Embed Size (px)
Citation preview

Triage Protein Fold PredictionHongxian He, Gregory McAllister, Temple F. Smith*BioMolecular Engineering Research Center, Biomedical Engineering Department, Boston University, Boston, Massachusetts
ABSTRACT We have constructed, in a com-pletely automated fashion, a new structure templatelibrary for threading that represents 358 distinct SCOPfolds where each model is mathematically repre-sented as a Hidden Markov model (HMM). Becausethe large number of models in the library can poten-tially dilute the prediction measure, a new triagemethod for fold prediction is employed. In the firststep of the triage method, the most probable struc-tural class is predicted using a set of manually con-structed, high-level, generalized structural HMMs thatrepresent seven general protein structural classes:all-�, all-�, �/�, ���, irregular small metal-binding,transmembrane �-barrel, and transmembrane �-he-lical. In the second step, only those fold modelsbelonging to the determined structural class areselected for the final fold prediction. This triagemethod gave more predictions as well as more cor-rect predictions compared with a simple predictionmethod that lacks the initial classification step. Twodifferent schemes of assigning Bayesian model pri-ors are presented and discussed. Proteins 2002;48:654-663. © 2002 Wiley-Liss, Inc.© 2002 Wiley-Liss, Inc.
Key words: protein fold prediction; threading; HMM;DSM; triage
INTRODUCTION
Threading methods have proven to be powerful tools forprotein fold recognition, especially when there is no detect-able sequence similarity to a protein of known structure.Generally these methods search a set of structural tem-plates, which are abstract descriptions of the three-dimensional coordinates of known structures, for an opti-mal sequence-structure alignment.1–6 This alignment isevaluated by means of an empirical energy function orprobabilistic scoring scheme where it is presumed that themaximum score identifies the optimal alignment to a givenstructural model. It is generally assumed that the modelhaving the highest maximum alignment score predicts thecorrect fold. Since the correct prediction is possible only ifan appropriate structural model is present in the library,such methods require the construction of a comprehensivelibrary of structural templates.
A threading method previously developed in our centeruses discrete state-space models (DSMs) to representtertiary structural classes.7,8 DSMs are mathematicallyrepresented as hidden Markov models (HMMs) but differin that the model parameters are designed with ourknowledge of protein structures, as opposed to being
trained on a particular training set, as is customary inHMM construction.9,10 A predefined set of DSMs wasconstructed manually following the classification of struc-tural domains by Jane Richardson11 and represents twenty-four structural classes, called macro-classes, such as4-helix bundle proteins and TIM-barrel proteins. Macro-classes were grouped under four major folding super-classes of globular single-domain proteins: �, �, �/�, andirregular. The tertiary structure prediction is made byfinding the macro-class (and super-class) with the highestscore, which is expressed as the posterior probability of agiven macro-class for the given sequence. The macro-classprobabilities are obtained by summing the probabilities ofall DSMs belonging to the given macro-class, and thesuper-classes probabilities are obtained by summing upthe probabilities of all underlying macro-classes. Theprobability of each individual DSM for a given sequence iscomputed using a Bayesian formula.7
The large number of currently known structures presentin the PDB database12 renders the manual construction ofmodels impractical. We have, therefore, built a new DSMlibrary according to the SCOP database13 via an auto-mated procedure similar to that of Bienkowska et al.14
Each DSM in the library, representing a distinct SCOPfold, was automatically constructed from the determinedatomic coordinates of a representative structure from thePDB database, along with a set of generalized rules. Thereliance of the structural prediction on a model libraryimposes several problems. First, the large number ofmodels increases the likelihood that the correct model willexhibit a small posterior probability since the calculationinvolves the normalization over all the models in thelibrary. Second, the calculation of posterior probabilitiesalways results in one model being the most probable, evenif the appropriate structural model is not present in thelibrary. In order to reduce these problems, we have deviseda triage method that first classifies the protein sequenceinto a general structural class prior to searching the fullDSM library. This approach relies on a set of manuallyconstructed, high-level generic DSMs that model majorstructural classes, such as all-�, all-�, �/�, and membrane
Grant sponsor: Department of Energy; Grant number: DE-FG02-98ER62558.
*Correspondence to: Temple F. Smith, BioMolecular EngineeringResearch Center, Biomedical Engineering Department, Boston Univer-sity, 36 Cummington St., Boston, MA 02215.E-mail: [email protected]
Received 10 September 2001; Accepted 2 May 2002
Published online 00 Month 2002 in Wiley InterScience(www.interscience.wiley.com). DOI: 10.1002/prot.10194
PROTEINS: Structure, Function, and Genetics 48:654-663 (2002)
© 2002 WILEY-LISS, INC.

proteins. This two-step method eliminated many of theincompatible models from the model library before the foldposterior probability distribution is calculated, thereforeincreasing the chance that the correct structural templatewill be identified. It also helps to reduce the number ofpotential false positives by filtering out the models that donot fall into the same structural class through the initialclassification.
METHODSPosterior Model Probability
Given a set of N structural models, S � Mi (i � 1, . . ., N),each of which defines a structural hypothesis, the goal ofstructure prediction is to find the best hypothesis toexplain the given sequence. This is quantified by theposterior probability P(Mi�seq) computed according toBayes’ rule:
P�Mi�seq� �P�seq�Mi�P�Mi�
�k � 1
N
P�seq�Mk�P�Mk�
, (1)
where N is the total number of models in the library.P(seq�Mi), the total probability that the observed se-
quence would have been generated by model Mi, is calcu-lated using an optimal filtering algorithm or the forwardalgorithm.15,16 This algorithm differs from the more com-monly used Viterbi algorithm in that it sums over allpossible paths through the model as opposed to only theoptimal path. Here, P(Mi) is the prior probability of modelMi. The method of assigning priors, P(Mi), will be dis-cussed later.
We apply a conservative binary decision rule for predic-tion: if a model has a posterior probability greater than 0.5,a prediction is made.
Automated Construction of a PDB Domain DSMLibrary
A discrete state-space model (DSM) is a linear represen-tation of structural states that correspond to the structuralpositions of a protein. A structural state is defined by thetype of secondary structure (helix, strand, loop, or turn)and degree of solvent exposure (buried, partially-buried, orexposed). Each state is associated with a characteristicemission or occupancy probability distribution over the 20amino acids derived from the statistical analysis of a largeset of representative protein structures (unpublished data).Structural states are connected to form a Markov chainwhere the state transition probabilities are designed tomodel the important constraints on secondary structureimposed by the specific tertiary structure being modeled.
