 min
before nicotine and remained throughout the stimulation. α7-nAChRs were stimulated
by application of choline (1 mM; Cho; dark grey bars) (Quarta et al., 2009).
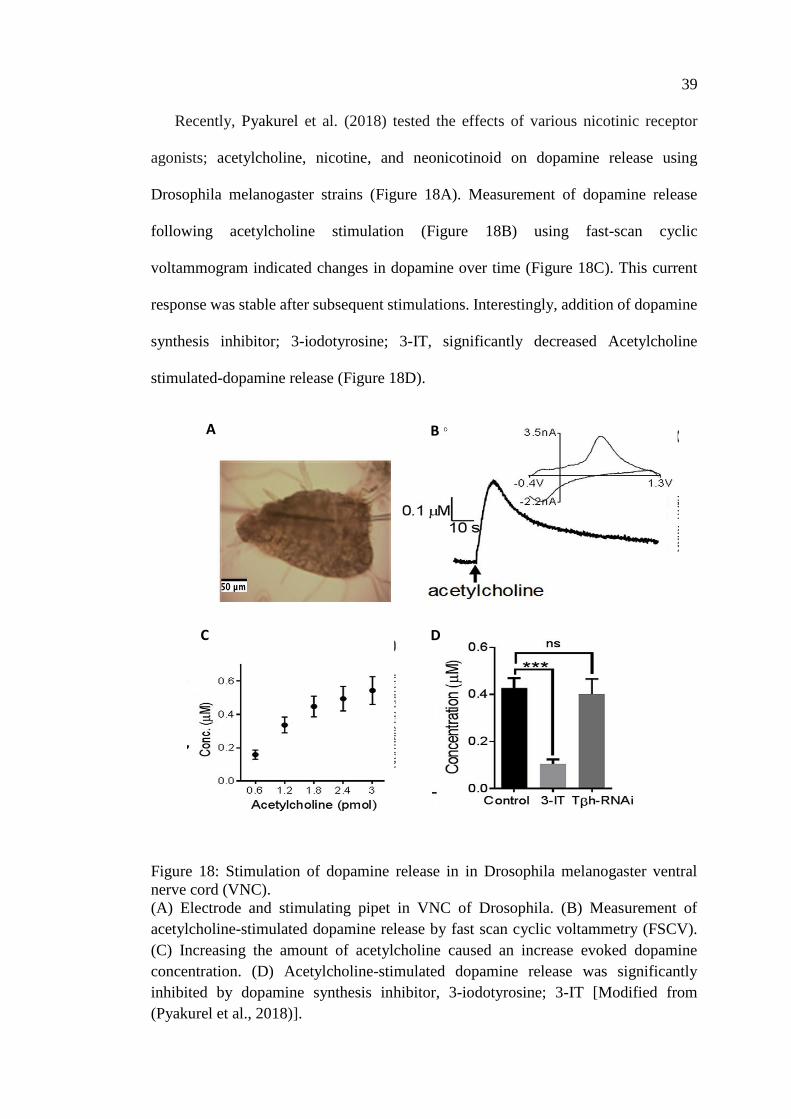
39
Recently, Pyakurel et al. (2018) tested the effects of various nicotinic receptor
agonists; acetylcholine, nicotine, and neonicotinoid on dopamine release using
Drosophila melanogaster strains (Figure 18A). Measurement of dopamine release
following acetylcholine stimulation (Figure 18B) using fast-scan cyclic
voltammogram indicated changes in dopamine over time (Figure 18C). This current
response was stable after subsequent stimulations. Interestingly, addition of dopamine
synthesis inhibitor; 3-iodotyrosine; 3-IT, significantly decreased Acetylcholine
stimulated-dopamine release (Figure 18D).
Figure 18: Stimulation of dopamine release in in Drosophila melanogaster ventral
nerve cord (VNC).
(A) Electrode and stimulating pipet in VNC of Drosophila. (B) Measurement of
acetylcholine-stimulated dopamine release by fast scan cyclic voltammetry (FSCV).
(C) Increasing the amount of acetylcholine caused an increase evoked dopamine
concentration. (D) Acetylcholine-stimulated dopamine release was significantly
inhibited by dopamine synthesis inhibitor, 3-iodotyrosine; 3-IT [Modified from
(Pyakurel et al., 2018)].
D C
A B
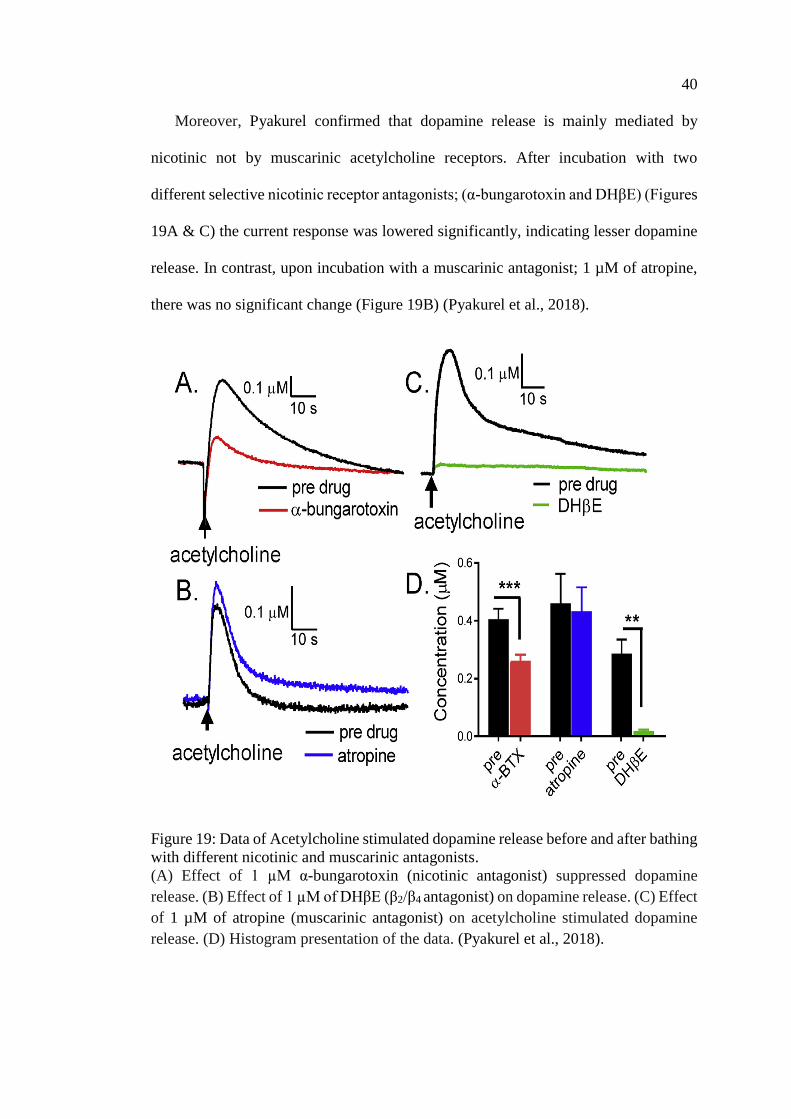
40
Moreover, Pyakurel confirmed that dopamine release is mainly mediated by
nicotinic not by muscarinic acetylcholine receptors. After incubation with two
different selective nicotinic receptor antagonists; (α-bungarotoxin and DHβE) (Figures
19A & C) the current response was lowered significantly, indicating lesser dopamine
release. In contrast, upon incubation with a muscarinic antagonist; 1 µM of atropine,
there was no significant change (Figure 19B) (Pyakurel et al., 2018).
Figure 19: Data of Acetylcholine stimulated dopamine release before and after bathing
with different nicotinic and muscarinic antagonists.
(A) Effect of 1 µM α-bungarotoxin (nicotinic antagonist) suppressed dopamine
release. (B) Effect of 1 µM of DHβE (β2/β4 antagonist) on dopamine release. (C) Effect
of 1 µM of atropine (muscarinic antagonist) on acetylcholine stimulated dopamine
release. (D) Histogram presentation of the data. (Pyakurel et al., 2018).

41
1.4.2 Immune Modulation via Nicotinic Receptors
Activation of the immune system in the CNS takes place in various neurological
disorders such as in; stroke, neurodegenerative diseases, spinal cord injury, multiple
sclerosis, and brain injury (Long-Smith et al., 2009; Napoli and Neumann, 2010;
Prokop et al., 2013; Yenari et al., 2010). In the CNS, the innate immune system is
represented by a type of macrophage, the microglia, a member of the
reticuloendothelial system and a resident immune cell in CNS. Under normal
physiological conditions, microglia are inactive, with a small cell body and highly
ramified branching processes. In response to injury or pathogen invasion, microglia
transform into active phagocytic microglia helping with the clearance of aggregated
proteins and cell debris (Stence et al., 2001). Chronic activation of microglia leads to
noxious effects on neurons by the release of pro-inflammatory molecules and, thus,
contributing to the pathophysiology of neurodegenerative diseases (Figure 20A)
(Gehrmann et al., 1995; Liu and Hong, 2003).
Figure 20: The role of microglia in health and disease.
(A) Unchallenged microglia perform functions in the healthy CNS including
phagocytosis of normal apoptotic debris, secretion of trophic factors (IGF-1, TGFβ),
and synaptic maintenance. (B) Challenged microglia respond to CNS damage
secreting pro-inflammatory factors (TNFα, IL-1β and ROS). When these mediators
persist unchecked, neurodegeneration can ensue (Derecki et al., 2013).
A B
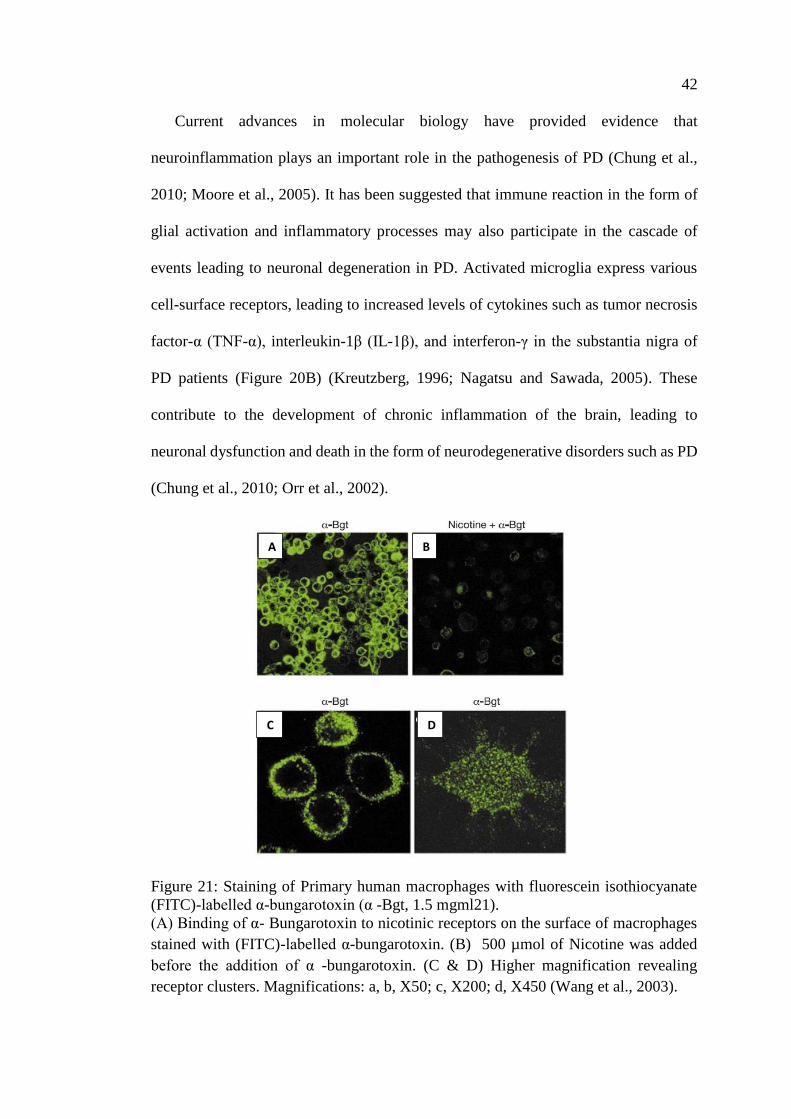
42
Current advances in molecular biology have provided evidence that
neuroinflammation plays an important role in the pathogenesis of PD (Chung et al.,
2010; Moore et al., 2005). It has been suggested that immune reaction in the form of
glial activation and inflammatory processes may also participate in the cascade of
events leading to neuronal degeneration in PD. Activated microglia express various
cell-surface receptors, leading to increased levels of cytokines such as tumor necrosis
factor-α (TNF-α), interleukin-1β (IL-1β), and interferon-γ in the substantia nigra of
PD patients (Figure 20B) (Kreutzberg, 1996; Nagatsu and Sawada, 2005). These
contribute to the development of chronic inflammation of the brain, leading to
neuronal dysfunction and death in the form of neurodegenerative disorders such as PD
(Chung et al., 2010; Orr et al., 2002).
Figure 21: Staining of Primary human macrophages with fluorescein isothiocyanate
(FITC)-labelled α-bungarotoxin (α -Bgt, 1.5 mgml21).
(A) Binding of α- Bungarotoxin to nicotinic receptors on the surface of macrophages
stained with (FITC)-labelled α-bungarotoxin. (B) 500 µmol of Nicotine was added
before the addition of α -bungarotoxin. (C & D) Higher magnification revealing
receptor clusters. Magnifications: a, b, X50; c, X200; d, X450 (Wang et al., 2003).
A B
C D

43
Neuronal nAChRs have been shown to regulate inflammation, in particular via the
α7-nAChR activation in microglia, this is known as ‘cholinergic anti-inflammatory
pathway’. In his letter to Nature journal, Wang first established the relationship
between the cholinergic and immune systems (Figure 21) (Wang et al., 2003).
A year later, Shytle and collaborators demonstrated the expression of α7-nAChRs
on microglia (Figure 22A) and their results suggested that α7-nAChRs play important
roles in the neuroinflammatory processes (Figure 22B) (Shytle et al., 2004). Consistent
with these findings, it has been demonstrated that nicotine has a neuroprotective effect
on dopaminergic neurons via an anti-inflammatory mechanism mediated by the
modulation of microglial activation (Park et al., 2007). Both Shytle and Park
documented that nicotine treatment decreased microglial activation, with significant
reduction of the bacterial cell wall endotoxin LPS-induced TNF- α release (Park et al.,
2007; Shytle et al., 2004).
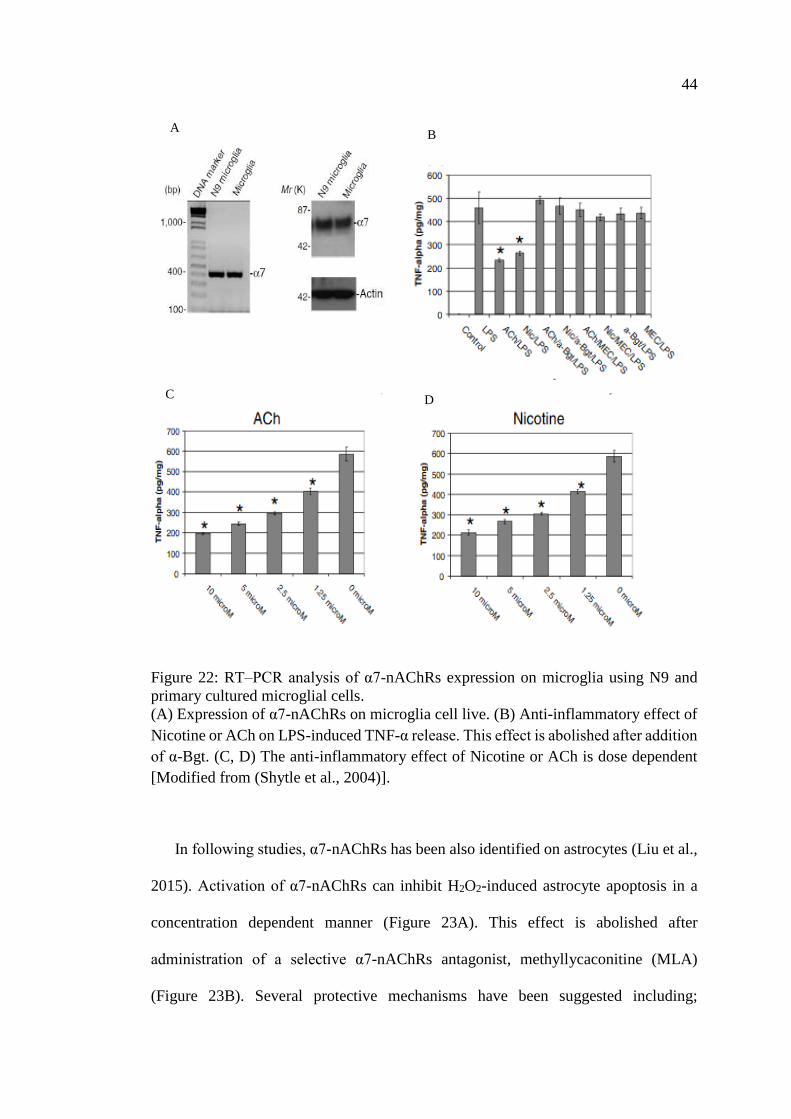
44
Figure 22: RT–PCR analysis of α7-nAChRs expression on microglia using N9 and
primary cultured microglial cells.
(A) Expression of α7-nAChRs on microglia cell live. (B) Anti-inflammatory effect of
Nicotine or ACh on LPS-induced TNF-α release. This effect is abolished after addition
of α-Bgt. (C, D) The anti-inflammatory effect of Nicotine or ACh is dose dependent
[Modified from (Shytle et al., 2004)].
In following studies, α7-nAChRs has been also identified on astrocytes (Liu et al.,
2015). Activation of α7-nAChRs can inhibit H2O2-induced astrocyte apoptosis in a
concentration dependent manner (Figure 23A). This effect is abolished after
administration of a selective α7-nAChRs antagonist, methyllycaconitine (MLA)
(Figure 23B). Several protective mechanisms have been suggested including;
A
C
B
D
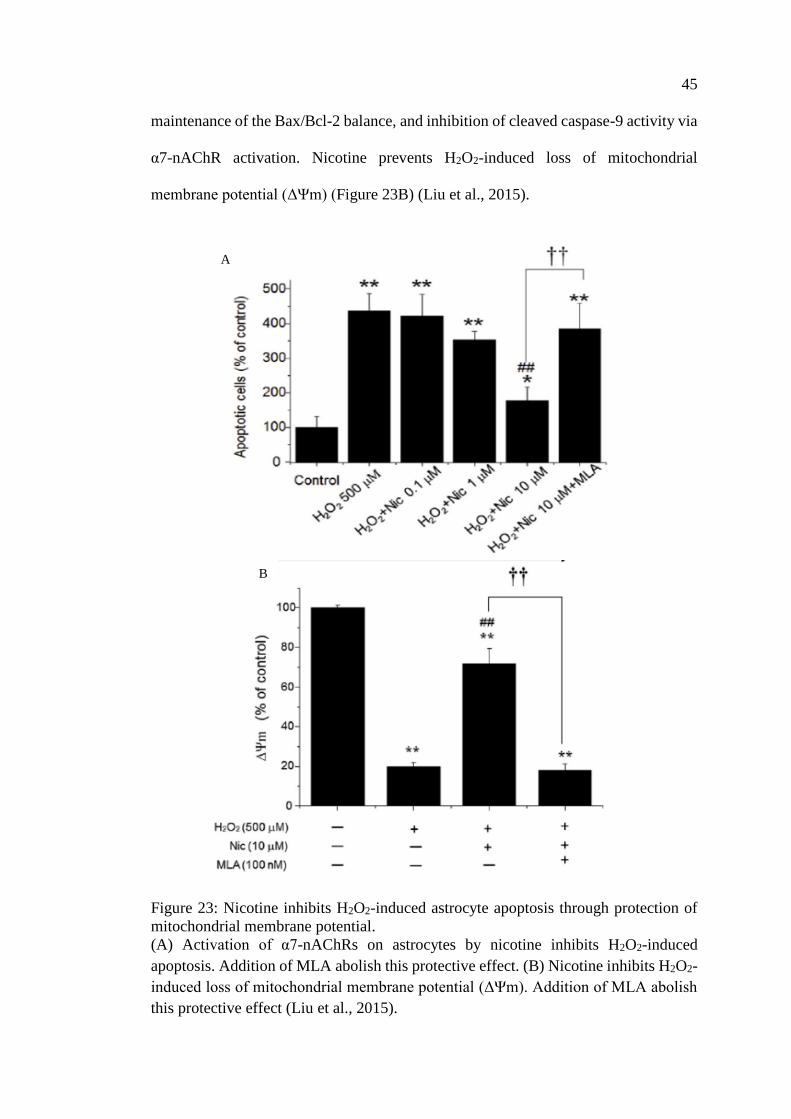
45
maintenance of the Bax/Bcl-2 balance, and inhibition of cleaved caspase-9 activity via
α7-nAChR activation. Nicotine prevents H2O2-induced loss of mitochondrial
membrane potential (ΔΨm) (Figure 23B) (Liu et al., 2015).
Figure 23: Nicotine inhibits H2O2-induced astrocyte apoptosis through protection of
mitochondrial membrane potential.
(A) Activation of α7-nAChRs on astrocytes by nicotine inhibits H2O2-induced
apoptosis. Addition of MLA abolish this protective effect. (B) Nicotine inhibits H2O2-
induced loss of mitochondrial membrane potential (ΔΨm). Addition of MLA abolish
this protective effect (Liu et al., 2015).
A
B

46
1.4.3 Effect of Nicotinic Receptors on L-Dopa Induced Dyskinesia
L-dopa-induced dyskinesia (LID) which occurs in majority of patients on prolonged
L-dopa treatment (Ahlskog and Muenter, 2001), can be very debilitating and impairs
patient’s quality of life. Indeed, LID represent one of the major drawbacks of L-dopa
treatment (Calabresi et al., 2008; Carta et al., 2008; Fahn, 2008; Fox et al., 2008;
Jenner, 2008).
The pathophysiological mechanism behind LID is not fully understood, but it is
primarily due to the altered handling of levodopa presynaptically as well as dopamine
receptors hypersensitization postsynaptically (Bastide et al., 2015).
Several pharmacological approaches to treat LID have been implicated. For
example the use of amantadine, a nonselective NMDA receptor antagonist has been
suggested (Blanchet et al., 1996) but its effect is limited, short-lived and associated
with many adverse effects (Sawada et al., 2010; Thomas et al., 2004; Verhagen
Metman et al., 1998)Reduction of L-dopa daily dose has also been considered as a line
of LID treatment, but this might lead to the re-emergence of PD symptoms (Goetz et
al., 2005; Tambasco et al., 2012). The surgical approach has been also suggested for
the treatment of LID, namely, deep brain stimulation (DBS). Deep brain stimulation is
based on targeting the nuclei of selective deep brain structures such as; the internal
segment of the globus pallidus (GPi), subthalamic nucleus (STN), and thalamus by
high-frequencies electrical stimuli. This is based on interruption and/or modulation of
the neuronal signaling of the structures within the basal ganglia (Brown and Eusebio,
2008; Obeso and Lanciego, 2011). Though it is an effective method of treatment, it
needs careful selection and screening of patients with dyskinesia with quality of life

47
severely affected, with on–off fluctuations, and treatment-resistant tremor. It is an
invasive procedure with considerable side effects (Okun, 2012).
The demand for safe and non-invasive approaches in treating LID is crucial. Many
laboratories are targeting different neurotransmitter systems in the brain, such as; the
serotonergic, glutamatergic, GABAergic, adrenergic, cannabinoid, opioid, adenosine
and other neurotransmitter systems to test the effect of pharmacological agents on
different neurotransmitter systems. Any one of these has the potential to improve
dyskinesias, in fact some laboratories have shown promising results (Blandini and
Armentero, 2012; Brotchie et al., 2005; Cenci and Lundblad, 2007, 2006; Fox et al.,
2009; Linazasoro et al., 2008; Olanow et al., 2006; Quik et al., 2008; Samadi et al.,
2006; Sgambato-Faure and Cenci, 2012).
One of the proposed strategies against LID is to target neuronal nicotinic
acetylcholine receptors. Accumulating evidence suggests that nicotine and nAChR can
reduce the abnormal involuntary movements or dyskinesias complicating L-dopa
treatment (Hickey and Stacy, 2013; Kerr, 2010; Quik et al., 2014, 2012). Pioneering
studies were done on MPTP-induced Parkinson in monkeys to investigate the effect of
long-term nicotine treatment against nigrostriatal damage in non-human primates
where they exhibited parkinsonian motor symptoms very similar to those in
Parkinson’s disease patients and develop abnormal involuntary movements after L-
dopa treatment analogous to those in L-dopa-treated Parkinson’s disease patients. The
results demonstrated the effectiveness of nicotine treatment in PD model (Quik et al.,
2006). This was followed by number of studies by Quik and his colleagues where they
could demonstrate that nicotinic administration to L-dopa-treated animal models led
to ~60% decrease in LIDs (Figure 24). Nicotine reduced LIDs -whether given orally
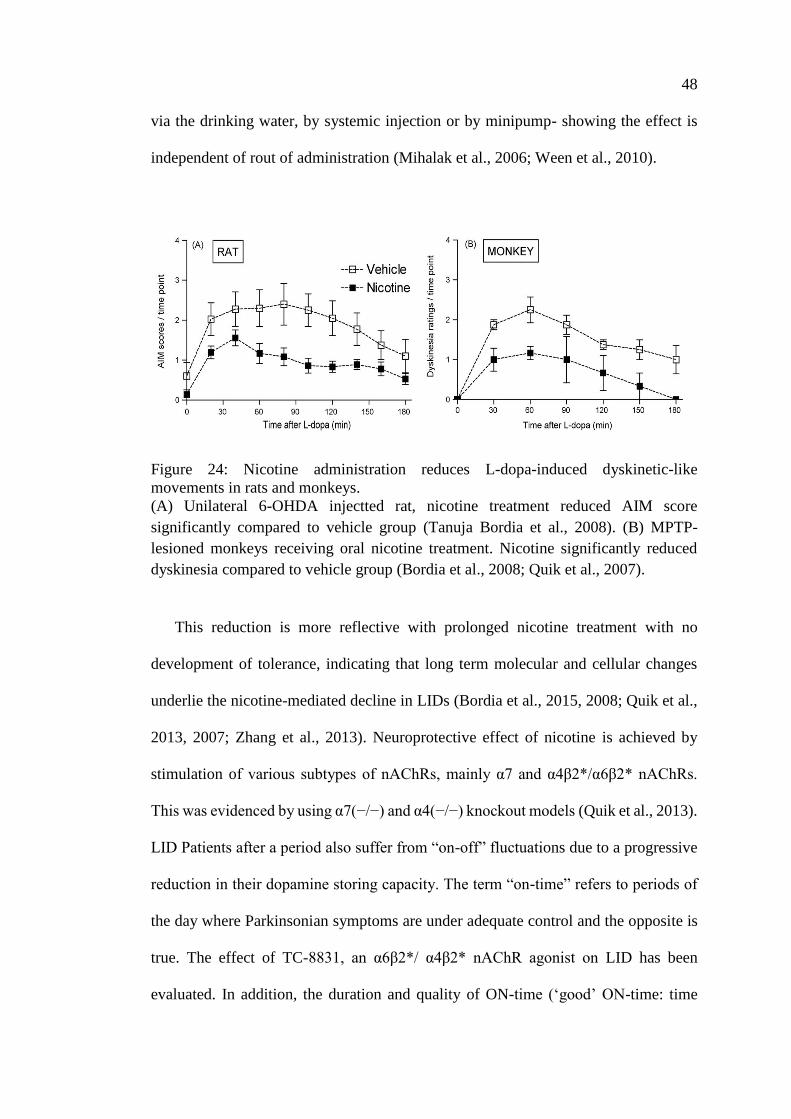
48
via the drinking water, by systemic injection or by minipump- showing the effect is
independent of rout of administration (Mihalak et al., 2006; Ween et al., 2010).
Figure 24: Nicotine administration reduces L-dopa-induced dyskinetic-like
movements in rats and monkeys.
(A) Unilateral 6-OHDA injectted rat, nicotine treatment reduced AIM score
significantly compared to vehicle group (Tanuja Bordia et al., 2008). (B) MPTP-
lesioned monkeys receiving oral nicotine treatment. Nicotine significantly reduced
dyskinesia compared to vehicle group (Bordia et al., 2008; Quik et al., 2007).
This reduction is more reflective with prolonged nicotine treatment with no
development of tolerance, indicating that long term molecular and cellular changes
underlie the nicotine-mediated decline in LIDs (Bordia et al., 2015, 2008; Quik et al.,
2013, 2007; Zhang et al., 2013). Neuroprotective effect of nicotine is achieved by
stimulation of various subtypes of nAChRs, mainly α7 and α4β2*/α6β2* nAChRs.
This was evidenced by using α7(−/−) and α4(−/−) knockout models (Quik et al., 2013).
LID Patients after a period also suffer from “on-off” fluctuations due to a progressive
reduction in their dopamine storing capacity. The term “on-time” refers to periods of
the day where Parkinsonian symptoms are under adequate control and the opposite is
true. The effect of TC-8831, an α6β2*/ α4β2* nAChR agonist on LID has been
evaluated. In addition, the duration and quality of ON-time (‘good’ ON-time: time

49
without disabling dyskinesia, ‘bad’ on-time: time with disabling dyskinesia) was
studied and compared to amantadine. TC-8831caused a significant reduction in the
duration of ‘bad’ ON-time (62%) as well as severity of LID (reduced chorea and
dystonia) with minimal effect on L-DOPA anti-parkinsonian benefits, in a dose
dependent manner. Parallel to TC-88331, amantadine reduced ‘bad’ ON-time by up to
61% but total ON-time has dropped by up to 23% (Figure 25) (Johnston et al., 2013).
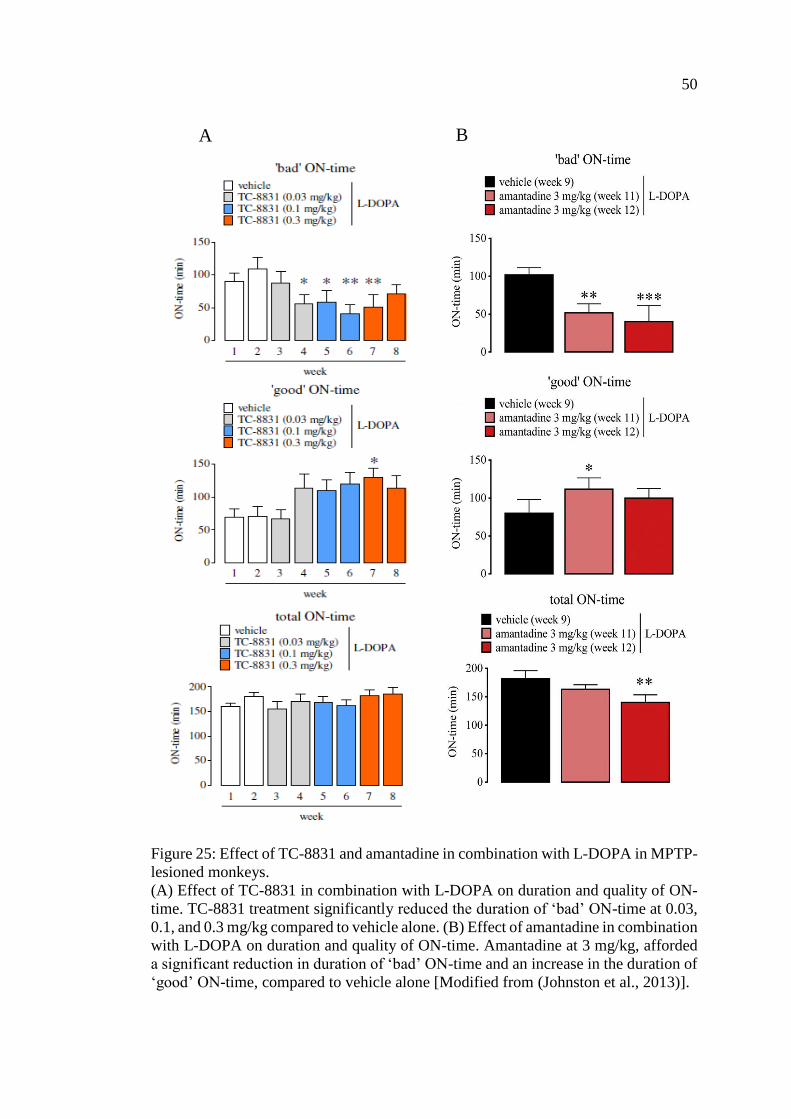
50
Figure 25: Effect of TC-8831 and amantadine in combination with L-DOPA in MPTP-
lesioned monkeys.
(A) Effect of TC-8831 in combination with L-DOPA on duration and quality of ON-
time. TC-8831 treatment significantly reduced the duration of ‘bad’ ON-time at 0.03,
0.1, and 0.3 mg/kg compared to vehicle alone. (B) Effect of amantadine in combination
with L-DOPA on duration and quality of ON-time. Amantadine at 3 mg/kg, afforded
a significant reduction in duration of ‘bad’ ON-time and an increase in the duration of
‘good’ ON-time, compared to vehicle alone [Modified from (Johnston et al., 2013)].
A B
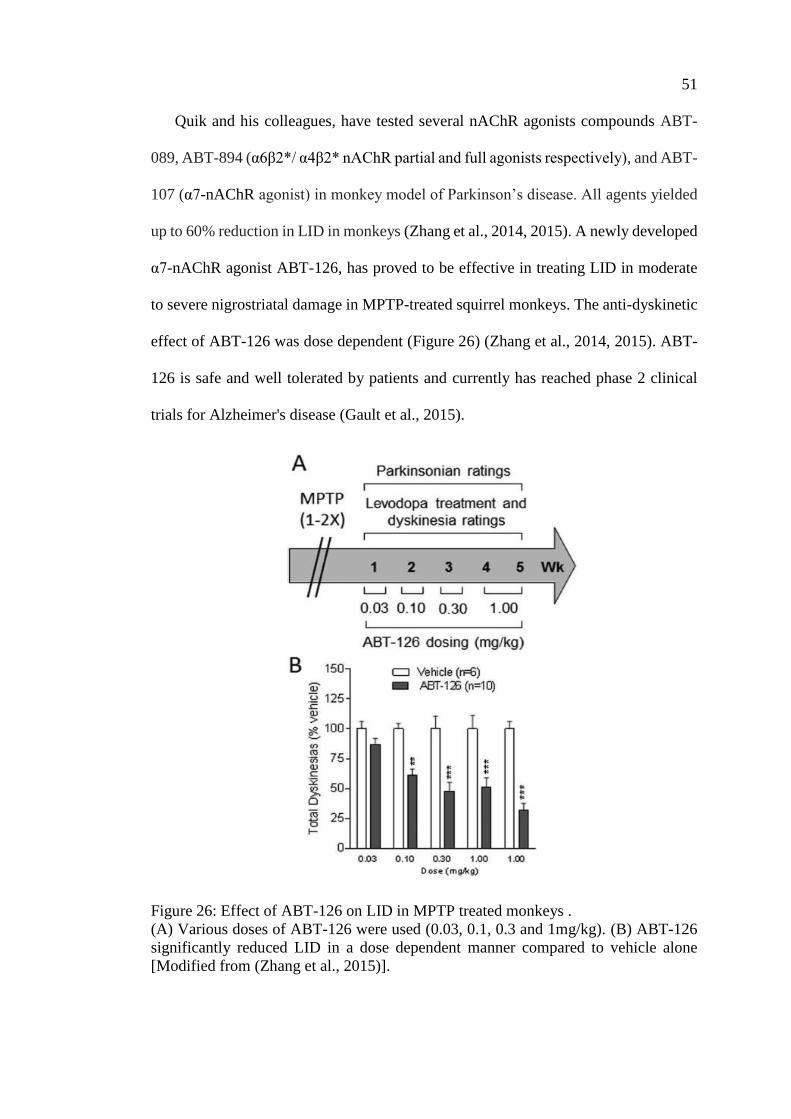
51
Quik and his colleagues, have tested several nAChR agonists compounds ABT-
089, ABT-894 (α6β2*/ α4β2* nAChR partial and full agonists respectively), and ABT-
107 (α7-nAChR agonist) in monkey model of Parkinson’s disease. All agents yielded
up to 60% reduction in LID in monkeys (Zhang et al., 2014, 2015). A newly developed
α7-nAChR agonist ABT-126, has proved to be effective in treating LID in moderate
to severe nigrostriatal damage in MPTP-treated squirrel monkeys. The anti-dyskinetic
effect of ABT-126 was dose dependent (Figure 26) (Zhang et al., 2014, 2015). ABT-
126 is safe and well tolerated by patients and currently has reached phase 2 clinical
trials for Alzheimer's disease (Gault et al., 2015).
Figure 26: Effect of ABT-126 on LID in MPTP treated monkeys .
(A) Various doses of ABT-126 were used (0.03, 0.1, 0.3 and 1mg/kg). (B) ABT-126
significantly reduced LID in a dose dependent manner compared to vehicle alone
[Modified from (Zhang et al., 2015)].

52
1.4.4 Molecular Neuroprotective Mechanisms of Nicotinic Acetylcholine
Receptors
Epidemiological and experimental evidence indicate that nicotine is protective for
the vulnerable dopamine neurons in Parkinson disease. The molecular mechanism
involves alterations in calcium (Ca2+) signaling, although calcium independent
nAChR-mediated mechanisms (cytosolic Ca2+) have also been reported (Dajas-
Bailador and Wonnacott, 2004; Picciotto and Zoli, 2008; Shimohama, 2009; Ward et
al., 2008).
Stimulation of nAChR mediates several intracellular changes, namely protein
kinases such as; protein kinase A (PKA), extracellular signal-regulated mitogen-
activated protein kinase (ERK/MAPK), calcium-calmodulin-dependent protein kinase
(CaM) and phosphatidylinositol 3-kinase (PI3K)/Akt-or protein kinase B-dependent
signaling (Toulorge et al., 2011). Modulation of protein kinases are all calcium
dependent. On the other hand, calcium independent modulation involves modifications
in the JAK2 (Janus kinase 2)/PI3K and/or JAK2/STAT3 (signal transducer and
activator of transcription 3) pathways (Hosur and Loring, 2011; Kawamata and
Shimohama, 2011). These pathways are also known as survival pathways leading to
cell survival by modulating different downstream signaling cascades such as; caspase
activity (3, 8 and 9), cell survival proteins such as Bcl-2 (B-cell lymphoma 2) and Bcl-
x, NFκB, CREB (cAMP response element-binding), and other molecular components
(Figure 27). Also it has been reported that nicotine-induced changes in basic fibroblast
growth factor-2 (FGF-2), brain-derived neurotrophic factor (BDNF) and nerve growth
factor (NGF) in brain dopaminergic and other regions attenuate neuronal damage
(Belluardo et al., 2000; Formaggio et al., 2010; Massey et al., 2006; Zhou et al., 2004).
In addition to neuroprotective role of nicotine- nAChRs stimulation, other mechanisms

53
are involved in its neuroprotective effect of nicotine, including: reduction in
mitochondrial complex 1 activity, inhibition of reactive oxygen species generation,
oxidative or anti-oxidative potential and radical scavenging properties (Cormier et al.,
2003; Ferger et al., 1998; Newman et al., 2002; Xie et al., 2005). Overall, all these data
suggest the involvement of multiple molecular transduction mechanisms in nicotinic
receptor-mediated neuroprotection under various pathological conditions.
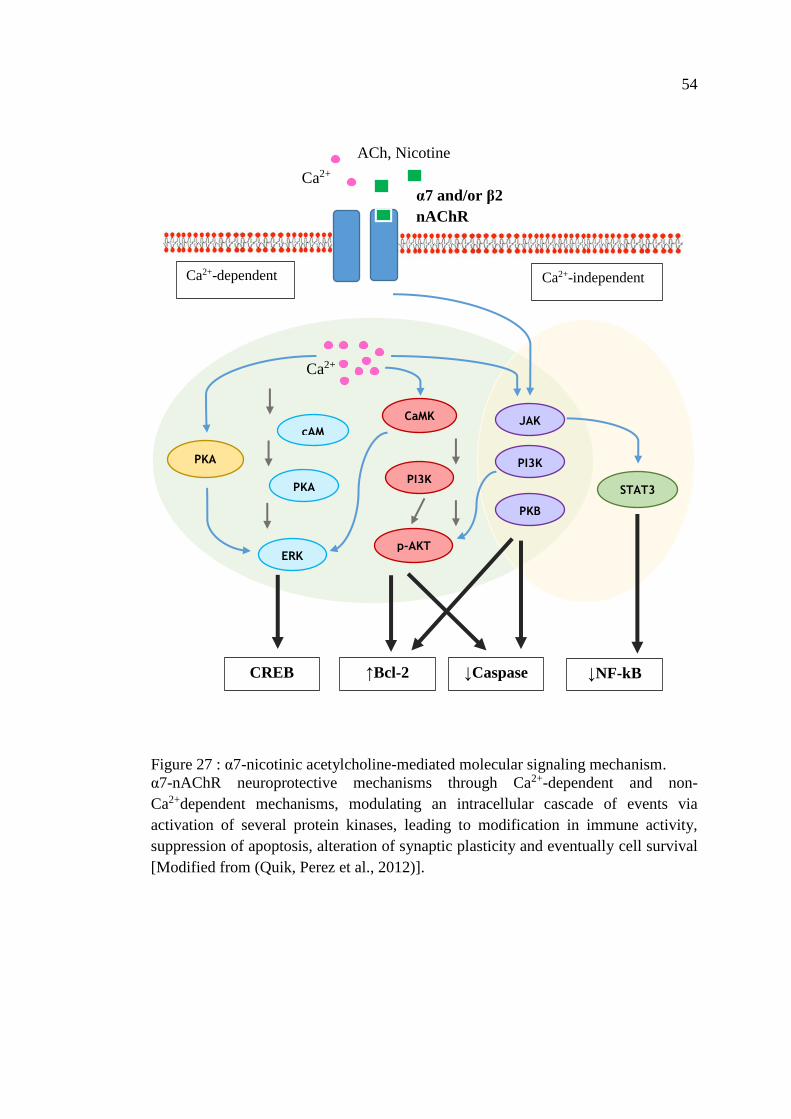
54
Figure 27 : α7-nicotinic acetylcholine-mediated molecular signaling mechanism.
α7-nAChR neuroprotective mechanisms through Ca2+-dependent and non-
Ca2+dependent mechanisms, modulating an intracellular cascade of events via
activation of several protein kinases, leading to modification in immune activity,
suppression of apoptosis, alteration of synaptic plasticity and eventually cell survival
[Modified from (Quik, Perez et al., 2012)].
JAK2
PI3K
PKB
STAT3
CaMK
PI3K PKA
PKA
cAMP
ERK p-AKT
Ca2+
α7 and/or β2
nAChR
ACh, Nicotine
Ca2+
CREB ↑Bcl-2 ↓Caspase ↓NF-kB
Ca2+-dependent Ca2+-independent

55
1.5 Animal Models of Parkinson’s Disease
The use of animal models has contributed significantly to a better understanding
of disease process and made a significant breakthrough in development of new
therapeutic strategies specially in LID and surgery. An ideal animal model of PD
should include both pathological and clinical manifestation (motor and non-motor
symptoms), involving central and peripheral nervous system, dopaminergic and non-
dopaminergic neurons. Unfortunately, none of the available models includes the full
picture of PD, still they can replicate many of the disease characteristics. Experimental
models can be categorized into two main classes: toxic and genetic.
1.5.1 The Neurotoxin Model
Many pharmacological and toxic agents have been implicated in primates as well
as in rodents and other mammals. Though neurotoxic models considered to be the best
for testing nigrostriatal dopaminergic degeneration, some drawbacks need to be
mentioned: the rapidly progressive dopaminergic degeneration (within days – weeks)
compared to the disease progression in human taking years to develop, most of the
animal models lack the pathological hallmark; Lewy bodies, and motor/behavioral
abnormalities (Blesa and Przedborski, 2014).
1.5.1.1 Classical Model: 6-Hydroxydopamine
6-Hydroxydopamine (6-OHDA) is a catecholaminergic neurotoxin that
selectively damages dopaminergic and noradrenergic neurons (Luthman et al., 1989).
This model of “Chemical Denervation” was first established by Ungerstedt in 1968
and proved to knock out 60 – 90 % of tyrosine hydroxylase (TH)-positive neurons in
SNpc. 6-hydroxydopamine is a photosensitive compound and can be rapidly oxidized

56
if exposed to light, hence it should be protected from light exposure, also its dissolved
in ascorbic acid to protect it from oxidation (Bové and Perier, 2012). As 6-OHDA
cannot cross blood-brain barrier, it should be injected directly (mostly as unilateral
injection; hemiparkinsonian model) into the targeted area; medial forebrain bundle
(MFB), substantia nigra pars compacta (SNpc), striatum or intraventricular
administration (Blandini et al., 2008; Rodríguez Díaz et al., 2001).
Since it’s a hydroxylated analog of dopamine (Figure 28), 6-OHDA is taken up by
the neurons via dopamine transporters (DAT) and accumulate in the cytosol where it
gets oxidized leading to ROS formation and cytotoxicity due to oxidative stress (Figure
29) (Blum et al., 2001; Graham, 1978; Rangel-Barajas et al., 2015; Saner and Thoenen,
1971). Proteinaceous aggregates and Lewy-like inclusions are not produced in this
type of PD (Blesa and Przedborski, 2014).
Figure 28: Chemical structures of 6-hydroxydopamine (6-OHDA) and dopamine
(Bové and Perier, 2012)

57
For the following reasons, it was decided to use the animal model of PD in which
multiple injections of 6-OHDA into the striatum was employed: 1) Striatal nerve
terminals are more sensitive to 6-OHDA toxicity than the axon and cell bodies (Bruyn,
1983; Malmfors and Sachs, 1968). 2) Progressive and less extensive lesion are more
relevant to the pathophysiology of PD (Cannon and Greenamyre, 2010). 3) Striatal
injection produces also non-motor symptoms of the disease; cognitive, psychiatric, and
GI symptoms (Branchi et al., 2008; Tadaiesky et al., 2008). 4) Including wider and
larger area of striatum can increase the success rate (Tieu, 2011). 5) It provides a well-
characterized behavioral, biochemical, and pathological feature of the disease which
is correlated tightly with the number of injection sites (Rosenblad et al., 1999). 6) Any
model that induce a predictable lesion over a long period of time will provide a greater
opportunity for neuroprotective strategies to succeed, and striatum is therefore an ideal
target to test for neuroprotection (Kirik et al., 1998).
Following unilateral 6-OHDA injection, drug-induced rotation test (drug:
dopamine receptor agonist; apomorphine or dopamine releasing compound such as
amphetamine) are performed to assess the extent of motor impairment. Systemic
injection of any of these drugs will result in asymmetrical rotation. Number of turns
are counted to ipsilateral side in case of intraperitoneal injection of amphetamine or
contralateral side in case of subcutaneous injection of apomorphine (Dunnett and
Torres, 2011; Hefti et al., 1980b; Ungerstedt and Arbuthnott, 1970).
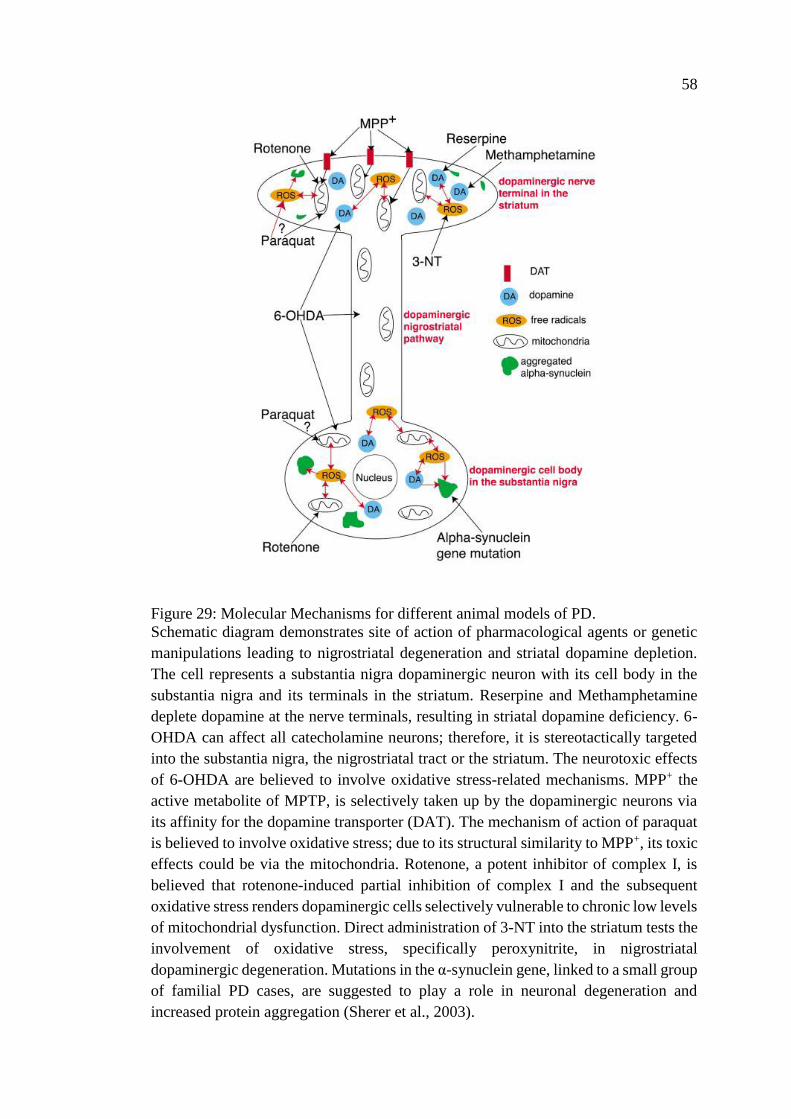
58
Figure 29: Molecular Mechanisms for different animal models of PD.
Schematic diagram demonstrates site of action of pharmacological agents or genetic
manipulations leading to nigrostriatal degeneration and striatal dopamine depletion.
The cell represents a substantia nigra dopaminergic neuron with its cell body in the
substantia nigra and its terminals in the striatum. Reserpine and Methamphetamine
deplete dopamine at the nerve terminals, resulting in striatal dopamine deficiency. 6-
OHDA can affect all catecholamine neurons; therefore, it is stereotactically targeted
into the substantia nigra, the nigrostriatal tract or the striatum. The neurotoxic effects
of 6-OHDA are believed to involve oxidative stress-related mechanisms. MPP+ the
active metabolite of MPTP, is selectively taken up by the dopaminergic neurons via
its affinity for the dopamine transporter (DAT). The mechanism of action of paraquat
is believed to involve oxidative stress; due to its structural similarity to MPP+, its toxic
effects could be via the mitochondria. Rotenone, a potent inhibitor of complex I, is
believed that rotenone-induced partial inhibition of complex I and the subsequent
oxidative stress renders dopaminergic cells selectively vulnerable to chronic low levels
of mitochondrial dysfunction. Direct administration of 3-NT into the striatum tests the
involvement of oxidative stress, specifically peroxynitrite, in nigrostriatal
dopaminergic degeneration. Mutations in the α-synuclein gene, linked to a small group
of familial PD cases, are suggested to play a role in neuronal degeneration and
increased protein aggregation (Sherer et al., 2003).

59
The site of 6-OHDA injection significantly affects the characteristics and extent of
neurodegeneration (Agid et al., 1973; Przedborski et al., 1995). Injection of 6-OHDA
into substantia nigra, cause a very rapid degeneration process of dopaminergic neurons
(up to 90% loss) to take place within hours (12 hours) producing a complete damage
in the nigrostriatal pathway, followed by loss of striatal terminals in 2 to 3 days (Faull
and Laverty, 1969; Jeon et al., 1995), this is similar to end-stage PD. In the medial
forebrain bundle (MFB) model, striatal terminals are degenerated before dopaminergic
neurons (80 - 90% loss) producing a near complete damage in the nigrostriatal pathway
within 3 to 4 days after injection which is equivalent to end-stage in PD. In the
consecutive 3 weeks, dopamine content is totally lost in the striatum. In comparison to
substantia nigra and medial forebrain bundle, 6-OHDA injection into striatum,
produces slow, (in 3 weeks’ time) progressive, and partial loss of dopaminergic
neurons and damage in the nigrostriatal pathway in a retrograde fashion which is more
relevant to PD (Przedborski et al., 1995; Sauer and Oertel, 1994). The percent of
dopaminergic cell death is dose-dependent and can vary from 30 to 75% depending on
number of injection sites. Up to four injections can be delivered into the striatum (Kirik
et al., 1998). This model is characterized by producing non-motor symptoms;
cognitive, psychiatric, and gastrointestinal dysfunction (Branchi et al., 2008; Cannon
and Greenamyre, 2010). Therefore, this model was employed in this study.

60
1.5.1.2 Gold Standard: MPTP
The 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model was discovered
accidentally in California 1983 in intravenous drug abusers using an analogue of the
synthetic opioid; meperidine which was contaminated with MPTP, patients showed
some movement abnormalities (bradykinesia) similar to PD (Davis et al., 1979;
Langston et al., 1983). L-dopa was successful in treating these patients and
confirmatory postmortem studies demonstrated the loss of nigrostriatal structures in
the brain of same patients (Davis et al., 1979; Langston et al., 1999). MPTP is a
lipophilic compound that can cross blood-brain barrier, once inside the brain it can be
taken up by the astrocytes and metabolized by MAO-B enzyme into its active toxic
form 1-methyl-4-phenylpyridinium MPP+ (Figure 30). The active metabolite MPP+
then can be taken up by the neighboring dopaminergic neurons via dopamine
transporters (DAT) and stored in the vesicles by vesicular monoamine Transporter
(VMAT) displacing and releasing dopamine from the storing vesicles (Figure 29).
Released dopamine intracellularly can form toxic compounds such as; DOPAL
(Panneton et al., 2010) where it can be exposed to superoxide radical (5-cysteinyl-DA)
and hydroxyl radical (6-OHDA). Still, MPP+ can be more damaging by inhibiting
complex I causing to mitochondrial electron chain inhibition leading to energy failure
and oxidative stress and dopaminergic cell death (Mizuno et al., 1987; Nicklas et al.,
1985). MPTP causes severe damage to nigrostriatal pathway with significant loss of
dopaminergic neurons in the striatum and SNpc (Dauer and Przedborski, 2003).

