Embed Size (px)
Citation preview

SÍNDROME DE PRADER-WILLI
Trastorno congénito No heredado
No relacionado con sexo, raza o condición de
vida
Incidencia 1 por cada 10,000 nacidos
Por deleciones en una región concreta del
brazo largo del cromosoma 15 del
padre

CausaPérdida de los genes de a región 15q11-q13 del padre, los procedentes de la madre con inactivados por imprinting.
AUSENCIA DE LA FUNCIÓN DE LOS GENES DE DICHA REGIÓN.• Deleción de novo de la copia paterna
en la división celular.• La copia de la madre se inactiva por imprinting.
70%
• Ambas copias de la región provienen de la madre por alteración en la división celular Disomía uniparental materna.
• El imprinting desactiva ambas copias.
25%
• Debido a una alteración del imprinting los genes del padre son identificados como procedentes de la madre y son inactivados.
• HEREDADA
3-4%

Manifestaciones clínicasPeriodo fetal y
neonatal
• Movimientos fetales disminuidos
• Problemas de la alimentación
• Llanto débil o ausente
• Hipotonía axial (nuca&tronco) , distonía en extremidades
• Saliva espesa• Hipoplasia genital, criptorquidia
• Peso y talla bajos
Lactante y niño pequeño
• Falta de medro• Retraso del desarrollo psicomotor y del lenguaje
• Rasgos faciales característicos• Pelo claro• Ojos azules
Escolar
• Apetito voraz, obesidad
• Talla corta (respuesta disminuida GH), manos y pies pequeños, escoliosis (genu valgo&pies planos)
• Rascado descontrolado, autolesiones
• Caries• Somnolencia diurna excesiva
• Sensibilidad alterad a la temperatura
• Estrabismo
Adolescente
• Cataplejía, pseudocrisis
• Desarrollo sexual secundario incompleto
• Carácter obsesivo, problemas comportamentales
• Incapacidad de independencia personal
• Hipogonadismo
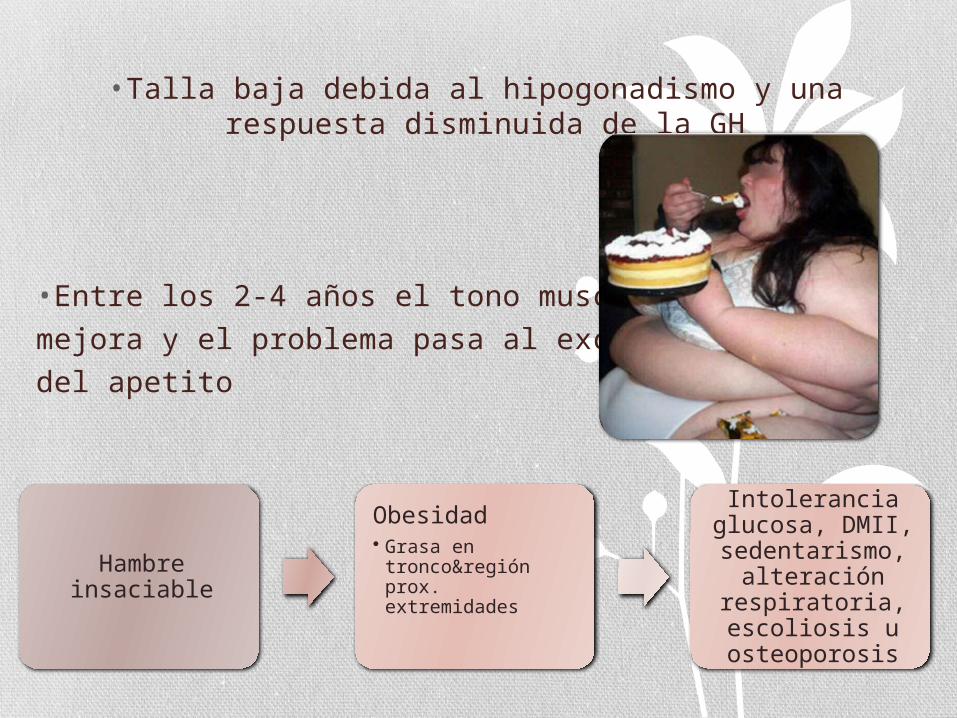
• Talla baja debida al hipogonadismo y una respuesta disminuida de la GH
• Entre los 2-4 años el tono muscular
mejora y el problema pasa al exceso
del apetito
Hambre insaciable
Obesidad • Grasa en
tronco®ión prox. extremidades
Intolerancia glucosa, DMII, sedentarismo,
alteración respiratoria, escoliosis u osteoporosis