In our automated model generation,14 each DSM wasconstructed from a solved protein structure in a hierarchi-cal fashion. First, secondary structural elements (SSE)were determined using the DSSP program17 with thedegree of solvent exposure for each residue position calcu-lated according to Eisenberg et al.18 Second, each SSE wasmapped into a DSM module, which is a sequence ofstructural states defined by the determined secondary
structure and calculated degree of solvent exposure. Addi-tional structural states and state-to-state transitions wereadded at both ends of each module. This allows for lengthvariations (extension/deletion by one or two residues)while maintaining the minimum length of a secondarystructure element: two residues for a strand and fiveresidues for a helix. Finally, these secondary structuralmodules were connected by loop modules according to theirtopological order in the native protein structure. The typeof loop module was determined by the geometrical distancebetween the ends of two consecutive SSEs.
The set of representative structures was selected accord-ing to the SCOP classification (Release 1.48). The level ofclassification in which we are interested is the “fold,”which is defined as a group of structural domains havingthe same major secondary structural elements with thesame topology. All structures having repeated or inter-twined domains, in addition to all membrane proteins,were excluded, resulting in 358 unique SCOP folds. Forevery chosen SCOP fold, a structure was selected fromeach of its constituent “families” for a total of 881 represen-tatives. For structural folds whose PDB records includemultiple subunits, domains, or bound cofactors, multipleDSMs were constructed accounting for differing solventexposure profiles corresponding to the presence or absenceof additional elements. There are, therefore, multiplemodels representing each SCOP fold. The final libraryconsists of 1,282 models, denoted as PDB domain DSMs.
Generic DSMs
As a feature of our triage method, a set of generic DSMswas designed to represent all known gross structuralclasses. These models represent the general structuralfeatures of a particular class while allowing a large rangeof anticipated secondary structure variations. Seven ge-neric DSMs have been manually built for the followingstructural classes, where class 1–5 refer to soluble proteinsonly: (1) all-� proteins having only �-helical secondarystructure, (2) all-� proteins having mainly �-strand second-ary structure, (3) �/� proteins having alternating �-helicaland �-strand secondary structures, (4) ��� proteins hav-ing local clusters of �-helices and �-strands segregatedalong the primary sequence, (5) irregular proteins havingfew secondary structure elements and a high content ofdisulfide bonds or metal ligands, (6) transmembrane �-bar-rel proteins, and (7) transmembrane �-helical proteins.These classes are considered to be mutually exclusive andexhaustive.
Generic All-� DSM
Figure 1 illustrates the model structure of the genericall-� DSM. Basic secondary structure modules have beencombined to form larger secondary structure complexes,called plexes for short. Each oval represents either asecondary structure module or a plex. The overall modelconsists of two identical �-plexes, each constructed from a�-helix module (amphipathic or buried) followed by a turnor loop module. A secondary structure module contains aseries of structural states that represent structural posi-
TRIAGE PROTEIN FOLD PREDICTION 655

tions in a protein. The arrows indicate the transitions fromone structural state or module to another with the transi-tion probabilities assigned based on our knowledge of therelative frequencies of these secondary structure elementsin all-� proteins. For example, there are no all-� proteinswith fewer than two helices, amphipathic helices are morecommon than completely buried helices in globular pro-teins, and turns occur more often between helices thanloops. We explicitly model the length distribution for thesemodules according to the typical lengths observed in suchproteins. The �-helix module has a uniform length distribu-tion between 5 and 30 residues and exponential distribu-tion over a length greater than 30 residues. The loop has auniform length distribution between 5 and 20 residues andan exponential distribution for more than 20 residues. Theturn can be 2-, 3-, or 4-residues in length with equalprobability.
The generic DSM shown in Figure 1 differs from atypical DSM, which is called a left-to-right model or aBakis model.19 When a left-to-right model is used togenerate a state sequence, the state index increases orstays the same as time (or sequence position in the DSManalysis) increases. A DSM representing a protein struc-ture proceeds from its N-terminal to its C-terminal, i.e.,the structural states are numbered from the amino end tothe carboxy end of the protein structure. A sequence isalways filtered through the model from the first structuralstate, corresponding to the N-terminal residue position, tothe last state, corresponding to the C-terminal residueposition. Such a model imposes a strict probabilistic lengthdistribution including a minimum length that can bethreaded onto or generated by the model. However, ourgeneric model requires, by definition, the accommodationof sequences of near arbitrary length. We achieved thisgoal by introducing a special state, the end state, and theend symbol. The latter is an artificial symbol placed at theend of the query sequence as the last amino acid. The end
state is positioned after the last plex in the model and hasa transition path back to the first state in the last plex witha probability of 1 (Fig. 1A). This state plays a dual role inthe filtering process. During iteration of the forwardfiltering process, this end state is equivalent to a loopstate, emitting the 20 amino acids according to the sameamino acid emission probability distribution. When theend symbol is filtered through the end state, it acts as if itsemission probabilities were zero for all the 20 amino acidsand one for the end symbol. Since all of the other states inthe DSM have zero emission probabilities for the endsymbol, this forces the end of the sequence to be aligned tothe end of the model.
Generic All-� DSM
The generic all-� model is similar in architecture to theall-� model, but with the �-plexes replaced by �-plexes(Fig. 2). Additionally, a minimum of four �-plexes isrequired, there is a slightly higher probability for a buriedstrand, and the connecting region between adjacent strandscan be a short helix, in addition to a loop or turn. The�-strand module has a uniform length distribution be-tween 3 to 14 residues. The loop module is the same asused in the generic all-� model.
Generic �/� DSM.
Figure 3 depicts the generic �/� model, which is de-signed to allow an alternating helix/strand architecturewhile permitting two consecutive repeats of each withsmall probabilities. It is more probable to (1) find a strandat the N-terminal of such proteins, and (2) have twoconsecutive strands than two consecutive helices. Modulelengths in this model have a more constrained distribu-tion: the lengths of the strands are uniformly distributedbetween 3 to 10 residues and the lengths of the helices areuniformly distributed between 4 to 23 residues. Note thatthe model has two end states but the computation of modellikelihood remains the same.