61
Figure 30: Chemical structures of MPTP and MPP+
(Bové and Perier, 2012)
MPTP is a toxic compound in a wide range of species including mice, monkeys,
sheep, dogs, guinea pigs, cats, but not rats which were found to be resistant to this of
toxicity (Bezard et al., 1998; Giovanni et al., 1994; Przedborski et al., 2001). Mouse
model is a practical and can provide a model of genetic mutation compared to the gold
standard-monkey model which needs highly trained lab workers (Giovanni et al.,
1994). MPTP model also lack the pathological hallmark of PD disease; Lewy bodies,
but in the other hand some reports have investigated the expression of α-synuclein
(Dauer et al., 2002; Halliday et al., 2009; Purisai et al., 2005; Shimoji et al., 2005; Vila
et al., 2000).
1.5.1.3 Rotenone
Rotenone is a natural compound found in plants that belong to family of
Leguminosa (Tieu, 2011). It has been used mainly in farming as a pesticide. Rotenon
model was developed for the first time by Greenamyre and his colleagues where the
administered rotenone chronically using low-dose treatment in rats (Betarbet et al.,
2000; Cannon et al., 2009; Sherer et al., 2003). Due its high lipophilicity, it crosses
blood-brain barrier and enter almost all cells independent of any transporter. Similar
to MPTP cytotoxic mechanism, rotenone also inhibits complex I with the
consequential oxidative stress and neuronal cell death (Parker et al., 1989; Schapira et

62
al., 1989). Interestingly, chronic systemic administration of rotenone to rats replicates
all pathological features of PD, namely; proteinaceous aggregate, Lewy-like bodies,
α-synuclein, behavioral abnormalities, and oxidative stress (Greenamyre et al., 2010;
Sherer et al., 2003). Rotenone can be administered by different routes of
administration, mainly intraperitoneal daily injection (Cannon et al., 2009),
subcutaneous, or intravenous (Fleming et al., 2004). Most recently, it has been tested
in mice via chronic intragastric rout or administration and direct stereotaxic injection
or infusion into the brain (Alam and Schmidt, 2004; Pan-Montojo et al., 2010). A
major drawback of this model is the high variability between different animals with
high mortality which may reach up to 50% (Blesa and Przedborski, 2014; Emborg,
2004). Moreover, trials to induce PD using rotenone in other species like mice or
monkeys were unsuccessful, and not even in humans (Blesa et al., 2012).
1.5.1.4 Paraquat
Paraquat (N,N’-dimethyl-4-4’-bipiridinium) is a divalent cation compound that has
a structure similarity with MPTP (Figure 29) (Snyder and D’Amato, 1985). Originally,
it was used as a herbicide that has been forbidden in European Union after 2007 (Bové
and Perier, 2012). Due to its hydrophilicity, paraquat uses neutral amino acid transport
mechanism to penetrate blood-brain barrier. Once inside the brain paraquat causes
cytotoxicity by inducing redox cycling with a cellular diaphorase in form of NADPH
oxidase and nitric oxide synthase leading ROS production such as superoxide radical,
hydrogen peroxide and the hydroxyl radical causing damage of cellular lipids,
proteins, DNA and RNA (Day et al., 1999). Injection of paraquat in mice causes dose-
and age-dependent nigral damage (Brooks et al., 1999; McCormack et al., 2002;
Thiruchelvam et al., 2003). Paraquat causes not only brain toxicity but multi-organ

63
toxicity; lung, liver, and kidney, which may result in death after acute exposure in
human (Grant et al., 1980; Hughes, 1988). Several reports established the presence of
α-synuclein aggregates and Lewy bodies in this model (Fernagut et al., 2007; Mak et
al., 2010; Manning-Bog et al., 2002).
Other neurotoxic models
Several other models gave have been used to induce nigrostriatal degeneration and
dopaminergic cell death. However, those models are not in common use.
1.5.1.5 Reserpine
Reserpine was one of the earliest pharmacological agents that have been used to induce
Parkinson’s disease in rodents, after the observation that it induces movement
abnormalities; akinesia, in injected animals (Carlsson et al., 1957). Remarkably, these
symptoms could be reversed by L-dopa treatment (Carlsson, 1959). Carlsson
demonstrated that reserpine depletes monoamines in general in the brains of injected
animals. Reserpine model has been reproduced also in other mammals such as; cats,
rabbits, guinea pigs, and monkeys (Bezard et al., 1998). Reserpine is not only
reversible but also non-specific; it mediates it’s action by hindering catecholamine
storage in synaptic vesicles through ATP- and magnesium mechanism (Bezard et al.,
1998), such mechanism has no direct relation to dopamine and not toxic to nigrostriatal
pathway. The aforementioned findings have weakened the use of this model.
1.5.1.6 α-Methyl-Para-Tyrosine
This reversible pharmacological agent is an enzyme inhibitor, it inhibits the
enzyme tyrosine hydroxylase in dopamine synthesis process, blocking dopamine
synthesis (Corrodi and Hanson, 1966; Spector et al., 1965). α-methyl-p-tyrosine has

64
the same weakness points of reserpine being reversible, non-specific, and not
triggering neurodegeneration in nigrostriatal pathway (Tieu, 2011).
1.5.1.7 Amphetamines
A pharmacological group of compounds that belongs to psychostimulant drugs,
has been found to be highly neurotoxic and producing behavioral and structural
alteration similar to PD (Cadet et al., 2007; Thrash et al., 2009). Amphetamine
derivatives such as: methamphetamine (METH) and 3,4-
methylenedioxymethamphetamine (MDMA), p-chloroamphetamine (PCA), and
fenfluramine are toxic to serotoninergic and dopaminergic terminals in the striatum,
nucleus accumbens, and frontal cortex (Hess et al., 1990; Krasnova and Cadet, 2009),
but SN and VTA neuronal cell bodies are affected only at higher doses (Sonsalla et al.,
1996; Trulson et al., 1985).
1.5.2 Genetic Models
Genetic animal models of PD have been designed on previously recognized targets
to better study the disease process and therapy (Bezard and Przedborski, 2011;
Meredith et al., 2008), noting that none of such genetic mutations are expressed in
humans. Moreover, most of the genetic models don’t exhibit the behavioral
phenotypes and neurodegeneration of PD (Dawson et al., 2010). For instance, many
studies have reported the presence of mitochondrial dysfunction (Exner et al., 2012;
Matsui et al., 2014; Morais et al., 2014), ubiquitin-proteasome dysfunction (Dantuma
and Bott, 2014), ROS formation (Gandhi et al., 2009; Joselin et al., 2012; Ottolini et
al., 2013). nevertheless, none of the studies could demonstrate significant changes in

65
dopaminergic neurons (Andres-Mateos et al., 2007; Goldberg et al., 2003; Hinkle et
al., 2012; Sanchez et al., 2014). Here are the commonly used transgenic models of PD:
1.5.2.1 α-synuclein
α-syn gene is the core component of Lewy bodies (LB), and a type of missense
mutations of α-syn. It is also called Park1. Up to date, two mutations in the gene
(A53T, A30P) identified and causes a dominantly-inherited form of familial PD
(Goedert et al., 2013; Krüger et al., 1998). This model generates a transgenic mice with
typical behavioral and motor phenotype, but with no effect on DA neurons (Abeliovich
et al., 2000; Thomas et al., 2011). Conversely, studies that have been done in
Drosophila with a mutant α-syn gene, showed a reduced TH expression with a
significant dopaminergic nigral cell loss, and inclusion bodies (Feany and Bender,
2000). With all these controversies, hence the role of α-syn gene is still to be
determined.
1.5.2.2 LRKK2
LRRK2 (leucine rich repeat kinase 2) gene is localized to membranes.
Mutation in LRKK2 causes an autosomal dominant inherited form of PD (Healy et al.,
2008). Several mutations have been identified (Rudenko and Cookson, 2014), but all
with no considerable disruption of SN dopaminergic neurons (Wang et al., 2008).
1.5.2.3 PINK1
PINK1 (phosphatase and tensin homolog- induced novel kinase 1) gene is
localized to the mitochondria, causing autosomal recessive PD, and it’s also called
PARK6 (Scarffe et al., 2014). In this form of mutation there is an age-dependent

66
moderate loss of dopaminergic neurons which is associated with overexpression of α-
syn (Oliveras-Salvá et al., 2014). This model shares a common phenotypes similarity
with those of Parkin KO and DJ-1 KO mice.
1.5.2.4 Parkin
Parkin is an autosomal recessive mutation, accounting for 50% of familial
cases of the disease and 20% of the young onset PD cases (Lücking et al., 2000;
Periquet et al., 2003). Parkin KO mice show reduced DA level with minimal or no
behavioral abnormalities (Itier et al., 2003; Kitada et al., 2009). One of this gene
mutations; Parkin-Q311X-DAT-BAC mice displays age-dependent motor impairment
along with loss of dopaminergic terminals in striatum and degeneration of SN cell
bodies (Lu et al., 2009).
1.5.2.5 DJ-1
DJ-1 mutation is an autosomal recessive and early-onset for the disease
(Puschmann, 2013). This form of genetic mutation results in motor and behavioral
abnormalities, reduced DA level, but no effect on dopaminergic neuronal cells in SN
(Goldberg et al., 2005; Kim et al., 2005).

67
1.6 Fundamental Methods of Assessing Structure and Function of Nigrostriatal
Pathway
Assessment of neuropathological process may vary from one study to another.
However, when using neurotoxic model, and planning to investigate
neurodegeneration or neuroprotection, like in our case- it is essential to study
nigrostriatal pathway integrity and function.
1.6.1 Dopaminergic Neurons in the Substantia Nigra Pars Compacta
Substantia nigra pars compacta (SNpc) is a very rich area in dopaminergic neurons,
its estimated that a mouse brain contains around 8000 ∼ 14,000 dopaminergic neurons,
and this is variable from one strain to another (Zaborszky and Vadasz, 2001). The
density of dopaminergic neurons is not distributed homogenously in SN being denser
in rostral region. In Figure 31, coronal section of SN from caudal to rostral regions
show variable distribution of the cells. Therefore, to sample the populations from all
regions of SN we should choose an unbiased quantification method ensuring sampling
of all regions at systemic section intervals. Currently, the gold standard method is to
use unbiased stereological cell counting with an optical fractionator system (West et
al., 1996, 1993, 1991).
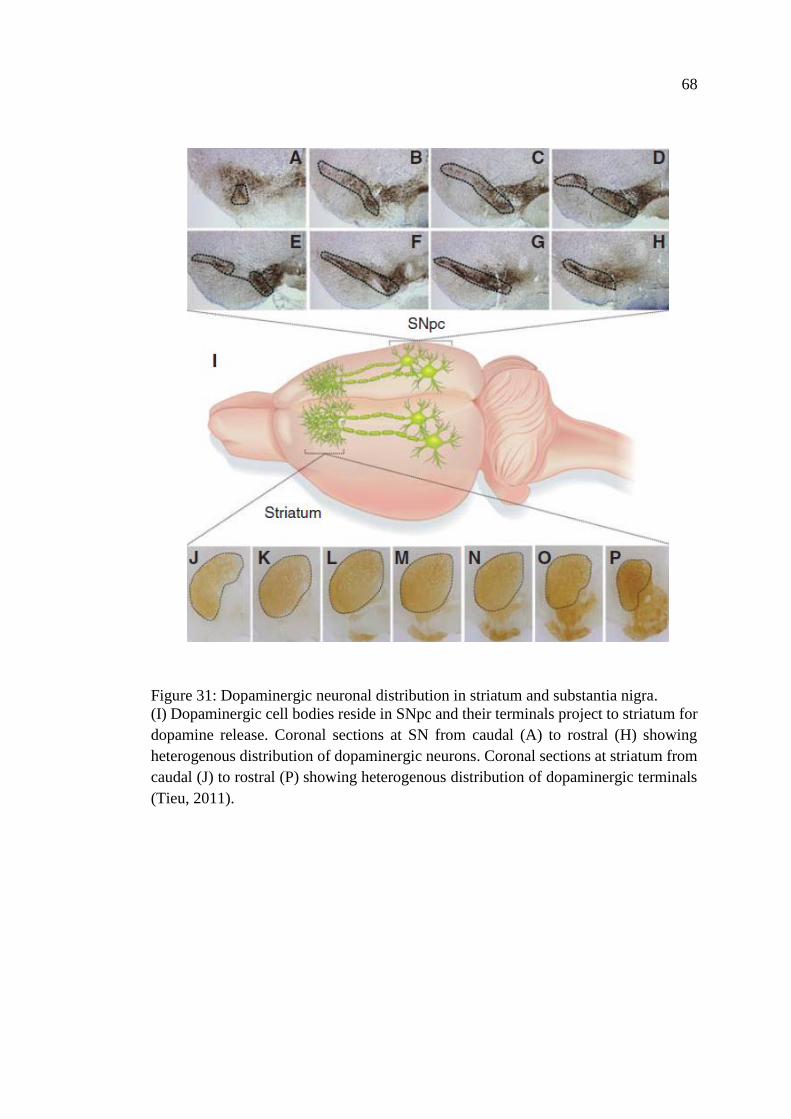
68
Figure 31: Dopaminergic neuronal distribution in striatum and substantia nigra.
(I) Dopaminergic cell bodies reside in SNpc and their terminals project to striatum for
dopamine release. Coronal sections at SN from caudal (A) to rostral (H) showing
heterogenous distribution of dopaminergic neurons. Coronal sections at striatum from
caudal (J) to rostral (P) showing heterogenous distribution of dopaminergic terminals
(Tieu, 2011).

69
1.6.2 Dopaminergic Terminals in the Striatum
The density of dopaminergic terminals in the striatum correlates with the cell
bodies they originate from in the SN, hence, the distribution of striatal terminals would
vary moving from caudal to rostral (Bockaert et al., 1976; Widmann and Sperk, 1986).
Immunohistochemical reaction of TH activity is a good measure of quantification.
Another way of assessing dopamine terminals level is immunoblotting technique using
TH and DA markers.
1.6.3 Striatal Dopamine
All the above-mentioned forms of assessments are assessing the structural integrity
of nigrostriatal pathway, its important also to assess the function of this pathway. This
can be done by measuring striatal dopamine release and its reflection on motor activity
and behavior. Measuring of striatal dopamine release can be performed by using high-
performance liquid chromatography (HPLC) with an electrochemical detector.
1.6.4 Lewy Body Aggregates
Lewy bodies are considered the pathological hallmark in PD. There are several
methods for detection of this mis-/or unfolded protein aggregates. An easy straight
forward method is to use an antibody against α-synuclein (Beach et al., 2008).
1.6.5 Behavioral/Motor Assessment
As we mentioned earlier, dopamine depletion is reflected on animals’ behavior.
There are several behavioral tests to assess the locomotor activities of animal models,
such as; rotarod, drug-induced rotation, limb use asymmetry, forelimb placing test,
swim test, adhesive removal test, and pole test (Meredith and Kang, 2006).

70
Chapter 2: Aims and Objectives
The main goal of in this study was to investigate the effect of curcumin application
on the functional properties of human neuronal 7 nicotinic acetylcholine receptors
(nAChRs). This work was divided into two main parts:
2.1 In-vitro Electrophysiological Study
o In the first part of the study, human 7-nAChR were expressed homomerically in a
Xenopus oocyte expression system to investigate:
1. The effect of curcumin on the functional properties of 7-nAChRs.
2. Determination of the voltage-dependence of curcumin action on the α7-
nAChRs, and the role of endogenously expressed Ca2+ activated Chloride
channel and the role of pertussis toxin sensitive G proteins in mediating the
effect of curcumin on the α7-nAChR.
3. The effect of curcumin on other nAChRs subunits & ligand gated ion channels.
4. The effect of other curcumin analogues and metabolites on 7-nAChRs.
5. The binding affinity of curcumin and its derivatives with 7-nAChRs.
2.2 In-vivo Study
o To establish the neuroprotective effect of curcumin in animal model of Parkinson’s
disease by investigating:
1. Improvement of abnormal motor behavior of the animals.
2. Effect of curcumin on the survival of dopaminergic striatal nerve terminals.
3. Effect of curcumin on the survival of dopaminergic neurons in SNpc.
71
o To test whether the neuroprotective effect of curcumin in animal model of PD is
mediated through an α7-nAChRs.

72
Chapter 3: Materials and Methods
3.1 Electrophysiological In-vitro Study
Materials
3.1.1 Female Xenopus Oocytes
Mature female African clawed frogs (Xenopus laevis) were purchased from
Xenopus Express, Haute-Loire, France (Figure 32). They were housed in in a water-
tank (dimensions; 32 cm width, 130 cm length, and 66 cm height) filled with
dechlorinated water. The room temperature was maintained at 19-21°C with a 12/12-
hour light/dark cycle. Frogs were fed twice a week with food pellets, supplied by
Xenopus Express, France. Tank-water was changed twice a week. Animal care and
handling experiments conducted in this study were in accordance with institutional
guidelines and approved by the Animal Ethic Committee of the CMHS/UAEU.
Figure 32: An adult female Xenopus laevis (Professor Murat Oz’s laboratory)
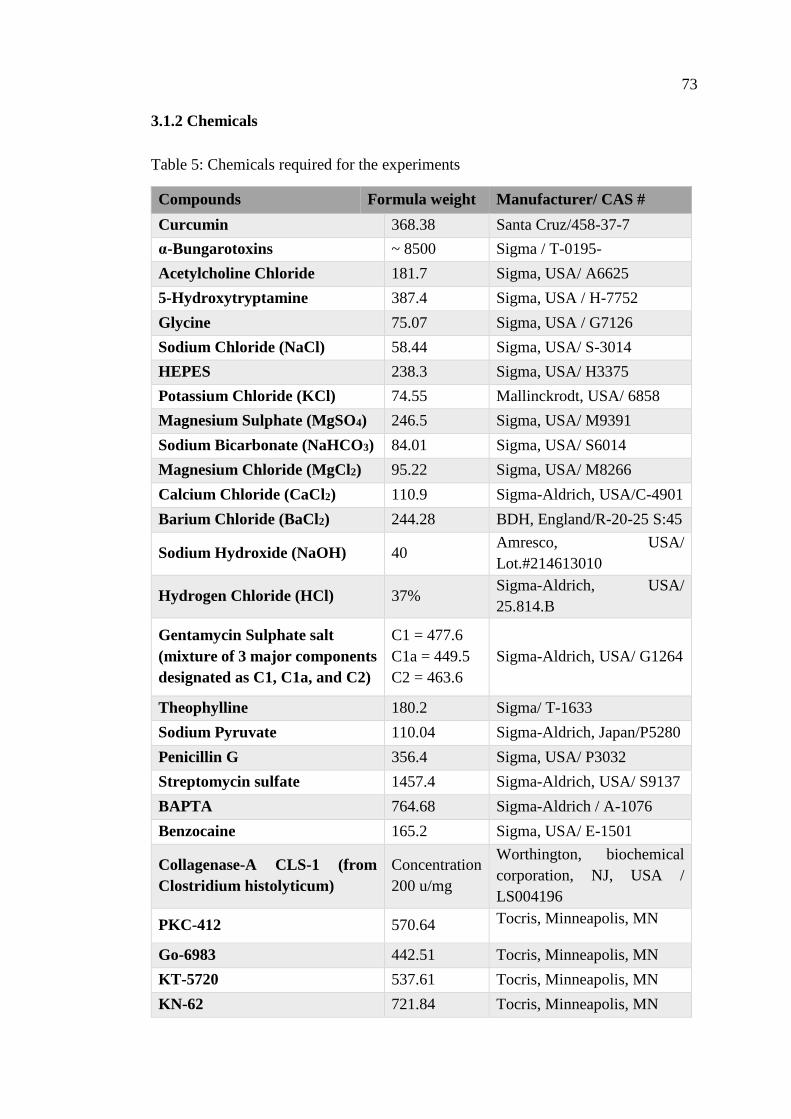
73
3.1.2 Chemicals
Table 5: Chemicals required for the experiments
Compounds Formula weight Manufacturer/ CAS #
Curcumin 368.38 Santa Cruz/458-37-7
α-Bungarotoxins ~ 8500 Sigma / T-0195-
Acetylcholine Chloride 181.7 Sigma, USA/ A6625
5-Hydroxytryptamine 387.4 Sigma, USA / H-7752
Glycine 75.07 Sigma, USA / G7126
Sodium Chloride (NaCl) 58.44 Sigma, USA/ S-3014
HEPES 238.3 Sigma, USA/ H3375
Potassium Chloride (KCl) 74.55 Mallinckrodt, USA/ 6858
Magnesium Sulphate (MgSO4) 246.5 Sigma, USA/ M9391
Sodium Bicarbonate (NaHCO3) 84.01 Sigma, USA/ S6014
Magnesium Chloride (MgCl2) 95.22 Sigma, USA/ M8266
Calcium Chloride (CaCl2) 110.9 Sigma-Aldrich, USA/C-4901
Barium Chloride (BaCl2) 244.28 BDH, England/R-20-25 S:45
Sodium Hydroxide (NaOH) 40 Amresco, USA/
Lot.#214613010
Hydrogen Chloride (HCl) 37% Sigma-Aldrich, USA/
25.814.B
Gentamycin Sulphate salt
(mixture of 3 major components
designated as C1, C1a, and C2)
C1 = 477.6
C1a = 449.5
C2 = 463.6
Sigma-Aldrich, USA/ G1264
Theophylline 180.2 Sigma/ T-1633
Sodium Pyruvate 110.04 Sigma-Aldrich, Japan/P5280
Penicillin G 356.4 Sigma, USA/ P3032
Streptomycin sulfate 1457.4 Sigma-Aldrich, USA/ S9137
BAPTA 764.68 Sigma-Aldrich / A-1076
Benzocaine 165.2 Sigma, USA/ E-1501
Collagenase-A CLS-1 (from
Clostridium histolyticum)
Concentration
200 u/mg
Worthington, biochemical
corporation, NJ, USA /
LS004196
PKC-412 570.64 Tocris, Minneapolis, MN
Go-6983 442.51 Tocris, Minneapolis, MN
KT-5720 537.61 Tocris, Minneapolis, MN
KN-62 721.84 Tocris, Minneapolis, MN
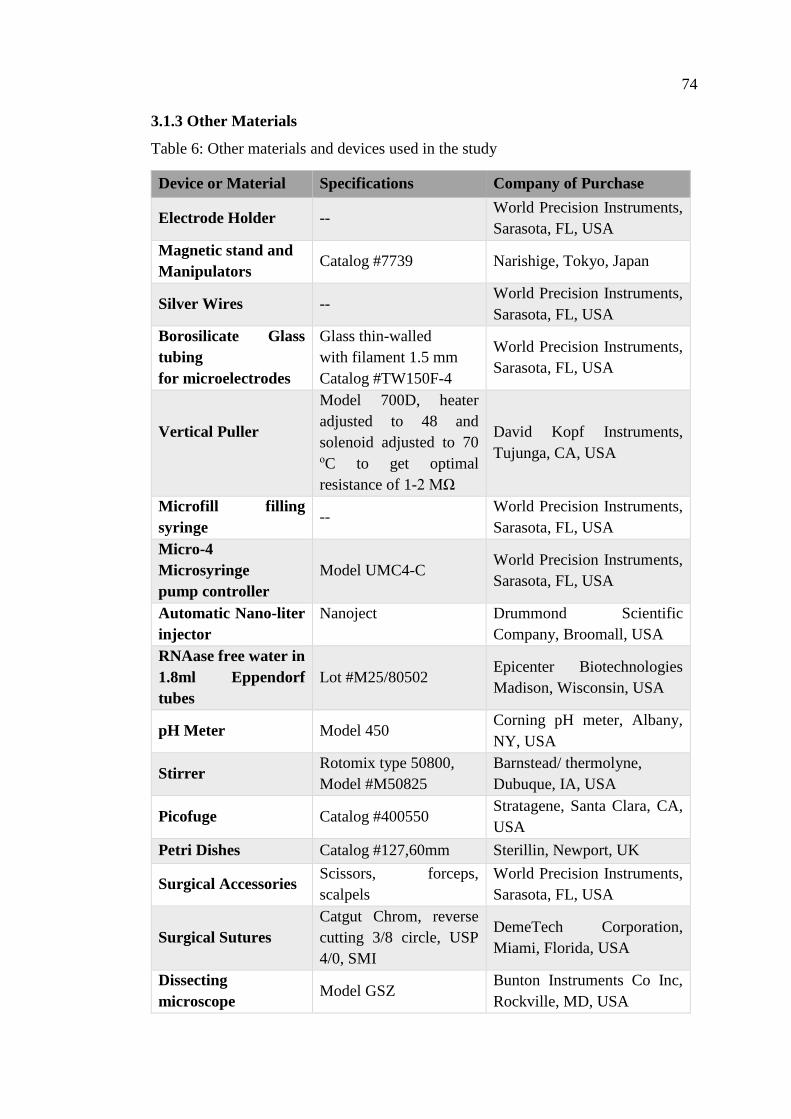
74
3.1.3 Other Materials
Table 6: Other materials and devices used in the study
Device or Material Specifications Company of Purchase
Electrode Holder -- World Precision Instruments,
Sarasota, FL, USA
Magnetic stand and
Manipulators Catalog #7739 Narishige, Tokyo, Japan
Silver Wires -- World Precision Instruments,
Sarasota, FL, USA
Borosilicate Glass
tubing
for microelectrodes
Glass thin-walled
with filament 1.5 mm
Catalog #TW150F-4
World Precision Instruments,
Sarasota, FL, USA
Vertical Puller
Model 700D, heater
adjusted to 48 and
solenoid adjusted to 70 οC to get optimal
resistance of 1-2 MΩ
David Kopf Instruments,
Tujunga, CA, USA
Microfill filling
syringe --
World Precision Instruments,
Sarasota, FL, USA
Micro-4
Microsyringe
pump controller
Model UMC4-C World Precision Instruments,
Sarasota, FL, USA
Automatic Nano-liter
injector
Nanoject
Drummond Scientific
Company, Broomall, USA
RNAase free water in
1.8ml Eppendorf
tubes
Lot #M25/80502 Epicenter Biotechnologies
Madison, Wisconsin, USA
pH Meter Model 450 Corning pH meter, Albany,
NY, USA
Stirrer Rotomix type 50800,
Model #M50825
Barnstead/ thermolyne,
Dubuque, IA, USA
Picofuge Catalog #400550 Stratagene, Santa Clara, CA,
USA
Petri Dishes Catalog #127,60mm Sterillin, Newport, UK
Surgical Accessories Scissors, forceps,
scalpels
World Precision Instruments,
Sarasota, FL, USA
Surgical Sutures
Catgut Chrom, reverse
cutting 3/8 circle, USP
4/0, SMI
DemeTech Corporation,
Miami, Florida, USA
Dissecting
microscope Model GSZ
Bunton Instruments Co Inc,
Rockville, MD, USA

75
3.1.4 Experimental Setup
The experimental setup for electrophysiological recordings using the two electrode
voltage-clamp is shown in Figure 33 and 34. Two-electrode voltage clamp (TEVC)
technique was applied using a GeneClamp-500B amplifier (Axon Instruments,
Molecular Devices, Inc., Sunnyvale, CA, USA), as described previously (Ashoor et
al., 2013) recording setup included magnetic holding devices (Kanetec USA
Corporation, Bensenville, IL, USA), two manual micromanipulators (M33;
Märzhäuser, Wetzlar, Germany) and head-stages for voltage (HS-2A Headstage, Gain
1 MG, Axon Instruments, Molecular Devices, Inc., Sunnyvale, CA, USA) and current
(HS-2A Headstage, Gain 10MG) were attached to the manipulators. The two glass
electrodes were inserted in electrode holders and then connected to the head-stages.
Micromanipulators were used to control the electrodes and impale the oocyte.
The perfusion apparatus consisted of perfusion tubes and bottles containing
extracellular solutions connected to the recording bath by silicon tubing (Cole Parmer
Instrument Company, I.D. 1/16 inch, O.D. 1/8 inch and WALL1/32, Vernon Hills,
Illinois, USA). Flow rate of perfusion was set to 3 - 5 ml/minute. A multichannel
perfusion system was used for drug applications included tubing (C-Flex tubing, Cole-
Parmer Instrument Company, I.D. 1/32 inch, O.D. 3/32 inch and WALL1/32 inch,
Vernon Hills, Illinois, USA), 50 mL glass syringes, and coupling devices. The drug
application system was based on gravity flow by means of a micropipette that was set
at a distance of about 2-3 mm from the oocyte position in the perfusion
chamber/recording chamber (Warner Instruments LLC, Hamden, CT, UK) designed
for placing oocyte to be impaled with the microelectrodes (Figure 34). The perfusion

76
solution in the recording chamber was removed by using a glass suction tube
connected to an adjustable vacuum source and collected in the waste tank.
Figure 33: Two-electrode voltage-clamp (TEVC) recording set-up from Xenopus
oocytes.
The oocyte was placed in the recording chamber and continuously bathed with
physiological solution. The oocyte membrane was penetrated with two
microelectrodes one for voltage sensing and the other for current injection. Drugs and
compounds were applied using gravity-based multichannel application system, at the
time of drug application the perfusion was stopped. TEVC is achieved using
Genclamp500 amplifier interfaced to a PC computer equipped with
electrophysiological software for data acquisition (Professor Murat Oz’s laboratory).

77
Figure 34: The oocyte impaled with two microelectrodes.
Illustration of the plastic chamber. The oocyte was placed in the recording chamber
and perfused continuously during the experiment (Professor Murat Oz’s Laboratory).
A fiber optic light source was used for illumination of the recording chamber (Fiber
Lite, High Intensity Illuminator Series 180, Dolan-Jenner Industries Inc. Boxborough,
MA, USA). A Low-power stereo-dissection microscope was used for visual
observation of the recording chamber (Olympus, Tokyo, Japan, SZ-STB1, 100 AL0.5
X, WD186). Computer set up for data acquisition consisted of a Compaq computer,
(Compaq Corporation, Wynyard, UK) and analog-digital converter, BNC 2081
(National Instruments, Austin Texas, USA).

78
Methods
3.1.5 Preparation of Required Solutions
3.1.5.1 Modified Barth’s Solution
Modified Barth’s Solution (MBS) was used during oocytes isolation process. The
composition of MBS is shown in table below:
a) Calcium free MBS solution:
Table 7: Calcium free MBS solution composition
Compound Concentration
(mM)
1x
(Weight in grams)
10x
(Weight in grams)
NaCl 88 5.14 51.4
HEPES 10 2.38 23.8
NaHCO3 2.1 0.2 2
KCl 1 0.075 0.75
MgSO4 0.8 0.2 2
The above components were dissolved in distilled water to get total volume of 1 L and
the pH was adjusted to 7.5 using NaOH.
b) Calcium containing MBS solution:
Similar to previous composition, with the addition of 0.22 g and 2.2 g of CaCl2
(2mM) to make stock solutions of 1x and 10x respectively. All compounds were
dissolved in distilled water to get total volume of 1 L and the pH was adjusted to 7.5
3.1.5.2 Oocyte Storage Solution
The storage solution was prepared as following: 1) 100 ml of 10x Calcium
containing MBS solution; 2) 900 ml Distilled water; 3) Antibiotic materials shown in
(Table 8) were dissolved in the previous solution; 5) pH was adjusted to 7.5; 6) The
mixture was filtered and stored in a sterile container.
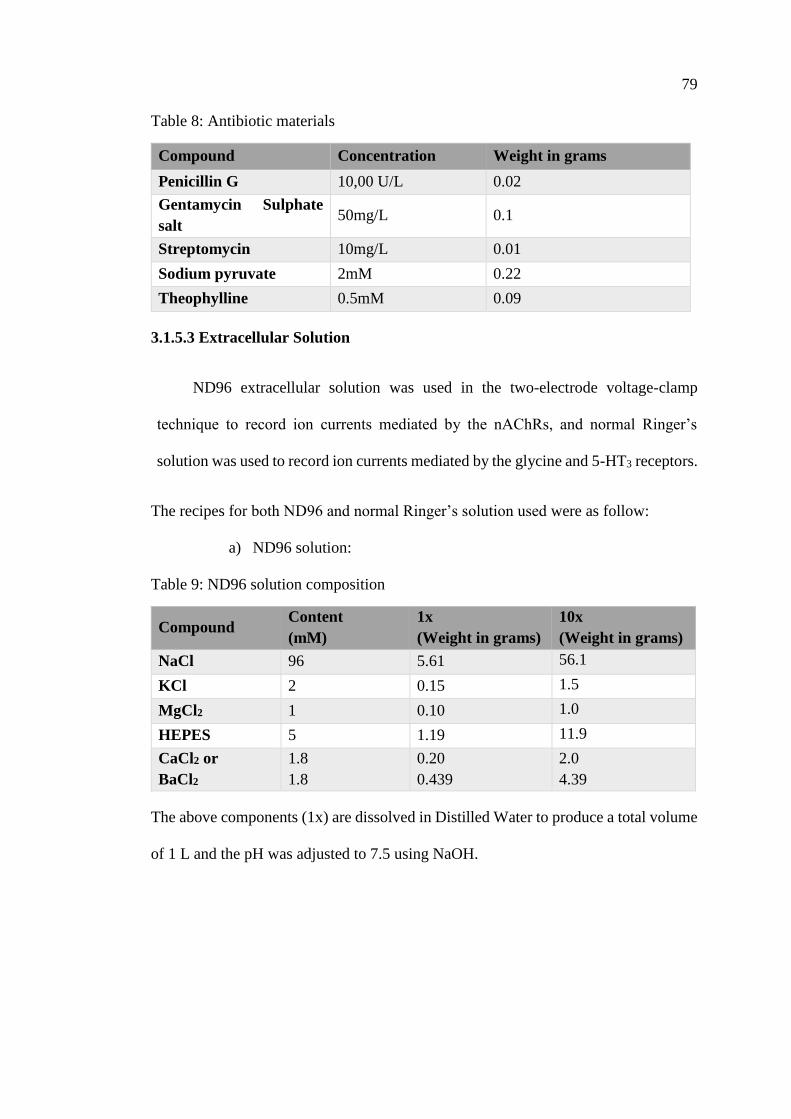
79
Table 8: Antibiotic materials
Compound Concentration Weight in grams
Penicillin G 10,00 U/L 0.02
Gentamycin Sulphate
salt 50mg/L 0.1
Streptomycin 10mg/L 0.01
Sodium pyruvate 2mM 0.22
Theophylline 0.5mM 0.09
3.1.5.3 Extracellular Solution
ND96 extracellular solution was used in the two-electrode voltage-clamp
technique to record ion currents mediated by the nAChRs, and normal Ringer’s
solution was used to record ion currents mediated by the glycine and 5-HT3 receptors.
The recipes for both ND96 and normal Ringer’s solution used were as follow:
a) ND96 solution:
Table 9: ND96 solution composition
Compound Content
(mM)
1x
(Weight in grams)
10x
(Weight in grams)
NaCl 96 5.61 56.1
KCl 2 0.15 1.5
MgCl2 1 0.10 1.0
HEPES 5 1.19 11.9
CaCl2 or
BaCl2
1.8
1.8
0.20
0.439
2.0
4.39
The above components (1x) are dissolved in Distilled Water to produce a total volume
of 1 L and the pH was adjusted to 7.5 using NaOH.
80
b) Normal Ringer’s solution:
Table 10: Normal Ringer’s solution composition
Compound Content
(mM)
1x
(Weight in grams)
10x
(Weight in grams)
NaCl 115 6.72 67.2
KCl 1 0.074 0.74
CaCl2 1.8 0.199 1.99
HEPES 10 2.38 23.8
The above components (1x) were dissolved in distilled water to get total volume of 1
L and the pH was adjusted to 7.4 using NaOH.
3.1.6 Drug Application
Stock solutions of the test compounds were prepared in ND96 solution when
recording ion currents mediated by the nAChRs (or in normal Ringer’s solution when
recording ion currents mediated by glycine and serotonin receptors) using the
following formula:
Weight (mg) = (MW) x (Volume (L)) x (concentration (mM))
Further dilutions were prepared using the Charles equation:
C1 x V1 = C2 x V2
Where,
C1 = concentration of stock solution
V1 = volume of stock solution to be used
C2 = desired concentration to be prepared
V2 = desired volume to be prepared
Stock solutions and required dilutions were prepared freshly before
starting the experiments.

81
3.1.7 Isolation and Maintenance of Oocyte from Xenopus Laevis
Xenopus laevis female frogs were anesthetized in 1L of 0.03% w/v benzocaine
solution, prepared by dissolving 300 mg of ethyl p-aminobenzoate in 15 mL of 70 %
ethanol and then adding that to 1L of cold tap water. The end point of anesthesia was
determined by failure to respond to noxious stimuli induced by pinching of the lower
limbs. Under described conditions, usually 5-10 minutes to achieve full anesthesia, the
anesthetized frog was placed on crushed ice covered with a wet paper towel to avoid
skin from drying out during surgery while maintaining low core body temperature
during surgery. A small incision of about 1.5 cm length was made through the
epidermal layer and in the inner muscular layer of the lower abdominal area slightly
to the left or right of the midline, and a similar cut. Using sterilized forceps, one to
two ovarian lobes (small clumps of oocytes) were removed and placed in a petri dish
containing Ca2+- free MBS (Figure 35).
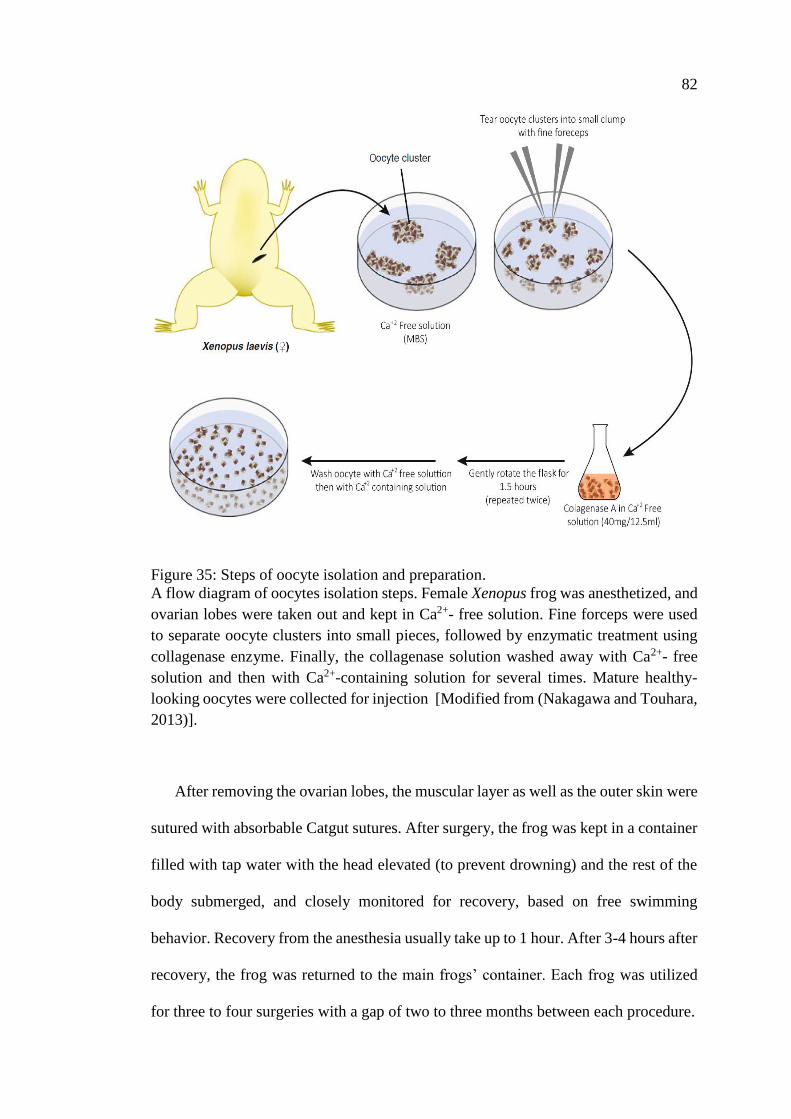
82
Figure 35: Steps of oocyte isolation and preparation.
A flow diagram of oocytes isolation steps. Female Xenopus frog was anesthetized, and
ovarian lobes were taken out and kept in Ca2+- free solution. Fine forceps were used
to separate oocyte clusters into small pieces, followed by enzymatic treatment using
collagenase enzyme. Finally, the collagenase solution washed away with Ca2+- free
solution and then with Ca2+-containing solution for several times. Mature healthy-
looking oocytes were collected for injection [Modified from (Nakagawa and Touhara,
2013)].
After removing the ovarian lobes, the muscular layer as well as the outer skin were
sutured with absorbable Catgut sutures. After surgery, the frog was kept in a container
filled with tap water with the head elevated (to prevent drowning) and the rest of the
body submerged, and closely monitored for recovery, based on free swimming
behavior. Recovery from the anesthesia usually take up to 1 hour. After 3-4 hours after
recovery, the frog was returned to the main frogs’ container. Each frog was utilized
for three to four surgeries with a gap of two to three months between each procedure.

83
3.1.8 Oocyte Preparation
The preparation of oocytes was performed according to procedures described earlier
(Oz et al., 2003). Briefly, using fine forceps, the inner ovarian epithelium, theca, and
follicular layers were removed as much as possible to produce smaller clusters of
oocytes (Figure 36).
The resulting oocyte clusters were then treated with collagenase A solution
(collagenase enzyme solution was prepared by dissolving 80 mg of Collagenase, type
A, in 25 ml of Ca2+ Free MBS solution). The oocyte clusters were incubated in a small
conical flask containing 12.5 ml collagenase solution with constant stirring (60-80
rotations/minute) at room temperature for 1.5 hour. After that, collagenase solution
was replaced with fresh collagenase solution 12.5 ml and kept in stirring for another
1.5 hour. Finally, oocytes were washed gently; 5 times using Ca2+ Free MBS, then 5
times with Ca2+ containing MBS solution. Subsequently, oocytes were transferred to
a petri dish filled with Ca2+ containing MBS for oocyte selection.
Only healthy-looking mature oocytes (stage V-VI) were selected under a dissecting
microscope (Bunton Instruments Co Inc., Model GSZ, Rockville, MD, U.S.A.). Stage
V and VI oocytes characterized by larger size with 1.0 mm - 1.2 mm diameter, rounded
shape, and clear dark brown animal pole and yellow vegetal pole divisions (Figures 36
& 37) were selected. The oocytes were subsequently maintained in MBS at 18 °C and
used within 5 - 7 days.
Throughout the electrophysiological experiments, we employed a Xenopus oocyte
expression system for the following reasons: 1) methods for harvesting and
maintaining oocytes are well established and straightforward. As a result, high
numbers of cells with desirable expression levels were routinely available. 2) the

84
oocytes are freshly derived from living frogs, thus they can be treated as primary cell
culture. 3) the two-electrode voltage clamp technique is a relatively less laborious and
a high-yield electrophysiological assay system. 4) the two-electrode voltage clamp
technique is stable for many hours, making it an ideal method for experiments
requiring long-recording times.
Figure 36: Frog’s ovarian lobe.
(A) An ovarian lobe, containing oocytes at different developmental stages. (B)
Separating oocytes by removal of epithelial and follicular layers manually with fine
forceps. Isolated stage V and VI oocyte of Xenopus laevis after collagenase treatment.
A
B

85
Figure 37: Stages of oocyte development.
Oocytes vary in size according to maturation stage. Oocyte size range from 100 – 1300
µM (Wozniak et al., 2018).
3.1.9 Synthesis of cRNA
The cDNA clone of human α7-nAChR was kindly provided by Dr. J. Lindstorm
(University of Pennsylvania, PA, U.S.A.). Capped cRNA transcripts were synthesized
in vitro using a mMESSAGE mMESSAGE kit (Ambion, Austin, TX, U.S.A.) and
analyzed on 1.2% formaldehyde agarose gel to check the size and quality of the
transcripts (Figure 38).
Human α7-nAChR mRNA was prepared by in vitro transcription and confirmed
by gel analysis. Restriction enzyme (Xbal) was used to digest the cDNA of human α7
nAChR, and it was cleaned by the Qiagen kit. Linearized plasmid cDNA was
transcribed in vitro by SP6 RNA polymerase to produce α7-nACh receptor RNA using
a mMESSAGE mMACHINE kit. This RNA was cleaned and purified by
phenol:chloroform extraction and ethanol precipitation. The quantity of RNA was
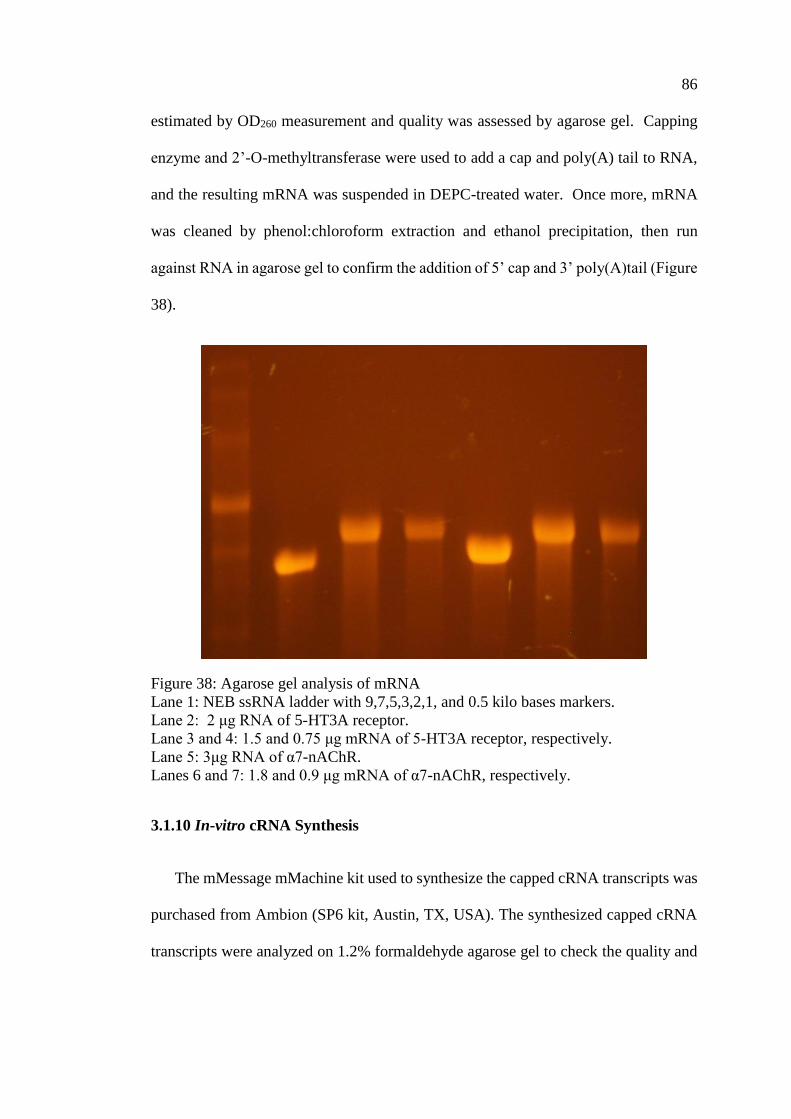
86
estimated by OD260 measurement and quality was assessed by agarose gel. Capping
enzyme and 2’-O-methyltransferase were used to add a cap and poly(A) tail to RNA,
and the resulting mRNA was suspended in DEPC-treated water. Once more, mRNA
was cleaned by phenol:chloroform extraction and ethanol precipitation, then run
against RNA in agarose gel to confirm the addition of 5’ cap and 3’ poly(A)tail (Figure
38).
Figure 38: Agarose gel analysis of mRNA
Lane 1: NEB ssRNA ladder with 9,7,5,3,2,1, and 0.5 kilo bases markers.
Lane 2: 2 μg RNA of 5-HT3A receptor.
Lane 3 and 4: 1.5 and 0.75 μg mRNA of 5-HT3A receptor, respectively.
Lane 5: 3μg RNA of α7-nAChR.
Lanes 6 and 7: 1.8 and 0.9 μg mRNA of α7-nAChR, respectively.
3.1.10 In-vitro cRNA Synthesis
The mMessage mMachine kit used to synthesize the capped cRNA transcripts was
purchased from Ambion (SP6 kit, Austin, TX, USA). The synthesized capped cRNA
transcripts were analyzed on 1.2% formaldehyde agarose gel to check the quality and

87
size of the transcripts. The cDNA clones of human α7-nAChR was provided by Dr. J.
Lindstorm (University of Pennsylvania, PAM, USA).
3.1.11 Microinjection of cRNA into Oocytes
The mRNA concentration of synthesized human α7-nAChRs and other subunits
used in this study are shown in (Table 11). They were stored as 1μl aliquots in freezer
at -80o C. Once the oocytes were prepared and sorted, only one RNA aliquot was
transferred in an ice bucket to the laboratory. To maintain a RNase free environment
while working with cRNA, the tube was centrifuged using a microcentrifuge at 1200
rpm for 1- 3 min, and then by using a sterile pipette and RNase/DNase free pipette tips
(Denville Scientific Inc., Metuchen, NJ, USA), and resulting cRNA pellet was diluted
to a final concentration of 10 ng/50 nl in RNase-/DNase- free water. Only 3 to 4 μL of
diluted cRNA was used for each batch of oocytes, and the remaining cRNA was
returned to the freezer. A vertical puller (Model 700 D, David Kopf Instruments
Tujunga, CA, USA) was used for making glass microelectrodes with fine needle
shaped tip from autoclaved glass capillaries (World Precision Instruments, Sarasota,
FL, USA). The tip of each glass microelectrode was broken by applying gentle
pressure using a fine pair of forceps (Fine Science Tools Inc., Vancouver, Canada)
under a dissecting microscope (Bunton Instruments Co., Rockville, MD, USA).
The glass needle was backfilled with mineral oil (Sigma, St. Louis, MO, USA) using
a 1 ml glass syringe. Then the glass needle was fitted into a microdispenser connected
to the micromanipulator (Figures 39 & 40).

88
Table 11: The initial concentration of all subunits
Receptor Subunit Main stock conc. (µg/µl)
nACh receptors
α7 3.7
α3 2.43
α4 2.15
β2 1.66
β4 1.71
Glycine receptor
α1 2.18
α2 1.678
α3 1.595
5-HT3 receptor A 2.1
Subsequently, 3 to 4 μL of diluted cRNA or distilled water was dropped on the
center of a mineral oil drop placed on parafilm (American National Can Co.,
Greenwich, CT, USA). By focusing on the drop under the dissecting microscope, the
tip of the glass needle was placed at the center of the droplet, and the aqueous phase
was carefully withdrawn into the glass needle using the withdrawal option on the
microdispenser controller. After loading the sample, the sorted oocytes were arranged
in a U-shaped pattern in a small petri dish, containing MBS, with mesh-bottom to hold
the oocytes in place during the microinjection procedure. Each oocyte was impaled by
the glass needle and gently injected with 50 nL of cRNA solution or distilled water
using the microinjector (driven by a Micro-4, micro syringe pump controller).

89
Figure 39: Microelectrode set-up for cRNA injection.
Schematic presentation of microinjector set-up. The glass microelectrode backfilled
with cRNA solution for oocyte injection (Bianchi, 2006; Nakagawa and Touhara,
2013).
Figure 40: cRNA injection set-up (Professor Murat Oz’s Laboratory)
Low-power stereo micro-scope
Volume control
Microinjecto

90
Following the injection, the oocytes were stored in 25 ml petri dishes filled with
oocyte storage solution and incubated at 18ºC. In initial experiments, the oocytes were
divided into two groups; oocytes that were injected with cRNA encoding the human
α7-nAChR, and oocytes that were injected with distilled water (control). Xenopus
oocytes were used in the electrophysiology study 2-3 days after microinjection to allow
maximal expression of receptors. Healthy oocytes were transferred to fresh dishes with
new storage solution daily. A bottle of storage solution was stored in the incubator
with the oocytes. The oocytes were used for about 7-10 days.
3.1.12 Two Electrode Voltage Clamp
The voltage clamp technique is a method that allows ion flow across the cell
membrane to be measured as an electric current while the transmembrane potential is
held constant (clamped) with a feedback amplifier. Functional properties of ion
channels expressed in Xenopus oocytes can be studied effectively using the two-
microelectrode voltage clamp (TEVC). In TEVC, two intracellular microelectrodes
were employed, a voltage sensor electrode and a current injection electrode to maintain
the transmembrane potential at the desired/command voltage. The voltage electrode
was connected to the membrane potential amplifier to measure the membrane potential
(Vm). The membrane potential as measured by the voltage-sensing electrode and a high
input impedance amplifier was compared with a command voltage, and the difference
is brought to zero by a feedback amplifier. The injected current by the amplifier
provides a measure of the total membrane current (Im).

91
Figure 41: Schematic of glass microelectrode assembly.
Glass microelectrode mounted on electrode holder containing a coated silver wire
(AgCl2) to allow signal transmission. Microelectrode was filled with electrolyte
solution; potassium chloride (KCl). Glass microelectrode was used to measure the
intracellular voltage difference.
The experimental setup for TEVC is depicted in Figure 33. At the beginning of the
experiments, perfusion containers and application systems were filled with the
appropriate solutions and allowed to run through the connected tubes. For each
experiment, a single oocyte was placed in the recording chamber and continuously
perfused with ND96 solution at a rate of 3 - 5 ml/minute. The oocyte was impaled
with two glass microelectrodes prepared using a vertical microelectrode puller (heater
and solenoid values were adjusted to a setting of 50 and 70, respectively; David Kopf
Instruments Tujunga, CA, USA) and filled with 3 M KCl solution (Figure 41). Using
the micromanipulator, the tip of the two electrodes were dipped into the bath solution,
and the voltage of both electrodes was adjusted to 0 mV. Then, the animal pole (the
dark pole) of the oocyte was impaled with the two microelectrodes (Figure 42). Pipette
resistances ranged from 0.5-1.8 MΩ. The tip of the electrode was visualized using the
microscope. After both electrodes were inserted, the amplifier was set in voltage-
clamp mode, and the clamp-voltage was set on -70 mV. Drugs were applied by a
Pin connector
KCl filled microelectrode
KCl fluid
Silver wire
Ag/AgCl pellet
Glass microelectrode

92
gravity-based multichannel application system via a micropipette positioned about 2-
3 mm from the impaled oocyte. Perfusion of the ND96 was stopped immediately
before the drug application and started immediately after drug application.
Figure 42: Schematic illustration of two-electrode voltage clamp setup using Xenopus
oocytes.
The membrane of the oocyte at the animal pole was penetrated with two
microelectrodes; one for voltage sensing and the other for current injection, while the
transmembrane potential was held constant with the feedback amplifier [Modified
from (Nakagawa and Touhara, 2013)].
Throughout the experiment oocytes were voltage clamped at a holding potential of
-70 mV (command potential) using a Geneclamp 500 amplifier (Axon Instruments,
Molecular Devices, Inc, Sunnyvale, CA, USA). Current responses were recorded and
stored digitally for further analysis using Strathclyde Electrophysiology Software,
WinWCP V4.0.8/ WinEDR V3.7.1 (University of Strathclyde, Glasgow, UK).