• El desarrollo puberal no alcanza un estadio adulto, es habitual la amenorea primaria.
No son fértiles.
• Alteración de la arquitectura del sueño• Retraso del comienzo• Despertares frecuentes• Aumento número de ciclos REM-NO REM• Fragmentación sueño REM

• Trastornos del sueño relacionados con la obesidad y con una disfunción hipotalámica• Ronquido
• Taquipnea
• Apnea del sueño
• Anomalías congénitas• Hexadactilia
• Displasia de caderas
• Malformaciones de los pies
• Craneosintosis
• Reflujo urinario
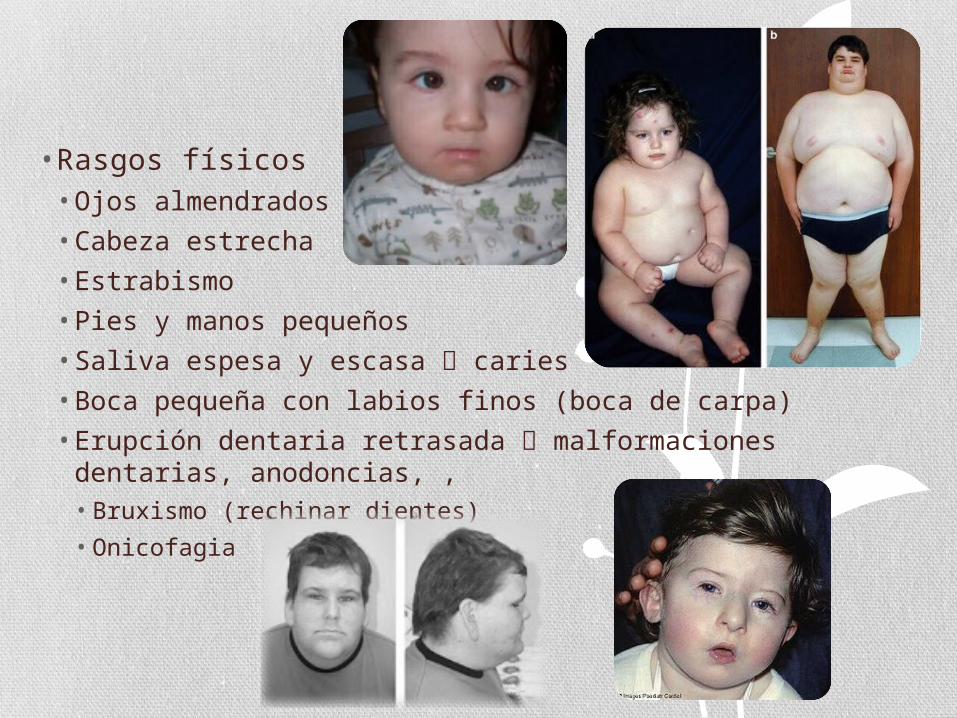
• Rasgos físicos• Ojos almendrados• Cabeza estrecha• Estrabismo• Pies y manos pequeños• Saliva espesa y escasa caries• Boca pequeña con labios finos (boca de carpa) • Erupción dentaria retrasada malformaciones dentarias,
anodoncias, , • Bruxismo (rechinar dientes)
• Onicofagia

Características cognitivas• CI varía entre 30-105
• Presentan discapacidad intelectual de ligera a moderada• Carencia estrategias de solución de problemas
• Frágil metacognición• Abstracción inefectiva
• Razonamiento inferencial deficiente• Pobre aplicación de reglas
Porcentaje Discapacidad intelectual
C.I.
5% C.I. normal >85
27% C.I. límite 70-85
34% R.M. leve 55-69
Lenguaje y hablaDebido a hipotonía. Varia tipo
y severidad
Pobreza del
vocabulario
Primeras palabras >2 años
Grafía de mala
calidad

