
Faraday Discuss. Chem. Soc., 1983, 75, 141-153
The Correspondence Principle and Intramolecular Dynamics
BY ERIC J. HELLER
Los Alamos National Laboratory, University of California, Los Alamos, New Mexico 87544, U.S.A.
Received 10th Jaiiuary, 1983
It is the purpose of this paper to describe some recent developments in the semiclassical approximations to quantum vibrationaI dynamics. In section 1 we describe a new and very simple method for finding uniformized semicIassica1 wavefunctions. The wavefunctions are given expIicitly as contractions over Gaussian functions, with the parameters of the Gaussians chosen according to an arbitrary classical trajectory. The semiclassical functions moreover are highly suitable for use as basis functions in ab initio work. In section 2 we present new resuIts for the quantum dynamics of a highly anharmonic classically chaotic system with an infinite number of quantum bound states. The results show the utility of the spectral criterion we have been advocating as a measure of phase-space flow in molecular systems, and they show some interesting quantum effects. Also, we show that the classical and quantum dynamics of a local-mode C-H stretch agree extremely well as to the fraction of available phase space covered in the course of the subsequent dynamics. In quantum- mechanical systems the optical spectrum gives a direct measure of this fraction.
1. A SIMPLE QUANTIZATION SCHEME FOR WAVEFUNCTIONS
We have two previous publications on finding multidimensional, uniformized semiclassical wavefunctions.'*2 The first made use of methods developed by Sorbie and Handy for finding semiclassical eigenvalues. Our idea was to.run the trajectory that semiclassically quantized a dynamical system, and to allow this trajectory to " guide " a Gaussian wavepacket (or, equivalently, a coherent state) as in our previous
The wavepacket was taken to be " frozen " about the trajectory, i.e. it did not spread, but followed the trajectory (both in position and momentum). Then, using the idea that
we replaced the exact quantum dynamics of v/,(x) by the moving frozen Gaussian (FG), used the classical action plus Maslov corrections for the phase of the FG, and used the semiclassical energy E of Sorbie and Handy in the Fourier transform. This technique worked extremely well (fig. l), but required that the quantizing trajectories be found. This is a tedious procedure at best, and made calculations in three and more dimensions seem forboding.
led us to conclude that the success of the Sorbie-Handy method for eigenvalues was surprising in view of the fact that it was not a topologically " proper " quantization in many cases. Instead of holding the iV topologically invariant action integrals to integer-plus-Maslov corrections, the Sorbie and Handy (S.H.) method held only the sum of the actions to the correct value,
Discussions with Marcus and Noid
Publ
ishe
d on
01
Janu
ary
1983
. Dow
nloa
ded
by U
nive
rsita
t Pol
itècn
ica
de V
alèn
cia
on 2
1/10
/201
4 22
:54:
02.
View Article Online / Journal Homepage / Table of Contents for this issue

142 CORRESPONDENCE PRINCIPLE AND INTRAMOLECULAR KINETICS
while making approximations to the individual actions. Yet we found the S.H. method worked quite well. This led us to investigate its success further, and we came upon a general technique for using arbitrary trajectories to find energies and semi- classical wavefunctions of comparable quality.2 It turned out that the S.H. method is one of a class of extrapoZation methods that work away from the true E.B.K. quantizing trajectories, but extrapolate to the E.B.K. values. This realization freed us from having to make the search for the quantized actions in the first place, but did
Fig. 1. (left) Semiclassical wavefunctions generated as a superposition of Gaussian wave- packets along a classical trajectory. (right) Numerically converged ab initio wavefunction
corresponding to the semiclassical state on the left.
require us to find the value of the independent actions for the (arbitrary) trajectories used to quantize the system. We found a new way of getting these true actions by running trajectories nearby the arbitrary trajectory. This arbitrary trajectory necessarily falls somewhere in action space near E.B.K. quantized actions, and a simple linear extrapolation to the corresponding energies and wavefunctions of those states was satisfactory. As the extrapolation distance in action space increases, the accuracy naturally decreases, but several energies and wavefunctions can be obtained from a single trajectory. The method for finding the actions is, quite importantly, independent of the topology of the trajectory. This means that classical resonances of any order present no difficulties. (Chaotic trajectories are not quantizable, how- ever, by this method.)
Here we report a similar but greatly simplified procedure to obtain semiclassical energies and uniformized semiclassical wavefunctions.6 The method works even in the quasi-integrable (partly chaotic) domain. To derive it we return to eqn (l), and project both sides of the equation into some state, Ip), which for the moment is arbitrary
The amplitude (plyE) will be non-zero only if a correct quantum eigenvalue E is used in eqn (2). We again use FGs to approximate ly,, call this ly;". This gives
This equation is none other than that used previously to obtain molecular spectra, using FG dynamic^.^.^ The idea is to examine the FG spectrum and use the energies
Publ
ishe
d on
01
Janu
ary
1983
. Dow
nloa
ded
by U
nive
rsita
t Pol
itècn
ica
de V
alèn
cia
on 2
1/10
/201
4 22
:54:
02.
