Embed Size (px)
Citation preview

The Spectrum of Subclinical Best Vitelliform MacularDystrophy in Subjects with Mutations in BEST1 Gene
Giuseppe Querques,1 Jennyfer Zerbib,1,2 Rossana Santacroce,3 Maurizio Margaglione,3
Nathalie Delphin,2 Lea Querques,1 Jean-Michel Rozet,2 Josseline Kaplan,2
and Eric H. Souied1,4
PURPOSE. To describe the morphologic and functional charac-teristics of subclinical Best vitelliform macular dystrophy(VMD) in subjects with mutation in the BEST1 gene.
METHODS. Best-corrected visual acuity (BCVA), funduscopicappearance, fundus autofluorescence (FAF), spectral-domainoptical coherence tomography (SD-OCT), and electro-oculog-raphy (EOG) were assessed in 23 consecutive subjects fromnine unrelated families with known mutations in the BEST1gene (eight distinct BEST1 mutations).
RESULTS. Six subjects were identified with BEST1 mutations(three male, three female; aged 8 to 30 years) without clinicallydetectable (subclinical) Best VMD (absence of both symptomsand funduscopic lesions). All six subjects showed 20/20 BCVAand normal FAF findings. In these 6 of 26 subjects from fivedifferent families, we found five distinct mutations in theBEST1 gene. In three (six eyes) out of these six subjects withBEST1 gene mutations (two families: p.G15D; p.A243V), SD-OCT showed overall normal findings. In the other three sub-jects (six eyes) with BEST1 gene mutations (three families:p.V9A; p.R92C; p.I230T), we found, on SD-OCT, a thicker andmore reflective appearance of the layer between the retinalpigment epithelium and the interface of inner segments andouter segments of the photoreceptor (the Verhoeff’s mem-brane). EOG showed a reduced light-peak:dark-trough ratio in5 of 12 eyes. Changes on SD-OCT were present in the absenceof EOG abnormalities (two of six eyes), and vice versa (one ofsix eyes).
CONCLUSIONS. Subclinical Best VMD (absence of both symptomsand funduscopic lesions) in subjects with BEST1 mutation mayvary from the absence of any morphologic and functionalabnormalities to the presence of specific SD-OCT and EOGchanges. (Invest Ophthalmol Vis Sci. 2011;52:4678–4684) DOI:10.1167/iovs.10-6500
Vitelliform macular dystrophy (VMD) (OMIM 153,700), alsocalled Best disease,1 has an autosomal dominant pattern of
inheritance with very variable penetrance and expressivity.BEST1 (chromosome 11q12-q13)2 is the only gene virtuallyinvolved in all Best VMD cases. It encodes a 68 kDa proteincalled bestrophin-1,3 which is localized to the basolateralplasma membrane of the retinal pigment epithelium (RPE) andappears to exhibit properties of Ca��–activated Cl- channels.4
Nearly all BEST1 mutations causing Best VMD affect singleamino acids, at one of 66 different positions in bestrophin-1.
Best VMD is a clinically heterogenous and pleomorphicdisease, having a bimodal onset distribution with one maxi-mum peak before puberty and a second following puberty andextending through the fifth decade of life.5–7
The onset of Best VMD is characterized by symptoms ofmetamorphopsia, blurred vision, and decrease of central vi-sion. At the fundus a well-circumscribed 0.5- to 2-disc–diame-ter “egg-yolk” lesion within the macula may be observed.8 Thisrepresents the vitelliform stage of the disease, the second offive progressive stages defined on the basis of fundus exami-nation. This early stage of the disease may be followed by thepseudohypopyon third stage, the vitelliruptive fourth stage,and the atrophic/fibrotic fifth stage. The subclinical form of thedisease, or previtelliform stage, represents the first stage and ischaracterized by absence of symptoms and normal macula orsubtle RPE alterations on fundus examination.
An abnormal electro-oculogram (EOG)9–11 with a reducedor nondetectable light-peak:dark-trough ratio (� 1.55), a block-age effect of fluorescein in the choroid,12 and evidence ofincreased autofluorescence by the yellow vitelliform lesions7
are considered the main diagnostic criteria in the clinical diag-nosis of Best VMD.11,12 Several authors have recently high-lighted the usefulness of optical coherence tomography (OCT)in the diagnosis of Best VMD to be the demonstration of thevitelliform material that accumulates in the subretinal spaceand on the outer retinal surface.13–16 However, no data havebeen published so far on the different functional and morpho-logic aspects of the subclinical form of the disease (the previ-telliform stage).
Our purpose was to analyze the functional and morphologiccharacteristics in asymptomatic subclinical subjects issuingfrom Best VMD families carriers of mutations in the BEST1gene.
METHODS
Nine Best VMD families segregating BEST1 mutations,17 ascertained atthe Creteil University Eye Clinic and at the Foggia University Eye Clinic,were included in this study. Informed consent was obtained accordingto a Paris XII University and a Foggia University Institutional ReviewBoard–approved protocol. This study has been performed in accor-dance with the ethical standards set forth in the 1964 Declaration ofHelsinki.
From the 1Department of Ophthalmology, Hopital Intercommunalde Creteil, University Paris Est Creteil, Creteil, France; 2Department ofGenetics, Necker Hospital, University Paris V, Paris, France; 3Depart-ment of Genetics, Ospedali Riuniti, University of Foggia, Foggia, Italy;and 4Unite Fonctionnelle de Recherche Clinique, Creteil, France.
Presented in part as a poster at the American Academy of Oph-thalmology Annual Meeting, Chicago, October 14–19, 2010.
Supported by the Centre de Reference Maladie Rare (CRMR)program.
Submitted for publication August 31, 2010; revised October 27,2010, and January 5 and February 14, 2011; accepted March 6, 2011.