Características conductuales• Sus limitaciones cognitivas tienen impacto sobre su conducta. Por su deficit de procesamiento secuencial da reacciones de ansiedad, frustración, rigidez, irritabilidad
Infancia
• Rabietas• Se molestan con
facilidad• Extravertidos• Tercos
Adolescencia
• Interacción social pobre• Obsesiones/
perseverancia• Hurtos• Agresividad• Comen de más
Edad adulta
• Interacción social pobre• Trastornos obsesivos• Explosiones• Hurtos• Mentiras• Agresividad• Síntomas psicóticos• Depresión, tristeza• Ansiedad• Comen de más
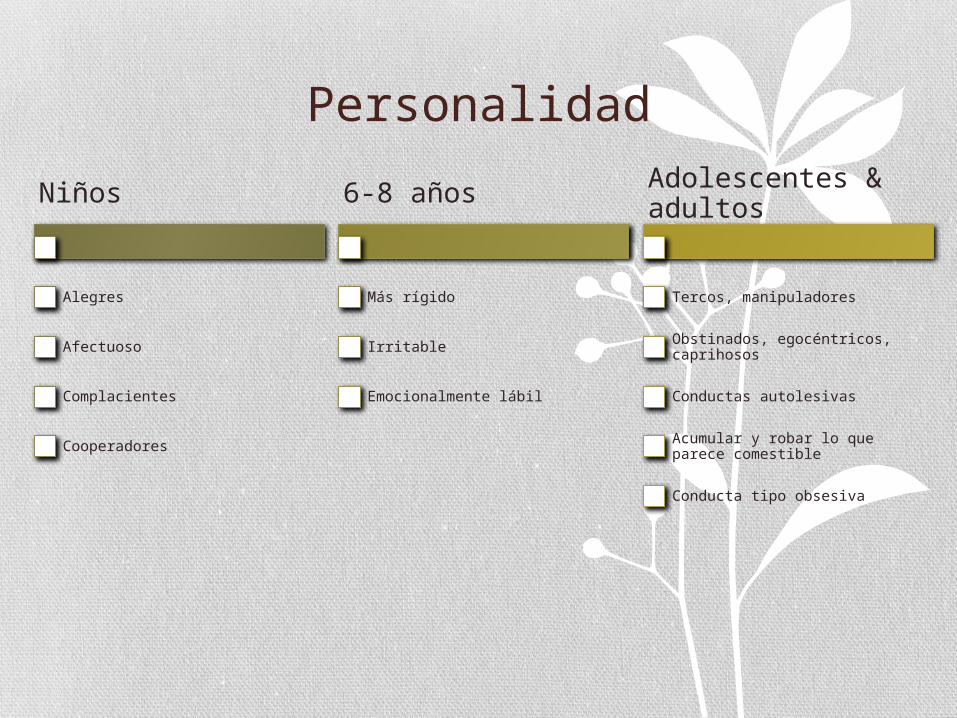
Personalidad
Niños
Alegres
Afectuoso
Complacientes
Cooperadores
6-8 años
Más rígido
Irritable
Emocionalmente lábil
Adolescentes & adultos
Tercos, manipuladores
Obstinados, egocéntricos, caprihosos
Conductas autolesivas
Acumular y robar lo que parece comestible
Conducta tipo obsesiva

