Embed Size (px)
Citation preview

ANALYTICAL
Analytical Biochemistry 325 (2004) 68–76
BIOCHEMISTRY
www.elsevier.com/locate/yabio
Use of protein biotinylation in vivofor chromatin immunoprecipitation
Antoine Viens, Undine Mechold, Heike Lehrmann, Annick Harel-Bellan,and Vasily Ogryzko*
Laboratoire Oncog�ense, Diff�erenciation et Transduction du Signal, Institut Andr�e Lwoff, 7 rue Guy Moquet, 94800 Villejuif, France
Received 4 June 2003
Abstract
We describe a system designed to express biotinylated proteins in mammalian cells in vivo and its application to the study of
protein–DNA interactions in vivo by chromatin immunoprecipitation (ChIP). The system is based on coexpression of the target
protein fused to a short biotin acceptor domain together with the biotinylating enzyme BirA from Escherichia coli. The superior
strength of the biotin–avidin interaction allows one to employ more stringent washing conditions in the ChIP protocol, resulting in a
better signal/noise ratio.
� 2003 Elsevier Inc. All rights reserved.
Keywords: Epitope tagging; Biotinylation; Chromatin immunoprecipitation; Protein–DNA interaction
Protein–DNA interactions play major roles in the
regulation of gene activity and other functions of the
genome. A number of methods, such as electropho-
retic gel mobility shift assay (EMSA)1 [1,2] and
others, have been developed to study different aspects
of protein–DNA interactions. However, in most ofthese approaches, the DNA–protein interaction is
studied in vitro, out of the context of chromatin and
the nuclear environment, which certainly limits their
value.
Chromatin immunoprecipitation (ChIP) [3,4] is a
method designed to analyze the protein–DNA interac-
tions in a more physiological context, as it detects them
directly in the living cell. In the past several years it has
* Corresponding author. Fax: +33-1-49-58-34-01.
E-mail address: [email protected] (V. Ogryzko).1 Abbreviations used: EMSA, electrophoretic gel mobility shelt
assay; ChIp chromatin immunoprecipitation; DMEM, Dulbecco�smodified Eagle�s medium; FBS, fetal bovine serum; FCS, fetal calf
serum; NP-40, Nonedet p-40; PBS, phosphate-buffered saline; GEP,
green fluorescent protein; FITC, fluorescein isothiocyanate; BAD,
biotin acceptor domain; ORF, open reading frame; IRES, internal
ribosome entrance sequence.
0003-2697/$ - see front matter � 2003 Elsevier Inc. All rights reserved.
doi:10.1016/j.ab.2003.10.015
been widely used to study the in vivo association of a
particular DNA sequence with regulatory proteins and
posttranslationally modified histones. This method has
its own limitations. In particular, it requires antibodies
of a quality that is sufficient for a specific interaction and
at the same time can tolerate stringent binding andwashing conditions.
The interaction between biotin and streptavidin is one
of the strongest known noncovalent interactions [5].
Many commercially available reagents for purification
and detection of biotinylated macromolecules have been
developed. Correspondingly, we wanted to take advan-
tage of this interaction, in the particular application of
ChIP, as it would permit more stringent washing con-ditions than those usually utilized in ChIP experiments.
Here, we describe a system for expression of biotinylated
proteins of choice in mammalian cells in vivo. We
demonstrate that, in our system, the chromatin immu-
noprecipitation experiments can be performed under
conditions more stringent than usual, which significantly
improves the signal to noise ratio. We believe that ChIP
with in vivo biotinylated proteins could have manyapplications relevant for the study of protein–DNA
interactions in vivo.

A. Viens et al. / Analytical Biochemistry 325 (2004) 68–76 69
Materials and methods
Stable cell line generation and transient transfection
All cells were grown in Dulbecco�s modified
Eagle�s medium (DMEM, Gibco) with 10% fetal
bovine serum (FCS, PAN Biotech). For transient
transfection, a standard calcium phosphate precipita-
tion method was used, and the cells were analyzed 1or 2 days after transfection. In some experiments we
also used Polyfect (Qiagen). For retroviral transduc-
tion, the packaging cell line Phenix-E was transfected
with the plasmid DNA of the retroviral vector; 2
days later the retroviral supernatant was harvested,
filter-sterilized, and added to the HeLa or HEK 293
cells. Two days afterward, the transduced cells were
sorted using anti-IL2R antibody coupled to magneticaffinity beads [6].
Western analysis
Total cell extract for the analysis was prepared
using RIPA buffer (50mM Tris–HCl, pH 8.0, 50mM
NaCl, 1% NP-40, 0.5% Na-deoxyholate, 0.1% SDS,
1mM EDTA). First, cells were washed with phos-phate-buffered saline (PBS) and then incubated with
RIPA buffer directly in the six well plates for 5min.