View Article Online
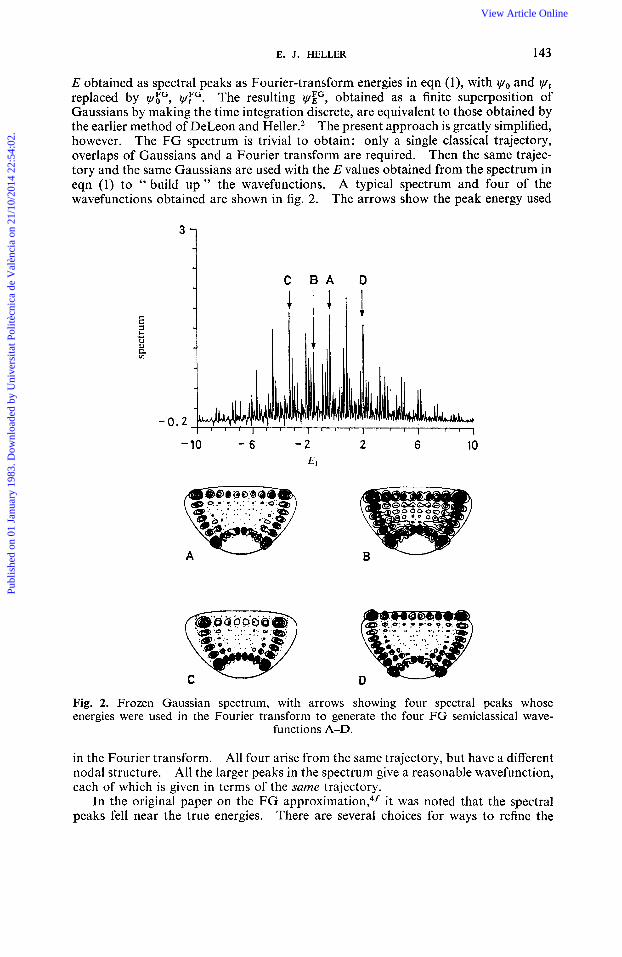
E. J. HELLER 143
E obtained as spectral peaks as Fourier-transform energies in eqn (l), with yo and yt replaced by y;", I,$". The resulting YE", obtained as a finite superposition of Gaussians by making the time integration discrete, are equivalent to those obtained by the earlier method of DeLeon and Heller.2 The present approach is greatly simplified, however. The FG spectrum is trivial to obtain: only a single classical trajectory, overlaps of Gaussians and a Fourier transform are required. Then the same trajec- tory and the same Gaussians are used with the E values obtained from the spectrum in eqn (1) to " build up " the wavefunctions. A typical spectrum and four of the wavefunctions obtained are shown in fig. 2. The arrows show the peak energy used
3 1 C B A D
-10 - 6 - 2 2 6 10 El
Fig. 2. Frozen Gaussian spectrum, with arrows showing four spectral peaks whose energies were used in the Fourier transform to generate the four FG semiclassical wave-
functions A-D.
in the Fourier transform. All four arise from the same trajectory, but have a different nodal structure. All the larger peaks in the spectrum give a reasonable wavefunction, each of which is given in terms of the same trajectory.
In the original paper on the FG a p p r o ~ i m a t i o n , ~ ~ it was noted that the spectral peaks fell near the true energies. There are several choices for ways to refine the
Publ
ishe
d on
01
Janu
ary
1983
. Dow
nloa
ded
by U
nive
rsita
t Pol
itècn
ica
de V
alèn
cia
on 2
1/10
/201
4 22
:54:
02.
View Article Online

1 44 CORRESPONDENCE PRINCIPLE AND 1NTRAMOLECULAR KINETICS
phase of ly,"", and we see from eqn (1) and (3) that we can multiply yr" by e-l(dEE'), and shift the Fourier transform energy E and dE, without changing the wavefunction t,uEc. Thus the semiclassical energies will vary according to the convention used for the phase. One convention, however, gives energies identical to those of ref, (2) without having to know the actions. The classical frequencies of motion are needed, however. These are usually easy to obtain by Fourier transform of a classical variable. Details may be found in ref. (6).
The eigenvalues, and indeed the contracted-Gaussian-sum wavefunctions are of poorer quality than if the true E.B.K. quantizing trajectories were used. However, the important advantages of this method are (1) its extreme ease of implementation and (2) the ability to use the semiclassical, Gaussian-contracted wavefunction as basis functions for solving the time-independent Schrodinger equation. These functions start out very close to true eigcnfunctions, even for very anharmonic potentials. Then a very small set of these can be used to converge on the true eigenfunctions. We have just started to explore the possibilities of this " contracted-classical Gaussian basis set " idea.