93
3.1.13 Parameters Tested by Electrophysiological Recording
The oocytes were voltage-clamped at a holding potential of -70 mV using a
GeneClamp-500 amplifier (Axon Instruments, Molecular Devices, Inc., Sunnyvale,
CA, USA), and current response induced by application of 100 µM of acetylcholine
chloride (ACh) was recorded digitally on an IBM\PC.
3.1.13.1 Concentration Response Curve (EC50 determination)
For α7-nAChR, three to five recordings of ion current induced by ACh (100µM)
were measured with 5 min intervals of wash out with ND96 solution. The average of
stable readings was calculated and considered as control reading. After obtaining the
control readings, different concentrations of curcumin were routinely applied, and
100µM ACh solution applied at the end of 5 min intervals which also included same
concentration of curcumin. The current induced by ACh + curcumin application was
recorded, and the average of 2-3 readings were calculated to determine the effect of
curcumin. Thereafter, drug application was stopped, and the oocytes were washed with
ND96 alone to obtain the recovery readings (100 µM ACh alone).
Concentration-response data for each oocyte were normalized to the maximum
current produced with 100 µM ACh for that oocyte, and percentage of potentiation
was calculated by dividing the average of drugs-induced currents by the control values
obtained before drug application. Curcumin was used at varying concentrations to
construct dose response curves. For each concentration of curcumin, averages of 5-6
oocytes were used. The concentration of curcumin which produced a 60 % potentiation
of ACh-induced currents (IC50) was obtained by nonlinear curve-fitting and regression
fits (logistic equation) using computer statistical software v 8.5 (Origin Lab Corp.,
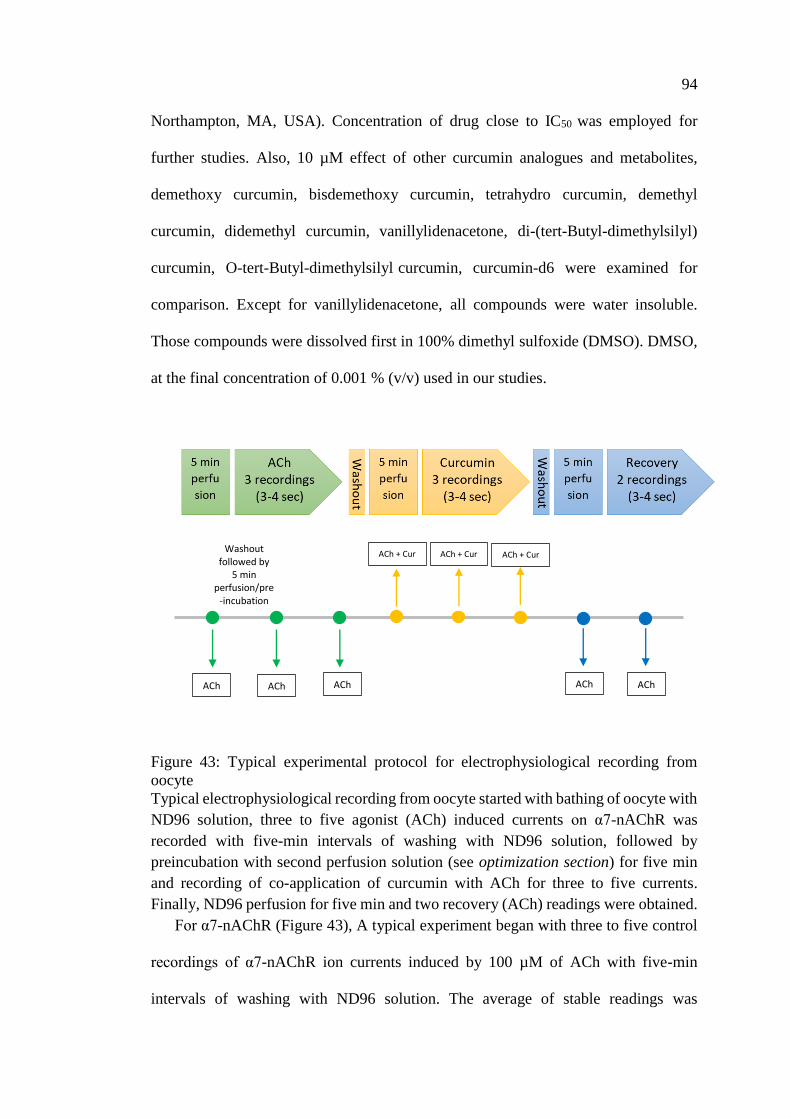
94
Northampton, MA, USA). Concentration of drug close to IC50 was employed for
further studies. Also, 10 µM effect of other curcumin analogues and metabolites,
demethoxy curcumin, bisdemethoxy curcumin, tetrahydro curcumin, demethyl
curcumin, didemethyl curcumin, vanillylidenacetone, di-(tert-Butyl-dimethylsilyl)
curcumin, O-tert-Butyl-dimethylsilyl curcumin, curcumin-d6 were examined for
comparison. Except for vanillylidenacetone, all compounds were water insoluble.
Those compounds were dissolved first in 100% dimethyl sulfoxide (DMSO). DMSO,
at the final concentration of 0.001 % (v/v) used in our studies.
Figure 43: Typical experimental protocol for electrophysiological recording from
oocyte
Typical electrophysiological recording from oocyte started with bathing of oocyte with
ND96 solution, three to five agonist (ACh) induced currents on α7-nAChR was
recorded with five-min intervals of washing with ND96 solution, followed by
preincubation with second perfusion solution (see optimization section) for five min
and recording of co-application of curcumin with ACh for three to five currents.
Finally, ND96 perfusion for five min and two recovery (ACh) readings were obtained.
For α7-nAChR (Figure 43), A typical experiment began with three to five control
recordings of α7-nAChR ion currents induced by 100 µM of ACh with five-min
intervals of washing with ND96 solution. The average of stable readings was
Washout followed by
5 min perfusion/pre
-incubation
ACh ACh ACh ACh ACh
ACh + Cur ACh + Cur ACh + Cur

95
calculated as the control value. Following the control recordings, the oocyte was
perfused for a total of 15 minutes with the selected concentration of tested compound;
Curcumin or any of its derivatives. The average three responses at the end of the 15
min drug application were calculated to determine the effect of the compound.
Subsequently, the application of drug was stopped, and the oocytes were washed with
ND96 alone to obtain the recovery readings (100 µM ACh alone).
3.1.13.2 Effect of Curcumin on α7-nAChR
Time-course of the onset of curcumin and the vehicle applications (0.01% DMSO)
on the maximal amplitudes of ACh-induced currents were investigated. Curcumin (10
µM) was co-applied in presence of 100 µM ACh for 5 min without any pre-application
(0 pre-application time), and then washed out with ND96 to obtain the recovery
readings (ACh alone). The time course of curcumin potentiation was further tested by
comparing the effect of varying the curcumin pre-application time on ACh-induced
currents.
3.1.13.3 Ca2+ Contribution to Observed Drug Action
In this series of experiments, 50 nL of BAPTA (1,2-bis(o-aminophenoxy) ethane-
N,N,N',N'-tetraacetic acid) stock solution (100 mM) was injected into each oocyte
(Sands, Costa & Patrick, 1993) to rule out the influence of the endogenously expressed
Ca2+ activated Cl- channels (CaCCs) in the plasma membrane of Xenopus oocytes.
Stock solution of BAPTA was prepared in distilled-water and the pH was adjusted to
7.4 by CsOH. Following BAPTA-injections, oocytes were kept in ND96 for 5 to 10
min and placed in a ND96 solution containing Ba2+ instead of Ca2+(prepared ND96
solution contained 1.8 mM BaCl2 instead of 1.8 mM CaCl2). In order to determine the

96
contribution of intracellular Ca2+ levels, the effect of the tested compound on the ACh-
induced currents in BAPTA injected oocytes were investigated in either Ca2+ or Ba2+
containing ND96 solutions, and the extent of drug potentiation was compared.
3.1.13.4 Voltage-Dependency of Drug Action
Voltage-dependence of the compound's action was determined by holding the
membrane potential at different values (ranging from -120 mV to 20 mV) for 30 s. At
each point, membrane potential was returned to -70 mV, and subsequent readings were
taken every five minutes. During these experiments, ACh was used at a concentration
of 100 μM. Current-voltage (I-V) relationships for ACh-induced currents were
determined in the absence and presence of 10 μM tested compound.
3.1.13.5 Competitive and Non-competitive Inhibition
Concentration-response curves for ACh were determined by using increasing ACh
concentrations starting from 1 μM to 1 mM. The effects of Curcumin on different
concentrations of ACh-induced currents were determined in the absence and presence
of this compound. During these experiments, oocytes were voltage-clamped at a
holding potential of −70 mV. For any set of concentrations of ACh, experiments were
repeated in 6-8 different oocytes and percent of potentiation was calculated as
described earlier.

97
3.1.13.6 Effect of Curcumin on the Other Members of Cys-loop Family of
Ligand-Gated Ion Channel
The α7-nAChR belongs to the Cys-loop family of ligand-gated ion channels.
Therefore, the effect of curcumin was investigated on the activity of other members of
Cys-loop family of ligand gated ion channel. Ion current induced by 100 µM ACh, 1
µM 5-HT, and 30 µM Gly in oocytes injected cRNAs of nicotinic, serotonin type3 and
glycine receptors, respectively. Current sizes were compared in the presence and
absence of 10 µM curcumin. The average of 2-3 readings in the presence curcumin
were calculated to determine the effect of curcumin. Subsequently, drug application
was stopped, and the oocytes were washed with ND96/Ringer solution. Percentage of
potentiation was calculated by dividing the average of currents in the presence of
curcumin by the control values obtained before drug application.
3.1.13.7 Radioligand Binding
Oocytes were injected with 5 ng of human α7-nAChR cRNA, and the functional
expression of the receptors was tested by electrophysiology on day 3. Isolation of
oocyte membranes was carried out by modification of a method described previously
(Oz et al., 2004). In brief, oocytes (200–300 oocytes per assay) were suspended
(approximately 20 ml/oocyte) in a homogenization buffer containing 10mMHEPES,
1mMEDTA, 0.1 mM phenylmethane sulfonyl fluoride (PMSF), 0.02% NaN3, and 50
mg/ml bacitracin (pH 7.4) at 4°C on ice and homogenized using a motorized Teflon
homogenizer (six strokes, 15 seconds each at high speed). The homogenate was
centrifuged for 10 minutes at 800 rpm. The supernatant was collected, and the pellet
was resuspended in homogenization buffer and recentrifuged at 800 rpm for 10
minutes. Supernatants were then combined and centrifuged for 1 hour at 36,000 rpm.

98
The membrane pellet was resuspended in homogenization buffer and used for the
binding studies. Binding assays were performed in 500 ml of 10mMHEPES (pH 7.4)
containing 50 ml of oocyte preparation and 0.1–5 nM [125I] α-bungarotoxin (2200
Ci/mmol; PerkinElmer, Inc., Waltham, MA). Nonspecific binding was determined
using 10 mM α-bungarotoxin. Oocyte membranes were incubated with [125I]α-
bungarotoxin in the absence and presence of drugs for 1 hour at room temperature (22–
24°C). The radioligand was separated by rapid filtration onto GF/C filters presoaked
in 0.2% polyethyleneimine. Filters were then washed with two 5-ml washes of ice-
cold HEPES buffer, and the radioactivity was determined by counting samples in a
Beckman Gamma-300 g-counter (Beckman Coulter, Inc., Indianapolis, IN).
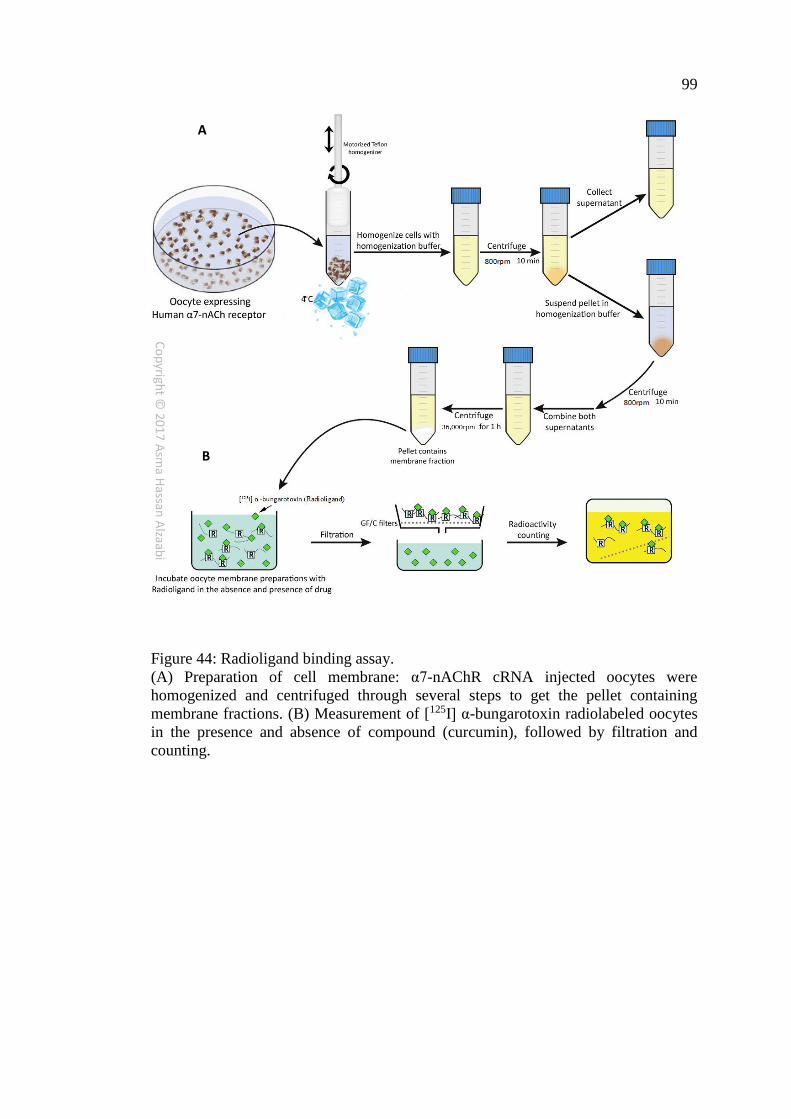
99
Figure 44: Radioligand binding assay.
(A) Preparation of cell membrane: α7-nAChR cRNA injected oocytes were
homogenized and centrifuged through several steps to get the pellet containing
membrane fractions. (B) Measurement of [125I] α-bungarotoxin radiolabeled oocytes
in the presence and absence of compound (curcumin), followed by filtration and
counting.
A
B

100
3.1.14 Statistical Analysis
In oocyte experiments, average values were calculated as the mean ± S.E.M.
Statistical significance was analyzed using Student’s t test or analysis of variance
(ANOVA) as indicated. Concentration-response curves were obtained by fitting the
data to the logistic equation
y= Emax/[1 + (x/EC50)-n]
Where x and y are concentration and response, respectively; Emax is the maximal
response; EC50 is the half-maximal concentration; and n is the slope factor (apparent
Hill coefficient).

101
3.2 Molecular Docking Experiments
The NMR structure of the human α7-nAChR transmembrane domain was obtained
from the protein data bank (“wwPDB: Worldwide Protein Data Bank,” n.d.) (PDB:
2MAW (Bondarenko et al., 2014)). The transmembrane domain structure prepared via
the Protein Preparation Wizard (Sastry et al., 2013) in the Maestro program (“Maestro
11 | Schrödinger,” n.d.); mainly to assign protonation states on ionizable groups and
to set up partial charges on the protein atoms. Brief energy minimization was employed
to relief any existing clashes between protein residues. Residues believed to play a role
in type II PAM binding were used to define the binding site for docking; which
included the whole intra-cavity of the α7-nAChR transmembrane domain. Then, a grid
box was created using the Receptor Grid Generation module in Glide (“Small-
Molecule Drug Discovery Suite | Schrödinger,” n.d.).
All ligands were created using Maestro (“Maestro 11 | Schrödinger,” n.d.) and
prepared using LigPrep (“LigPrep | Schrödinger,” n.d.) in order to give partial charges
to ligand atoms, assign protonation states on ionizable groups, and generate a single
low energy conformation for each ligand using the OPLS forcefield. Using Glide
(“Small-Molecule Drug Discovery Suite | Schrödinger,” n.d.), all prepared ligands
were docked into the previously prepared intra-cavity of the α7-nAChR
transmembrane where the extra-precision (XP) Algorithm was employed for
conformational sampling. Subsequently, docked poses were scored via Glide_XP
which contains terms for hydrogen bonding, electrostatic interactions, van der Waals
interactions, desolvation penalty and intra-ligand contact penalty (Friesner et al.,
2006).

102
3.3 In-vivo Study: Animal Model of Parkinson’s Disease
3.3.1 Animals
A total of 50 adult male Wistar rats weighing between 220 and 250 grams at the
beginning of the study were used. Four or five rats were housed in a large cage with
free access to rat chow and water under a 12:12 h light–dark cycle, at a room
temperature (22 °C). All experimental procedures were approved by the Animal Ethics
Committee of the CMHS, UAE University and were performed in accordance with the
guidelines of the European Communities Council directive of 24 November 1986
(86/609/EEC).
The rats were randomly assigned into Five groups: 1) Vehicle treated group
(ascorbic acid injection into the right striatum, and received daily oral gavage carboxy
methyl cellulose (CMC) n= 6, 2) 6-OHDA treated group (6-OHDA injection into right
striatum) n= 8, 3) 6-OHDA + curcumin pre- and post-treatment [intra-gastric oral
gavage curcumin (200 mg/kg) once a day for four weeks in total (2 weeks before and
2 weeks after surgery), with 6-OHDA lesioning at the end of week 2 of curcumin
treatment) n= 8, 4) 6-OHDA + curcumin + MLA (the same as group 3, with the
addition of MLA I.P injection 10 min before curcumin administration) n= 9, and 5) 6-
OHDA + MLA (the same as group 2, with the addition of MLA I.P injection 10 min
before apomorphine-induced rotation testing), n=8.
3.3.2 Drugs
6-hydroxydopamine hydrochloride, apomorphine hydrochloride, and curcumin,
were purchased from Sigma-Aldrich (Sigma Chemicals Co.; St. Louis, MO), and MLA
was purchased from Abcam, USA.

103
6-OHDA-HCl with the purity ≥97%, was dissolved in ice-cold 0.01% ascorbate in
0.9% normal saline and used within 2 hours after preparation. Apomorphine-HCL with
the purity ≥98.5%, was dissolved in 0.1 ascorbic acid in saline and prepared on
demand. Curcumin was suspended in 0.5% sodium carboxy methyl cellulose (CMC)
and 50 µL of (10 M) NaOH and prepared on daily bases. MLA was dissolved in normal
saline and preserved in +4℃.
3.3.3 Surgical Procedure
All rats were deeply anesthetized using an equal mixture of ketamine
hydrochloride (80 mg/kg, Pantex Holland B.V., Holland) and xylazine Hydrochloride
(20 mg/kg, Troy Laboratory PTY Limited, NSW, Australia) administered
intraperitoneally. To lesion the nigrostriatal pathway, the heads of the animals were
shaved and placed into a Stoelting stereotaxic frame and a unilateral hole on the right-
side of the skull was made aiming at the striatum (Figure 46). The rats were divided
into Five groups. The first group (n=8) received unilateral injections of 6-OHDA, at
three distinct locations within the caudate-putamen (7 µg in each site). The neurotoxin
6-OHDA-HCl (Sigma Chemicals Co.; St. Louis, MO) was dissolved in ice-cold 0.01%
ascorbate in 0.9% normal saline and used within 2 hours. Three intrastriatal 6-OHDA
injections were performed using pulled glass micropipette with an outer diameter of
approximately 50 µm. Seven µg 6-OHDA (dissolved in 2 µl) was injected at each site,
using the following coordinates; AP: +1.0, -0.1, -1.2 / ML: -3.0, -3.7, -4.5 / DV: -5.0,
-5.0, -5.0) (Figure 47). The tooth bar of the stereotaxic frame was fixed at 0.0 relative
to bregma (Paxinos and Watson, 2004). 6-OHDA was injected at a rate of 1 µl/min,
and the injection micropipette was left in place for an additional 3 minutes to prevent
backflow. All rats were allowed to recover for 3 weeks before behavioral testing.
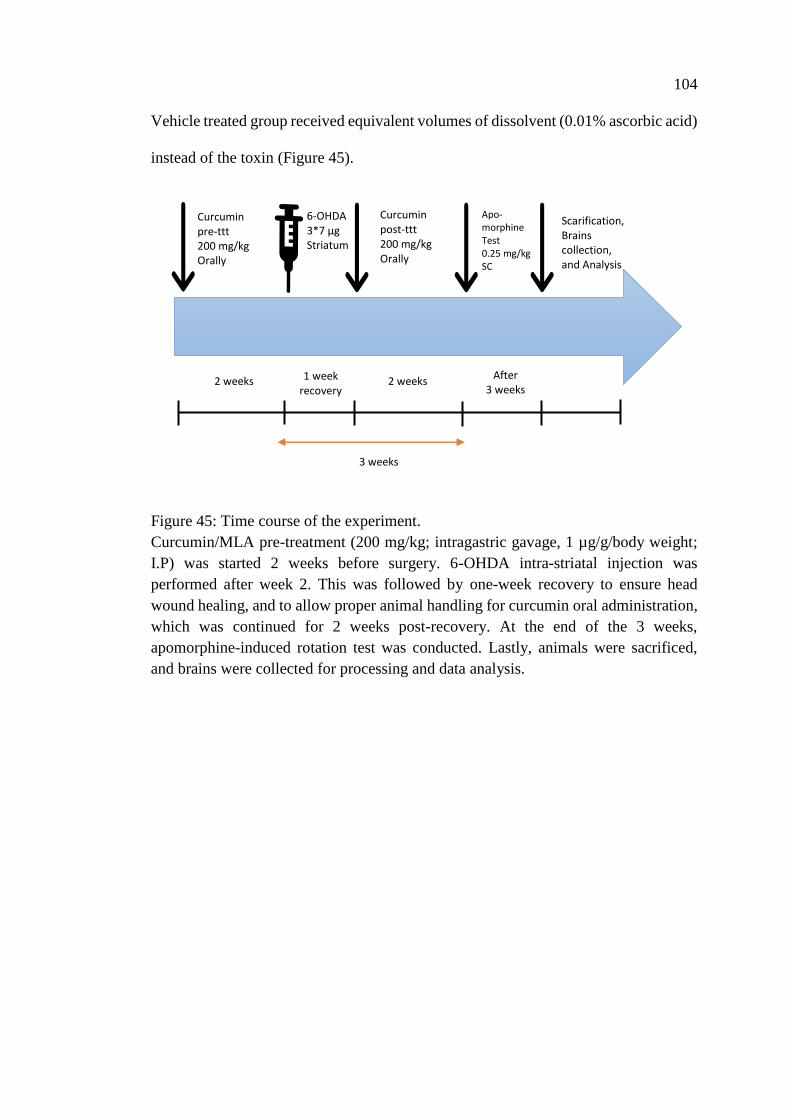
104
Vehicle treated group received equivalent volumes of dissolvent (0.01% ascorbic acid)
instead of the toxin (Figure 45).
Figure 45: Time course of the experiment.
Curcumin/MLA pre-treatment (200 mg/kg; intragastric gavage, 1 µg/g/body weight;
I.P) was started 2 weeks before surgery. 6-OHDA intra-striatal injection was
performed after week 2. This was followed by one-week recovery to ensure head
wound healing, and to allow proper animal handling for curcumin oral administration,
which was continued for 2 weeks post-recovery. At the end of the 3 weeks,
apomorphine-induced rotation test was conducted. Lastly, animals were sacrificed,
and brains were collected for processing and data analysis.
Scarification, Brains collection, and Analysis
Apo-morphine Test 0.25 mg/kg SC
Curcumin pre-ttt 200 mg/kg Orally
Curcumin post-ttt 200 mg/kg
Orally
6-OHDA 3*7 µg Striatum
2 weeks 2 weeks 1 week recovery
After 3 weeks
3 weeks

105
Figure 46: Stereotaxic surgery to lesion nigrostriatal pathway.
The head of the animal was placed into a Stoelting stereotaxic frame and a unilateral
hole on the rite of the skull was made. Three intrastriatal 6-OHDA injections were
performed using 3 coordinated relative to the bregma (see Figure 47), according to the
rat brain atlas of Paxinos and Watson, 2004.
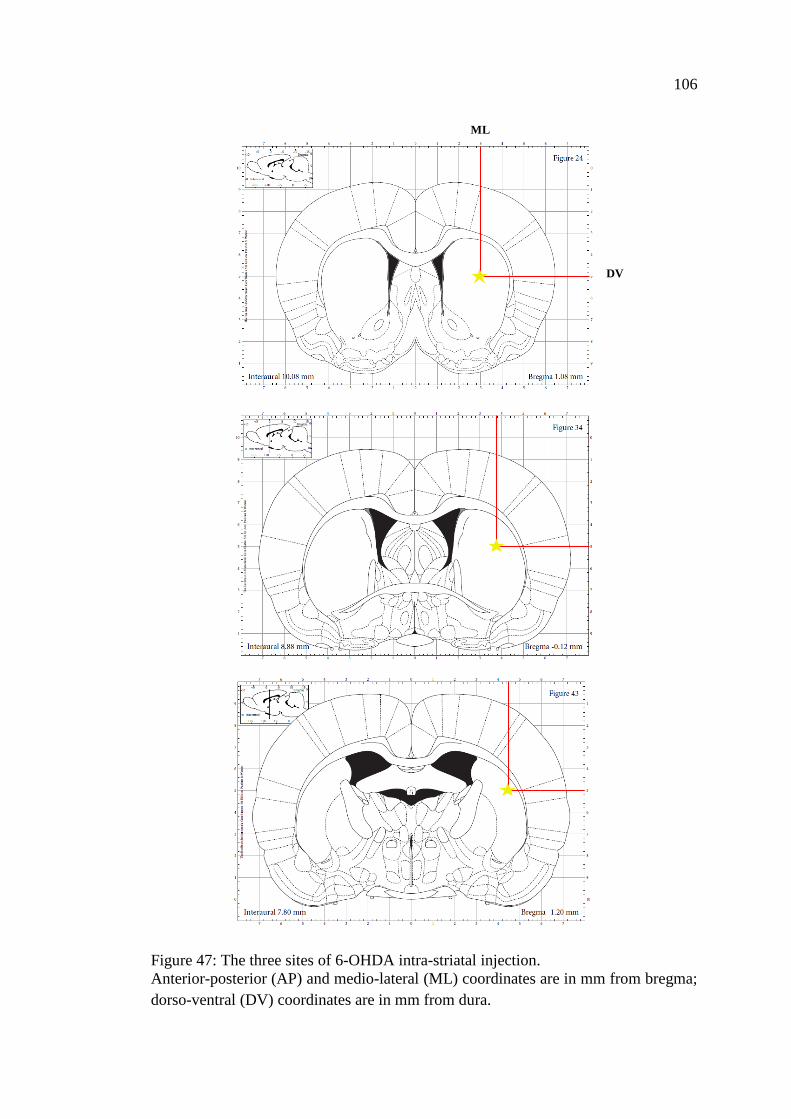
106
Figure 47: The three sites of 6-OHDA intra-striatal injection.
Anterior-posterior (AP) and medio-lateral (ML) coordinates are in mm from bregma;
dorso-ventral (DV) coordinates are in mm from dura.
ML
DV

107
3.3.4 Apomorphine-Induced Rotational Behavior
Three weeks after surgery, drug-induced rotational behavior was monitored in rounded
bowl (42cm wide at top and 22 cm deep) (Truong et al., 2006). Animals were injected
with 0.25 mg/kg apomorphine-HCL (Kirik et al., 1998) (Sigma - Aldrich) (dissolved
in 0.1 ascorbic acid in saline) subcutaneously or 0.1 ascorbic acid alone for vehicle
treated group, and rotational asymmetry was monitored. Directly after injection of
vehicle or drug, the animals were allowed to acclimatize in the bowl for 5-min. Turns
of 360° in the clockwise and counter-clockwise directions were continuously recorded
for 30 min. Net rotational asymmetry score was expressed as full body turns/min.
Direction contralateral to the lesion was considered as positive. Rats that exhibited 5-
7 full turns/min were included in the study as Parkinsonian animals (Kozlowski et al.,
2004; Metz and Whishaw, 2002; Shimizu et al., 2008). All rotational data were
expressed as means of the full turns per minute ± SEM. The data were expressed as
the net (contralateral minus ipsilateral turns) rotations/min.
3.3.5 Histology
Animals were euthanized with an over dose of urethane 25% (2 ml/200 g of animal
weight) injected intraperitoneally and perfused transcardially through the ascending
aorta with 50 ml of phosphate buffered saline (PBS), followed by 500 ml of 4%
paraformaldehyde in 0.1 M phosphate buffer (PB, pH 7.4) over 20 min. The brains
were removed, post-fixed in the same fixative solution for 4 hours and stored in 30%
sucrose and kept until they had sunk before being sectioned at cryostat. Coronal
sections (40 µm thickness) of striatum and substantia nigra were collected serially and
processed for detection of TH immunoreactivity using the avidin–biotin-complex
(ABC) method, as previously described (Shehab et al., 2015, 2003).

108
Immunohistochemical staining for tyrosine hydroxylase
Immunohistochemical staining was performed on free-floating sections. For this
purpose, brain sections were first rinsed three times with PBS solution followed by
incubation in a 50% ethanol for 30 min to increase the penetration of the antibodies
(Llewellyn-Smith et al., 1992). After rinsing in PBS, the sections were then incubated
overnight at room temperature in primary antibody solution containing 1% bovine
serum albumin in PBS containing Triton-X-100. The primary antibodies used for
immunohistochemical staining were raised in rabbit against tyrosine hydroxylase (TH)
as a marker for dopaminergic neurons, AB152, Millipore; at 1:1000 dilution). On the
second day, the sections were rinsed in PBS and then incubated with biotinylated goat
anti-rabbit IgG (Jackson, 1:500) for 1 h then in extravidin-peroxidase conjugate
(Sigma, 1:1000) for another hour. After rinsing, to visualize any TH+
immunoreactivity the sections were incubated for 5–8 min in a solution of 25 mg
diaminobenzidine (DAB) in 50 ml 0.1 M phosphate buffer (PB, pH 7.4) with 7.5 μl
hydrogen peroxide (30%) and 1 ml nickel chloride (3.5%) added to intensify the
reaction. Finally, the sections were rinsed in PB and mounted on gelatin-coated slides.
After drying in air the sections were dehydrated in graded alcohol, cleared in xylene
and mounted with DPX mounting media. All antibodies were diluted in PBS
containing 0.3% Triton.
3.3.6 Measurement of Striatal Fiber Density
Striatal TH+ immunoreactive fibers were captured with Nikon microscope (Nikon,
Japan) equipped with DS-Ri2 camera. Using Image J software (National Institutes of
Health, USA), optical densitometry of TH+ stained striatal terminals was measured.

109
The average optical density over the entire area of the striatum for four striatal
sections in each animal was quantified: rostral (+1.56 mm from Bregma), middle 1
(+0.72 mm from Bregma), middle 2 (+0.12 mm from Bregma), and caudal (−0.24 mm
from Bregma) in each animal. To compensate for differences in background coloring
between the slides, the optical density was subtracted from values obtained in the
cortex of each sample. The data were expressed as a percentage of the fiber density on
the lesioned side to the non-lesioned side.
3.3.7 Stereological Analysis
Substantia nigra cell counts
The total number of TH+ neurons in the SNpc in both hemispheres was estimated
with an unbiased stereology using the optical fractionator (West, 1999),
StereoInvestigator, (MicroBrightField, Colchester, VT, USA). The final number of
animals included in stereological analysis was as follow: Vehicle treated = 6[6], 6-
OHDA = 8[9], 6-OHDA+Cur = 8[8], 6-OHDA+Cur+MLA = 8[8], 6-OHDA+MLA =
8[8] (numbers in square parenthesis represent the number of animals that were used
for the behavioral testing). To estimate the number of TH+ cell numbers in the SNpc,
the borders defining the SNpc were delineated by using a low-power objective lens
(5X; S Plan) on referral to anatomical morphology. The counting was done using a
63X Plan-Apo oil objective to generate counting areas of 106X106 µm. A counting
frame (3184µm2) was placed randomly on the first counting area and systematically
moved through all counting areas. The section thickness was estimated to be 25±5 µm,
after dehydration and coverslipping, in different animals. Guard volumes of 2 µm were
excluded from each surface to avoid the problem of lost caps. The sampling interval
in the X-Y axis was adjusted so that at least 100 cells were counted for each region of

110
interest. The coefficient of error due to the estimation was calculated according to
(Gundersen & Jensen, 1987) and values < 0.10 was accepted. For each animal the
optical density was measured at three sections at three different rostrocaudal levels
according to the atlas of Paxinos & Watson (2004) over the whole substantia nigra: (i)
AP 4.8; (ii) AP 5.6; (iii) AP 6.2, relative to the bregma. The data represents the percent
of surviving nigral TH+ neurons per level as well as the total survival analysis of the
three rostrocaudal levels of the lesioned (right) side in comparison with the intact (left)
side.
3.3.8 Statistical Analysis
ANOVA with post hoc Bonferroni test was used to analyze differences in
behavioral tests and cell numbers between groups. Paired t test was used to compare
Apomorphine-induced rotation with and without MLA post-injection. Results were
expressed as means ± SEM.

111
Chapter 4: Results
4.1 Results
4.1.1 Effects of Curcumin on α7-nicotinic Acetylcholine Receptors
Application of 100 μM ACh for 3 - 4 seconds activated fast inward currents that
desensitized rapidly in oocytes injected with cRNA encoding the α7-subunit of human
nAChR (Figure 48A). In addition, ACh-induced currents were inhibited completely
with 100 nM α-bungarotoxin (Figures 48A & B), indicating that these currents are
mediated by the activation of α7-nAChRs. Bath application of curcumin (100 µM) for
5 minutes did not produce detectable currents in oocytes expressing α7-nAChRs (n =
8 oocytes).
The effect of curcumin was tested on ion currents induced with ACh (100 µM). An
effect of 10-minute curcumin (1 μM) application on α7-nAChR–mediated currents is
shown in Figure 49A. Time courses of effects of curcumin or vehicle (0.1% DMSO)
applications on the maximal amplitudes of ACh-induced currents are presented in
Figure 49B. Curcumin caused a significant potentiation of the current, which was
partially reversed during a 10–15-minute washout. In the absence of curcumin, vehicle
(0.1% DMSO) alone did not alter the amplitude of the ACh-induced current, further
suggesting that curcumin acts on nAChRs (Figure 49A, controls vs. curcumin
treatment group at 10 minutes of exposure, ANOVA, n = 5–7; P < 0.05).
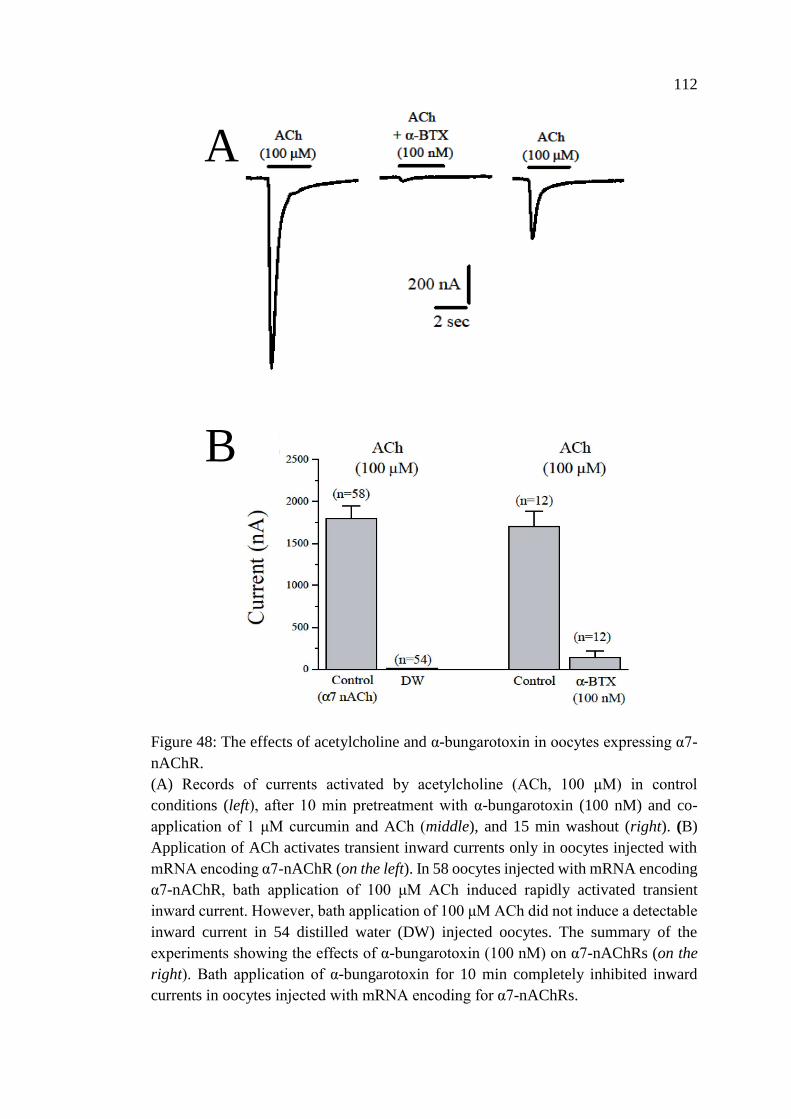
112
Figure 48: The effects of acetylcholine and α-bungarotoxin in oocytes expressing α7-
nAChR.
(A) Records of currents activated by acetylcholine (ACh, 100 μM) in control
conditions (left), after 10 min pretreatment with α-bungarotoxin (100 nM) and co-
application of 1 μM curcumin and ACh (middle), and 15 min washout (right). (B)
Application of ACh activates transient inward currents only in oocytes injected with
mRNA encoding α7-nAChR (on the left). In 58 oocytes injected with mRNA encoding
α7-nAChR, bath application of 100 μM ACh induced rapidly activated transient
inward current. However, bath application of 100 μM ACh did not induce a detectable
inward current in 54 distilled water (DW) injected oocytes. The summary of the
experiments showing the effects of α-bungarotoxin (100 nM) on α7-nAChRs (on the
right). Bath application of α-bungarotoxin for 10 min completely inhibited inward
currents in oocytes injected with mRNA encoding for α7-nAChRs.
B
A
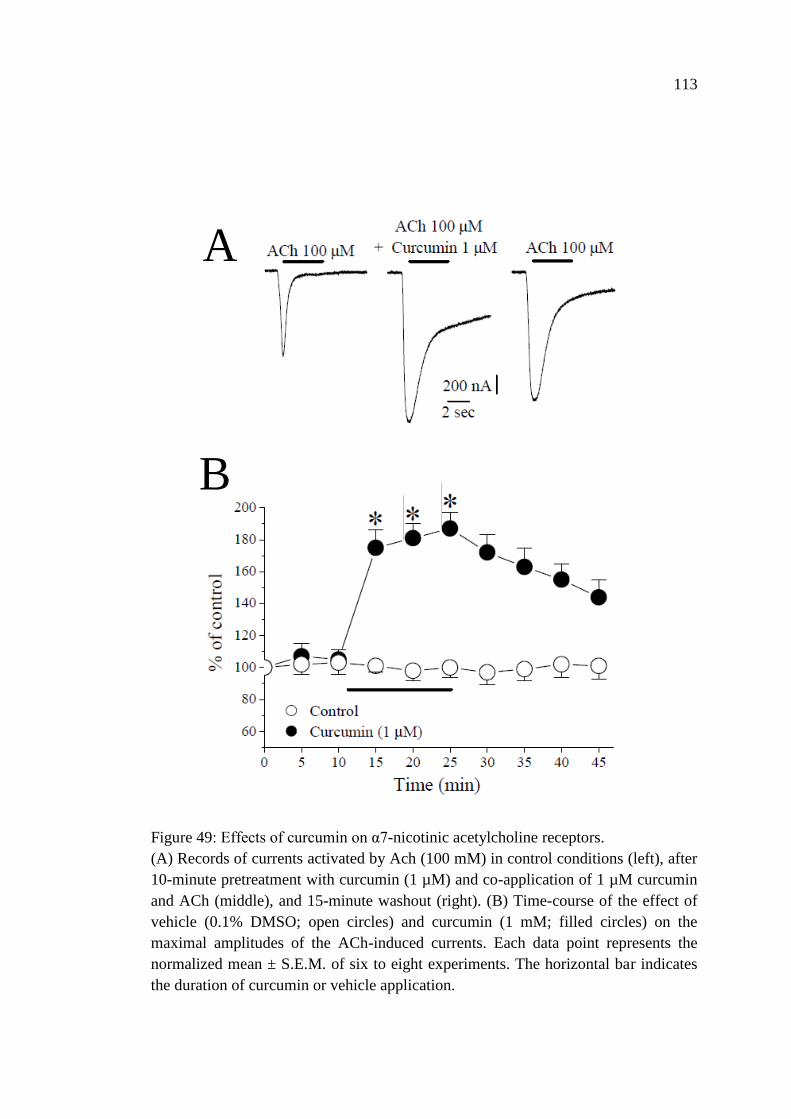
113
Figure 49: Effects of curcumin on α7-nicotinic acetylcholine receptors.
(A) Records of currents activated by Ach (100 mM) in control conditions (left), after
10-minute pretreatment with curcumin (1 µM) and co-application of 1 µM curcumin
and ACh (middle), and 15-minute washout (right). (B) Time-course of the effect of
vehicle (0.1% DMSO; open circles) and curcumin (1 mM; filled circles) on the
maximal amplitudes of the ACh-induced currents. Each data point represents the
normalized mean ± S.E.M. of six to eight experiments. The horizontal bar indicates
the duration of curcumin or vehicle application.
C
D B
A

114
4.1.2 Concentration Response Curve
The potentiating effect of curcumin was significantly dependent on the application
mode. For example, without preincubation, coapplication of curcumin (1 μM) and
ACh (100 μM) did not alter the amplitudes of maximal currents (Figure 50A).
However, when oocytes were preincubated with curcumin, the drug was found to
potentiate maximal ACh-induced currents in a time-dependent manner, reaching a
maximal level within 5 minutes with a half-time (τ1/2) of 1.6 minutes (Figure 50A).
Since the magnitude of the curcumin effect was time-dependent, 10-minute curcumin
application time was used routinely to ensure equilibrium conditions. Curcumin was
found to upregulate the function of α7-nAChR in a concentration-dependent manner
with EC50 and slope values of 0.21 ± 0.14 µM and 1.6, respectively (Figure 50B).
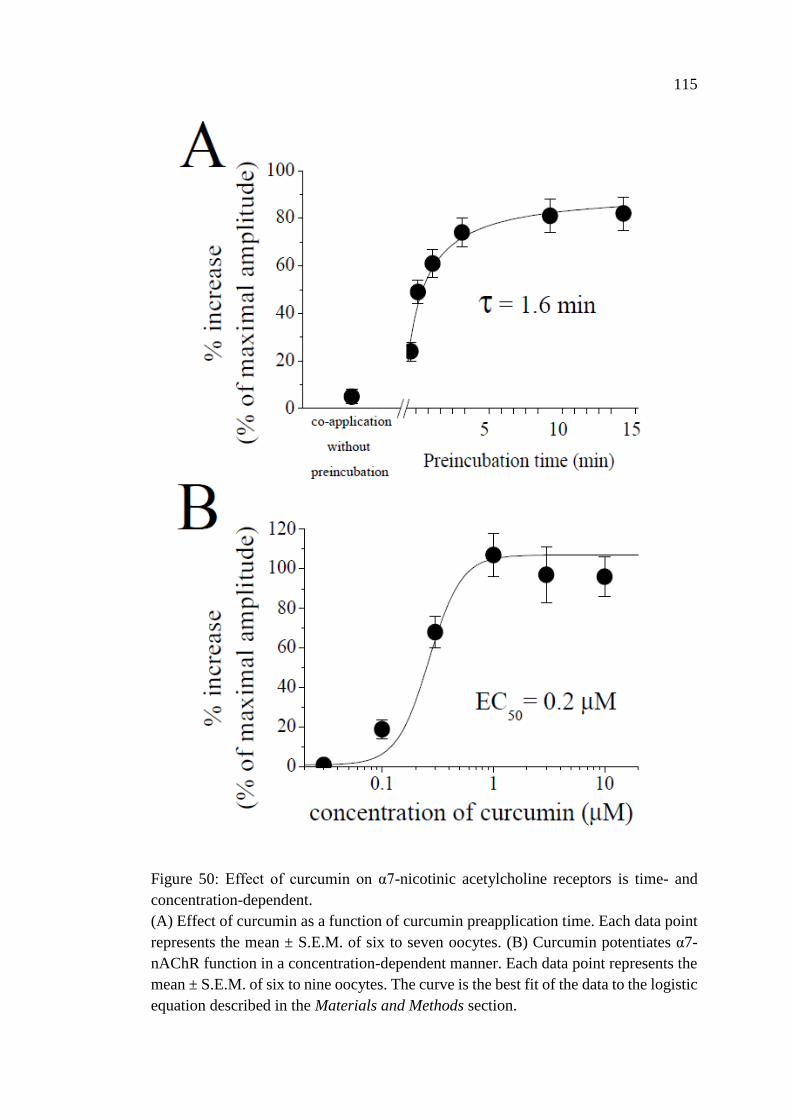
115
Figure 50: Effect of curcumin on α7-nicotinic acetylcholine receptors is time- and
concentration-dependent.
(A) Effect of curcumin as a function of curcumin preapplication time. Each data point
represents the mean ± S.E.M. of six to seven oocytes. (B) Curcumin potentiates α7-
nAChR function in a concentration-dependent manner. Each data point represents the
mean ± S.E.M. of six to nine oocytes. The curve is the best fit of the data to the logistic
equation described in the Materials and Methods section.

116
4.1.3 Effects of Curcumin on α7-nAChRs are not Mediated by G-proteins
As shown in Figures 49B & 50B, regulation of α7-nAChR function by curcumin
occurs gradually, reaching steady-state levels within a few minutes of curcumin
application. Therefore, it is possible that activation of second messenger pathways by
G-protein-coupled receptors (Liu et al., 2013; Pérez-Lara et al., 2011; Yang et al.,
2015) is involved in curcumin regulation of α7-nAChRs. Thus, we investigated the
effects of pretreatments with NEM (10 mM, 50 nl, 30-minute preincubation time), a
sulfhydryl-alkylating agent that blocks G-protein-effector interactions by alkylating α-
subunits of PTX-sensitive GTP binding protein (Oz and Renaud, 2002), and GDPβS
(10 mM, 50 nl, 30-minute preincubation time), an agent that inhibits binding of GTP
to the α-subunit of G-proteins (Oz et al., 1998). Treatments with NEM and GDPβS did
not alter the extent of curcumin potentiation of α7-nAChR (Figure 51A).
Similarly, pretreatment with PTX (5 µg/ml, 50 nl, 30-minute preincubation time),
toxin that inhibits the α-subunit of Gi/o proteins, did not reverse the potentiating effect
of curcumin (Figure 51B).
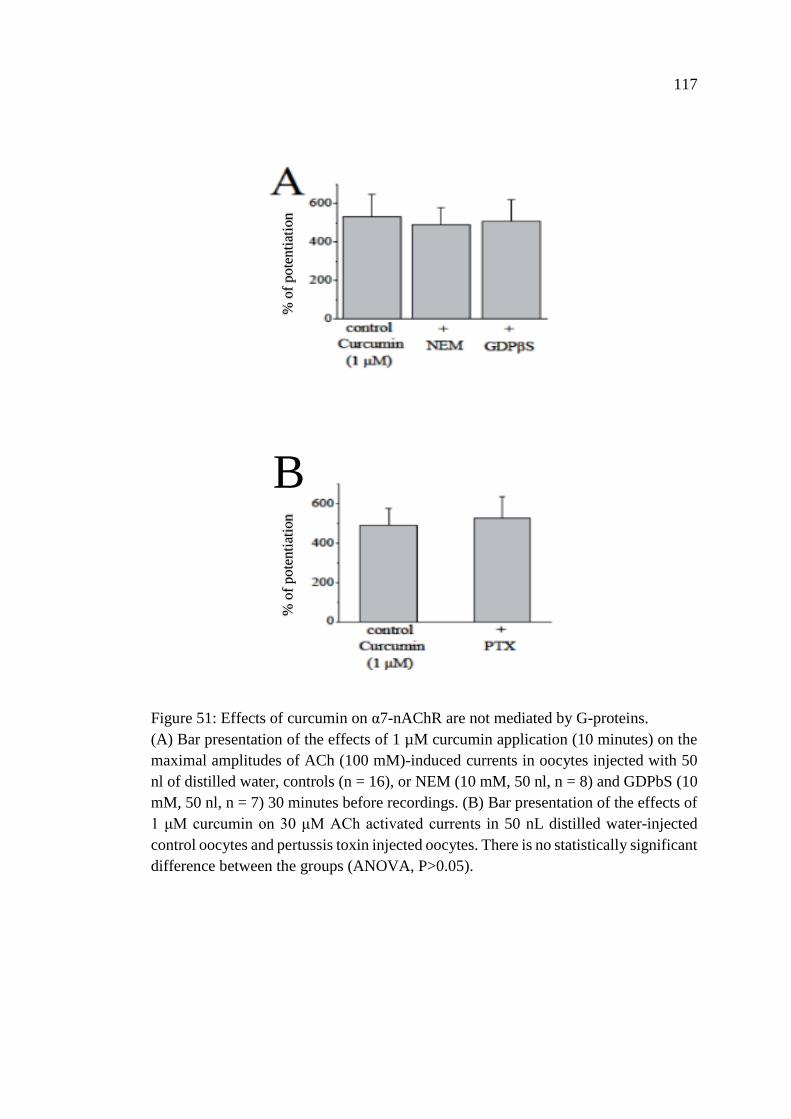
117
Figure 51: Effects of curcumin on α7-nAChR are not mediated by G-proteins.
(A) Bar presentation of the effects of 1 µM curcumin application (10 minutes) on the
maximal amplitudes of ACh (100 mM)-induced currents in oocytes injected with 50
nl of distilled water, controls (n = 16), or NEM (10 mM, 50 nl, n = 8) and GDPbS (10
mM, 50 nl, n = 7) 30 minutes before recordings. (B) Bar presentation of the effects of
1 μM curcumin on 30 μM ACh activated currents in 50 nL distilled water-injected
control oocytes and pertussis toxin injected oocytes. There is no statistically significant
difference between the groups (ANOVA, P>0.05).
B
% o
f pote
nti
atio
n
% o
f p
ote
nti
atio
n

118
4.1.4 Effects of Curcumin on α7-nAChRs are not Mediated by Protein Kinases
We also investigated the involvement of protein kinases A and C, and Ca2+-
calmodulin–dependent kinase (CaM-kinase) in curcumin potentiation of α7-nAChRs.
For this purpose, the effects of curcumin were tested in oocytes pretreated with PKC-
412 (nonspecific kinase inhibitor, 10 µM for 30 minutes pretreatment), Go-6983
(specific protein kinase C inhibitor, 10 µM for 30 minutes), KT-5720 (specific protein
kinase A inhibitor, 10 µM for 30 minutes), and KN-62 (specific inhibitor of CaM-
kinase II, 50 µM for 30 min). Curcumin continued to upregulate nicotinic receptor–
mediated currents in oocytes pretreated with kinase inhibitors (Figures 53A & B).

119
Figure 52: Effects of curcumin on α7-nAChR are not mediated by prtein kinases.
(A) Bar presentation of the effects of 1 μM curcumin on α7-nAChR–mediated currents
in oocytes pretreated with vehicle (0.01% DMSO, n = 5) or PKC-412 (PKC; 10 mM,
30-minute pretreatment, n = 7), or Go-6983 (GO; 10 mM, 30-minute pretreatment, n
= 6). (B) Bar presentation of the effects of 1 mM curcumin on a7-nAChR–mediated
currents in oocytes pretreated with vehicle (0.01% DMSO, n = 7) or KT-5720 (KT; 10
mM, 30-minute pretreatment, n = 7) and KN-62 (KN; 50 mM, 30-minute pretreatment,
n = 6).
B
A
% o
f pote
nti
atio
n
% o
f p
ote
nti
atio
n

120
4.1.5 Effects of Curcumin on α7-nAChRs are not Dependent on Intracellular Ca2+
Activation of α7-nAChRs allows sufficient Ca2+ entry to activate endogenous
Ca2+-dependent Cl− channels in Xenopus oocytes (Sands et al., 1993; Uteshev, 2012).
Therefore, it was important to determine whether the effect of curcumin was exerted
on nAChR–mediated currents or on Cl− currents induced by Ca2+ entry. For this
reason, we injected the Ca2+ chelator BAPTA into oocytes and replaced extracellular
Ca2+ with Ba2+ which can pass through α7-nAChRs but causes less activation of Ca2+-
dependent Cl− channels (Sands et al., 1993). Under these conditions, we tested the
effect of curcumin in a solution containing 2 mM Ba2+ in BAPTA-injected oocytes.
Curcumin (1 µM) produced the same level of potentiation (195 ± 18 in controls vs.
210 ± 22 in BAPTA-injected oocytes; ANOVA, P > 0.05; n = 6 to 7) on ACh-induced
currents in BAPTA-injected oocytes when currents were recorded in Ca2+-free solution
containing 2 mM Ba2+ (Figure 53A). It is important to mention that in the oocyte
expression system, curcumin-induced changes in nicotinic receptor–mediated currents
can be attributable to Ca2+-activated Cl− channels and concomitant alterations in the
holding currents. However, in control experiments, curcumin (100 µM for 10 minutes)
did not change the magnitudes of holding currents in oocytes voltage clamped at −70
mV (n = 7), indicating that intracellular Ca2+ levels were not altered by curcumin.
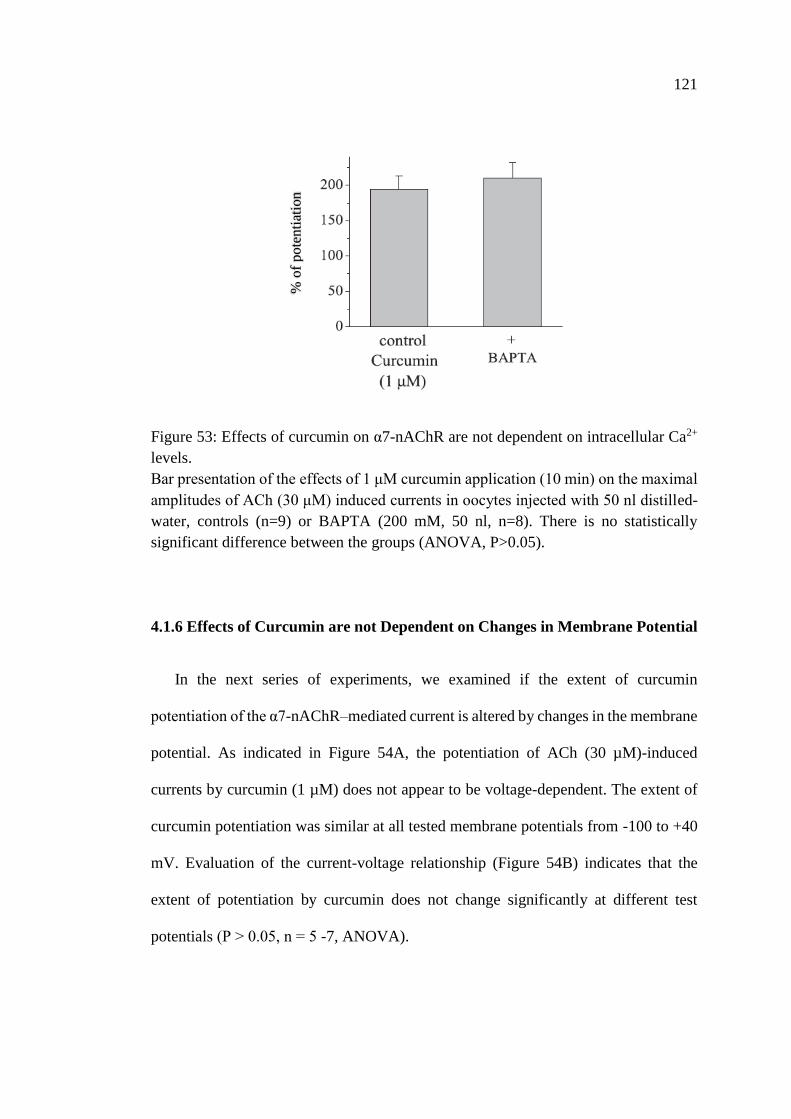
121
Figure 53: Effects of curcumin on α7-nAChR are not dependent on intracellular Ca2+
levels.
Bar presentation of the effects of 1 μM curcumin application (10 min) on the maximal
amplitudes of ACh (30 μM) induced currents in oocytes injected with 50 nl distilled-
water, controls (n=9) or BAPTA (200 mM, 50 nl, n=8). There is no statistically
significant difference between the groups (ANOVA, P>0.05).
4.1.6 Effects of Curcumin are not Dependent on Changes in Membrane Potential
In the next series of experiments, we examined if the extent of curcumin
potentiation of the α7-nAChR–mediated current is altered by changes in the membrane
potential. As indicated in Figure 54A, the potentiation of ACh (30 µM)-induced
currents by curcumin (1 µM) does not appear to be voltage-dependent. The extent of
curcumin potentiation was similar at all tested membrane potentials from -100 to +40
mV. Evaluation of the current-voltage relationship (Figure 54B) indicates that the
extent of potentiation by curcumin does not change significantly at different test
potentials (P ˃ 0.05, n = 5 -7, ANOVA).
B
% o
f p
ote
nti
atio
n
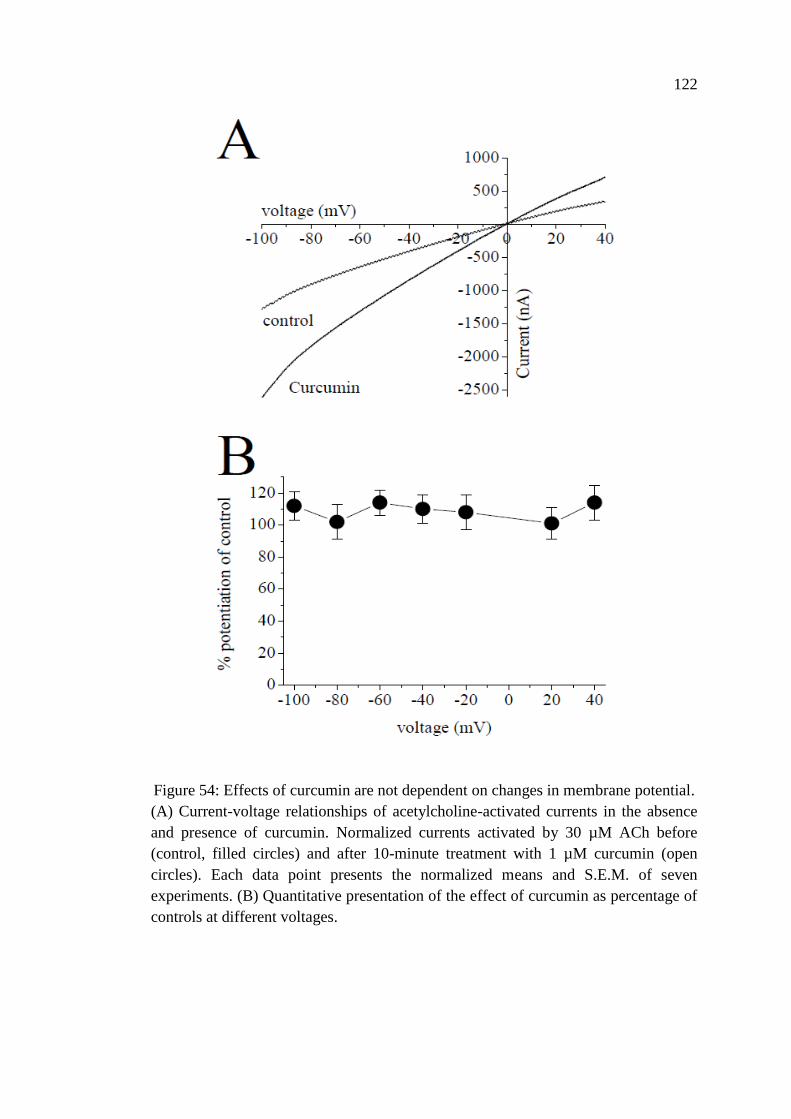
122
Figure 54: Effects of curcumin are not dependent on changes in membrane potential.
(A) Current-voltage relationships of acetylcholine-activated currents in the absence
and presence of curcumin. Normalized currents activated by 30 µM ACh before
(control, filled circles) and after 10-minute treatment with 1 µM curcumin (open
circles). Each data point presents the normalized means and S.E.M. of seven
experiments. (B) Quantitative presentation of the effect of curcumin as percentage of
controls at different voltages.