Disclosure: G. Querques, None; J. Zerbib, None; R. Santacroce,None; M. Margaglione, None; N. Delphin, None; L. Querques,None; J.-M. Rozet, None; J. Kaplan, None; E.H. Souied, None
Corresponding author: Giuseppe Querques, Department of Oph-thalmology, University of Paris XII, Centre Hospitalier Intercommunalde Creteil, 40 Avenue de Verdun, 94000 Creteil, France;[email protected].
Retina
Investigative Ophthalmology & Visual Science, June 2011, Vol. 52, No. 74678 Copyright 2011 The Association for Research in Vision and Ophthalmology, Inc.
Downloaded From: http://iovs.arvojournals.org/pdfaccess.ashx?url=/data/journals/iovs/933461/ on 05/24/2018

In each family, affected and unaffected relatives who agreed toparticipate to the study were screened for the BEST1 mutation iden-tified in the BEST VMD proband using direct sequencing as previouslydescribed.17
All individuals included in this study underwent a complete oph-thalmologic examination, including assessment of best-corrected visualacuity (BCVA) measured at 4 m with standard Early Treatment DiabeticRetinopathy Study charts, fundus biomicroscopy, color photography ofthe fundus (TRC-50 retinal camera; Topcon, Tokyo, Japan), fundusautofluorescence (FAF) frames (Heidelberg Retina Angiograph II;Heidelberg Engineering, Heidelberg, Germany), and red free and fluo-rescein angiography frames (Topcon TRC-50; Heidelberg Retina Angio-graph II). Recordings of EOG were done according to the InternationalSociety for Clinical Electrophysiology of Vision standard. OCT exami-nation was performed with time-domain (TD)-OCT (OCT 3000 Stratus;Humphrey-Zeiss, San Leandro, CA) or spectral domain (SD)-OCT (HD-OCT, OCT 4000 Cirrus, Humphrey-Zeiss; Spectralis SD-OCT, Heidel-berg Engineering). All scans were positioned within the macular areaand throughout the vitelliform lesions, based on color fundus photog-raphy and FAF. Fundus pictures and SD-OCT scans were analyzed and
interpreted independently by three retinal specialists (GQ, JZ, andEHS). Disagreement regarding interpretation of the different featureswas resolved by open adjudication.
The clinical diagnosis of Best VMD (� second stage) was based onthe presence, on fundus examination, of one or multiple subfovealvitelliform lesions in at least one eye and on the evidence of autofluo-rescence by the yellow vitelliform lesions. The subclinical form of thedisease represents the first stage of Best VMD (the previtelliform stage),which is defined by absence of symptoms and normal macula or subtleRPE alterations on fundus examination. In this study, subjects werediagnosed with the subclinical form of the disease if they were asymp-tomatic and did not show subfoveal lesions or FAF changes.
On EOG, a reduced light-peak:dark-trough ratio � 1.55 was con-sidered abnormal (as per our laboratory protocol). Ratios � 1.55 wereconsidered as normal values.
RESULTS
A total of 23 subjects (11 male and 12 female; mean age, 26.9years; range, 3 to 70 years), from nine unrelated families with
FIGURE 1. Pedigree of the studiedfamilies. Here all subjects with thesubclinical form of Best vitelliformmacular dystrophy are presented inwhite because of the absence ofsymptoms. In our previous analysis17
subjects CT03 and CT14 were pre-sented in black because of explora-tion abnormalities (OCT and EOG).
IOVS, June 2011, Vol. 52, No. 7 Subclinical Best VMD 4679
Downloaded From: http://iovs.arvojournals.org/pdfaccess.ashx?url=/data/journals/iovs/933461/ on 05/24/2018
TA
BLE
1.
Sum
mar
yo
fC
linic
alFi
nd
ings
and
Pro
ban
ds
BE