Generic ��� DSM and ��� DSM
The ��� structural class has segregated helical andstrand regions within its structure and often within theprimary sequence as well. We have designed two DSMs torepresent the two principle types of ��� structures: the
Fig. 1. Schematic of the generic model for all-� proteins. All paths arelabeled with transition probabilities from one structural module to another.A: The DSM organization at the �-plex level of detail. All the �-plexes areidentical. The circle labeled with ES is the end state. B: Each �-plexconsists of an �-helix (amphipathic or buried) followed by the connectingloop or turn. C: The structure of the loop module. Each circle represents aloop state and all the states are identical. The numbers in the circle are theindices of the states.
Fig. 2. Schematic of the generic model for all-� proteins. A: The DSMorganization at the �-plex level. All the �-plexes are identical. B: Each�-plex consists of a �-strand (amphipathic or buried) and connectingregion, which can be a loop, turn, or short helix.
656 H. HE ET AL.

generic ��� model allows a �-cluster (minimum of 4helices) followed by a �-cluster, while the generic ���allows a �-cluster followed by an �-cluster. Both modelsrepresent two separate structural classes since they areindependent of each other in the model space (Fig. 4).
There are a few protein folds in this category that we didnot attempt to model. One example is a protein fold with a�-cluster - �-cluster - �-cluster topology. A model for thisfold will have many helical elements and will be expectedto overlap with the all-� model to a large extent. In fact,most of these proteins can usually be further divided intoseparate domains after visual inspection.
Generic DSM for irregular proteins
The generic model for small, irregular/metal-bindingproteins consists mainly of loops and turns. The amino-acid emission probabilities for the loop state in this modeldiffer from those for loop states in other generic models inthat they have much higher probabilities for the aminoacids histidine and cysteine (Fig. 5).
Generic DSM for transmembrane �-barrel proteins
Transmembrane �-barrel proteins, which are found inthe outer membranes of bacteria, mitochondria, and chlo-roplasts, fall into three main categories of solved structure:outer membrane proteins, outer membrane phospholipaseA, and porins.20 The size of these proteins ranges fromsmall eight-stranded to large twenty-four-stranded �-bar-rels. There are three common features for this type ofprotein: (1) the number of �-strands is even, (2) strandconnections on the periplasmic side of the membrane aregenerally short turns, while on the external side they aregenerally long loops or very short helices, and (3) trans-membrane �-strands are amphipathic, composed of alter-native polar (inside barrel) and non-polar (outside barrel)residues.
The generic model for this class has an architecture verysimilar to the generic all-� model with the principledifferences accounting for the features as described above.The minimum number of strands is eight and an evennumber of strands is required. The residue positions at thebarrel inside and outside are modeled by two specialstructural states whose emission probabilities were pre-calculated for this class of proteins.
Generic DSM for transmembrane �-helical proteins
A generic DSM representing �-helical membrane-spanning proteins was designed based on known struc-tural features of these proteins. It contains transmem-brane helical regions connected by intervening regionsallowing loops, turns, and helices. The structural modulefor the transmembrane helical region consists of thehydrophobic core region flanked by membrane interfaceregions at both ends (Fig. 6). Two structural states weredefined to model the membrane core and interface. Theiremission probability distributions were calculated from astatistical analysis of a set of 56 known transmembraneproteins from ABC transporter complexes extracted fromthe BMERC all-genome database (http://bmerc-www.bu.edu/information/all_genomes.shtml). The transmem-brane helix has a uniform length distribution between 20and 38 residues, corresponding to the approximate lengthof a helix needed to span a typical bilayer.21
Models were also constructed at the fold level for �-heli-cal transmembrane proteins. Due to the paucity of mem-brane protein structures in the PDB, the automatedmethod cannot be relied on for a complete fold coverage ofthis class. Therefore, 56 individual models were manuallydesigned for proteins containing between 5 and 12 trans-membrane helical regions with the intervening regionsallowing loops, turns, or helices.22 These models do notrepresent the SCOP folds and are classified based on the
Fig. 3. Schematic of the generic model for �/� proteins. Each ��-plexconsists of a �-helix followed by a �-strand. Each ��-plex consists of a�-strand followed by a �-helix. An ��-plex contains two consecutivehelices. In a ��-plex or ��-plex, there is a parallel path with the helix (notshown), allowing a loop to replace the helix between two adjacent strandswith a small probability. All the �-helix modules are identical, as are the�-strand modules.
Fig. 4. Schematic of the generic model for ��� proteins. A: Generic��� model contains an �-cluster followed by a �-cluster. B: Generic ���model contains a �-cluster followed by an �-cluster. C: The structure of a�-cluster, which generates a subsequence of 3 to 7 strands with equalprobabilities. Similarly, an �-cluster generates a subsequence of 4 to 7helices with equal probabilities.
Fig. 5. Schematic of the generic model for irregular/metal-bindingproteins. Each Iplex is composed of a loop followed by a turn module.
TRIAGE PROTEIN FOLD PREDICTION 657

number of transmembrane helices being modeled. Thesespecific models can be used to predict individual transmem-brane regions for a given protein sequence using thesmoothing algorithm, as explained.16,8,22
Triage Method for Fold PredictionStep 1. Protein structure classification
The first step of the triage method is to attempt toidentify the most probable structural class of a querysequence using only the set of generic DSMs. The modellikelihood is computed for each generic model using theoptimal filtering algorithm. The posterior probability ofeach model is then calculated according to Eq. 1 with equalmodel prior probabilities. This prior assignment modelsthe minimum bias for structural class. Our aim is toreduce the number of PDB domain DSMs to be consideredfor subsequent fold prediction.
Step 2. Protein fold prediction
All of the PDB domain DSMs are considered for eachstructural class for which the generic model has a poste-rior probability greater than 0.3 (an empirical value). Themethod of categorizing the PDB domain DSMs into differ-ent structural classes will be discussed later. If the struc-tural class for a query sequence is uniquely determined tobe a transmembrane �-barrel protein, no further analysisis made. Otherwise the fold prediction is made based onthe set of selected PDB domain models.
Consistent with previous DSM analysis,7 we considereach PDB domain model as a structural hypothesis andcompute the probability that the primary sequence camefrom each of the candidate hypotheses, P(M�seq), using themodel priors and likelihoods (Eq. 1). Fold prediction ismade according to the probabilities of the SCOP folds,which are obtained by summing the probabilities of allPDB domain models that belong to the given fold (union ofhypotheses):
Formula 1 (hierarchical scheme):
P�foldi�seq� � �Mε foldi
P�M�seq�,1 � i � Nf,Mε�Mj�
for: 1 � j � Nm, (2)
where Nf is the number of unique folds being consideredand Nm is the total number of PDB domain DSMs beingconsidered.
