Embed Size (px)
Citation preview


Es una enfermedad congénita que se caracteriza porque los huesos de las personas que la sufren se
rompen muy fácilmente, tras un traumatismo mínimo e incluso sin causa aparente.
La matriz ósea contiene fibrillas anormales de colágeno tipo III y V. Los cristales de hidroxiapatita
que se depositan en la matriz no están bien alineados con respecto al eje de las fibrillas.
Se debe a la insuficiente y/o defectuosa formación del colágeno del cuerpo, como consecuencia de un
fallo genético.
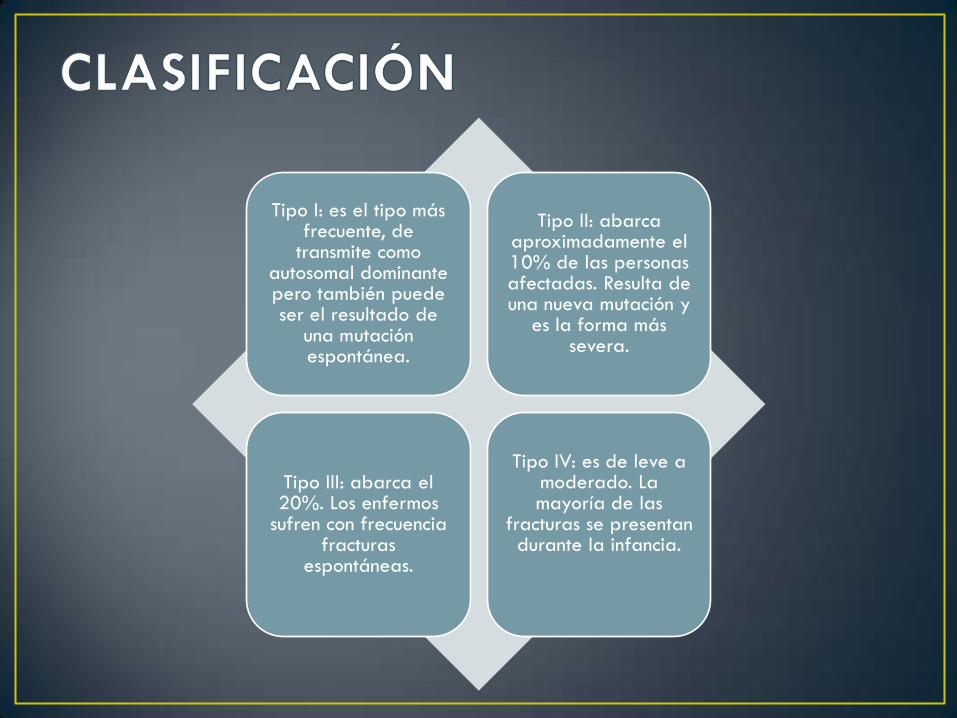
Tipo I: es el tipo más frecuente, de
transmite como autosomal dominante pero también puede ser el resultado de
una mutación espontánea.
Tipo II: abarca aproximadamente el 10% de las personas afectadas. Resulta de una nueva mutación y
es la forma más severa.
Tipo III: abarca el 20%. Los enfermos
sufren con frecuencia fracturas
espontáneas.
Tipo IV: es de leve a moderado. La mayoría de las
fracturas se presentan durante la infancia.

La compone una triada:
• Fragilidad ósea.
• Escleróticas azules.
• Sordera prematura.
Además:
• Fractura
• Deformidad de las extremidades o extremidades cortas.
• Cifosis.
• Cifoescoliosis.
• Baja estatura.
• Deformidades dentales.
• Puente nasal bajo.

Se hace por medio de estudios de colágeno que se realizan con una biopsia de perforación. Se aprecia una disminución del colágeno Tipo I
(que forma las laminillas óseas a nivel de la piel) y mayor proporción del colágeno Tipo III, en todos los tipos de osteogénesis imperfecta.
Los rayos x se convierten en una prueba frecuente y necesaria de asistencia al
diagnóstico y tratamiento.

La buena nutrición y el ejercicio supervisado
La fisioterapia y la rehabilitación pueden ser muy beneficiosas.
El implante de varillas metálicas en los huesos pueden ayudar a su fortalecimiento y a prevenir deformidades.
Bifosfonatos en los niños con Osteogénesis Imperfecta se está investigando en la actualidad con algunos resultados prometedores.
Otras incluyen el trasplante de médula ósea, el uso de la hormona del crecimiento y la terapia genética también se están investigando

• Es una enfermedad resultado de la pérdida temporal o
permanente de la entrada de sangre en los huesos.
• Sin sangre, el tejido óseo muere y causa que el hueso colapse.
• Si el proceso involucra los huesos cerca de una
articulación, normalmente lleva al colapso de la superficie de
la articulación.