123
4.1.7 Effects of Curcumin at Different Concentrations of Acetylcholine
In the next series of experiments, we attempted to test the effects of curcumin at
different ACh concentrations. Traces of low ACh (10 µM)-induced currents after 10-
minute treatment with 1 µM curcumin are presented in Figure 55A. At low ACh
(10 µM) concentrations, curcumin caused approximately 11- to 12-fold increase of
ACh-induced currents with an EC50 of 58 nM (Figure 55B). Our experiments indicated
that the extent of curcumin potentiation decreased significantly with increasing
concentrations of ACh (Figure 56A). Concentration-response curves for ACh in the
absence and presence of 1 µM curcumin are presented in Figure 56B. In the presence
of 1 µM curcumin, the maximal ACh response increased by 60%–70% of controls (n =
6–8). In the absence and presence of curcumin, the EC50 values for ACh were 107 ±
18 and 63 ± 16 μM, and slope values were 2.2 ± 0.4 and 1.9 ± 0.3, respectively (n =
6–7).
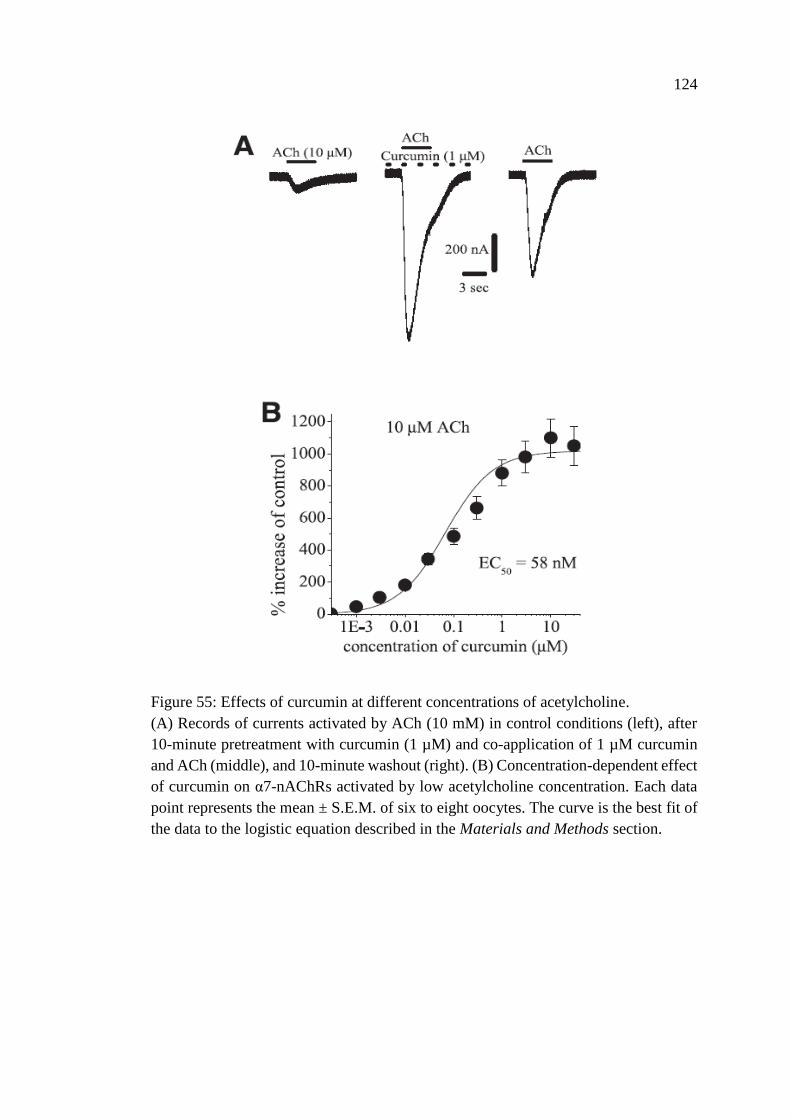
124
Figure 55: Effects of curcumin at different concentrations of acetylcholine.
(A) Records of currents activated by ACh (10 mM) in control conditions (left), after
10-minute pretreatment with curcumin (1 µM) and co-application of 1 µM curcumin
and ACh (middle), and 10-minute washout (right). (B) Concentration-dependent effect
of curcumin on α7-nAChRs activated by low acetylcholine concentration. Each data
point represents the mean ± S.E.M. of six to eight oocytes. The curve is the best fit of
the data to the logistic equation described in the Materials and Methods section.
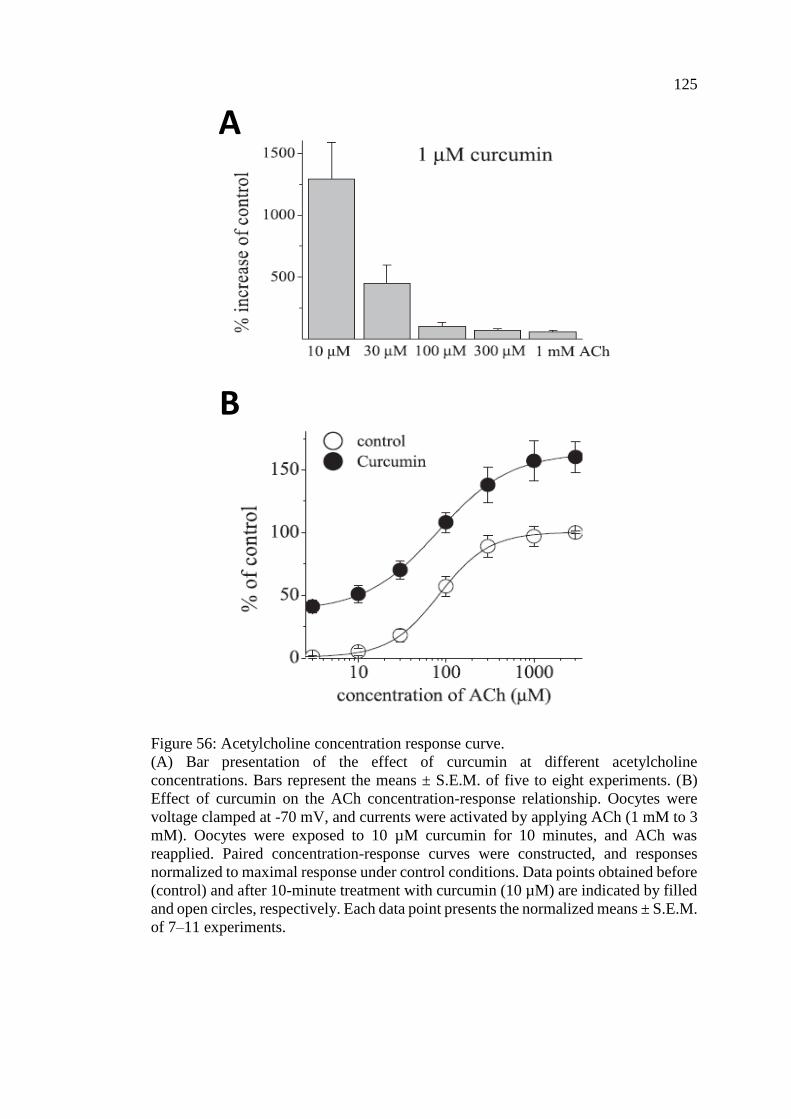
125
Figure 56: Acetylcholine concentration response curve.
(A) Bar presentation of the effect of curcumin at different acetylcholine
concentrations. Bars represent the means ± S.E.M. of five to eight experiments. (B)
Effect of curcumin on the ACh concentration-response relationship. Oocytes were
voltage clamped at -70 mV, and currents were activated by applying ACh (1 mM to 3
mM). Oocytes were exposed to 10 µM curcumin for 10 minutes, and ACh was
reapplied. Paired concentration-response curves were constructed, and responses
normalized to maximal response under control conditions. Data points obtained before
(control) and after 10-minute treatment with curcumin (10 µM) are indicated by filled
and open circles, respectively. Each data point presents the normalized means ± S.E.M.
of 7–11 experiments.
B
A

126
4.1.8 Effects of Curcumin on the Desensitization of Nicotinic Receptors
The results of functional studies indicate that curcumin significantly decreases
desensitization of currents mediated by the activation of α7-nAChRs. Normalized and
superimposed current traces in the absence or presence of 1 μM curcumin are
presented in Figure 57A. A summary of the results showing the effect of curcumin
(1 μM) on the half decay times of the α7-nAChR–mediated currents is shown in Figure
57B. In the absence and presence of curcumin, means of half decay times were 208 ±
42 and 643 ± 85 seconds, respectively (paired t test; n = 8; P ˂ 0.01). These findings
suggest that curcumin causes a significant decrease of α7-nAChR desensitization. We
further investigated whether curcumin could convert α7-nAChRs that were already
desensitized by a concentration of an agonist back to conducting state. We tested the
effect of a bath application of curcumin (10 µM) on the α7-nAChRs that was
desensitized by 100 µM nicotine application for 25–30 seconds (n = 6). As illustrated
in Figure 57C, subsequent addition of curcumin resulted in activation of a sustained
inward current that was reversed during washout.
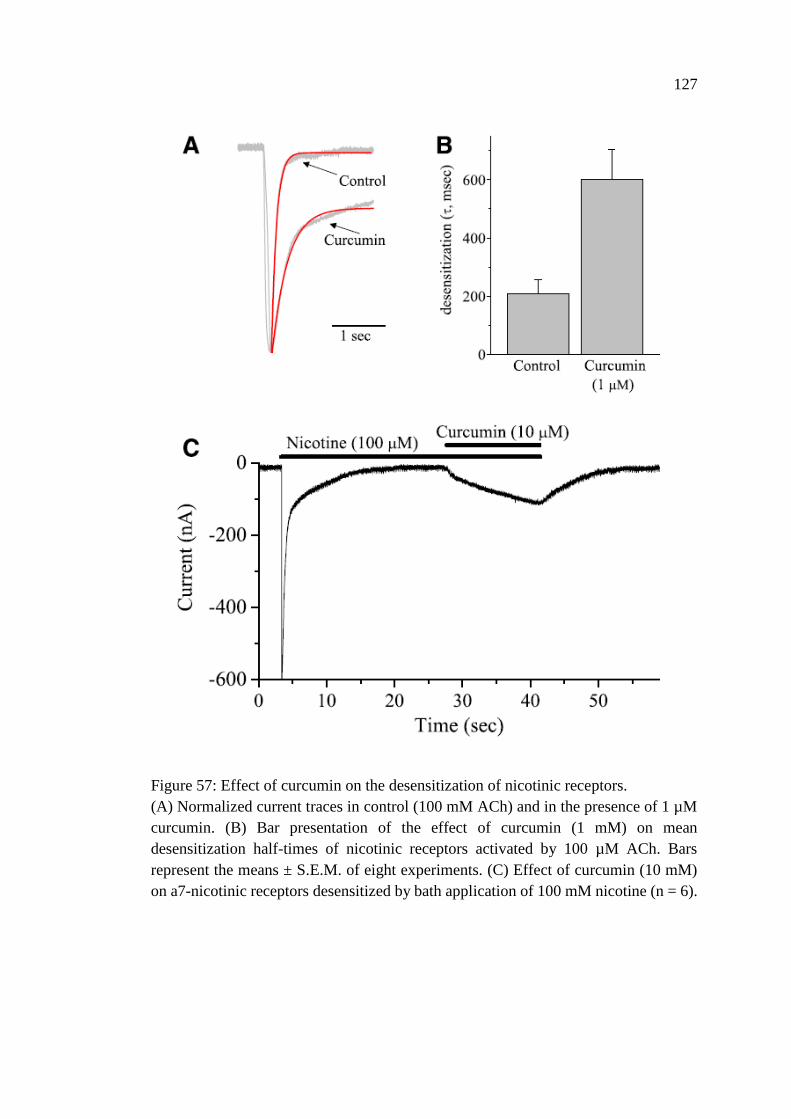
127
Figure 57: Effect of curcumin on the desensitization of nicotinic receptors.
(A) Normalized current traces in control (100 mM ACh) and in the presence of 1 µM
curcumin. (B) Bar presentation of the effect of curcumin (1 mM) on mean
desensitization half-times of nicotinic receptors activated by 100 µM ACh. Bars
represent the means ± S.E.M. of eight experiments. (C) Effect of curcumin (10 mM)
on a7-nicotinic receptors desensitized by bath application of 100 mM nicotine (n = 6).

128
4.1.9 Effects of Curcumin on the Specific Binding of [125I]α-bungarotoxin
[125I]α-bungarotoxin competes with ACh, an endogenous activator of α7-nAChRs,
by binding to the ACh binding site on the receptor (Albuquerque et al., 2009). For this
reason, the effect of curcumin was investigated on the specific binding of [125I]α-
bungarotoxin. Equilibrium curves for the binding of [125I]α-bungarotoxin in the
presence and absence (controls) of curcumin are presented in Figure 58A. Maximum
binding activities (Bmax) of [125I]α-bungarotoxin were 3.61 ± 0.37 and 3.47 ± 0.41
pM/mg (means ± S.E.M.) for controls and curcumin-treated preparations, respectively.
The apparent affinities (KD) of the receptor for [125I]α-bungarotoxin were 0.87 ± 0.26
and 0.67 ± 0.23 pM for controls and curcumin, respectively. There was no statistically
significant difference between controls and curcumin-treated groups with respect
to KD and Bmax values (P > 0.05, ANOVA, n = 6–7), suggesting that curcumin does
not compete with α-bungarotoxin at the same binding site. Curcumin up to a
concentration of 100 μM did not cause a significant change on the specific binding of
[125I]α-bungarotoxin (Figure 58B).
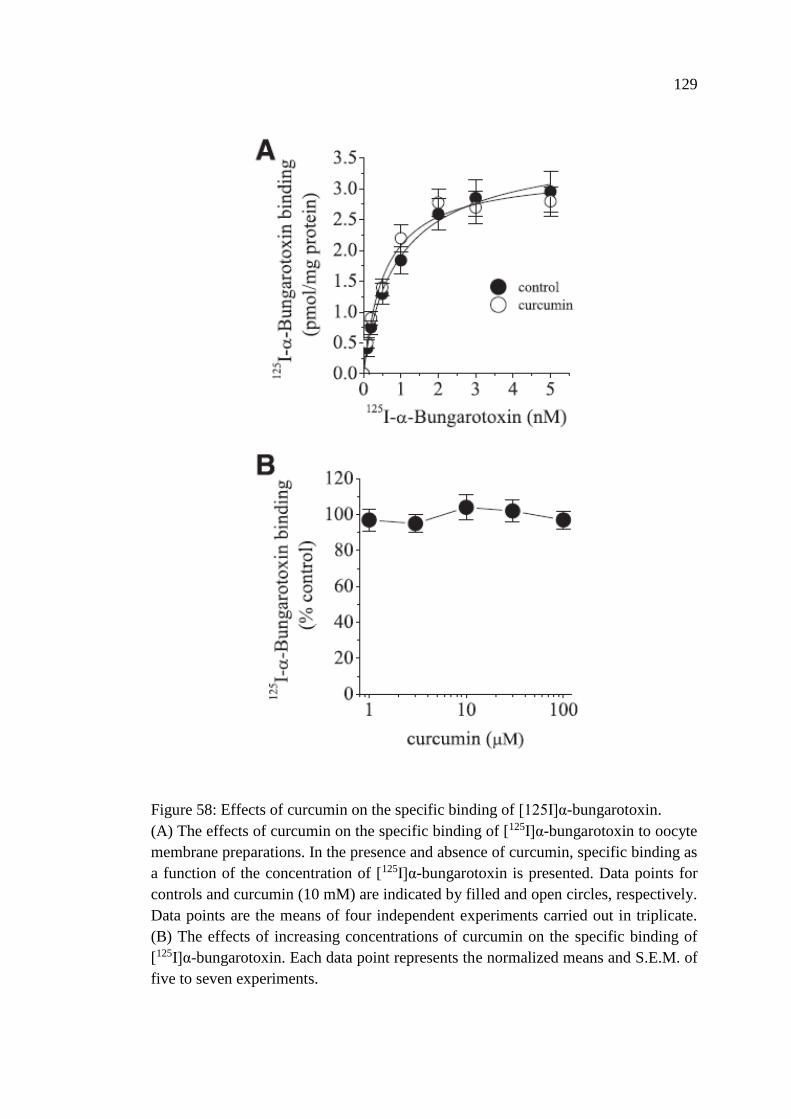
129
Figure 58: Effects of curcumin on the specific binding of [125I]α-bungarotoxin.
(A) The effects of curcumin on the specific binding of [125I]α-bungarotoxin to oocyte
membrane preparations. In the presence and absence of curcumin, specific binding as
a function of the concentration of [125I]α-bungarotoxin is presented. Data points for
controls and curcumin (10 mM) are indicated by filled and open circles, respectively.
Data points are the means of four independent experiments carried out in triplicate.
(B) The effects of increasing concentrations of curcumin on the specific binding of
[125I]α-bungarotoxin. Each data point represents the normalized means and S.E.M. of
five to seven experiments.

130
4.1.10 Effects of Curcumin on the Current Mediated by Different Nicotinic
Receptor Subunits and Other Members of Ligand-Gated Ion Channels
The effect of curcumin on the functional properties of other neuronal nAChR
subtypes was also examined. Application of curcumin (10 µM for 15 minutes) did not
cause alterations of ACh (100 µM)-induced currents mediated by different subtype
combinations of nicotinic receptors expressed in oocytes (Figure 59A). Similarly,
curcumin (10 µM for 15 minutes) did not cause significant changes on the amplitudes
of currents mediated by 5-HT3A (1 µM 5-HT) subunit and glycine receptors (30 µM
glycine; mediated by α1β1, α2β1, and α3β1 subunit combinations) (Figure 59B).
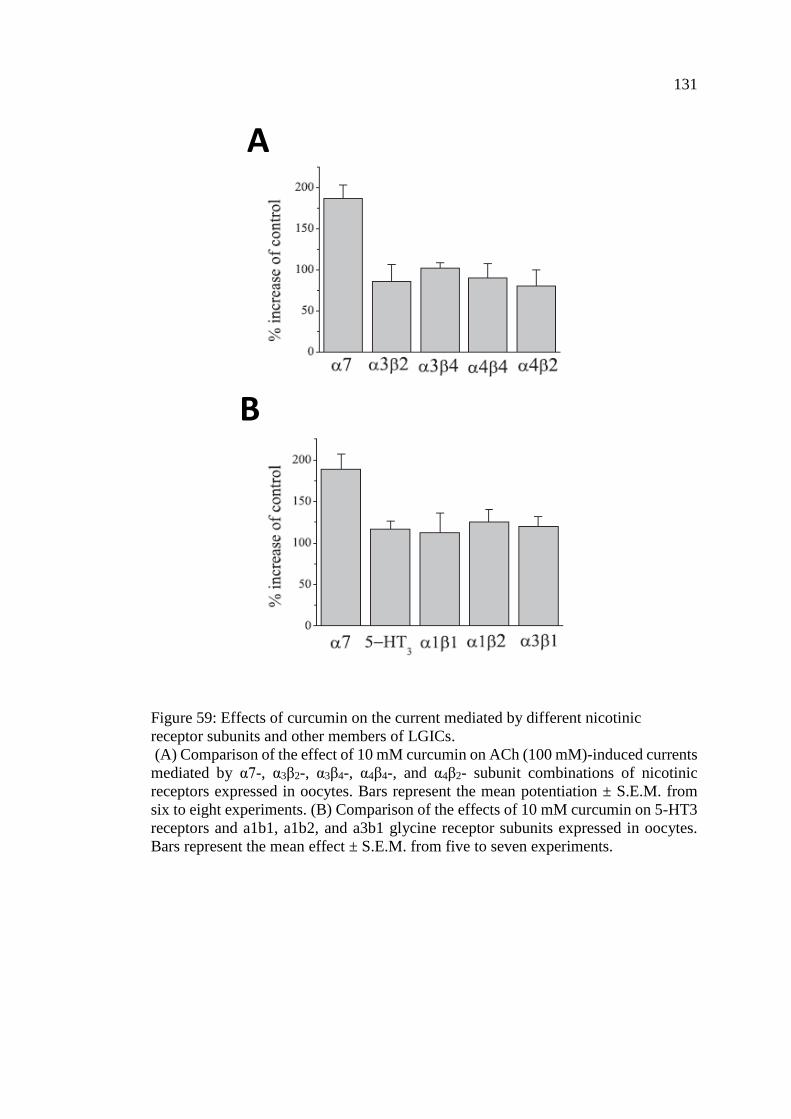
131
Figure 59: Effects of curcumin on the current mediated by different nicotinic
receptor subunits and other members of LGICs.
(A) Comparison of the effect of 10 mM curcumin on ACh (100 mM)-induced currents
mediated by α7-, α3β2-, α3β4-, α4β4-, and α4β2- subunit combinations of nicotinic
receptors expressed in oocytes. Bars represent the mean potentiation ± S.E.M. from
six to eight experiments. (B) Comparison of the effects of 10 mM curcumin on 5-HT3
receptors and a1b1, a1b2, and a3b1 glycine receptor subunits expressed in oocytes.
Bars represent the mean effect ± S.E.M. from five to seven experiments.
B
A

132
4.1.11 Effects of Other Curcumin’s Analogues and Metabolites on the Current
Mediated by α7 Nicotinic Acetylcholine Receptors
The following experiment was done to screen the effects of various selected
curcumin’s analogues and metabolites at 10 µM concentration, on the function of α7-
nAChR. The effects of the two curcumin’s analogues; demethoxycurcumin (DMC),
bisdemethoxycurcumin (BDMC), and seven curcumin’s metabolites;
tetrahydrocurcumin, demethylcurcumin, didemethylcurcumin, vanillylidenacetone,
di-(tert-Butyl-dimethylsilyl) curcumin, O-tert-Butyl-dimethylsilyl curcumin, and
curcumin-d6.
Both curcumin analogues; DMC and BDMC potentiated ACh (100 µM)-induced
currents but lesser than curcumin potentiation (133.3 ± 10.5, 132.3 ± 8.7, and 181.0 ±
11.3 respectively, n=6, ANOVA) (Figure 60A).
Moreover, curcumin showed maximum stimulation of 100 µM ACh-induced
current through α7-nAChR-expressing oocytes compared to all other screened
metabolites (181.0 ± 11.3, n=6) (Figure 60B) and therefore it was selected for further
in-vivo study. Summary of the effects of 15 minutes bath applications of curcumin
analogues and metabolites on the ACh-induced ion currents are shown in Figure 60A
and B.
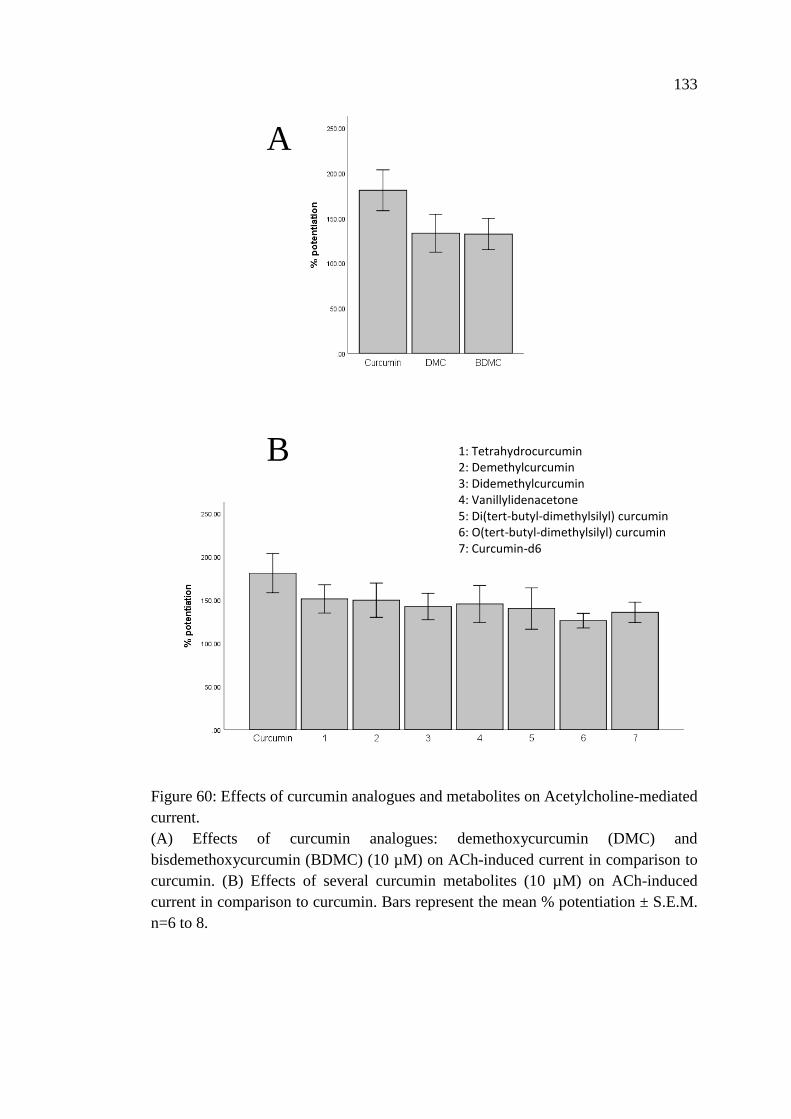
133
Figure 60: Effects of curcumin analogues and metabolites on Acetylcholine-mediated
current.
(A) Effects of curcumin analogues: demethoxycurcumin (DMC) and
bisdemethoxycurcumin (BDMC) (10 µM) on ACh-induced current in comparison to
curcumin. (B) Effects of several curcumin metabolites (10 µM) on ACh-induced
current in comparison to curcumin. Bars represent the mean % potentiation ± S.E.M.
n=6 to 8.
A
B 1: Tetrahydrocurcumin 2: Demethylcurcumin 3: Didemethylcurcumin 4: Vanillylidenacetone 5: Di(tert-butyl-dimethylsilyl) curcumin 6: O(tert-butyl-dimethylsilyl) curcumin 7: Curcumin-d6

134
4.1.12 Docking of Curcumin and Curcumin Derivatives into the Human α7-
nAChR Transmembrane Domain
Docking simulations generated interesting binding modes (Figure 61) for
curcumins with remarkable docking scores, as shown in and Table 12. Curcumin
obtained excellent binding energy, scoring as low Glide-XP score as -10.53 kcal/mol.
All ligands were able to score better binding energies than the known type II PAMs
PNU-120596 (-6.29 kcal/mol) and most of them scored better than TQS (-8.33
kcal/mol). As shown in Figure 61, curcumin had the optimum shape complementarity
to fill the intra-cavity of the α7-nAChR transmembrane domain, despite its large size.
Curcumin was able to form multiple hydrogen bonding interactions with the side
chains of Ser249 and Thr289 as well as the backbone amide of Lys239. Additionally,
its aromatic ring was involved in a cation-π interaction with the protonated amine of
Lys239 and in a π-π interaction with the aromatic ring of Phe230 (Figure 61).
Curcumin binding was also potentiated by forming extensive hydrophobic interactions
with the side chains of the surrounding residues (e.g. Ile222, Ser223 and Ala226).

135
Table 12: Binding energies of curcumin and curcumin derivatives, generated from
their docking into the human α7-nAChR transmembrane domain, along with the
docking scores of two known type II PAMs.
Ligand name Glide-XP (kcal/mol)
Curcumin -10.53
Demethyl curcumin -11.28
Didemethyl curcumin -11.68
Tetrahydro curcumin 10.33
Demethoxy curcumin -9.55
Vanillylidenacetone -6.40
Bisdemethoxy curcumin -8.80
Di-(tert-Butyl-dimethylsilyl) curcumin -6.34
O-tert-Butyl-dimethylsilyl curcumin -9.2
TQS -8.33
PNU-120596 -6.29
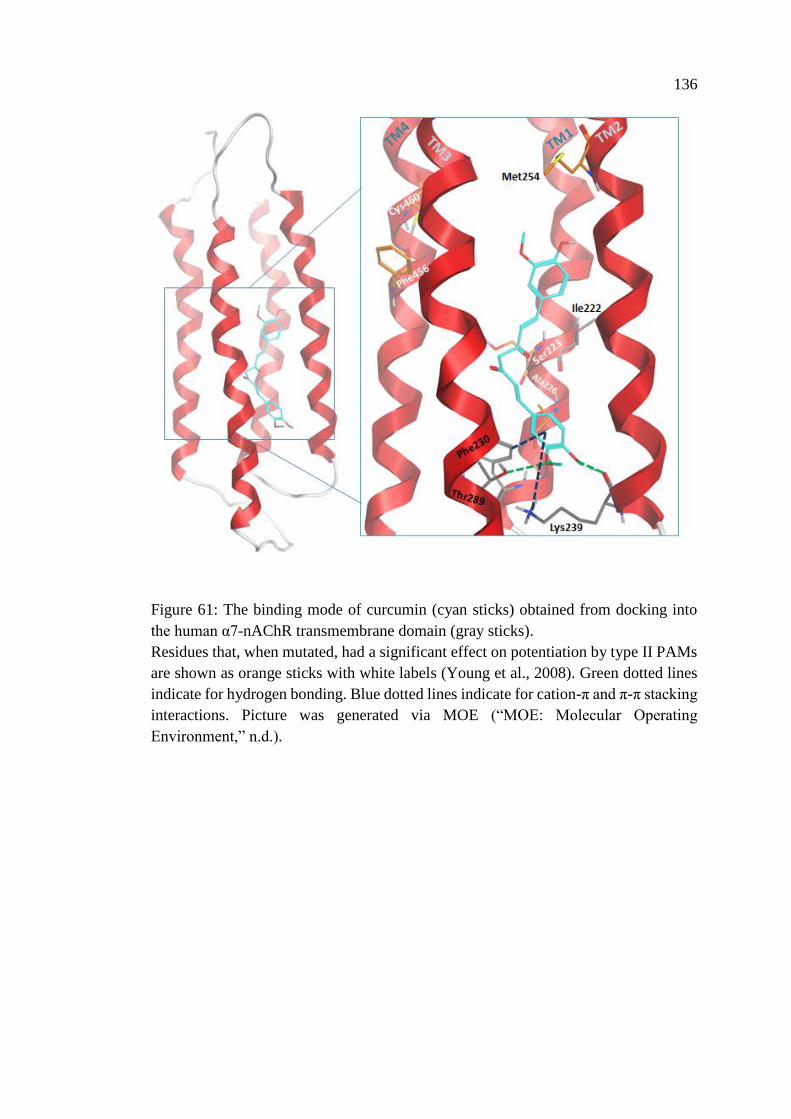
136
Figure 61: The binding mode of curcumin (cyan sticks) obtained from docking into
the human α7-nAChR transmembrane domain (gray sticks).
Residues that, when mutated, had a significant effect on potentiation by type II PAMs
are shown as orange sticks with white labels (Young et al., 2008). Green dotted lines
indicate for hydrogen bonding. Blue dotted lines indicate for cation-π and π-π stacking
interactions. Picture was generated via MOE (“MOE: Molecular Operating
Environment,” n.d.).

137
4.2 In-vivo Results
The neuroprotective effects of curcumin, given orally (200 mg/kg) two weeks pre-
and post-surgically were determined by behavioral and morphological analysis. The
assessment of motor function was performed three weeks after surgery. This was
followed by brain sectioning and immunohistochemistry processing.
4.2.1 Apomorphine-Induced Rotation Test
Deficits in the motor function were clearly observed in the 6-OHDA-treated rats. The
injection of apomorphine in these rats provoked a strong contralateral turning response
with the average of 257.8 ± 23.4 turns/ 30 min in the 6-OHDA group in comparison
with the vehicle treated group where turns in both sides almost negligible (8.9 ± 5.0
turns/ 30 min, ANOVA, P˂0.000). Statistical analysis showed that curcumin
administration has improved motor performance in the 6-OHDA+Cur group as the
turning response was significantly lower compared to 6-OHDA treated group (126.9
± 23.8 turns/ 30 min, ANOVA, P˂ 0.002). However, administration of α7-nAChRs
blocker; MLA abrogated the neuroprotective effect of curcumin in 6-
OHDA+Cur+MLA treated group in comparison with 6-OHDA+Cur group (226.9 ±
23.8 turns/ 30 min, ANOVA, P˂ 0.039). The aim of having 6-OHDA+MLA animal
group is to investigate if there is any effect for MLA per se. Therefore, MLA I.P
injection preceded apomorphine S.C injection by 10 min in 6-OHDA+MLA treated
group. The MLA treated group did not show any significant statistical difference from
6-OHDA or from 6-OHDA+Cur+MLA group (231.7 ± 30.2 turns/ 30 min, ANOVA,
P value in between groups = 1.00) (Figure 62). MLA itself didn’t have any effect on
turning response as indicated by motor assessment performed before and after MLA
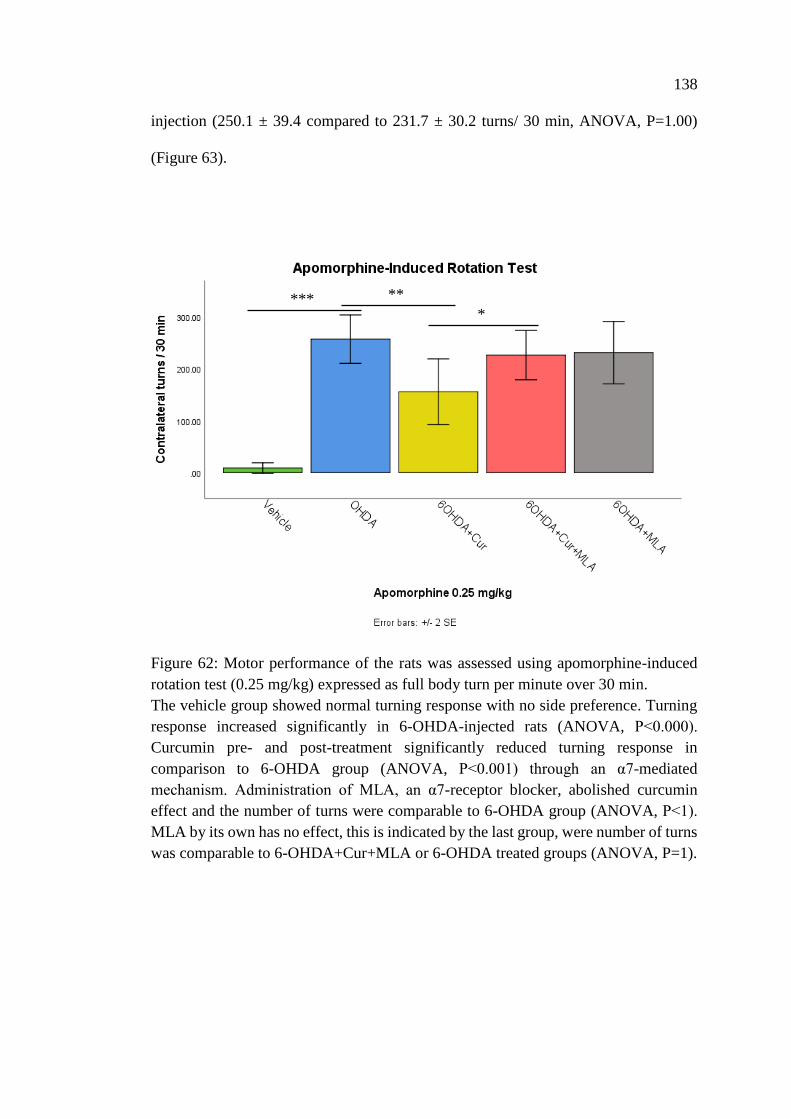
138
injection (250.1 ± 39.4 compared to 231.7 ± 30.2 turns/ 30 min, ANOVA, P=1.00)
(Figure 63).
Figure 62: Motor performance of the rats was assessed using apomorphine-induced
rotation test (0.25 mg/kg) expressed as full body turn per minute over 30 min.
The vehicle group showed normal turning response with no side preference. Turning
response increased significantly in 6-OHDA-injected rats (ANOVA, P˂0.000).
Curcumin pre- and post-treatment significantly reduced turning response in
comparison to 6-OHDA group (ANOVA, P˂0.001) through an α7-mediated
mechanism. Administration of MLA, an α7-receptor blocker, abolished curcumin
effect and the number of turns were comparable to 6-OHDA group (ANOVA, P˂1).
MLA by its own has no effect, this is indicated by the last group, were number of turns
was comparable to 6-OHDA+Cur+MLA or 6-OHDA treated groups (ANOVA, P=1).
*** **
*
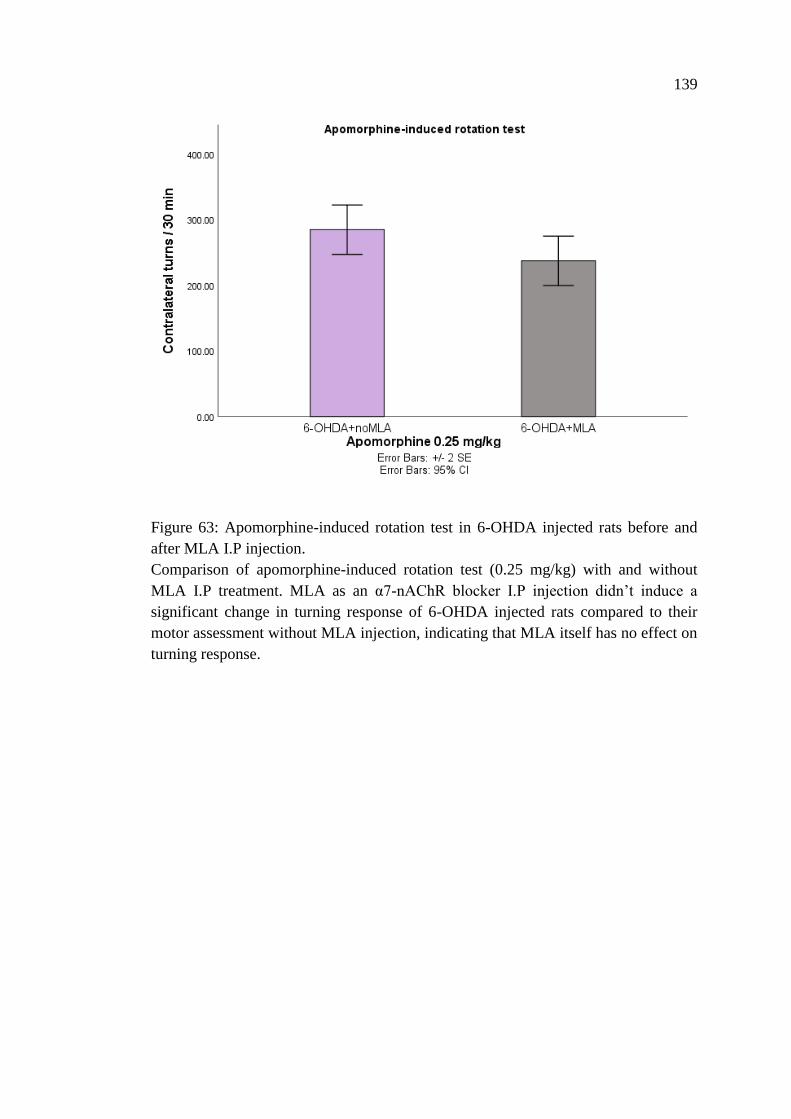
139
Figure 63: Apomorphine-induced rotation test in 6-OHDA injected rats before and
after MLA I.P injection.
Comparison of apomorphine-induced rotation test (0.25 mg/kg) with and without
MLA I.P treatment. MLA as an α7-nAChR blocker I.P injection didn’t induce a
significant change in turning response of 6-OHDA injected rats compared to their
motor assessment without MLA injection, indicating that MLA itself has no effect on
turning response.

140
4.2.2 Morphological Analysis
4.2.2.1 Effects on Tyrosine Hydroxylase-Positive Striatal Innervation
The extent of denervation caused by the intrastriatal 6-OHDA injection was
analyzed by TH-immunohistochemistry in serial coronal sections throughout the
rostro-caudal extent of the striatum. Quantitative assessment of striatal. TH fiber
density was obtained by optical density measurements at four defined rostro-caudal
levels, as shown in Figure 64.
Vehicle treated rats had no significant difference between both lesioned and non-
lesioned side throughout the four rostocaudal levels of the striatum (98.29±5.9 /
ANOVA). In contrast, multiple 6-OHDA-injection resulted in an extensive
denervation in the striatum diffusing all over the four rostocaudal levels compared to
vehicle treated group (7.14±3.2 / ANOVA, P˂0.000). In third group of rats, pre- and
post-surgical curcumin treatment was administered orally on daily bases for four
weeks as described earlier in Methods section. Curcumin treatment has a restorative
effect on TH+ innervated fibers compared to 6-OHDA treated group (32.46±4.2 /
ANOVA, P˂0.011). To investigate whether the effect of curcumin was mediated via
α7-nAChRs or no, we treated the rats with a selective α7-nAChRs blocker; MLA (I.P)
prior to curcumin daily treatment. Blocking α7-nAChR reversed the neuroprotective
effect of curcumin throughout the four levels of striatum in 6-OHDA+Cur+MLA
treated rats compared to 6-OHDA+Cur group (4.81±1.85 / ANOVA, P˂0.005), with
no statistical difference from 6-OHDA group (P˃1.000). When the same amount of
toxin was injected to 6-OHDA+MLA, with MLA injection before motor assessment,
MLA alone showed no effect on striatal innervation (7.79±0.9 / ANOVA), compared
to 6-OHDA or 6-OHDA+Cur+MLA (P˃1.00) (Figure 65).
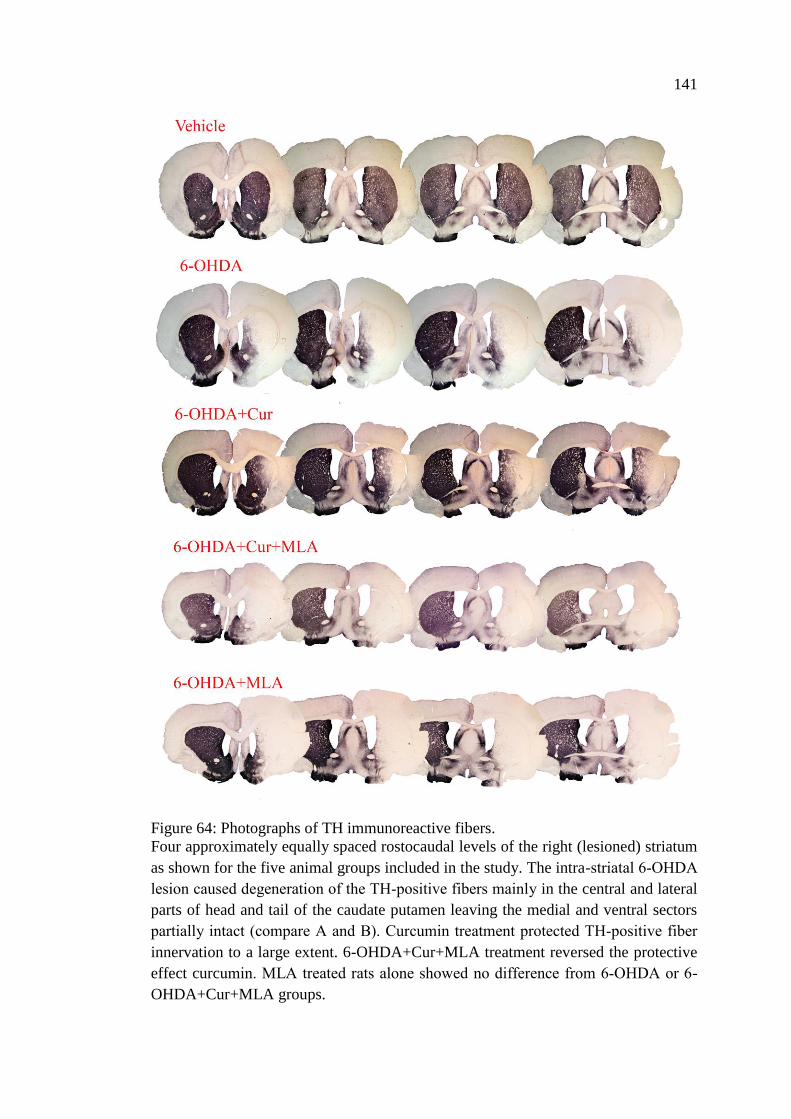
141
Figure 64: Photographs of TH immunoreactive fibers.
Four approximately equally spaced rostocaudal levels of the right (lesioned) striatum
as shown for the five animal groups included in the study. The intra-striatal 6‐OHDA
lesion caused degeneration of the TH‐positive fibers mainly in the central and lateral
parts of head and tail of the caudate putamen leaving the medial and ventral sectors
partially intact (compare A and B). Curcumin treatment protected TH‐positive fiber
innervation to a large extent. 6-OHDA+Cur+MLA treatment reversed the protective
effect curcumin. MLA treated rats alone showed no difference from 6‐OHDA or 6-
OHDA+Cur+MLA groups.
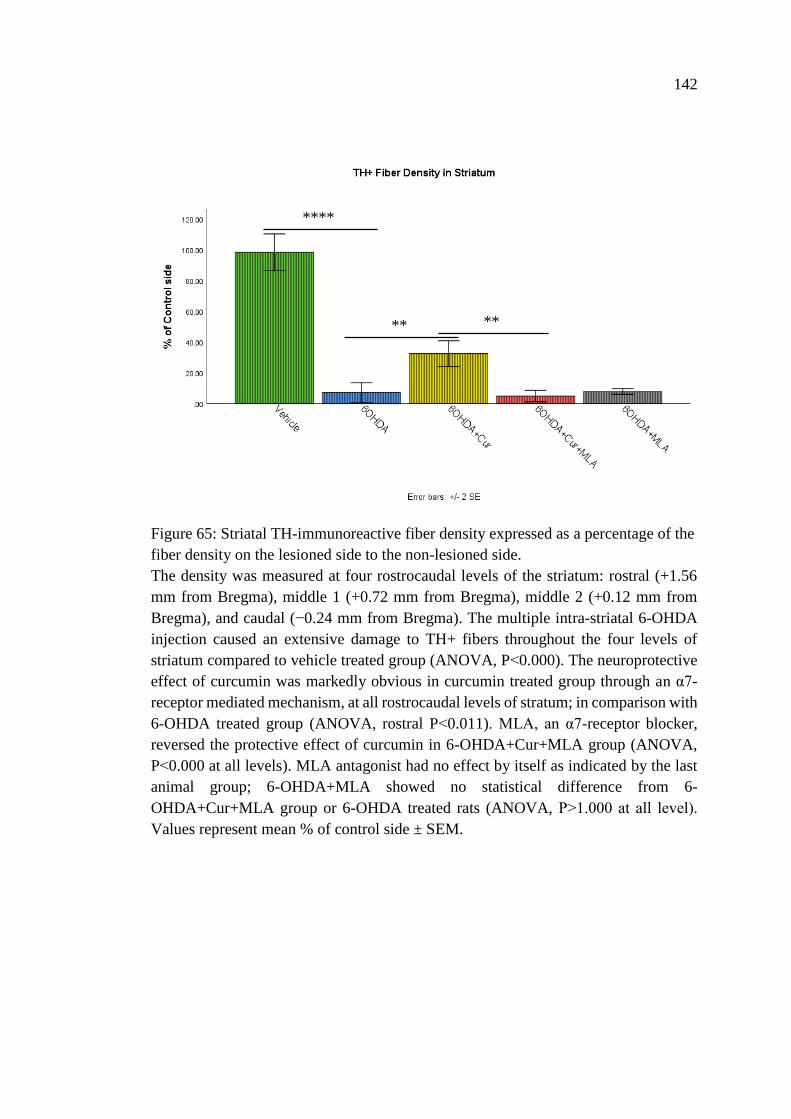
142
Figure 65: Striatal TH-immunoreactive fiber density expressed as a percentage of the
fiber density on the lesioned side to the non-lesioned side.
The density was measured at four rostrocaudal levels of the striatum: rostral (+1.56
mm from Bregma), middle 1 (+0.72 mm from Bregma), middle 2 (+0.12 mm from
Bregma), and caudal (−0.24 mm from Bregma). The multiple intra-striatal 6-OHDA
injection caused an extensive damage to TH+ fibers throughout the four levels of
striatum compared to vehicle treated group (ANOVA, P˂0.000). The neuroprotective
effect of curcumin was markedly obvious in curcumin treated group through an α7-
receptor mediated mechanism, at all rostrocaudal levels of stratum; in comparison with
6-OHDA treated group (ANOVA, rostral P˂0.011). MLA, an α7-receptor blocker,
reversed the protective effect of curcumin in 6-OHDA+Cur+MLA group (ANOVA,
P˂0.000 at all levels). MLA antagonist had no effect by itself as indicated by the last
animal group; 6-OHDA+MLA showed no statistical difference from 6-
OHDA+Cur+MLA group or 6-OHDA treated rats (ANOVA, P˃1.000 at all level).
Values represent mean % of control side ± SEM.
****
** **

143
4.2.2.2 Effects on Tyrosine Hydroxylase-Positive Neurons in Substantia Nigra
The total number of SN TH+ neurons were assessed bilaterally (using the left un-
lesioned side as a control) in each animal by the unbiased stereological analysis using
the optical fractionator principle. The counted region included substantia nigra pars
compacta. The number of TH positive neurons in three different levels of SN (caudal,
middle, and rostral) were counted for each animal; to overcome several anatomical and
technical confounding factors such as; non-homogeneous distribution of dopaminergic
neurons, variability between animals, variability in the immunohistochemical staining
from one batch of specimens to another. The intra-striatal 6-OHDA injection caused a
substantial loss of SN cells with sparing of VTA TH+ neurons. There was no
significant difference bilaterally in vehicle treated group at the three levels; caudal,
middle, rostral, and total survival SN cell analysis [rostral: 100.3±6.2, middle:
94.0±6.6, caudal: 105.6±5.0, total: 99.7±2.5 / ANOVA]. However, on day 21 after 6-
OHDA lesioning, the number of the TH-positive neurons were markedly decreased on
the lesioned side compared with vehicle treated group at all rostrocaudal levels
[ANOVA, rostral: 11.3±2.9 (P˂0.000), middle: 10.2±2.0 (P˂0.000), caudal: 8.9±2.6
(P˂0.000), total: 9.9±1.9 (P˂0.000)]. Curcumin pre- and post-treatment had a
neuroprotective property and could significantly restore 6-OHDA damaging effect on
dopaminergic neurons in comparison with 6-OHDA treated animals at caudal, middle,
rostral and total survival SN cell count in 6-OHDA+Cur treated group [rostral: caudal;
[ANOVA, rostral: 30.2±5.1 (P˂0.003), middle: 39.2±6.8 (P˂0.000), caudal: 42.5±4.6
(P˂0.000), total: 32.0±4.7 (P˂0.000)]. In contrast, blocking of α7-nAChR with MLA
I.P injection prior to curcumin oral administration significantly reversed the
neuroprotective effect of curcumin at all the three levels in 6-OHDA+Cur+MLA
treated group [ANOVA, rostral: 6.7±0.9 (P˂0.000), middle: 12.3±2.6 (P˂0.000),