The extract was harvested and spun at high speed on
a microcentrifuge. SDP–PAGE separation, transfer to
a nitrocellulose membrane, blocking, incubation with
antibody, and electrochemiluminescence detection
were performed according to a standard protocol,
except that for the detection of biotinylated proteins,500mM NaCl was added to the washing buffer
(PBS+0.1% Tween). The anti-GFP antibodies were
purchased from Clontech, the HP1 antibodies were
from Euromedex, and the streptavidin–HPO conju-
gate was from Sigma.
Immunofluorescence
NIH3T3 cells were grown and transfected with
Polyfect on coverslips. The day before fixation, 0.2%
biotin was added to the medium. Fixation was ac-
complished with 4% formaldehyde in PBS for 15min at4 �C. Fixed cells were permeabilized with 0.5% Triton
for 15min, blocked with 1% BSA and 1% FBS, and
incubated with anti-HP1c antibodies. They were then
washed with PBS and incubated with secondary anti-
body conjugated to rhodamine. For detection of bio-
tinylated proteins, the cells were incubated with
streptavidin–FITC for 1 h at 37 �C, and then washed
with PBS. The cells were observed using a fluorescencemicroscope, and images were acquired using a charge-
coupled device camera.
Chromatin immunoprecipitation
NIH 3T3 cells from a six-well plate well or a 10-cm
dish at 50–70% confluency were transfected with Poly-
fect (Qiagen). The next day the cells were split (to keep
them asynchronous) and put into DMEM with 10%
FBS and 0.2mg/L biotin. The next day this medium was
removed and replaced by fresh DMEM, 10% FBS
without biotin. Formaldehyde (Sigma, Cat. No. F-8775)was added to a final concentration of 1%. Cross-linking
was performed at 37 �C for 10min and stopped by ad-
dition of glycine to a final concentration of 0.125M,
followed by 5-min incubation at room temperature.
Fixed cells were washed twice with cold PBS and har-
vested in cold PBS containing protease inhibitors. Cells
were pelleted by centrifugation, suspended in 1mL of
SDS buffer (50mM Tris at pH 8.1, 0.5% SDS, 100mMNaCl, 5mM EDTA and protease inhibitors), and in-
cubated for 10min on ice. Cells were then pelleted by
centrifugation, resuspended in 400 lL of IP buffer (0.3%
SDS, 1.1% Triton X-100, 1.2mM EDTA, 16.7mM Tris
at pH 8.1, 167mM NaCl, and protease inhibitors), and
disrupted by sonication, yielding genomic DNA frag-
ments of a size of 100–500 bp. For each immunopre-
cipitation, 50 lg of chromatin was diluted to a finalvolume of 300 lL in IP buffer. Chromatin was precle-
ared for 1 h by addition of 40 lL of blocked protein A
beads (Pierce, Cat. No. 20334) (50% slurry proteinA–
agarose, 3mg/mL BSA, 0.1mg/mL salmon sperm DNA,
in IP buffer). Samples were next incubated for 3 h with
40 lL of streptavidin-coated magnetic particles (Pro-
mega, Cat. No. Z5481). Beads were then washed twice
with 2% SDS (or with 100mM NaOH, or 3M guanidin–Cl, or 8M urea, or 0.5% SDS+100mM NaOH, or 5M
NaCl) followed by three washes with LiCl wash buffer
(100mM Tris at pH 8.0, 500mM LiCl, 1% NP-40, 1%
deoxycholic acid), or five washes with the LiCl wash
buffer (normal wash [7]). Samples were then decross-
linked by an overnight incubation at 67 �C in 60 lL of
300mM NaCl solution. Proteinase K and 5X Proteinase
K buffer (50mM Tris at pH 7.5, 25mM EDTA, 1.25%SDS) were then added to the samples. After a 2-h in-
cubation at 45 �C, DNA from the samples was purified
using Qiagen miniprep columns (Cat. No. 27106). Pu-
rified DNA was recovered in 50 lL of water. In addition
to the precipitated samples, 5% of the input chromatin
for each sample was decross-linked, processed, and an-
alyzed in the same way. For the immunoprecipitation
with anti-HA antibody (Roche, Cat. No.1867423), thesame procedure was used, except that biotin was omit-
ted from the medium, and chromatin was incubated
overnight with 1 lL of anti-HA antibody and then in-
cubated with 80 lL of blocked protein A beads. After
washing, the immunoprecipitate was eluted in 200 lLof elution buffer (50mM NaHCO3, 1% SDS) before
decross-linking.

70 A. Viens et al. / Analytical Biochemistry 325 (2004) 68–76
Quantitative PCR
One microliter of DNA was used in 20 ll of quanti-tative PCR (using a Quantitect SYBR Green PCR kit
from Qiagen) on the LightCycler (Roche). Each reaction
was performed in duplicate. The PCR primer sequences
were B1 50: gcc ggg tgt ggt ggc gca cac ctt t, B1 30: gagaca ggg ttt ctc tgt gta gcc ct, DHFR 50: gcg gag cct tag
ctg cac aa, DHFR 30: tac cag cct tca cgc tag ga, b-actin50: acc gag cgt ggc tac agc tt, b-actin 30: tgg ccg tca ggc
agc tca ta, GAPDH 50: cca atg tgt ccg tcg tgg atc t, and
GAPDH 30: gtt gaa gtc gca gga gac acc. The linearity of
analysis was confirmed by using serial sample dilutions
(data not shown). All PCRs gave a unique product of
expected size, as confirmed by agarose gel analysis. For
each sample the value of the signal was calculated as a
percentage of input. Then the signal to noise ratio wascalculated taking the value of a GFP sample as a non-
specific background (noise).