ST1
Mu
tati
on
s
Sub
ject
sM
uta
tio
nP
osi
tio
nM
isse
nse
Eff
ect
Age
,Se
xA
geo
fO
nse
t(y
)
Lesi
on
Typ
eB
CV
A
Co
mp
lica
tio
ns
RE
LER
ELE
FG01
(Fam
ilyFG
I)C
�T
728
het
ero
zygo
us
Exo
n7
A24
3V49
,M41
Atr
op
hy
Atr
op
hy
20/1
2520
/160
—FG
02(F
amily
FGI)
C�
T72
8h
eter
ozy
gou
sEx
on
7A
243V
45,F
37V
itel
liru
pti
veV
itel
liru
pti
ve20
/25
20/2
5—
FG03
(Fam
ilyFG
I)C
�T
728
het
ero
zygo
us
Exo
n7
A24
3V75
,M67
Pse
ud
oh
ypo
pio
nV
itel
liru
pti
ve20
/50
20/1
25—
FG04
(Fam
ilyFG
I)C
�T
728
het
ero
zygo
us
Exo
n7
A24
3V13
,F—
No
ne
No
ne
20/2
020
/20
—FG
05(F
amily
FGI)
C�
T72
8h
eter
ozy
gou
sEx
on
7A
243V
17,F
—N
on
eN
on
e20
/20
20/2
0—
FG06
(Fam
ilyFG
II)
G�
A27
5h
eter
ozy
gou
sEx
on
4R
92H
16,F
11Fi
bro
sis
Fib
rosi
s20
/160
20/1
60C
NV
RLE
FG07
(Fam
ilyFG
II)
G�
A27
5h
eter
ozy
gou
sEx
on
4R
92H
3,M
2V
itel
lifo
rmV
itel
lifo
rm20
/32
20/3
2—
FG08
(Fam
ilyFG
III)
G�
A44
het
ero
zygo
us
Exo
n2
G15
D3,
F2
Vit
ellif
orm
Vit
ellif
orm
20/2
520
/25
—FG
09(F
amily
FGII
I)G
�A
44h
eter
ozy
gou
sEx
on
2G
15D
30,M
—N
on
eN
on
e20
/20
20/2
0—
CT
01(F
amily
CT
I)C
�T
274
het
ero
zygo
us
Exo
n4
R92
C8,
M8
Pre
-vit
ellif
orm
Pre
-vit
ellif
orm
20/2
020
/20
CT
02(F
amily
CT
I)C
�T
274
het
ero
zygo
us
Exo
n4
R92
C14
,M8
Fib
rosi
sFi
bro
sis
20/5
020
/40
CN
VR
LEC
T03
(Fam
ilyC
TII
)T
�C
689
het
ero
zygo
us
Exo
n7
I230
T11
,M10
Pre
-vit
ellif
orm
Pre
-vit
ellif
orm
20/2
020
/25
—C
T04
(Fam
ilyC
TII
)T
�C
689
het
ero
zygo
us
Exo
n7
I230
T42
,F41
Mu
ltif
oca
lM
ult
ifo
cal
20/3
220
/25
—C
T05
(Fam
ilyC
TII
)T
�C
689
het
ero
zygo
us
Exo
n7
I230
T9,
M6
Vit
ellir
up
tive
Vit
ellir
up
tive
20/1
2520
/125
—C
T06
(Fam
ilyC
TII
I)C
�T
272
het
ero
zygo
us
Exo
n4
T91
I44
,M36
Atr
op
hy
Atr
op
hy
20/1
2520
/40
—C
T07
(Fam
ilyC
TII
I)C
�T
272
het
ero
zygo
us
Exo
n4
T91
I19
,F11
Fib
rosi
sFi
bro
sis
20/2
0020
/40
CN
VR
EC
T08
(Fam
ilyC
TIV
)A
�G
10h
eter
ozy
gou
sEx
on
2T
4A27
,F20
Atr
op
hy
No
ne
20/5
020
/25
—C
T09
(Fam
ilyC
TIV
)A
�G
10h
eter
ozy
gou
sEx
on
2T
4A23
,F16
Pse
ud
oh
ypo
pio
nA
tro
ph
y20
/32
20/5
0C
NV
LEC
T10
(Fam
ilyC
TV
)C
�T
73h
eter
ozy
gou
sEx
on
2R
25W
10,F
9V
itel
liru
pti
veFi
bro
sis
20/2
020
/200
—C
T11
(Fam
ilyC
TV
)C
�T
73h
eter
ozy
gou
sEx
on
2R
25W
36,F
30V
itel
liru
pti
veV
itel
liru
pti
ve20
/63
20/6
3—
CT
12(F
amily
CT
V)
C�
T73
het
ero
zygo
us
Exo
n2
R25
W70
,M60
Pse
ud
oh
ypo
pio
nN
on
e20
/50
20/2
0—
CT
13(F
amily
CT
VI)
T�
C26
het
ero
zygo
us
Exo
n2
V9A
44,M
7A
tro
ph
yFi
bro
sis
20/5
020
/200
—C
T14
(Fam
ilyC
TV
I)T
�C
26h
eter
ozy
gou
sEx
on
2V
9A12
,F12
Pre
-vit
ellif
orm
Pre
-vit
ellif
orm
20/2
020
/20
—
BC
VA
,b
est
corr
ecte
dvi
sual
acu
ity;
CN
V,
cho
roid
aln
eova
scu
lari
zati
on
;F,
fem
ale;
LE,
left
eye;
M,
mal
e;R
E,ri
ght
eye.
4680 Querques et al. IOVS, June 2011, Vol. 52, No. 7
Downloaded From: http://iovs.arvojournals.org/pdfaccess.ashx?url=/data/journals/iovs/933461/ on 05/24/2018
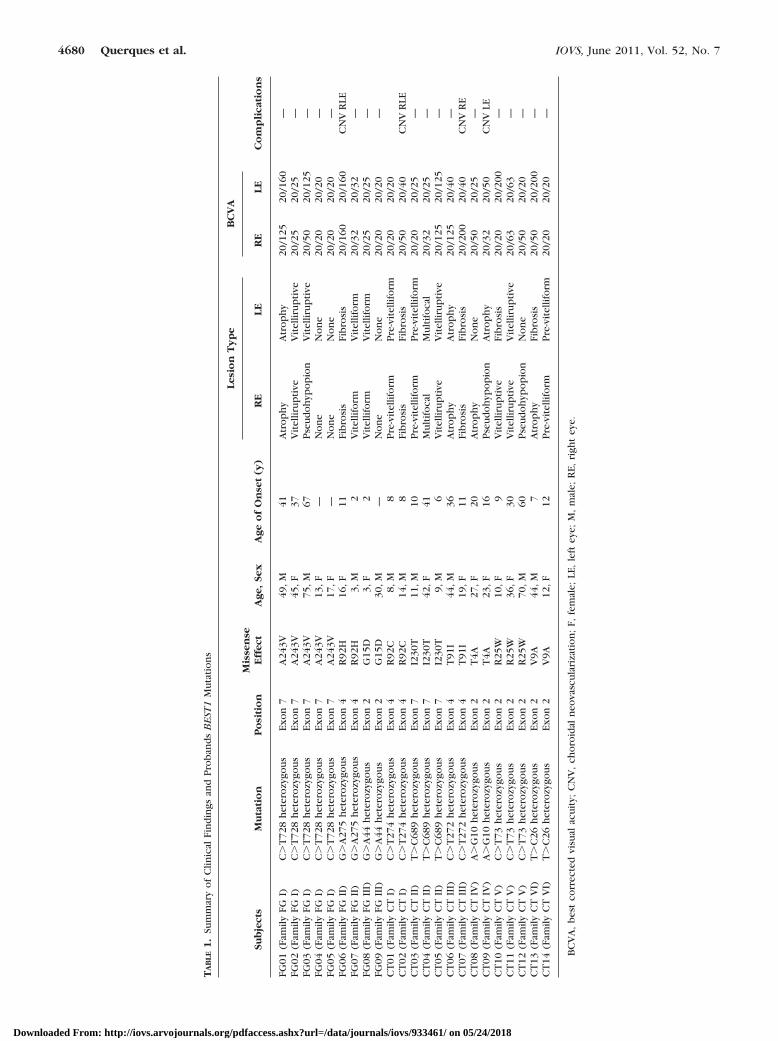
known mutations in the BEST1 gene and presenting at leastone family member affected with Best VMD, were included inthis study (Fig. 1; Table 1). Many affected and unaffectedrelatives of Best VMD patients (for a total of 63 relatives) wereexcluded from the study because they were not screened formutations in the BEST1 gene; of note, only few (12 out of 63unscreened relatives) were diagnosed with Best VMD (Fig. 1).Two unrelated individuals screened for mutations were alsoexcluded from the study because they did not carry any BEST1 mutation; they had no clinically detectable Best VMD withnormal SD-OCT and EOG findings.
