N
L
Pdw
Eda
D
TaTtac
taHnl
ophnoctd
ealbrt
gH
cc
2
eurología. 2013;28(9):584—593
NEUROLOGÍA
www.elsevier.es/neurologiaaaddtm
msanfcpb(
aatas
l
gstsfs
fiifp
Pgw
ETTERS TO THE EDITOR
rogressive myoclonus epilepsies:escription of a case of Lafora diseaseith autopsy�
pilepsia mioclónica progresiva: Descripcióne un caso de enfermedad de Lafora conutopsia
ear Editor
he term ‘progressive myoclonus epilepsy’ (PME) refers ton array of clinical entities with heterogeneous causes.hey are characterised by different types of epilep-ic seizures (mainly myoclonic), intellectual impairment,nd other clinical manifestations mainly involving theerebellum.1,2
We present the case of a 15-year-old male whose gesta-ion and birth were uneventful. Psychomotor developmentnd educational level were also normal until the age of 11.e had a healthy brother 3 years older. Their parents wereot consanguineous, although both were from the same vil-age of some 1000 inhabitants.
At the age of 12, the patient began to suffer episodesf disorientation lasting a few seconds, which were inter-reted as absence seizures. During the following months,e presented several generalised tonic-clonic seizures. Cra-ial CT yielded normal results; EEG revealed overall slowingf the background activity plus some diffuse spike-waveomplexes. Intermittent light stimulation generated a pho-oparoxysmal response at low frequencies. Epileptic activityid not increase during stages of drowsiness.
He was initially diagnosed with juvenile myoclonuspilepsy and treated with valproic acid (1500 mg per day)ssociated with clonazepam (40 mg per day). Given theack of response to medication, doctors added phenobar-ital (90 mg per day) and ethosuximide (1000 mg per day) toeduce generalised seizures and absence seizures, respec-ively.
During the following 2 years, the patient’s epilepsy pro-ressed unfavourably with increasingly frequent seizures.is different types of epileptic seizures were classified as
� Please cite this article as: Jiménez Caballero PE. Epilepsia mio-lónica progresiva: Descripción de un caso de enfermedad de Laforaon autopsia. Neurología. 2013;28:584—586.
eaofaps
173-5808/$ – see front matter © 2012 Sociedad Española de Neurología.
typical absence seizures, multifocal cortical myoclonus,nd generalised tonic-clonic seizures. Doctors also observedeclining academic performance with multiple cognitiveeficits mainly affecting visuospatial and literacy abili-ies. The patient was finally diagnosed with progressiveyoclonus epilepsy based on the above symptoms.General physical examination revealed no cutaneous stig-
ata (phacomatosis), visceromegalies, or retinal cherry-redpots. Neurological examination revealed bradypsychia andmnestic deficit for recent events. Cranial nerves wereormal, except for horizontal nystagmus with quick phaseollowing the direction of the gaze. There were no relevanthanges in the motor system or in sensitivity. The patientresented truncal ataxia, tremor in both hands that coulde increased voluntarily, dysarthria, and bilateral dysmetriafinger-to-nose test).
Analytical tests, including a haemogram, renal, liver,nd thyroid profiles, copper, ceruloplasmin, creatine kinase,ntineuronal antibodies, and baseline and post-exercise lac-ate levels, all yielded normal results. Blood and urine aminocid levels were normal. Results from the lysosomal enzymetudy were also unremarkable.
Brain MRI showed moderate overall cerebral and cerebel-ar atrophy.
The EEG showed slow background activity witheneralised epileptiform discharges manifesting aspike/polyspike wave complexes of variable durationhat persisted throughout the reading. Intermittent lighttimulation generated a photoparoxysmal response at lowrequencies. Epileptic activity did not increase duringtages of drowsiness.
Striated muscle biopsy revealed no structural changes;bre diameters were moderately variable. The basophilic
ntermyofibrillar network was increased by multiples fibresorming small round basophilic deposits, which were PAS-ositive according to enzymatic oxidation methods.