Returning to the spectrum seen in fig. 2, we note it is best to pick the large peaks in the spectrum as Fourier energies in eqn (1). The physical reason for this i s easily understood, If the initial Gaussian yrc has a large component in a particular eigen- state, that means the action variables of the Gaussian match those of the eigenstate, i.e. the guiding trajectory of the Gaussian looks like the eigenstate itself. In such a case the Gaussian superpositions can readily represent the eigenstates. Tho further away in action space that the eigenstate is from the guiding trajectory, the smaller the intensity of the spectral peak. To get all the eigenstates we need to move the guiding trajectory to several new spots in action space.
Finding the eigenvalues by this method is not really the point, however.
The basis functions are roughly of the quality shown in fig. 1 !
2. QUANTUM AND CLASSICAL ENERGY TRANSFER
For some time now, we have advocated a spectral intensity criterion 9-12 for measuring the flow of probability through the available quantum mechanical phase space. The basic ideas follow. An absorption or emission spectrum is a measurable. Any spectrum with a spread in energy corresponds to some non-stationary state, i.e. let the spectrum be
where p i are the spectral intensities, determined by
where pa is the spectral non-stationary state and II,Y,,) are the eigenstates of the system. In an electronic absorption, for example, qa would typically correspond to the ground vibrational state multiplied by the transition m~rnent .~ ' -~. Thus va is a displaced wave- packet on the upper electronic potential surface. Other qa values may be produced by varying the experimental conditions, or may be produced at will by theorists trying to understand the dynamics of a particular potential surface. In any case, the distri- bution ofpi values contains much information about the dynamics of va. Indeed, the P,* values may be used to calculate the fraction of available phase space covered in the course of the dynamics of q 3 , . I 2 By " available " phase space, we mean all those states or cells in phase space whose total energy E and energy dispersion AE match or are consistent with the mean energy E and spread AE of the state qa. This is the direct
Publ
ishe
d on
01
Janu
ary
1983
. Dow
nloa
ded
by U
nive
rsita
t Pol
itècn
ica
de V
alèn
cia
on 2
1/10
/201
4 22
:54:
02.
View Article Online

E. f. HELLER 145
analogue of classical phase-space flow. Because energy is conserved in both classical and quantum mechanics, the " available " phase is restricted by energy conservation.
A totally chaotic, or R.R.K.M. molecule, would be expected to cover 100% of its available phase space; a quasiperiodic molecule much less. However, the amount of phase space covered depends not only on the intrinsic dynamics but also on the initial state ya. Some states qa might already be distributed through much of phase space (like the metal filaments in an old-style Aash bulb) while other states pa might be more compact. Clearly, a filamentary pa is, in general, expected to cuver more phase space than the compact one, given the same dynamics. So two factors emerge as important: (1) the intrinsic dynamics and (2) the states qa used to test the dynamics (in the case of theorists) or the spectroscopic states va (avaiIable experimentally).
The key to understanding the importance of spectral intensities in elucidating energy transfer lies in the two formulae
and
where P(alb) = P(bla) is the time-averaged probability of starting in the state qa and being found later in the state q b , i.e.
Thus by measuring a spectrum (to high resolution) we can say how much time qa spends in the vicinity of its own birthplace [i.e. P(ala), eqn (6)], or we can say where in phase space it goes [P(alb) for all b].
Note that for a = b in eqn (8), the integrand is the often studied autocorrelation function P ( t ) = ](~l,lq,(,)>l*.~~.'~,~~ Contrary to the impression which seems to exist. the fundamental quantity is not the functional form ofP(t), but rather its average area:
There is a very simple interpretation for P(a\a). Suppose qa is an eigenstate. Then P(a]a) = 1. Suppose pa is an equal-amplitude sum of No eigenstates. Then
Suppose Pb is one of the eigenstates composing pa. Then also
P(alb) = I/N,,.
Publ
ishe
d on
01
Janu
ary
1983
. Dow
nloa
ded
by U
nive
rsita
t Pol
itècn
ica
de V
alèn
cia
on 2
1/10
/201
4 22
:54:
02.