El alcoholismo, uso excesivo de esteroides, síndrome de descompresión, compresión
vascular,hipertensión, vasculitis, trombosis,
daño por radiación, anemia falciforme y por la Enfermedad
de Gaucher.
ENFERMEDADES ASOCIADAS
• Gota
• Arterioesclerosis
• Diabetes
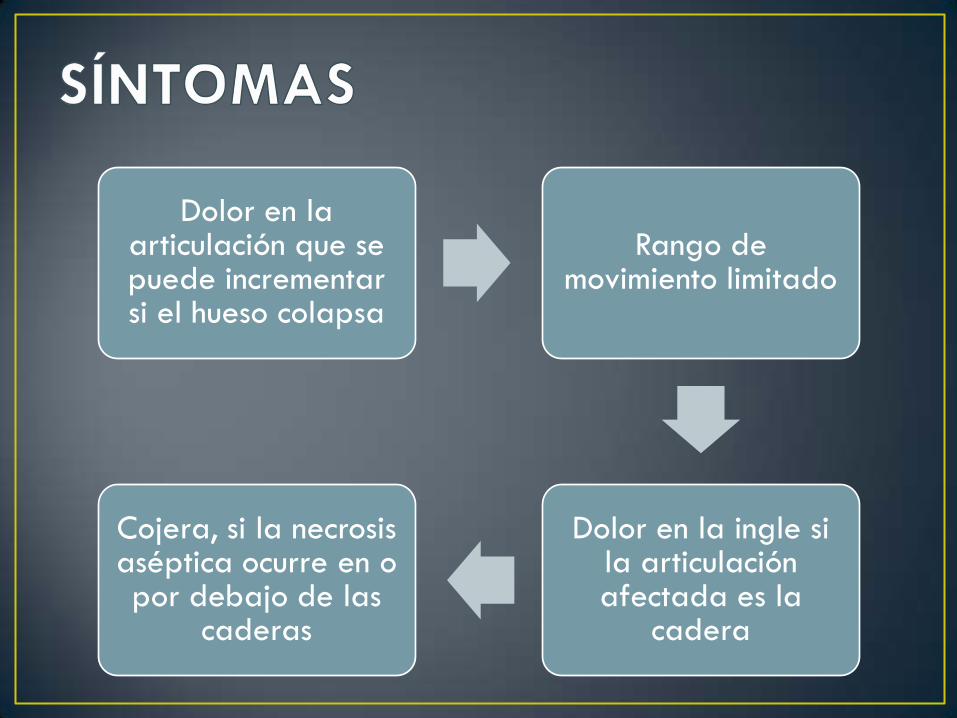
Dolor en la articulación que se puede incrementar si el hueso colapsa
Rango de movimiento limitado
Dolor en la ingle si la articulación afectada es la
cadera
Cojera, si la necrosis aséptica ocurre en o por debajo de las
caderas
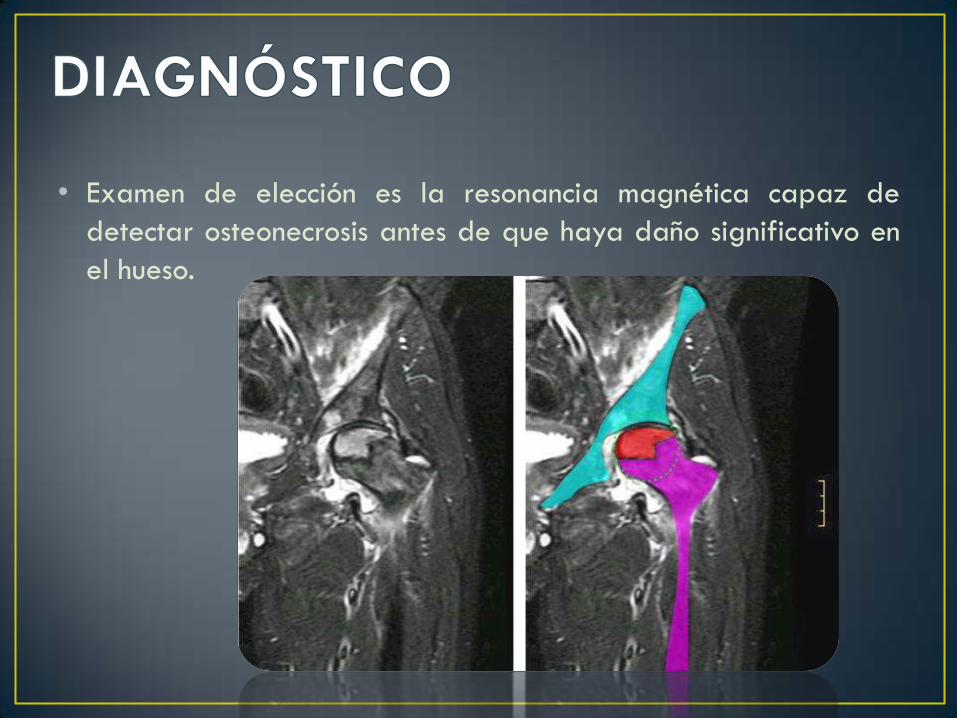
• Examen de elección es la resonancia magnética capaz de
detectar osteonecrosis antes de que haya daño significativo en
el hueso.