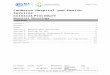
Axillary skin biopsies (Fig. 1A) showed rounded intenselyAS-positive formations in the epithelial cells of the apocrinelands and in the ducts of the eccrine glands. Both biopsiesere compatible with Lafora disease.
Disease progression was aggressive, with multiple gen-ralised tonic-clonic, myoclonic, and partial seizuresccompanied by visual symptoms that persisted in spitef treatment with several drug combinations. He lost
unctional abilities to the point of becoming completely dis-bled; nasogastric feeding was required since his frequentalatal myoclonias provoked difficulty swallowing. He pre-ented bladder and bowel incontinence and tetraparesis,Published by Elsevier España, S.L. All rights reserved.

LETTERS TO THE EDITOR
Figure 1 (A) Axillary skin biopsy (PAS stain, 2 × 400 magnifi-cation) showing round-shaped intensely PAS-positive formationsin the epithelial cells of the apocrine glands and in the ductsof the eccrine glands which correspond to Lafora bodies. (B)Heart (PAS stain, 1 × 200 magnification). Multiple Lafora bodiesin myocytes.
ar
oc
crcM
edarE
adsayec(g
bociin
acctdd
tcwsmbles
mb
eb
ooasc
585
nd became confined to bed and armchair. He died of aspi-ation pneumonia 8 years after disease onset.
Autopsy revealed typical Lafora bodies in several areasf the central nervous system (especially the thalamus anderebellum), the liver, and the heart (Fig. 1B).
The most frequent causes of PME affecting most of theases are Unverricht-Lundborg disease, Lafora disease, neu-onal ceroid lipofuscinoses, sialidosis, and mitochondrialytopathies (myoclonus epilepsy with ragged red fibres,ERRF) (Table 1).
Dr Rodríguez Lafora was the first to describe Lafora dis-ase in 1911.3 It consists of a degenerative and progressiveisorder of the central nervous system with a recessiveutosomal inheritance pattern. This disease presents no sex-elated differences and it is predominantly found in southernuropean countries.
Lafora disease is clinically characterised by gener-lised tonic-clonic seizures, myoclonias, progressive mentalecline, and pyramidal, extrapyramidal, and cerebellarigns.4 It appears at the end of childhood or duringdolescence (6 to 20 years) and it leads to death 10ears after the onset of the first symptoms. The dis-ase is caused by a homozygous EPM2 mutation linked tohromosome 6q23-25, which codifies tyrosine phosphataselaforin), a protein involved in the metabolic control oflycogen.5
From a histological point of view, it is characterisedy the presence of intracytoplasmic inclusion bodies inrgans such as the liver, heart, and brain. They are espe-ially common in biopsies of axillary skin.6 These bodiesnclude glucose polymers (polyglucosan) and their presencen an axillary skin biopsy is considered nearly pathog-omonic.
Myoclonias become continuous during waking hours; theyre resistant to antiepileptic medication and usually asso-iated with occipital lobe seizure. Occipital seizures areharacterised by simple visual hallucinations that are some-imes complex. These hallucinations are typical of Laforaisease. The onset of myoclonias coincides with progressiveeterioration of cortical function and ataxia.
On rare occasions, electroencephalographic manifes-ations may appear prior to symptom onset. They areharacterised by increasingly frequent spike- or polyspike-ave paroxysms. Subsequently, the baseline record becomes
lower and more disorganised. Paroxysms caused by inter-ittent light stimulation grow more frequent and graduallyecome continuous; photoparoxysmal response is typical atow frequencies.7 Unlike in juvenile myoclonus epilepsy,pileptic anomalies do not increase during sleep in initialtages of Lafora disease.