View Article Online
146 CORRESPONDENCE PRINCIPLE AND INTRAMOLECULAR KINETICS
Suppose qc is in a normalized state constructed out of the No vn, but otherwise arbitrary. Then
P(alc) = l /No. (12) Thus qa spends (I/N,)th of its time in the state qa, or (l/N,)th of its time in any state pb or pc, which is made up of one or more of the No states that have intensity in the spectrum of QI,. The spectrum is
where w,, = EJh. It is as if pa has No cells to visit in phase space, and it spends equal time in each cell. It does not matter how we divide up the cells, as long as each cell has unit volume and lies within the space of No states. The point of eqn (10)-(13) is that
P(ala) 5 (no. of phase space cells visited by ya)-I = 2 (p3' n
In general we have learned to deal with non-equal amplit~des,~- '~ but the interpret- ation, eqn (14), remains the same. Eqn (14) is paramount to the realization that
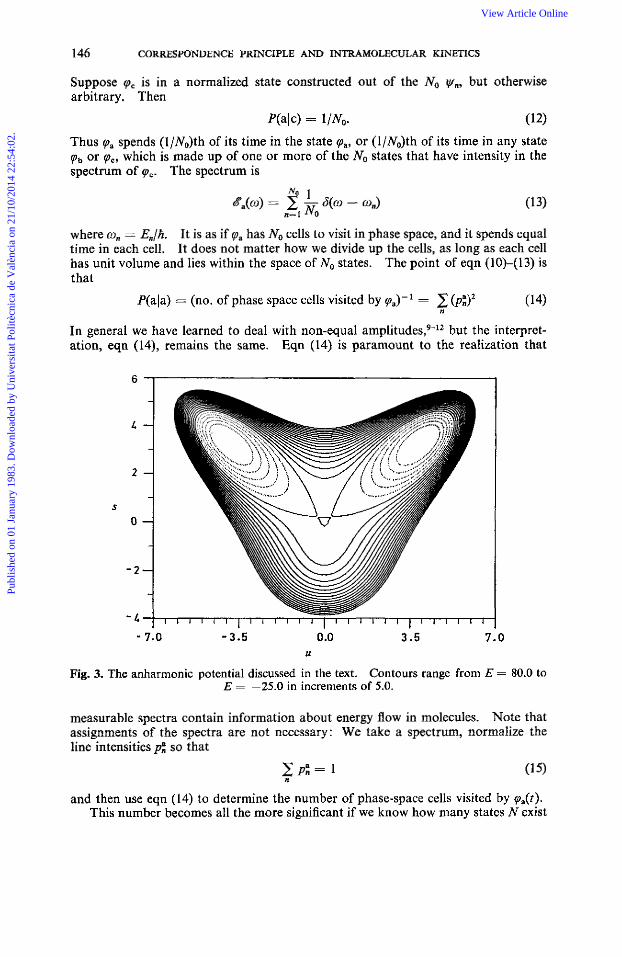
" 7.0 - 3.5 0.0 3.5 7.0 U
Fig. 3. The anharmonic potential discussed in the text. Contours range from E = 80.0 to E = -25.0 in increments of 5.0,
measurable spectra contain information about energy flow in molecules. Note that assignments of the spectra are not necessary: We take a spectrum, normalize the line intensities pi so that
and then use eqn (14) to determine the number of phase-space cells visited by y,(t). This number becomes all the more significant if we know how many states N exist
Publ
ishe
d on
01
Janu
ary
1983
. Dow
nloa
ded
by U
nive
rsita
t Pol
itècn
ica
de V
alèn
cia
on 2
1/10
/201
4 22
:54:
02.
View Article Online
E. J. HELLER 147
6
4- -
- 2 -
- s
0-
- 2 -
-
-
within a range AE of the mean energy E of the state qa (where AE is the energy dis- persion of a given pa). That is, if the No states seen in the spectrum are only a subset of those states available in the same energy regime, then we can say that pa has visited only a fraction of the available phase-space cells. This fraction is simply
F = NolN
6 -
4-
2 -
0-
- 2 -
-
-
where
PSTo(ala) = 1/N. (17)
STO stands for stochastic, because if P(ala) = PSTo(aIa), the state pa visits all N available cells. All we need to determine N is the energy uncertainty AE of qa, and the density of states B(E) of the system, so that
N = D(E)AE. (1 8) Note that in a given spectrum a Iot of missing lines (i.e. low or zero spectral intensity in certain eigenstates) implies No > N . Crudely speaking, if we look at a spectrum and see No lines in a range AE, but there are N eigenstates in AE, then F is just the
6 ,
0
- 2 :i -4 I
6 1
:1 0
- 6 -4 - 2 0 2 4 6 - 6 - 4 - 2 0 2 4 6
- 4 4 1 - 4 1 1 - 6 -4 - 2 0 2 4 6 -6 - 4 - 2 0 2 4 6
U 11
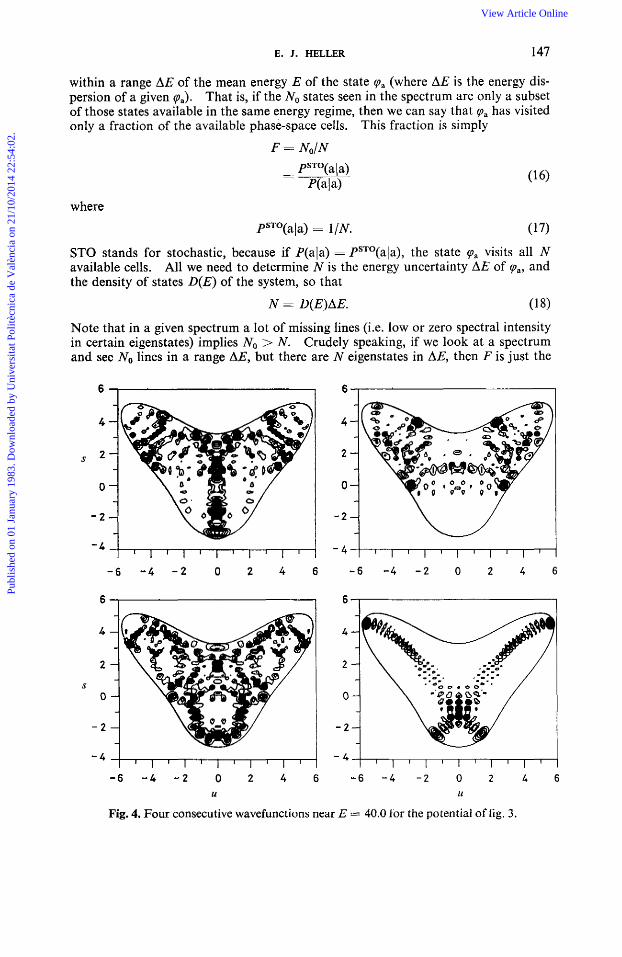
Fig. 4. Four consecutive wavefunctions near E == 40.0 for the potential of fig. 3.