The model priors, P(M), are assigned in a hierarchicalmanner as in White et al.7 The underlying assumption isthat all models at the same hierarchical level are exclu-sive, all-inclusive, and equally possible. The SCOP hier-archy is used here. This approach has also been used byBienkowska et al.14 for a small set of automaticallygenerated DSMs. We denote this approach as the hierarchi-cal scheme.
Here we propose an additional approach to fold predic-tion. Because all the models under a specific SCOP foldrepresent the same fold, i.e., they are overlapping models,it is not proper to treat them as individual hypotheses inthe Bayesian formula. Instead, we view the hypothesisspace as composed of distinct SCOP folds, that is, a givensequence is considered as coming from one, and only one,of the SCOP folds. Since there is more than one PDBdomain DSM that represents a given fold, we only selectthe constituent DSM with the highest model likelihood torepresent the fold and compute the probability of theSCOP fold using Bayes’ rule:
Formula 2 (equal-fold-probability scheme):
P� foldi�seq� �P�seq� foldi�P� foldi�
�k � 1
Nf
P�seq� foldk�P� foldk�
. (3)
Prior probabilities are uniformly assigned to each foldselected, P(foldk) � 1/Nf, to minimize the bias for differentfolds. P(seq�foldi) is the model likelihood of the representa-tive DSM for foldi:
P�seq� foldi� � max�P�seq�M�, M foldi�,1 � i � Nf. (4)
We name this approach as the equal-fold-probabilityscheme.
Finally, fold prediction is made if the most probableSCOP fold has a posterior probability, P(fold�seq), greaterthan 0.5 according to either Formula 1 (Eq. 2) or Formula 2(Eq. 3).
Categorization of PDB Domain DSMs
The success of the triage method will largely depend onthe success of the initial classification. The grouping of thePDB domain DSMs into different generic DSM classesshould be consistent with the classification of a givensequence, thus allowing the correct domain model to beselected in the subsequent fold prediction.
Initially, the library of PDB domain models was orga-nized according to the SCOP classification, so it seems tobe straightforward to group them into different structuralclasses according to their SCOP class assignment. How-ever, in the top-level of triage, the structural classesdefined by our generic DSMs do not universally correspondto the SCOP classification. The generic DSM design empha-sizes the composition of secondary structure elements and,very importantly, their topological order, while the SCOP
Fig. 6. Schematic of the generic model for the transmembrane helicalproteins. A: The model contains a transmembrane helix followed by eithera loop, turn, or non-membrane-spanning helix. B: The structural modulefor the transmembrane helix, where the membrane core region is flankedby the interface regions.
658 H. HE ET AL.
classification is mainly based on the spatial arrangementof secondary structure elements as judged by humanexperts. It is, thus, not surprising that the structuralclassification using our generic DSMs does not alwaysagree with the SCOP classification (unpublished data). Wehave, therefore, categorized each PDB domain DSM intothe predicted structural class(es) (for which the genericmodel has a posterior probability greater than 0.3) basedon its template PDB sequence.
Using the above procedure for the 1,282 PDB domainDSMs in the library, 1,047 (82%) were mapped into asingle class, 233 (18%) into two classes, and two into threestructural classes. The classification of the model librarycan be found at http://bmerc-www.bu.edu/hxian/triage/.Table I shows the number of PDB domain models in eachstructural class. Note that the structural classes listed inTable I are slightly different from those introduced in theabove section. This difference will be explained in Resultsin addition to the three misclassifications that resulted inclassifying six domain models into the transmembrane�-helical class.
RESULTSProbabilistic Overlap Between Generic DSMs
Each DSM can be viewed as a sequence generator thatcan generate a string of amino acids with a certainprobability. The generation procedure draws amino acidsrandomly based on the state transition probability matrixand emission probability matrix of a particular model. Wehave used this random sequence generation method tomeasure the overlap or degree of independence betweengeneric DSMs, as described below.
To validate the use of generic DSMs for the initialclassification of triage, we measured the overlap betweenthese models through the following simulation. Two hun-dred artificial amino acid sequences were generated fromeach generic model. Each of the 200 sequences was thenthreaded through the full set of generic models and themodel posterior probabilities were computed. If a model
has a posterior probability above 0.5 for a sequence, aprediction was recorded. The results are shown in Table II.Generic models for irregular proteins and transmembraneproteins show little overlap with other generic models. Theall-� model and all-� model are also clearly separated fromeach other, although a small overlap is found between theall-� and �/�, and between the all-� and �/� model. A largeoverlap is found between the all-� and ��� model, andbetween all-� model and ��� model. When we combinedeach of the above pairs of overlapping models into a singleclass, mainly-� and mainly-�, better results were ob-tained. To compute the posterior probability for mainly-�or mainly-� class, the likelihood for the class takes thehigher likelihood value of the two generic models from thatclass. The overlap between different structural classesafter the above adjustment is shown in Table III.
Protein Fold Prediction
We first tested the triage method in the self-threadingusing the set of 882 template proteins. The results of foldprediction are shown in Table IV. To illustrate the triagemethod, we compare it with a simple prediction method,where the query sequence is filtered through every modelin the library and the posterior probabilities are calculatedusing all the models. Furthermore, for each of the abovetwo methods, we test the two formulas of computing thefold probabilities according to Eq. 2 and Eq. 3, respectively.