Results
Description of the pBBH and CMV.BBH vectors
The principle of in vivo biotinylation is based on
coexpression of the biotinylation enzyme BirA from
Escherichia coli together with the protein of interest
fused to a short peptide, the biotin acceptor domain
(BAD), shown to be an efficient target for BirA [8].
Fig. 1 shows a map of the retroviral vector pBBH that
was generated in our laboratory to express proteins
biotinylated in vivo.The principal feature of our design is the placing of
the BirA ORF on the same vector downstream of the
gene of interest. Its expression from the same mRNA is
ensured by an internal ribosome entrance sequence
(IRES). The rationale for this particular design is to
provide for a high local concentration of the BirA en-
zyme in the vicinity of the protein of interest, which
would ensure efficient biotinylation with minimalamounts of the BirA enzyme. To minimize the vector
size, we used as an IRES a short sequence (designated
ShIRES) containing a 9-nt sequence complementary to
18S rRNA [9].
We generated versions of the vector that can be used
for either N-terminal or C-terminal tagging. We also
added a His tag for double-epitope tag purification. In
addition to the pBBHN and pBBHC vectors for gen-eration of stable cell lines based on the retroviral
MMLV backbone, we constructed vectors for transient
transfection, with transcription driven by a strong CMV
promoter (CMV.BBHN and CMV.BBHC, for N- and
C-terminal tagging correspondingly).
An additional feature of our vectors is the third ORF
encoding a selection marker, IL2R [10], which is under
control of a second IRES (EMC IRES). This tricistronicdesign allows us to minimize the number of steps and
reagents necessary to generate a required cell line.
100% efficiency of biotinylation of a target protein
To determine how efficiently a target protein could be
biotinylated in our system, we cloned the eGFP gene
(Clontech) in the retroviral vector pBBHN and gener-ated a HeLa cell line stably transduced with this con-
struct. As a control to test the requirement for BirA
enzyme, we also generated HeLa cells transduced with
pBH.GFP (this vector expresses the BAD.GFP fusion,
but it does not encode BirA). The state of the GFP in
these cells was monitored by Western blotting with anti-
GFP antibodies. As seen from Fig. 2A (lane 2 versus
lane 1), when BirA is expressed in the cells (pBBH.GFPtransduced cells), the BAD.GFP fusion protein migrates
noticeably slower compared to the same protein from
the cells that do not express BirA (pBH.GFP transduced
cells). To confirm that this change in migration is due to
biotinylation of the protein, we took advantage of the
fact that streptavidin–biotin binding resists conditions
of SDS–PAGE electrophoresis. Preincubation of the
sample with 1 lg of streptavidin before SDS–PAGEcaused a further, more dramatic shift in the GFP mo-
bility (lane 3). Since no residual fast-migrating band
remained after the streptavidin supershift, we conclude
that 100% of the GFP is biotinylated under these con-
ditions. Addition of streptavidin to the sample from the
pBH.GFP transduced cells (not expressing BirA) does
not cause a shift in mobility (not shown). The biotiny-
lated status of GFP from the pBBH.GFP transducedcells was also confirmed by probing the Western blot
with a peroxydase-conjugated streptavidin (Fig. 2B).
Similar tests with a GFP fusion having the BAD
domain at its C terminus also show 100% biotinylation
(Fig. 2A, lanes 5 and 6). The CMV.BBH vectors for
transient transfection were tested and gave 100% bioti-
nylated GFP (lanes 7–9 for CMV.BBHN; CMV.BBHC
not shown).
Intracellular localization of proteins is not generally
disturbed by their biotinylation in vivo
Epitope-tagging of proteins can lead to artifacts, as in
some cases it can disturb the protein�s properties [11]. Todetermine whether, in addition to this general concern,
biotinylation presents any specific problem for all pro-teins, we tested the intracellular localization of several
biotinylated proteins with a known distribution in the
nucleus.
We transiently transfected NIH3T3 cells with vectors
expressing biotinylated HP1a, HP1b, and HP1c, the
mammalian homologues of the Drosophila heterochro-
matin protein HP1. To follow the nuclear localization of

Fig. 1. Vectors for in vivo biotinylation. (A) Map of the retroviral vector pBBHN. The principal components (BAD domain, BirA, IL2R, and
IRESes) and restriction sites are shown. (B) Sequences of the BAD domain, the 6XHis tag, and the multiple cloning sites for the N-terminal fusion
and C-terminal fusion vectors, correspondingly. The amino acid sequence of the tags and the restriction sites are underlined.