Brain MRI shows no relevant changes in initial and inter-ediate stages of the disease; final stages are characterisedy cerebral and cerebellar atrophy.8
In theory, doctors can offer genetic counselling andstablish a prenatal diagnosis when the genetic anomaly haseen detected in a family member.9
In conclusion, doctors should assign a suspected diagnosisf Lafora disease when a young patient (in late childhood
r adolescence) begins experiencing myoclonias followed bytaxia and progressive cognitive decline with no evidence oftructural changes in neuroimaging tests and no metabolichanges in the analytical study.10
586 LETTERS TO THE EDITOR
Table 1 Differential diagnosis of progressive myoclonic epilepsies.
Progressivemyoclonic epilepsy
Inheritance Age at onset Indicative symptoms Pathological patterns Gene
Unverricht-Lundborgdisease
AR 6—15 Slow progression, mildand late-onsetcerebellar impairment;absence of dementia
None CSTB (PME)
Lafora disease RA 6—19 Visual symptoms Polyglucosan inclusions(Lafora bodies)
EMP2A EMP2B
MERRF Maternal Any age Lactic acidosis Ragged red fibres MT-TK (tRNALys)Neuronal ceroid
lipofuscinosesAR or AD Variable Macular degeneration
and visual impairment(except in the adultform)
Lipopigment deposits:granular osmiophilic,curvilinear, orfingerprint-like shapes
Various
Sialidosis AR 8—15 Gradual cerebellarchanges; maculopathywith cherry-red spot
Oligosaccharidedeficiency in urine andneuraminidase
NEU1
onus
R
1
P
Cáceres, Spain
AD: autosomal dominant; AR: autosomal recessive; MERRF: myocl
eferences
1. Visorte A, Sardinas N, Esteban EM, Vargas-Díaz J, Novoa-López L,Rojas-Massippe E, et al. Epilepsia mioclónica progresiva: carac-terización clínica de 18 pacientes. Rev Neurol. 1999;29:102—4.
2. Berkovic SF, Andermann F, Carperter S, Wolfe LS. Progressivemyoclonus epilepsies: specific causes and diagnosis. N Engl JMed. 1986;315:296—305.
3. Pozo Alonso AJ, Pozo Lauzan D, Pozo Alonso D. Epilepsiasmioclónicas en el nino y el adolescente. Rev Cubana Pediatr.2001;73:186—93.
4. López-Meza EG, Cerda-Téllez F, Alanis-Guevara IM, FérnandezGonzález-Aragón MC, Ruano-Caldera LA. Estado epiléptico noconvulsivo asociado a enfermedad de Lafora: presentación dedos casos. Rev Neurol. 2003;37:945—7.
5. Serratosa JM, Gómez-Garre P, Gallardo ME, Anta B, de BernabéDB, Lindhout D, et al. A novel protein tyrosine phosphatase geneis mutated in progressive mioclonus epilepsy of the Lafora type(EMP2). Hum Mol Genet. 1999;8:345—52.
6. Férnandez-Armayor V, Moreno JM, Martín A, García ML,Revilla B, Moreno JL. Lafora y la neuropatología. Rev Neurol.1997;25:2036—9.
E
8
deficiency in fibroblasts
epilepsy with ragged red fibres.
7. Kobayashi K, Iyoda K, Ohtsuka Y, Ohtahara S, Yamada M.Longitudinal clinicoelectrophysiologic study of a case ofLafora disease proven by skin biopsy. Epilepsia. 1990;31:194—201.
8. Dapena M, Castilla J, Prieto JM, Lema M, Vadillo FJ, Noya M.Enfermedad de Lafora. Anatomía patológica y resonancia mag-nética. Rev Neurol. 1991;6:631—2.
9. Lesca G, Boutry-Kryza N, de Toffol B, Milh M, SteschenkoD, Lemesle-Martin M, et al. Novel mutations in EPM2A andNHLRC1 widen the spectrum of Lafora disease. Epilepsia.2010;51:1691—8.
0. Monaghan TS, Delanty N. Lafora disease: epidemiology,pathophysiology and management. CNS Drugs. 2010;24:549—61.
.E. Jiménez Caballero
Servicio de Neurología, Hospital San Pedro de Alcántara,
-mail address: [email protected]
February 2012 22 April 2012
Recommended