Among the 23 patients harboring a BEST1 mutation (Table1), 17 patients presented with either bilateral (15 of 17) orunilateral (2 of 17) clinically detectable Best VMD, while sixpatients had 20 of 20 BCVA and normal funduscopic and FAFfindings (subclinical Best VMD). These six carrier individuals(three male, three female; aged 8 to 30 years) belonged to fivefamilies, each of which segregated a different BEST1 mutation(Table 2).
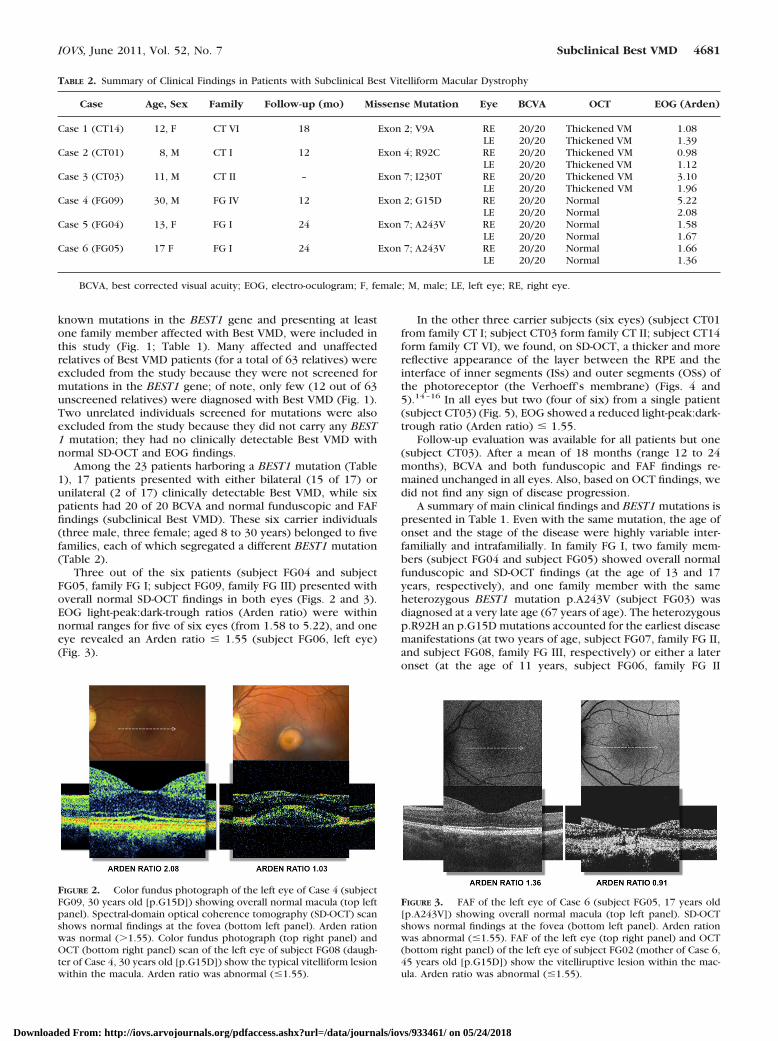
Three out of the six patients (subject FG04 and subjectFG05, family FG I; subject FG09, family FG III) presented withoverall normal SD-OCT findings in both eyes (Figs. 2 and 3).EOG light-peak:dark-trough ratios (Arden ratio) were withinnormal ranges for five of six eyes (from 1.58 to 5.22), and oneeye revealed an Arden ratio � 1.55 (subject FG06, left eye)(Fig. 3).
In the other three carrier subjects (six eyes) (subject CT01from family CT I; subject CT03 form family CT II; subject CT14form family CT VI), we found, on SD-OCT, a thicker and morereflective appearance of the layer between the RPE and theinterface of inner segments (ISs) and outer segments (OSs) ofthe photoreceptor (the Verhoeff’s membrane) (Figs. 4 and5).14–16 In all eyes but two (four of six) from a single patient(subject CT03) (Fig. 5), EOG showed a reduced light-peak:dark-trough ratio (Arden ratio) � 1.55.
Follow-up evaluation was available for all patients but one(subject CT03). After a mean of 18 months (range 12 to 24months), BCVA and both funduscopic and FAF findings re-mained unchanged in all eyes. Also, based on OCT findings, wedid not find any sign of disease progression.
