Embed Size (px)
Citation preview

Immunodeficiency disorders
Combined immunodeficiencies
9/8/2014

Combined Immunodeficiencies
Combined immunodeficiencies /combined immunity deficiency are immunodeficiency disorders that involve multiple components of the immune system, including both humoral immunity and cell-mediated immunity

Severe combined immunodeficiencies
Combined immunodeficiency do not characteristically lead to death from overwhelming infection in the first year of life.
Mutations of a particular gene may lead to SCID or to milder combined immunodeficiency, depending upon whether the gene defect is fully penetrant or on the functional consequences of the specific mutation: amorphic (complete defect) or hypomorphic (partial defect).

In SCID neither the T cells nor the B cells work properly.
Babies with SCID can't produce IgG,so once the IgG from the mother has gone, they easily get the types of infections that antibodies are good at preventing.

Bubble Boy Disease
SCID is often called “bubble boy disease”. SCID became widely known during the 1970′s and 80′s, when the world learned of David Vetter, a boy with X-linked SCID, who lived for 12 years in a plastic, germ-free bubble.


Types
Typical SCID - describes cases with fewer than 300 autologous T cells /L.
Leaky SCID – describes cases due to incomplete mutation(s) in a typical SCID gene, T cells ranging from 300–1,500/L and may have a later age of onset of clinical symptoms.
Variant SCID – describes cases with no known gene defect and a persistence of 300–1,500 T cells/L that have impaired function.

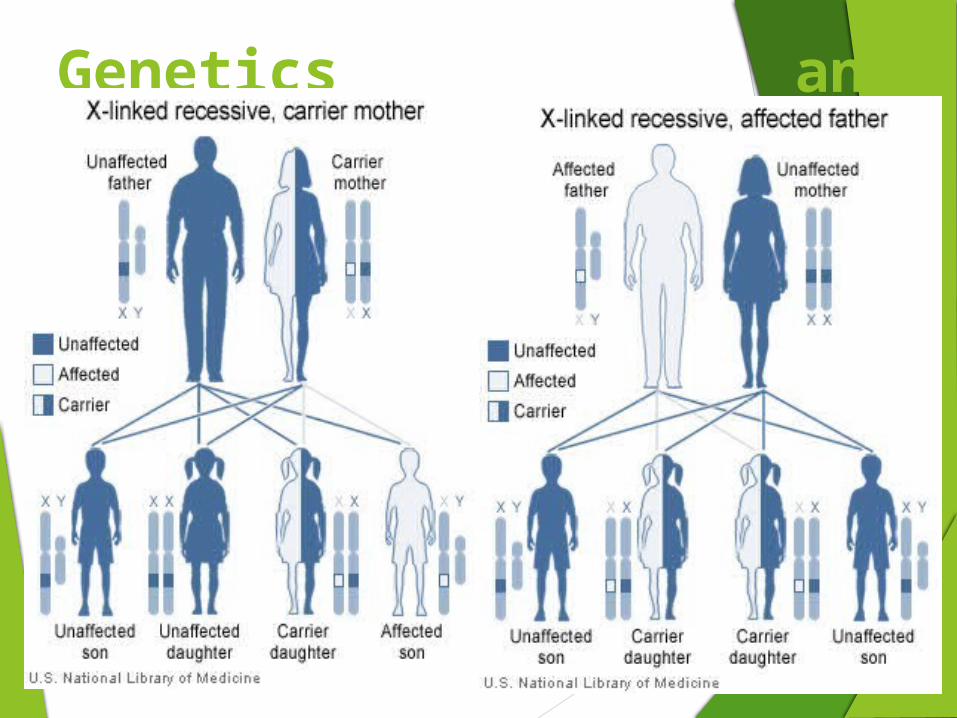
SCID is usually an inherited disorder X-linked - This means that it only affects boys but is
transmitted by their mothers, who are called 'carriers'. A daughter of a carrier mother has a 50% chance of being a carrier herself. Each son of a carrier mother has a 50% chance of being affected by the disorder.
Autosomal recessive disorders - In this situation both parents are carriers and each child, whether a girl or a boy, has a 25% chance of being affected. Sometimes the autosomal recessive form of the disease is caused by a deficiency of an enzyme called adenosine deaminase, which is found by means of a special blood test.
Genetics and Inheritance:

