Embed Size (px)
Citation preview

THE NEUROMUSCULAR
JUNCTION DISORDERS
part 1
Dr Sudhakar Marella

PARTS OF NMJ
The anatomy of NMJ consist of following parts:
1. Pre-synaptic membrane
2. Synaptic cleft
3. Post-Synaptic membrane
4. Contractile apparatus
• The nerve is separated from the surface of the
muscle by a gap of about 20nm called junctional
cleft.
• Presynaptic membrane contains prejunctional
acetylcholine receptors and active zone.
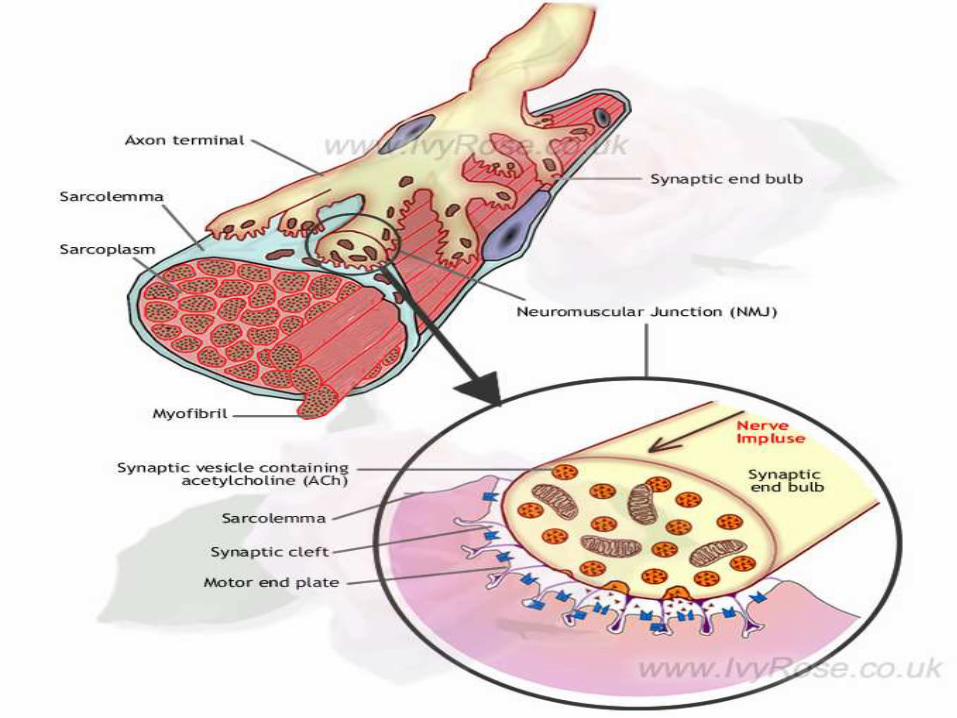

Synaptic cleft: Lies Between the muscle endplate
and nerve terminal which are held in tight alignment by
basal lamina.
Post synaptic membrane – acetylcholine
receptors: At the post synaptic membrane the area
overlying the nerve terminal is called muscle end plate.
The membrane here is thrown into primary and
secondary clefts.
• At the shoulder of these clefts numerous acetylcholine
receptors are present.

The acetylcholine receptors are nicotinic and are of
following types
a. Junctional or mature
b. Extra junctional or immature
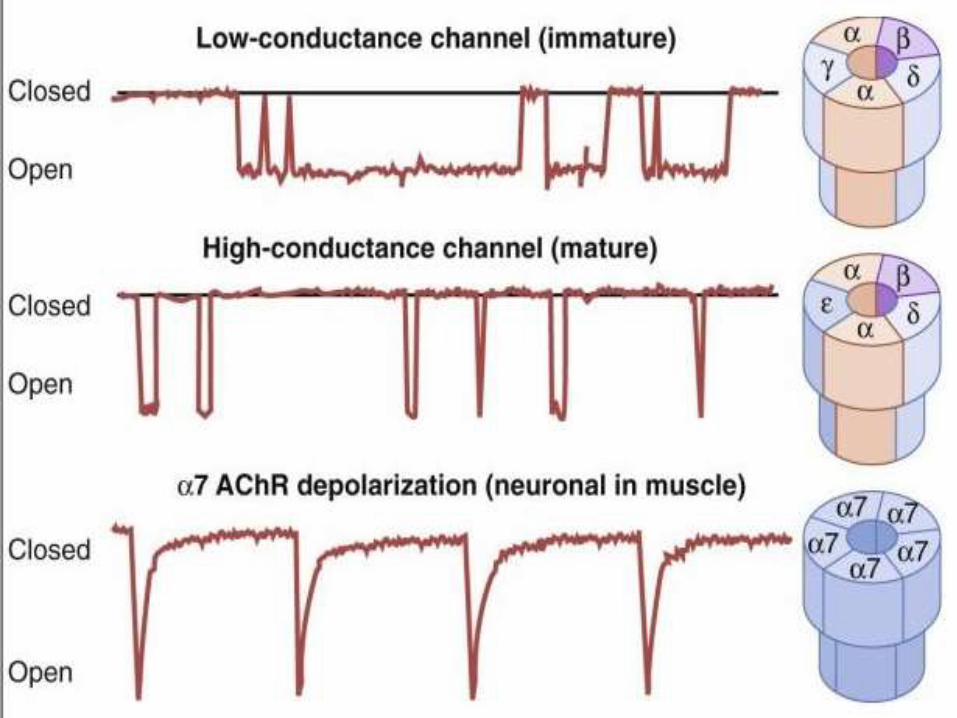

• Nicotinic receptors are broadly classified into two
subtypes based on their primary sites of expression:
1. muscle-type nicotinic receptors and
2. neuronal-type nicotinic receptors.
• In the muscle-type receptors, found at the neuromuscular
junction, receptors are either the embryonic form,
composed of α1, β1, δ, and γ subunits in a 2:1:1:1 ratio,
or the adult form composed of α1, β1, δ, and ε subunits
in a 2:1:1:1 ratio
• The neuronal subtypes are various homomeric or
heteromeric combinations of twelve different nicotinic
receptor subunits: α2 through α10 and β2 through β4

Acetyl choline receptors/Post junctional receptors:
• Present in the post junctional membrane of the motor
end plate & are of nicotinic type. These receptors exist
in pairs.
• It consists of protein made up of 1000 amino acids,
made up of 5 protein subunits designated as alpha, beta,
delta and epsilon joined to form a channel that
penetrates through and projects on each side of the
membrane.
• An average human end plate contains about 15 to 40
million acetylcholine receptors