A. Viens et al. / Analytical Biochemistry 325 (2004) 68–76 71
these proteins, we used two complementary approaches:
staining with streptavidin–FITC to detect biotinylated
proteins and staining with monoclonal antibodies spe-
cific for different HP1 proteins. All HP1s showed clear
colocalization with heterochromatin by both kinds of
staining (Fig. 3A). Intriguingly, HP1c, believed to belocalized in a different manner, showed a pattern un-
distinguishable from those of the other HP1s, even in
untransfected cells (Fig. 3B). On the other hand, the
staining pattern of HP1a was dependent on the antibody
used (Fig. 3B, compare 2HP-269 and 2HP-1H5, see
Discussion).
To rule out the unlikely possibility that all biotiny-
lated proteins are recruited into heterochromatin, we
tested several proteins known to show a different local-
ization pattern. Two examples presented here are
SUMO1 and HMG14. As seen from Fig. 3C, they show
staining distinct from that of HP1 proteins. Thus, bio-tinylation does not target every protein to heterochro-
matin, and the localization of proteins by our technique
should reflect their physiological localization.
Thus, at least by the criterion of intracellular locali-
zation, in vivo biotinylation does not appear to disturb
protein function. Admittedly, one cannot exclude the
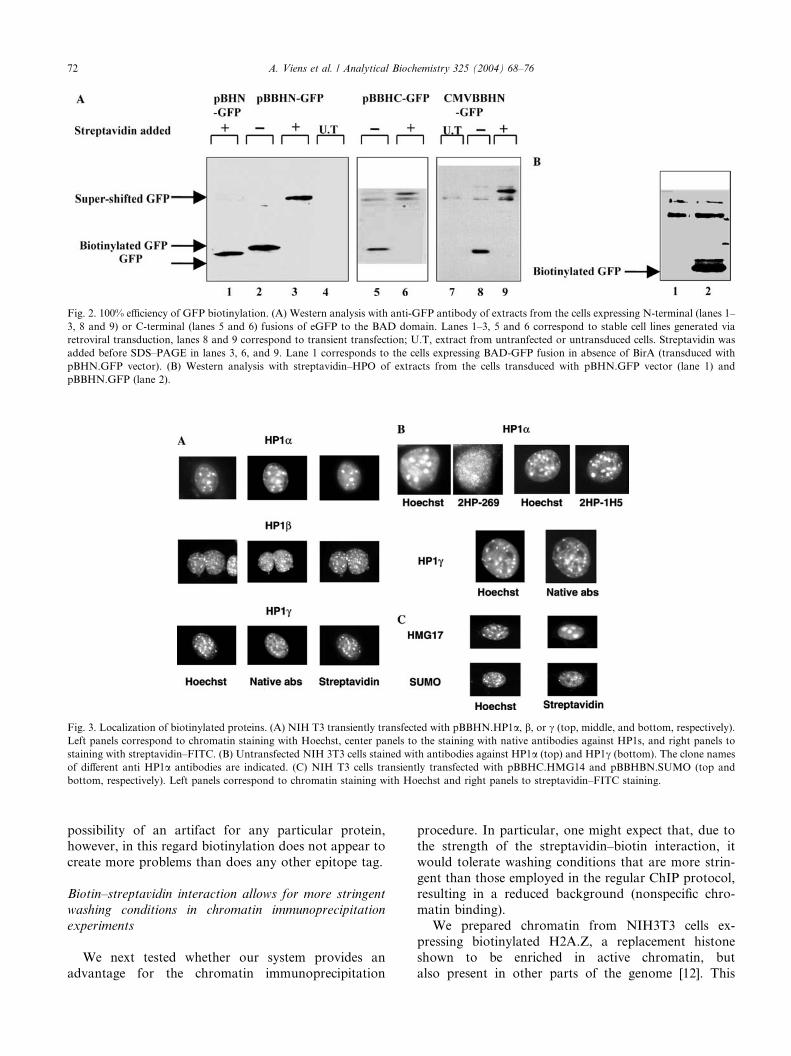
Fig. 2. 100% efficiency of GFP biotinylation. (A) Western analysis with anti-GFP antibody of extracts from the cells expressing N-terminal (lanes 1–
3, 8 and 9) or C-terminal (lanes 5 and 6) fusions of eGFP to the BAD domain. Lanes 1–3, 5 and 6 correspond to stable cell lines generated via
retroviral transduction, lanes 8 and 9 correspond to transient transfection; U.T, extract from untranfected or untransduced cells. Streptavidin was
added before SDS–PAGE in lanes 3, 6, and 9. Lane 1 corresponds to the cells expressing BAD-GFP fusion in absence of BirA (transduced with
pBHN.GFP vector). (B) Western analysis with streptavidin–HPO of extracts from the cells transduced with pBHN.GFP vector (lane 1) and
pBBHN.GFP (lane 2).