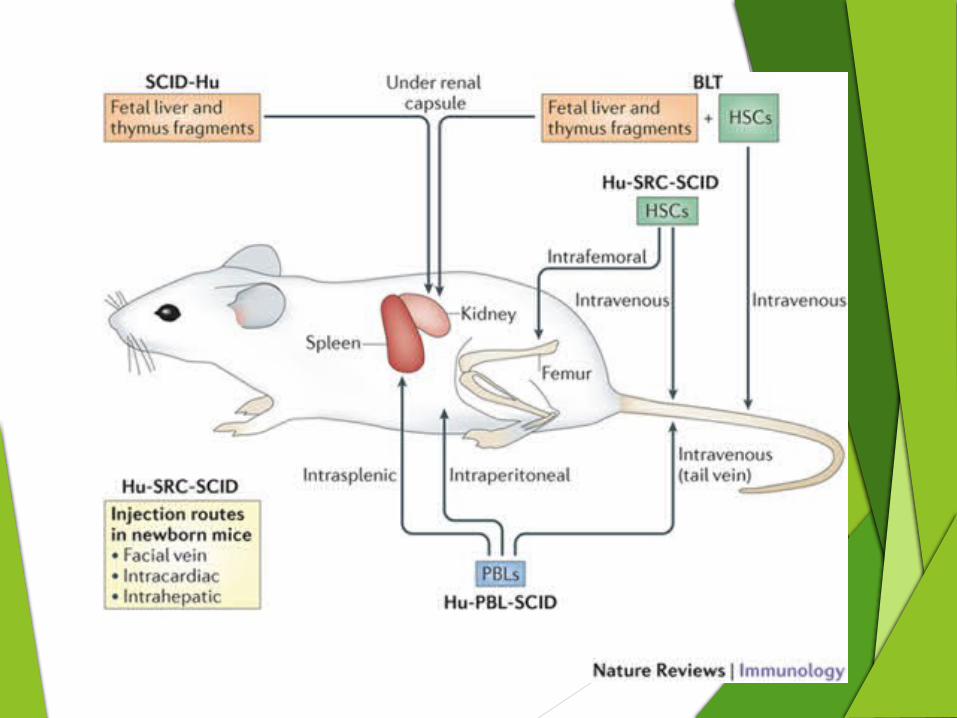
SCID Mice
In 1983, Melvin & Gayle Bosma & their colleagues describes Autosomal Recessive Mutation.
• They designated the trait SCID as it similar to human severe combined immunodeficiency.
• SCID mouse have early B & T lineage cells but absence of lymphoid cells in thymus, spleen, lymph node, & gut tissue.
Cells other than lymphocytes develop normally in SCID mouse.
The precursor T & B cell in SCID mouse unable to differentiate into mature functional B & T lymphocytes.
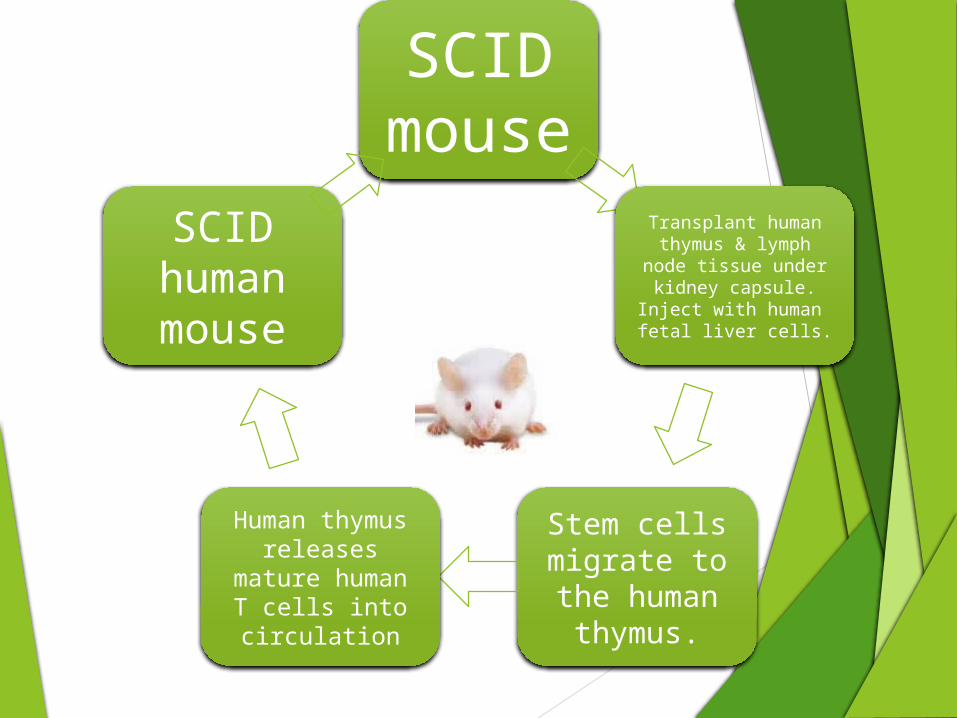
SCID mouse
Transplant human thymus & lymph
node tissue under kidney capsule.
Inject with human fetal liver cells.
Stem cells migrate to the human
thymus.
Human thymus releases mature human T cells into circulation
SCID human mouse