Publ
ishe
d on
01
Janu
ary
1983
. Dow
nloa
ded
by U
nive
rsita
t Pol
itècn
ica
de V
alèn
cia
on 2
1/10
/201
4 22
:54:
02.
View Article Online
148 CORRESPONDENCE PRINCIPLE AND INTRAMOLECULAR KINETICS
ratio given by eqn (16). These arguments lead to the conclusion that a completely chaotic system in quantum mechanics ought to have every line present in a spectrum, as dense a spectrum as the density of states permits. The sparser the spectrum, the fewer phase-space cells sampled by the initial state.
energy
l-T-T-l
0 20 40 60 80
9 .Q
4 . 5
'00 0.0
- 4 . 5
-9.0
-4 - 2 0 2 4 S
- 7 - 6 - 5 - 4 - 3 -2 - 3 0 NFCF
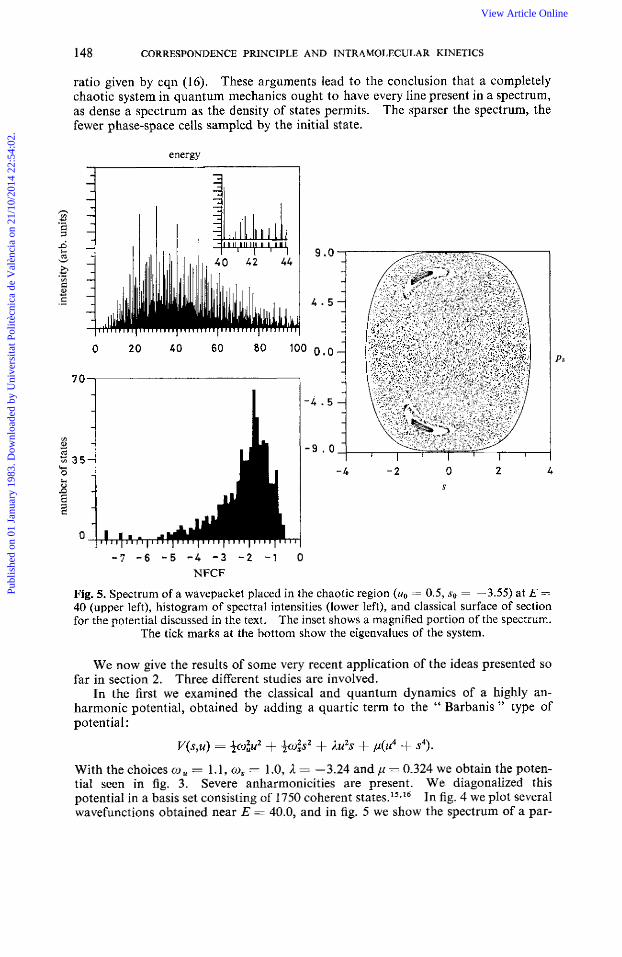
Fig. 5. Spectrum of a wavepacket placed in the chaotic region (uo = 0.5, so = -3.55) at E =1
40 (upper left), histogram of spectral intensities (lower left), and classical surface of section for the potential discussed in the text. The inset shows a magnified portion of the spectrum.
The tick marks at the bottom show the eigenvalues of the system.
We now give the results of some very recent application of the ideas presented so far in section 2.
In the first we examined the classical and quantum dynamics of a highly an- harmonic potential, obtained by adding a quartic term to the " Barbanis " type of pot en t ial :
Three different studies are involved.
V(s,u) = +w:u2 + +wfs2 + Au2s + p(u4 + d). With the choices ci), = 1 .1 , us = 1 .O, 1 = -3.24 and p = 0.324 we obtain the poten- tial seen in fig. 3. Severe anharmonicities are present. We diagonalized this potential in a basis set consisting of 1750 coherent states.'5v'6 In fig. 4 we plot several wavefunctions obtained near E = 40.0, and in fig. 5 we show the spectrum of a par-
Publ
ishe
d on
01
Janu
ary
1983
. Dow
nloa
ded
by U
nive
rsita
t Pol
itècn
ica
de V
alèn
cia
on 2
1/10
/201
4 22
:54:
02.