Among the 882 template proteins, 703 were uniquelyclassified, 154 classified into two classes, and one intothree classes. On average, there were 672 PDB domainmodels being considered in the final fold prediction foreach sequence. This number is only half of the size of thePDB domain model library. The triage method, as com-pared to the simple method, gave many more predictions(hits) using either the hierarchical scheme (Formula 1) orthe equal-fold-probability scheme (Formula 2), when using0.5 as the cutoff for the fold probability. This cutoff reflectsour confidence level for prediction. The triage method alsogave more correct predictions (true hits), resulting in a
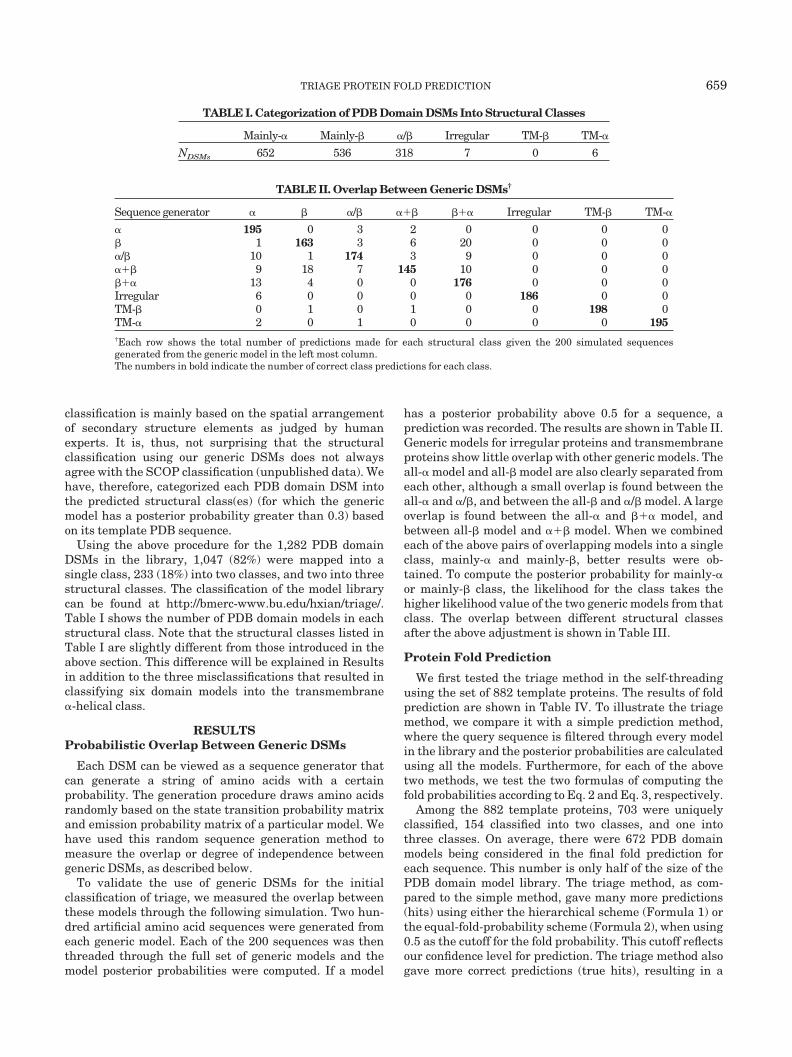
TABLE I. Categorization of PDB Domain DSMs Into Structural Classes
Mainly-� Mainly-� �/� Irregular TM-� TM-�
NDSMs 652 536 318 7 0 6
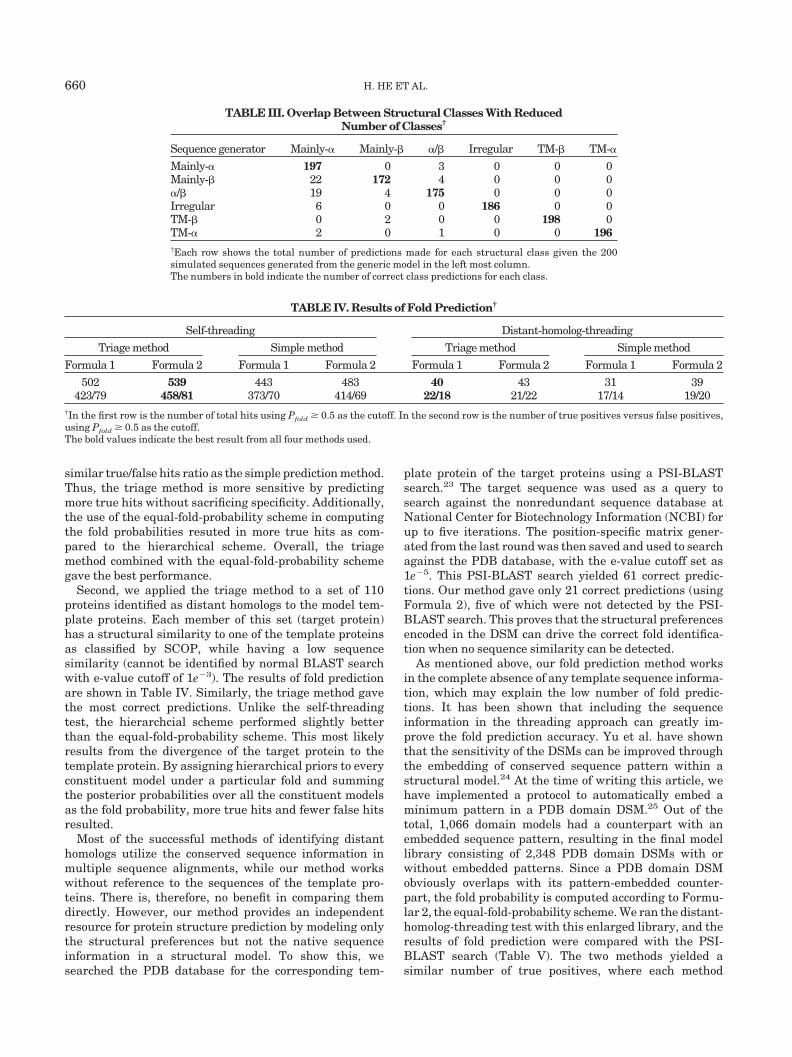
TABLE II. Overlap Between Generic DSMs†
Sequence generator � � �/� ��� ��� Irregular TM-� TM-�
� 195 0 3 2 0 0 0 0� 1 163 3 6 20 0 0 0�/� 10 1 174 3 9 0 0 0��� 9 18 7 145 10 0 0 0��� 13 4 0 0 176 0 0 0Irregular 6 0 0 0 0 186 0 0TM-� 0 1 0 1 0 0 198 0TM-� 2 0 1 0 0 0 0 195†Each row shows the total number of predictions made for each structural class given the 200 simulated sequencesgenerated from the generic model in the left most column.The numbers in bold indicate the number of correct class predictions for each class.
TRIAGE PROTEIN FOLD PREDICTION 659
similar true/false hits ratio as the simple prediction method.Thus, the triage method is more sensitive by predictingmore true hits without sacrificing specificity. Additionally,the use of the equal-fold-probability scheme in computingthe fold probabilities resuted in more true hits as com-pared to the hierarchical scheme. Overall, the triagemethod combined with the equal-fold-probability schemegave the best performance.