Defects that underlie SCID:
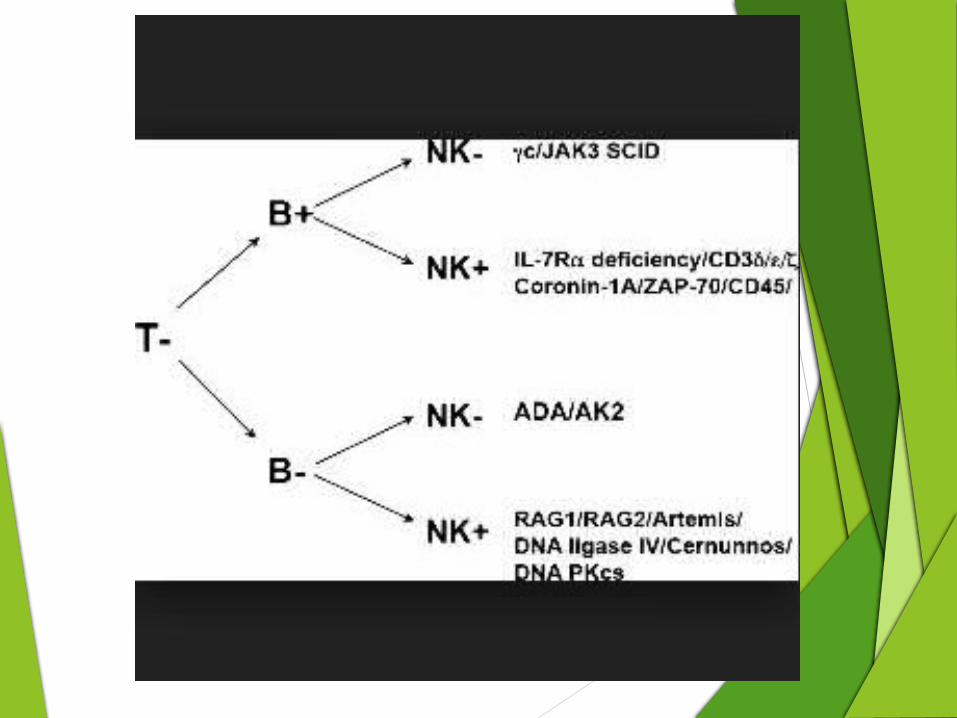

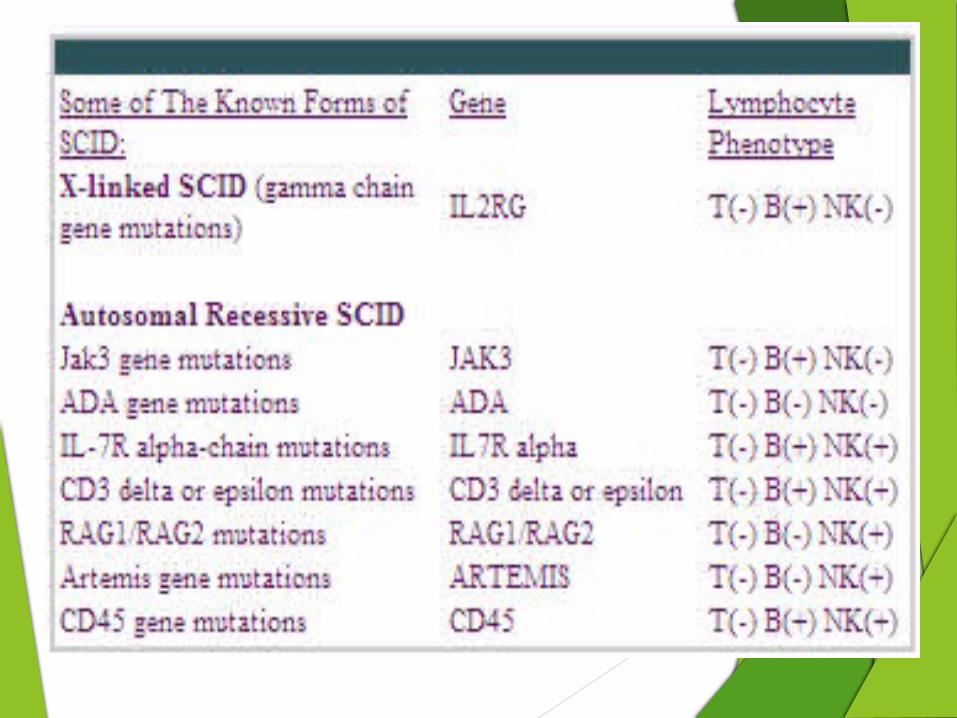
Deficiency of the Common Gamma Chain of the T-Cell Receptor (X-SCID) The most common form of SCID
A mutation in a gene on the X chromosome that encodes a component shared by the T-cell growth factor receptor and other growth factor receptors.
This component is referred to as γc, for common gamma chain.
Mutations in this gene result in very low T-lymphocyte and NK-lymphocyte counts, but the B-lymphocyte count is high .
Despite the high number of B-lymphocytes, there is no B-lymphocyte function since the B-cells have abnormal receptors for growth factors on their cell surfaces.
Only males have this type of SCID, but females may carry the gene and have a 1 in 2 chance (50%) of passing it on to each son as well as a 1 in 2 chance of passing the carrier state on to each daughter