• Each receptor has central funnel shaped core which is
an ion channel, 4 nm in diameter at entrance narrowing
to less than 0.7nm within the membrane.
• The receptor is 11 nm in length and extends 2nm into
the cytoplasm of the muscle cell.
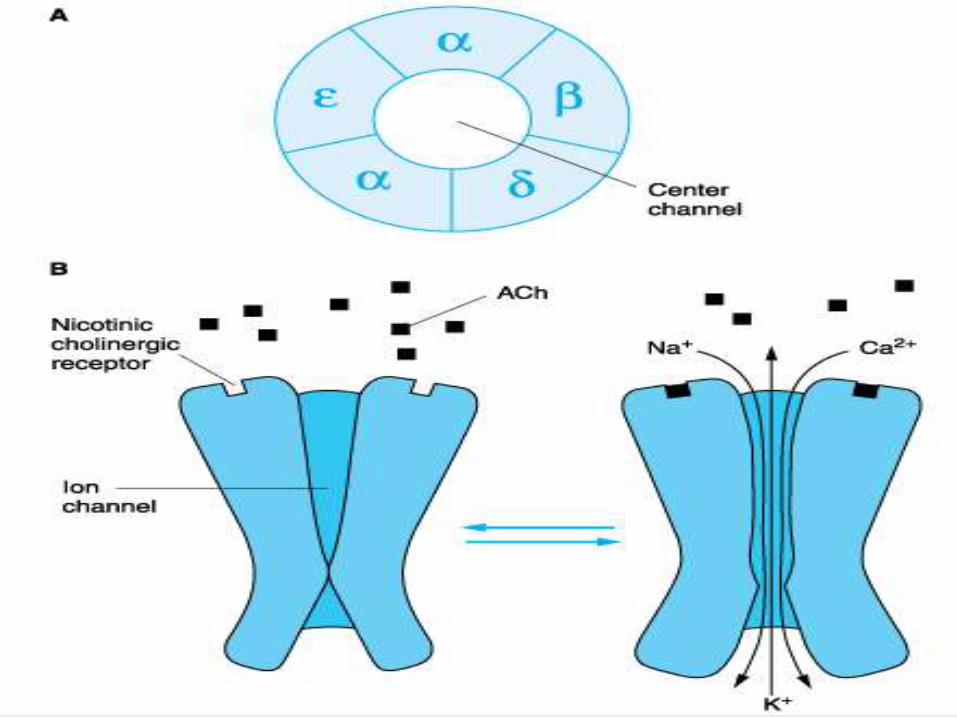
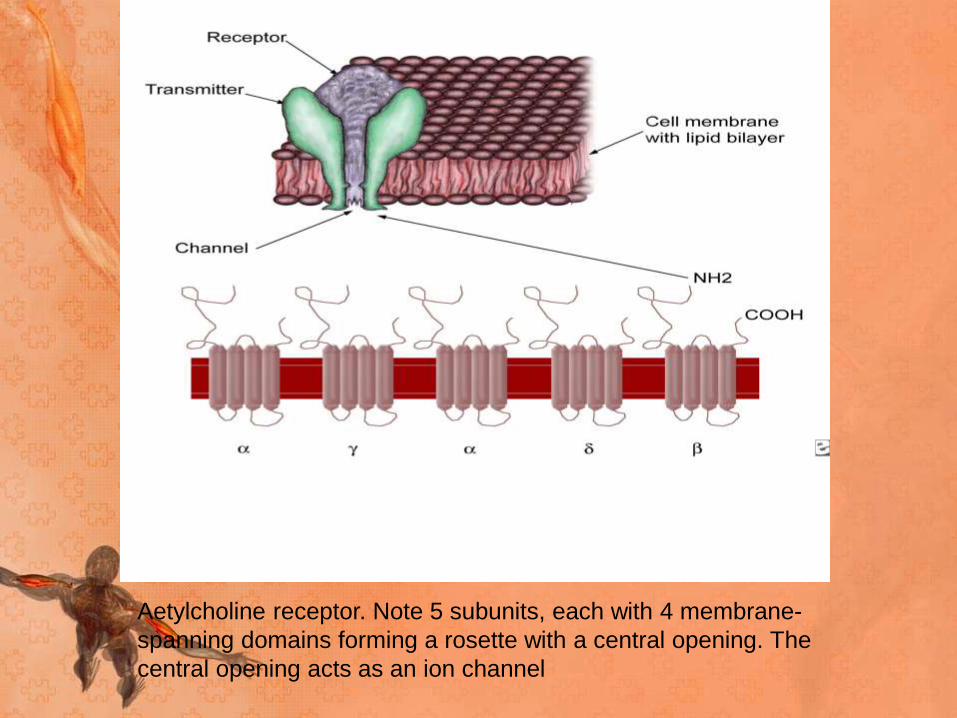
Aetylcholine receptor. Note 5 subunits, each with 4 membrane-
spanning domains forming a rosette with a central opening. The
central opening acts as an ion channel

When acetylcholine receptors bind to the pentamericcomplex, they induce a conformational change in the proteins of the alpha subunits which opens the channel and occurs only if it binds to both the alpha binding sites.
For ions to pass through the channel both the gates should be open.
Cations flow through the open channel, sodium and calcium in and potassium out, thus generating end plate potential.
Na ions are attracted to the inside of the cell which induces depolarisation.

PREJUNCTIONAL RECEPTORS
• These are nicotinic receptors that control ion channel
specific for calcium which is essential for synthesis and
mobilization of acetylcholine.
• They contain protein subunits that are blocked by non
depolarising muscle relaxants resulting in fade and
exhaustion.
• They are also blocked by aminoglycosides and
polymyxin antibiotics

EXTRAJUNCTIONAL RECEPTOR
• These tend to be concentrated around the end plate,
where they mix with post junctional receptors but may be
found anywhere on the muscle membrane. In them, the
adult epsilon subunit is replaced by the fetal gamma
subunit.
• They are not found in normal active muscle, but appear
very rapidly after injury or whenever muscle activity has
ended.
• They can appear within 18hrs of injury and an altered
response to neuromuscular blocking drugs can be
detected in 24hrs of the insult.

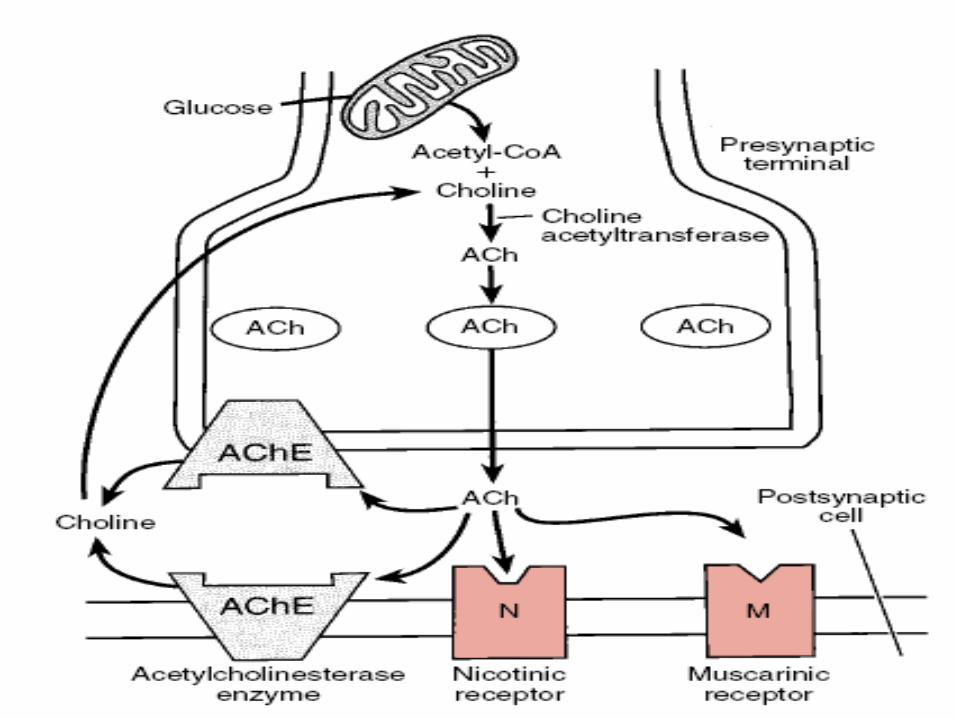
Ach (Synthesis, storage, release)
• Synthesized in the Presynaptic terminal from substrate
Choline and Acetyl CoA.
CHAT
CHOLINE + ACETYL CoA ACETYL CHOLINE
COMT
50% Carrier Facilitated Transport Release
CHOLINE + ACETYL CoA ACETYL CHOLINE
Synaptic Cleft

• Different pools of acetylcholine in the nerve terminal
have variable availability for release
a) The immediately releasable stores, VP2:
Responsible for the maintainance of transmitter release
under conditions of low nerve activity. 1% of vesicles
b) The reserve pool, VP1: Released in response to nerve
impulses. 80% of vesicles
c) The stationary store: The remainder of the vesicles.