144
caudal: 12.9±1.3 (P˂0.000), total: 10.3±1.4 (P˂0.000)], supporting that curcumin
neuroprotective effect is mediated via α7-nAChR. The SN cell count in 6-
OHDA+MLA animal group were comparable to 6-OHDA+Cur+MLA or 6-OHDA
groups with no statistical difference between the three groups at all levels [ANOVA,
rostral: 6.7±1.9, middle: 7.0±1.2, caudal: 8.0±3.3, total: 7.1±2.0 (P˃1.0 at all levels)],
as it was not expected from this group to have any change at cellular level and the main
target of this group was to test the behavioral variation due to MLA administration.
Noting that, in all sections, the SN on the un-lesioned left side, both the
morphology and the number of TH-positive neurons remained unchanged (Figures 66
& 67).
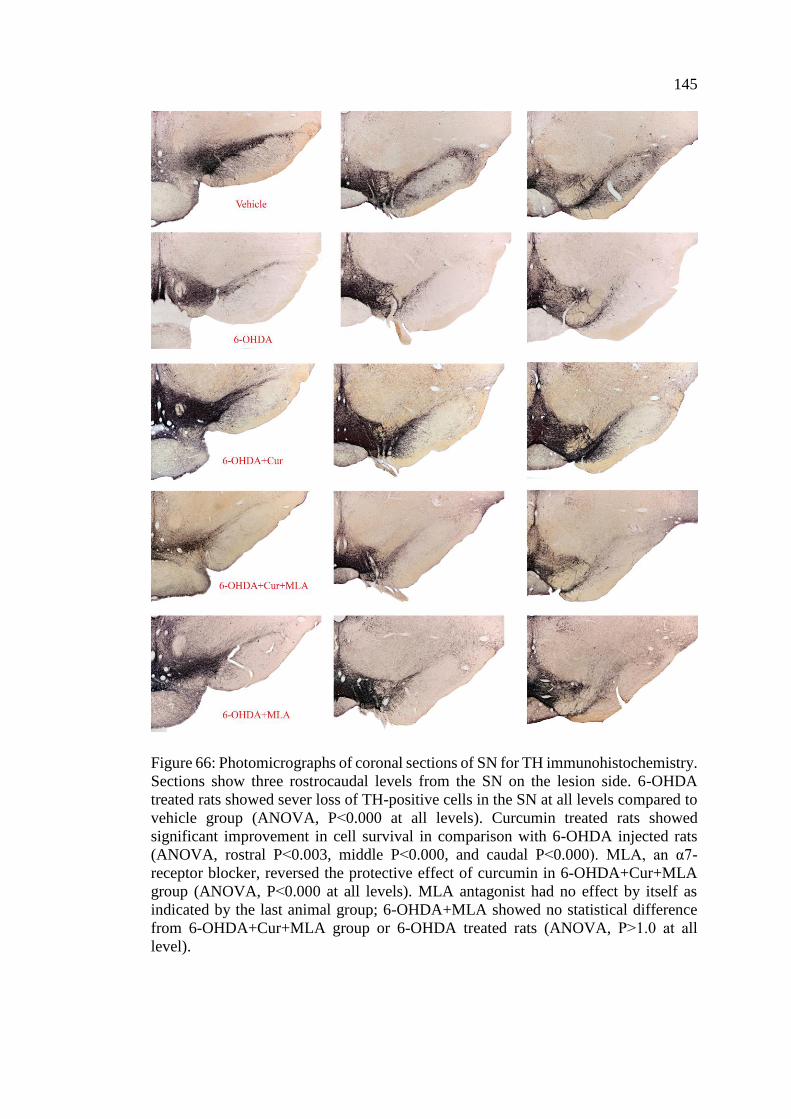
145
Figure 66: Photomicrographs of coronal sections of SN for TH immunohistochemistry.
Sections show three rostrocaudal levels from the SN on the lesion side. 6-OHDA
treated rats showed sever loss of TH-positive cells in the SN at all levels compared to
vehicle group (ANOVA, P˂0.000 at all levels). Curcumin treated rats showed
significant improvement in cell survival in comparison with 6-OHDA injected rats
(ANOVA, rostral P˂0.003, middle P˂0.000, and caudal P˂0.000). MLA, an α7-
receptor blocker, reversed the protective effect of curcumin in 6-OHDA+Cur+MLA
group (ANOVA, P˂0.000 at all levels). MLA antagonist had no effect by itself as
indicated by the last animal group; 6-OHDA+MLA showed no statistical difference
from 6-OHDA+Cur+MLA group or 6-OHDA treated rats (ANOVA, P˃1.0 at all
level).
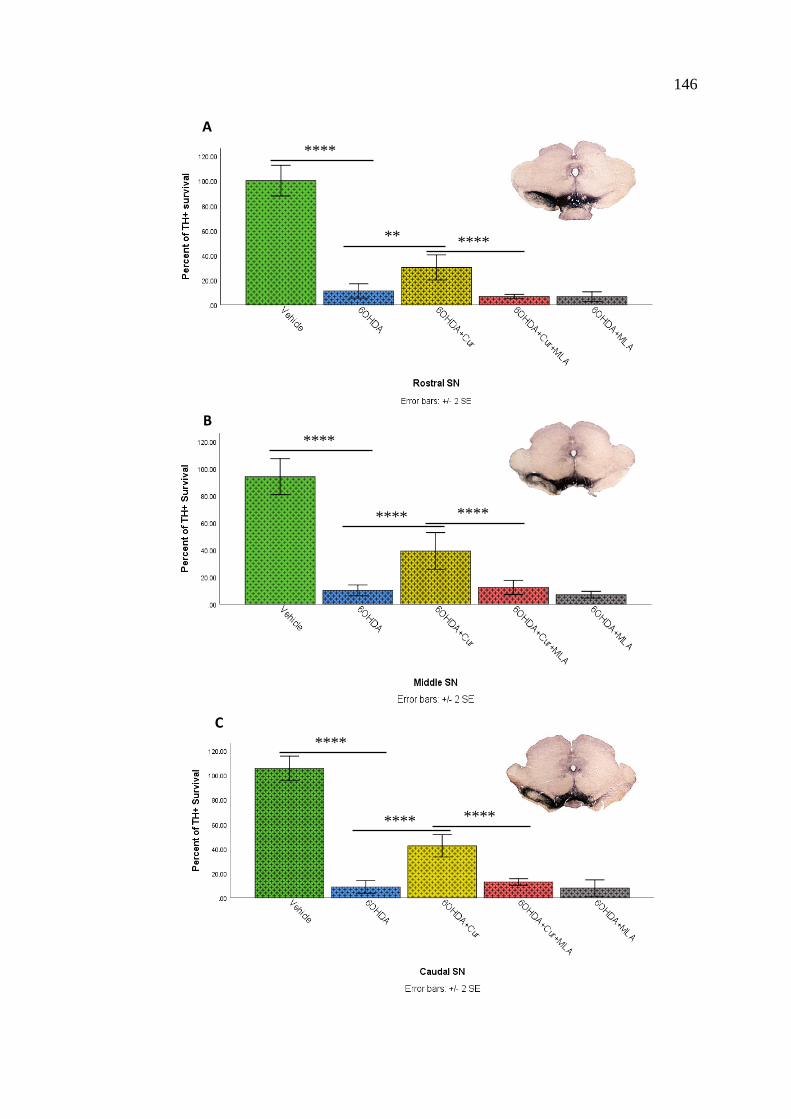
146
A
****
** ****
B
****
**** ****
****
**** ****
C
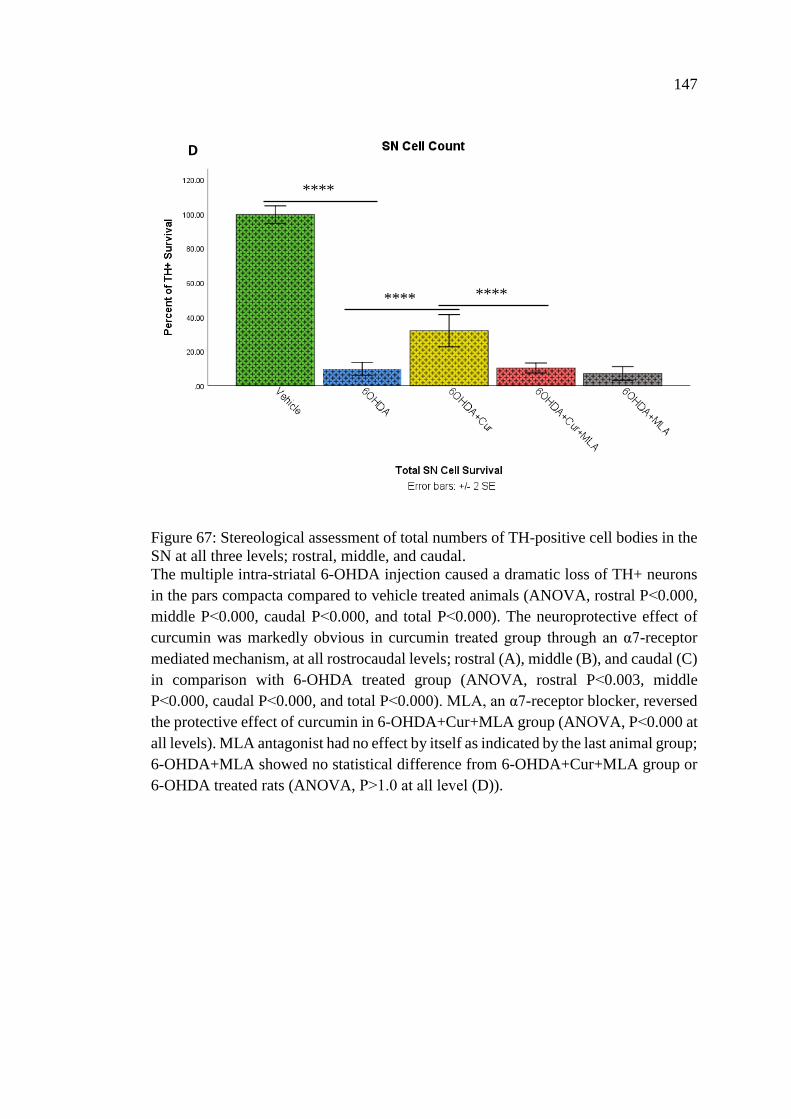
147
Figure 67: Stereological assessment of total numbers of TH-positive cell bodies in the
SN at all three levels; rostral, middle, and caudal.
The multiple intra-striatal 6-OHDA injection caused a dramatic loss of TH+ neurons
in the pars compacta compared to vehicle treated animals (ANOVA, rostral P˂0.000,
middle P˂0.000, caudal P˂0.000, and total P˂0.000). The neuroprotective effect of
curcumin was markedly obvious in curcumin treated group through an α7-receptor
mediated mechanism, at all rostrocaudal levels; rostral (A), middle (B), and caudal (C)
in comparison with 6-OHDA treated group (ANOVA, rostral P˂0.003, middle
P˂0.000, caudal P˂0.000, and total P˂0.000). MLA, an α7-receptor blocker, reversed
the protective effect of curcumin in 6-OHDA+Cur+MLA group (ANOVA, P˂0.000 at
all levels). MLA antagonist had no effect by itself as indicated by the last animal group;
6-OHDA+MLA showed no statistical difference from 6-OHDA+Cur+MLA group or
6-OHDA treated rats (ANOVA, P˃1.0 at all level (D)).
D
**** ****
****

148
Chapter 5: Discussion
5.1 Discussion
In the present study, electrophysiological, in silico (computational), behavioral,
and morphological analyses were used to provide evidence that curcumin (i)
upregulates the function of human α7-nAChRs expressed in Xenopus oocytes and (ii)
reverses neurodegeneration in rat models of 6-OHDA of PD through an α7-nAChR
mechanism.
5.1.1 Effects of Curcumin on α7-Nicotinic Acetylcholine Receptor
Many researchers have extensively studied curcumin in recent years. However,
molecular and cellular targets mediating the pharmacological actions of curcumin
remain largely unknown. In this study, the effects of curcumin on the functional
properties of α7-nicotinic acetylcholine receptor have been investigated, using the two-
electrode voltage clamp technique. The effect of curcumin was tested on ion currents
induced with acetylcholine. Curcumin caused a significant potentiation of the current,
which was partially reversed during washout.
The potentiation effect of curcumin was significantly dependent on the mode of
application. For example, without preincubation, the co-application of curcumin and
ACh did not alter the amplitude of maximal currents. Whereas, preincubation of
curcumin caused an increase in the extent of α7-nAChRs potentiation. It is likely that
this preincubation time is crucial for oocyte preparation to adapt the effect of curcumin
on the expressed α7-nAChRs by the oocytes. This is because the preincubation timing
allows longer timeframe for the compound with the same experimental dose used,
eventually resulting in a more potent effect.

149
The time course of the effect of curcumin on the maximal amplitudes of ACh-
induced currents was relatively slow which might indicate the possible interaction of
curcumin with the lipid membrane. The enhancement of α7-nAChR function by
curcumin is reversible and occurs in a time- and concentration-dependent manner.
Moreover, other curcumin derivatives have been screened and as expected, they
showed potentiation effects on α7-nAChRs, although to a lesser extent in comparison
to curcumin. Being products of curcumin themselves, curcumin metabolites had a
comparable potentiation effects on the receptor function.
The potentiation effect of curcumin was also obvious on other subtypes of nAChRs
and the other members of ligand gated ion channels; serotonin and glycine receptors.
Curcumin was found less effective on other nicotinic receptor subunit combinations
and other members of ligand-gated ion channels.
5.1.2 Effects of Curcumin on α7-Nicotinic Receptor are not Mediated by G-
proteins and Protein Kinases, and are not Dependent on Intracellular
Ca2+ Levels, and Membrane Potential
A relatively slow time course of curcumin effect and the results of earlier studies
on curcumin modulation of various second messenger pathways and kinases
(Mahmmoud, 2007; Takikawa et al., 2013) suggest that activation of G-protein-
coupled receptors (Liu et al., 2013; Pérez-Lara et al., 2011; Yang et al., 2015) and/or
kinase-mediated phosphorylation is involved in curcumin-induced upregulation of α7-
nAChRs (Talwar and Lynch, 2014; Zhang et al., 1995). However, neither treatments
with established kinase inhibitors nor pharmacological disruption of G-protein activity
reversed curcumin potentiation of α7-nAChRs, suggesting that curcumin acts directly
on ion channel-receptor complex. Furthermore, the enhancement of α7-nAChR
function by curcumin is not altered by changes in intracellular Ca2+ levels, or

150
membrane potential. This was evidenced by the same level of curcumin potentiation
at all tested membrane potentials.
In Xenopus oocytes, activation of α7-nAChRs, due to their high Ca2+ permeability,
allows sufficient Ca2+ entry to activate endogenous Ca2+-dependent Cl− channels
(Hartzell et al., 2005; Sands et al., 1993). Ca2+-activated Cl− channels are highly
sensitive to intracellular Ca2+ levels [KD of Ca2+-activated Cl−channels for Ca2+ is less
than 1 µM; see review by (Hartzell et al., 2005)], and alterations in intracellular
Ca2+ levels would be reflected by changes in the holding current under voltage-clamp
conditions. Curcumin has been reported to alter Ca2+ homeostasis in various cell types
(Dyer et al., 2002; Ibrahim et al., 2011; Moustapha et al., 2015; Wang et al., 2012).
Therefore, the direct actions of curcumin on Ca2+-dependent Cl− channels may
contribute to the observed effects of curcumin on ACh-activated currents in this
expression system. However, in Xenopus oocytes injected with BAPTA and recorded
in a solution containing 2 mM Ba2+, co-application of curcumin did not cause
alterations in baseline or holding currents and continued to potentiate α7-nAChR–
mediated ion currents after the chelation of intracellular Ca2+ by BAPTA, suggesting
that Ca2+-dependent Cl− channels were not involved in curcumin potentiation of
nicotinic responses. In addition, the reversal potential in solutions containing Ba2+ was
not altered in the presence of curcumin, suggesting that the potentiation by curcumin
is not due to alterations in the Ca2+ permeability of the α7-nAChR-channel complex.
Negative results in BAPTA calcium chelating experiment; may advocate a second
review of experimental set-up including solutions. More importantly, it would have
been appropriate to test BAPTA effectiveness on any other well-established
intracellular Ca2+-dependent pathway as a positive control study before starting
oocytes’ experiments.

151
5.1.3 Effects of Curcumin at Different Concentrations of Acetylcholine
Previous studies have demonstrated that curcumin acts on several integral
membrane proteins, including enzymes, transporters, and ion channels (Li et al., 2017;
X. Zhang et al., 2014), T-type Ca2+channels in bovine adrenal cells (Enyeart et al.,
2009); IC50 = 10–20 µM), L-type Ca2+ channels in hippocampal neurons (Liu et al.,
2013); IC50 ≈ 10 µM), TREK-1 K+ channels (Enyeart et al., 2008); IC50 = 0.9 µM),
Kv1.4 K+ channels (Liu et al., 2006) in bovine adrenal cells, Kv1.4. K+ channels (Lian
et al., 2013); IC50 = 4.2 µM) in human T-lymphocytes, ERG K+ channels (Hu et al.,
2012), IC50= 5.5 µM; (Choi et al., 2013), IC50 = 10.6 µM; (Banderali et al., 2011),
IC50 = 2 µM), and K+ channels in rabbit coronary arterial smooth muscle cells (Hong
et al., 2013); IC50 = 1.1 µM). In addition to voltage-dependent conductance, curcumin
has also been shown to act on transient-receptor potential receptors (Yeon et al., 2010;
Zhi et al., 2013). In this study, curcumin was applied in the concentration range of 1
nM to 100 µM, and it was found that it can enhance the effects of ACh on the function
of α7-nAChRs in a concentration-dependent manner, with EC50 values ranging from
58 nM to several micromolar. The concentration of curcumin in plasma and its ability
to pass the blood-brain barrier following oral and intravenous administration have been
studied previously (Anand et al., 2007). When curcumin was given orally at a dose of
2 g/kg to rats, a maximum serum concentration of 1.35 µg/ml ± 0.23 μg/mL was
attained at time 0.83 h (Shoba et al., 1998). Since curcumin is a highly lipophilic
compound with a logP (octanol–water partition coefficient) value of 3.3
(https://pubchem.ncbi.nlm.nih.gov/compound/curcumin#section=Top), its membrane
concentration is expected to be considerably higher than blood levels. Therefore, the
functional modulation of α7-nAChRs demonstrated in this study can be

152
pharmacologically relevant. Because of curcumin’s lipophilicity, it would be
interesting to test the effects of curcumin with a different application such as
intracellular injection allowing direct interaction with its transmembrane binding site.
As mentioned earlier, the roles of G-proteins, kinases, and intracellular Ca2+ levels
in curcumin actions were excluded in our functional and pharmacological studies.
Further experiments indicated that the extent of curcumin potentiation decreased
significantly with increasing concentrations of ACh suggesting that curcumin acts
through a non-competitive mechanism of action.
5.1.4 Effects of Curcumin on the Specific Binding of [125I]α-bungarotoxin
By definition, an antagonist is a compound that upon binding to the receptor, have
no effect on their own but rather block the action of an endogenous or exogenous
agonist (Williams and Raddatz, 2006). There are two types of antagonist; competitive
antagonist where the antagonist and the agonist bind to the same active binding site,
so both are competing to bind to the same receptor. While non-competitive antagonist
binds to a different site other than the active site and block the action of the agonist
(Swinney, 2004).
Our results demonstrated the negative correlation between an increase in ACh
concentrations and the extent of curcumin potentiation, and this assumption was
supported by competition radioligand binding experiments. Notably, binding of α-
bungarotoxin, a competitive antagonist of ACh, was not altered in the presence of
curcumin, suggesting that curcumin does not interact with the ACh binding site in the
receptor.

153
5.1.5 Effects of Curcumin on Desensitization of Nicotinic Receptors
It is likely that curcumin, a highly lipophilic agent, first dissolves into the lipid
membrane and then diffuses into a non-annular lipid space to potentiate the function
of the ion channel-receptor complex. Consistent with this idea, the effect of curcumin
on α7-nAChR reached a maximal level within 5–10 minutes of application, suggesting
that the binding site(s) for these allosteric modifiers is located inside the lipid
membrane and requires a relatively slow (in minutes) time course to modulate the
function of the receptor. It is likely that these hydrophobic agents affect the energy
requirements for gating-related conformational changes in ligand-gated ion channels
(Spivak et al., 2007).
α7-nAChRs recovery from desensitization is mainly dependent on agonist potency,
concentration, and duration of exposure. Therefore, for desensitization experiment we
have used a stronger and highly selective nicotinic agonist; nicotine with a dose of 100
µM and much longer exposure time (25 - 30 sec). Contrarily, ACh was used in all our
experiments. This is because ACh mimics the endogenously secreted agonist in its fast
receptor activation of both muscarinic and nicotinic, rapid degradation, and short
response duration.
The hallmark charactarestics of α7-nAChRs include its high permeability to Ca2+,
allowing huge amount of Ca2+ influx into the cell which may have a cytotoxic effect
(Guerra-Álvarez et al., 2015). However, the fast kinetics of α7-nAChRs exemplified
in fast desensitization and brief opening duration may be the reason behind avoidance
of cell toxicity.
It is plausible that curcumin acts as an allosteric modulator for various receptors
and ion channels at the lipid membrane, accounting for some of its pharmacological

154
actions in animal studies (Zhang et al., 2014). Allosteric modulators alter the
functional properties of ligand-gated ion channels by interacting with sites that are
topographically distinct from the ligand binding sites [see review by (Onaran and
Costa, 2009)]. Two different types of positive allosteric modulator (PAM) have been
postulated (Chatzidaki and Millar, 2015; Uteshev, 2014). Whereas type I enhances
agonist-induced currents without affecting macroscopic current kinetics, type II PAMs
delay desensitization and reactivate desensitized receptors. Further analysis of the
curcumin effect indicated that curcumin significantly (more than 3-fold) decreased
desensitization of the receptor and reactivate the already-desensitized nAChR to its
conducting state suggesting that curcumin acts as a type II PAM.
Rapid desensitization of homomeric α7-nAChRs can be tested under different
conditions, by assembling α7 subunit with different stoichiometries; namely β2,
forming functional heteromeric receptor subtype. It is expected that β2 subunit would
result in a slower receptor kinetics. α7β2 receptors have been detected in various brain
areas (Liu et al., 2012, 2009; Moretti et al., 2014; Thomsen et al., 2015; Zoli et al.,
2015). Interestingly, α7β2 has been shown to play a role in the neuropathy of
Alzheimer’s disease and is highly susceptible to inhibition by
the volatile anesthetic isoflurane (Liu et al., 2009; Mowrey et al., 2013).
5.1.6 Docking of Curcumin and Curcumin Derivatives into the Human α7-
nAChR Transmembrane Domain
All of these indications together, have guided us towards molecular docking
studies to identify the exact binding site of curcumin -as a type II positive allosteric
modulator- in relation to α7-nAChRs. The binding site of type II positive allosteric
modulators (PAMs) were proposed to be within the transmembrane domain of α7-
nAChRs (Gill et al., 2011; Young et al., 2008). Several pieces of evidence have

155
indicated that amino acids within TM1-TM3 are responsible for the potentiation effect
induced by PAMs. Out of these residues, mutations of two particular residues Ala225
(in TM1) or Met253 (in TM2) had the greatest influence on α7-nAChR potentiation
by the known type II PAM, PNU-120596 (Young et al., 2008). Corradi and Bouzat
(2016) claimed that PNU-120695 is the most efficacious type II PAM and, therefore
it has been used along with TQS as reference compounds in docking experiments.
Like other type II PAMs, the intra-cavity of transmembrane α7-nAChR domain
could be proposed as the binding site of curcumin and its derivatives. Fortunately,
Bondarenko et al. have recently discovered the structure of the human α7-nAChR
transmembrane domain, intra-cavity of which was used for curcumin docking
(Bondarenko et al., 2014).
Interestingly, all of the interacting residues of curcumin are located in TM1, TM2
and TM3 which are known for their importance for type II PAM binding. Also, many
of those residues, that when mutated play an important role in type II PAM activity,
appear to be in close proximity to curcumin; most importantly Ala226 which had direct
contacts with the ligand carbons. It is highly favorable that mutation at those specific
residues lining the cavity would affect curcumin interaction with the receptor.
Furthermore it has been postulated that this intracavity is a common PAM modulatory
site within the pLGIC superfamily, allowing wide variety of compounds to mediate an
allosteric effect (Corradi et al., 2011; Jayakumary et al., 2010; Nury et al., 2011;
Sauguet et al., 2014).
To sum up, curcumin and curcumin derivatives have what it takes to fit nicely into
the α7-nAChR transmembrane domain intra-cavity, the proposed binding site of type
II PAMs.

156
5.1.7 Neuroprotective Properties of Curcumin in Parkinson’s Disease
Neurodegenerative diseases such as Alzheimer and Parkinson’s disease result from
loss of neurons as a sequel of their dysfunction. As discussed earlier, degenerative
changes in PD occur due to three main mechanisms; mitochondrial dysfunction,
oxidative stress, and modifications in protein handling, affecting all cellular functions
(Franco-Iborra et al., 2016; Olanow and McNaught, 2011; Schapira and Jenner, 2011).
A crucial unmet demand in the management of Parkinson’s disease is the discovery
of new approaches that could slow down, stop, or even reverse the process of
neurodegeneration. Several pieces of experimental evidence have revealed that the
cholinergic system is a potential pharmacological target for the treatment of PD (Guan
et al., 2002; Quik et al., 2012). Nicotine, a selective agonist of α7-nAChRs, enhances
the dopaminergic system in the striatum in animal models of PD (O’Neill et al., 2002;
Picciotto and Zoli, 2008; Quik and Wonnacott, 2011). The widespread distribution of
various subtypes of nicotinic receptors in the brain contribute significantly to nicotinic
receptor-mediated neuroprotection mechanisms. Several in-vitro studies using primary
cultures from different brain regions such as striatal, nigral, cortical, or neuronal cell
lines have demonstrated that pre-treatment with nAChR agonists have a
neuroprotective activity against toxic insults via α7 or α4β2* nAChRs mediated
mechanisms (Bordia et al., 2015; Gatto et al., 2004; O’Neill et al., 2002; Picciotto and
Zoli, 2008; Quik and Kulak, 2002; Roncarati et al., 2009; Ward et al., 2008; Yang et
al., 2017). Thus, drugs/compounds that can modulate/regulate α7-nAChRs may
possibly have a neuroprotective effect against 6-OHDA induced toxicity, which leads
to dopaminergic neuronal loss.

157
Our in-vitro results demonstrated vividly that curcumin enhances the effects of
ACh on the function of α7-nAChRs in a concentration-dependent manner, and that
curcumin significantly decreases desensitization of the receptor leading the proposal
that it acts as a type II PAM. These results suggest that curcumin may play a key role
in brain regions affected by 6-OHDA in animal model of PD. We therefore tested this
effect after systemic in-vivo administration in rats. α7-nAChRs are widely distributed
throughout the brain in neuronal and non-neuronal immune cells, such as microglia
and astrocytes (Liu et al., 2015; Park et al., 2007; Shytle et al., 2004; Wang et al.,
2003). α7-nAChR expressed on immune cells are involved in the initiation,
maintenance, and resolution of inflammation, and modulate neuro-inflammatory
processes. In addition, α7-nAChRs are expressed on microglia regulating
inflammatory factors in the CNS (Bagdas et al., 2018).
A recent systematic literature review involving 13 studies of different PD animal
models, between the periods 2005 to 2014. All these studies except one, elaborated on
the anti-oxidant, anti-inflammatory, and anti-apoptotic properties of curcumin (Figure
68) and proved its neuroprotective activity and ability to improve neurological
functions in different animal models of PD (Wang et al., 2017).
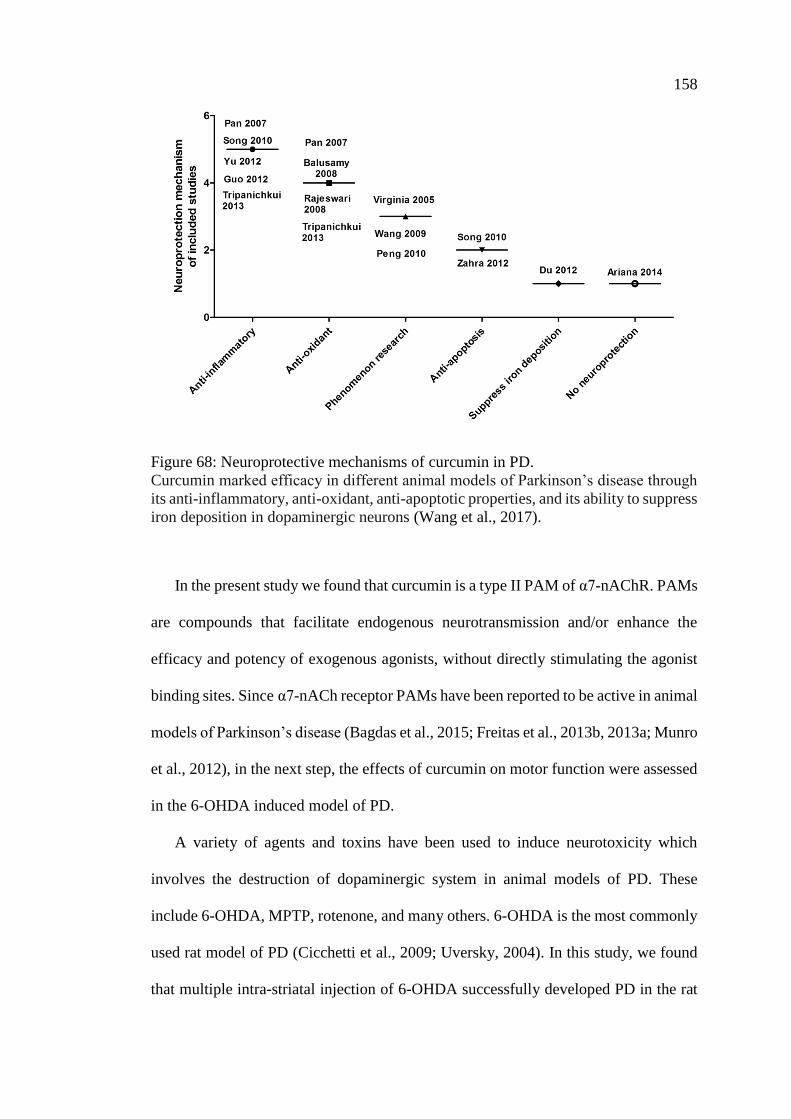
158
Figure 68: Neuroprotective mechanisms of curcumin in PD.
Curcumin marked efficacy in different animal models of Parkinson’s disease through
its anti-inflammatory, anti-oxidant, anti-apoptotic properties, and its ability to suppress
iron deposition in dopaminergic neurons (Wang et al., 2017).
In the present study we found that curcumin is a type II PAM of α7-nAChR. PAMs
are compounds that facilitate endogenous neurotransmission and/or enhance the
efficacy and potency of exogenous agonists, without directly stimulating the agonist
binding sites. Since α7-nACh receptor PAMs have been reported to be active in animal
models of Parkinson’s disease (Bagdas et al., 2015; Freitas et al., 2013b, 2013a; Munro
et al., 2012), in the next step, the effects of curcumin on motor function were assessed
in the 6-OHDA induced model of PD.
A variety of agents and toxins have been used to induce neurotoxicity which
involves the destruction of dopaminergic system in animal models of PD. These
include 6-OHDA, MPTP, rotenone, and many others. 6-OHDA is the most commonly
used rat model of PD (Cicchetti et al., 2009; Uversky, 2004). In this study, we found
that multiple intra-striatal injection of 6-OHDA successfully developed PD in the rat

159
as evidenced by the loss of TH-immunoreactive cells, a marker of dopaminergic
neurons and motor dysfunction of the SNpc. The mortality rate of around 6.8 % in this
study which was calculated from the loss of one rat from 6-OHDA group and two rats
from 6-OHDA+Cur group was relatively low.
The drug-induced rotation test is the gold standard assessment of unilateral 6-
hydroxydopamine (6-OHDA) lesions. The direction of rotation differs according to
the drug used. Amphetamine, a dopamine releasing compound, induces striatal
dopamine release, and also inhibits its reuptake-, thus strongly activating DA release
in the intact side. Therefore, amphetamine induces animals to rotate in a direction
ipsilateral to the lesion side which has weak or no DA to be stimulated (Figure 69)
(Schwarting and Huston, 1996; Ungerstedt, 1971). In contrast, Apomorphine is a direct
DA agonist and has a postsynaptic mechanism of action. Because of apomorphine's
predominant postsynaptic mechanism of action, it stimulates post-synaptic D1 and D2
receptors preferentially in the lesioned side which become supersensitive to
apomorphine stimulation, hence, a dose of 0.25 mg/kg of apomorphine induces a
contralateral rotation in 6-OHDA treated animals (Figure 69) (Hefti et al., 1980a;
Hudson et al., 1993). Previous studies have shown that intra-striatal injection of 6-
OHDA causes retrograde degeneration of SN dopaminergic neurons, resulting in
depletion of striatal dopamine (Singh et al., 2003). Consistent with previous studies,
we found that 21 days after 6-OHDA treatment, rats displayed significant impairment
in the motor functions.
Apomorphine-induced rotation test was used to assess motor abnormalities
between different groups as it induces contralateral rotations only in rats with sever
DA depletion (≥ 75 – 95%) (Schwarting and Huston, 1996). This test is a reliable,
objective, and closely related to the degree of nigrostriatal dysfunction as well as DA
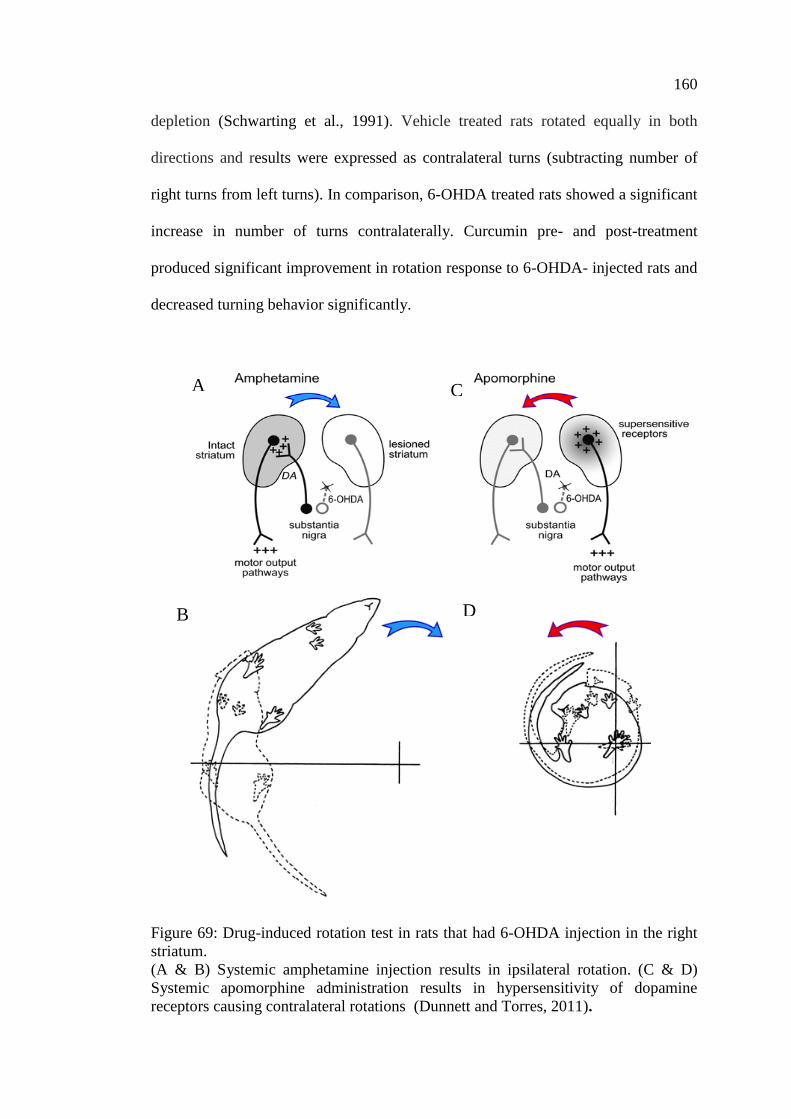
160
depletion (Schwarting et al., 1991). Vehicle treated rats rotated equally in both
directions and results were expressed as contralateral turns (subtracting number of
right turns from left turns). In comparison, 6-OHDA treated rats showed a significant
increase in number of turns contralaterally. Curcumin pre- and post-treatment
produced significant improvement in rotation response to 6-OHDA- injected rats and
decreased turning behavior significantly.
Figure 69: Drug-induced rotation test in rats that had 6-OHDA injection in the right
striatum.
(A & B) Systemic amphetamine injection results in ipsilateral rotation. (C & D)
Systemic apomorphine administration results in hypersensitivity of dopamine
receptors causing contralateral rotations (Dunnett and Torres, 2011).
A
B
C
D

161
Based upon our findings that curcumin treatment significantly decreased turning
response in 6-OHDA treated rats, we investigated the neuroprotective effects of
curcumin against neuronal loss.
According to the study of Dadhaniya et al. (2011), the LD50 of the preparation in
rats as well as in mice was found to be greater than 2000 mg/kg body weight after 90
days treatment protocol. They concluded that no Observed-Adverse-Effect Level
(NOAEL) for the standardized novel curcumin preparation is determined as 750
mg/kg/day, (the highest dose tested) (Dadhaniya et al., 2011). To study the effect of
curcumin at the cellular level, a non-toxic dose of curcumin (200 mg/kg) was used.
Thus, our dose for the in-vivo study is considered safe. Indeed, it is equivalent to 2
g/day in human and very close to the recommended dose suggested by Perkins et al.
(2002).
The present study was designed to assess the ability of oral curcumin
administration to preserve the integrity of nigrostriatal dopaminergic system (cell
bodies, striatal terminals, and motor behavior). The total number of TH+ neurons in
the SNpc of both hemispheres was evaluated using an unbiased stereology technique.
In 6-OHDA+Cur treated group, curcumin significantly reduced the 6-OHDA-induced
degenerative effect on SNpc dopaminergic neuronal cells, indicating that curcumin has
a neuroprotective effect on DA neuronal damage by 6-OHDA. In the next step of the
study, the involvement of α7-nAChRs in curcumin neuroprotection was investigated.
The results demonstrated that neuroprotective effect of curcumin is mediated via α7-
nAChRs as demonstrated in 6-OHDA+Cur+MLA group. In these animals the number
of TH positive neurons in SN were comparable to 6-OHDA group with no significance
difference between both groups. The restorative effect of curcumin was distinctly
reflected on denervated TH+ DA striatal fibers, as curcumin significantly protected the

162
striatal fibers from the 6-OHDA toxin through an α7-nAChRs mediated mechanism as
indicated by the abolished effect of curcumin after the use of MLA in 6-
OHDA+Cur+MLA group. Taken together, our data suggest that, curcumin improved
turning response, striatal fiber density, and SNpc TH-positive neuronal cells, are
considered as a good index for curcumin to have an anti-Parkinsonian effect.
α7-nAChR is considered to be a potential pharmacological target for several
neurological disorders, such as Parkinson’s disease, Alzheimer's disease, and
Schizophrenia. Currently, a number of α7-nAChR agonists and modulators are under
clinical trials (Yang et al., 2017). In case of Parkinson’s disease and nicotinic
receptors, several clinical trials have been conducted as shown in Table 13.
Keeping in view that many clinical trials in which α7-nAChR agonists were used
(e.g.: Tropisetron, EVP-6124) have been either suspended or terminated. This could
be attributed to the lack of high selectivity indicated by cross-activity with other LGIC,
(for example: 5-HT3 receptors) (Huang et al., 2014; Macor et al., 2001).
In comparison, α7-nAChR PAMs, presented very positive and promising results,
especially PNU-120695; a type II PAM which has proved to have a pro-cognitive
effect in rodents and non-human primates but failed in clinical trials for being too-
potent drug causing a very high calcium influx and potential cytotoxic effect (Callahan
et al., 2013; Ng et al., 2007).
As a basic rule in research: “safety remains the most important starting point and
efficacy becomes a matter of validation”, the need of safe and effective approach in
regards of α7-nAChR is mandatory. Curcumin, as demonstrated in our research to be
a type II PAM, is a natural compound with a high safety profile with no reported
toxicity (Lao et al., 2006) has undergone several clinical trials for treatment of

163
neurodegenerative disorders (Table 14) (e.g.: Alzheimer’s disease, cognitive
impairment).
Overall, current findings of clinical trials on nicotinic receptors and Parkinson’s
disease or curcumin and neuro-degenerative disorders such as Parkinson’s disease are
very promising, but further pre-clinical studies and clinical trials are needed to improve
curcumin’s bioavailability and define its hidden targets of curcumin.
Our findings are in agreement with many other previous studies carried out, where
motor, cellular, and biochemical alterations in PD rats have been improved by
curcumin and its derivatives (Agrawal et al., 2012; Khuwaja et al., 2011; Singh and
Kumar, 2017; Song et al., 2016; Tripanichkul and Jaroensuppaperch, 2012, 2013;
Yang et al., 2014; Zbarsky et al., 2005), and our findings suggest a new mechanism
for curcumin-induced neuroprotection.
Figure 70 illustrates the possible mechanisms by which curcumin may act through
α7-nAChR to protect against the neurotoxic effects in an animal model of Parkinson’s
disease. The results of in-vitro, in silico, and in-vivo experiments of this study suggest
that increasing Ca2+ influx through curcumin α7-nAChR potentiation may have a
neuroprotective mechanism in neuronal and non-neuronal cells via various
intracellular mechanisms.
The lipid signaling cascade initiated by PKC, via phosphorylation of
phosphatidylinositol 3-kinase (PI3K/Akt), is credited with modulating the activities of
neuroprotective and apoptotic factors, such as Bcl-2 and caspases, respectively. In
addition to to their neuronal expression, α7-nAChRs are expressed on microglia and
astrocytes, playing a major role in immune response via “cholinergic anti-
inflammatory pathway”, activation of α7-nAChR and increase in IC Ca2+
concentration modulate Janus kinase 2 (JAK2) and/or signal transducer and activator

164
of transcription 3 (STAT3), ending up with upregulation of protein kinase B (PKB)
leading to inhibition of nuclear factor-kB (NFκB). IC Ca2+ trigger protein kinase A
(PKA) and/or calcium-calmodulin-dependent protein kinase (CaMK) which in turn
trigger extracellular signal-regulated mitogen-activated protein kinase (ERK/MAPK)
pathway. ERK/MAPK signaling is a crucial event in cell survival pathway via
upregulation of the cellular transcription factor; cAMP response element-binding
(CREB), increasing gene expression of tyrosine hydroxylase, and enhancing dopamine
release. As an end result, maintaining cell viability through α7-nAChR activation
induce dopamine release from synaptic vesicles via Ca2+-dependent facilitation
mechanism.
Collectively, all or some of these factors may contribute to decrease apoptosis,
modify immune response, and alter synaptic plasticity enhancing neuronal protection
and survival (Hosur and Loring, 2011; Picciotto and Zoli, 2008; Quik and Kulak, 2002;
Ward et al., 2008).
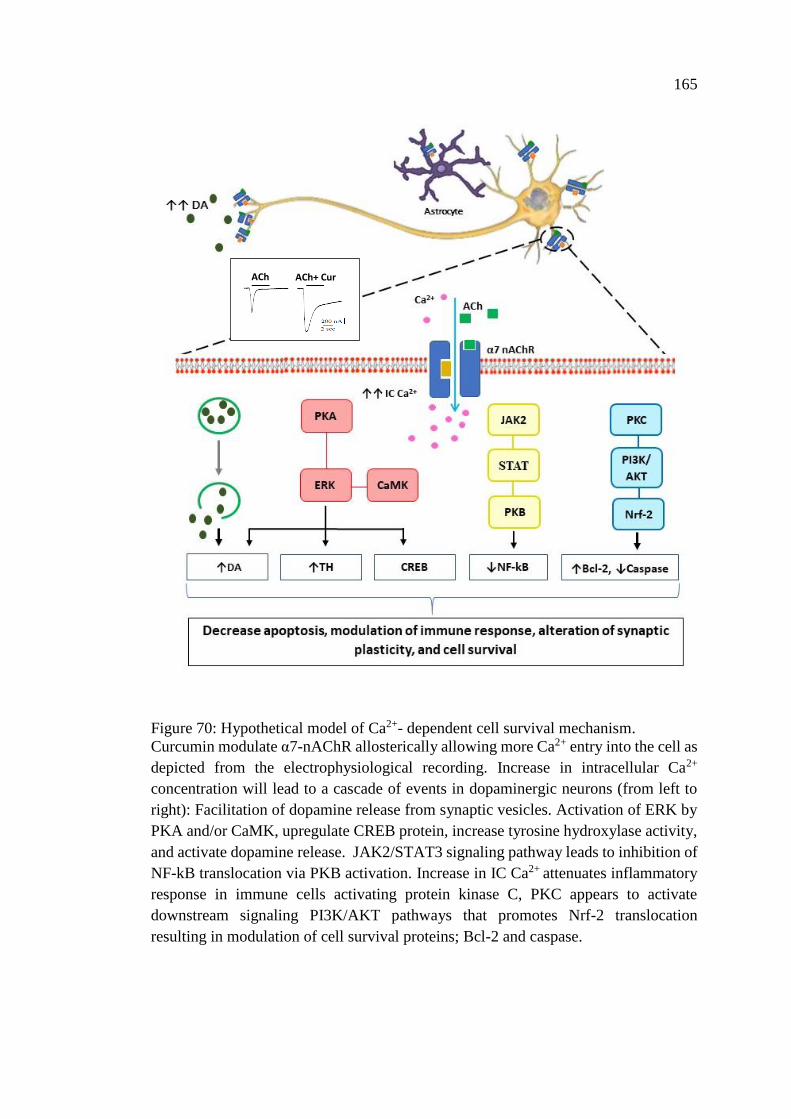
165
Figure 70: Hypothetical model of Ca2+- dependent cell survival mechanism.
Curcumin modulate α7-nAChR allosterically allowing more Ca2+ entry into the cell as
depicted from the electrophysiological recording. Increase in intracellular Ca2+
concentration will lead to a cascade of events in dopaminergic neurons (from left to
right): Facilitation of dopamine release from synaptic vesicles. Activation of ERK by
PKA and/or CaMK, upregulate CREB protein, increase tyrosine hydroxylase activity,
and activate dopamine release. JAK2/STAT3 signaling pathway leads to inhibition of
NF-kB translocation via PKB activation. Increase in IC Ca2+ attenuates inflammatory
response in immune cells activating protein kinase C, PKC appears to activate
downstream signaling PI3K/AKT pathways that promotes Nrf-2 translocation
resulting in modulation of cell survival proteins; Bcl-2 and caspase.
ACh ACh+ Cur
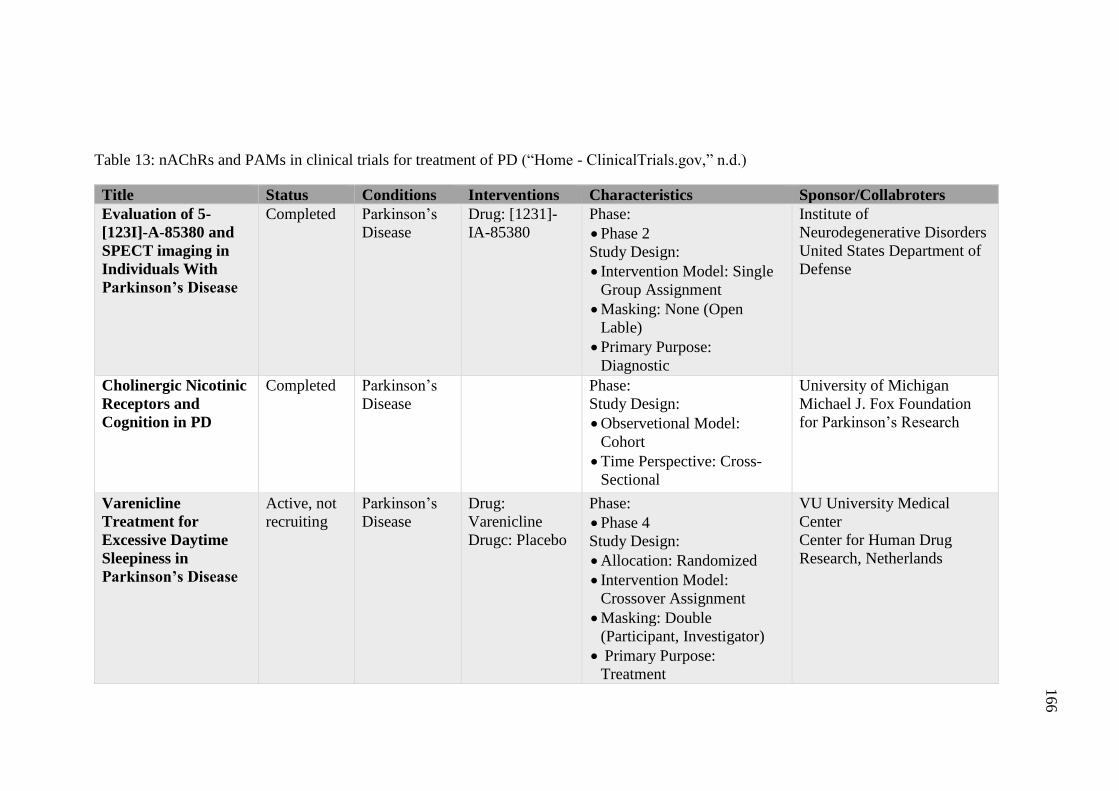
166
Table 13: nAChRs and PAMs in clinical trials for treatment of PD (“Home - ClinicalTrials.gov,” n.d.)
Title Status Conditions Interventions Characteristics Sponsor/Collabroters
Evaluation of 5-
[123I]-A-85380 and
SPECT imaging in
Individuals With
Parkinson’s Disease
Completed Parkinson’s
Disease
Drug: [1231]-
IA-85380
Phase:
• Phase 2
Study Design:
• Intervention Model: Single
Group Assignment
• Masking: None (Open
Lable)
• Primary Purpose:
Diagnostic
Institute of
Neurodegenerative Disorders
United States Department of
Defense
Cholinergic Nicotinic
Receptors and
Cognition in PD
Completed Parkinson’s
Disease
Phase:
Study Design:
• Observetional Model:
Cohort
• Time Perspective: Cross-
Sectional
University of Michigan
Michael J. Fox Foundation
for Parkinson’s Research
Varenicline
Treatment for
Excessive Daytime
Sleepiness in
Parkinson’s Disease
Active, not
recruiting
Parkinson’s
Disease
Drug:
Varenicline
Drugc: Placebo
Phase:
• Phase 4
Study Design:
• Allocation: Randomized
• Intervention Model:
Crossover Assignment
• Masking: Double
(Participant, Investigator)
• Primary Purpose:
Treatment
VU University Medical
Center
Center for Human Drug
Research, Netherlands
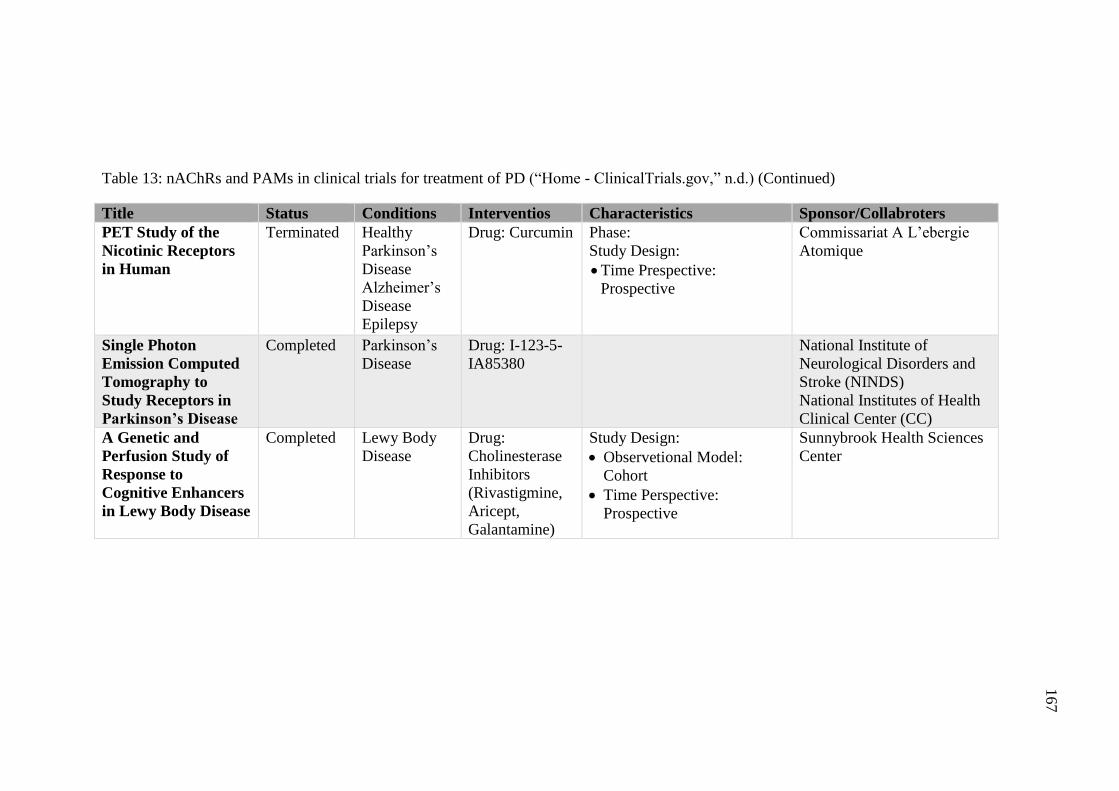
167
Table 13: nAChRs and PAMs in clinical trials for treatment of PD (“Home - ClinicalTrials.gov,” n.d.) (Continued)
Title Status Conditions Interventios Characteristics Sponsor/Collabroters
PET Study of the
Nicotinic Receptors
in Human
Terminated Healthy
Parkinson’s
Disease
Alzheimer’s
Disease
Epilepsy
Drug: Curcumin Phase:
Study Design:
• Time Prespective:
Prospective
Commissariat A L’ebergie
Atomique
Single Photon
Emission Computed
Tomography to
Study Receptors in
Parkinson’s Disease
Completed Parkinson’s
Disease
Drug: I-123-5-
IA85380
National Institute of
Neurological Disorders and
Stroke (NINDS)
National Institutes of Health
Clinical Center (CC)
A Genetic and
Perfusion Study of
Response to
Cognitive Enhancers
in Lewy Body Disease
Completed Lewy Body
Disease
Drug:
Cholinesterase
Inhibitors
(Rivastigmine,
Aricept,
Galantamine)
Study Design:
• Observetional Model:
Cohort
• Time Perspective:
Prospective
Sunnybrook Health Sciences
Center
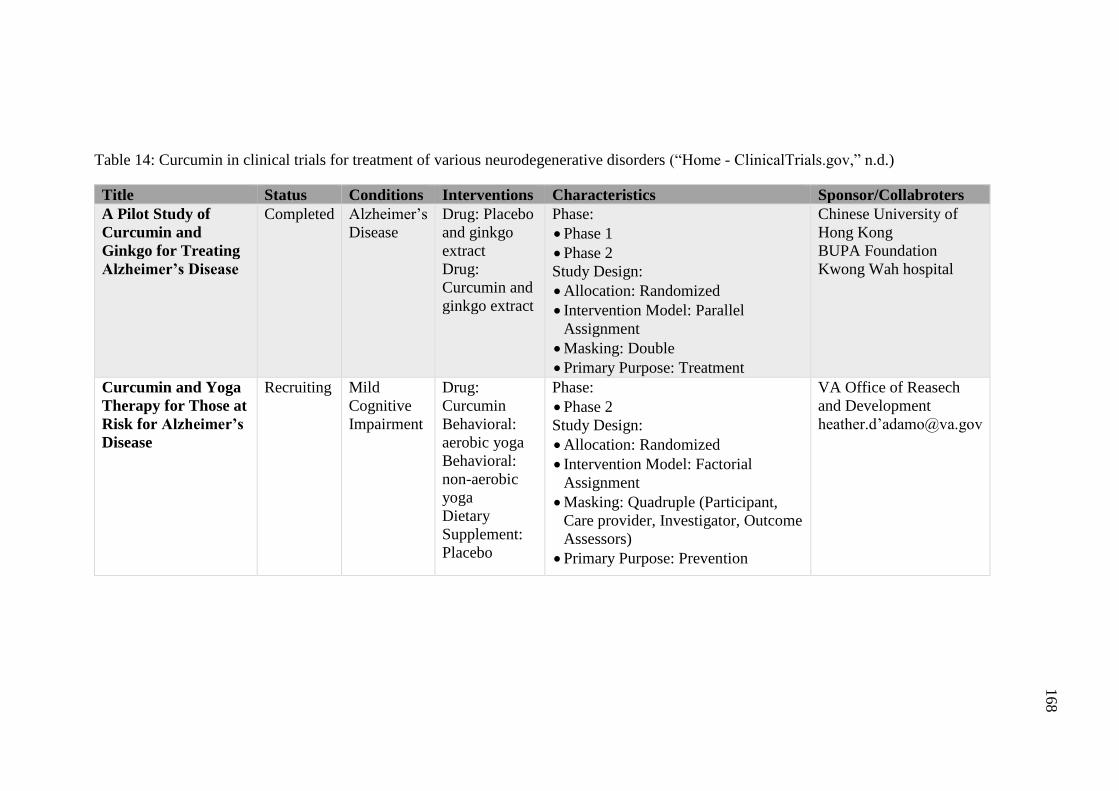
168
Table 14: Curcumin in clinical trials for treatment of various neurodegenerative disorders (“Home - ClinicalTrials.gov,” n.d.)
Title Status Conditions Interventions Characteristics Sponsor/Collabroters
A Pilot Study of
Curcumin and
Ginkgo for Treating
Alzheimer’s Disease
Completed Alzheimer’s
Disease
Drug: Placebo
and ginkgo
extract
Drug:
Curcumin and
ginkgo extract
Phase:
• Phase 1
• Phase 2
Study Design:
• Allocation: Randomized
• Intervention Model: Parallel
Assignment
• Masking: Double
• Primary Purpose: Treatment
Chinese University of
Hong Kong
BUPA Foundation
Kwong Wah hospital
Curcumin and Yoga
Therapy for Those at
Risk for Alzheimer’s
Disease
Recruiting Mild
Cognitive
Impairment
Drug:
Curcumin
Behavioral:
aerobic yoga
Behavioral:
non-aerobic
yoga
Dietary
Supplement:
Placebo
Phase:
• Phase 2
Study Design:
• Allocation: Randomized
• Intervention Model: Factorial
Assignment
• Masking: Quadruple (Participant,
Care provider, Investigator, Outcome
Assessors)
• Primary Purpose: Prevention
VA Office of Reasech
and Development
heather.d’[email protected]
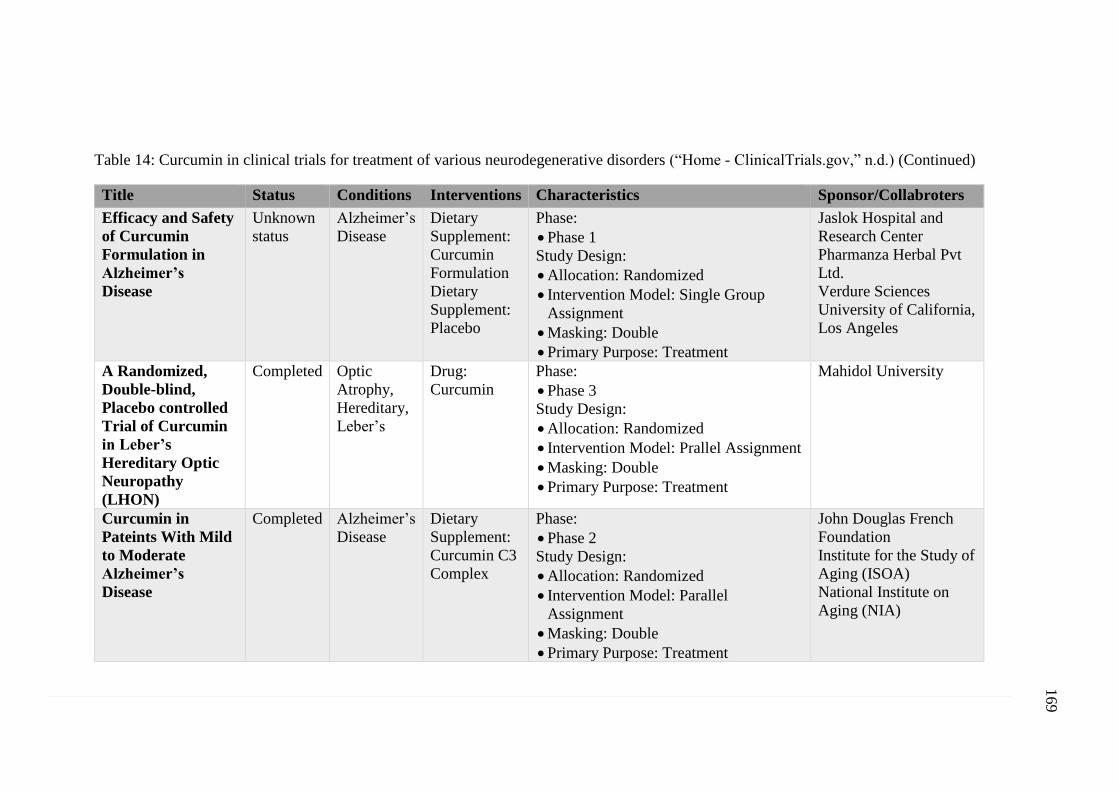
169
Table 14: Curcumin in clinical trials for treatment of various neurodegenerative disorders (“Home - ClinicalTrials.gov,” n.d.) (Continued)
Title Status Conditions Interventions Characteristics Sponsor/Collabroters
Efficacy and Safety
of Curcumin
Formulation in
Alzheimer’s
Disease
Unknown
status
Alzheimer’s
Disease
Dietary
Supplement:
Curcumin
Formulation
Dietary
Supplement:
Placebo
Phase:
• Phase 1
Study Design:
• Allocation: Randomized
• Intervention Model: Single Group
Assignment
• Masking: Double
• Primary Purpose: Treatment
Jaslok Hospital and
Research Center
Pharmanza Herbal Pvt
Ltd.
Verdure Sciences
University of California,
Los Angeles
A Randomized,
Double-blind,
Placebo controlled
Trial of Curcumin
in Leber’s
Hereditary Optic
Neuropathy
(LHON)
Completed Optic
Atrophy,
Hereditary,
Leber’s
Drug:
Curcumin
Phase:
• Phase 3
Study Design:
• Allocation: Randomized
• Intervention Model: Prallel Assignment
• Masking: Double
• Primary Purpose: Treatment
Mahidol University
Curcumin in
Pateints With Mild
to Moderate
Alzheimer’s
Disease
Completed Alzheimer’s
Disease
Dietary
Supplement:
Curcumin C3
Complex
Phase:
• Phase 2
Study Design:
• Allocation: Randomized
• Intervention Model: Parallel
Assignment
• Masking: Double
• Primary Purpose: Treatment
John Douglas French
Foundation
Institute for the Study of
Aging (ISOA)
National Institute on
Aging (NIA)