Adenosine Deaminase Deficiency
Adenosine deaminase catalyzes conversion of adenosine to inosine, and its deficiency results in accumulation of adenosine, which interferes with purine metabolism and DNA synthesis.
ADA is essential for the metabolic function of a variety of body cells but especially T-cells.
The absence of this enzyme leads to an accumulation of toxic metabolic by-products within lymphocytes that cause the cells to die.
Babies with this type of SCID have the lowest total lymphocyte counts of all, and T, B and NK-lymphocyte counts are all very low. This form of SCID is inherited as an autosomal recessive trait. boys and girls can be affected

Lack of the ADA enzyme also leads to neurological problems such as
-cognitive impairment
-hearing and visual impairment
-low muscle tone and movement disorders.
The neurological problems are not fully curable by bone marrow transplantation

Deficiency of the Alpha Chain of the IL-7 Receptor This form of SCID is due to mutations in a gene that encodes another
growth factor receptor component, the alpha chain of the IL-7 receptor (IL-7Rα).
Infants with this type have B- cells and NK-cells, but no T-cells.
However, the B-cells do not work because of the lack of T-cells. IL-7Rα deficiency is the third most common cause of SCID accounting for 11% of SCID cases.
It is inherited as an autosomal recessive trait.
Both boys and girls can be affected.

Deficiency of Janus Kinase 3 It is a defect in the body's cytokine receptors and their signaling.
JAK3 encodes Janus kinase 3, a tyrosine kinase that belongs to the Janus family.
JAK3 functions in signal transduction and interacts with members of the STAT family.
This enzyme is necessary for function γc.
Thus, when T, B and NK-lymphocyte counts are done, infants with they are T-, B+, NK-.
Since this form of SCID is inherited as an autosomal recessive trait both boys and girls can be affected.

Deficiencies of CD3 Chains Three other forms of SCID are due to mutations in the genes that
encode three of the individual protein chains that make up another component of a group of molecules on the surface of T-lymphocytes, the T-cell receptor complex, CD3.
These SCID-causing gene mutations result in deficiencies of CD3δ, ε or ζ chains.
These deficiencies are also inherited as autosomal recessive traits

Deficiency of CD45
CD45 (lymphocyte common antigen) is a receptor-linked protein tyrosine phosphatase that is expressed on all leucocytes, and which plays a crucial role in the function of these cells.
It is is necessary for T-cell function.
This deficiency is also inherited as an autosomal recessive trait.
Mutation is due to a large deletion at one allele and a point mutation at the other.
The population of T lymphocytes is diminished and unresponsive to mitogen stimulation.
The level of B lymphocyte numbers, serum immunoglobulin decreased with age.