Second, we applied the triage method to a set of 110proteins identified as distant homologs to the model tem-plate proteins. Each member of this set (target protein)has a structural similarity to one of the template proteinsas classified by SCOP, while having a low sequencesimilarity (cannot be identified by normal BLAST searchwith e-value cutoff of 1e3). The results of fold predictionare shown in Table IV. Similarly, the triage method gavethe most correct predictions. Unlike the self-threadingtest, the hierarchcial scheme performed slightly betterthan the equal-fold-probability scheme. This most likelyresults from the divergence of the target protein to thetemplate protein. By assigning hierarchical priors to everyconstituent model under a particular fold and summingthe posterior probabilities over all the constituent modelsas the fold probability, more true hits and fewer false hitsresulted.
Most of the successful methods of identifying distanthomologs utilize the conserved sequence information inmultiple sequence alignments, while our method workswithout reference to the sequences of the template pro-teins. There is, therefore, no benefit in comparing themdirectly. However, our method provides an independentresource for protein structure prediction by modeling onlythe structural preferences but not the native sequenceinformation in a structural model. To show this, wesearched the PDB database for the corresponding tem-
plate protein of the target proteins using a PSI-BLASTsearch.23 The target sequence was used as a query tosearch against the nonredundant sequence database atNational Center for Biotechnology Information (NCBI) forup to five iterations. The position-specific matrix gener-ated from the last round was then saved and used to searchagainst the PDB database, with the e-value cutoff set as1e5. This PSI-BLAST search yielded 61 correct predic-tions. Our method gave only 21 correct predictions (usingFormula 2), five of which were not detected by the PSI-BLAST search. This proves that the structural preferencesencoded in the DSM can drive the correct fold identifica-tion when no sequence similarity can be detected.
As mentioned above, our fold prediction method worksin the complete absence of any template sequence informa-tion, which may explain the low number of fold predic-tions. It has been shown that including the sequenceinformation in the threading approach can greatly im-prove the fold prediction accuracy. Yu et al. have shownthat the sensitivity of the DSMs can be improved throughthe embedding of conserved sequence pattern within astructural model.24 At the time of writing this article, wehave implemented a protocol to automatically embed aminimum pattern in a PDB domain DSM.25 Out of thetotal, 1,066 domain models had a counterpart with anembedded sequence pattern, resulting in the final modellibrary consisting of 2,348 PDB domain DSMs with orwithout embedded patterns. Since a PDB domain DSMobviously overlaps with its pattern-embedded counter-part, the fold probability is computed according to Formu-lar 2, the equal-fold-probability scheme. We ran the distant-homolog-threading test with this enlarged library, and theresults of fold prediction were compared with the PSI-BLAST search (Table V). The two methods yielded asimilar number of true positives, where each method
TABLE III. Overlap Between Structural Classes With ReducedNumber of Classes†
Sequence generator Mainly-� Mainly-� �/� Irregular TM-� TM-�
Mainly-� 197 0 3 0 0 0Mainly-� 22 172 4 0 0 0�/� 19 4 175 0 0 0Irregular 6 0 0 186 0 0TM-� 0 2 0 0 198 0TM-� 2 0 1 0 0 196†Each row shows the total number of predictions made for each structural class given the 200simulated sequences generated from the generic model in the left most column.The numbers in bold indicate the number of correct class predictions for each class.
TABLE IV. Results of Fold Prediction†
Self-threading Distant-homolog-threading
Triage method Simple method Triage method Simple method
Formula 1 Formula 2 Formula 1 Formula 2 Formula 1 Formula 2 Formula 1 Formula 2
502 539 443 483 40 43 31 39423/79 458/81 373/70 414/69 22/18 21/22 17/14 19/20
†In the first row is the number of total hits using Pfold � 0.5 as the cutoff. In the second row is the number of true positives versus false positives,using Pfold � 0.5 as the cutoff.The bold values indicate the best result from all four methods used.
660 H. HE ET AL.

correctly predicted some unique proteins. Of the 55 truepositives identified by pattern-embedded DSMs, twelvewere not found by PSI-BLAST.
Classification Between Transmembrane andSoluble Proteins
We took all the non-identical transmembrane proteinsfrom the PDB to test the ability of the set of generic DSMsto differentiate soluble from transmembrane proteins.This set includes 16 transmembrane �-helical proteinsand 12 transmembrane �-barrel proteins. The 882 tem-plate soluble proteins were used as negative controls.Table VI summarizes the results of structural classifica-tion between the soluble proteins and transmembraneproteins.
Among the 882 template proteins, only one (SCOP ID:1uag_1) was uniquely predicted to be a transmembranehelical protein. This sequence is the N-terminal domain(residue 1-93) of a D-glutamate ligase and has a stretch of20 hydrophobic residues in a helix, which possibly explainsthe misclassification. Only one transmembrane helicalprotein (SCOP ID: 1eulA) was misclassified as a solubleprotein. The structure reveals that it has two large in-serted globular domains (143 and 444 residues) betweenadjacent transmembrane helices; thus, it is not surprisingthat a generic model for soluble proteins had a higherprobability (Note: we are not currently modeling theglobular domains that often appear within the adjacentmembrane-spanning helices in the transmembrane helicalprotein model). There are also two cases where the tem-plate protein was classified into both the transmembranehelical protein structural class and a soluble structuralclass with low probabilities. Since their correspondingdomain models are not uniquely classified into the struc-tural class of transmembrane helical proteins, these mis-classifications were ignored.