170
Chapter 6: Conclusions
To our knowledge, this is the first study to establish that:
• Curcumin selectively enhances the effect of ACh on the function of α7-
nAChRs in a concentration dependent manner.
• The effects of curcumin on α7-nicotinic receptor are not mediated by G-
proteins and protein kinases, and are not dependent on intracellular Ca2+ levels,
and membrane potential.
• Curcumin does not interact with ACh binding to receptors.
• Curcumin significantly decreased desensitization of the receptor and
reactivates the already-desensitized nAChR back to conducting state.
• Curcumin and curcumin derivatives fit nicely into the α7-nAChR
transmembrane domain intra-cavity, the proposed binding site of type II
PAMs.
• Curcumin pre- and post-treatment significantly improved motor function in the
6-OHDA induced model of PD.
• Curcumin pre- and post-treatment attenuated the toxic effects of 6-OHDA on
the reduction level of striatal dopamine.
• Curcumin treatment protected dopaminergic neuronal cells in SNpc from 6-
OHDA toxicity, suggesting that curcumin produced significant restoration of
dopamine in the nigrostriatal pathway.

171
Limitations and Future Directions
1- One of the limitations in this study was the lack of positive control in BAPTA
and protein kinase experiments. Testing BAPTA effectiveness on any well-
established intracellular Ca2+-dependent pathway, as a positive control study
before starting oocytes’ experiments, would have provided further evidence
that Ca2+-activated Cl- channels plays no role in curcumin α7-nAChR
activation, and the same applies for protein kinase experiments.
2- Curcumin solubility: DMSO was used as curcumin dissolvent in in-vitro study
with a final concentration of 0.001%. However, in in-vivo study higher doses
of curcumin and DMSO injected intraperitoneally were required. This method
resulted in a high mortality rate. Therefore, it was decided to use different
dissolvent (CMC) and different route of administration (intragastric).
3- Our results from molecular docking experiment demonstrated that curcumin
performed better than PNU-120695 which is considered as a reference
compound for PAM type II. Therefore, it would be very informative to test it
in-vivo and perform a comparative study in PD animal model.
4- Parkinson’s disease is neurodegenerative disorder, where inflammation and
oxidative stress plays a major role in the pathophysiology of the disease. Thus,
biochemical analysis and western blotting of anti-inflammatory, and anti-
oxidant biomarkers would have provided valuable information and should be
the next step.

172
References
Abeliovich, A., Schmitz, Y., Fariñas, I., Choi-Lundberg, D., Ho, W.H., Castillo, P.E.,
Shinsky, N., Verdugo, J.M., Armanini, M., Ryan, A., Hynes, M., Phillips, H.,
Sulzer, D., Rosenthal, A., 2000. Mice lacking alpha-synuclein display
functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252.
Aggarwal, B.B., Harikumar, K.B., 2009. Potential therapeutic effects of curcumin,
the anti-inflammatory agent, against neurodegenerative, cardiovascular,
pulmonary, metabolic, autoimmune and neoplastic diseases. Int. J. Biochem.
Cell Biol. 41, 40–59. https://doi.org/10.1016/j.biocel.2008.06.010
Aggarwal, B.B., Sundaram, C., Malani, N., Ichikawa, H., 2007. Curcumin: the Indian
solid gold. Adv. Exp. Med. Biol. 595, 1–75. https://doi.org/10.1007/978-0-
387-46401-5_1
Aggarwal, B.B., Sung, B., 2009. Pharmacological basis for the role of curcumin in
chronic diseases: an age-old spice with modern targets. Trends Pharmacol.
Sci. 30, 85–94. https://doi.org/10.1016/j.tips.2008.11.002
Agid, Y., Javoy, F., Glowinski, J., Bouvet, D., Sotelo, C., 1973. Injection of 6-
hydroxydopamine into the substantia nigra of the rat. II. Diffusion and
specificity. Brain Res. 58, 291–301.
Agrawal, S.S., Gullaiya, S., Dubey, V., Singh, V., Kumar, A., Nagar, A., Tiwari, P.,
2012. Neurodegenerative Shielding by Curcumin and Its Derivatives on Brain
Lesions Induced by 6-OHDA Model of Parkinson’s Disease in Albino Wistar
Rats. Cardiovasc. Psychiatry Neurol. 2012, 942981.
https://doi.org/10.1155/2012/942981
Ahlskog, J.E., Muenter, M.D., 2001. Frequency of levodopa-related dyskinesias and
motor fluctuations as estimated from the cumulative literature. Mov Disord
16, 448–58.
Alam, M., Schmidt, W.J., 2004. L-DOPA reverses the hypokinetic behaviour and
rigidity in rotenone-treated rats. Behav. Brain Res. 153, 439–446.
https://doi.org/10.1016/j.bbr.2003.12.021
Albuquerque, E. X., Pereira, E.F., Alkondon, M., Rogers, S.W., 2009. Mammalian
nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89,
73–120. https://doi.org/10.1152/physrev.00015.2008
Allam, M.F., Campbell, M.J., Hofman, A., Del Castillo, A.S., Fernandez-Crehuet
Navajas, R., 2004. Smoking and Parkinson’s disease: systematic review of

173
prospective studies. Mov Disord 19, 614–21.
https://doi.org/10.1002/mds.20029
Ammon, H.P., Wahl, M.A., 1991. Pharmacology of Curcuma longa. Planta Med. 57,
1–7. https://doi.org/10.1055/s-2006-960004
Anand, P., Kunnumakkara, A.B., Newman, R.A., Aggarwal, B.B., 2007.
Bioavailability of curcumin: problems and promises. Mol. Pharm. 4, 807–
818. https://doi.org/10.1021/mp700113r
Anand, P., Thomas, S.G., Kunnumakkara, A.B., Sundaram, C., Harikumar, K.B.,
Sung, B., Tharakan, S.T., Misra, K., Priyadarsini, I.K., Rajasekharan, K.N.,
Aggarwal, B.B., 2008. Biological activities of curcumin and its analogues
(Congeners) made by man and Mother Nature. Biochem. Pharmacol. 76,
1590–1611. https://doi.org/10.1016/j.bcp.2008.08.008
Andres-Mateos, E., Perier, C., Zhang, L., Blanchard-Fillion, B., Greco, T.M.,
Thomas, B., Ko, H.S., Sasaki, M., Ischiropoulos, H., Przedborski, S.,
Dawson, T.M., Dawson, V.L., 2007. DJ-1 gene deletion reveals that DJ-1 is
an atypical peroxiredoxin-like peroxidase. Proc. Natl. Acad. Sci. U. S. A.
104, 14807–14812. https://doi.org/10.1073/pnas.0703219104
Antonini, A., Abbruzzese, G., Ferini-Strambi, L., Tilley, B., Huang, J., Stebbins,
G.T., Goetz, C.G., Barone, P., MDS-UPDRS Italian Validation Study Group,
Bandettini di Poggio, M., Fabbrini, G., Di Stasio, F., Tinazzi, M., Bovi, T.,
Ramat, S., Meoni, S., Pezzoli, G., Canesi, M., Martinelli, P., Maria Scaglione,
C.L., Rossi, A., Tambasco, N., Santangelo, G., Picillo, M., Morgante, L.,
Morgante, F., Quatrale, R., Sensi, M., Pilleri, M., Biundo, R., Nordera, G.,
Caria, A., Pacchetti, C., Zangaglia, R., Lopiano, L., Zibetti, M., Zappia, M.,
Nicoletti, A., Quattrone, A., Salsone, M., Cossu, G., Murgia, D., Albanese,
A., Del Sorbo, F., 2013. Validation of the Italian version of the Movement
Disorder Society--Unified Parkinson’s Disease Rating Scale. Neurol. Sci.
Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 34, 683–687.
https://doi.org/10.1007/s10072-012-1112-z
Aoki, H., Takada, Y., Kondo, S., Sawaya, R., Aggarwal, B.B., Kondo, Y., 2007.
Evidence that curcumin suppresses the growth of malignant gliomas in vitro
and in vivo through induction of autophagy: role of Akt and extracellular
signal-regulated kinase signaling pathways. Mol. Pharmacol. 72, 29–39.
https://doi.org/10.1124/mol.106.033167
Arun, N., Nalini, N., 2002. Efficacy of turmeric on blood sugar and polyol pathway
in diabetic albino rats. Plant Foods Hum. Nutr. Dordr. Neth. 57, 41–52.

174
Ashoor, A., Nordman, J.C., Veltri, D., Yang, K.-H.S., Al Kury, L., Shuba, Y.,
Mahgoub, M., Howarth, F.C., Sadek, B., Shehu, A., Kabbani, N., Oz, M.,
2013. Menthol binding and inhibition of α7-nicotinic acetylcholine receptors.
PloS One 8, e67674. https://doi.org/10.1371/journal.pone.0067674
Aum, D.J., Tierney, T.S., 2018. Deep brain stimulation: foundations and future
trends. Front. Biosci. Landmark Ed. 23, 162–182.
Babu, P.S., Srinivasan, K., 1995. Influence of dietary curcumin and cholesterol on
the progression of experimentally induced diabetes in albino rat. Mol. Cell.
Biochem. 152, 13–21.
Bacchelli, E., Battaglia, A., Cameli, C., Lomartire, S., Tancredi, R., Thomson, S.,
Sutcliffe, J.S., Maestrini, E., 2015. Analysis of CHRNA7 rare variants in
autism spectrum disorder susceptibility. Am. J. Med. Genet. A. 167A, 715–
723. https://doi.org/10.1002/ajmg.a.36847
Bagchi, D., Chaudhuri, S., Sardar, S., Choudhury, S., Polley, N., Lemmens, P., Pal,
S.K., 2015. Modulation of stability and functionality of a phyto-antioxidant
by weakly interacting metal ions: curcumin in aqueous solution. RSC Adv. 5,
102516–102524. https://doi.org/10.1039/C5RA21593E
Bagdas, D., Gurun, M.S., Flood, P., Papke, R.L., Damaj, M.I., 2018. New Insights on
Neuronal Nicotinic Acetylcholine Receptors as Targets for Pain and
Inflammation: A Focus on α7 nAChRs. Curr. Neuropharmacol. 16, 415–425.
https://doi.org/10.2174/1570159X15666170818102108
Bagdas, D., Targowska-Duda, K.M., López, J.J., Perez, E.G., Arias, H.R., Damaj,
M.I., 2015. The Antinociceptive and Antiinflammatory Properties of 3-furan-
2-yl-N-p-tolyl-acrylamide, a Positive Allosteric Modulator of α7 Nicotinic
Acetylcholine Receptors in Mice. Anesth. Analg. 121, 1369–1377.
https://doi.org/10.1213/ANE.0000000000000902
Banderali, U., Belke, D., Singh, A., Jayanthan, A., Giles, W.R., Narendran, A., 2011.
Curcumin blocks Kv11.1 (erg) potassium current and slows proliferation in
the infant acute monocytic leukemia cell line THP-1. Cell. Physiol. Biochem.
Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 28, 1169–1180.
https://doi.org/10.1159/000335850
Bastide, M.F., Meissner, W.G., Picconi, B., Fasano, S., Fernagut, P.-O., Feyder, M.,
Francardo, V., Alcacer, C., Ding, Y., Brambilla, R., Fisone, G., Jon Stoessl,
A., Bourdenx, M., Engeln, M., Navailles, S., De Deurwaerdère, P., Ko,
W.K.D., Simola, N., Morelli, M., Groc, L., Rodriguez, M.-C., Gurevich,
E.V., Quik, M., Morari, M., Mellone, M., Gardoni, F., Tronci, E., Guehl, D.,
Tison, F., Crossman, A.R., Kang, U.J., Steece-Collier, K., Fox, S., Carta, M.,

175
Angela Cenci, M., Bézard, E., 2015. Pathophysiology of L-dopa-induced
motor and non-motor complications in Parkinson’s disease. Prog. Neurobiol.
132, 96–168. https://doi.org/10.1016/j.pneurobio.2015.07.002
Bayet-Robert, M., Kwiatkowski, F., Leheurteur, M., Gachon, F., Planchat, E., Abrial,
C., Mouret-Reynier, M.-A., Durando, X., Barthomeuf, C., Chollet, P., 2010.
Phase I dose escalation trial of docetaxel plus curcumin in patients with
advanced and metastatic breast cancer. Cancer Biol. Ther. 9, 8–14.
Beach, T.G., White, C.L., Hamilton, R.L., Duda, J.E., Iwatsubo, T., Dickson, D.W.,
Leverenz, J.B., Roncaroli, F., Buttini, M., Hladik, C.L., Sue, L.I., Noorigian,
J.V., Adler, C.H., 2008. Evaluation of alpha-synuclein immunohistochemical
methods used by invited experts. Acta Neuropathol. (Berl.) 116, 277–288.
https://doi.org/10.1007/s00401-008-0409-8
Belcaro, G., Hosoi, M., Pellegrini, L., Appendino, G., Ippolito, E., Ricci, A., Ledda,
A., Dugall, M., Cesarone, M.R., Maione, C., Ciammaichella, G., Genovesi,
D., Togni, S., 2014. A controlled study of a lecithinized delivery system of
curcumin (Meriva®) to alleviate the adverse effects of cancer treatment.
Phytother. Res. PTR 28, 444–450. https://doi.org/10.1002/ptr.5014
Belluardo, N., Mudo, G., Blum, M., Fuxe, K., 2000. Central nicotinic receptors,
neurotrophic factors and neuroprotection. Behav Brain Res 113, 21–34.
Benabid, A.L., Chabardes, S., Mitrofanis, J., Pollak, P., 2009. Deep brain stimulation
of the subthalamic nucleus for the treatment of Parkinson’s disease. Lancet
Neurol. 8, 67–81. https://doi.org/10.1016/S1474-4422(08)70291-6
Berardelli, A., Rothwell, J.C., Thompson, P.D., Hallett, M., 2001. Pathophysiology
of bradykinesia in Parkinson’s disease. Brain J. Neurol. 124, 2131–2146.
Bertrand, D., Bertrand, S., Ballivet, M., 1992. Pharmacological properties of the
homomeric alpha 7 receptor. Neurosci. Lett. 146, 87–90.
Betarbet, R., Sherer, T.B., MacKenzie, G., Garcia-Osuna, M., Panov, A.V.,
Greenamyre, J.T., 2000. Chronic systemic pesticide exposure reproduces
features of Parkinson’s disease. Nat. Neurosci. 3, 1301–1306.
https://doi.org/10.1038/81834
Bezard, E., Imbert, C., Gross, C.E., 1998. Experimental models of Parkinson’s
disease: from the static to the dynamic. Rev. Neurosci. 9, 71–90.
Bezard, E., Przedborski, S., 2011. A tale on animal models of Parkinson’s disease.
Mov. Disord. Off. J. Mov. Disord. Soc. 26, 993–1002.
https://doi.org/10.1002/mds.23696

176
Bianchi, L., 2006. Heterologous expression of C. elegans ion channels in Xenopus
oocytes. WormBook. https://doi.org/10.1895/wormbook.1.117.1
Blaha, C.D., Allen, L.F., Das, S., Inglis, W.L., Latimer, M.P., Vincent, S.R., Winn,
P., 1996. Modulation of dopamine efflux in the nucleus accumbens after
cholinergic stimulation of the ventral tegmental area in intact,
pedunculopontine tegmental nucleus-lesioned, and laterodorsal tegmental
nucleus-lesioned rats. J Neurosci 16, 714–22.
Blaha, C.D., Winn, P., 1993. Modulation of dopamine efflux in the striatum
following cholinergic stimulation of the substantia nigra in intact and
pedunculopontine tegmental nucleus-lesioned rats. J Neurosci 13, 1035–44.
Blanchet, P.J., Metman, L.V., Mouradian, M.M., Chase, T.N., 1996. Acute
pharmacologic blockade of dyskinesias in Parkinson’s disease. Mov Disord
11, 580–1. https://doi.org/10.1002/mds.870110516
Blandini, F., Armentero, M.-T., 2012. New pharmacological avenues for the
treatment of L-DOPA-induced dyskinesias in Parkinson’s disease: targeting
glutamate and adenosine receptors. Expert Opin. Investig. Drugs 21, 153–
168. https://doi.org/10.1517/13543784.2012.651457
Blandini, F., Armentero, M.-T., Martignoni, E., 2008. The 6-hydroxydopamine
model: news from the past. Parkinsonism Relat. Disord. 14 Suppl 2, S124-
129. https://doi.org/10.1016/j.parkreldis.2008.04.015
Blesa, J., Phani, S., Jackson-Lewis, V., Przedborski, S., 2012. Classic and new
animal models of Parkinson’s disease. J. Biomed. Biotechnol. 2012, 845618.
https://doi.org/10.1155/2012/845618
Blesa, J., Przedborski, S., 2014. Parkinson’s disease: animal models and
dopaminergic cell vulnerability. Front. Neuroanat. 8, 155.
https://doi.org/10.3389/fnana.2014.00155
Blesa, J., Trigo-Damas, I., Quiroga-Varela, A., Jackson-Lewis, V.R., 2015.
Oxidative stress and Parkinson’s disease. Front. Neuroanat. 9.
https://doi.org/10.3389/fnana.2015.00091
Blum, D., Torch, S., Lambeng, N., Nissou, M., Benabid, A.L., Sadoul, R., Verna,
J.M., 2001. Molecular pathways involved in the neurotoxicity of 6-OHDA,
dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s
disease. Prog. Neurobiol. 65, 135–172.
Bockaert, J., Premont, J., Glowinski, J., Thierry, A.M., Tassin, J.P., 1976.
Topographical distribution of dopaminergic innervation and of dopaminergic

177
receptors in the rat striatum. II. Distribution and characteristics of dopamine
adenylate cyclase--interaction of d-LSD with dopaminergic receptors. Brain
Res. 107, 301–315.
Bondarenko, V., Mowrey, D.D., Tillman, T.S., Seyoum, E., Xu, Y., Tang, P., 2014.
NMR structures of the human α7 nAChR transmembrane domain and
associated anesthetic binding sites. Biochim. Biophys. Acta 1838, 1389–
1395. https://doi.org/10.1016/j.bbamem.2013.12.018
Bordia, T., Campos, C., Huang, L., Quik, M., 2008. Continuous and intermittent
nicotine treatment reduces L-3,4-dihydroxyphenylalanine (L-DOPA)-induced
dyskinesias in a rat model of Parkinson’s disease. J Pharmacol Exp Ther 327,
239–47. https://doi.org/10.1124/jpet.108.140897
Bordia, T., McGregor, M., Papke, R.L., Decker, M.W., McIntosh, J.M., Quik, M.,
2015. The alpha7 nicotinic receptor agonist ABT-107 protects against
nigrostriatal damage in rats with unilateral 6-hydroxydopamine lesions. Exp
Neurol 263, 277–84. https://doi.org/10.1016/j.expneurol.2014.09.015
Bové, J., Perier, C., 2012. Neurotoxin-based models of Parkinson’s disease.
Neuroscience 211, 51–76. https://doi.org/10.1016/j.neuroscience.2011.10.057
Braak, H., Braak, E., 2000. Pathoanatomy of Parkinson’s disease. J. Neurol. 247
Suppl 2, II3-10. https://doi.org/10.1007/PL00007758
Braak, H., Braak, E., Yilmazer, D., de Vos, R.A., Jansen, E.N., Bohl, J., Jellinger, K.,
1994. Amygdala pathology in Parkinson’s disease. Acta Neuropathol. (Berl.)
88, 493–500.
Braak, H., Del Tredici, K., Rüb, U., de Vos, R.A.I., Jansen Steur, E.N.H., Braak, E.,
2003. Staging of brain pathology related to sporadic Parkinson’s disease.
Neurobiol. Aging 24, 197–211.
Braak, H., Ghebremedhin, E., Rüb, U., Bratzke, H., Del Tredici, K., 2004. Stages in
the development of Parkinson’s disease-related pathology. Cell Tissue Res.
318, 121–134. https://doi.org/10.1007/s00441-004-0956-9
Branchi, I., D’Andrea, I., Armida, M., Cassano, T., Pèzzola, A., Potenza, R.L.,
Morgese, M.G., Popoli, P., Alleva, E., 2008. Nonmotor symptoms in
Parkinson’s disease: investigating early-phase onset of behavioral
dysfunction in the 6-hydroxydopamine-lesioned rat model. J. Neurosci. Res.
86, 2050–2061. https://doi.org/10.1002/jnr.21642

178
Brooks, A.I., Chadwick, C.A., Gelbard, H.A., Cory-Slechta, D.A., Federoff, H.J.,
1999. Paraquat elicited neurobehavioral syndrome caused by dopaminergic
neuron loss. Brain Res. 823, 1–10.
Brotchie, J., Jenner, P., 2011. New approaches to therapy. Int Rev Neurobiol 98,
123–50. https://doi.org/10.1016/b978-0-12-381328-2.00005-5
Brotchie, J.M., Lee, J., Venderova, K., 2005. Levodopa-induced dyskinesia in
Parkinson’s disease. J Neural Transm Vienna 112, 359–91.
https://doi.org/10.1007/s00702-004-0251-7
Brouet, I., Ohshima, H., 1995. Curcumin, an anti-tumour promoter and anti-
inflammatory agent, inhibits induction of nitric oxide synthase in activated
macrophages. Biochem. Biophys. Res. Commun. 206, 533–540.
Brown, P., Eusebio, A., 2008. Paradoxes of functional neurosurgery: clues from
basal ganglia recordings. Mov Disord 23, 12–20; quiz 158.
https://doi.org/10.1002/mds.21796
Bruyn, G.W., 1983. Handbook of chemical neuroanatomy, vol. 1 (methods in
chemical neuroanatomy): By A. Björklund and T. Hökfelt (eds.), 560 pages,
Elsevier Biomedical Press, Amsterdam, 1983, US$ 140.50, Dfl 330.00. J.
Neurol. Sci. 62, 358. https://doi.org/10.1016/0022-510X(83)90212-5
Cadet, J.L., Krasnova, I.N., Jayanthi, S., Lyles, J., 2007. Neurotoxicity of substituted
amphetamines: molecular and cellular mechanisms. Neurotox. Res. 11, 183–
202.
Calabresi, P., Di Filippo, M., Ghiglieri, V., Picconi, B., 2008. Molecular mechanisms
underlying levodopa-induced dyskinesia. Mov Disord 23 Suppl 3, S570-9.
https://doi.org/10.1002/mds.22019
Callahan, P.M., Hutchings, E.J., Kille, N.J., Chapman, J.M., Terry, A.V., 2013.
Positive allosteric modulator of α7 nicotinic-acetylcholine receptors, PNU-
120596 augments the effects of donepezil on learning and memory in aged
rodents and non-human primates. Neuropharmacology 67, 201–212.
https://doi.org/10.1016/j.neuropharm.2012.10.019
Cannon, J.R., Greenamyre, J.T., 2010. Neurotoxic in vivo models of Parkinson’s
disease recent advances. Prog. Brain Res. 184, 17–33.
https://doi.org/10.1016/S0079-6123(10)84002-6
Cannon, J.R., Tapias, V., Na, H.M., Honick, A.S., Drolet, R.E., Greenamyre, J.T.,
2009. A highly reproducible rotenone model of Parkinson’s disease.
Neurobiol. Dis. 34, 279–290.

179
Cao, L., Liu, J., Zhang, L., Xiao, X., Li, W., 2016. Curcumin inhibits H2O2-induced
invasion and migration of human pancreatic cancer via suppression of the
ERK/NF-κB pathway. Oncol. Rep. 36, 2245–2251.
https://doi.org/10.3892/or.2016.5044
Carlsson, A., 1959. The occurrence, distribution and physiological role of
catecholamines in the nervous system. Pharmacol. Rev. 11, 490–493.
Carlsson, A., Lindqvist, M., Magnusson, T., 1957. 3,4-Dihydroxyphenylalanine and
5-hydroxytryptophan as reserpine antagonists. Nature 180, 1200.
Carroll, R.E., Benya, R.V., Turgeon, D.K., Vareed, S., Neuman, M., Rodriguez, L.,
Kakarala, M., Carpenter, P.M., McLaren, C., Meyskens, F.L., Brenner, D.E.,
2011. Phase IIa clinical trial of curcumin for the prevention of colorectal
neoplasia. Cancer Prev. Res. Phila. Pa 4, 354–364.
https://doi.org/10.1158/1940-6207.CAPR-10-0098
Carta, M., Carlsson, T., Munoz, A., Kirik, D., Bjorklund, A., 2008. Serotonin-
dopamine interaction in the induction and maintenance of L-DOPA-induced
dyskinesias. Prog Brain Res 172, 465–78. https://doi.org/10.1016/s0079-
6123(08)00922-9
Cenci, M.A., Lundblad, M., 2007. Ratings of L-DOPA-induced dyskinesia in the
unilateral 6-OHDA lesion model of Parkinson’s disease in rats and mice.
Curr Protoc Neurosci Chapter 9, Unit 9.25.
https://doi.org/10.1002/0471142301.ns0925s41
Cenci, M.A., Lundblad, M., 2006. Post- versus presynaptic plasticity in L-DOPA-
induced dyskinesia. J Neurochem 99, 381–92. https://doi.org/10.1111/j.1471-
4159.2006.04124.x
Changeux, J.-P., 2010. Allosteric receptors: from electric organ to cognition. Annu.
Rev. Pharmacol. Toxicol. 50, 1–38.
https://doi.org/10.1146/annurev.pharmtox.010909.105741
Changeux, J.P., Kasai, M., Lee, C.Y., 1970. Use of a snake venom toxin to
characterize the cholinergic receptor protein. Proc. Natl. Acad. Sci. U. S. A.
67, 1241–1247.
Chatzidaki, A., Millar, N.S., 2015. Allosteric modulation of nicotinic acetylcholine
receptors. Biochem. Pharmacol. 97, 408–417.
https://doi.org/10.1016/j.bcp.2015.07.028
Chen, A., Xu, J., Johnson, A.C., 2006. Curcumin inhibits human colon cancer cell
growth by suppressing gene expression of epidermal growth factor receptor

180
through reducing the activity of the transcription factor Egr-1. Oncogene 25,
278–287. https://doi.org/10.1038/sj.onc.1209019
Chen, X., Zong, C., Gao, Y., Cai, R., Fang, L., Lu, J., Liu, F., Qi, Y., 2014.
Curcumol exhibits anti-inflammatory properties by interfering with the JNK-
mediated AP-1 pathway in lipopolysaccharide-activated RAW264.7 cells.
Eur. J. Pharmacol. 723, 339–345.
https://doi.org/10.1016/j.ejphar.2013.11.007
Cheng, A.L., Hsu, C.H., Lin, J.K., Hsu, M.M., Ho, Y.F., Shen, T.S., Ko, J.Y., Lin,
J.T., Lin, B.R., Ming-Shiang, W., Yu, H.S., Jee, S.H., Chen, G.S., Chen,
T.M., Chen, C.A., Lai, M.K., Pu, Y.S., Pan, M.H., Wang, Y.J., Tsai, C.C.,
Hsieh, C.Y., 2001. Phase I clinical trial of curcumin, a chemopreventive
agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 21,
2895–2900.
Cheng, K.K., Yeung, C.F., Ho, S.W., Chow, S.F., Chow, A.H.L., Baum, L., 2013.
Highly stabilized curcumin nanoparticles tested in an in vitro blood-brain
barrier model and in Alzheimer’s disease Tg2576 mice. AAPS J. 15, 324–
336. https://doi.org/10.1208/s12248-012-9444-4
Cho, J.-W., Lee, K.-S., Kim, C.-W., 2007. Curcumin attenuates the expression of IL-
1beta, IL-6, and TNF-alpha as well as cyclin E in TNF-alpha-treated HaCaT
cells; NF-kappaB and MAPKs as potential upstream targets. Int. J. Mol. Med.
19, 469–474.
Choi, S.W., Kim, K.S., Shin, D.H., Yoo, H.Y., Choe, H., Ko, T.H., Youm, J.B., Kim,
W.K., Zhang, Y.H., Kim, S.J., 2013. Class 3 inhibition of hERG K+ channel
by caffeic acid phenethyl ester (CAPE) and curcumin. Pflugers Arch. 465,
1121–1134. https://doi.org/10.1007/s00424-013-1239-7
Chung, K.K., Zhang, Y., Lim, K.L., Tanaka, Y., Huang, H., Gao, J., Ross, C.A.,
Dawson, V.L., Dawson, T.M., 2001. Parkin ubiquitinates the alpha-
synuclein-interacting protein, synphilin-1: implications for Lewy-body
formation in Parkinson disease. Nat. Med. 7, 1144–1150.
https://doi.org/10.1038/nm1001-1144
Chung, Y.C., Ko, H.W., Bok, E., Park, E.S., Huh, S.H., Nam, J.H., Jin, B.K., 2010.
The role of neuroinflammation on the pathogenesis of Parkinson’s disease.
BMB Rep 43, 225–32.
Cicchetti, F., Drouin-Ouellet, J., Gross, R.E., 2009. Environmental toxins and
Parkinson’s disease: what have we learned from pesticide-induced animal
models? Trends Pharmacol. Sci. 30, 475–483.
https://doi.org/10.1016/j.tips.2009.06.005

181
Clementi, F., Fornasari, D., Gotti, C., 2000. Neuronal nicotinic receptors, important
new players in brain function. Eur J Pharmacol 393, 3–10.
Cohly, H.H., Taylor, A., Angel, M.F., Salahudeen, A.K., 1998. Effect of turmeric,
turmerin and curcumin on H2O2-induced renal epithelial (LLC-PK1) cell
injury. Free Radic. Biol. Med. 24, 49–54.
Cole, G.M., Teter, B., Frautschy, S.A., 2007. Neuroprotective effects of curcumin.
Adv. Exp. Med. Biol. 595, 197–212. https://doi.org/10.1007/978-0-387-
46401-5_8
Conney, A.H., Lysz, T., Ferraro, T., Abidi, T.F., Manchand, P.S., Laskin, J.D.,
Huang, M.T., 1991. Inhibitory effect of curcumin and some related dietary
compounds on tumor promotion and arachidonic acid metabolism in mouse
skin. Adv. Enzyme Regul. 31, 385–396.
Connolly, B.S., Lang, A.E., 2014. Pharmacological treatment of Parkinson disease: a
review. JAMA 311, 1670–1683. https://doi.org/10.1001/jama.2014.3654
Cormier, A., Morin, C., Zini, R., Tillement, J.P., Lagrue, G., 2003. Nicotine protects
rat brain mitochondria against experimental injuries. Neuropharmacology 44,
642–52.
Corradi, J., Andersen, N., Bouzat, C., 2011. A novel mechanism of modulation of 5-
HT₃A receptors by hydrocortisone. Biophys. J. 100, 42–51.
https://doi.org/10.1016/j.bpj.2010.10.046
Corradi, J., Bouzat, C., 2016. Understanding the Bases of Function and Modulation
of α7 Nicotinic Receptors: Implications for Drug Discovery. Mol. Pharmacol.
90, 288–299. https://doi.org/10.1124/mol.116.104240
Corrodi, H., Hanson, L.C., 1966. Central effects of an inhibitor of tyrosine
hydroxylation. Psychopharmacologia 10, 116–125.
Couturier, S., Bertrand, D., Matter, J.M., Hernandez, M.C., Bertrand, S., Millar, N.,
Valera, S., Barkas, T., Ballivet, M., 1990. A neuronal nicotinic acetylcholine
receptor subunit (alpha 7) is developmentally regulated and forms a homo-
oligomeric channel blocked by alpha-BTX. Neuron 5, 847–856.
Creţu, E., Trifan, A., Vasincu, A., Miron, A., 2012. Plant-derived anticancer agents -
curcumin in cancer prevention and treatment. Rev. Med. Chir. Soc. Med. Nat.
Iasi 116, 1223–1229.
Dadhaniya, P., Patel, C., Muchhara, J., Bhadja, N., Mathuria, N., Vachhani, K., Soni,
M.G., 2011. Safety assessment of a solid lipid curcumin particle preparation:
acute and subchronic toxicity studies. Food Chem. Toxicol. Int. J. Publ. Br.

182
Ind. Biol. Res. Assoc. 49, 1834–1842.
https://doi.org/10.1016/j.fct.2011.05.001
Dajas-Bailador, F., Wonnacott, S., 2004. Nicotinic acetylcholine receptors and the
regulation of neuronal signalling. Trends Pharmacol Sci 25, 317–24.
https://doi.org/10.1016/j.tips.2004.04.006
Dani, J.A., 2001. Overview of nicotinic receptors and their roles in the central
nervous system. Biol Psychiatry 49, 166–74.
Dantuma, N.P., Bott, L.C., 2014. The ubiquitin-proteasome system in
neurodegenerative diseases: precipitating factor, yet part of the solution.
Front. Mol. Neurosci. 7, 70. https://doi.org/10.3389/fnmol.2014.00070
Darvesh, A.S., Carroll, R.T., Bishayee, A., Novotny, N.A., Geldenhuys, W.J., Van
der Schyf, C.J., 2012. Curcumin and neurodegenerative diseases: a
perspective. Expert Opin. Investig. Drugs 21, 1123–1140.
https://doi.org/10.1517/13543784.2012.693479
Dauer, W., Kholodilov, N., Vila, M., Trillat, A.-C., Goodchild, R., Larsen, K.E.,
Staal, R., Tieu, K., Schmitz, Y., Yuan, C.A., Rocha, M., Jackson-Lewis, V.,
Hersch, S., Sulzer, D., Przedborski, S., Burke, R., Hen, R., 2002. Resistance
of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc.
Natl. Acad. Sci. U. S. A. 99, 14524–14529.
https://doi.org/10.1073/pnas.172514599
Dauer, W., Przedborski, S., 2003. Parkinson’s disease: mechanisms and models.
Neuron 39, 889–909.
Davis, G.C., Williams, A.C., Markey, S.P., Ebert, M.H., Caine, E.D., Reichert, C.M.,
Kopin, I.J., 1979. Chronic Parkinsonism secondary to intravenous injection of
meperidine analogues. Psychiatry Res. 1, 249–254.
Dawson, T.M., Ko, H.S., Dawson, V.L., 2010. Genetic animal models of Parkinson’s
disease. Neuron 66, 646–661. https://doi.org/10.1016/j.neuron.2010.04.034
Day, B.J., Patel, M., Calavetta, L., Chang, L.-Y., Stamler, J.S., 1999. A mechanism
of paraquat toxicity involving nitric oxide synthase. Proc. Natl. Acad. Sci. U.
S. A. 96, 12760–12765.
de Lau, L.M.L., Breteler, M.M.B., 2006. Epidemiology of Parkinson’s disease.
Lancet Neurol. 5, 525–535. https://doi.org/10.1016/S1474-4422(06)70471-9
De Smet, F., Christopoulos, A., Carmeliet, P., 2014. Allosteric targeting of receptor
tyrosine kinases. Nat Biotechnol 32, 1113–20.
https://doi.org/10.1038/nbt.3028

183
Del Tredici, K., Braak, H., 2016. Review: Sporadic Parkinson’s disease:
development and distribution of α-synuclein pathology. Neuropathol. Appl.
Neurobiol. 42, 33–50. https://doi.org/10.1111/nan.12298
Deodhar, S.D., Sethi, R., Srimal, R.C., 1980. Preliminary study on antirheumatic
activity of curcumin (diferuloyl methane). Indian J. Med. Res. 71, 632–634.
Derecki, N.C., Cronk, J.C., Kipnis, J., 2013. The role of microglia in brain
maintenance: implications for Rett syndrome. Trends Immunol 34, 144–50.
https://doi.org/10.1016/j.it.2012.10.002
Descarries, L., Mechawar, N., 2000. Ultrastructural evidence for diffuse transmission
by monoamine and acetylcholine neurons of the central nervous system. Prog
Brain Res 125, 27–47. https://doi.org/10.1016/s0079-6123(00)25005-x
Dikshit, M., Rastogi, L., Shukla, R., Srimal, R.C., 1995. Prevention of ischaemia-
induced biochemical changes by curcumin & quinidine in the cat heart.
Indian J. Med. Res. 101, 31–35.
Doherty, K.M., van de Warrenburg, B.P., Peralta, M.C., Silveira-Moriyama, L.,
Azulay, J.-P., Gershanik, O.S., Bloem, B.R., 2011. Postural deformities in
Parkinson’s disease. Lancet Neurol. 10, 538–549.
https://doi.org/10.1016/S1474-4422(11)70067-9
Doty, R.L., Deems, D.A., Stellar, S., 1988. Olfactory dysfunction in parkinsonism: a
general deficit unrelated to neurologic signs, disease stage, or disease
duration. Neurology 38, 1237–1244.
Dunnett, S.B., Torres, E.M., 2011. Rotation in the 6-OHDA-Lesioned Rat, in:
Animal Models of Movement Disorders, Neuromethods. Humana Press, pp.
299–315. https://doi.org/10.1007/978-1-61779-298-4_15
Dyer, J.L., Khan, S.Z., Bilmen, J.G., Hawtin, S.R., Wheatley, M., Javed, M. -u.-H.,
Michelangeli, F., 2002. Curcumin: a new cell-permeant inhibitor of the
inositol 1,4,5-trisphosphate receptor. Cell Calcium 31, 45–52.
Elbaz, A., Moisan, F., 2008. Update in the epidemiology of Parkinson’s disease. Curr
Opin Neurol 21, 454–60. https://doi.org/10.1097/WCO.0b013e3283050461
Emborg, M.E., 2004. Evaluation of animal models of Parkinson’s disease for
neuroprotective strategies. J. Neurosci. Methods 139, 121–143.
https://doi.org/10.1016/j.jneumeth.2004.08.004
Enyeart, J.A., Liu, H., Enyeart, J.J., 2009. Curcumin inhibits ACTH- and angiotensin
II-stimulated cortisol secretion and Ca(v)3.2 current. J. Nat. Prod. 72, 1533–
1537. https://doi.org/10.1021/np900227x

184
Enyeart, J.A., Liu, H., Enyeart, J.J., 2008. Curcumin inhibits bTREK-1 K+ channels
and stimulates cortisol secretion from adrenocortical cells. Biochem.
Biophys. Res. Commun. 370, 623–628.
https://doi.org/10.1016/j.bbrc.2008.04.001
Epelbaum, R., Schaffer, M., Vizel, B., Badmaev, V., Bar-Sela, G., 2010. Curcumin
and gemcitabine in patients with advanced pancreatic cancer. Nutr. Cancer
62, 1137–1141. https://doi.org/10.1080/01635581.2010.513802
Exner, N., Lutz, A.K., Haass, C., Winklhofer, K.F., 2012. Mitochondrial dysfunction
in Parkinson’s disease: molecular mechanisms and pathophysiological
consequences. EMBO J. 31, 3038–3062.
https://doi.org/10.1038/emboj.2012.170
Fagerström, K.O., Pomerleau, O., Giordani, B., Stelson, F., 1994. Nicotine may
relieve symptoms of Parkinson’s disease. Psychopharmacology (Berl.) 116,
117–119.
Fahn, S., 2010. Parkinson’s disease: 10 years of progress, 1997-2007. Mov Disord 25
Suppl 1, S2-14. https://doi.org/10.1002/mds.22796
Fahn, S., 2008. How do you treat motor complications in Parkinson’s disease:
Medicine, surgery, or both? Ann Neurol 64 Suppl 2, S56-64.
https://doi.org/10.1002/ana.21453
Faull, R.L., Laverty, R., 1969. Changes in dopamine levels in the corpus striatum
following lesions in the substantia nigra. Exp. Neurol. 23, 332–340.
Feany, M.B., Bender, W.W., 2000. A Drosophila model of Parkinson’s disease.
Nature 404, 394–398. https://doi.org/10.1038/35006074
Ferger, B., Spratt, C., Earl, C.D., Teismann, P., Oertel, W.H., Kuschinsky, K., 1998.
Effects of nicotine on hydroxyl free radical formation in vitro and on MPTP-
induced neurotoxicity in vivo. Naunyn Schmiedebergs Arch Pharmacol 358,
351–9.
Fernagut, P.O., Hutson, C.B., Fleming, S.M., Tetreaut, N.A., Salcedo, J., Masliah, E.,
Chesselet, M.F., 2007. Behavioral and histopathological consequences of
paraquat intoxication in mice: effects of alpha-synuclein over-expression.
Synap. N. Y. N 61, 991–1001. https://doi.org/10.1002/syn.20456
Ferreira, J.J., Katzenschlager, R., Bloem, B.R., Bonuccelli, U., Burn, D., Deuschl,
G., Dietrichs, E., Fabbrini, G., Friedman, A., Kanovsky, P., Kostic, V.,
Nieuwboer, A., Odin, P., Poewe, W., Rascol, O., Sampaio, C., Schüpbach,
M., Tolosa, E., Trenkwalder, C., Schapira, A., Berardelli, A., Oertel, W.H.,

185
2013. Summary of the recommendations of the EFNS/MDS-ES review on
therapeutic management of Parkinson’s disease. Eur. J. Neurol. 20, 5–15.
https://doi.org/10.1111/j.1468-1331.2012.03866.x
Fleming, S.M., Zhu, C., Fernagut, P.-O., Mehta, A., DiCarlo, C.D., Seaman, R.L.,
Chesselet, M.-F., 2004. Behavioral and immunohistochemical effects of
chronic intravenous and subcutaneous infusions of varying doses of rotenone.
Exp. Neurol. 187, 418–429. https://doi.org/10.1016/j.expneurol.2004.01.023
Flor, P.J., Acher, F.C., 2012. Orthosteric versus allosteric GPCR activation: the great
challenge of group-III mGluRs. Biochem Pharmacol 84, 414–24.
https://doi.org/10.1016/j.bcp.2012.04.013
Formaggio, E., Fazzini, F., Dalfini, A.C., Di Chio, M., Cantu, C., Decimo, I., Fiorini,
Z., Fumagalli, G., Chiamulera, C., 2010. Nicotine increases the expression of
neurotrophin receptor tyrosine kinase receptor A in basal forebrain
cholinergic neurons. Neuroscience 166, 580–9.
https://doi.org/10.1016/j.neuroscience.2009.12.073
Forster, G.L., Blaha, C.D., 2003. Pedunculopontine tegmental stimulation evokes
striatal dopamine efflux by activation of acetylcholine and glutamate
receptors in the midbrain and pons of the rat. Eur J Neurosci 17, 751–62.
Fox, S.H., Chuang, R., Brotchie, J.M., 2009. Serotonin and Parkinson’s disease: On
movement, mood, and madness. Mov. Disord. Off. J. Mov. Disord. Soc. 24,
1255–1266. https://doi.org/10.1002/mds.22473
Fox, S.H., Chuang, R., Brotchie, J.M., 2008. Parkinson’s disease–opportunities for
novel therapeutics to reduce the problems of levodopa therapy. Prog Brain
Res 172, 479–94. https://doi.org/10.1016/s0079-6123(08)00923-0
Franco-Iborra, S., Vila, M., Perier, C., 2016. The Parkinson Disease Mitochondrial
Hypothesis: Where Are We at? Neurosci. Rev. J. Bringing Neurobiol. Neurol.
Psychiatry 22, 266–277. https://doi.org/10.1177/1073858415574600
Freitas, K., Carroll, F.I., Damaj, M.I., 2013a. The antinociceptive effects of nicotinic
receptors α7-positive allosteric modulators in murine acute and tonic pain
models. J. Pharmacol. Exp. Ther. 344, 264–275.
https://doi.org/10.1124/jpet.112.197871
Freitas, K., Ghosh, S., Ivy Carroll, F., Lichtman, A.H., Imad Damaj, M., 2013b.
Effects of α7 positive allosteric modulators in murine inflammatory and
chronic neuropathic pain models. Neuropharmacology 65, 156–164.
https://doi.org/10.1016/j.neuropharm.2012.08.022

186
Friesner, R.A., Murphy, R.B., Repasky, M.P., Frye, L.L., Greenwood, J.R., Halgren,
T.A., Sanschagrin, P.C., Mainz, D.T., 2006. Extra precision glide: docking
and scoring incorporating a model of hydrophobic enclosure for protein-
ligand complexes. J. Med. Chem. 49, 6177–6196.
https://doi.org/10.1021/jm051256o
Gandhi, S., Wood-Kaczmar, A., Yao, Z., Plun-Favreau, H., Deas, E., Klupsch, K.,
Downward, J., Latchman, D.S., Tabrizi, S.J., Wood, N.W., Duchen, M.R.,
Abramov, A.Y., 2009. PINK1-associated Parkinson’s disease is caused by
neuronal vulnerability to calcium-induced cell death. Mol. Cell 33, 627–638.
https://doi.org/10.1016/j.molcel.2009.02.013
Garcea, G., Berry, D.P., Jones, D.J.L., Singh, R., Dennison, A.R., Farmer, P.B.,
Sharma, R.A., Steward, W.P., Gescher, A.J., 2005. Consumption of the
putative chemopreventive agent curcumin by cancer patients: assessment of
curcumin levels in the colorectum and their pharmacodynamic consequences.
Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res.
Cosponsored Am. Soc. Prev. Oncol. 14, 120–125.
Garcea, G., Jones, D.J.L., Singh, R., Dennison, A.R., Farmer, P.B., Sharma, R.A.,
Steward, W.P., Gescher, A.J., Berry, D.P., 2004. Detection of curcumin and
its metabolites in hepatic tissue and portal blood of patients following oral
administration. Br. J. Cancer 90, 1011–1015.
https://doi.org/10.1038/sj.bjc.6601623
Gatto, G.J., Bohme, G.A., Caldwell, W.S., Letchworth, S.R., Traina, V.M., Obinu,
M.C., Laville, M., Reibaud, M., Pradier, L., Dunbar, G., Bencherif, M., 2004.
TC-1734: an orally active neuronal nicotinic acetylcholine receptor
modulator with antidepressant, neuroprotective and long-lasting cognitive
effects. CNS Drug Rev. 10, 147–166.
Gault, L.M., Ritchie, C.W., Robieson, W.Z., Pritchett, Y., Othman, A.A., Lenz, R.A.,
2015. A phase 2 randomized, controlled trial of the α7 agonist ABT-126 in
mild-to-moderate Alzheimer’s dementia. Alzheimers Dement. N. Y. N 1, 81–
90. https://doi.org/10.1016/j.trci.2015.06.001
Gazewood, J.D., Richards, D.R., Clebak, K., 2013a. Parkinson disease: an update.
Am. Fam. Physician 87, 267–273.
Gazewood, J.D., Richards, D.R., Clebak, K., 2013b. Parkinson Disease: An Update.
Am. Fam. Physician 87, 267–273.
Gehrmann, J., Matsumoto, Y., Kreutzberg, G.W., 1995. Microglia: intrinsic
immuneffector cell of the brain. Brain Res Brain Res Rev 20, 269–87.