• Each vesicle contains approx 12,000 molecules of
acetylcholine, which are loaded into the vesicles by an
active transport process in the vesicle membrane
involving a magnesium dependent H+ pump ATPase.
• Contents of a single vesicle constitute a quantum of
acetylcholine.
• Release of acetylcholine may be
a) Spontaneous or
b) In response to a nerve impulse.

• When a nerve impulse invades the nerve terminal,
calcium channels in the nerve terminal membrane are
opened up.
• Calcium enters the nerve terminal and there is calcium
dependant synchronous release of the contents from 50-
100 vesicles.
• The number of quanta released by each nerve impulse is
very sensitive to extracellular ionized calcium
concentrations.
• Increased calcium concentration results in increased
quanta released.

• If calcium is not present, depolarization of the nerve,
even by electrical stimulation, will not produce release
of transmitter.
• Doubling the extracellular calcium results in a 16-fold
increase in the quantal content of an end plate potential.
• The calcium current persists until the membrane
potential is returned to normal by outward fluxes of
potassium from inside the nerve cell.
• Thus, the calcium current can be prolonged by
potassium channel blockers (e.g., 4-aminopyridine, and
tetraethylammonium), which slow or prevent potassium
efflux out the nerve

• Once the contents have been discharged, they are
rapidly refilled from the reserve stores.
• The reserve vesicles are anchored to actin fibrils in the
cytoskeleton, by vesicular proteins called synapsins
• Some calcium that enters the axoplasm, on the arrival of
the nerve impulse binds to calmodulin, which activates
protein kinase-2 which phosphorylates synapsins, which,
in turn dissociates the vesicle from the actin fibrils
allowing it to move forward to the release site.

• Docking of the vesicle and subsequent discharge of
acetylcholine by exocytosis, involves several other
proteins.
• Membrane protein called SNAREs ( Soluble N-
ethylmatrimide sensitive attachment proteins) are
involved in fusion, docking, and release of acetylcholine
at the active zone.
• SNARE includes – synaptic vesicle protein
synaptobrevin, synataxin and SNAP-25.
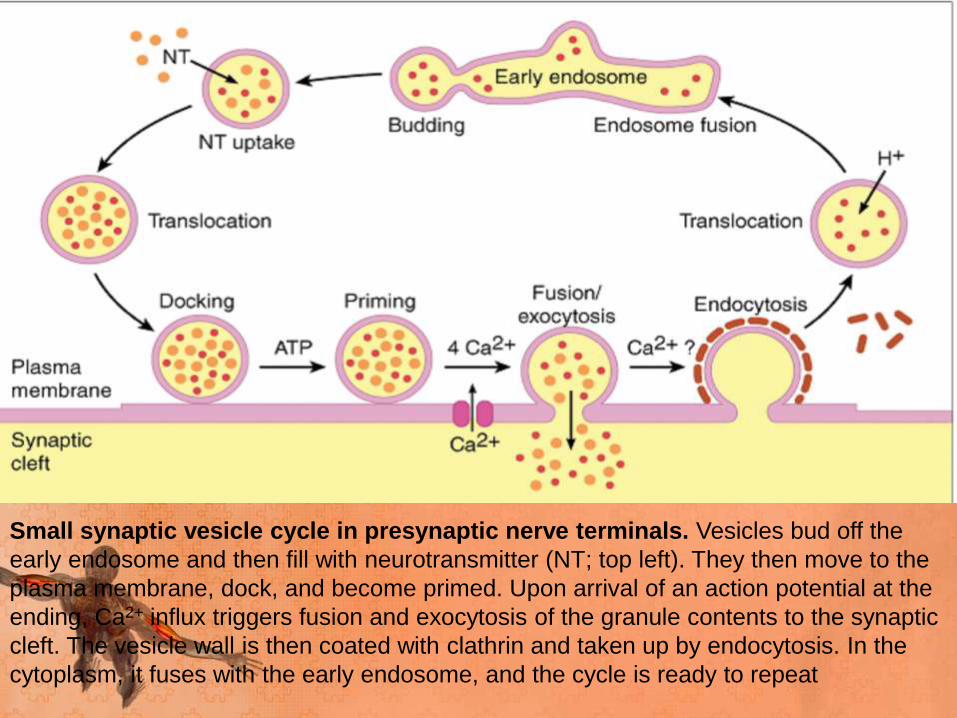
Small synaptic vesicle cycle in presynaptic nerve terminals. Vesicles bud off the
early endosome and then fill with neurotransmitter (NT; top left). They then move to the
plasma membrane, dock, and become primed. Upon arrival of an action potential at the
ending, Ca2+ influx triggers fusion and exocytosis of the granule contents to the synaptic
cleft. The vesicle wall is then coated with clathrin and taken up by endocytosis. In the
cytoplasm, it fuses with the early endosome, and the cycle is ready to repeat
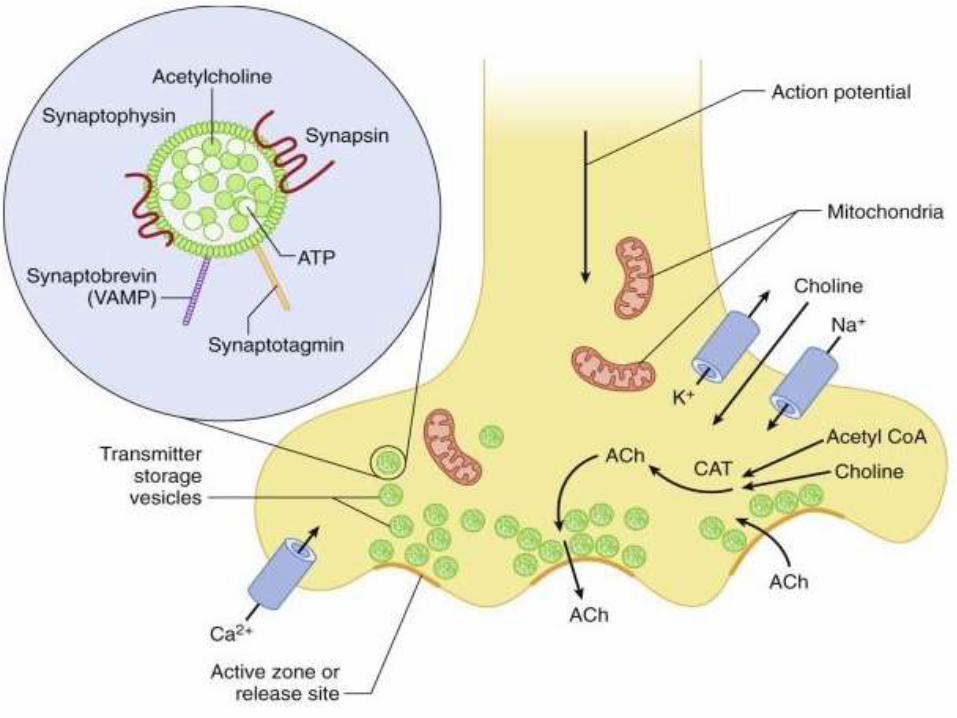