Secondary Structure Prediction
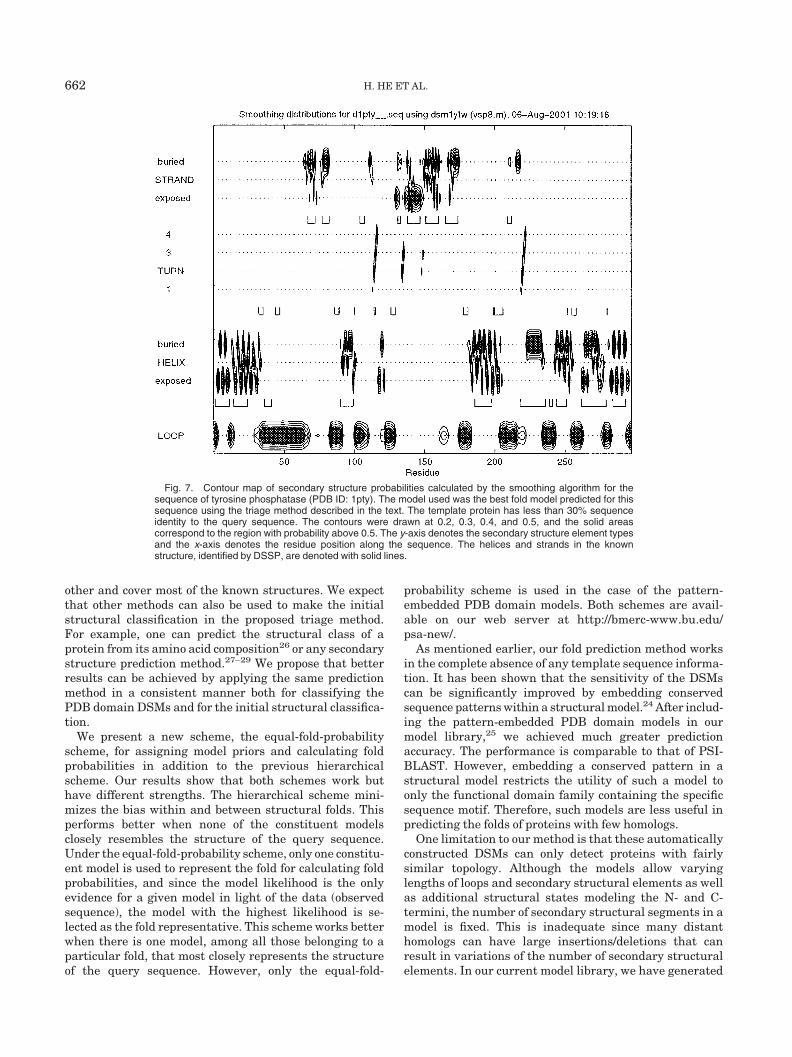
Once the most probable PDB domain model (the one thatrepresents the given fold) is determined for a given se-quence, it can be used to compute the probability of each
residue being in a particular secondary structure element.These probabilities are calculated using the smoothing orforward-backward algorithm.7,16 The smoothing algo-rithm computes the probability of each residue being in astructural state given the entire sequence. The probabilityof a residue being in a secondary structure element is thenobtained by summing the probabilities of the residue beingin all the structural states belonging to the secondarystructure element. The result can be visualized in acontour map, as shown in Figure 7. Since each PDBdomain model represents a real PDB structure with thestructural states corresponding to the structural positionsin the structure, the smoothing results can be used toguide the alignment of the query sequence to the PDBstructure. This sequence-structure alignment can thenprovide a starting point for a detailed modeling for thequery sequence.
DISCUSSION
We present an extension of the DSM approach7,8 thatprovides a greatly expanded structural template librarycovering most of the known structural folds, as classifiedby the SCOP database. This involves two major innova-tions: first, an automated procedure for model generationand, second, a triage method for protein fold predictionthat is applicable to any such large model library.
The automated method of model generation constructs astructural DSM directly from a protein structure depositedin the PDB. It inherits many advantages of the DSMdesign methodology. It requires only a single structure tobuild the model and thus is able to construct a structuralmodel for a protein with no or few homologs. In contrast,the conventional profile HMM construction usually re-quires about 20 homologous sequences to train the model. 9,10
The DSM design also explicitly incorporates expert knowl-edge into the model design.14 The automation of thismethod gives us the ability to expand the library as eachnew structure is identified.
The triage method relies on a set of manually designedhigh-level or generic models. These generic models areused to classify a given sequence into its probable struc-tural class before the fold prediction is made, improvingthe chance that the correct fold can be identified fromwithin the large model library. This approach is particu-larly useful when using a Bayesian or similar likelihoodevaluation approach.
Since our fold prediction method uses a Bayesian estima-tion approach, which will always predict one model as themost probable, this requires the model library to be ascomplete as possible. However, this very completeness canresult in a small posterior probability for the correct modelwhen all PDB domain models in the library are used forcalculating the posterior probabilities. The triage methodlimits this effect by reducing the final number of PDBdomain models used in computing the posterior probabili-ties. This reduction in the model set also reduces thecomputational cost. To ensure that the set of generic DSMsis suitable for the initial structural classification, we haveshown that these generic models are exclusive from each
TABLE V. Results of Fold Prediction UsingPattern-Embedded DSMs†
Pattern-embedded DSMs
PSI-BLASTTriage method Simple method
55/12 54/10 61/16
The numbers shown here are the number of true hits vs.false hits using Pfold � 0.5 as the cutoff.
TABLE VI. Results of Structural Classification
Structural classification
Soluble protein TM-� protein TM-� protein
Soluble proteins (882) 881 1 0TM-� (12) 0 0 12TM-� (16) 1 15 0
TRIAGE PROTEIN FOLD PREDICTION 661
other and cover most of the known structures. We expectthat other methods can also be used to make the initialstructural classification in the proposed triage method.For example, one can predict the structural class of aprotein from its amino acid composition26 or any secondarystructure prediction method.27–29 We propose that betterresults can be achieved by applying the same predictionmethod in a consistent manner both for classifying thePDB domain DSMs and for the initial structural classifica-tion.
We present a new scheme, the equal-fold-probabilityscheme, for assigning model priors and calculating foldprobabilities in addition to the previous hierarchicalscheme. Our results show that both schemes work buthave different strengths. The hierarchical scheme mini-mizes the bias within and between structural folds. Thisperforms better when none of the constituent modelsclosely resembles the structure of the query sequence.Under the equal-fold-probability scheme, only one constitu-ent model is used to represent the fold for calculating foldprobabilities, and since the model likelihood is the onlyevidence for a given model in light of the data (observedsequence), the model with the highest likelihood is se-lected as the fold representative. This scheme works betterwhen there is one model, among all those belonging to aparticular fold, that most closely represents the structureof the query sequence. However, only the equal-fold-
probability scheme is used in the case of the pattern-embedded PDB domain models. Both schemes are avail-able on our web server at http://bmerc-www.bu.edu/psa-new/.
As mentioned earlier, our fold prediction method worksin the complete absence of any template sequence informa-tion. It has been shown that the sensitivity of the DSMscan be significantly improved by embedding conservedsequence patterns within a structural model.24 After includ-ing the pattern-embedded PDB domain models in ourmodel library,25 we achieved much greater predictionaccuracy. The performance is comparable to that of PSI-BLAST. However, embedding a conserved pattern in astructural model restricts the utility of such a model toonly the functional domain family containing the specificsequence motif. Therefore, such models are less useful inpredicting the folds of proteins with few homologs.