A summary of main clinical findings and BEST1 mutations ispresented in Table 1. Even with the same mutation, the age ofonset and the stage of the disease were highly variable inter-familially and intrafamilially. In family FG I, two family mem-bers (subject FG04 and subject FG05) showed overall normalfunduscopic and SD-OCT findings (at the age of 13 and 17years, respectively), and one family member with the sameheterozygous BEST1 mutation p.A243V (subject FG03) wasdiagnosed at a very late age (67 years of age). The heterozygousp.R92H an p.G15D mutations accounted for the earliest diseasemanifestations (at two years of age, subject FG07, family FG II,and subject FG08, family FG III, respectively) or either a lateronset (at the age of 11 years, subject FG06, family FG II
TABLE 2. Summary of Clinical Findings in Patients with Subclinical Best Vitelliform Macular Dystrophy
Case Age, Sex Family Follow-up (mo) Missense Mutation Eye BCVA OCT EOG (Arden)
Case 1 (CT14) 12, F CT VI 18 Exon 2; V9A RE 20/20 Thickened VM 1.08LE 20/20 Thickened VM 1.39
Case 2 (CT01) 8, M CT I 12 Exon 4; R92C RE 20/20 Thickened VM 0.98LE 20/20 Thickened VM 1.12
Case 3 (CT03) 11, M CT II – Exon 7; I230T RE 20/20 Thickened VM 3.10LE 20/20 Thickened VM 1.96
Case 4 (FG09) 30, M FG IV 12 Exon 2; G15D RE 20/20 Normal 5.22LE 20/20 Normal 2.08
Case 5 (FG04) 13, F FG I 24 Exon 7; A243V RE 20/20 Normal 1.58LE 20/20 Normal 1.67
Case 6 (FG05) 17 F FG I 24 Exon 7; A243V RE 20/20 Normal 1.66LE 20/20 Normal 1.36
BCVA, best corrected visual acuity; EOG, electro-oculogram; F, female; M, male; LE, left eye; RE, right eye.
FIGURE 2. Color fundus photograph of the left eye of Case 4 (subjectFG09, 30 years old [p.G15D]) showing overall normal macula (top leftpanel). Spectral-domain optical coherence tomography (SD-OCT) scanshows normal findings at the fovea (bottom left panel). Arden rationwas normal (�1.55). Color fundus photograph (top right panel) andOCT (bottom right panel) scan of the left eye of subject FG08 (daugh-ter of Case 4, 30 years old [p.G15D]) show the typical vitelliform lesionwithin the macula. Arden ratio was abnormal (�1.55).
FIGURE 3. FAF of the left eye of Case 6 (subject FG05, 17 years old[p.A243V]) showing overall normal macula (top left panel). SD-OCTshows normal findings at the fovea (bottom left panel). Arden rationwas abnormal (�1.55). FAF of the left eye (top right panel) and OCT(bottom right panel) of the left eye of subject FG02 (mother of Case 6,45 years old [p.G15D]) show the vitelliruptive lesion within the mac-ula. Arden ratio was abnormal (�1.55).
IOVS, June 2011, Vol. 52, No. 7 Subclinical Best VMD 4681
Downloaded From: http://iovs.arvojournals.org/pdfaccess.ashx?url=/data/journals/iovs/933461/ on 05/24/2018

[p.R92H]), or even no disease manifestation (at the age of 30years, subject FG09, family FG III [p.G15D]), respectively. Infamily CT I, the heterozygous p.R92H mutation resulted inbilateral CNV development in subject CT02 (14 years old) andin previtelliform Best VMD in subject CT01 (8 years old).Subject CT04 (family CTII), heterozygous for the p.I230T,presented bilateral multifocal Best VMD features on fundusexamination. Interestingly, one of her sons with the sameheterozygous BEST1 mutation (subject CT05) presented thevitelliruptive stage of the disease in both eyes at the age of nineyears, and another of her sons with the same heterozygousBEST1 mutation (subject CT03) presented the previtelliformstage of the disease in both eyes at the age of 11 years. In familyCT VI, the heterozygous p.V9A mutation resulted in bilateralend stage in both eyes in subject CT13 (44 years old) and inprevitelliform Best VMD in subject CT14 (12 years old). Twounrelated subjects carrying different mutations of exon 2 pre-sented unilateral Best VMD (subject CT08 [p.T4A] and subjectCT12 [p.R25W] from family CT IV and family CT V, respec-tively).
DISCUSSION
Incomplete penetrance and expressivity are well-known fea-tures in Best VMD disease. The large variability within andbetween families of the clinical expression of BEST1 mutationsranging, in our series, from severe Best VMD to subclinicaldisease is consistent with previous reports.17–32 Recently, Lac-assagne et al.28 found not only a considerable intrafamilialphenotypic variability in patients carrying the p.S144G muta-tion, but even absence of pathologic phenotype in a patientcarrying the isolated p.Y5X mutation in the BEST1 gene. Age ofonset may also vary greatly among patients with Best VMD29;however, a recent report highlighted how age of onset may beconsidered a major criterion to distinguish Best VMD fromother similar macular dystrophies not strictly related to muta-tions in the BEST1 gene.30 In this context of broad phenotypicvariability, even with a single BEST1 mutation, fundus auto-fluorescence, optical coherence tomography, and EOG arevaluable noninvasive techniques for phenotyping and fol-low-up of Best VMD patients.31,32
In the current series, among BEST1 mutations carriers diag-nosed with subclinical Best VMD (n � 6), half presented with
and half without morphologic and/or functional alterations onSD-OCT and EOG, respectively. It is interesting to note thatpatients without EOG and SD-OCT alterations were youngsubjects (FG04 13 years old, FG05 17 years old, and FG09 30years old). However, the absence of EOG and SD-OCT altera-tions in young subjects was not strictly related to the genotype.In fact, all five BEST1 mutations affected amino acid residues ofdistinct functional regions of bestrophin.