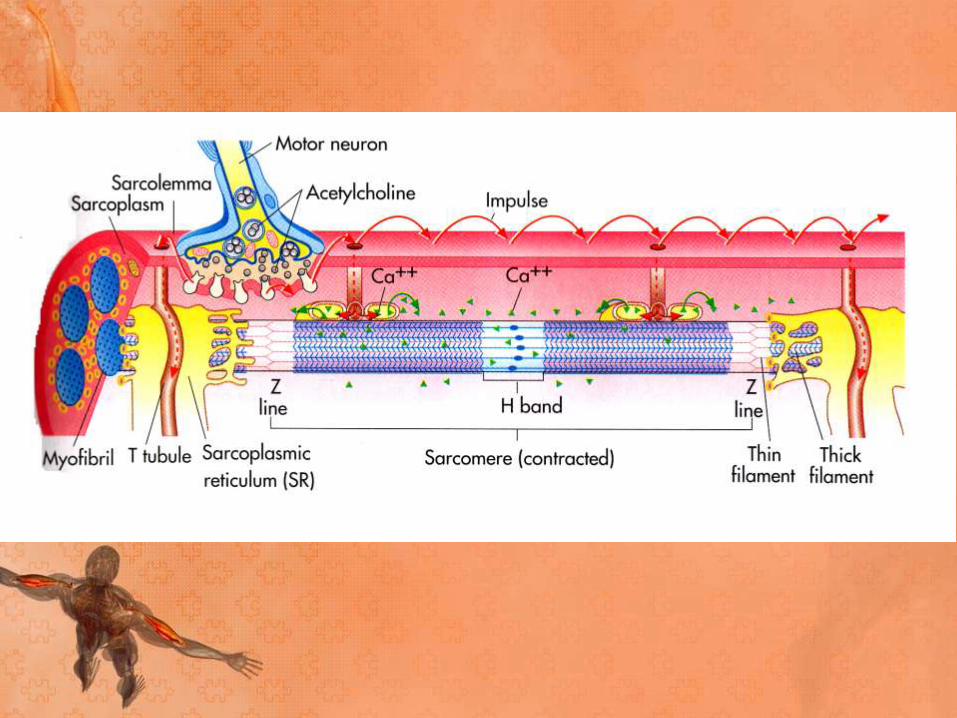
• The released acetylcholine diffuses to the muscle type
nicotinic acetylcholine receptors which are concentrated
at the tops of junctional folds of membrane of the motor
end plate.
• Binding of acetylcholine to these receptors increases Na
and K conductance of membrane and resultant influx of
Na produces a depolarising potential, end plate potential.
• The current created by the local potential depolarise the
adjacent muscle membrane to firing level.

• Acetylcholine is then removed by acetylcholinesterase
from synaptic cleft, which is present in high
concentration at NMJ.
• Action potential generated on either side of end plate
and are conducted away from end plate in both
directions along muscle fiber.
• The muscle action potential in turn initiates muscle
contraction

STRUCTURE OF NA CHANNEL
• This Na channel is cylindrical
• Has membrane protein
• Its two ends act as gates
• Both should be open to allow passage of ions.
• Voltage dependent gate is closed in resting state and
opens only on application of a depolarising voltage,
remains open as long as the voltage persists
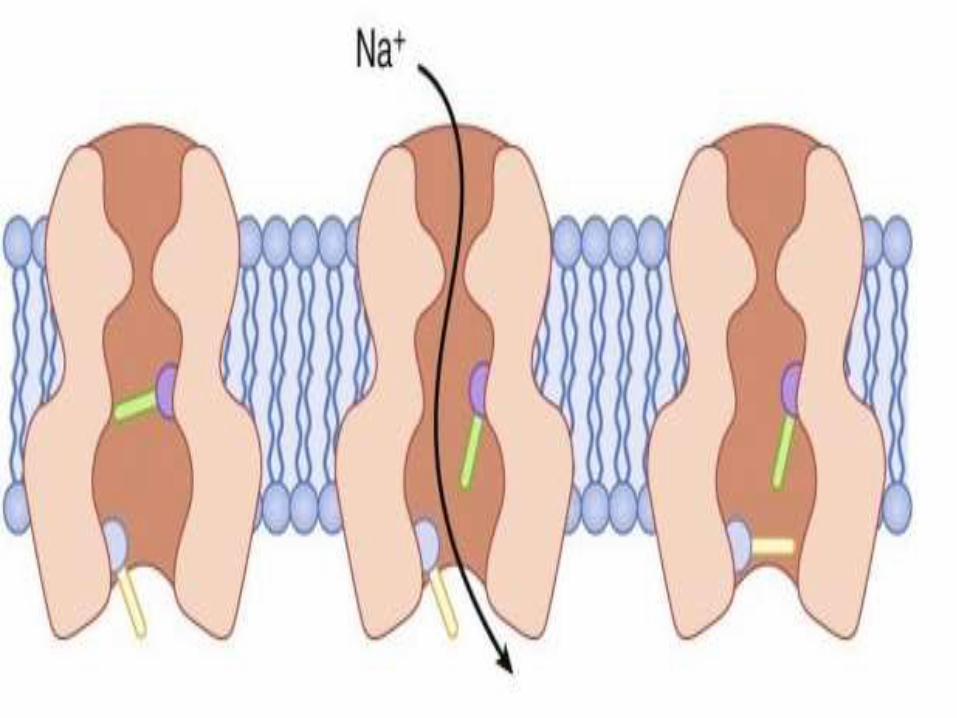

• The time dependent gate is normally open at rest closing
a few milliseconds after the voltage gate opens and
remains closed as long as the voltage gate is open
• It reopens after the voltage gate closes.
• The channel is patent, allowing sodium ions only when
the gates are open.

POSSIBLE CONFIGURATION OF Na
CHANNELS• Resting state: Voltage gate closed
Time gate open
Channel closed
• Depolarization: Voltage gate open
Time gate open
Channel open
• With in a few milliseconds: Voltage gate open
Time gate closed
Channel closed
• End of depolarization: Voltage gate closed
Time gate open
Channel closed
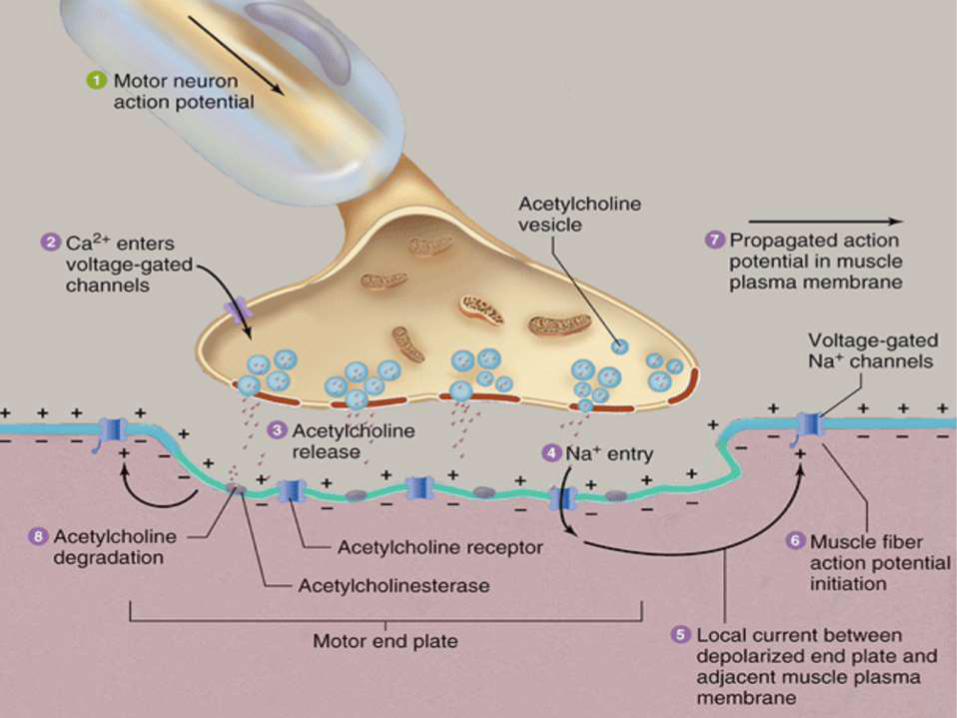