View Article Online
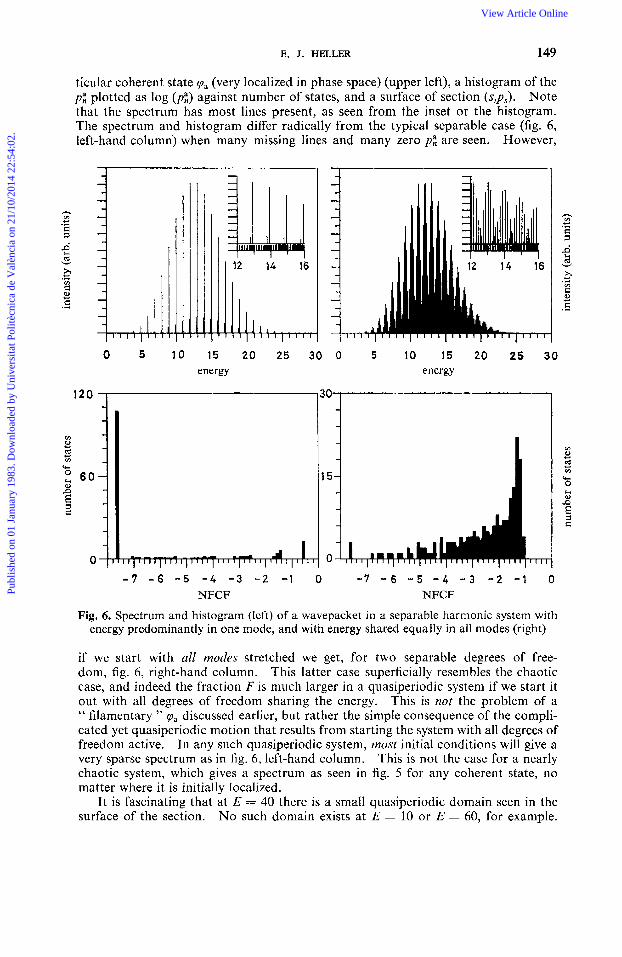
E. J. HELLER 149
ticular coherent state !pa (very localized in phase space) (upper left), a histogram of the p i plotted as log (pi) against number of states, and a surface of section (s,ps). Note that the spectrum has most lincs present, as seen from the inset or the histogram. The spectrum and histogram differ radically from the typical separable case (fig. 6, left-hand column) when many missing lines and many zero p i are seen. However,
t
15
u 12 14 16
L I 4 1 71 I
20 25 30 0 5 10 15 20 25 30 energy
120 I
x
energy
- 7 - 6 -5 - 4 - 3 - 2 - 1 0 -7 - 6 - 5 - 4 - 3 - 2 - 1 0 NFCF NFCF
Fig. 6. Spectrum and histogram (left) of a wavepacket in a separable harmonic system with energy predominantly in one rnodc, and with energy shared equally in all modes (right)
if we start with all modes stretched we get, for two separable degrees of free- dom, fig. 6, right-hand column. This latter case superficially resembles the chaotic case, and indeed the fraction F is much larger in a quasiperiodic system if we start it out with all degrees of freedom sharing the energy, This is not the problem of a " filamentary " pa discussed earlicr, but rather the simple consequence of the compli- cated yet quasiperiodic motion that results from starting the system with all degrees of freedom active. In any such quasiperiodic system, most initial conditions will give a very sparse spectrum as in fig. 6, left-hand column. This is not the case for a nearly chaotic system, which gives a spectrum as seen in fig. 5 for any coherent state, no matter where it is initially localized.
It is fascinating that at E = 40 there is a small quasiperiodic domain seen in the surface of the section. No such domain exists at E = 10 or E = 60, for example,
Publ
ishe
d on
01
Janu
ary
1983
. Dow
nloa
ded
by U
nive
rsita
t Pol
itècn
ica
de V
alèn
cia
on 2
1/10
/201
4 22
:54:
02.