El más común es del reemplazo total de la cadera, por un implante prostético.
Un nuevo y más prometedor tratamiento donde no se remueve todo el hueso, sino que se preserva. En éste sólo la cabeza del fémur es retirada a
diferencia del reemplazo completo donde todo el cuello es retirado.
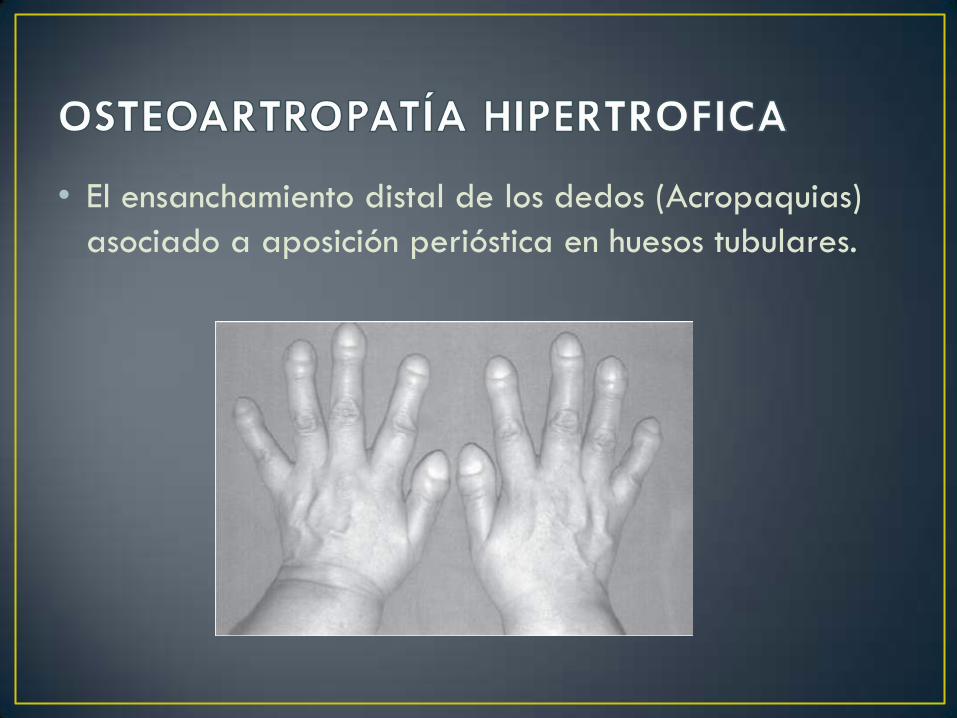
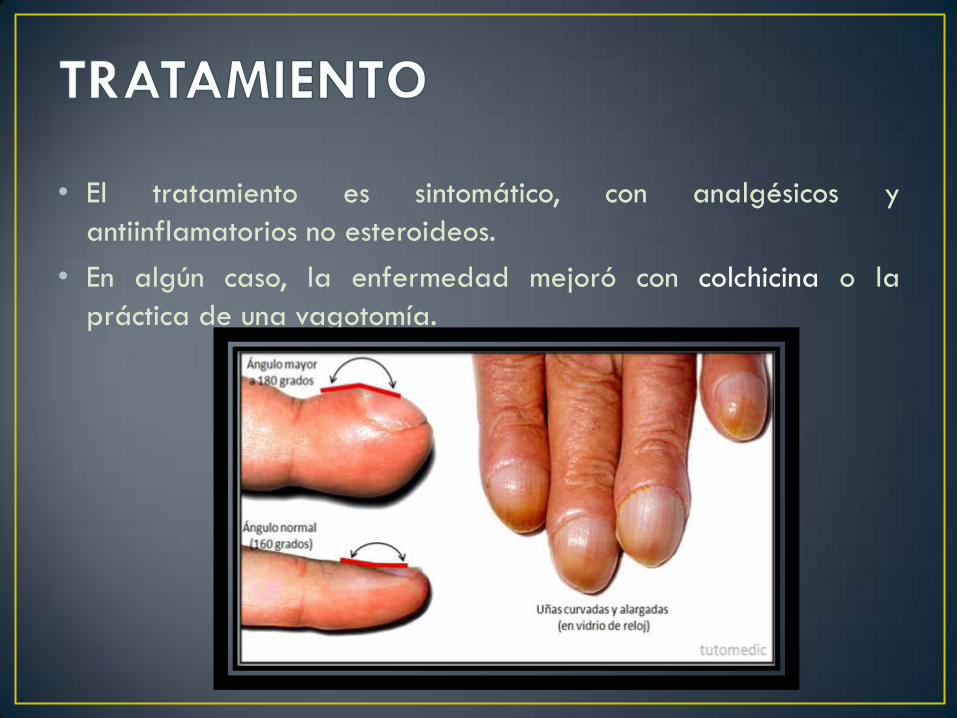
• El ensanchamiento distal de los dedos (Acropaquias)
asociado a aposición perióstica en huesos tubulares.