Fig. 3. Localization of biotinylated proteins. (A) NIH T3 transiently transfected with pBBHN.HP1a, b, or c (top, middle, and bottom, respectively).
Left panels correspond to chromatin staining with Hoechst, center panels to the staining with native antibodies against HP1s, and right panels to
staining with streptavidin–FITC. (B) Untransfected NIH 3T3 cells stained with antibodies against HP1a (top) and HP1c (bottom). The clone names
of different anti HP1a antibodies are indicated. (C) NIH T3 cells transiently transfected with pBBHC.HMG14 and pBBHBN.SUMO (top and
bottom, respectively). Left panels correspond to chromatin staining with Hoechst and right panels to streptavidin–FITC staining.
72 A. Viens et al. / Analytical Biochemistry 325 (2004) 68–76
possibility of an artifact for any particular protein,
however, in this regard biotinylation does not appear to
create more problems than does any other epitope tag.
Biotin–streptavidin interaction allows for more stringent
washing conditions in chromatin immunoprecipitation
experiments
We next tested whether our system provides an
advantage for the chromatin immunoprecipitation
procedure. In particular, one might expect that, due to
the strength of the streptavidin–biotin interaction, it
would tolerate washing conditions that are more strin-
gent than those employed in the regular ChIP protocol,resulting in a reduced background (nonspecific chro-
matin binding).
We prepared chromatin from NIH3T3 cells ex-
pressing biotinylated H2A.Z, a replacement histone
shown to be enriched in active chromatin, but
also present in other parts of the genome [12]. This
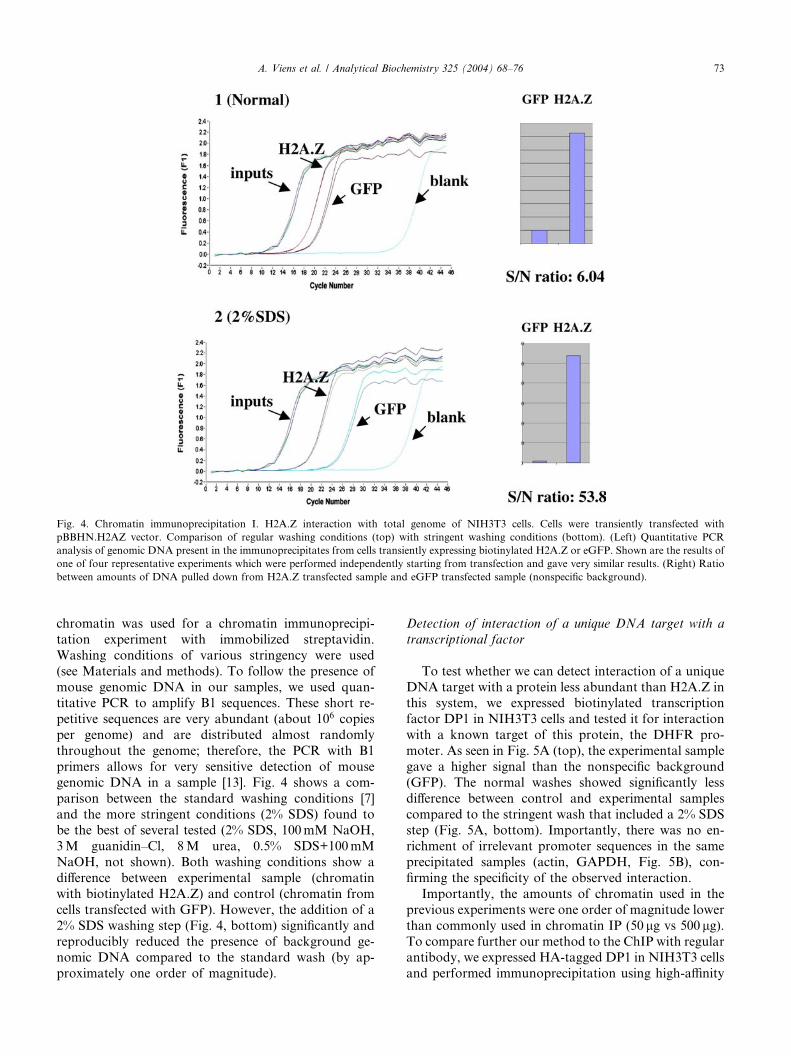
Fig. 4. Chromatin immunoprecipitation I. H2A.Z interaction with total genome of NIH3T3 cells. Cells were transiently transfected with
pBBHN.H2AZ vector. Comparison of regular washing conditions (top) with stringent washing conditions (bottom). (Left) Quantitative PCR
analysis of genomic DNA present in the immunoprecipitates from cells transiently expressing biotinylated H2A.Z or eGFP. Shown are the results of
one of four representative experiments which were performed independently starting from transfection and gave very similar results. (Right) Ratio
between amounts of DNA pulled down from H2A.Z transfected sample and eGFP transfected sample (nonspecific background).