Three out of six BEST1 mutations carriers presented onSD-OCT a bilateral thicker and more reflective appearance ofthe layer between the RPE and the IS/OS interface (Verhoeff’smembrane). At first sight, this finding could seem in contrast tothe normal localization of bestrophin to the basal aspect of theRPE. In fact, one would expect that the vitelliform materialwould accumulate in the RPE. Indeed, Arnold et al.33 in theirclinicopathological report found that the predominant findingwas a collection of extracellular material beneath the sensoryretina and proposed that this material was derived internallyfrom photoreceptor outer segments and externally from theRPE, the latter first undergoing hypertrophy and then disrup-tion and attenuation. Moreover, in contrast to previous studiesthat demonstrated massive lipofuscin accumulation in the RPE,Mullins et al.26 reported one patient in whom the RPE ap-peared histologically healthy in some regions of the maculathat exhibited loss of photoreceptors. Based on immunofluo-rescence studies, they proposed that the possible mistargetingof bestrophin may result in a harmful alteration of the ionicmilieu of the subretinal space and contribute to the type ofphotoreceptor cell loss observed histologically. Therefore, wehypothesize that the lesions, at the previtelliform stage, liebeneath the sensory retina and consist of mainly photoreceptordebris, possibly as result of faulty phagocytosis by the RPE,mixed with pigment granules liberated as the RPE undergoesdisruption.
Interestingly, no strict correlation between electrophysio-logical alterations and morphologic changes was evidenced.However, EOG was altered in most eyes with SD-OCT altera-tions (four of six) and in only one eye without morphologicchanges (one of six). These data suggest a trend toward animpaired function, as evaluated by EOG (light-peak:dark-troughratio � 1.55), in the presence of morphologic changes (thick-ening of the Verhoeff’s membrane), yet subclinical (normalBCVA and fundus findings). These data also suggest an overallgood sensitivity for SD-OCT to detect or exclude the presenceof subclinical Best VMD.
FIGURE 5. Color fundus photograph of the right eye of Case 3(subject CT03, 11 years old [p.I230T]) showing overall normal macula(top left panel). SD-OCT scan shows a thicker and more reflectiveappearance of the Verhoeff’s membrane (bottom left panel). Ardenratio was normal (�1.55). Color fundus photograph (top right panel)and SD-OCT (bottom right panel) scan of the right eye of subject CT05(brother of Case 4, 9 years old [p.I230T]) show the vitelliruptive lesionwithin the macula. Arden ratio was abnormal (�1.55).
FIGURE 4. FAF of the right eye of Case 2 (subject CT01, 8 years old[p.R92C]) showing overall normal macula (top left panel). SD-OCTshows a thicker and more reflective appearance of the Verhoeff’smembrane (bottom left panel). Arden ratio was abnormal (�1.55). FAFof the right eye (top right panel) and SD-OCT (bottom right panel) ofthe right eye of subject CT02 (brother of Case 2, 14 years old [p.R92C])show the fibrotic lesion within the macula. Arden ratio was abnormal(�1.55).
4682 Querques et al. IOVS, June 2011, Vol. 52, No. 7
Downloaded From: http://iovs.arvojournals.org/pdfaccess.ashx?url=/data/journals/iovs/933461/ on 05/24/2018

Even though our study was not designed to investigatedisease progression, we had the opportunity to follow up fiveof six of the BEST1 carrier subjects with subclinical BEST VMDover a period of 18 months (mean). BCVA, funduscopic, andOCT findings remained unchanged in all eyes on examination.We acknowledge that this follow-up time is short and probablynot enough with respect to disease progression. However,considering patient management and counseling, it is meaning-ful that subclinical form of the disease may not progress at leastin a short-time period.
Our study has several limitations. Many relatives (12 af-fected and 51 unaffected) of Best VMD patients were notscreened for the BEST1 mutation and thus were excluded fromthe study: This may represent an ascertainment bias of thecurrent analysis. Moreover, in some Best VMD patients we usedTD-OCT that is not very suitable to pick up slight changes.However, given that all subclinical Best VMD patients havebeen investigated by means of SD-OCT, and that TD-OCT hasbeen used only in patients with clinically detectable Best VMD(disease stage 2 to 5), the different resolutions of the twodevices (TD-OCT versus SD-OCT) may have not influenced ourdata. Finally, we considered light-peak:dark-trough ratio � 1.55as normal EOG, while other authors use � 1.85 as normal,1.3–1.85 as mildly reduced, and �1.3 as severely reduced.However, also considering light-peak:dark-trough ratio � 1.85as normal, some BEST1 carrier subjects with subclinical BESTVMD, with and without thickening of the Verhoeff’s mem-brane, still presented normal EOG (two of six subjects, 4 of 12eyes).
A broad phenotypic variability may be observed in associa-tion with specific BEST1 mutations. In dominant heterozygousBest VMD, the variable phenotype is highlighted in the presentstudy as well as in other studies.17–32 The high phenotypicvariability is not unique to BEST1-related disease and may alsobe seen in other autosomal dominantly inherited retinal dis-eases, such as those caused by peripherin or RDS mutations.34
Different mutations might cause Best VMD by differentmechanisms. Some mutations in the BEST1 gene may lead to amore severely affected RPE and formation of extramacularvitelliform lesions26,35,36; however, there seems to be no clearpattern relating type of BEST1 mutation to severity of clinicalexpression.31 Compound heterozygous, biallelic recessive, orhomozygous dominant mutations in BEST1 may confer a par-ticularly severe phenotype, featuring widespread retinal degen-eration, in addition to Best VMD.31,37,38 Patch-clamp studiesshowed a reduced channel function, which was restituted aftercotransfection with wild-type bestrophin, consistent with aloss of (channel) function mechanism of disease.37 However,biological events other than regulation of ion flow in the RPE,such as ceramide accumulation, may be involved in the bestro-phin-associated disease process.39 Presently unknown addi-tional genetic and environmental modifying factors may exerttheir influence on the phenotypic outcome.