Primaria puede presentarse de forma aislada (idiopática) o ser familia.
Secundaria puede ser localizada o generalizada y se asocia a múltiples enfermedades. En niños se trata sobre todo de cardiopatías congénitas, infecciones pulmonares y metástasis pulmonares de osteosarcoma.
La mayoría de los pacientes no refieren síntomas (excepto los
relacionados con su enfermedad de base).
Otros pueden presentar dolor óseo en extremidades inferiores
acentuado por la posición en declive y que se alivia al elevar los miembros. En algunos casos
hay derrame articular.
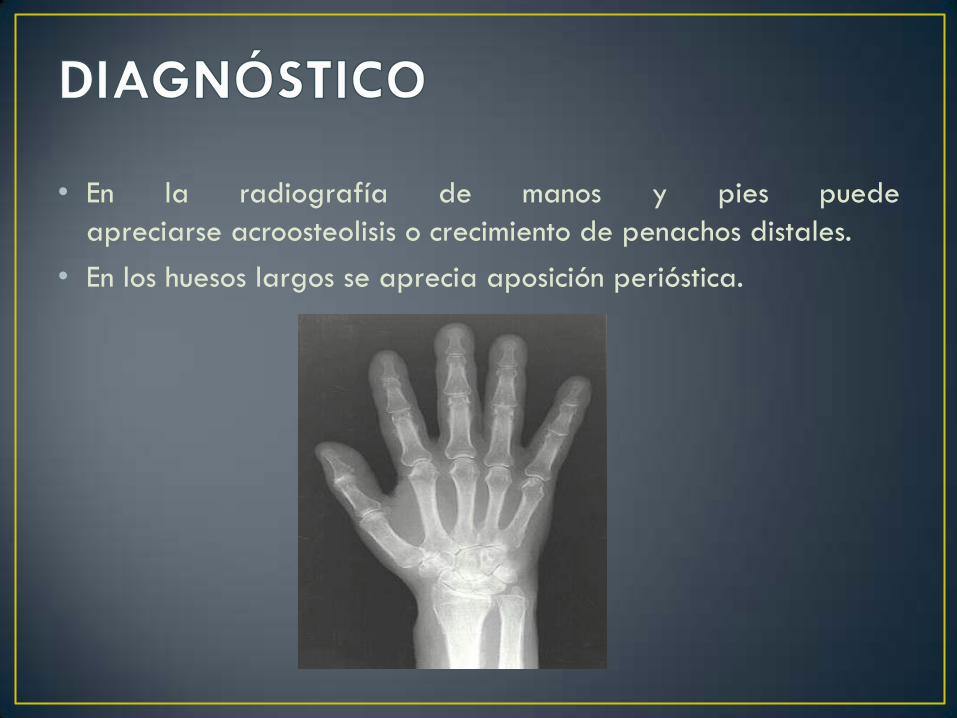
• En la radiografía de manos y pies puede
apreciarse acroosteolisis o crecimiento de penachos distales.
• En los huesos largos se aprecia aposición perióstica.
• El tratamiento es sintomático, con analgésicos y
antiinflamatorios no esteroideos.
• En algún caso, la enfermedad mejoró con colchicina o la
práctica de una vagotomía.
• Es un trastorno óseo circunscrito
que suele afectar zonas difusas
del esqueleto y que se debe a un
mayor remodelamiento del hueso
• Es iniciado por una hiperactividad
en la resorción ósea osteoclástica,
a la que le sigue un aumento
compensador de la formación de
hueso nuevo osteoblástico.
• Se desconocen las causas de la
enfermedad de Paget, pero las
evidencias apoyan tanto causas
genéticas como víricas
• Los patrones familiares de la
enfermedad en varios parientes a
gran escala son compatibles con un
patrón autosómico dominante y
una penetrancia variable en la
herencia.