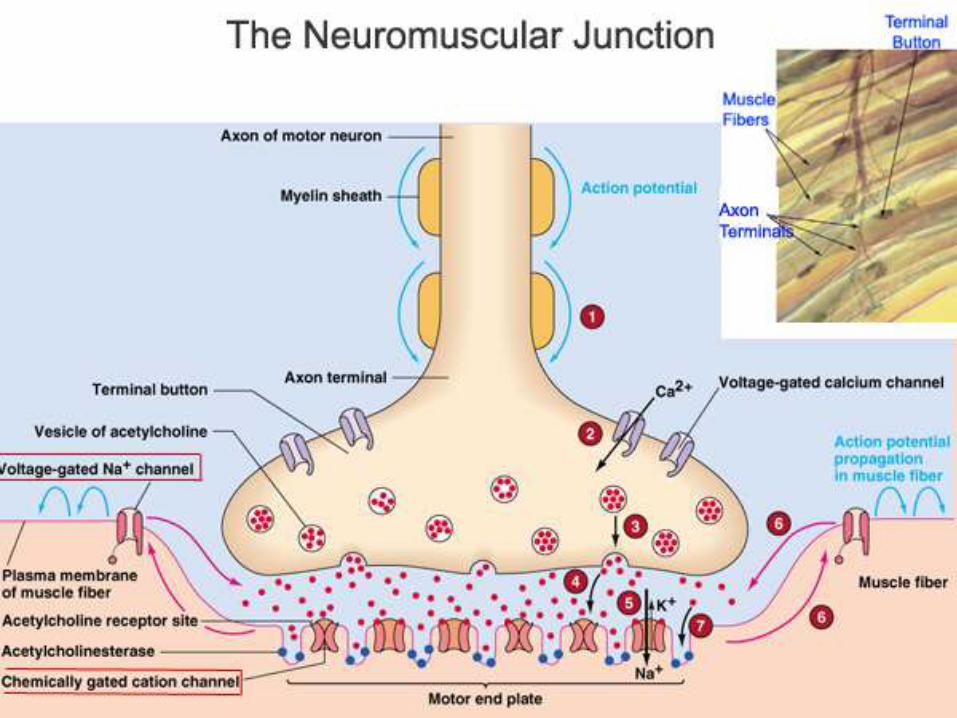
Major Events in Neuromuscular
Transmission
• Motor neuron depolarization causes action potential to travel down
the nerve fiber to the NMJ
• Depolarization of the axon terminal causes an influx of Ca2+ which
triggers fusion of the synaptic vesicles and release of
neurotransmitter (ACh)
• ACh diffuses across the synaptic cleft and binds to post-synaptic
ACh receptor (AChR) located on the muscle fiber at the motor
end-plate
• Binding of ACh to AChRs opens the channels causing an influx of
Na, depolarization of the sarcolemma that travels down the t-
tubules and ultimately causes the release of Ca2+ from the
sarcoplasmic reticulum - CONTRACTION.
• Unbound ACh in synaptic cleft defuses away or is hydrolyzed
(inactivated) by AChE

Major disorders of the
neuromuscular junction
1. Acquired Myasthenic Syndromes
2. Hereditary and Congenital Myasthenic
Syndromes

Acquired Myasthenic Syndromes
Presynaptic• Botulism
• Lambert-Eaton myasthenic syndrome
Synaptic• Insecticides
Postsynaptic• Myasthenia gravis
• Snake venom toxins

Hereditary and Congenital
Myasthenic Syndromes
Presynaptic
• Episodic apnea
• Paucity of synaptic vesicles
Synaptic
• AChE deficiency
Postsynaptic
• Slow channel syndrome
• Fast channel syndrome
• Primary AChR deficiency
• Rapsyn deficiency
• Plectin deficiency

Myasthenia Gravis
• Myasthenia gravis was first recognised as a distinct clinical entity by Thomas Willis, a 17th century Oxford physician,
• The first modern description was made in 1877 by Samuel Wilks, a London physician.
• The first reasonably complete accounts were those of Wilhelm Erb (1878), who characterized the disease as a bulbar palsy without an anatomic lesion, and of Samuel Goldflam (1893); for many years thereafter, the disorder was referred to as the Erb-Goldflam syndrome.
• Jolly (1895) was the first to use the name myasthenia gravis, to which he added the term pseudoparalytica to indicate the lack of structural changes at autopsy.

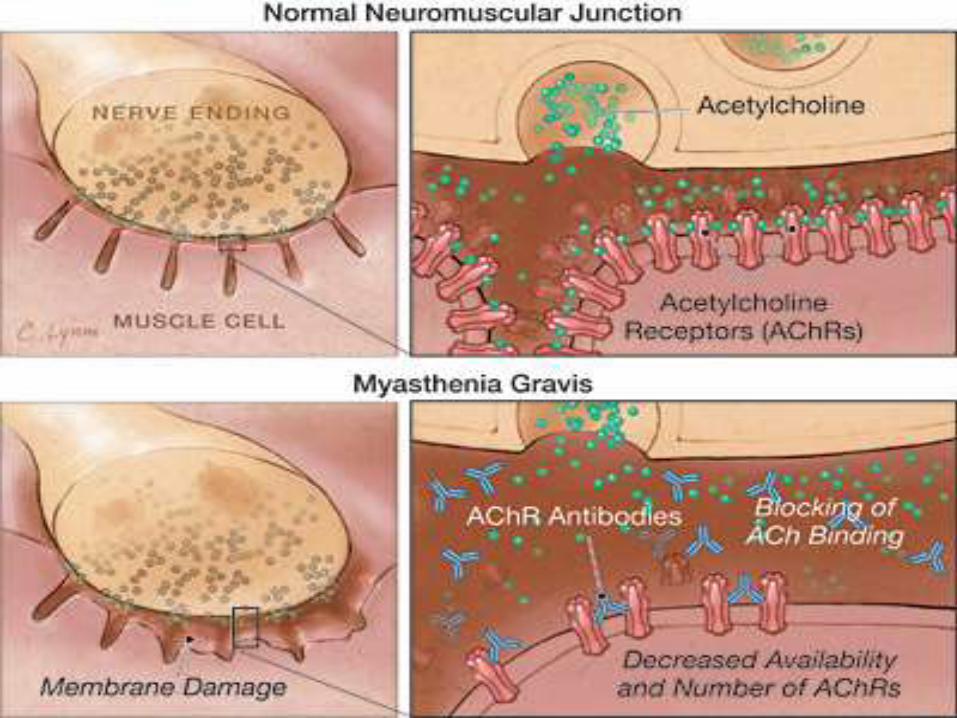
• In MG, the fundamental defect is a decrease in the number of available AChRs at the postsynaptic muscle membrane.
• In addition, the postsynaptic folds are flattened, or "simplified."
• These changes result in decreased efficiency of neuromuscular transmission.
• Therefore, although ACh is released normally, it produces small end-plate potentials that may fail to trigger muscle action potentials.
• Failure of transmission at many neuromuscular junctions results in weakness of muscle contraction.
• The amount of ACh released per impulse normally
declines on repeated activity (termed presynaptic
rundown).
• In the myasthenic patient, the decreased efficiency of
neuromuscular transmission combined with the normal
rundown results in the activation of fewer and fewer
muscle fibers by successive nerve impulses and hence
increasing weakness, or myasthenic fatigue.
• This mechanism also accounts for the decremental
response to repetitive nerve stimulation seen on
electrodiagnostic testing