diagnostico
Los criterios para el Dx se basan en las
características clínicas y
citogenéticas del síndrome agrupadas
en criterios principales y secundarios
• Para establecer el Dx de SPW en niños <3años se requieren 5 pts, de los cuales 4 deben pertenecer a criterios principales
• En >3 años se necesitan 8 pts de los que al menos 5 corresponden a criterios principales
Deben tenerse en cuenta otras características como:• Alto umbral de dolor• Dificultad para vomitar• Temperatura inestable• Sensibilidad alterada a la
temperatura• Escoliosis• Adrenarquia precoz• Osteoporosis• Entre otros
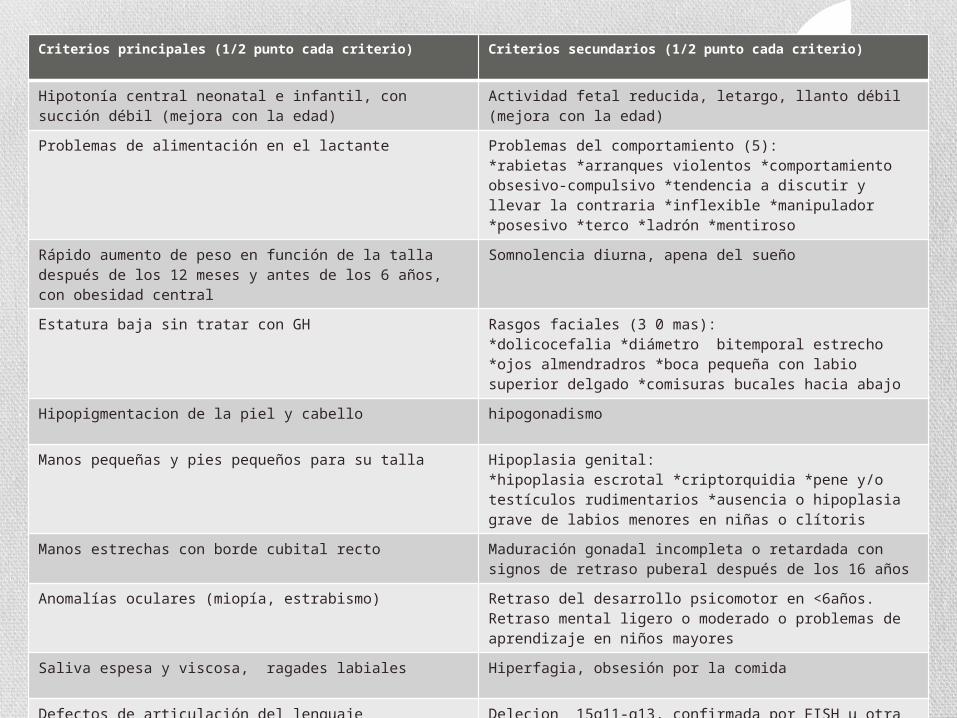
Criterios principales (1/2 punto cada criterio) Criterios secundarios (1/2 punto cada criterio)
Hipotonía central neonatal e infantil, con succión débil (mejora con la edad)
Actividad fetal reducida, letargo, llanto débil (mejora con la edad)
Problemas de alimentación en el lactante Problemas del comportamiento (5):*rabietas *arranques violentos *comportamiento obsesivo-compulsivo *tendencia a discutir y llevar la contraria *inflexible *manipulador *posesivo *terco *ladrón *mentiroso
Rápido aumento de peso en función de la talla después de los 12 meses y antes de los 6 años, con obesidad central
Somnolencia diurna, apena del sueño
Estatura baja sin tratar con GH Rasgos faciales (3 0 mas): *dolicocefalia *diámetro bitemporal estrecho *ojos almendradros *boca pequeña con labio superior delgado *comisuras bucales hacia abajo
Hipopigmentacion de la piel y cabello hipogonadismo
Manos pequeñas y pies pequeños para su talla Hipoplasia genital:*hipoplasia escrotal *criptorquidia *pene y/o testículos rudimentarios *ausencia o hipoplasia grave de labios menores en niñas o clítoris
Manos estrechas con borde cubital recto Maduración gonadal incompleta o retardada con signos de retraso puberal después de los 16 años
Anomalías oculares (miopía, estrabismo) Retraso del desarrollo psicomotor en <6años. Retraso mental ligero o moderado o problemas de aprendizaje en niños mayores
Saliva espesa y viscosa, ragades labiales Hiperfagia, obsesión por la comida
Defectos de articulación del lenguaje Delecion 15q11-q13, confirmada por FISH u otra anomalía como disomia uniparental materna
Rascarse las heridas o auto-provocarlas