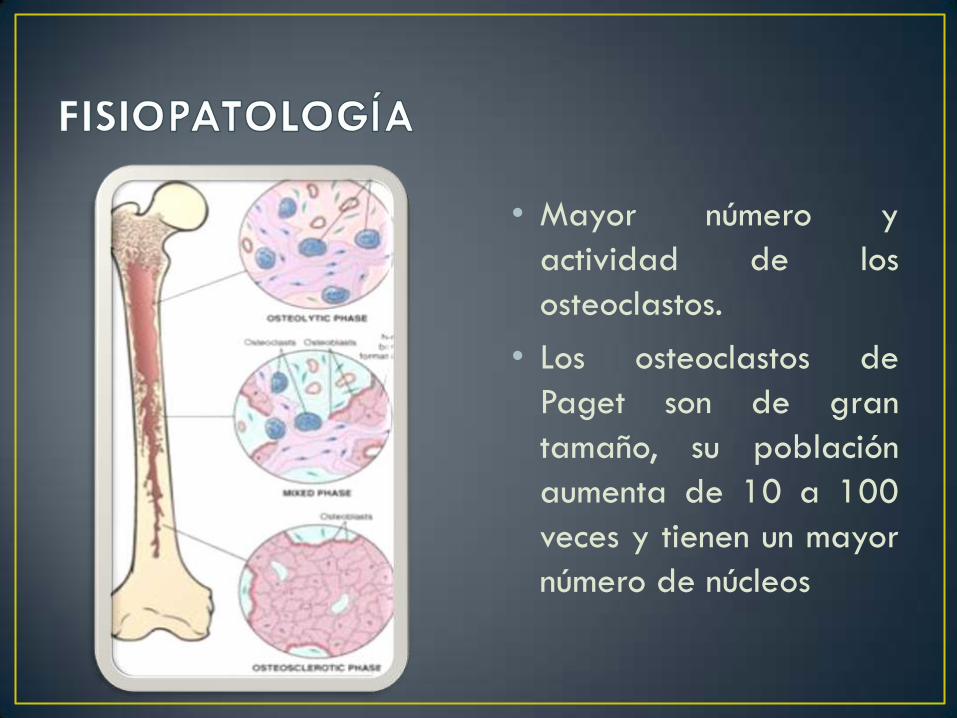
• Mayor número y
actividad de los
osteoclastos.
• Los osteoclastos de
Paget son de gran
tamaño, su población
aumenta de 10 a 100
veces y tienen un mayor
número de núcleos
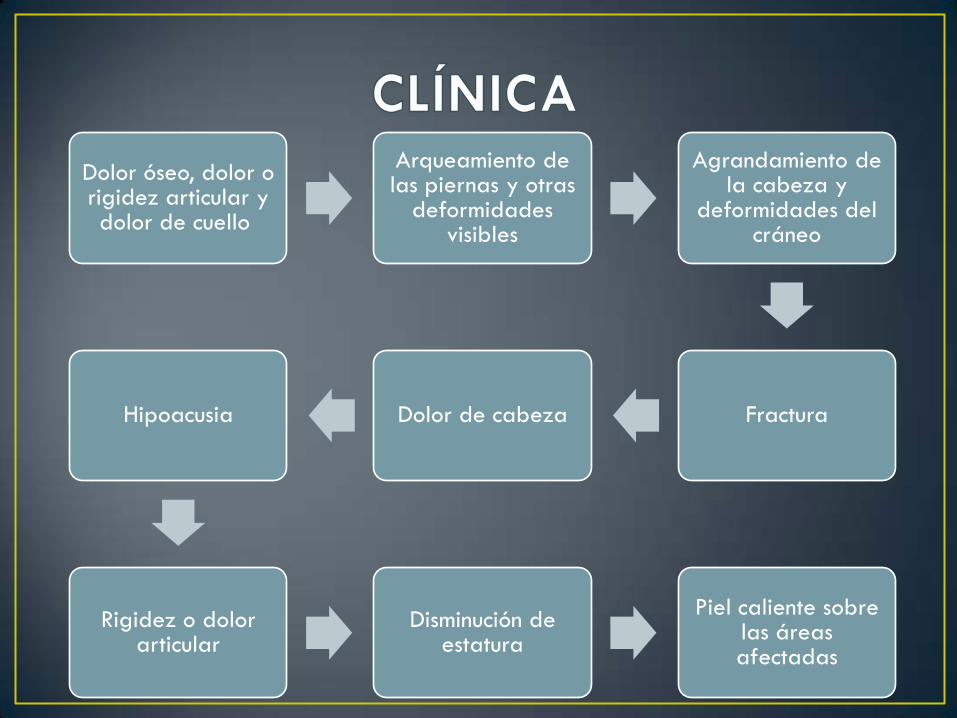
Dolor óseo, dolor o rigidez articular y dolor de cuello
Arqueamiento de las piernas y otras
deformidades visibles
Agrandamiento de la cabeza y
deformidades del cráneo
FracturaDolor de cabezaHipoacusia