A. Viens et al. / Analytical Biochemistry 325 (2004) 68–76 73
chromatin was used for a chromatin immunoprecipi-
tation experiment with immobilized streptavidin.
Washing conditions of various stringency were used
(see Materials and methods). To follow the presence of
mouse genomic DNA in our samples, we used quan-titative PCR to amplify B1 sequences. These short re-
petitive sequences are very abundant (about 106 copies
per genome) and are distributed almost randomly
throughout the genome; therefore, the PCR with B1
primers allows for very sensitive detection of mouse
genomic DNA in a sample [13]. Fig. 4 shows a com-
parison between the standard washing conditions [7]
and the more stringent conditions (2% SDS) found tobe the best of several tested (2% SDS, 100mM NaOH,
3M guanidin–Cl, 8M urea, 0.5% SDS+100mM
NaOH, not shown). Both washing conditions show a
difference between experimental sample (chromatin
with biotinylated H2A.Z) and control (chromatin from
cells transfected with GFP). However, the addition of a
2% SDS washing step (Fig. 4, bottom) significantly and
reproducibly reduced the presence of background ge-nomic DNA compared to the standard wash (by ap-
proximately one order of magnitude).
Detection of interaction of a unique DNA target with a
transcriptional factor
To test whether we can detect interaction of a unique
DNA target with a protein less abundant than H2A.Z inthis system, we expressed biotinylated transcription
factor DP1 in NIH3T3 cells and tested it for interaction
with a known target of this protein, the DHFR pro-
moter. As seen in Fig. 5A (top), the experimental sample
gave a higher signal than the nonspecific background
(GFP). The normal washes showed significantly less
difference between control and experimental samples
compared to the stringent wash that included a 2% SDSstep (Fig. 5A, bottom). Importantly, there was no en-
richment of irrelevant promoter sequences in the same
precipitated samples (actin, GAPDH, Fig. 5B), con-
firming the specificity of the observed interaction.
Importantly, the amounts of chromatin used in the
previous experiments were one order of magnitude lower
than commonly used in chromatin IP (50 lg vs 500 lg).To compare further our method to the ChIP with regularantibody, we expressed HA-tagged DP1 in NIH3T3 cells
and performed immunoprecipitation using high-affinity
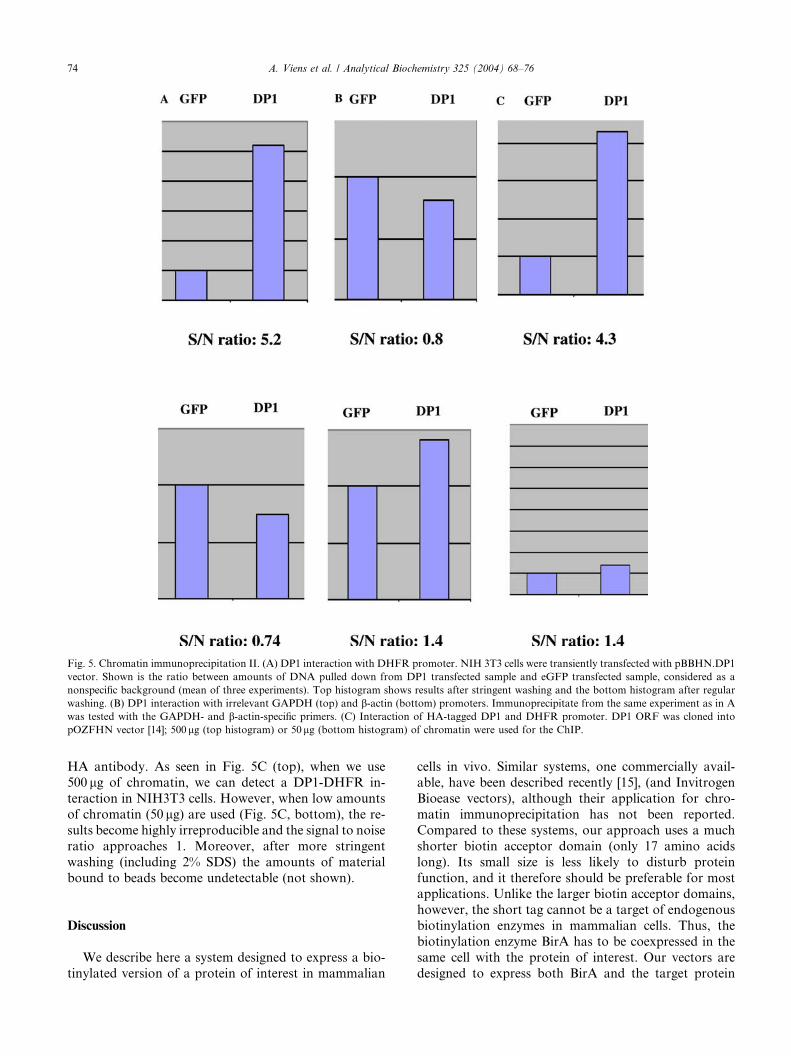
Fig. 5. Chromatin immunoprecipitation II. (A) DP1 interaction with DHFR promoter. NIH 3T3 cells were transiently transfected with pBBHN.DP1
vector. Shown is the ratio between amounts of DNA pulled down from DP1 transfected sample and eGFP transfected sample, considered as a
nonspecific background (mean of three experiments). Top histogram shows results after stringent washing and the bottom histogram after regular
washing. (B) DP1 interaction with irrelevant GAPDH (top) and b-actin (bottom) promoters. Immunoprecipitate from the same experiment as in A
was tested with the GAPDH- and b-actin-specific primers. (C) Interaction of HA-tagged DP1 and DHFR promoter. DP1 ORF was cloned into
pOZFHN vector [14]; 500lg (top histogram) or 50lg (bottom histogram) of chromatin were used for the ChIP.