187
Gerfen, C.R., Herkenham, M., Thibault, J., 1987. The neostriatal mosaic: II. Patch-
and matrix-directed mesostriatal dopaminergic and non-dopaminergic
systems. J Neurosci 7, 3915–34.
Ghalaut, V.S., Sangwan, L., Dahiya, K., Ghalaut, P.S., Dhankhar, R., Saharan, R.,
2012. Effect of imatinib therapy with and without turmeric powder on nitric
oxide levels in chronic myeloid leukemia. J. Oncol. Pharm. Pract. Off. Publ.
Int. Soc. Oncol. Pharm. Pract. 18, 186–190.
https://doi.org/10.1177/1078155211416530
Gill, J.K., Savolainen, M., Young, G.T., Zwart, R., Sher, E., Millar, N.S., 2011.
Agonist activation of alpha7 nicotinic acetylcholine receptors via an allosteric
transmembrane site. Proc Natl Acad Sci U A 108, 5867–72.
https://doi.org/10.1073/pnas.1017975108
Giovanni, A., Sieber, B.A., Heikkila, R.E., Sonsalla, P.K., 1994. Studies on species
sensitivity to the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-
tetrahydropyridine. Part 1: Systemic administration. J. Pharmacol. Exp. Ther.
270, 1000–1007.
Goedert, M., Spillantini, M.G., Del Tredici, K., Braak, H., 2013. 100 years of Lewy
pathology. Nat. Rev. Neurol. 9, 13–24.
https://doi.org/10.1038/nrneurol.2012.242
Goel, A., Aggarwal, B.B., 2010. Curcumin, the golden spice from Indian saffron, is a
chemosensitizer and radiosensitizer for tumors and chemoprotector and
radioprotector for normal organs. Nutr. Cancer 62, 919–930.
https://doi.org/10.1080/01635581.2010.509835
Goel, A., Kunnumakkara, A.B., Aggarwal, B.B., 2008. Curcumin as “Curecumin”:
from kitchen to clinic. Biochem. Pharmacol. 75, 787–809.
https://doi.org/10.1016/j.bcp.2007.08.016
Goetz, C.G., Fahn, S., Martinez-Martin, P., Poewe, W., Sampaio, C., Stebbins, G.T.,
Stern, M.B., Tilley, B.C., Dodel, R., Dubois, B., Holloway, R., Jankovic, J.,
Kulisevsky, J., Lang, A.E., Lees, A., Leurgans, S., LeWitt, P.A., Nyenhuis,
D., Olanow, C.W., Rascol, O., Schrag, A., Teresi, J.A., Van Hilten, J.J.,
LaPelle, N., 2007. Movement Disorder Society-sponsored revision of the
Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Process, format,
and clinimetric testing plan. Mov. Disord. Off. J. Mov. Disord. Soc. 22, 41–
47. https://doi.org/10.1002/mds.21198
Goetz, C.G., Poewe, W., Rascol, O., Sampaio, C., 2005. Evidence-based medical
review update: pharmacological and surgical treatments of Parkinson’s

188
disease: 2001 to 2004. Mov. Disord. Off. J. Mov. Disord. Soc. 20, 523–539.
https://doi.org/10.1002/mds.20464
Goldberg, M.S., Fleming, S.M., Palacino, J.J., Cepeda, C., Lam, H.A., Bhatnagar, A.,
Meloni, E.G., Wu, N., Ackerson, L.C., Klapstein, G.J., Gajendiran, M., Roth,
B.L., Chesselet, M.-F., Maidment, N.T., Levine, M.S., Shen, J., 2003. Parkin-
deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic
neurons. J. Biol. Chem. 278, 43628–43635.
https://doi.org/10.1074/jbc.M308947200
Goldberg, M.S., Pisani, A., Haburcak, M., Vortherms, T.A., Kitada, T., Costa, C.,
Tong, Y., Martella, G., Tscherter, A., Martins, A., Bernardi, G., Roth, B.L.,
Pothos, E.N., Calabresi, P., Shen, J., 2005. Nigrostriatal dopaminergic
deficits and hypokinesia caused by inactivation of the familial Parkinsonism-
linked gene DJ-1. Neuron 45, 489–496.
https://doi.org/10.1016/j.neuron.2005.01.041
Gorell, J.M., Rybicki, B.A., Johnson, C.C., Peterson, E.L., 1999. Smoking and
Parkinson’s disease: a dose-response relationship. Neurology 52, 115–9.
Grady, S.R., Salminen, O., Laverty, D.C., Whiteaker, P., McIntosh, J.M., Collins,
A.C., Marks, M.J., 2007. The subtypes of nicotinic acetylcholine receptors on
dopaminergic terminals of mouse striatum. Biochem Pharmacol 74, 1235–46.
https://doi.org/10.1016/j.bcp.2007.07.032
Graham, D.G., 1978. Oxidative pathways for catecholamines in the genesis of
neuromelanin and cytotoxic quinones. Mol. Pharmacol. 14, 633–643.
Grant, H., Lantos, P.L., Parkinson, C., 1980. Cerebral damage in paraquat poisoning.
Histopathology 4, 185–195.
Greenamyre, J.T., Cannon, J.R., Drolet, R., Mastroberardino, P.-G., 2010. Lessons
from the rotenone model of Parkinson’s disease. Trends Pharmacol. Sci. 31,
141–142; author reply 142-143. https://doi.org/10.1016/j.tips.2009.12.006
Grosset, D.G., Macphee, G.J.A., Nairn, M., Guideline Development Group, 2010.
Diagnosis and pharmacological management of Parkinson’s disease:
summary of SIGN guidelines. BMJ 340, b5614.
https://doi.org/10.1136/bmj.b5614
Guan, Z.Z., Nordberg, A., Mousavi, M., Rinne, J.O., Hellstrom-Lindahl, E., 2002.
Selective changes in the levels of nicotinic acetylcholine receptor protein and
of corresponding mRNA species in the brains of patients with Parkinson’s
disease. Brain Res 956, 358–66.

189
Guerra-Álvarez, M., Moreno-Ortega, A.J., Navarro, E., Fernández-Morales, J.C.,
Egea, J., López, M.G., Cano-Abad, M.F., 2015. Positive allosteric modulation
of alpha-7 nicotinic receptors promotes cell death by inducing Ca(2+) release
from the endoplasmic reticulum. J. Neurochem. 133, 309–319.
https://doi.org/10.1111/jnc.13049
Gupta, S.C., Patchva, S., Koh, W., Aggarwal, B.B., 2012. Discovery of curcumin, a
component of golden spice, and its miraculous biological activities. Clin.
Exp. Pharmacol. Physiol. 39, 283–299. https://doi.org/10.1111/j.1440-
1681.2011.05648.x
Halliday, G., Herrero, M.T., Murphy, K., McCann, H., Ros-Bernal, F., Barcia, C.,
Mori, H., Blesa, F.J., Obeso, J.A., 2009. No Lewy pathology in monkeys with
over 10 years of severe MPTP Parkinsonism. Mov. Disord. Off. J. Mov.
Disord. Soc. 24, 1519–1523. https://doi.org/10.1002/mds.22481
Hartzell, C., Putzier, I., Arreola, J., 2005. Calcium-activated chloride channels.
Annu. Rev. Physiol. 67, 719–758.
https://doi.org/10.1146/annurev.physiol.67.032003.154341
Hasima, N., Aggarwal, B.B., 2012. Cancer-linked targets modulated by curcumin.
Int. J. Biochem. Mol. Biol. 3, 328–351.
Healy, D.G., Falchi, M., O’Sullivan, S.S., Bonifati, V., Durr, A., Bressman, S., Brice,
A., Aasly, J., Zabetian, C.P., Goldwurm, S., Ferreira, J.J., Tolosa, E., Kay,
D.M., Klein, C., Williams, D.R., Marras, C., Lang, A.E., Wszolek, Z.K.,
Berciano, J., Schapira, A.H.V., Lynch, T., Bhatia, K.P., Gasser, T., Lees,
A.J., Wood, N.W., International LRRK2 Consortium, 2008. Phenotype,
genotype, and worldwide genetic penetrance of LRRK2-associated
Parkinson’s disease: a case-control study. Lancet Neurol. 7, 583–590.
https://doi.org/10.1016/S1474-4422(08)70117-0
Hefti, F., Melamed, E., Sahakian, B.J., Wurtman, R.J., 1980a. Circling behavior in
rats with partial, unilateral nigro-striatal lesions: effect of amphetamine,
apomorphine, and DOPA. Pharmacol. Biochem. Behav. 12, 185–188.
Hefti, F., Melamed, E., Wurtman, R.J., 1980b. Partial lesions of the dopaminergic
nigrostriatal system in rat brain: biochemical characterization. Brain Res.
195, 123–137.
Hendrickson, L.M., Guildford, M.J., Tapper, A.R., 2013. Neuronal nicotinic
acetylcholine receptors: common molecular substrates of nicotine and alcohol
dependence. Front Psychiatry 4, 29. https://doi.org/10.3389/fpsyt.2013.00029

190
Hess, A., Desiderio, C., McAuliffe, W.G., 1990. Acute neuropathological changes in
the caudate nucleus caused by MPTP and methamphetamine:
immunohistochemical studies. J. Neurocytol. 19, 338–342.
Hickey, P., Stacy, M., 2013. AAV2-neurturin (CERE-120) for Parkinson’s disease.
Expert Opin Biol Ther 13, 137–45.
https://doi.org/10.1517/14712598.2013.754420
Hinkle, K.M., Yue, M., Behrouz, B., Dächsel, J.C., Lincoln, S.J., Bowles, E.E.,
Beevers, J.E., Dugger, B., Winner, B., Prots, I., Kent, C.B., Nishioka, K., Lin,
W.-L., Dickson, D.W., Janus, C.J., Farrer, M.J., Melrose, H.L., 2012. LRRK2
knockout mice have an intact dopaminergic system but display alterations in
exploratory and motor co-ordination behaviors. Mol. Neurodegener. 7, 25.
https://doi.org/10.1186/1750-1326-7-25
Hirayama, M., 2006. Sweating dysfunctions in Parkinson’s disease. J. Neurol. 253
Suppl 7, VII42-47. https://doi.org/10.1007/s00415-006-7010-7
Hisahara, S., Shimohama, S., 2011. Dopamine Receptors and Parkinson’s Disease.
Int. J. Med. Chem. 2011. https://doi.org/10.1155/2011/403039
Hoehle, S.I., Pfeiffer, E., Sólyom, A.M., Metzler, M., 2006. Metabolism of
curcuminoids in tissue slices and subcellular fractions from rat liver. J. Agric.
Food Chem. 54, 756–764. https://doi.org/10.1021/jf058146a
Holder, G.M., Plummer, J.L., Ryan, A.J., 1978. The metabolism and excretion of
curcumin (1,7-bis-(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione)
in the rat. Xenobiotica Fate Foreign Compd. Biol. Syst. 8, 761–768.
Home - ClinicalTrials.gov [WWW Document], n.d. URL https://clinicaltrials.gov/
(accessed 12.15.18).
Hong, D.H., Son, Y.K., Choi, I.-W., Park, W.S., 2013. The inhibitory effect of
curcumin on voltage-dependent K+ channels in rabbit coronary arterial
smooth muscle cells. Biochem. Biophys. Res. Commun. 430, 307–312.
https://doi.org/10.1016/j.bbrc.2012.10.132
Hosur, V., Loring, R.H., 2011. alpha4beta2 nicotinic receptors partially mediate anti-
inflammatory effects through Janus kinase 2-signal transducer and activator
of transcription 3 but not calcium or cAMP signaling. Mol Pharmacol 79,
167–74. https://doi.org/10.1124/mol.110.066381
Hu, C.-W., Sheng, Y., Zhang, Q., Liu, H.-B., Xie, X., Ma, W.-C., Huo, R., Dong, D.-
L., 2012. Curcumin inhibits hERG potassium channels in vitro. Toxicol. Lett.
208, 192–196. https://doi.org/10.1016/j.toxlet.2011.11.005

191
Huang, H.C., Jan, T.R., Yeh, S.F., 1992. Inhibitory effect of curcumin, an anti-
inflammatory agent, on vascular smooth muscle cell proliferation. Eur. J.
Pharmacol. 221, 381–384.
Huang, M., Felix, A.R., Flood, D.G., Bhuvaneswaran, C., Hilt, D., Koenig, G.,
Meltzer, H.Y., 2014. The novel α7 nicotinic acetylcholine receptor agonist
EVP-6124 enhances dopamine, acetylcholine, and glutamate efflux in rat
cortex and nucleus accumbens. Psychopharmacology (Berl.) 231, 4541–4551.
https://doi.org/10.1007/s00213-014-3596-0
Hudson, J.L., van Horne, C.G., Strömberg, I., Brock, S., Clayton, J., Masserano, J.,
Hoffer, B.J., Gerhardt, G.A., 1993. Correlation of apomorphine- and
amphetamine-induced turning with nigrostriatal dopamine content in
unilateral 6-hydroxydopamine lesioned rats. Brain Res. 626, 167–174.
Hughes, A.J., Daniel, S.E., Kilford, L., Lees, A.J., 1992. Accuracy of clinical
diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of
100 cases. J. Neurol. Neurosurg. Psychiatry 55, 181–184.
Hughes, J.T., 1988. Brain damage due to paraquat poisoning: a fatal case with
neuropathological examination of the brain. Neurotoxicology 9, 243–248.
Huot, P., Johnston, T.H., Koprich, J.B., Fox, S.H., Brotchie, J.M., 2013. The
pharmacology of L-DOPA-induced dyskinesia in Parkinson’s disease.
Pharmacol Rev 65, 171–222. https://doi.org/10.1124/pr.111.005678
Hurst, R., Rollema, H., Bertrand, D., 2013. Nicotinic acetylcholine receptors: from
basic science to therapeutics. Pharmacol Ther 137, 22–54.
https://doi.org/10.1016/j.pharmthera.2012.08.012
Ibrahim, A., El-Meligy, A., Lungu, G., Fetaih, H., Dessouki, A., Stoica, G.,
Barhoumi, R., 2011. Curcumin induces apoptosis in a murine mammary
gland adenocarcinoma cell line through the mitochondrial pathway. Eur. J.
Pharmacol. 668, 127–132. https://doi.org/10.1016/j.ejphar.2011.06.048
Iravani, M.M., McCreary, A.C., Jenner, P., 2012. Striatal plasticity in Parkinson’s
disease and L-dopa induced dyskinesia. Park. Relat Disord 18 Suppl 1, S123-
5. https://doi.org/10.1016/s1353-8020(11)70038-4
Itier, J.-M., Ibanez, P., Mena, M.A., Abbas, N., Cohen-Salmon, C., Bohme, G.A.,
Laville, M., Pratt, J., Corti, O., Pradier, L., Ret, G., Joubert, C., Periquet, M.,
Araujo, F., Negroni, J., Casarejos, M.J., Canals, S., Solano, R., Serrano, A.,
Gallego, E., Sanchez, M., Denefle, P., Benavides, J., Tremp, G., Rooney,
T.A., Brice, A., Garcia de Yebenes, J., 2003. Parkin gene inactivation alters

192
behaviour and dopamine neurotransmission in the mouse. Hum. Mol. Genet.
12, 2277–2291. https://doi.org/10.1093/hmg/ddg239
Jankovic, J., 2008. Parkinson’s disease: clinical features and diagnosis. J. Neurol.
Neurosurg. Psychiatry 79, 368–376.
https://doi.org/10.1136/jnnp.2007.131045
Jayakumary, M., Jayadevan, S., Ranade, A.V., Mathew, E., 2010. Prevalence and
pattern of dokha use among medical and allied health students in Ajman,
United Arab Emirates. Asian Pac J Cancer Prev 11, 1547–9.
Jenner, P., 2008. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev
Neurosci 9, 665–77. https://doi.org/10.1038/nrn2471
Jensen, A.A., Frolund, B., Liljefors, T., Krogsgaard-Larsen, P., 2005. Neuronal
nicotinic acetylcholine receptors: structural revelations, target identifications,
and therapeutic inspirations. J Med Chem 48, 4705–45.
https://doi.org/10.1021/jm040219e
Jeon, B.S., Jackson-Lewis, V., Burke, R.E., 1995. 6-Hydroxydopamine lesion of the
rat substantia nigra: time course and morphology of cell death. Neurodegener.
J. Neurodegener. Disord. Neuroprotection Neuroregeneration 4, 131–137.
Jeong, G.S., Oh, G.S., Pae, H.-O., Jeong, S.-O., Kim, Y.-C., Shin, M.-K., Seo, B.Y.,
Han, S.Y., Lee, H.S., Jeong, J.-G., Koh, J.-S., Chung, H.-T., 2006.
Comparative effects of curcuminoids on endothelial heme oxygenase-1
expression: ortho-methoxy groups are essential to enhance heme oxygenase
activity and protection. Exp. Mol. Med. 38, 393–400.
https://doi.org/10.1038/emm.2006.46
Joe, B., Vijaykumar, M., Lokesh, B.R., 2004. Biological properties of curcumin-
cellular and molecular mechanisms of action. Crit. Rev. Food Sci. Nutr. 44,
97–111. https://doi.org/10.1080/10408690490424702
Johnston, T.H., Huot, P., Fox, S.H., Koprich, J.B., Szeliga, K.T., James, J.W., Graef,
J.D., Letchworth, S.R., Jordan, K.G., Hill, M.P., Brotchie, J.M., 2013. TC-
8831, a nicotinic acetylcholine receptor agonist, reduces L-DOPA-induced
dyskinesia in the MPTP macaque. Neuropharmacology 73, 337–347.
https://doi.org/10.1016/j.neuropharm.2013.06.005
Jones, A.K., Buckingham, S.D., Sattelle, D.B., 2010. Proteins interacting with
nicotinic acetylcholine receptors: expanding functional and therapeutic
horizons. Trends Pharmacol. Sci. 31, 455–462.
https://doi.org/10.1016/j.tips.2010.07.001

193
Joselin, A.P., Hewitt, S.J., Callaghan, S.M., Kim, R.H., Chung, Y.-H., Mak, T.W.,
Shen, J., Slack, R.S., Park, D.S., 2012. ROS-dependent regulation of Parkin
and DJ-1 localization during oxidative stress in neurons. Hum. Mol. Genet.
21, 4888–4903. https://doi.org/10.1093/hmg/dds325
Jost, W.H., 2003. Autonomic dysfunctions in idiopathic Parkinson’s disease. J.
Neurol. 250 Suppl 1, I28-30. https://doi.org/10.1007/s00415-003-1105-z
Jurado-Coronel, J.C., Avila-Rodriguez, M., Capani, F., Gonzalez, J., Moran, V.E.,
Barreto, G.E., 2016. Targeting the Nicotinic Acetylcholine Receptors
(nAChRs) in Astrocytes as a Potential Therapeutic Target in Parkinson’s
Disease. Curr. Pharm. Des. 22, 1305–1311.
Jurenka, J.S., 2009. Anti-inflammatory properties of curcumin, a major constituent of
Curcuma longa: a review of preclinical and clinical research. Altern. Med.
Rev. J. Clin. Ther. 14, 141–153.
Kalappa, B.I., Sun, F., Johnson, S.R., Jin, K., Uteshev, V.V., 2013. A positive
allosteric modulator of α7 nAChRs augments neuroprotective effects of
endogenous nicotinic agonists in cerebral ischaemia. Br. J. Pharmacol. 169,
1862–1878. https://doi.org/10.1111/bph.12247
Kalf, J.G., Bloem, B.R., Munneke, M., 2012. Diurnal and nocturnal drooling in
Parkinson’s disease. J. Neurol. 259, 119–123. https://doi.org/10.1007/s00415-
011-6138-2
Kalf, J.G., Borm, G.F., de Swart, B.J., Bloem, B.R., Zwarts, M.J., Munneke, M.,
2011. Reproducibility and validity of patient-rated assessment of speech,
swallowing, and saliva control in Parkinson’s disease. Arch. Phys. Med.
Rehabil. 92, 1152–1158. https://doi.org/10.1016/j.apmr.2011.02.011
Kawamata, J., Shimohama, S., 2011. Stimulating nicotinic receptors trigger multiple
pathways attenuating cytotoxicity in models of Alzheimer’s and Parkinson’s
diseases. J Alzheimers Dis 24 Suppl 2, 95–109. https://doi.org/10.3233/jad-
2011-110173
Kerr, D.S., 2010. Treatment of mitochondrial electron transport chain disorders: a
review of clinical trials over the past decade. Mol Genet Metab 99, 246–55.
https://doi.org/10.1016/j.ymgme.2009.11.005
Khatri, D.K., Juvekar, A.R., 2016. Neuroprotective effect of curcumin as evinced by
abrogation of rotenone-induced motor deficits, oxidative and mitochondrial
dysfunctions in mouse model of Parkinson’s disease. Pharmacol. Biochem.
Behav. 150–151, 39–47. https://doi.org/10.1016/j.pbb.2016.09.002

194
Khuwaja, G., Khan, M.M., Ishrat, T., Ahmad, A., Raza, S.S., Ashafaq, M., Javed, H.,
Khan, M.B., Khan, A., Vaibhav, K., Safhi, M.M., Islam, F., 2011.
Neuroprotective effects of curcumin on 6-hydroxydopamine-induced
Parkinsonism in rats: behavioral, neurochemical and immunohistochemical
studies. Brain Res. 1368, 254–263.
https://doi.org/10.1016/j.brainres.2010.10.023
Kim, R.H., Smith, P.D., Aleyasin, H., Hayley, S., Mount, M.P., Pownall, S.,
Wakeham, A., You-Ten, A.J., Kalia, S.K., Horne, P., Westaway, D., Lozano,
A.M., Anisman, H., Park, D.S., Mak, T.W., 2005. Hypersensitivity of DJ-1-
deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and
oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 102, 5215–5220.
https://doi.org/10.1073/pnas.0501282102
Kim, S.G., Veena, M.S., Basak, S.K., Han, E., Tajima, T., Gjertson, D.W., Starr, J.,
Eidelman, O., Pollard, H.B., Srivastava, M., Srivatsan, E.S., Wang, M.B.,
2011. Curcumin treatment suppresses IKKβ kinase activity of salivary cells
of patients with head and neck cancer: a pilot study. Clin. Cancer Res. Off. J.
Am. Assoc. Cancer Res. 17, 5953–5961. https://doi.org/10.1158/1078-
0432.CCR-11-1272
King, J.R., Nordman, J.C., Bridges, S.P., Lin, M.-K., Kabbani, N., 2015.
Identification and Characterization of a G Protein-binding Cluster in α7
Nicotinic Acetylcholine Receptors. J. Biol. Chem. 290, 20060–20070.
https://doi.org/10.1074/jbc.M115.647040
Kinon, B.J., Millen, B.A., Zhang, L., McKinzie, D.L., 2015. Exploratory analysis for
a targeted patient population responsive to the metabotropic glutamate 2/3
receptor agonist pomaglumetad methionil in schizophrenia. Biol. Psychiatry
78, 754–762. https://doi.org/10.1016/j.biopsych.2015.03.016
Kirik, D., Rosenblad, C., Björklund, A., 1998. Characterization of behavioral and
neurodegenerative changes following partial lesions of the nigrostriatal
dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. Exp.
Neurol. 152, 259–277. https://doi.org/10.1006/exnr.1998.6848
Kiso, Y., Suzuki, Y., Watanabe, N., Oshima, Y., Hikino, H., 1983. Antihepatotoxic
principles of Curcuma longa rhizomes. Planta Med. 49, 185–187.
Kitada, T., Pisani, A., Karouani, M., Haburcak, M., Martella, G., Tscherter, A.,
Platania, P., Wu, B., Pothos, E.N., Shen, J., 2009. Impaired dopamine release
and synaptic plasticity in the striatum of parkin-/- mice. J. Neurochem. 110,
613–621. https://doi.org/10.1111/j.1471-4159.2009.06152.x

195
Kiuchi, F., Goto, Y., Sugimoto, N., Akao, N., Kondo, K., Tsuda, Y., 1993.
Nematocidal activity of turmeric: synergistic action of curcuminoids. Chem.
Pharm. Bull. (Tokyo) 41, 1640–1643.
Kozlowski, D.A., Miljan, E.A., Bremer, E.G., Harrod, C.G., Gerin, C., Connor, B.,
George, D., Larson, B., Bohn, M.C., 2004. Quantitative analyses of
GFRalpha-1 and GFRalpha-2 mRNAs and tyrosine hydroxylase protein in the
nigrostriatal system reveal bilateral compensatory changes following
unilateral 6-OHDA lesions in the rat. Brain Res. 1016, 170–181.
https://doi.org/10.1016/j.brainres.2004.05.003
Krasnova, I.N., Cadet, J.L., 2009. Methamphetamine toxicity and messengers of
death. Brain Res. Rev. 60, 379–407.
https://doi.org/10.1016/j.brainresrev.2009.03.002
Kreutzberg, G.W., 1996. Microglia: a sensor for pathological events in the CNS.
Trends Neurosci 19, 312–8.
Krüger, R., Kuhn, W., Müller, T., Woitalla, D., Graeber, M., Kösel, S., Przuntek, H.,
Epplen, J.T., Schöls, L., Riess, O., 1998. Ala30Pro mutation in the gene
encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108.
https://doi.org/10.1038/ng0298-106
Kunnumakkara, A.B., Bordoloi, D., Padmavathi, G., Monisha, J., Roy, N.K., Prasad,
S., Aggarwal, B.B., 2016. Curcumin, the golden nutraceutical: multitargeting
for multiple chronic diseases. Br. J. Pharmacol.
https://doi.org/10.1111/bph.13621
Kuttan, R., Sudheeran, P.C., Josph, C.D., 1987. Turmeric and curcumin as topical
agents in cancer therapy. Tumori 73, 29–31.
Lahrmann, H., Cortelli, P., Hilz, M., Mathias, C.J., Struhal, W., Tassinari, M., 2006.
EFNS guidelines on the diagnosis and management of orthostatic
hypotension. Eur. J. Neurol. 13, 930–936. https://doi.org/10.1111/j.1468-
1331.2006.01512.x
Langley, J.N., 1905. On the reaction of cells and of nerve-endings to certain poisons,
chiefly as regards the reaction of striated muscle to nicotine and to curari. J.
Physiol. 33, 374–413.
Langston, J.W., Ballard, P., Tetrud, J.W., Irwin, I., 1983. Chronic Parkinsonism in
humans due to a product of meperidine-analog synthesis. Science 219, 979–
980.

196
Langston, J.W., Forno, L.S., Tetrud, J., Reeves, A.G., Kaplan, J.A., Karluk, D., 1999.
Evidence of active nerve cell degeneration in the substantia nigra of humans
years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann.
Neurol. 46, 598–605.
Lao, C.D., Ruffin, M.T., Normolle, D., Heath, D.D., Murray, S.I., Bailey, J.M.,
Boggs, M.E., Crowell, J., Rock, C.L., Brenner, D.E., 2006. Dose escalation of
a curcuminoid formulation. BMC Complement. Altern. Med. 6, 10.
https://doi.org/10.1186/1472-6882-6-10
Le Novere, N., Zoli, M., Changeux, J.P., 1996. Neuronal nicotinic receptor alpha 6
subunit mRNA is selectively concentrated in catecholaminergic nuclei of the
rat brain. Eur J Neurosci 8, 2428–39.
Lewis, A.S., van Schalkwyk, G.I., Bloch, M.H., 2017. Alpha-7 nicotinic agonists for
cognitive deficits in neuropsychiatric disorders: A translational meta-analysis
of rodent and human studies. Prog. Neuropsychopharmacol. Biol. Psychiatry
75, 45–53. https://doi.org/10.1016/j.pnpbp.2017.01.001
Li, C., Zhang, Y., Su, T., Feng, L., Long, Y., Chen, Z., 2012. Silica-coated flexible
liposomes as a nanohybrid delivery system for enhanced oral bioavailability
of curcumin. Int. J. Nanomedicine 7, 5995–6002.
https://doi.org/10.2147/IJN.S38043
Li, Y., Revalde, J., Paxton, J.W., 2017. The effects of dietary and herbal
phytochemicals on drug transporters. Adv. Drug Deliv. Rev. 116, 45–62.
https://doi.org/10.1016/j.addr.2016.09.004
Lian, Y.-T., Yang, X.-F., Wang, Z.-H., Yang, Yong, Yang, Ying, Shu, Y.-W.,
Cheng, L.-X., Liu, K., 2013. Curcumin serves as a human kv1.3 blocker to
inhibit effector memory T lymphocyte activities. Phytother. Res. PTR 27,
1321–1327. https://doi.org/10.1002/ptr.4863
LigPrep | Schrödinger [WWW Document], n.d. URL
https://www.schrodinger.com/ligprep (accessed 1.15.17).
Limtrakul, P., Lipigorngoson, S., Namwong, O., Apisariyakul, A., Dunn, F.W., 1997.
Inhibitory effect of dietary curcumin on skin carcinogenesis in mice. Cancer
Lett. 116, 197–203.
Linazasoro, G., Van Blercom, N., Ugedo, L., Ruiz Ortega, J.A., 2008.
Pharmacological treatment of Parkinson’s disease: life beyond dopamine
D2/D3 receptors? J Neural Transm Vienna 115, 431–41.
https://doi.org/10.1007/s00702-007-0852-z

197
Lindstrom, J.M., 2003. Nicotinic acetylcholine receptors of muscles and nerves:
comparison of their structures, functional roles, and vulnerability to
pathology. Ann N Acad Sci 998, 41–52.
Liu, B., Hong, J.S., 2003. Role of microglia in inflammation-mediated
neurodegenerative diseases: mechanisms and strategies for therapeutic
intervention. J Pharmacol Exp Ther 304, 1–7.
https://doi.org/10.1124/jpet.102.035048
Liu, H., Danthi, S.J., Enyeart, J.J., 2006. Curcumin potently blocks Kv1.4 potassium
channels. Biochem. Biophys. Res. Commun. 344, 1161–1165.
https://doi.org/10.1016/j.bbrc.2006.04.020
Liu, K., Gui, B., Sun, Y., Shi, N., Gu, Z., Zhang, T., Sun, X., 2013. Inhibition of L-
type Ca(2+) channels by curcumin requires a novel protein kinase-theta
isoform in rat hippocampal neurons. Cell Calcium 53, 195–203.
https://doi.org/10.1016/j.ceca.2012.11.014
Liu, Q., Huang, Y., Shen, J., Steffensen, S., Wu, J., 2012. Functional α7β2 nicotinic
acetylcholine receptors expressed in hippocampal interneurons exhibit high
sensitivity to pathological level of amyloid β peptides. BMC Neurosci. 13,
155. https://doi.org/10.1186/1471-2202-13-155
Liu, Q., Huang, Y., Xue, F., Simard, A., DeChon, J., Li, G., Zhang, J., Lucero, L.,
Wang, M., Sierks, M., Hu, G., Chang, Y., Lukas, R.J., Wu, J., 2009. A novel
nicotinic acetylcholine receptor subtype in basal forebrain cholinergic
neurons with high sensitivity to amyloid peptides. J. Neurosci. Off. J. Soc.
Neurosci. 29, 918–929. https://doi.org/10.1523/JNEUROSCI.3952-08.2009
Liu, Y., Zeng, X., Hui, Y., Zhu, C., Wu, J., Taylor, D.H., Ji, J., Fan, W., Huang, Z.,
Hu, J., 2015. Activation of α7 nicotinic acetylcholine receptors protects
astrocytes against oxidative stress-induced apoptosis: implications for
Parkinson’s disease. Neuropharmacology 91, 87–96.
https://doi.org/10.1016/j.neuropharm.2014.11.028
Llewellyn-Smith, I.J., Phend, K.D., Minson, J.B., Pilowsky, P.M., Chalmers, J.P.,
1992. Glutamate-immunoreactive synapses on retrogradely-labelled
sympathetic preganglionic neurons in rat thoracic spinal cord. Brain Res. 581,
67–80.
Long-Smith, C.M., Sullivan, A.M., Nolan, Y.M., 2009. The influence of microglia
on the pathogenesis of Parkinson’s disease. Prog Neurobiol 89, 277–87.
https://doi.org/10.1016/j.pneurobio.2009.08.001

198
Low, V., Ben-Shlomo, Y., Coward, E., Fletcher, S., Walker, R., Clarke, C.E., 2015.
Measuring the burden and mortality of hospitalisation in Parkinson’s disease:
A cross-sectional analysis of the English Hospital Episodes Statistics
database 2009-2013. Parkinsonism Relat. Disord. 21, 449–454.
https://doi.org/10.1016/j.parkreldis.2015.01.017
Lu, X.-H., Fleming, S.M., Meurers, B., Ackerson, L.C., Mortazavi, F., Lo, V.,
Hernandez, D., Sulzer, D., Jackson, G.R., Maidment, N.T., Chesselet, M.-F.,
Yang, X.W., 2009. Bacterial artificial chromosome transgenic mice
expressing a truncated mutant parkin exhibit age-dependent hypokinetic
motor deficits, dopaminergic neuron degeneration, and accumulation of
proteinase K-resistant alpha-synuclein. J. Neurosci. Off. J. Soc. Neurosci. 29,
1962–1976. https://doi.org/10.1523/JNEUROSCI.5351-08.2009
Lücking, C.B., Dürr, A., Bonifati, V., Vaughan, J., De Michele, G., Gasser, T.,
Harhangi, B.S., Meco, G., Denèfle, P., Wood, N.W., Agid, Y., Brice, A.,
French Parkinson’s Disease Genetics Study Group, European Consortium on
Genetic Susceptibility in Parkinson’s Disease, 2000. Association between
early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J.
Med. 342, 1560–1567. https://doi.org/10.1056/NEJM200005253422103
Luthman, J., Fredriksson, A., Sundström, E., Jonsson, G., Archer, T., 1989. Selective
lesion of central dopamine or noradrenaline neuron systems in the neonatal
rat: motor behavior and monoamine alterations at adult stage. Behav. Brain
Res. 33, 267–277.
Macleod, A.D., Taylor, K.S.M., Counsell, C.E., 2014. Mortality in Parkinson’s
disease: a systematic review and meta-analysis. Mov. Disord. Off. J. Mov.
Disord. Soc. 29, 1615–1622. https://doi.org/10.1002/mds.25898
Macor, J.E., Gurley, D., Lanthorn, T., Loch, J., Mack, R.A., Mullen, G., Tran, O.,
Wright, N., Gordon, J.C., 2001. The 5-HT3 antagonist tropisetron (ICS 205-
930) is a potent and selective alpha7 nicotinic receptor partial agonist.
Bioorg. Med. Chem. Lett. 11, 319–321.
Maestro 11 | Schrödinger [WWW Document], n.d. URL
https://www.schrodinger.com/maestro (accessed 1.15.17).
Mahmmoud, Y.A., 2007. Modulation of protein kinase C by curcumin; inhibition
and activation switched by calcium ions. Br. J. Pharmacol. 150, 200–208.
https://doi.org/10.1038/sj.bjp.0706970
Mak, S.K., McCormack, A.L., Manning-Bog, A.B., Cuervo, A.M., Di Monte, D.A.,
2010. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 285,
13621–13629. https://doi.org/10.1074/jbc.M109.074617

199
Malmfors, T., Sachs, C., 1968. Degeneration of adrenergic nerves produced by 6-
hydroxydopamine. Eur. J. Pharmacol. 3, 89–92.
Mameli-Engvall, M., Evrard, A., Pons, S., Maskos, U., Svensson, T.H., Changeux,
J.P., Faure, P., 2006. Hierarchical control of dopamine neuron-firing patterns
by nicotinic receptors. Neuron 50, 911–21.
https://doi.org/10.1016/j.neuron.2006.05.007
Manikandan, P., Sumitra, M., Aishwarya, S., Manohar, B.M., Lokanadam, B.,
Puvanakrishnan, R., 2004. Curcumin modulates free radical quenching in
myocardial ischaemia in rats. Int. J. Biochem. Cell Biol. 36, 1967–1980.
https://doi.org/10.1016/j.biocel.2004.01.030
Manning-Bog, A.B., McCormack, A.L., Li, J., Uversky, V.N., Fink, A.L., Di Monte,
D.A., 2002. The herbicide paraquat causes up-regulation and aggregation of
alpha-synuclein in mice: paraquat and alpha-synuclein. J. Biol. Chem. 277,
1641–1644. https://doi.org/10.1074/jbc.C100560200
Mansvelder, H.D., Keath, J.R., McGehee, D.S., 2002. Synaptic mechanisms underlie
nicotine-induced excitability of brain reward areas. Neuron 33, 905–19.
Marcus, M.M., Björkholm, C., Malmerfelt, A., Möller, A., Påhlsson, N.,
Konradsson-Geuken, Å., Feltmann, K., Jardemark, K., Schilström, B.,
Svensson, T.H., 2016. Alpha7 nicotinic acetylcholine receptor agonists and
PAMs as adjunctive treatment in schizophrenia. An experimental study. Eur.
Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 26, 1401–
1411. https://doi.org/10.1016/j.euroneuro.2016.07.004
Marubio, L.M., del Mar Arroyo-Jimenez, M., Cordero-Erausquin, M., Lena, C., Le
Novere, N., de Kerchove d’Exaerde, A., Huchet, M., Damaj, M.I., Changeux,
J.P., 1999. Reduced antinociception in mice lacking neuronal nicotinic
receptor subunits. Nature 398, 805–10. https://doi.org/10.1038/19756
Marubio, L.M., Gardier, A.M., Durier, S., David, D., Klink, R., Arroyo-Jimenez,
M.M., McIntosh, J.M., Rossi, F., Champtiaux, N., Zoli, M., Changeux, J.P.,
2003. Effects of nicotine in the dopaminergic system of mice lacking the
alpha4 subunit of neuronal nicotinic acetylcholine receptors. Eur J Neurosci
17, 1329–37.
Massey, K.A., Zago, W.M., Berg, D.K., 2006. BDNF up-regulates alpha7 nicotinic
acetylcholine receptor levels on subpopulations of hippocampal interneurons.
Mol Cell Neurosci 33, 381–8. https://doi.org/10.1016/j.mcn.2006.08.011

200
Matsui, H., Uemura, N., Yamakado, H., Takeda, S., Takahashi, R., 2014. Exploring
the pathogenetic mechanisms underlying Parkinson’s disease in medaka fish.
J. Park. Dis. 4, 301–310. https://doi.org/10.3233/JPD-130289
McCormack, A.L., Thiruchelvam, M., Manning-Bog, A.B., Thiffault, C., Langston,
J.W., Cory-Slechta, D.A., Di Monte, D.A., 2002. Environmental risk factors
and Parkinson’s disease: selective degeneration of nigral dopaminergic
neurons caused by the herbicide paraquat. Neurobiol. Dis. 10, 119–127.
McNally, S.J., Harrison, E.M., Ross, J.A., Garden, O.J., Wigmore, S.J., 2007.
Curcumin induces heme oxygenase 1 through generation of reactive oxygen
species, p38 activation and phosphatase inhibition. Int. J. Mol. Med. 19, 165–
172.
Meredith, G.E., Kang, U.J., 2006. Behavioral models of Parkinson’s disease in
rodents: A new look at an old problem. Mov. Disord. 21, 1595–1606.
https://doi.org/10.1002/mds.21010
Meredith, G.E., Sonsalla, P.K., Chesselet, M.-F., 2008. Animal models of
Parkinson’s disease progression. Acta Neuropathol. (Berl.) 115, 385–398.
https://doi.org/10.1007/s00401-008-0350-x
Metz, G.A., Whishaw, I.Q., 2002. Drug-induced rotation intensity in unilateral
dopamine-depleted rats is not correlated with end point or qualitative
measures of forelimb or hindlimb motor performance. Neuroscience 111,
325–336.
Mihalak, K.B., Carroll, F.I., Luetje, C.W., 2006. Varenicline is a partial agonist at
alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol
Pharmacol 70, 801–5. https://doi.org/10.1124/mol.106.025130
Miledi, R., Potter, L.T., 1971. Acetylcholine receptors in muscle fibres. Nature 233,
599–603.
Millar, N.S., Gotti, C., 2009. Diversity of vertebrate nicotinic acetylcholine
receptors. Neuropharmacology 56, 237–246.
https://doi.org/10.1016/j.neuropharm.2008.07.041
Mineur, Y.S., Picciotto, M.R., 2008. Genetics of nicotinic acetylcholine receptors:
Relevance to nicotine addiction. Biochem Pharmacol 75, 323–33.
https://doi.org/10.1016/j.bcp.2007.06.010
Mizuno, Y., Sone, N., Saitoh, T., 1987. Effects of 1-methyl-4-phenyl-1,2,3,6-
tetrahydropyridine and 1-methyl-4-phenylpyridinium ion on activities of the

201
enzymes in the electron transport system in mouse brain. J. Neurochem. 48,
1787–1793.
MOE: Molecular Operating Environment [WWW Document], n.d. URL
http://www.chemcomp.com/MOE-Molecular_Operating_Environment.htm
(accessed 1.12.17).
Monderer, R., Thorpy, M., 2009. Sleep disorders and daytime sleepiness in
Parkinson’s disease. Curr. Neurol. Neurosci. Rep. 9, 173–180.
Moore, D.J., West, A.B., Dawson, V.L., Dawson, T.M., 2005. Molecular
pathophysiology of Parkinson’s disease. Annu Rev Neurosci 28, 57–87.
https://doi.org/10.1146/annurev.neuro.28.061604.135718
Morais, V.A., Haddad, D., Craessaerts, K., De Bock, P.-J., Swerts, J., Vilain, S.,
Aerts, L., Overbergh, L., Grünewald, A., Seibler, P., Klein, C., Gevaert, K.,
Verstreken, P., De Strooper, B., 2014. PINK1 loss-of-function mutations
affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling.
Science 344, 203–207. https://doi.org/10.1126/science.1249161
Moretti, M., Zoli, M., George, A.A., Lukas, R.J., Pistillo, F., Maskos, U., Whiteaker,
P., Gotti, C., 2014. The novel α7β2-nicotinic acetylcholine receptor subtype
is expressed in mouse and human basal forebrain: biochemical and
pharmacological characterization. Mol. Pharmacol. 86, 306–317.
https://doi.org/10.1124/mol.114.093377
Morgante, L., Morgante, F., Moro, E., Epifanio, A., Girlanda, P., Ragonese, P.,
Antonini, A., Barone, P., Bonuccelli, U., Contarino, M.F., Capus, L.,
Ceravolo, M.G., Marconi, R., Ceravolo, R., D’Amelio, M., Savettieri, G.,
2007. How many parkinsonian patients are suitable candidates for deep brain
stimulation of subthalamic nucleus? Results of a questionnaire. Parkinsonism
Relat. Disord. 13, 528–531. https://doi.org/10.1016/j.parkreldis.2006.12.013
Motterlini, R., Foresti, R., Bassi, R., Green, C.J., 2000. Curcumin, an antioxidant and
anti-inflammatory agent, induces heme oxygenase-1 and protects endothelial
cells against oxidative stress. Free Radic. Biol. Med. 28, 1303–1312.
Moustapha, A., Pérétout, P.A., Rainey, N.E., Sureau, F., Geze, M., Petit, J.-M.,
Dewailly, E., Slomianny, C., Petit, P.X., 2015. Curcumin induces crosstalk
between autophagy and apoptosis mediated by calcium release from the
endoplasmic reticulum, lysosomal destabilization and mitochondrial events.
Cell Death Discov. 1, 15017. https://doi.org/10.1038/cddiscovery.2015.17
Mowrey, D.D., Liu, Q., Bondarenko, V., Chen, Q., Seyoum, E., Xu, Y., Wu, J.,
Tang, P., 2013. Insights into distinct modulation of α7 and α7β2 nicotinic

202
acetylcholine receptors by the volatile anesthetic isoflurane. J. Biol. Chem.
288, 35793–35800. https://doi.org/10.1074/jbc.M113.508333
Munro, G., Hansen, R., Erichsen, H., Timmermann, D., Christensen, J., Hansen, H.,
2012. The α7 nicotinic ACh receptor agonist compound B and positive
allosteric modulator PNU-120596 both alleviate inflammatory hyperalgesia
and cytokine release in the rat. Br. J. Pharmacol. 167, 421–435.
https://doi.org/10.1111/j.1476-5381.2012.02003.x
Nagatsu, T., Sawada, M., 2005. Inflammatory process in Parkinson’s disease: role
for cytokines. Curr Pharm Des 11, 999–1016.
Nakagawa, T., Touhara, K., 2013. Functional Assays for Insect Olfactory Receptors
in Xenopus Oocytes, in: Pheromone Signaling, Methods in Molecular
Biology. Humana Press, Totowa, NJ, pp. 107–119.
https://doi.org/10.1007/978-1-62703-619-1_8
Napoli, I., Neumann, H., 2010. Protective effects of microglia in multiple sclerosis.
Exp Neurol 225, 24–8. https://doi.org/10.1016/j.expneurol.2009.04.024
Newman, M.B., Arendash, G.W., Shytle, R.D., Bickford, P.C., Tighe, T., Sanberg,
P.R., 2002. Nicotine’s oxidative and antioxidant properties in CNS. Life Sci
71, 2807–20.
Ng, H.J., Whittemore, E.R., Tran, M.B., Hogenkamp, D.J., Broide, R.S., Johnstone,
T.B., Zheng, L., Stevens, K.E., Gee, K.W., 2007. Nootropic alpha7 nicotinic
receptor allosteric modulator derived from GABAA receptor modulators.
Proc. Natl. Acad. Sci. U. S. A. 104, 8059–8064.
https://doi.org/10.1073/pnas.0701321104
Nicklas, W.J., Vyas, I., Heikkila, R.E., 1985. Inhibition of NADH-linked oxidation
in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the
neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 36,
2503–2508.
Nicoletti, A., Pugliese, P., Nicoletti, G., Arabia, G., Annesi, G., Mari, M.D.,
Lamberti, P., Grasso, L., Marconi, R., Epifanio, A., Morgante, L., Cozzolino,
A., Barone, P., Torchia, G., Quattrone, A., Zappia, M., 2010. Voluptuary
habits and clinical subtypes of Parkinson’s disease: the FRAGAMP case-
control study. Mov Disord 25, 2387–94. https://doi.org/10.1002/mds.23297
Nirmala, C., Puvanakrishnan, R., 1996a. Protective role of curcumin against
isoproterenol induced myocardial infarction in rats. Mol. Cell. Biochem. 159,
85–93.

203
Nirmala, C., Puvanakrishnan, R., 1996b. Effect of curcumin on certain lysosomal
hydrolases in isoproterenol-induced myocardial infarction in rats. Biochem.
Pharmacol. 51, 47–51.
Nishikawa, T., Takashima, M., Toru, M., 1983. Increased [3H]kainic acid binding in
the prefrontal cortex in schizophrenia. Neurosci. Lett. 40, 245–250.
Nordberg, A., 1992. Neuroreceptor changes in Alzheimer disease. Cerebrovasc Brain
Metab Rev 4, 303–28.
Nosalski, R., Guzik, T.J., 2017. Perivascular adipose tissue inflammation in vascular
disease. Br. J. Pharmacol. https://doi.org/10.1111/bph.13705
Noyce, A.J., Bestwick, J.P., Silveira-Moriyama, L., Hawkes, C.H., Giovannoni, G.,
Lees, A.J., Schrag, A., 2012. Meta-analysis of early nonmotor features and
risk factors for Parkinson disease. Ann Neurol 72, 893–901.
https://doi.org/10.1002/ana.23687
Nury, H., Van Renterghem, C., Weng, Y., Tran, A., Baaden, M., Dufresne, V.,
Changeux, J.-P., Sonner, J.M., Delarue, M., Corringer, P.-J., 2011. X-ray
structures of general anaesthetics bound to a pentameric ligand-gated ion
channel. Nature 469, 428–431. https://doi.org/10.1038/nature09647
Nys, M., Kesters, D., Ulens, C., 2013. Structural insights into Cys-loop receptor
function and ligand recognition. Biochem. Pharmacol. 86, 1042–1053.
https://doi.org/10.1016/j.bcp.2013.07.001
Obeso, J.A., Lanciego, J.L., 2011. Past, present, and future of the pathophysiological
model of the Basal Ganglia. Front Neuroanat 5, 39.
https://doi.org/10.3389/fnana.2011.00039
Okun, M.S., 2012. Deep-brain stimulation for Parkinson’s disease. N. Engl. J. Med.
367, 1529–1538. https://doi.org/10.1056/NEJMct1208070
Olanow, C.W., McNaught, K., 2011. Parkinson’s disease, proteins, and prions:
milestones. Mov. Disord. Off. J. Mov. Disord. Soc. 26, 1056–1071.
https://doi.org/10.1002/mds.23767
Olanow, C.W., Obeso, J.A., Stocchi, F., 2006. Continuous dopamine-receptor
treatment of Parkinson’s disease: scientific rationale and clinical implications.
Lancet Neurol 5, 677–87. https://doi.org/10.1016/s1474-4422(06)70521-x
Olincy, A., Blakeley-Smith, A., Johnson, L., Kem, W.R., Freedman, R., 2016. Brief
Report: Initial Trial of Alpha7-Nicotinic Receptor Stimulation in Two Adult
Patients with Autism Spectrum Disorder. J. Autism Dev. Disord. 46, 3812–
3817. https://doi.org/10.1007/s10803-016-2890-6

204
Oliveras-Salvá, M., Macchi, F., Coessens, V., Deleersnijder, A., Gérard, M., Van der
Perren, A., Van den Haute, C., Baekelandt, V., 2014. Alpha-synuclein-
induced neurodegeneration is exacerbated in PINK1 knockout mice.
Neurobiol. Aging 35, 2625–2636.
https://doi.org/10.1016/j.neurobiolaging.2014.04.032
Onaran, H.O., Costa, T., 2009. Allosteric coupling and conformational fluctuations
in proteins. Curr. Protein Pept. Sci. 10, 110–115.
O’Neill, M.J., Murray, T.K., Lakics, V., Visanji, N.P., Duty, S., 2002. The role of
neuronal nicotinic acetylcholine receptors in acute and chronic
neurodegeneration. Curr. Drug Targets CNS Neurol. Disord. 1, 399–411.
Ono, K., Hasegawa, K., Naiki, H., Yamada, M., 2004. Curcumin has potent anti-
amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. J.
Neurosci. Res. 75, 742–750. https://doi.org/10.1002/jnr.20025
Onofrj, M., Thomas, A., Bonanni, L., 2007. New approaches to understanding
hallucinations in Parkinson’s disease: phenomenology and possible origins.
Expert Rev. Neurother. 7, 1731–1750.
https://doi.org/10.1586/14737175.7.12.1731
Orr, C.F., Rowe, D.B., Halliday, G.M., 2002. An inflammatory review of
Parkinson’s disease. Prog Neurobiol 68, 325–40.
Ottolini, D., Calì, T., Negro, A., Brini, M., 2013. The Parkinson disease-related
protein DJ-1 counteracts mitochondrial impairment induced by the tumour
suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria
tethering. Hum. Mol. Genet. 22, 2152–2168.
https://doi.org/10.1093/hmg/ddt068
Oz, M., Melia, M.T., Soldatov, N.M., Abernethy, D.R., Morad, M., 1998. Functional
coupling of human L-type Ca2+ channels and angiotensin AT1A receptors
coexpressed in xenopus laevis oocytes: involvement of the carboxyl-terminal
Ca2+ sensors. Mol. Pharmacol. 54, 1106–1112.
Oz, M., Renaud, L.P., 2002. Angiotensin AT(1)-receptors depolarize neonatal spinal
motoneurons and other ventral horn neurons via two different conductances.
J. Neurophysiol. 88, 2857–2863. https://doi.org/10.1152/jn.00978.2001
Oz, M., Ravindran, A., Diaz-Ruiz, O., Zhang, L., Morales, M., 2003. The
endogenous cannabinoid anandamide inhibits alpha7 nicotinic acetylcholine
receptor-mediated responses in Xenopus oocytes. J. Pharmacol. Exp. Ther.
306, 1003–1010. https://doi.org/10.1124/jpet.103.049981

205
Oz, M., Zakharova, I., Dinc, M., Shippenberg, T., 2004. Cocaine inhibits
cromakalim-activated K+ currents in follicle-enclosed Xenopus oocytes.
Naunyn. Schmiedebergs Arch. Pharmacol. 369, 252–259.
https://doi.org/10.1007/s00210-003-0838-9
Pan, M.H., Huang, T.M., Lin, J.K., 1999. Biotransformation of curcumin through
reduction and glucuronidation in mice. Drug Metab. Dispos. Biol. Fate Chem.
27, 486–494.
Pan-Montojo, F., Anichtchik, O., Dening, Y., Knels, L., Pursche, S., Jung, R.,
Jackson, S., Gille, G., Spillantini, M.G., Reichmann, H., Funk, R.H.W., 2010.
Progression of Parkinson’s disease pathology is reproduced by intragastric
administration of rotenone in mice. PloS One 5, e8762.
https://doi.org/10.1371/journal.pone.0008762
Panneton, W.M., Kumar, V.B., Gan, Q., Burke, W.J., Galvin, J.E., 2010. The
neurotoxicity of DOPAL: behavioral and stereological evidence for its role in
Parkinson disease pathogenesis. PloS One 5, e15251.
https://doi.org/10.1371/journal.pone.0015251
Parashos, S.A., Maraganore, D.M., O’Brien, P.C., Rocca, W.A., 2002. Medical
services utilization and prognosis in Parkinson disease: a population-based
study. Mayo Clin. Proc. 77, 918–925. https://doi.org/10.4065/77.9.918
Park, H.J., Lee, P.H., Ahn, Y.W., Choi, Y.J., Lee, G., Lee, D.Y., Chung, E.S., Jin,
B.K., 2007a. Neuroprotective effect of nicotine on dopaminergic neurons by
anti-inflammatory action. Eur J Neurosci 26, 79–89.
https://doi.org/10.1111/j.1460-9568.2007.05636.x
Parker, W.D., Boyson, S.J., Parks, J.K., 1989. Abnormalities of the electron transport
chain in idiopathic Parkinson’s disease. Ann. Neurol. 26, 719–723.
https://doi.org/10.1002/ana.410260606
Parkinson, J., 2002. An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin.
Neurosci. 14, 223–236; discussion 222. https://doi.org/10.1176/jnp.14.2.223
Patrick, J., Ballivet, M., Boas, L., Claudio, T., Forrest, J., Ingraham, H., Mason, P.,
Stengelin, S., Ueno, S., Heinemann, S., 1983. Molecular cloning of the
acetylcholine receptor. Cold Spring Harb. Symp. Quant. Biol. 48 Pt 1, 71–78.
Paulo, J.A., Brucker, W.J., Hawrot, E., 2009. Proteomic analysis of an alpha7
nicotinic acetylcholine receptor interactome. J. Proteome Res. 8, 1849–1858.
https://doi.org/10.1021/pr800731z