Rigidez o dolor articular
Disminución de estatura
Piel caliente sobre las áreas afectadas
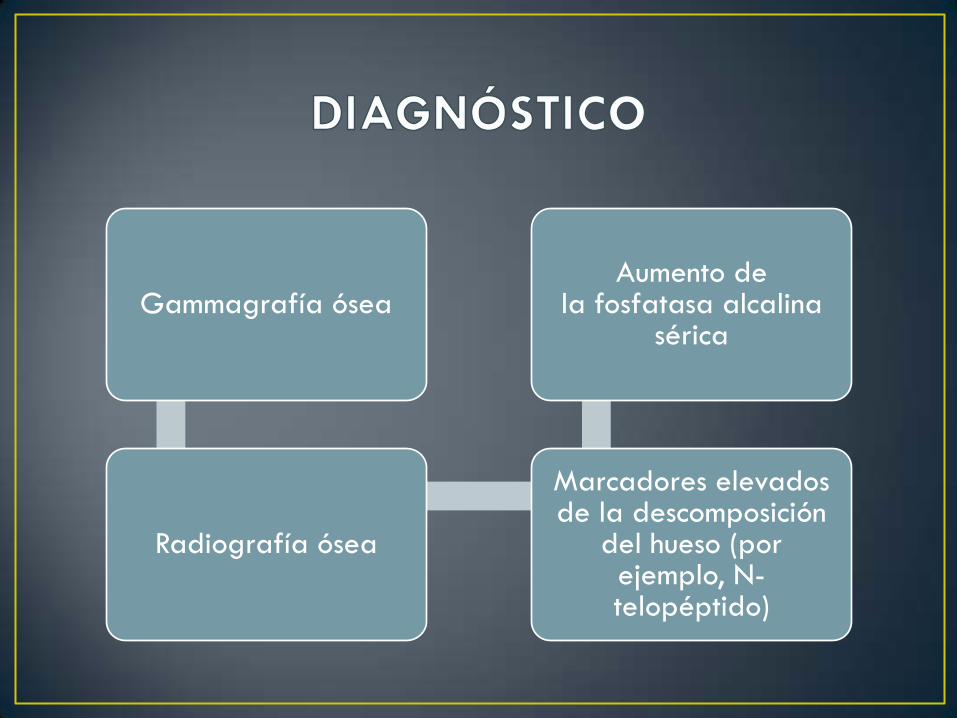
Gammagrafía ósea
Radiografía ósea
Marcadores elevados de la descomposición
del hueso (por ejemplo, N-telopéptido)
Aumento de la fosfatasa alcalina
sérica

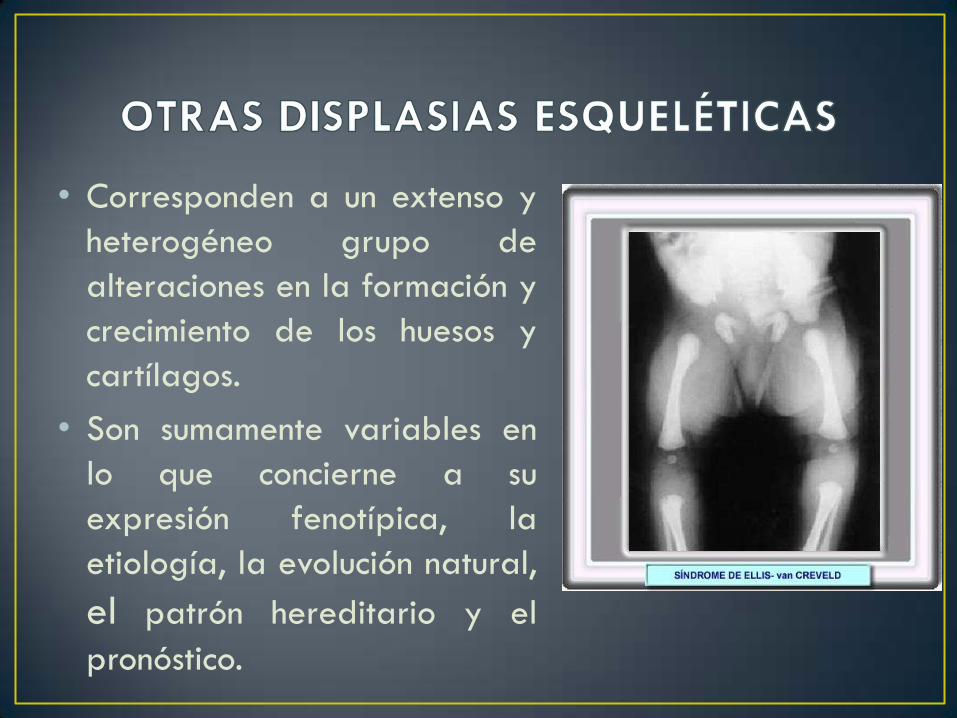
• Corresponden a un extenso y
heterogéneo grupo de
alteraciones en la formación y
crecimiento de los huesos y
cartílagos.