The well-known variability of clinical expression of BEST1mutations within and between families,17–32 together with ourfindings on the extremely variable expressivity of subclinicalBest VMD, are of particular interest in the era of assistedreproduction.40 Because prenatal diagnosis is difficult to offerin a disease with a variable expressivity and with knownsubclinical forms, preimplantation diagnosis is questionable,especially in individuals with subclinical forms, which have arisk to transmit the disease to descendants.
The evidence of specific SD-OCT and EOG changes in 50%of BEST1 carriers with no manifest Best VMD symptoms orfunduscopic lesions may be of help in families in which themutation cannot be identified (for instance, mutations lying inunscreened BEST1 regions). These findings may indeed be
valuable to detect asymptomatic carriers, and we proposethem as a suitable follow-up.
To date, the Human Gene Database (HGMD http://www.hgmd.cf.ac.uk/ac/all.php) reports the identification of 123 dif-ferent BEST1 mutations, the very large majority of which aremissense mutations or small indel that do not alter the readingframe. The nature of these mutations is consistent with thenotion that Best disease-causing mutations in bestrophin-1 leadto a loss of Cl� channel function with a dominant negativeeffect. However, few heterozygous null alleles have been re-ported,41,42 suggesting that, yet infrequent, bestrophin-1 hap-loinsufficiency may cause the disease. Furthermore, mutationsof BEST1 splicing regulators have been demonstrated to causedramatic phenotypes, including vitreoretinochoriopathy andnanophthalmos.43 These data, along with the very large vari-ability of clinical presentation of Best disease, sometimeswithin families, suggest that both the nature of mutations andmodifying factors may contribute to the phenotypic variability.It would be interesting to assess in future studies the conse-quence of missense mutations with variable clinical effect onthe Cl� channel function to precisely determine which of themare loss-of-function mutations. Moreover, when high-through-put sequencing will become available, it would be of majorinterest to perform association studies in Bestrophin-1 carrierpatients to identify polymorphisms involved in the modulationof the phenotype.
References
1. Best F. Uber eine hereditare Maculaafektion; Beitrage zur Vererg-slehre. Zschr Augenheilk. 1905;13:199–212.
2. Stone EM, Nichols BE, Streb LM, et al. Genetic linkage of vitelliformmacular degeneration Best’s disease to chromosome 11q13. NatGenet. 1992; 246–250.
3. Petrukhin K, Koisti MJ, Bakall B, et al. Identification of the generesponsible for Best macular dystrophy. Nat Genet. 1998;19:241–247.
4. Sun H, Tsunenari T, Yau KW, Nathans J. The vitelliform maculardystrophy protein defines a new family of chloride channels. ProcNatl Acad Sci U S A. 2002;99:4008–4013.
5. Nordstrom S, Barkman Y. Hereditary maculardegeneration (HMD)in 246 cases traced to one gene-source in central Sweden. Here-ditas. 1977;84:163–176.
6. Deutman AF, Hoyng CB. Macular dystrophies. In: Ryan SJ, ed.Retina, 3rd ed. St Louis: Mosby, 2001;1210–1257.
7. Souied EH, Querques G, Coscas G, Soubrane G. Retinal degenera-tions and dystrophies. In: Saxena S, Meredith TA, eds. OpticalCoherence Tomography in Retinal Diseases. New Delhi: Jaypee,2005;221–250.
8. Mohler CW, Fine SL. Long-term evaluation of patients with Best’svitelliform dystrophy. Ophthalmology. 1981;88:688–692.
9. Deutman AF. Electro-oculography in families with vitelliform dys-trophy of the fovea. Detection of the carrier state. Arch Ophthal-mol. 1969;81:305–316.
10. Cross HE, Bard L. Electro-oculography in Best’s macular dystrophy.Am J Ophthalmol. 1974;77:46–50.
11. Krill AE, Morse PA, Potts AM, Klien BA. Hereditary vitelliruptivemacular degeneration. Am J Ophthalmol. 1966;61:1405–1415.
12. Godel V, Chaine G, Regenbogen L, Coscas G. Best’s vitelliformmacular dystrophy. Acta Ophthalmol Suppl. 1986;175:1–31.
13. Spaide RF, Noble K, Morgan A, Freund KB. Vitelliform maculardystrophy. Ophthalmology. 2006;113:1392–1400.
14. Querques G, Regenbogen M, Quijano C, Delphin N, Soubrane G,Souied EH. High definition optical coherence tomography featuresin vitelliform macular dystrophy. Am J Ophthalmol. 2008;146:501–507.
15. Querques G, Regenbogen M, Soubrane G, Souied EH. High-resolu-tion spectral domain optical coherence tomography findings inmultifocal vitelliform macular dystrophy. Surv Ophthalmol. 2009;54:311–316.
IOVS, June 2011, Vol. 52, No. 7 Subclinical Best VMD 4683
Downloaded From: http://iovs.arvojournals.org/pdfaccess.ashx?url=/data/journals/iovs/933461/ on 05/24/2018

16. Ferrara DC, Costa RA, Tsang S, Calucci D, Jorge R, Freund KB.Multimodal fundus imaging in Best vitelliform macular dystrophy.Graefes Arch Clin Exp Ophthalmol. 2010;248:1377–1386.
17. Querques G, Zerbib J, Santacroce R, et al. Functional and clinicaldata of Best vitelliform macular dystrophy patients with mutationsin BEST1 gene. Mol Vis. 2009;15:2960–2972.