Other deficiencies that causes SCID

• Defects in ZAP-70 may have normal levels of immunoglobulin and CD4 lymphocytes, but their CD4 T cells are nonfunctional.
• Deficiency in the enzyme purine nucleoside phosphorylase (PNP) Impairment of this enzyme causes elevated dGTP levels resulting in T-cell toxicity and deficiency.
• Defect in the gene for the cell-surface phosphatase CD45 defect caused lack of αβT-cells. failure to transcribe the genes that encode class II MHC

Bare-lymphocyte syndrome:
Condition caused by mutations in certain genes of the major histocompatibility complex.
Without these molecules, the patient’s lymphocytes cannot participate in cellular interactions with T helper cells.
This includes defective interaction between a 5’ promoter sequence of the gene for the class II MHC molecule and a DNA-binding protein necessary for gene transcription.

Reticular dysgenesis :
• Is a rare genetic disorder of the bone marrow resulting in complete absence of granulocytes and decreased number of abnormal lymphocytes.
• Production of red blood cells (erythrocytes) and megakaryocytes (platelet precursors) is not affected.
• There is also poor development of the secondary lymphoid organ.
• The cause of reticular dysgenesis is the inability of granulocyte precursors to form granules secondary to mitochondrial adenylate kinase 2 malfunction

Clinical Presentation of Severe Combined Immune Deficiency
Children with SCID may develop infections caused by organisms or vaccines.
Among the most dangerous is an organism called Pneumocystis jiroveci, which can cause a rapidly fatal pneumonia (PCP) if not diagnosed and treated promptly.
Another dangerous organism is the chicken pox virus (varicella).
In the patient with SCID, chicken pox can be fatal because it does not resolve and can progress to cause infection in the lungs, liver and brain.
Cytomegalovirus (CMV), which nearly all people carry in their salivary glands, may cause fatal pneumonia in patients with SCID

• Other dangerous viruses for patients with SCID are the cold sore virus (Herpes simplex), adenovirus, para influenza 3, Epstein-Barr virus (EBV, the infectious mononucleosis virus), polioviruses, measles virus (rubella) and rotavirus.
• Fungal (yeast) infections in patients with SCID may be very difficult to treats such as oral thrush.
• Candida pneumonia, abscesses, esophageal infection or even meningitis may develop in patients with SCID.
• Persistent diarrhea, resulting in growth failure or mal absorption, is a common problem in children with SCID.
• Patients with SCID may also have a rash that is mistakenly diagnosed as eczema, but is actually caused by a reaction of the mother’s T-cells (that entered the SCID baby’s circulation before birth) against the baby’s tissues. This reaction is called graft-versus-host disease (GVHD).

Diagnosis
The average lymphocyte count for patients with all types of SCID is 1,500 lymphocytes (per cubic millimeter)
The most definitive test to examine the function of the T-lymphocytes is to place blood lymphocytes in culture tubes, treat them with various stimulants and then, incubate them for several days.
Normal T-lymphocytes react to these stimulants by undergoing cell division.
In contrast, lymphocytes from patients with SCID usually do not react to these stimuli

The diagnosis of SCID can also be made before the baby is born.
This can be done by molecular testing of cells from a chorionic villous sampling (CVS) or from an amniocentesis, where a small amount of amniotic fluid (which contains fetal cells) is removed from the uterine cavity.

Tests for Diagnosis
Tests used to help diagnose an immunodeficiency disorder may include:
1. Complement levels in the blood, or other tests to measure substances released by the immune system
2. HIV test
3. Immunoglobulin levels in the blood
4. Protein electrophoresis (blood or urine)
5. T (thymus derived) lymphocyte count
6. White blood cell count
7. Bead Microarray for detection of SCID.(CD3 and CD45 )

Treatment
1. Bone marrow transplants may be used to treat certain immunodeficiency conditions.
2. Passive immunity may sometimes be recommended to prevent illness after you have been exposed to bacteria or other germs.
3. Patients with hypogammaglobulinemia are treated with immunoglobulin infusions through a vein. These infusions raise blood immunoglobulin levels and protect against many infections.

4. Deficiency of the enzyme Adenosine Deaminase can sometimes be treated by replacing the missing enzyme with injections of purified enzyme, which has been specially treated. This special treatment makes the enzyme last long enough in the blood for it to work.
5. These can be replaced by immunoglobulin replacement therapy.
6. Cord blood transplants may be an alternative to bone marrow transplants