Pathophysiology
• Binding of AChR antibodies to AChR results in impairment of neuromuscular transmission in several ways, including the following:
I. Cross-linking 2 adjacent AChRs with anti-AChR antibody, thus accelerating internalization and degradation of AChRmolecules
II. Causing complement-mediated destruction of junctionalfolds of the postsynaptic membrane
III. Blocking the binding of ACh to AChR
IV. Decreasing the number of AChRs at the NMJ by damaging the junctional folds on the postsynaptic membrane, thereby reducing the surface area available for insertion of newly synthesized AChRs

• Patients without anti-AChR antibodies are recognized
as having seronegative MG (SNMG).
• Many patients with SNMG have antibodies against
muscle-specific kinase (MuSK).
• MuSK plays a critical role in postsynaptic differentiation
and clustering of AChRs

Role of the thymus • The role of the thymus in the pathogenesis of MG is not entirely
clear, but 75% of patients with MG have some degree of thymus abnormality (eg, hyperplasia or thymoma).
• Histopathologic studies have shown prominent germinal centers.
• Epithelial myoid cells normally present in the thymus do resemble skeletal muscle cells and possess AChRs on their surface membrane. These cells may become antigenic and unleash an autoimmune attack on the muscular endplate AChRs by molecular mimicry
• Of patients with MG, 75% have thymic disease, 85% have thymichyperplasia, and 10-15% have thymoma.
• Extrathymic tumors may include small cell lung cancer and Hodgkin disease.
• Hyperthyroidism is present in 3-8% of patients with MG and has a particular association with ocular MG

• prevalence of 2–7 in 10,000.
• It affects individuals in all age groups, but peaks of
incidence occur in women in their twenties and
thirties and in men in their fifties and sixties.
• Overall, women are affected more frequently than
men, in a ratio of 3:2.

• The cardinal features are weakness and fatigability of
muscles
• The special vulnerability of certain muscles gives myasthenia
a characteristic stamp.
• Usually the eyelids and the muscles of the eyes—and
somewhat less often of the face, jaws, throat, and neck—are
the first to be affected.
• Infrequently the initial complaint is referable to the limbs.
• More specifically, the weakness of the levator palpebrae or
extraocular muscles is the initial manifestation of the disease
in about half the cases, and these muscles are involved
eventually in more than 90 percent
• Muscles of facial expression, mastication, swallowing,
and speech are affected in 80 percent of patients at
some time in the illness, and in 5 to 10 percent these
are the first or only muscles to be involved.
• Less frequent is an initial or early involvement of the
flexors and extensors of the neck, muscles of the
shoulder girdle, and flexors of the hips

• Of the trunk muscles, the erector spinae are the most frequently affected.
• In the most advanced cases, all muscles are weakened, including the diaphragmatic, abdominal, and intercostal muscles and even the external sphincters of the bladder and bowel.
• The involvement of any group of muscles closely parallels their degree of weakness early in the disease.
• The clinical rule holds that the proximal muscles are far more vulnerable than distal ones, as they are in almost all other forms of myopathy

• As the disease advances, it often spreads from the
cranial to the limb and axial muscles.
• The onset of weakness is usually insidious
• The disease remains exclusively ocular in only 16% of
patients.
• About 87% of patients have generalized disease
within 13 months after onset.
• In patients with generalized disease, the interval from
onset to maximal weakness is less than 36 months in
83% of patients.

• The other characteristic feature of myasthenic
weakness is its tendency to increase as the day wears
on or with repeated use of an affected muscle group,
but patients seldom volunteer this information.
• A few patients report being paradoxically worse on
awakening, especially if they have not received
medication during the night.
• Smooth and cardiac muscles are not involved and
other neural functions are preserved.
• The tendon reflexes are unaffected

• Exposure to bright sunlight, surgery, immunization,
emotional stress, menstruation, and physical factors
might trigger or worsen exacerbations.
• Intercurrent illness (eg, viral infection) or medication can
exacerbate weakness, quickly precipitating a myasthenic
crisis and rapid respiratory compromise.
• Spontaneous remissions are rare.
• Long and complete remissions are even less common.
• Most remissions with treatment occur during the first 3
years of disease

MGFA Clinical Classification
Class I MG :Any ocular muscle weakness
May have weakness of eye closure
All other muscle strength is normal
Class II MG :Mild weakness affecting other than ocular muscles
May also have ocular muscle weakness of any severity
Class IIa:
Predominantly affecting limb, axial muscles, or both
May also have lesser involvement of oropharyngeal muscles
Class IIb:
Predominantly affecting oropharyngeal, respiratory muscles, or both
May also have lesser or equal involvement of limb, axial muscles, or both

Class III MG :Moderate weakness affecting other than ocular muscles
May also have ocular muscle weakness of any severity
Class IIIa MG :
Predominantly affecting limb, axial muscles, or both
May also have lesser involvement of oropharyngeal muscles
Class IIIb :
Predominantly affecting oropharyngeal, respiratory muscles, or both
May also have lesser or equal involvement of limb, axial muscles, or both
Class IV MG :Severe weakness affecting other than ocular muscles
May also have ocular muscle weakness of any severity
Class IVa :
Predominantly affecting limb, axial muscles, or both
May also have lesser involvement of oropharyngeal muscles
Class IVb :
Predominantly affecting oropharyngeal, respiratory muscles, or both
May also have lesser or equal involvement of limb, axial muscles,
or both
Class V MG :
Defined by intubation, with or without mechanical ventilation, except
when used during routine postoperative management
Use of a feeding tube without intubation places the patient in class
IVb

Diagnosis of Myasthenia Gravis
History • Diplopia, ptosis, weakness
• Weakness in characteristic distribution
• Fluctuation and fatigue: worse with repeated activity, improved by rest
• Effects of previous treatments
Physical examination • Ptosis, diplopia
• Motor power survey: quantitative testing of muscle strength
• Forward arm abduction time (5 min)
• Vital capacity
• Absence of other neurologic signs