18. Lotery AJ, Munier FL, Fishman GA, et al. Allelic variation in theVMD2 gene in best disease and age-related macular degeneration.Invest Ophthalmol Vis Sci. 2000;41:1291–1296.
19. Kramer F, White K, Pauleikhoff D, et al. Mutations in the VMD2gene are associated with juvenile-onset vitelliform macular dystro-phy (Best disease) and adult vitelliform macular dystrophy but notage-related macular degeneration. Eur J Hum Genet. 2000;8:286–292.
20. Wabbels B, Preising MN, Kretschmann U, Demmler A, Lorenz B.Genotype-phenotype correlation and longitudinal course in tenfamilies with Best vitelliform macular dystrophy. Graefes ArchClin Exp Ophthalmol. 2006;244:1455–1466.
21. Weber BH, Walker D, Muller B. Molecular evidence for non-penetrance in Best’s disease. J Med Genet. 1994;31:388–392.
22. Marmorstein AD, Kinnick TR. Focus on molecules: bestrophin(Best-1). Exp Eye Res. 2006;85:423–424.
23. Renner AB, Tillack H, Kraus H, et al. Late onset is common in bestmacular dystrophy associated with VMD2 gene mutations. Oph-thalmology. 2005;112:586–592.
24. Boon CJF, Klevering BJ, den Hollander AI, et al. Clinical andgenetic heterogeneity in multifocal vitelliform dystrophy. ArchOphthalmol. 2007;125:1100–1106.
25. Bakall B, Marknell T, Ingvast S, et al. The mutation spectrum of thebestrophin protein-functional implications. Hum Genet. 1999;104:383–389.
26. Mullins RF, Oh KT, Heffron E, Hageman GS, Stone EM. Latedevelopment of vitelliform lesions and flecks in a patient with bestdisease: clinicopathologic correlation. Arch Ophthalmol. 2005;123:1588–1594.
27. Mullins RF, Kuehn MH, Faidley EA, Syed NA, Stone EM. Differentialmacular and peripheral expression of bestrophin in human eyesand its implication for best disease. Invest Ophthalmol Vis Sci.2007;48:3372–3380.
28. Lacassagne E, Dhuez A, Rigaudiere F, et al. Phenotypic variabilityin a French family with a novel mutation in the BEST1 genecausing multifocal best vitelliform macular dystrophy. Mol Vis.2011;17:309–322.
29. Booij JC, Boon CJ, van Schooneveld MJ, et al. Course of visualdecline in relation to the Best1 genotype in vitelliform maculardystrophy. Ophthalmology. 2010;117:1415–1422.
30. Meunier I, Senechal A, Dhaenens CM, et al. Systematic screening ofBEST1 and PRPH2 in juvenile and adult vitelliform maculardystrophies: a rationale for molecular analysis. Ophthalmology[Epub ahead of print].
31. Schatz P, Bitner H, Sander B, et al. Evaluation of macular structureand function by OCT and electrophysiology in patients with vitel-liform macular dystrophy due to mutations in BEST1. Invest Oph-thalmol Vis Sci. 2010;51:4754–4765.
32. Boon CJ, Theelen T, Hoefsloot EH, et al. Clinical and moleculargenetic analysis of best vitelliform macular dystrophy. Retina.2009;29:835–847.
33. Arnold JJ, Sarks JP, Killingsworth MC, et al. Adult vitelliformmacular degeneration: a clinicopathological study. Eye. 2003;17:717–726.
34. Boon CJF, den Hollander AI, Hoyng CB, Cremers FPM, KleveringBJ, Keunen JEE. The spectrum of retinal dystrophies caused bymutations in the peripherin/RDS gene. Prog Retin Eye Res. 2008;27:213–235.
35. Maruko I, Iida T, Spaide RF, Kishi S. Indocyanine green angiogra-phy abnormality of the periphery in vitelliform macular dystrophy.Am J Ophthalmol. 2006;141:976–978.
36. Miller SA. Multifocal Best’s vitelliform dystrophy. Arch Ophthal-mol. 1977;95:984–990.
37. Burgess R, Millar ID, Leroy BP, et al. Biallelic mutation of BEST1causes a distinct retinopathy in humans. Am J Hum Genet. 2008;82:19–31.
38. Schatz P, Ponjavic V, Andreasson S, Dahl N. Variant phenotype ofBest vitelliform macular dystrophy associated with compoundheterozygous mutations in VMD2. Ophthalmic Genet. 2006;27:51–56.
39. Xiao Q, Yu K, Cui YY, Hartzell HC. Dysregulation of humanbestrophin-1 by ceramideinduced dephosphorylation. J Physiol.2009;587:4379–4391.
40. Sohrab MA, Allikmets R, Guarnaccia MM, Smith RT. Preimplanta-tion genetic diagnosis for Stargardt disease. Am J Ophthalmol.2010;149:651–655.
41. Allikmets R, Seddon JM, Bernstein PS, et al. Evaluation of the Bestdisease gene in patients with age-related macular degeneration andother maculopathies. Hum Genet. 1999;104:449–453.
42. Kramer F, Mohr N, Kellner U, Rudolph G, Weber BH. Ten novelmutations in VMD2 associated with Best macular dystrophy(BMD). Hum Mutat. 2003;22:418.
43. Yardley J, Leroy BP, Hart-Holden N, et al. Mutations of VMD2splicing regulators cause nanophthalmos and autosomal dominantvitreoretinochoroidopathy (ADVIRC). Invest Ophthalmol Vis Sci.2004;45:3683–3689.
4684 Querques et al. IOVS, June 2011, Vol. 52, No. 7
Downloaded From: http://iovs.arvojournals.org/pdfaccess.ashx?url=/data/journals/iovs/933461/ on 05/24/2018