View Article Online
150 CORRESPONDENCE PRINCIPLE AND INTRAMOLECULAR KINETICS
t ' I 'i
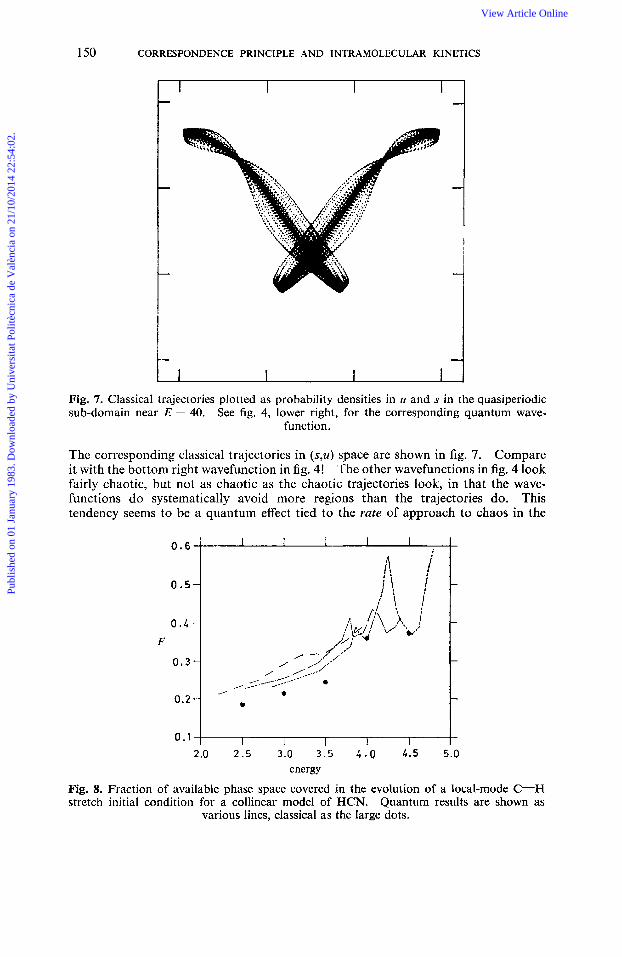
Fig. 7. Classical trajectories plotted as probability densities in u and s in the quasiperiodic sub-domain near E = 40. See fig, 4, lower right, for the corresponding quantum wave-
function.
The corresponding classical trajectories in (s,u) space are shown in fig. 7. Compare it with the bottom right wavefunction in fig. 4! The other wavefunctions in fig. 4 look fairly chaotic, but not as chaotic as the chaotic trajectories look, in that the wave- functions do systematically avoid more regions than the trajectories do. This tendency seems to be a quantum effect tied to the rute of approach to chaos in the
0.6-
0 . 5 -
0.4 - F
0.3-
0.2 -
0.1- 2.0 2 . 5 3.0 3.5 2.0 4.5 5.0
energy
Fig. 8. Fraction of available phase space covered in the evolution of a local-mode C-H stretch initial condition for a collinear model of HCN. Quantum results are shown as
various lines, classical as the large dots.
Publ
ishe
d on
01
Janu
ary
1983
. Dow
nloa
ded
by U
nive
rsita
t Pol
itècn
ica
de V
alèn
cia
on 2
1/10
/201
4 22
:54:
02.
View Article Online

E. J, HELLER 151
classical system. If the rate is slow (small Lyopanov exponent) the quantum rnech- anics may “ freeze ” at some time less than the time for complete mixing. This freezing effect is due to the finite energy-level spacing: after a time governed by the inverse of the smallest spacings, no ‘‘ new ” dynamics may occur quantum mechanic- ally. This freezing, discussed in ref. (9), may be classed as a quantum smoothing effect.17 It is consistent with the quasiperiodic state being so well reproduced.
Fig. 9. Wavefunctions showing tunnelling involving two very distinct types of wavefunctions, one involving mainly x-vibration (horizontal motion) and one y-vibration (vertical motion). For a small change in any parameter in the Hamiltonian these two states would become, separately, a vertical and a horizontal state. No single trajectory looks like the wave-
functions shown here.
The overall conclusion is that quantum mechanics is a little sluggish to respond to classical chaos, but the response is definitely there. This sluggishness has been seen before in the work of Hutchinson and Wyatt?
The second development concerns fig. 8, which shows the fraction F, determined quantum-mechanically as described above and in ref. (12) and (19), for local-mode C-H stretch initial conditions for a model HCN potential, as a function of energy of
Publ
ishe
d on
01
Janu
ary
1983
. Dow
nloa
ded
by U
nive
rsita
t Pol
itècn
ica
de V
alèn
cia
on 2
1/10
/201
4 22
:54:
02.
View Article Online

152 CORRESPONDENCE PRINCIPLE AND INTRAMOLECULAR KINETICS
the stretch. Without detailing the differences between the quantum calculations (denoted by lines), note the very good agreement with P determined from the same initial condition by classical mechanics (dots). Here we see classical and quantum mechanics agreeing very well on the dynamics of a local-mode stretch. The fraction F was obtained just from the spectrum. We emphasize it as available experimentally.
Finally, our third development has to do with quantum tunnelling. In the exten- sive wavefunction calculations we have performed recentIy we occasionally noticed a near degeneracy in the eigenvalue spectrum. Sometimes this was associated with a symmetry related tunnelling pair of' states, as discussed by Lawton and Child 2o and by us.21 Sometimes the states were not dynamically related in any way, either by symmetry or by a classical resonance condition, Two such eigenstates are shown in fig. 9. They represent tunnelling between two very different kinds of classical motion labelled crudely as vertica1 (V) and harizontaI (H). A sIight change of a parameter in the Hamiltonian would change this state (and its partner) to separate V and H states. The V and H states both look like classical trajectories and fig. 8 respresents tunnelling between these classical trajectories. In ref. (21b) we conjectured about the possible generic importance of such tunnelling.