Técnicas de dx genético
A partir de sangre periférica del paciente y de los padres
-Estudio citogenetico: análisis del cariotipo y se busca presencia de delecion en 15q11-q13 (FISH)
-Análisis molecular del ADN: por estudio de microsatelites para determinar la procedencia materna o paterna y la delecion -Análisis de metilación: identifica principales alteraciones asociadas a SPW y confirma el Dx cuando muestra el patrón materno
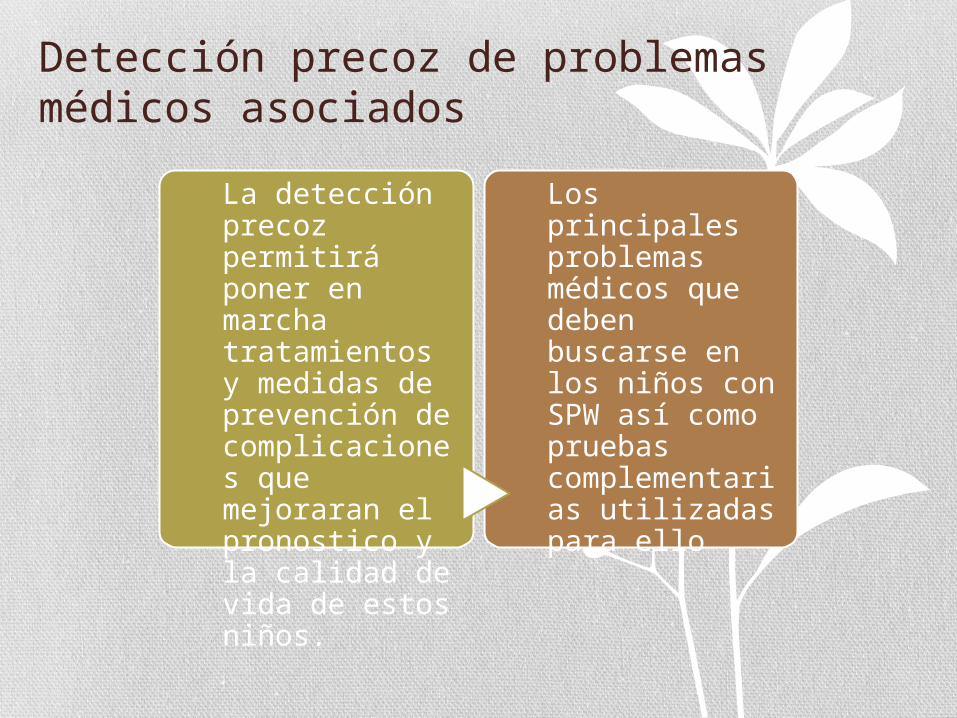
Detección precoz de problemas médicos asociados
La detección precoz permitirá poner en marcha tratamientos y medidas de prevención de complicaciones que mejoraran el pronostico y la calidad de vida de estos niños.
Los principales problemas médicos que deben buscarse en los niños con SPW así como pruebas complementarias utilizadas para ello