206
Pérez-Lara, A., Corbalán-García, S., Gómez-Fernández, J.C., 2011. Curcumin
modulates PKCα activity by a membrane-dependent effect. Arch. Biochem.
Biophys. 513, 36–41. https://doi.org/10.1016/j.abb.2011.06.010
Perez-Lloret, S., Nègre-Pagès, L., Ojero-Senard, A., Damier, P., Destée, A., Tison,
F., Merello, M., Rascol, O., COPARK Study Group, 2012. Oro-buccal
symptoms (dysphagia, dysarthria, and sialorrhea) in patients with Parkinson’s
disease: preliminary analysis from the French COPARK cohort. Eur. J.
Neurol. 19, 28–37. https://doi.org/10.1111/j.1468-1331.2011.03402.x
Periquet, M., Latouche, M., Lohmann, E., Rawal, N., De Michele, G., Ricard, S.,
Teive, H., Fraix, V., Vidailhet, M., Nicholl, D., Barone, P., Wood, N.W.,
Raskin, S., Deleuze, J.-F., Agid, Y., Dürr, A., Brice, A., French Parkinson’s
Disease Genetics Study Group, European Consortium on Genetic
Susceptibility in Parkinson’s Disease, 2003. Parkin mutations are frequent in
patients with isolated early-onset parkinsonism. Brain J. Neurol. 126, 1271–
1278.
Perkins, S., Verschoyle, R.D., Hill, K., Parveen, I., Threadgill, M.D., Sharma, R.A.,
Williams, M.L., Steward, W.P., Gescher, A.J., 2002. Chemopreventive
efficacy and pharmacokinetics of curcumin in the min/+ mouse, a model of
familial adenomatous polyposis. Cancer Epidemiol. Biomark. Prev. Publ.
Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 11, 535–540.
Pham-Huy, L.A., He, H., Pham-Huy, C., 2008. Free radicals, antioxidants in disease
and health. Int. J. Biomed. Sci. IJBS 4, 89–96.
Picciotto, M.R., Zoli, M., 2008. Neuroprotection via nAChRs: the role of nAChRs in
neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease.
Front Biosci 13, 492–504.
Pickel, V.M., Beckley, S.C., Joh, T.H., Reis, D.J., 1981. Ultrastructural
immunocytochemical localization of tyrosine hydroxylase in the neostriatum.
Brain Res 225, 373–85.
Poewe, W., 2006. The natural history of Parkinson’s disease. J. Neurol. 253 Suppl 7,
VII2-6. https://doi.org/10.1007/s00415-006-7002-7
Pont-Sunyer, C., Hotter, A., Gaig, C., Seppi, K., Compta, Y., Katzenschlager, R.,
Mas, N., Hofeneder, D., Brücke, T., Bayés, A., Wenzel, K., Infante, J., Zach,
H., Pirker, W., Posada, I.J., Álvarez, R., Ispierto, L., De Fàbregues, O.,
Callén, A., Palasí, A., Aguilar, M., Martí, M.J., Valldeoriola, F., Salamero,
M., Poewe, W., Tolosa, E., 2015. The onset of nonmotor symptoms in
Parkinson’s disease (the ONSET PD study). Mov. Disord. Off. J. Mov.
Disord. Soc. 30, 229–237. https://doi.org/10.1002/mds.26077

207
Posadas, I., Lopez-Hernandez, B., Cena, V., 2013. Nicotinic receptors in
neurodegeneration. Curr Neuropharmacol 11, 298–314.
https://doi.org/10.2174/1570159x11311030005
Postuma, R.B., Berg, D., Stern, M., Poewe, W., Olanow, C.W., Oertel, W., Obeso, J.,
Marek, K., Litvan, I., Lang, A.E., Halliday, G., Goetz, C.G., Gasser, T.,
Dubois, B., Chan, P., Bloem, B.R., Adler, C.H., Deuschl, G., 2015. MDS
clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. Off. J. Mov.
Disord. Soc. 30, 1591–1601. https://doi.org/10.1002/mds.26424
Prasad, S., Tyagi, A.K., Aggarwal, B.B., 2014. Recent developments in delivery,
bioavailability, absorption and metabolism of curcumin: the golden pigment
from golden spice. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 46, 2–18.
https://doi.org/10.4143/crt.2014.46.1.2
Priyadarsini, K.I., 2014. The chemistry of curcumin: from extraction to therapeutic
agent. Mol. Basel Switz. 19, 20091–20112.
https://doi.org/10.3390/molecules191220091
Priyadarsini, K.I., 2009. ChemInform Abstract: Photophysics, Photochemistry and
Photobiology of Curcumin: Studies from Organic Solutions, Biomimetics and
Living Cells. ResearchGate 40. https://doi.org/10.1002/chin.200951234
Prokop, S., Miller, K.R., Heppner, F.L., 2013. Microglia actions in Alzheimer’s
disease. Acta Neuropathol 126, 461–77.
Przedborski, S., Jackson-Lewis, V., Naini, A.B., Jakowec, M., Petzinger, G., Miller,
R., Akram, M., 2001. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-
tetrahydropyridine (MPTP): a technical review of its utility and safety. J.
Neurochem. 76, 1265–1274.
Przedborski, S., Levivier, M., Jiang, H., Ferreira, M., Jackson-Lewis, V., Donaldson,
D., Togasaki, D.M., 1995. Dose-dependent lesions of the dopaminergic
nigrostriatal pathway induced by intrastriatal injection of 6-
hydroxydopamine. Neuroscience 67, 631–647.
Purisai, M.G., McCormack, A.L., Langston, W.J., Johnston, L.C., Di Monte, D.A.,
2005. Alpha-synuclein expression in the substantia nigra of MPTP-lesioned
non-human primates. Neurobiol. Dis. 20, 898–906.
https://doi.org/10.1016/j.nbd.2005.05.028
Puschmann, A., 2013. Monogenic Parkinson’s disease and parkinsonism: clinical
phenotypes and frequencies of known mutations. Parkinsonism Relat. Disord.
19, 407–415. https://doi.org/10.1016/j.parkreldis.2013.01.020

208
Pyakurel, P., Shin, M., Venton, B.J., 2018. Nicotinic acetylcholine receptor (nAChR)
mediated dopamine release in larval Drosophila melanogaster. Neurochem.
Int. 114, 33–41. https://doi.org/10.1016/j.neuint.2017.12.012
Quarta, D., Naylor, C.G., Barik, J., Fernandes, C., Wonnacott, S., Stolerman, I.P.,
2009. Drug discrimination and neurochemical studies in alpha7 null mutant
mice: tests for the role of nicotinic alpha7 receptors in dopamine release.
Psychopharmacology (Berl.) 203, 399–410. https://doi.org/10.1007/s00213-
008-1281-x
Quik, M., Campos, C., Bordia, T., Strachan, J.P., Zhang, J., McIntosh, J.M.,
Letchworth, S., Jordan, K., 2013. alpha4beta2 Nicotinic receptors play a role
in the nAChR-mediated decline in L-dopa-induced dyskinesias in
parkinsonian rats. Neuropharmacology 71, 191–203.
https://doi.org/10.1016/j.neuropharm.2013.03.038
Quik, M., Cox, H., Parameswaran, N., O’Leary, K., Langston, J.W., Di Monte, D.,
2007. Nicotine reduces levodopa-induced dyskinesias in lesioned monkeys.
Ann Neurol 62, 588–96. https://doi.org/10.1002/ana.21203
Quik, M., Kulak, J.M., 2002. Nicotine and nicotinic receptors; relevance to
Parkinson’s disease. Neurotoxicology 23, 581–94.
Quik, M., O’Leary, K., Tanner, C.M., 2008. Nicotine and Parkinson’s disease:
implications for therapy. Mov Disord 23, 1641–52.
https://doi.org/10.1002/mds.21900
Quik, M., Parameswaran, N., McCallum, S.E., Bordia, T., Bao, S., McCormack, A.,
Kim, A., Tyndale, R.F., Langston, J.W., Di Monte, D.A., 2006. Chronic oral
nicotine treatment protects against striatal degeneration in MPTP-treated
primates. J Neurochem 98, 1866–75. https://doi.org/10.1111/j.1471-
4159.2006.04078.x
Quik, M., Perez, X.A., Bordia, T., 2012. Nicotine as a potential neuroprotective
agent for Parkinson’s disease. Mov Disord 27, 947–57.
https://doi.org/10.1002/mds.25028
Quik, M., Wonnacott, S., 2011. α6β2* and α4β2* nicotinic acetylcholine receptors as
drug targets for Parkinson’s disease. Pharmacol. Rev. 63, 938–966.
https://doi.org/10.1124/pr.110.003269
Quik, M., Zhang, D., Perez, X.A., Bordia, T., 2014. Role for the nicotinic cholinergic
system in movement disorders; therapeutic implications. Pharmacol Ther
144, 50–9. https://doi.org/10.1016/j.pharmthera.2014.05.004

209
Rachmawati, H., Edityaningrum, C.A., Mauludin, R., 2013. Molecular inclusion
complex of curcumin-β-cyclodextrin nanoparticle to enhance curcumin skin
permeability from hydrophilic matrix gel. AAPS PharmSciTech 14, 1303–
1312. https://doi.org/10.1208/s12249-013-0023-5
Rainey, N., Motte, L., Aggarwal, B.B., Petit, P.X., 2015. Curcumin hormesis
mediates a cross-talk between autophagy and cell death. Cell Death Dis. 6,
e2003. https://doi.org/10.1038/cddis.2015.343
Rajan, S., 2012.
https://commons.wikimedia.org/wiki/File:Lewy_bodies_(alpha_synuclein_in
clusions).svg
Rangel-Barajas, C., Coronel, I., Florán, B., 2015. Dopamine Receptors and
Neurodegeneration. Aging Dis. 6, 349–368.
https://doi.org/10.14336/AD.2015.0330
Rao, C.V., Rivenson, A., Simi, B., Reddy, B.S., 1995. Chemoprevention of colon
carcinogenesis by dietary curcumin, a naturally occurring plant phenolic
compound. Cancer Res. 55, 259–266.
Ravindran, J., Prasad, S., Aggarwal, B.B., 2009. Curcumin and cancer cells: how
many ways can curry kill tumor cells selectively? AAPS J. 11, 495–510.
https://doi.org/10.1208/s12248-009-9128-x
Ravindranath, V., Chandrasekhara, N., 1981. Metabolism of curcumin--studies with
[3H]curcumin. Toxicology 22, 337–344.
Reddy, A.C., Lokesh, B.R., 1994. Studies on the inhibitory effects of curcumin and
eugenol on the formation of reactive oxygen species and the oxidation of
ferrous iron. Mol. Cell. Biochem. 137, 1–8.
Reddy, A.C., Lokesh, B.R., 1992. Studies on spice principles as antioxidants in the
inhibition of lipid peroxidation of rat liver microsomes. Mol. Cell. Biochem.
111, 117–124.
Reijnders, J.S.A.M., Ehrt, U., Weber, W.E.J., Aarsland, D., Leentjens, A.F.G., 2008.
A systematic review of prevalence studies of depression in Parkinson’s
disease. Mov. Disord. Off. J. Mov. Disord. Soc. 23, 183–189; quiz 313.
https://doi.org/10.1002/mds.21803
Rizzone, M.G., Fasano, A., Daniele, A., Zibetti, M., Merola, A., Rizzi, L., Piano, C.,
Piccininni, C., Romito, L.M., Lopiano, L., Albanese, A., 2014. Long-term
outcome of subthalamic nucleus DBS in Parkinson’s disease: from the

210
advanced phase towards the late stage of the disease? Parkinsonism Relat.
Disord. 20, 376–381. https://doi.org/10.1016/j.parkreldis.2014.01.012
Robinson, T.P., Ehlers, T., Hubbard IV, R.B., Bai, X., Arbiser, J.L., Goldsmith, D.J.,
Bowen, J.P., 2003. Design, synthesis, and biological evaluation of
angiogenesis inhibitors: aromatic enone and dienone analogues of curcumin.
Bioorg. Med. Chem. Lett. 13, 115–117.
Rodríguez Díaz, M., Abdala, P., Barroso-Chinea, P., Obeso, J., González-Hernández,
T., 2001. Motor behavioural changes after intracerebroventricular injection of
6-hydroxydopamine in the rat: an animal model of Parkinson’s disease.
Behav. Brain Res. 122, 79–92.
Roncarati, R., Scali, C., Comery, T.A., Grauer, S.M., Aschmi, S., Bothmann, H.,
Jow, B., Kowal, D., Gianfriddo, M., Kelley, C., Zanelli, U., Ghiron, C.,
Haydar, S., Dunlop, J., Terstappen, G.C., 2009. Procognitive and
neuroprotective activity of a novel alpha7 nicotinic acetylcholine receptor
agonist for treatment of neurodegenerative and cognitive disorders. J.
Pharmacol. Exp. Ther. 329, 459–468. https://doi.org/10.1124/jpet.108.150094
Rosenblad, C., Kirik, D., Devaux, B., Moffat, B., Phillips, H.S., Björklund, A., 1999.
Protection and regeneration of nigral dopaminergic neurons by neurturin or
GDNF in a partial lesion model of Parkinson’s disease after administration
into the striatum or the lateral ventricle. Eur. J. Neurosci. 11, 1554–1566.
Rudenko, I.N., Cookson, M.R., 2014. Heterogeneity of leucine-rich repeat kinase 2
mutations: genetics, mechanisms and therapeutic implications. Neurother. J.
Am. Soc. Exp. Neurother. 11, 738–750. https://doi.org/10.1007/s13311-014-
0284-z
Samadi, P., Bedard, P.J., Rouillard, C., 2006. Opioids and motor complications in
Parkinson’s disease. Trends Pharmacol Sci 27, 512–7.
https://doi.org/10.1016/j.tips.2006.08.002
Sanchez, G., Varaschin, R.K., Büeler, H., Marcogliese, P.C., Park, D.S., Trudeau, L.-
E., 2014. Unaltered striatal dopamine release levels in young Parkin
knockout, Pink1 knockout, DJ-1 knockout and LRRK2 R1441G transgenic
mice. PloS One 9, e94826. https://doi.org/10.1371/journal.pone.0094826
Sands, S.B., Costa, A.C., Patrick, J.W., 1993. Barium permeability of neuronal
nicotinic receptor alpha 7 expressed in Xenopus oocytes. Biophys. J. 65,
2614–2621. https://doi.org/10.1016/S0006-3495(93)81296-7
Sandur, S.K., Pandey, M.K., Sung, B., Ahn, K.S., Murakami, A., Sethi, G.,
Limtrakul, P., Badmaev, V., Aggarwal, B.B., 2007. Curcumin,

211
demethoxycurcumin, bisdemethoxycurcumin, tetrahydrocurcumin and
turmerones differentially regulate anti-inflammatory and anti-proliferative
responses through a ROS-independent mechanism. Carcinogenesis 28, 1765–
1773. https://doi.org/10.1093/carcin/bgm123
Saner, A., Thoenen, H., 1971. Model experiments on the molecular mechanism of
action of 6-hydroxydopamine. Mol. Pharmacol. 7, 147–154.
Sastry, G.M., Adzhigirey, M., Day, T., Annabhimoju, R., Sherman, W., 2013.
Protein and ligand preparation: parameters, protocols, and influence on
virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234.
https://doi.org/10.1007/s10822-013-9644-8
Sauer, H., Oertel, W.H., 1994. Progressive degeneration of nigrostriatal dopamine
neurons following intrastriatal terminal lesions with 6-hydroxydopamine: a
combined retrograde tracing and immunocytochemical study in the rat.
Neuroscience 59, 401–415.
Sauguet, L., Shahsavar, A., Poitevin, F., Huon, C., Menny, A., Nemecz, À., Haouz,
A., Changeux, J.-P., Corringer, P.-J., Delarue, M., 2014. Crystal structures of
a pentameric ligand-gated ion channel provide a mechanism for activation.
Proc. Natl. Acad. Sci. U. S. A. 111, 966–971.
https://doi.org/10.1073/pnas.1314997111
Sawada, H., Oeda, T., Kuno, S., Nomoto, M., Yamamoto, K., Yamamoto, M.,
Hisanaga, K., Kawamura, T., 2010. Amantadine for dyskinesias in
Parkinson’s disease: a randomized controlled trial. PLoS One 5, e15298.
https://doi.org/10.1371/journal.pone.0015298
Sax’s Dangerous Properties of Industrial Materials, 5 Volume Set, 12th Edition
[WWW Document], n.d. . Wiley.com. URL https://www.wiley.com/en-
ae/Sax%27s_Dangerous_Properties_of_Industrial_Materials%2C_5+Volume
_Set%2C_12th_Edition-p-9780470623251 (accessed 12.3.18).
Scarffe, L.A., Stevens, D.A., Dawson, V.L., Dawson, T.M., 2014. Parkin and
PINK1: much more than mitophagy. Trends Neurosci. 37, 315–324.
https://doi.org/10.1016/j.tins.2014.03.004
Schapira, A.H., Cooper, J.M., Dexter, D., Jenner, P., Clark, J.B., Marsden, C.D.,
1989. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet
Lond. Engl. 1, 1269.
Schapira, A.H., Jenner, P., 2011. Etiology and pathogenesis of Parkinson’s disease.
Mov. Disord. Off. J. Mov. Disord. Soc. 26, 1049–1055.
https://doi.org/10.1002/mds.23732

212
Schwarting, R.K., Bonatz, A.E., Carey, R.J., Huston, J.P., 1991. Relationships
between indices of behavioral asymmetries and neurochemical changes
following mesencephalic 6-hydroxydopamine injections. Brain Res. 554, 46–
55.
Schwarting, R.K., Huston, J.P., 1996. The unilateral 6-hydroxydopamine lesion
model in behavioral brain research. Analysis of functional deficits, recovery
and treatments. Prog. Neurobiol. 50, 275–331.
Séguéla, P., Wadiche, J., Dineley-Miller, K., Dani, J.A., Patrick, J.W., 1993.
Molecular cloning, functional properties, and distribution of rat brain alpha 7:
a nicotinic cation channel highly permeable to calcium. J. Neurosci. Off. J.
Soc. Neurosci. 13, 596–604.
Sgambato-Faure, V., Cenci, M.A., 2012. Glutamatergic mechanisms in the
dyskinesias induced by pharmacological dopamine replacement and deep
brain stimulation for the treatment of Parkinson’s disease. Prog. Neurobiol.
96, 69–86. https://doi.org/10.1016/j.pneurobio.2011.10.005
Shehab, S. a. S., Spike, R.C., Todd, A.J., 2003. Evidence against cholera toxin B
subunit as a reliable tracer for sprouting of primary afferents following
peripheral nerve injury. Brain Res. 964, 218–227.
Shehab, S., Anwer, M., Galani, D., Abdulkarim, A., Al-Nuaimi, K., Al-Baloushi, A.,
Tariq, S., Nagelkerke, N., Ljubisavljevic, M., 2015. Anatomical evidence that
the uninjured adjacent L4 nerve plays a significant role in the development of
peripheral neuropathic pain after L5 spinal nerve ligation in rats. J. Comp.
Neurol. 523, 1731–1747. https://doi.org/10.1002/cne.23750
Sherer, T.B., Kim, J.H., Betarbet, R., Greenamyre, J.T., 2003. Subcutaneous
rotenone exposure causes highly selective dopaminergic degeneration and
alpha-synuclein aggregation. Exp. Neurol. 179, 9–16.
Shimizu, M., Miyazaki, I., Higashi, Y., Eslava-Alva, M.J., Diaz-Corrales, F.J.,
Asanuma, M., Ogawa, N., 2008. Specific induction of PAG608 in cranial and
spinal motor neurons of L-DOPA-treated parkinsonian rats. Neurosci. Res.
60, 355–363. https://doi.org/10.1016/j.neures.2007.12.006
Shimohama, S., 2009. Nicotinic receptor-mediated neuroprotection in
neurodegenerative disease models. Biol Pharm Bull 32, 332–6.
Shimoji, M., Zhang, L., Mandir, A.S., Dawson, V.L., Dawson, T.M., 2005. Absence
of inclusion body formation in the MPTP mouse model of Parkinson’s
disease. Brain Res. Mol. Brain Res. 134, 103–108.
https://doi.org/10.1016/j.molbrainres.2005.01.012

213
Shishodia, S., Singh, T., Chaturvedi, M.M., 2007. Modulation of transcription factors
by curcumin. Adv. Exp. Med. Biol. 595, 127–148.
https://doi.org/10.1007/978-0-387-46401-5_4
Shoba, G., Joy, D., Joseph, T., Majeed, M., Rajendran, R., Srinivas, P.S., 1998.
Influence of piperine on the pharmacokinetics of curcumin in animals and
human volunteers. Planta Med. 64, 353–356. https://doi.org/10.1055/s-2006-
957450
Shytle, R.D., Mori, T., Townsend, K., Vendrame, M., Sun, N., Zeng, J., Ehrhart, J.,
Silver, A.A., Sanberg, P.R., Tan, J., 2004. Cholinergic modulation of
microglial activation by alpha 7 nicotinic receptors. J Neurochem 89, 337–43.
https://doi.org/10.1046/j.1471-4159.2004.02347.x
Singh, A., Naidu, P.S., Kulkarni, S.K., 2003. Quercetin potentiates L-Dopa reversal
of drug-induced catalepsy in rats: possible COMT/MAO inhibition.
Pharmacology 68, 81–88. https://doi.org/10.1159/000069533
Singh, S., Kumar, P., 2017. Neuroprotective potential of curcumin in combination
with piperine against 6-hydroxy dopamine induced motor deficit and
neurochemical alterations in rats. Inflammopharmacology 25, 69–79.
https://doi.org/10.1007/s10787-016-0297-9
Sinkus, M.L., Graw, S., Freedman, R., Ross, R.G., Lester, H.A., Leonard, S., 2015.
The human CHRNA7 and CHRFAM7A genes: A review of the genetics,
regulation, and function. Neuropharmacology 96, 274–288.
https://doi.org/10.1016/j.neuropharm.2015.02.006
Small-Molecule Drug Discovery Suite | Schrödinger [WWW Document], n.d. URL
https://www.schrodinger.com/suites/small-molecule-drug-discovery-suite
(accessed 1.15.17).
Snyder, S.H., D’Amato, R.J., 1985. Predicting Parkinson’s disease. Nature 317, 198–
199.
Song, S., Nie, Q., Li, Z., Du, G., 2016. Curcumin improves neurofunctions of 6-
OHDA-induced parkinsonian rats. Pathol. Res. Pract. 212, 247–251.
https://doi.org/10.1016/j.prp.2015.11.012
Sonsalla, P.K., Jochnowitz, N.D., Zeevalk, G.D., Oostveen, J.A., Hall, E.D., 1996.
Treatment of mice with methamphetamine produces cell loss in the substantia
nigra. Brain Res. 738, 172–175.

214
Soung, Y.H., Chung, J., 2011. Curcumin inhibition of the functional interaction
between integrin α6β4 and the epidermal growth factor receptor. Mol. Cancer
Ther. 10, 883–891. https://doi.org/10.1158/1535-7163.MCT-10-1053
Spector, S., Sjoerdsma, A., Udenfriend, S., 1965. BLOCKADE OF ENDOGENOUS
NOREPINEPHRINE SYNTHESIS BY ALPHA-METHYL-TYROSINE,
AN INHIBITOR OF TYROSINE HYDROXYLASE. J. Pharmacol. Exp.
Ther. 147, 86–95.
Spivak, C.E., Lupica, C.R., Oz, M., 2007. The endocannabinoid anandamide inhibits
the function of alpha4beta2 nicotinic acetylcholine receptors. Mol.
Pharmacol. 72, 1024–1032. https://doi.org/10.1124/mol.107.036939
Sreejayan, N., Rao, M.N., 1996. Free radical scavenging activity of curcuminoids.
Arzneimittelforschung. 46, 169–171.
Srinivasan, M., 1972. Effect of curcumin on blood sugar as seen in a diabetic subject.
Indian J. Med. Sci. 26, 269–270.
Srivastava, R., Dikshit, M., Srimal, R.C., Dhawan, B.N., 1985. Anti-thrombotic
effect of curcumin. Thromb. Res. 40, 413–417.
Steinlein, O.K., Mulley, J.C., Propping, P., Wallace, R.H., Phillips, H.A., Sutherland,
G.R., Scheffer, I.E., Berkovic, S.F., 1995. A missense mutation in the
neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with
autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 11, 201–3.
https://doi.org/10.1038/ng1095-201
Stence, N., Waite, M., Dailey, M.E., 2001. Dynamics of microglial activation: a
confocal time-lapse analysis in hippocampal slices. Glia 33, 256–66.
Sullivan, J.P., Decker, M.W., Brioni, J.D., Donnelly-Roberts, D., Anderson, D.J.,
Bannon, A.W., Kang, C.H., Adams, P., Piattoni-Kaplan, M., Buckley, M.J.,
al, et, 1994. (+/-)-Epibatidine elicits a diversity of in vitro and in vivo effects
mediated by nicotinic acetylcholine receptors. J Pharmacol Exp Ther 271,
624–31.
Sun, F., Jin, K., Uteshev, V.V., 2013. A Type-II Positive Allosteric Modulator of α7
nAChRs Reduces Brain Injury and Improves Neurological Function after
Focal Cerebral Ischemia in Rats. PLoS ONE 8.
https://doi.org/10.1371/journal.pone.0073581
Swinney, D.C., 2004. Biochemical mechanisms of drug action: what does it take for
success? Nat. Rev. Drug Discov. 3, 801–808. https://doi.org/10.1038/nrd1500

215
Tadaiesky, M.T., Dombrowski, P.A., Figueiredo, C.P., Cargnin-Ferreira, E., Da
Cunha, C., Takahashi, R.N., 2008. Emotional, cognitive and neurochemical
alterations in a premotor stage model of Parkinson’s disease. Neuroscience
156, 830–840. https://doi.org/10.1016/j.neuroscience.2008.08.035
Takikawa, M., Kurimoto, Y., Tsuda, T., 2013. Curcumin stimulates glucagon-like
peptide-1 secretion in GLUTag cells via Ca2+/calmodulin-dependent kinase
II activation. Biochem. Biophys. Res. Commun. 435, 165–170.
https://doi.org/10.1016/j.bbrc.2013.04.092
Talwar, S., Lynch, J.W., 2014. Phosphorylation mediated structural and functional
changes in pentameric ligand-gated ion channels: implications for drug
discovery. Int. J. Biochem. Cell Biol. 53, 218–223.
https://doi.org/10.1016/j.biocel.2014.05.028
Tambasco, N., Simoni, S., Marsili, E., Sacchini, E., Murasecco, D., Cardaioli, G.,
Rossi, A., Calabresi, P., 2012. Clinical aspects and management of levodopa-
induced dyskinesia. Park. Dis. 2012, 745947.
https://doi.org/10.1155/2012/745947
Tanner, C.M., 2010. Advances in environmental epidemiology. Mov Disord 25
Suppl 1, S58-62. https://doi.org/10.1002/mds.22721
Tanner, C.M., Goldman, S.M., Aston, D.A., Ottman, R., Ellenberg, J., Mayeux, R.,
Langston, J.W., 2002. Smoking and Parkinson’s disease in twins. Neurology
58, 581–8.
Tasneem, A., Iyer, L.M., Jakobsson, E., Aravind, L., 2005. Identification of the
prokaryotic ligand-gated ion channels and their implications for the
mechanisms and origins of animal Cys-loop ion channels. Genome Biol. 6,
R4. https://doi.org/10.1186/gb-2004-6-1-r4
Thiruchelvam, M., McCormack, A., Richfield, E.K., Baggs, R.B., Tank, A.W., Di
Monte, D.A., Cory-Slechta, D.A., 2003. Age-related irreversible progressive
nigrostriatal dopaminergic neurotoxicity in the paraquat and maneb model of
the Parkinson’s disease phenotype. Eur. J. Neurosci. 18, 589–600.
Thomas, A., Iacono, D., Luciano, A.L., Armellino, K., Di Iorio, A., Onofrj, M.,
2004. Duration of amantadine benefit on dyskinesia of severe Parkinson’s
disease. J Neurol Neurosurg Psychiatry 75, 141–3.
Thomas, B., Mandir, A.S., West, N., Liu, Y., Andrabi, S.A., Stirling, W., Dawson,
V.L., Dawson, T.M., Lee, M.K., 2011. Resistance to MPTP-neurotoxicity in
α-synuclein knockout mice is complemented by human α-synuclein and

216
associated with increased β-synuclein and Akt activation. PloS One 6,
e16706. https://doi.org/10.1371/journal.pone.0016706
Thomsen, M.S., Zwart, R., Ursu, D., Jensen, M.M., Pinborg, L.H., Gilmour, G., Wu,
J., Sher, E., Mikkelsen, J.D., 2015. α7 and β2 Nicotinic Acetylcholine
Receptor Subunits Form Heteromeric Receptor Complexes that Are
Expressed in the Human Cortex and Display Distinct Pharmacological
Properties. PloS One 10, e0130572.
https://doi.org/10.1371/journal.pone.0130572
Thrash, B., Thiruchelvan, K., Ahuja, M., Suppiramaniam, V., Dhanasekaran, M.,
2009. Methamphetamine-induced neurotoxicity: the road to Parkinson’s
disease. Pharmacol. Rep. PR 61, 966–977.
Tieu, K., 2011. A guide to neurotoxic animal models of Parkinson’s disease. Cold
Spring Harb. Perspect. Med. 1, a009316.
https://doi.org/10.1101/cshperspect.a009316
Timofeeva, O.A., Levin, E.D., 2011. Glutamate and nicotinic receptor interactions in
working memory: importance for the cognitive impairment of schizophrenia.
Neuroscience 195, 21–36. https://doi.org/10.1016/j.neuroscience.2011.08.038
Toda, S., Miyase, T., Arichi, H., Tanizawa, H., Takino, Y., 1985. Natural
antioxidants. III. Antioxidative components isolated from rhizome of
Curcuma longa L. Chem. Pharm. Bull. (Tokyo) 33, 1725–1728.
Tolosa, E., Compta, Y., 2006. Dystonia in Parkinson’s disease. J. Neurol. 253 Suppl
7, VII7-13. https://doi.org/10.1007/s00415-006-7003-6
Toulorge, D., Guerreiro, S., Hild, A., Maskos, U., Hirsch, E.C., Michel, P.P., 2011.
Neuroprotection of midbrain dopamine neurons by nicotine is gated by
cytoplasmic Ca2+. Faseb J 25, 2563–73. https://doi.org/10.1096/fj.11-182824
Tripanichkul, W., Jaroensuppaperch, E., 2012. Curcumin protects nigrostriatal
dopaminergic neurons and reduces glial activation in 6-hydroxydopamine
hemiparkinsonian mice model. Int. J. Neurosci. 122, 263–270.
https://doi.org/10.3109/00207454.2011.648760
Tripanichkul, W., Jaroensuppaperch, E.-O., 2013. Ameliorating effects of curcumin
on 6-OHDA-induced dopaminergic denervation, glial response, and SOD1
reduction in the striatum of hemiparkinsonian mice. Eur. Rev. Med.
Pharmacol. Sci. 17, 1360–1368.
Trulson, M.E., Cannon, M.S., Faegg, T.S., Raese, J.D., 1985. Effects of chronic
methamphetamine on the nigral-striatal dopamine system in rat brain:

217
tyrosine hydroxylase immunochemistry and quantitative light microscopic
studies. Brain Res. Bull. 15, 569–577.
Truong, L., Allbutt, H., Kassiou, M., Henderson, J.M., 2006. Developing a
preclinical model of Parkinson’s disease: a study of behaviour in rats with
graded 6-OHDA lesions. Behav. Brain Res. 169, 1–9.
https://doi.org/10.1016/j.bbr.2005.11.026
Turek, J.W., Kang, C.H., Campbell, J.E., Arneric, S.P., Sullivan, J.P., 1995. A
sensitive technique for the detection of the alpha 7 neuronal nicotinic
acetylcholine receptor antagonist, methyllycaconitine, in rat plasma and
brain. J. Neurosci. Methods 61, 113–118.
Umana, I.C., Daniele, C.A., McGehee, D.S., 2013. Neuronal nicotinic receptors as
analgesic targets: it’s a winding road. Biochem Pharmacol 86, 1208–14.
https://doi.org/10.1016/j.bcp.2013.08.001
Ungerstedt, U., 1971. Striatal dopamine release after amphetamine or nerve
degeneration revealed by rotational behaviour. Acta Physiol. Scand. Suppl.
367, 49–68.
Ungerstedt, U., Arbuthnott, G.W., 1970. Quantitative recording of rotational
behavior in rats after 6-hydroxy-dopamine lesions of the nigrostriatal
dopamine system. Brain Res. 24, 485–493.
Uteshev, V.V., 2014. The therapeutic promise of positive allosteric modulation of
nicotinic receptors. Eur. J. Pharmacol. 727, 181–185.
https://doi.org/10.1016/j.ejphar.2014.01.072
Uteshev, V.V., 2012. α7 nicotinic ACh receptors as a ligand-gated source of Ca(2+)
ions: the search for a Ca(2+) optimum. Adv. Exp. Med. Biol. 740, 603–638.
https://doi.org/10.1007/978-94-007-2888-2_27
Uversky, V.N., 2004. Neurotoxicant-induced animal models of Parkinson’s disease:
understanding the role of rotenone, maneb and paraquat in
neurodegeneration. Cell Tissue Res. 318, 225–241.
https://doi.org/10.1007/s00441-004-0937-z
Verhagen Metman, L., Del Dotto, P., van den Munckhof, P., Fang, J., Mouradian,
M.M., Chase, T.N., 1998. Amantadine as treatment for dyskinesias and motor
fluctuations in Parkinson’s disease. Neurology 50, 1323–6.
Vila, M., Vukosavic, S., Jackson-Lewis, V., Neystat, M., Jakowec, M., Przedborski,
S., 2000. Alpha-synuclein up-regulation in substantia nigra dopaminergic

218
neurons following administration of the parkinsonian toxin MPTP. J.
Neurochem. 74, 721–729.
Virmani, T., Moskowitz, C.B., Vonsattel, J.-P., Fahn, S., 2015. Clinicopathological
characteristics of freezing of gait in autopsy-confirmed Parkinson’s disease.
Mov. Disord. Off. J. Mov. Disord. Soc. 30, 1874–1884.
https://doi.org/10.1002/mds.26346
Voorn, P., Vanderschuren, L.J., Groenewegen, H.J., Robbins, T.W., Pennartz, C.M.,
2004. Putting a spin on the dorsal-ventral divide of the striatum. Trends
Neurosci 27, 468–74. https://doi.org/10.1016/j.tins.2004.06.006
Wahlström, B., Blennow, G., 1978. A study on the fate of curcumin in the rat. Acta
Pharmacol. Toxicol. (Copenh.) 43, 86–92.
Wang, D., Tang, B., Zhao, G., Pan, Q., Xia, K., Bodmer, R., Zhang, Z., 2008.
Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival
of dopaminergic neurons. Mol. Neurodegener. 3, 3.
https://doi.org/10.1186/1750-1326-3-3
Wang, Hong, Yu, M., Ochani, M., Amella, C.A., Tanovic, M., Susarla, S., Li, J.H.,
Wang, Haichao, Yang, H., Ulloa, L., Al-Abed, Y., Czura, C.J., Tracey, K.J.,
2003. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator
of inflammation. Nature 421, 384–388. https://doi.org/10.1038/nature01339
Wang, W.-H., Chiang, I.-T., Ding, K., Chung, J.-G., Lin, W.-J., Lin, S.-S., Hwang,
J.-J., 2012. Curcumin-induced apoptosis in human hepatocellular carcinoma
j5 cells: critical role of ca(+2)-dependent pathway. Evid.-Based Complement.
Altern. Med. ECAM 2012, 512907. https://doi.org/10.1155/2012/512907
Wang, X.-S., Zhang, Z.-R., Zhang, M.-M., Sun, M.-X., Wang, W.-W., Xie, C.-L.,
2017. Neuroprotective properties of curcumin in toxin-base animal models of
Parkinson’s disease: a systematic experiment literatures review. BMC
Complement. Altern. Med. 17, 412. https://doi.org/10.1186/s12906-017-
1922-x
Ward, R.J., Lallemand, F., de Witte, P., Dexter, D.T., 2008. Neurochemical
pathways involved in the protective effects of nicotine and ethanol in
preventing the development of Parkinson’s disease: potential targets for the
development of new therapeutic agents. Prog Neurobiol 85, 135–47.
https://doi.org/10.1016/j.pneurobio.2008.03.003
Warpman, U., Nordberg, A., 1995. Epibatidine and ABT 418 reveal selective losses
of alpha 4 beta 2 nicotinic receptors in Alzheimer brains. Neuroreport 6,
2419–23.

219
Ween, H., Thorin-Hagene, K., Andersen, E., Gronlien, J.H., Lee, C.H.,
Gopalakrishnan, M., Malysz, J., 2010. Alpha3* and alpha 7 nAChR-mediated
Ca2+ transient generation in IMR-32 neuroblastoma cells. Neurochem Int 57,
269–77. https://doi.org/10.1016/j.neuint.2010.06.005
Weiland, S., Witzemann, V., Villarroel, A., Propping, P., Steinlein, O., 1996. An
amino acid exchange in the second transmembrane segment of a neuronal
nicotinic receptor causes partial epilepsy by altering its desensitization
kinetics. FEBS Lett 398, 91–6.
West, M.J., 1999. Stereological methods for estimating the total number of neurons
and synapses: issues of precision and bias. Trends Neurosci. 22, 51–61
West, M.J., Ostergaard, K., Andreassen, O.A., Finsen, B., 1996. Estimation of the
number of somatostatin neurons in the striatum: an in situ hybridization study
using the optical fractionator method. J. Comp. Neurol. 370, 11–22.
https://doi.org/10.1002/(SICI)1096-9861(19960617)370:1<11::AID-
CNE2>3.0.CO;2-O
West, M.J., 1993. New stereological methods for counting neurons. Neurobiol.
Aging 14, 275–285.
West, M.J., Slomianka, L., Gundersen, H.J., 1991. Unbiased stereological estimation
of the total number of neurons in thesubdivisions of the rat hippocampus
using the optical fractionator. Anat. Rec. 231, 482–497.
https://doi.org/10.1002/ar.1092310411
Widmann, R., Sperk, G., 1986. Topographical distribution of amines and major
amine metabolites in the rat striatum. Brain Res. 367, 244–249.
Williams, D.R., Lees, A.J., 2009. How do patients with parkinsonism present? A
clinicopathological study. Intern. Med. J. 39, 7–12.
https://doi.org/10.1111/j.1445-5994.2008.01635.x
Williams, D.R., Watt, H.C., Lees, A.J., 2006. Predictors of falls and fractures in
bradykinetic rigid syndromes: a retrospective study. J. Neurol. Neurosurg.
Psychiatry 77, 468–473. https://doi.org/10.1136/jnnp.2005.074070
Williams, M., Raddatz, R., 2006. Receptors as drug targets. Curr. Protoc. Pharmacol.
Chapter 1, Unit 1.1. https://doi.org/10.1002/0471141755.ph0101s32
Williams-Gray, C.H., Foltynie, T., Lewis, S.J.G., Barker, R.A., 2006. Cognitive
deficits and psychosis in Parkinson’s disease: a review of pathophysiology
and therapeutic options. CNS Drugs 20, 477–505.

220
Wirdefeldt, K., Adami, H.O., Cole, P., Trichopoulos, D., Mandel, J., 2011.
Epidemiology and etiology of Parkinson’s disease: a review of the evidence.
Eur J Epidemiol 26 Suppl 1, S1-58. https://doi.org/10.1007/s10654-011-
9581-6
Wolf, E., Seppi, K., Katzenschlager, R., Hochschorner, G., Ransmayr, G.,
Schwingenschuh, P., Ott, E., Kloiber, I., Haubenberger, D., Auff, E., Poewe,
W., 2010. Long-term antidyskinetic efficacy of amantadine in Parkinson’s
disease. Mov. Disord. Off. J. Mov. Disord. Soc. 25, 1357–1363.
https://doi.org/10.1002/mds.23034
Wozniak, K.L., Phelps, W.A., Tembo, M., Lee, M.T., Carlson, A.E., 2018. The
TMEM16A channel mediates the fast polyspermy block in Xenopus laevis. J.
Gen. Physiol. https://doi.org/10.1085/jgp.201812071
wwPDB: Worldwide Protein Data Bank [WWW Document], n.d. URL
http://www.wwpdb.org/ (accessed 1.15.17).
Xie, X., Tao, Q., Zou, Y., Zhang, F., Guo, M., Wang, Y., Wang, H., Zhou, Q., Yu,
S., 2011. PLGA nanoparticles improve the oral bioavailability of curcumin in
rats: characterizations and mechanisms. J. Agric. Food Chem. 59, 9280–9289.
https://doi.org/10.1021/jf202135j
Xie, Y.X., Bezard, E., Zhao, B.L., 2005. Investigating the receptor-independent
neuroprotective mechanisms of nicotine in mitochondria. J Biol Chem 280,
32405–12. https://doi.org/10.1074/jbc.M504664200
Yang, J., Song, S., Li, J., Liang, T., 2014. Neuroprotective effect of curcumin on
hippocampal injury in 6-OHDA-induced Parkinson’s disease rat. Pathol. Res.
Pract. 210, 357–362. https://doi.org/10.1016/j.prp.2014.02.005
Yang, J.-S., Seo, S.W., Jang, S., Jung, G.Y., Kim, S., 2012. Rational engineering of
enzyme allosteric regulation through sequence evolution analysis. PLoS
Comput. Biol. 8, e1002612. https://doi.org/10.1371/journal.pcbi.1002612
Yang, T., Xiao, T., Sun, Q., Wang, K., 2017. The current agonists and positive
allosteric modulators of α7 nAChR for CNS indications in clinical trials. Acta
Pharm. Sin. B 7, 611–622. https://doi.org/10.1016/j.apsb.2017.09.001
Yang, Y., Wu, X., Wei, Z., Dou, Y., Zhao, D., Wang, T., Bian, D., Tong, B., Xia,
Ying, Xia, Yufeng, Dai, Y., 2015. Oral curcumin has anti-arthritic efficacy
through somatostatin generation via cAMP/PKA and Ca(2+)/CaMKII
signaling pathways in the small intestine. Pharmacol. Res. 95–96, 71–81.
https://doi.org/10.1016/j.phrs.2015.03.016

221
Yenari, M.A., Kauppinen, T.M., Swanson, R.A., 2010. Microglial activation in
stroke: therapeutic targets. Neurotherapeutics 7, 378–91.
https://doi.org/10.1016/j.nurt.2010.07.005
Yeon, K.Y., Kim, S.A., Kim, Y.H., Lee, M.K., Ahn, D.K., Kim, H.J., Kim, J.S.,
Jung, S.J., Oh, S.B., 2010. Curcumin produces an antihyperalgesic effect via
antagonism of TRPV1. J. Dent. Res. 89, 170–174.
https://doi.org/10.1177/0022034509356169
Young, G.T., Zwart, R., Walker, A.S., Sher, E., Millar, N.S., 2008. Potentiation of
alpha7 nicotinic acetylcholine receptors via an allosteric transmembrane site.
Proc. Natl. Acad. Sci. U. S. A. 105, 14686–14691.
https://doi.org/10.1073/pnas.0804372105
Yu, S., Zheng, W., Xin, N., Chi, Z.-H., Wang, N.-Q., Nie, Y.-X., Feng, W.-Y.,
Wang, Z.-Y., 2010. Curcumin prevents dopaminergic neuronal death through
inhibition of the c-Jun N-terminal kinase pathway. Rejuvenation Res. 13, 55–
64. https://doi.org/10.1089/rej.2009.0908
Zaborszky, L., Vadasz, C., 2001. The midbrain dopaminergic system: anatomy and
genetic variation in dopamine neuron number of inbred mouse strains. Behav.
Genet. 31, 47–59.
Zbarsky, V., Datla, K.P., Parkar, S., Rai, D.K., Aruoma, O.I., Dexter, D.T., 2005.
Neuroprotective properties of the natural phenolic antioxidants curcumin and
naringenin but not quercetin and fisetin in a 6-OHDA model of Parkinson’s
disease. Free Radic. Res. 39, 1119–1125.
https://doi.org/10.1080/10715760500233113
Zhang, D., Bordia, T., McGregor, M., McIntosh, J.M., Decker, M.W., Quik, M.,
2014. ABT-089 and ABT-894 reduce levodopa-induced dyskinesias in a
monkey model of Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord.
Soc. 29, 508–517. https://doi.org/10.1002/mds.25817
Zhang, D., McGregor, M., Bordia, T., Perez, X.A., McIntosh, J.M., Decker, M.W.,
Quik, M., 2015. α7 nicotinic receptor agonists reduce levodopa-induced
dyskinesias with severe nigrostriatal damage. Mov. Disord. Off. J. Mov.
Disord. Soc. 30, 1901–1911. https://doi.org/10.1002/mds.26453
Zhang, D.-W., Fu, M., Gao, S.-H., Liu, J.-L., 2013. Curcumin and diabetes: a
systematic review. Evid.-Based Complement. Altern. Med. ECAM 2013,
636053. https://doi.org/10.1155/2013/636053
Zhang, L., Oz, M., Weight, F.F., 1995. Potentiation of 5-HT3 receptor-mediated
responses by protein kinase C activation. Neuroreport 6, 1464–1468.

222
Zhang, X., Chen, Q., Wang, Y., Peng, W., Cai, H., 2014. Effects of curcumin on ion
channels and transporters. Front. Physiol. 5, 94.
https://doi.org/10.3389/fphys.2014.00094
Zhi, L., Dong, L., Kong, D., Sun, B., Sun, Q., Grundy, D., Zhang, G., Rong, W.,
2013. Curcumin acts via transient receptor potential vanilloid-1 receptors to
inhibit gut nociception and reverses visceral hyperalgesia.
Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 25, e429-440.
https://doi.org/10.1111/nmo.12145
Zhou, F.M., Liang, Y., Dani, J.A., 2001. Endogenous nicotinic cholinergic activity
regulates dopamine release in the striatum. Nat Neurosci 4, 1224–9.
https://doi.org/10.1038/nn769
Zhou, F.M., Wilson, C.J., Dani, J.A., 2002. Cholinergic interneuron characteristics
and nicotinic properties in the striatum. J Neurobiol 53, 590–605.
https://doi.org/10.1002/neu.10150
Zhou, H., Beevers, C.S., Huang, S., 2011. Targets of curcumin. Curr. Drug Targets
12, 332–347.
Zhou, X., Nai, Q., Chen, M., Dittus, J.D., Howard, M.J., Margiotta, J.F., 2004. Brain-
derived neurotrophic factor and trkB signaling in parasympathetic neurons:
relevance to regulating alpha7-containing nicotinic receptors and synaptic
function. J Neurosci 24, 4340–50. https://doi.org/10.1523/jneurosci.0055-
04.2004
Zoli, M., Pistillo, F., Gotti, C., 2015. Diversity of native nicotinic receptor subtypes
in mammalian brain. Neuropharmacology 96, 302–11.
https://doi.org/10.1016/j.neuropharm.2014.11.003

223
List of Publications
El Nebrisi, E.G., Bagdas, D., Toma, W., Al Samri, H., Brodzik, A., Alkhlaif, Y.,
Yang, K.-H.S., Howarth, F.C., Damaj, I.M., Oz, M., 2018. Curcumin Acts as
a Positive Allosteric Modulator of α7-Nicotinic Acetylcholine Receptors and
Reverses Nociception in Mouse Models of Inflammatory Pain. J. Pharmacol.
Exp. Ther. 365, 190–200. https://doi.org/10.1124/jpet.117.245068
Nebrisi, E.E., Al Kury, L.T., Yang, K.-H.S., Jayaprakash, P., Howarth, F.C.,
Kabbani, N., Oz, M., 2018. Curcumin potentiates the function of human α7-
nicotinic acetylcholine receptors expressed in SH-EP1 cells. Neurochem. Int.
114, 80–84. https://doi.org/10.1016/j.neuint.2017.12.010
Oz, M., El Nebrisi, E.G., Yang, K.-H.S., Howarth, F.C., Al Kury, L.T., 2017.
Cellular and Molecular Targets of Menthol Actions. Front. Pharmacol. 8,
472. https://doi.org/10.3389/fphar.2017.00472
Sultan, A., Yang, K.-H.S., Isaev, D., Nebrisi, E.E., Syed, N., Khan, N., Howarth,
C.F., Sadek, B., Oz, M., 2017. Thujone inhibits the function of α7-nicotinic
acetylcholine receptors and impairs nicotine-induced memory enhancement
in one-trial passive avoidance paradigm. Toxicology 384, 23–32.
https://doi.org/10.1016/j.tox.2017.04.005

224
Appendix
Optimization
In-vitro
• Acetylcholine
- Stock solution of the Acetylcholine (100 µM) was prepared in ND96 solution
using the following formula:
Weight (mg) = (MW) x (Volume (L)) x (concentration (mM))
= 181.7 x 0.2 L x 0.100
= 3.6 mg of ACh in 200 ml ND96
- Further dilutions were prepared using the Charles equation:
C1 x V1 = C2 x V2
- For preparation of 10 µM ACh;
100 x V1 = 10 x 100 ml (ND96)
V1 = 10 ml of 100 µM stock solution plus 90 ml ND96
= 100 ml ACh solution of 10 µM concentration, divided into two parts:
50 ml for first application line, and the remaining 50 ml was for the preparation of
the second application line solution.
Stock solutions and required dilutions were prepared freshly before
starting the experiments.
ACh was applied every five minutes. Shorter intervals would lead to stimulation
the receptor during the desensitized state (the channel is insensitive to agonist
stimulus).

225
• Curcumin
Curcumin is a highly hydrophobic compound and is soluble only in organic solvents
e.g., dimethyl sulfoxide (DMSO).
Stock solution of the curcumin (10 µM) was prepared in DMSO (0.001%) using
the following formula:
Weight (mg) = (MW) x (Volume (L)) x (concentration (mM))
= 368.38 x 0.001 L x 10
= 3.68 mg of curcumin in 1 ml of DMSO
- Further dilutions were prepared using the Charles equation:
C1 x V1 = C2 x V2
- For preparation of 10 µM curcumin;
10 x V1 = 0.01 x 50 ml (ACh sol.)
V1 = 0.05 ml
= 50 µl of curcumin stock solution (10 µM) plus 50 ml ACh solution
prepared in the First step; second application line.
- First perfusion was ND96 physiological solution
- Second perfusion using curcumin stock solution (10 µM)
C1 x V1 = C2 x V2
10 x V1 = 0.01 x 600 ml ND96
V1 = 0.6 ml of curcumin stock solution (10 µM) plus 600 ml ND96.
Since the magnitude of the curcumin effect was time-dependent, 10-minute
curcumin application time was used routinely to ensure equilibrium conditions.
Same concept was applied for different concentrations of curcumin used in the study,
and also for other curcumin derivatives with the respect of the molecular weight of
each compound.

226
• Glycine
- Preparation of 30 µM from stock solution:
C1 x V1 = C2 x V2
100 x V1 = 0.03 x 100
V1 = 30 µl of Gly (100 mM) in 100 ml Ringer physiological solution.
50 ml for first application line, and the remaining 50 ml was for the preparation of
the second application line solution.
To prepare 10 µM from 100 mM stock curcumin:
C1 x V1 = C2 x V2
100 x V1 = 0.01 x 50
V1 = 5 µl of curcumin stock solution (100 mM) in 50 ml of Gly (30 µM) Ringer
solution, as a second application line.
- First perfusion was Ringer physiological solution
- Second perfusion using curcumin stock solution (10 µM)
C1 x V1 = C2 x V2
10 x V1 = 0.01 x 600 ml ND96
V1 = 0.6 ml of curcumin stock solution (10 µM) plus 600 ml Ringer solution.
• 5HT3
- Preparation of 1 µM from stock solution:
C1 x V1 = C2 x V2
100 x V1 = 0.001 x 100
V1 = 1 µl of 5HT (100 mM) in 100 ml Ringer physiological solution.
50 ml for first application line, and the remaining 50 ml was for the preparation of
the second application line solution.

227
- To prepare 10 µM from 100 mM stock curcumin:
C1 x V1 = C2 x V2
100 x V1 = 0.01 x 50
V1 = 5 µl of curcumin stock solution (100 mM) in 50 ml of Gly (30 µM) Ringer
solution, as a second application line.
- First perfusion was Ringer physiological solution
- Second perfusion using curcumin stock solution (10 µM)
C1 x V1 = C2 x V2
10 x V1 = 0.01 x 600 ml ND96
V1 = 0.6 ml of curcumin stock solution (10 µM) plus 600 ml Ringer solution.
• BAPTA
- Preparation of stock solution:
Weight (mg) = (MW) x (Volume (L)) x (concentration (mM))
= 476.23 x 0.001 x 100
= 47.623 mg BAPTA in 1 ml D/W
pH was adjusted to 7.4 by CsOH, and stored at +4℃
• Barium
- Preparation of Barium-containing solution:
ND96 solution but with substitution of BaCl2 (1.8 mM) instead of CaCL2 (1.8 mM).
• Protein Kinases Inhibitors
Stock solution of (100 µM) for each protein kinase inhibitor, to prepare the working
concentration of 10 µM using the formula:
C1 x V1 = C2 x V2

228
100 x V1 = 0.1 x 10
V1 = 0.01 ml
V1 = 10µM of stock solution plus 1 ml ND96 physiological solution for pre-
incubation.
Oocytes were pre-incubated for 30 minutes before electrophysiological recording.
• Nicotine
- Preparation of 100 mM stock solution:
Weight (mg) = (MW) x (Volume (L)) x (concentration (mM))
= 462.4 x 0.001x 100
= 46.24 mg in 1 ml D/W
- To prepare 100 µM from 100 mM stock solution of nicotine
C1 x V1 = C2 x V2
100 x V1= 0.1x100
V1= 0.1 ml of tock in 100 ml of D/W
In-vivo
• 6-hydroxydopamine (6-OHDA)
- Preparation of 0.9% NaCl:
0.9 NaCl in 100 ml H2O
- Preparation of 0.01% ascorbic acid solution:
10 mg of L-ascorbic acid in 0.9% NaCL
- Preparation of 6-OHDA injection solution:

229
Thirty-five milligram of 6-OHDA was dissolved in 1o ml of ice-cold 0.01%
ascorbate in 0.9% normal saline and aliquoted into 250 µl Eppendorf tubes and
kept in -40℃ until use.
• Curcumin
- Preparation of 0.5% CMC (vehicle):
0.5 gm CMC added to 100 ml distilled water, stirred, and heated up to 60℃ until it
dissolves completely.
- 200 mg of curcumin plus 50 µl of NaOH (10M) in 2 ml of 0.5% CMC.
• Methyllycaconitine (MLA)
Methyllycaconitine (MLA) is the most potent and selective competitive antagonist
of α7-nAChR (Turek et al., 1995). MLA was prepared in normal saline, no need for
pH adjustment, kept in +4℃ in fridge and its stable compound. MLA half-life is
around 19 min, and for in-vivo use its administered 10 min for maximal effect.
- Dose: 1 mg/kg/rat for 4 weeks, other research papers have used it 1-3 mg/kg,
but to avoid toxicity the daily dose should be 1/10th of the LD50 (LD50=5
mg/kg, 1/10th = 0.5 mg/kg), so we have used (1 mg/kg rat) = (1 µl/gram rat).
• Apomorphine
The dose of apomorphine that has been used in the study (0.25 mg/KG SC) (Kirik
et al., 1998) was significantly lower than the toxic dose (160 mg/kg) (“Sax’s
Dangerous Properties of Industrial Materials, 5 Volume Set, 12th Edition,” n.d.).
Apomorphine was dissolved in 0.1 ascorbic acid in saline and prepared on demand.
Animals were injected subcutaneously with apomorphine-HCL.










![Human a4b2 Nicotinic Acetylcholine Receptor as a Novel ......nicotine through the activation of nicotinic acetylcholine receptors (nAChRs) [22,23,24,25]. Previous studies indicate](https://img.pdfslide.net/doc/110x75/5f0f0a627e708231d442317c/human-a4b2-nicotinic-acetylcholine-receptor-as-a-novel-nicotine-through.jpg)









![18F]Flubatine as a novel α4β2 nicotinic acetylcholine](https://img.pdfslide.net/doc/110x75/629737326d4e5a451c0d4cae/18fflubatine-as-a-novel-42-nicotinic-acetylcholine-.jpg)