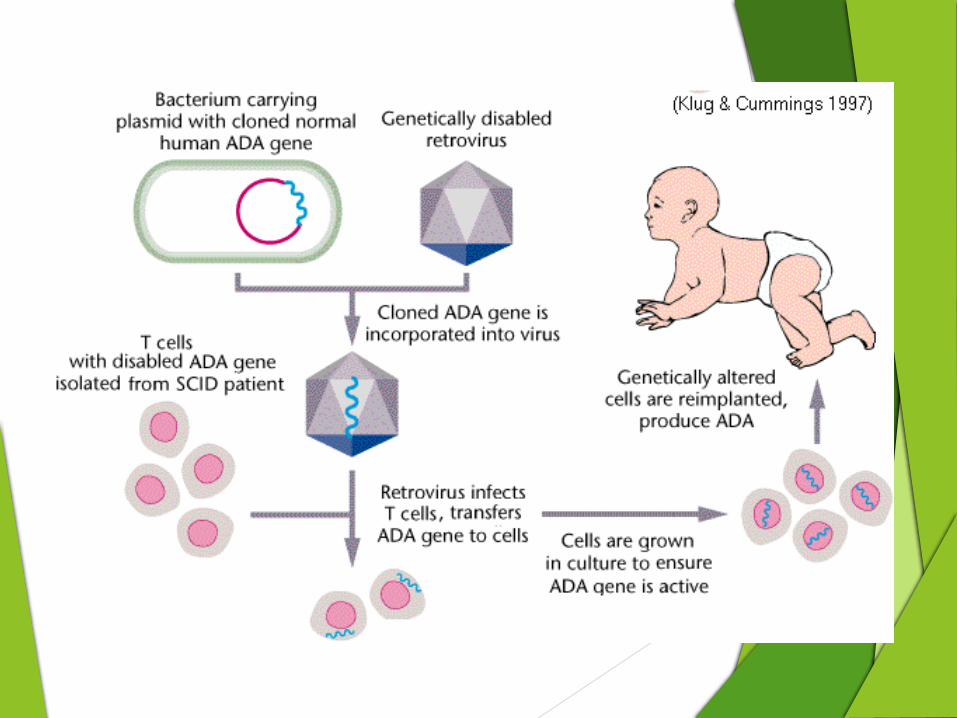
Gene therapy Recently gene therapy has been attempted as an alternative to the bone marrow transplant.
Transduction of the missing gene to hematopoietic stem cells using viral vectors is being tested in ADA SCID and X-linked SCID.
In 1990, four-year-old Ashanthi DeSilva became the first patient to undergo successful gene therapy.
Researchers collected samples of Ashanthi's blood, isolated some of her white blood cells, and used a virus to insert a healthy Adenosine Deaminase (ADA) gene into them.
These cells were then injected back into her body, and began to express a normal enzyme.
However, the concurrent treatment of ADA injections may impair the success of gene therapy, since transduced cells will have no selective advantage to proliferate if untransduced cells can survive in the presence of the injected ADA

Graft versus host disease
The bone marrow or cord blood stem cells need to come from a healthy donor with normal immune function.
The transplant cells are inevitably contaminated with T cells from the donor.
These T cells can recognise the foreignness of the patient's tissues and start to attack them (as would happen if the T cells were still in the donor and were called upon to reject an invader like a tumour, skin graft or infection).
This attack causes a condition called graft versus host disease (GVHD). A patient with GVHD might develop fever, measles like rash or diarrhoea and it can be very serious.

Prevention of GVHD(i) Selection of a donor with matching tissues (HLA matching)
(ii) T cell depletion of the donor marrow
(iii) Preventative drug treatment after the transplant (such as cyclosporine or methotrexate)

Prevention There is no known way to prevent congenital immunodeficiency
disorders. If you have a family history of immunodeficiency disorders, you might want to have genetic counselling.
Practicing safe sex and avoiding the sharing of body fluids may help prevent HIV infection and AIDS. Good nutrition may prevent acquired immunodeficiency caused by malnutrition.
Immune system disorders occur when the immune system does not fight tumours or harmful substances as it should. The immune response may be overactive or underactive.
Carrier detection and prenatal diagnosis is also available for adenosine deaminase deficient and some of the more recently identified forms of SCID.

References:
1. Kindt, Osborne, Goldsby, (2006), Kuby Immunology, 5th Ed., W. H. Freeman & Co.
2. Roitt Evan, Brostoff J. Male D. (1993) Immunology 6th Ed., Mosby & Co. London.
3. Patient and Family Handbook for The Primary Immune Diseases. Third Edition. 2001. Published by the Immune Deficiency Foundation:(http://primaryimmune.org/about-primary-immunodeficiencies/specific-disease-types)























![Immunodeficiencies [Autosaved]](https://img.pdfslide.net/doc/110x75/577cde971a28ab9e78af6d75/immunodeficiencies-autosaved.jpg)