• Son sumamente variables en
lo que concierne a su
expresión fenotípica, la
etiología, la evolución natural,
el patrón hereditario y el
pronóstico.
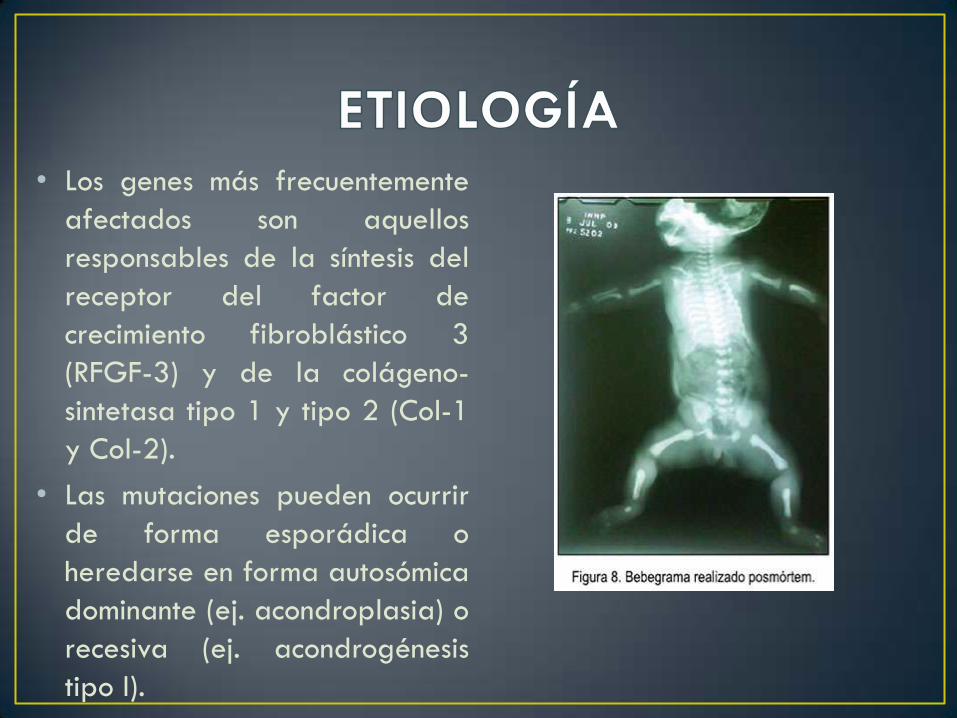
• Los genes más frecuentemente
afectados son aquellos
responsables de la síntesis del
receptor del factor de
crecimiento fibroblástico 3
(RFGF-3) y de la colágeno-
sintetasa tipo 1 y tipo 2 (Col-1
y Col-2).
• Las mutaciones pueden ocurrir
de forma esporádica o
heredarse en forma autosómica
dominante (ej. acondroplasia) o
recesiva (ej. acondrogénesis
tipo I).

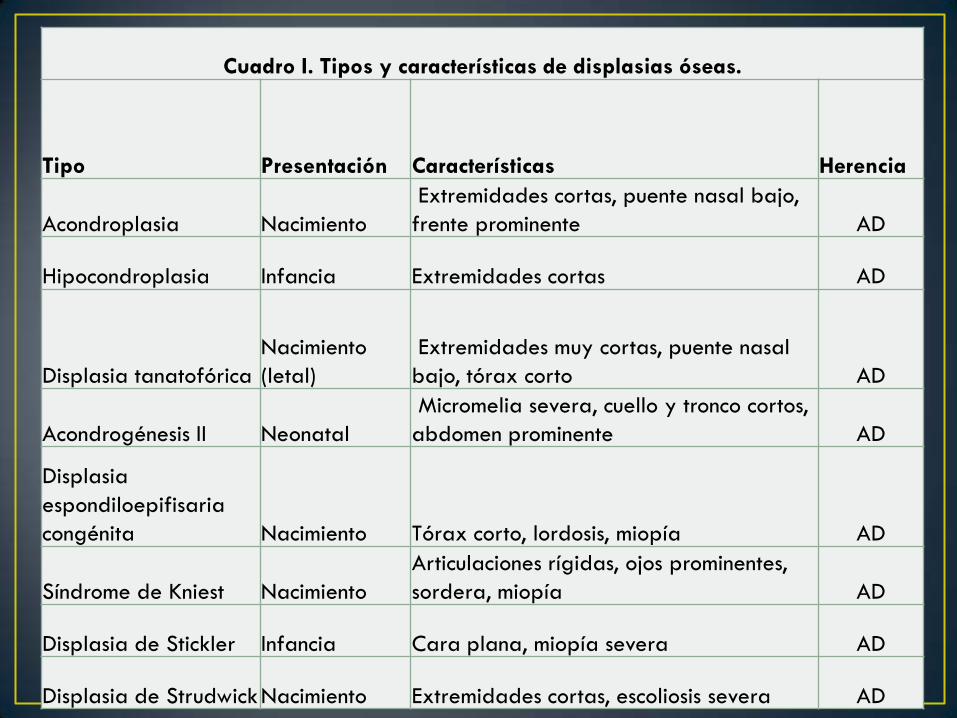
Cuadro I. Tipos y características de displasias óseas.