Laboratory testing
• Anti-AChR radioimmunoassay: 85% positive in generalized MG; 50% in ocular MG
• 40% of AChR antibody–negative patients with generalized MG have anti-MuSK antibodies.
• Repetitive nerve stimulation: decrement of >10% at 3 Hz: highly probable
• Single-fiber electromyography: blocking and jitter, with normal fiber density; confirmatory, but not specific
• Edrophonium chloride (Tensilon) 2 mg + 8 mg IV; highly probable diagnosis if unequivocally positive
• For ocular or cranial MG: exclude intracranial lesions by CT or MRI

Antibodies to Achr or Musk
• Anti-AChR antibodies are detectable in the serum of 85% of all myasthenic patients but in only about 50% of patients with weakness confined to the ocular muscles.
• The presence of anti-AChR antibodies is virtually diagnostic of MG, but a negative test does not exclude the disease.
• The measured level of anti-AChR antibody does not correspond well with the severity of MG
• In an individual patient, a treatment-induced fall in the antibody level often correlates with clinical improvement, while a rise in the level may occur with exacerbations.
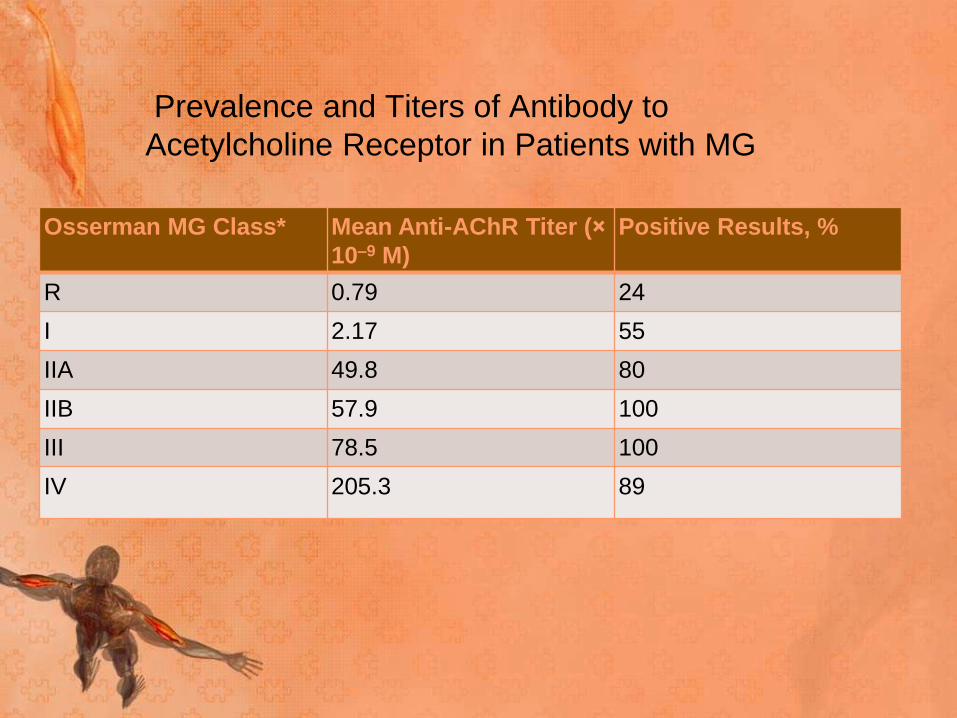
Prevalence and Titers of Antibody to
Acetylcholine Receptor in Patients with MG
Osserman MG Class* Mean Anti-AChR Titer (×
10–9 M)
Positive Results, %
R 0.79 24
I 2.17 55
IIA 49.8 80
IIB 57.9 100
III 78.5 100
IV 205.3 89

• Antibodies to MuSK have been found to be present in
40% of AChR antibody–negative patients with
generalized MG, and their presence is a useful
diagnostic test in these patients.
• MuSK antibodies are rarely present in AChR
antibody–positive patients or in patients with MG
limited to ocular muscles.
• These antibodies may interfere with clustering of
AChRs at neuromuscular junctions, as MuSK is
known to do during early development

• Anti-MuSK–positive individuals tend to have more
pronounced bulbar weakness and may have tongue
and facial atrophy.
• They may have neck, shoulder and respiratory
involvement without ocular weakness.
• They are also less likely to respond to acetylcholine
esterase (AChE) inhibitors, and their symptoms may
actually worsen with these medications

Anti–striated muscle antibody
• The anti–striated muscle (anti-SM) Ab test is also
important in patients with MG.
• Anti-SM Ab is present in about 84% of patients with
thymoma who are younger than 40 years and less often
in those without thymoma.
• Thus, a positive test result should prompt a search for
thymoma in patients younger than 40 years.
• In individuals older than 40 years, anti-SM Ab can be
present without thymoma

Antistriational antibody
• Serum from some patients with MG possesses antibodies that bind in a cross-striational pattern to skeletal and heart muscle tissue sections. These antibodies react with epitopes on the muscle protein titin and ryanodine receptors (RyR).
• Almost all patients with thymoma and MG, as well as half of the late-onset MG patients (onset at 50 years or later), manifest a broad striational antibody response.
• Striational antibodies are rarely found in anti-AChR–negative patients.
• They can be used as prognostic determinants in MG; as in all subgroups of MG, higher antibody titers are associated with more severe disease
• Because of a frequent association with thymoma, the presence of titin/RyR antibodies should arouse a strong suspicion of thymoma in a young patient with MG

Electrophysiologic Testing
The following 2 studies are commonly performed:
• Repetitive stimulation of a muscle at 2-3 Hz, also known as repetitive nerve stimulation (RNS)
• Single-fiber electromyography (SFEMG), aimed at evaluating neuromuscular block, jitter, and fiber density
• SFEMG is more sensitive than RNS in assessing MG.
• However, SFEMG is technically more difficult and much more dependent on the experience and skill of the testing physician.
• Consequently, RNS is the most frequently performed neurophysiologic test of neuromuscular transmission