One limitation to our method is that these automaticallyconstructed DSMs can only detect proteins with fairlysimilar topology. Although the models allow varyinglengths of loops and secondary structural elements as wellas additional structural states modeling the N- and C-termini, the number of secondary structural segments in amodel is fixed. This is inadequate since many distanthomologs can have large insertions/deletions that canresult in variations of the number of secondary structuralelements. In our current model library, we have generated
Fig. 7. Contour map of secondary structure probabilities calculated by the smoothing algorithm for thesequence of tyrosine phosphatase (PDB ID: 1pty). The model used was the best fold model predicted for thissequence using the triage method described in the text. The template protein has less than 30% sequenceidentity to the query sequence. The contours were drawn at 0.2, 0.3, 0.4, and 0.5, and the solid areascorrespond to the region with probability above 0.5. The y-axis denotes the secondary structure element typesand the x-axis denotes the residue position along the sequence. The helices and strands in the knownstructure, identified by DSSP, are denoted with solid lines.
662 H. HE ET AL.

models representing each SCOP family under each repre-sentative SCOP fold in order to capture the structuralvariations within each fold. However, this is limited by theavailable structural families within a particular fold.Another potential solution might be to generate consensusmodels allowing all the structural variations observed andanticipated within each SCOP fold, including variations inboth the number and length of secondary structural ele-ments. A second limitation results from the fact that manymodels in our current library are quite similar in terms ofthe linear sequence of secondary structural elements, thusthere is a potential for multiple models to overlap. As anexample, three SCOP folds, the NAD(P)-binding Rossmann-fold domain, the FAD/NAD(P)-binding domain, and thenucleotide-binding domain all adopt a three-layer, �/�/�structure with a central parallel �-sheet of 5/6 strands.Models generated from these similar proteins will overlap.The competition between similar models will result in thereduction of their individual posterior probabilities. Thisoverlap offers a possible explanation for the low number offold predictions in both the self-threading and the distant-homolog threading. The construction of a consensus modelis expected to ease this overlap problem.
ACKNOWLEDGMENTS
We thank Jadwiga Bienkowska for her helpful discus-sions about the model construction and Robert Rogers, Jr.,for setting up and maintaining the web server. We alsothank Nancy Sands for careful proofreading of the manu-script.
REFERENCES
1. Bowie JU, Clarke ND, Pabo CO, Sauer RT. Identification ofprotein folds: matching hydrophobicity patterns of sequence setswith solvent accessibility patterns of known structures. Proteins1990;7:257–264.
2. Bowie JU, Luthy R, Eisenberg D. A method to identify proteinsequences that fold into a known three-dimensional structure.Science 1991;253:164–253.
3. Jones DT, Taylor WR, Thornton JM. A new approach to proteinfold recognition. Nature 1992;358:86–89.
4. Sippl MJ, Weitckus S. Detection of native-like models for aminoacid sequences of unknown three-dimensional structure in a database of known protein conformations. Proteins 1992;13:258–271.
5. Godzik A, Kolinski A, Skolnick J. Topology fingerprint approach tothe inverse protein folding problem. J Mol Biol 1992;227:227–238.
6. Bryant SH, Lawrence CE. An empirical energy function forthreading protein sequence through the folding motif. Proteins1993;16:92–112.
7. White JV, Stultz CM, Smith TF. Protein classification by stochas-tic modeling and optimal filtering of amino acid sequences. MathBiosci 1994;119:35–75.
8. Stultz CM, White JV, Smith TF. Structural analysis based onstate-space modeling. Protein Sci 1993;2:305–314.
9. Krogh A, Brown M, Mian IS, Sjolander K, Haussler D. HiddenMarkov models in computational biology: applications to proteinmodeling. J Mol Biol 1994;235:1501–1531.
10. Eddy SR. Hidden Markov models. Curr Opin Struc Biol 1996;6:361–365.
11. Richardson JS. The anatomy and taxonomy of protein structures.Adv Protein Chem 1981;34:167–339.
12. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, WesissigH, Shindyalov IN, Bourne PE. The Protein Data Bank. NucleicAcids Res 2000;28:235–242.
13. Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a struc-tural classification of protein database for the investigation of thesequences and structures. J Mol Biol 1995;247:536–540.
14. Bienkowska JR, Yu L, Zarakhovich S, Rogers Jr RG, Smith TF.Protein fold recognition by total alignment probability. Protein Sci2000;40:451–462.
15. White JV. Modeling and filtering for discretely valued time series.In: Spall JC, editor. Bayesian analysis of time series and dynamicmodels. New York: Marcel Dekker. 1988. p 255–283.
16. Rabiner LR. A tutorial on hidden Markov models and selectedapplications in speech recognition. Proc Inst Electric Electron Eng1989;77:257–286.
17. Kabsch W, Sander C. Dictionary of protein secondary structure:pattern recognition of hydrogen-bonded and geometrical features.Biopolymers 1983;22:2577–2637.
18. Eisenberg D, MacLachlan AD. Solvation energy in protein foldingand binding. Nature Jan, 1986319:199–203.
19. Bakis R. Continuous speech word recognition via centisecondacoustic states. In: Proc ASA Meeting, Washington, DC. April,1976.
20. Schulz GE. �-Barrel membrane proteins. Curr Opin Struct Biol2000;10:443–447.
21. Engelman D, Steitz T, Goldman A. Identifying nonpolar transbi-layer helices in amino acid sequences of membrane proteins. AnnuRev Biophys Biophys Chem 1986;5:321.
22. McAllister GD. Modeling of transmembrane proteins using dis-crete state-space models. Master’s thesis, Boston University,Boston, MA, June 2001.
23. Altschul SF, Madden T, Schaffer A, Zhang J,-Zhang Z, Miller W,Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation ofprotein database search programs. Nucleic Acid Res 1997;25:3389–3402.
24. Yu L, White JV, Smith TF. A homology identification method thatcombines protein sequence and structure information. Protein Sci1998;7:2499–2510.
25. Bienkowska JR, He H, Smith TF. Automatic pattern embeddingin protein structure models. IEEE Intell Syst 2001;16:21–25.
26. Nakashima H, Nishikawa K, Ooi T. The folding type of a protein isrelevant to the amino acid composition. J Biochem Tokyo 1986;99:153–162.
27. Qian N, Sejnowski TJ. Predicting the secondary structure ofglobular proteins using neural network models. J Mol Biol 1988;202:865–884.
28. Rost B, Sander C. Improved prediction of protein secondarystructure by use of sequence profiles and neural networks. ProcNatl Acad Sci USA 1994;90:7558–7562.
29. Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 1999;292:195–202.
TRIAGE PROTEIN FOLD PREDICTION 663





