Tipo Presentación Características Herencia
Acondroplasia Nacimiento
Extremidades cortas, puente nasal bajo,
frente prominente AD
Hipocondroplasia Infancia Extremidades cortas AD
Displasia tanatofórica
Nacimiento
(letal)
Extremidades muy cortas, puente nasal
bajo, tórax corto AD
Acondrogénesis II Neonatal
Micromelia severa, cuello y tronco cortos,
abdomen prominente AD
Displasia
espondiloepifisaria
congénita Nacimiento Tórax corto, lordosis, miopía AD
Síndrome de Kniest Nacimiento
Articulaciones rígidas, ojos prominentes,
sordera, miopía AD
Displasia de Stickler Infancia Cara plana, miopía severa AD
Displasia de Strudwick Nacimiento Extremidades cortas, escoliosis severa AD
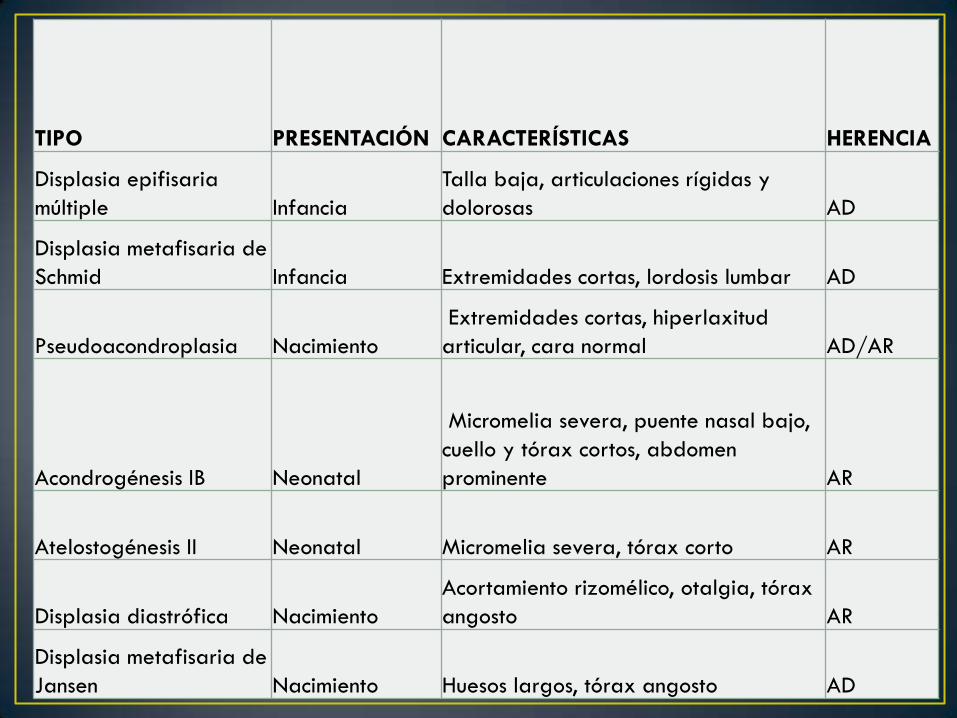
TIPO PRESENTACIÓN CARACTERÍSTICAS HERENCIA
Displasia epifisaria
múltiple Infancia
Talla baja, articulaciones rígidas y
dolorosas AD
Displasia metafisaria de
Schmid Infancia Extremidades cortas, lordosis lumbar AD
Pseudoacondroplasia Nacimiento
Extremidades cortas, hiperlaxitud
articular, cara normal AD/AR
Acondrogénesis IB Neonatal
Micromelia severa, puente nasal bajo,
cuello y tórax cortos, abdomen
prominente AR
Atelostogénesis II Neonatal Micromelia severa, tórax corto AR
Displasia diastrófica Nacimiento
Acortamiento rizomélico, otalgia, tórax
angosto AR
Displasia metafisaria de
Jansen Nacimiento Huesos largos, tórax angosto AD

No hay un tratamiento específico comprobado
Se pude utilizar el alargamiento óseo en los niños
de corta edad pero esto requiere de una total
disposicion por parte de los padres y el infante.






