74 A. Viens et al. / Analytical Biochemistry 325 (2004) 68–76
HA antibody. As seen in Fig. 5C (top), when we use
500 lg of chromatin, we can detect a DP1-DHFR in-
teraction in NIH3T3 cells. However, when low amounts
of chromatin (50 lg) are used (Fig. 5C, bottom), the re-
sults become highly irreproducible and the signal to noise
ratio approaches 1. Moreover, after more stringent
washing (including 2% SDS) the amounts of material
bound to beads become undetectable (not shown).
Discussion
We describe here a system designed to express a bio-
tinylated version of a protein of interest in mammalian
cells in vivo. Similar systems, one commercially avail-
able, have been described recently [15], (and Invitrogen
Bioease vectors), although their application for chro-
matin immunoprecipitation has not been reported.
Compared to these systems, our approach uses a much
shorter biotin acceptor domain (only 17 amino acids
long). Its small size is less likely to disturb protein
function, and it therefore should be preferable for mostapplications. Unlike the larger biotin acceptor domains,
however, the short tag cannot be a target of endogenous
biotinylation enzymes in mammalian cells. Thus, the
biotinylation enzyme BirA has to be coexpressed in the
same cell with the protein of interest. Our vectors are
designed to express both BirA and the target protein

A. Viens et al. / Analytical Biochemistry 325 (2004) 68–76 75
from the same mRNA. Under this cis arrangement, theenzyme is synthesized in close physical proximity to its
target, ensuring efficient biotinylation of the protein of
interest. Our attempts to coexpress BirA and the target
protein from different promoters (trans-biotinylation)
never led to 100% biotinylation (data not shown).
As demonstrated by immunofluorescence experi-
ments, in vivo biotinylation does not change the intra-
cellular localization of the proteins. Thus, in this respect,it does not appear to create any additional artifacts as
compared to other epitope tags. In the course of these
experiments, we also unexpectedly observed that HP1c,previously reported to be localized in euchromatin [16],
was distributed in NIH3T3 cells in a manner undistin-
guishable from that of HP1b and HP1a. This was the
case even with endogenous HP1c in untransfected cells
detected by antibody against the native protein. Sur-prisingly, we also found that the two different anti-HP1aantibodies that we tested (2HP-1H5 and 2HP-269) differ
dramatically in the immunofluorescence pattern ob-
tained. Their specificity was confirmed in independent
experiments with siRNA knockdowns. We believe that
the euchromatic staining of HP1a seen in our study with
the 2HP-269 antibody, or that of HP1c seen in previous
studies [16], most probably reflects the masking of theepitope recognized by particular antibodies in the con-
text of heterochromatin, rather than a true localization
to the euchromatic compartment.
The principal result of this work is the demonstration
that in vivo biotinylation allows the use of more strin-
gent washing conditions in chromatin immunoprecipi-
tation, compared to those employed in a regular ChIP
protocol. This significantly improves the signal to noiseratio, as judged by quantitative PCR analysis. As a re-
sult, our system should help to streamline the ChIP
procedure and make it more robust and economical.
Accordingly, with our system, chromatin from only
5� 106 transiently transfected cells was sufficient for two
to four immunoprecipitation experiments yielding a re-
liable difference between signal and background.
It is important to note that, due to the overexpressionof proteins during transient transfection, this particular
method might not adequately reflect the distribution of
native protein throughout the genome. Therefore, it is
not advisable to use transient transfection for identifi-
cation of the DNA targets of a particular protein. Stable
cell lines with proven low expression levels of the bio-
tinylated protein (5–10% of the endogenous levels)
should be preferred. Nevertheless, the transient trans-fection could still be useful in addressing many other
questions concerning protein–DNA interactions, mostly
concerning the requirements for binding of a protein to
a given genome site. In particular, one can compare
different mutated versions of a protein in their binding
to a given genomic target and determine which domains
and particular residues play an important role in the
interaction. Alternatively, one can probe the accessibil-ity of a particular chromatin site to a given protein
under different physiological conditions. Unlike other
methods used to study protein–DNA interactions, for
instance EMSA, the binding is detected in vivo, in the
context of chromatin and the nuclear environment, and
therefore the information obtained can be considered
more physiologically relevant.