problema Pruebas complementarias
Déficit de GH Test de provocación de secreción de GH, secreción nocturna de GH y niveles séricos de IGF-1 y IGFBP-3
Intolerancia a la glucosa DM tipo I
Niveles séricos de glucosa en ayunas, tolerancia oral a la glucosa y test de hemoglobina glucosilada
criptorquidia Ecografía abdominal, RM abdominal, niveles séricos de testosterona test de HCG
hipogonadismo Niveles séricos de testosterona (niños), estrógenos (niñas) y respuesta a LH/FSH
osteoporosis densitometria
Trastornos respiratorios de sueño
Monitorización de parámetros cardiorrespiratorios
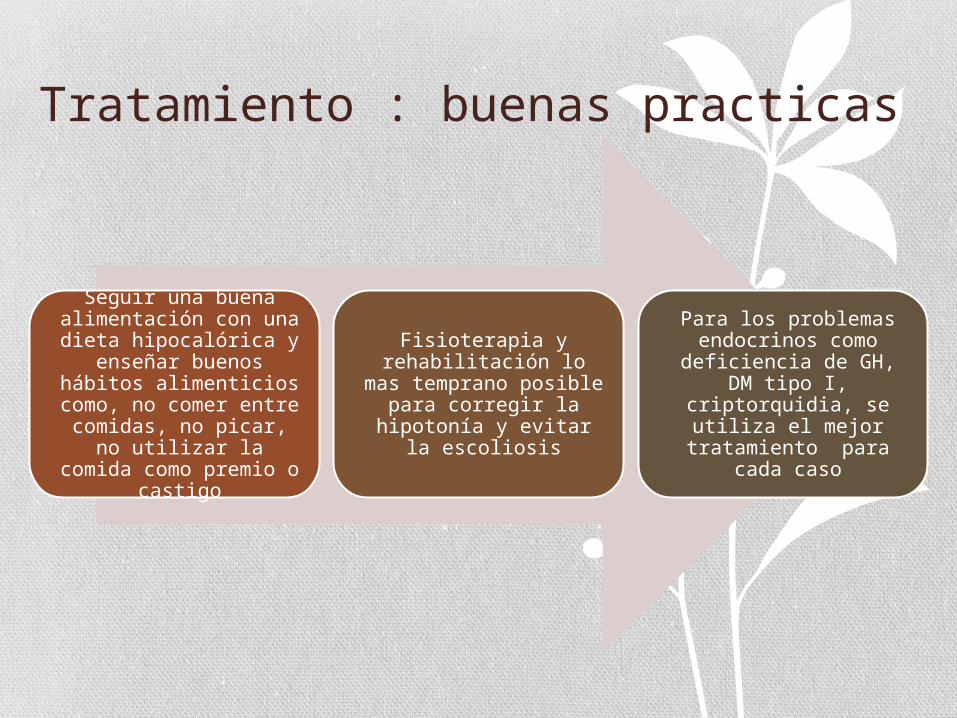
Tratamiento : buenas practicas
Seguir una buena alimentación con una dieta hipocalórica y
enseñar buenos hábitos alimenticios como, no
comer entre comidas, no picar, no utilizar la
comida como premio o castigo
Fisioterapia y rehabilitación lo mas
temprano posible para corregir la hipotonía y
evitar la escoliosis
Para los problemas endocrinos como
deficiencia de GH, DM tipo I, criptorquidia, se
utiliza el mejor tratamiento para cada
caso

CASO CLÍNICO

Al nacimiento• RN sexo masculino• Madre 27 años primer hijo• Adecuado control prenatal• Polihidramnios inductores para la maduración pulmonar iniciando trabajo de parto prematuro & sangrado transvaginal• Cesárea 32 semanas• Peso: 1,620 gramos• Apgar: 8-9• Piel clara, ojos azules, dolicocefalia, hipotonía muscular, criptorquidia bilateral• Se inició alimentación con sonda orogástrica

Primeras semanas
• Tono muscular disminuido y letargia• Perfil tiroideo normal• Sin succión aún a la tercera semana• Tamiz metabólico: conc alta de galactosa 6.8 U/g

Primer mes
• Peso 2,630 gramos• Toleraba succión por V.O.• Accuscreen con potenciales auditivos evocados con respuesta a partir de 60 dB• Cariotipo on microdeleción en 15q11-q13• Prueba FISH confirmó la microdeleción intersitical en los brazos largos y específicamente en la región 15q11-q13

Discusión
• Es la causa más común de obesdad de origen genético• Pocos niños se diagnostican en etapa neonatal• Edad media para la detección 36 meses• Es importante reconocer la disomía uniparental• Deleción proveniente del padre C.I. en el 60% >70• Disomía uniparental materna 9.5% con C.I. > 70

Conclusión• Este síndrome debe de ser considerado en el neonato que se presente con: • Hipotonía muscular• Pobre succión• Retraso en el desarrollo psicomotor y su crecimiento coporal• Características físicas del síndrome
Entre mas temprano se detecten los casos mejor será el pronóstico del niño.

Bibliografía• del barrio, jose. Sindrome de prader willi. file:///C:/Users/Usuario/Downloads/spw.pdf
• Hernandez, ricardo. Sindrome de prader willi en paciente con hipotonia muscular. file:///C:/Users/Usuario/Downloads/casoclinicospw.pdf





