The tunnelling has two immediate implications. One is that quantum energy flow can be enhanced over the classical, due to the tunnelling. Analysis of the HCN data l9 showed the enhancement of the quantum F seen in fig. 8, between the energies of 3.5 and 5.0, to be due to quantum-dynamical tunnelling. The second implication is that avoided crossings seen in the quantum eigenvaIue as a function of a parameter are due genericalIy to such quantum tunnelling, not to classical resonance conditions,22 We have seen many such avoidances, but none so far has been a clear-cut result of a classical resonance.
The work described herein is based on the results of coIlaboration with N. DeLeon R. Sundberg, E. Stechel and M Davis; it was performed under the auspices of the U.S. Department of Energy.
M. J. Davis and E. J. Heller, J . C h m . Phys., 1981, 75, 3916. N. DeLeon and E. J. Heller, J. Chew. Phys., in press. K. S. Sorbie and N. C. Handy, Mu/. Phys., 1977, 33, 1319; 1976, 32, 1327. (a) E. J. Heller, J . Chem. Phys., 1975, 62,1544; (b) 1978,68, 2066; ( r ) 1978,68, 3891 ; (a') 1976, 64, 63 ; (d) K. C . KuIander and E. J . HelIer, J. Chem. Phys., 1975, 69, 2439; (e) S. Y . Lee and E. J . Heller, J . Chern. P h y ~ , 1979, 71, 4777; (f) E. J. Heller, J. Chern. Phys., 1981, 75, 2923; (g) E. J. Heller, Arc. Chem Res., 1981, 14, 368; (A ) S. Y . Lee and E. J. Heller, J. Chem. Pkys. 1982, 76, 3035; (i) D. J. Tannor and E. 3. Heller, J. Chem. Phys., 1982, 77, 202. ' R. Marcus and D. Noid, personal communication. See also W. Eastes and R. A. Marcus,
J . Chem. Phys., 1974,61,4301; D. W. Noid and R. A. Marcus, J. Chem. Phys., 1975,62,2119; 1977, 67, 559. ' N. DeLeon and E. J. Heller, to be published. ' E. J. HelIer, J. Chem. Phys., 1981, 75, 2923.
The idea of Fourier transforming yr to get vE was used by M. Feit and J. Fleck, J. Cumput. P h y . ~ , 1982, 47, 412, using vt determined numerically on a grid in coordinate space. ' E. J. Heller, J. Chem. Phys., 1980, 72, 1337.
lo E. J. HeIIer and M. J. Davis, J . Phys. Chem., 1982, 86, 2118. M, J. Davis, E. B. Stechel, and E. J. Heller, Chem. Phys. Lett., 1980, 76, 21.
l2 M. J . Davis and E . J. Heller, to be published. P. Brumer and M. Shapiro, Chem. Phys. Left., 1980, 72, 528.
l4 M. Shapiru and P. Brumer, G e m . Phys. Lef t . , 1982, 90, 481 ; E. J. Heller and E. B. Stechet, Chem. Phys. Lett . , 1982, 90, 484.
l5 M. J. Davis and E. J. Heller, J. Chern. Phys., 1979, 71, 3383. The work described here is detailed in R. Sundberg and E. 3. Heller, to be published.
Publ
ishe
d on
01
Janu
ary
1983
. Dow
nloa
ded
by U
nive
rsita
t Pol
itècn
ica
de V
alèn
cia
on 2
1/10
/201
4 22
:54:
02.
View Article Online

E. J. HELLER 153
l7 M. V. Berry, N. L. Balaz and A, Voros, Ann. Phys, (N, Y.), 1979,122,26; M. V. Berry and M. Tabor, Proc. R. SOC. London, Ser. A , 1976,349, 101, J. S. Hutchinson and R. E. Wyatt, Phys. Rev. A , 1981, 83, 1567.
lished. l9 The work describcd in this paragraph is detailed in E. B. Stetchel and E. J. Heller, to be pub-
20 R. T. Lawton and M. S. Child, Mol. Phys., 1979, 37, 1799; 1980, 40, 733. 21 (a) M. J. Davis and E. J. Heller, J. Chem. Phys., 1981, 75, 246; (b) E, J . Heller and M. J . Davis,
J. Phys. Chem., 1981, 85, 307; (c) M. J. Davis, N. DeLeon and E. J. Heller, to be published. 22 D. W, Noid, M. L. Koszykowski, M. Tabor and R. A. Marcus, J . Chem. Phys., 1980,72,6169;
R. Ramaswamy and R. A. Marcus, J. Chenz. Phys., 1981, 74, 1379; 16, 1385.
Publ
ishe
d on
01
Janu
ary
1983
. Dow
nloa
ded
by U
nive
rsita
t Pol
itècn
ica
de V
alèn
cia
on 2
1/10
/201
4 22
:54:
02.
View Article Online
Recommended

