Compared to other epitope-tagging systems, bioti-nylation offers additional advantages, such as the wide
variety of commercially available reagents for detection
and purification of biotinylated macromolecules. These
reagents provide different choices of purification strat-
egies; for example, use of immobilized monomeric av-
idin allows for a gentle elution of biotinylated material,
compared to immobilized avidin or streptavidin. Thus,
the biotinylation system is also more flexible than otherepitope-tagging systems and should find many appli-
cations for the study of protein–DNA interaction
in vivo.
References
[1] M.A. Laniel, A. Beliveau, S.L. Guerin, Electrophoretic mobility
shift assays for the analysis of DNA-protein interactions, Meth-
ods Mol. Biol. 148 (2001) 13–30.
[2] L.D. Kerr, Electrophoretic mobility shift assay, Methods Enzy-
mol. 254 (1995) 619–632.
[3] M.H. Kuo, C.D. Allis, In vivo cross-linking and immunoprecip-
itation for studying dynamic Protein:DNA associations in a
chromatin environment, Methods 19 (3) (1999) 425–433.
[4] V. Orlando, Mapping chromosomal proteins in vivo by formal-
dehyde-crosslinked-chromatin immunoprecipitation, Trends Bio-
chem. Sci. 25 (3) (2000) 99–104.
[5] Y. Lindqvist, G. Schneider, Protein-biotin interactions, Curr.
Opin. Struct. Biol. Dec 6 (6) (1996) 798–803.
[6] V.V. Ogryzko, Y. Kotani, X. Zhang, R.L. Schiltz, T. Howard,
X.J. Yang, B.H. Howard, J. Qin, Y. Nakatani, Histone-like TAFs
within the PCAF histone acetylase complex, Cell 94 (1) (1998) 35–
44.
[7] A.S. Weinmann, P.J. Farnham, Identification of unknown target
genes of human transcription factors using chromatin immuno-
precipitation, Methods 26 (1) (2002) 37–47.
[8] P.J. Schatz, Use of peptide libraries to map the substrate
specificity of a peptide-modifying enzyme: a 13 residue consensus
peptide specifies biotinylation in Escherichia coli, Biotechnology
(NY) 11 (10) (1993) 1138–1143.
[9] S.A. Chappell, G.M. Edelman, V.P. Mauro, A 9-nt segment of a
cellular mRNA can function as an internal ribosome entry site
(IRES) and when present in linked multiple copies greatly
enhances IRES activity, Proc. Natl. Acad. Sci. USA 97 (4)
(2000) 1536–1541.
[10] R. Padmanabhan, C.D. Corsico, T.H. Howard, W. Holter, C.M.
Fordis, M. Willingham, B.H. Howard, Purification of transiently
transfected cells by magnetic affinity cell sorting, Anal. Biochem.
170 (2) (1988) 341–348.
[11] J.W. Jarvik, C.A. Telmer, Epitope tagging, Annu. Rev. Genet. 32
(1998) 601–618.
[12] T.J. Leach, M. Mazzeo, H.L. Chotkowski, J.P. Madigan, M.G.
Wotring, R.L. Glaser, Histone H2A.Z is widely but nonrandomly

76 A. Viens et al. / Analytical Biochemistry 325 (2004) 68–76
distributed in chromosomes of Drosophila melanogaster, J. Biol.
Chem. 275 (30) (2000) 23267–23272.
[13] M.E. Sifis, K. Both, L.A. Burgoyne, A more sensitive method for
the quantitation of genomic DNA by Alu amplification, J.
Forensic Sci. 47 (3) (2002) 589–592.
[14] T. Ikura, V.T. Ogryzko, M. Grigoriev, R. Groisman, J. Wang, M.
Horikoshi, R. Scully, J. Qin, Y. Nakatani, Involvement of the
TIP60 histone acetylase complex in DNA repair and apoptosis,
Cell 102 (4) (2000) 463–473.
[15] M.B. Parrott, M.A. Barry, Metabolic biotinylation of recombi-
nant proteins in mammalian cells and in mice, Mol. Ther. 1 (1)
(2000) 96–104.
[16] D.A. Horsley, A. Hutchings, G.W. Butcher, P.B. Singh,
M32, a murine homologue of Drosophila heterochroma-
tin protein 1 (HP1), localises to euchromatin within
interphase nuclei and is largely excluded from constitutive
heterochromatin, Cytogenet. Cell Genet. 73 (4) (1996)
308–311.
























![Research Paper JNK/AP1 Pathway Regulates MYC ...Chromatin immunoprecipitation assays (ChIP) ChIP analysis was performed as previously described [11]. Chromatin solutions were precipitated](https://img.pdfslide.net/doc/110x75/608625bcea8a6a2e9165f1fb/research-paper-jnkap1-pathway-regulates-myc-chromatin-immunoprecipitation-assays.jpg)