Repetitive nerve stimulation
• During low-frequency (1-5 Hz) RNS, the locally available acetylcholine (ACh) becomes depleted at all neuromuscular junctions (NMJs), and less is therefore available for immediate release. This results in smaller excitatory postsynaptic potentials (EPSPs).
• In patients without MG, all EPSPs exceed the threshold to generate an action potential (ie, there is a safety factor). No change in the summated compound muscle action potential (CMAP) is noted.
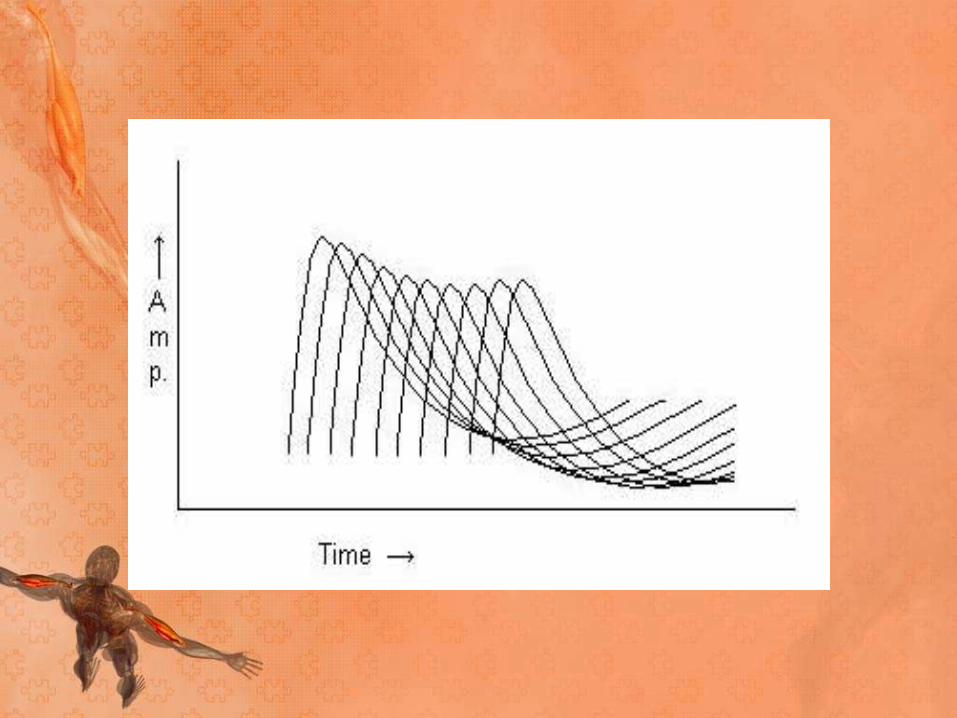
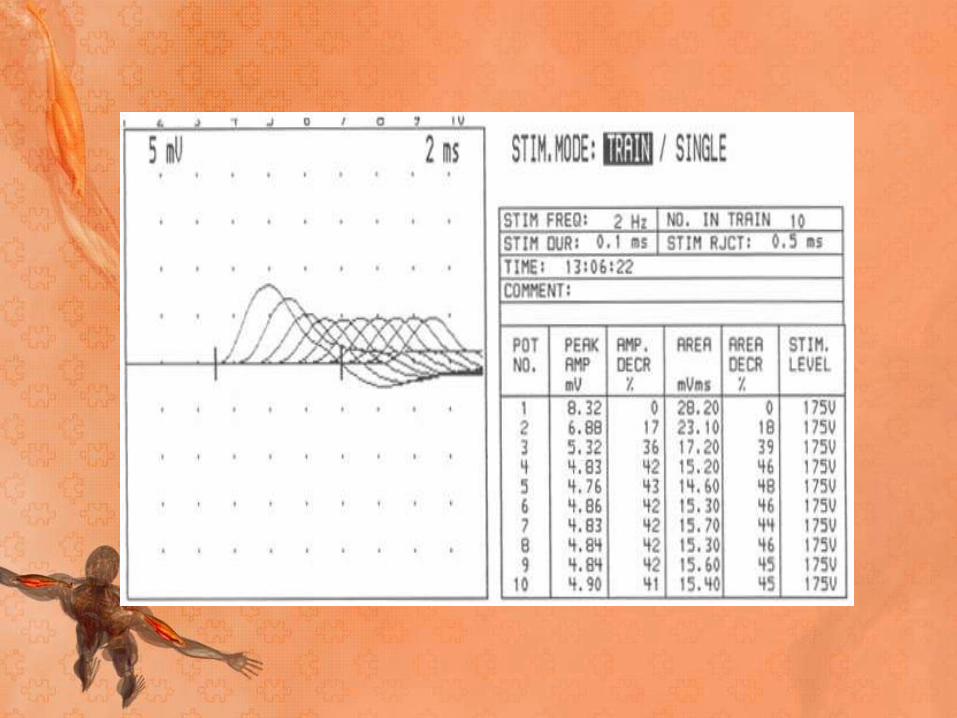
• In patients with MG, the number of AChRs is reduced, lowering the safety factor. During RNS, some EPSPs may not reach threshold, which means that no action potential is generated. This results in the decrement in the amplitude of the CMAP

• In patients with myasthenia gravis, this decremental
response usually has a maximum decrement at the
fourth or fifth response, followed by a tendency toward
repair
• A stimulation rate of 1-5 per second should result in a
10% or more decrease in amplitude by the fourth or
fifth action potential; any decrement over 10% is
considered abnormal.
• The most common employed stimulation rate is 3 Hz.

• Patients with MG rarely have a decreased response in
a clinically normal muscle.
• Thus, testing a proximal weak muscle gives a better
yield than testing a unaffected distal muscle, even if
the latter is technically easier.
• Testing a facial muscle (eg, the orbicularis oculi) is
useful because most patients suffer from eyelid
weakness or ptosis.
• RNS results are less likely to be positive in patients
with ocular MG

Factors affecting results
• Lower temperatures increase the amplitude of the
CMAPs.
• Patients with MG may report clinically significant
improvement in cold temp, and they typically report
worsening of ptosis in bright sunlight or on a warm day.
• Therefore, maintaining a constant and perhaps higher-
than-ambient temperature during RNS testing is
important to bring out abnormalities of NMJ function.
• The temperature of the skin overlying the tested muscle
should be at least 34°C.
• Administration of AChE inhibitors before testing may
mask the abnormality and consequently should be
avoided for at least 1 day beforehand
Single-fiber electromyography
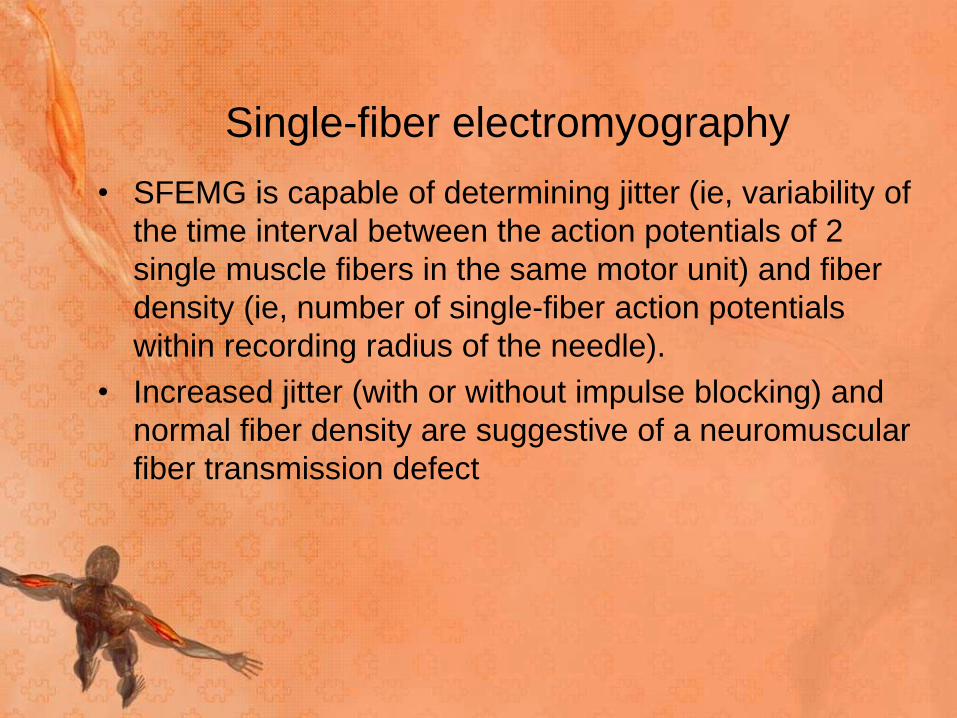
• SFEMG is capable of determining jitter (ie, variability of
the time interval between the action potentials of 2
single muscle fibers in the same motor unit) and fiber
density (ie, number of single-fiber action potentials
within recording radius of the needle).
• Increased jitter (with or without impulse blocking) and
normal fiber density are suggestive of a neuromuscular
fiber transmission defect
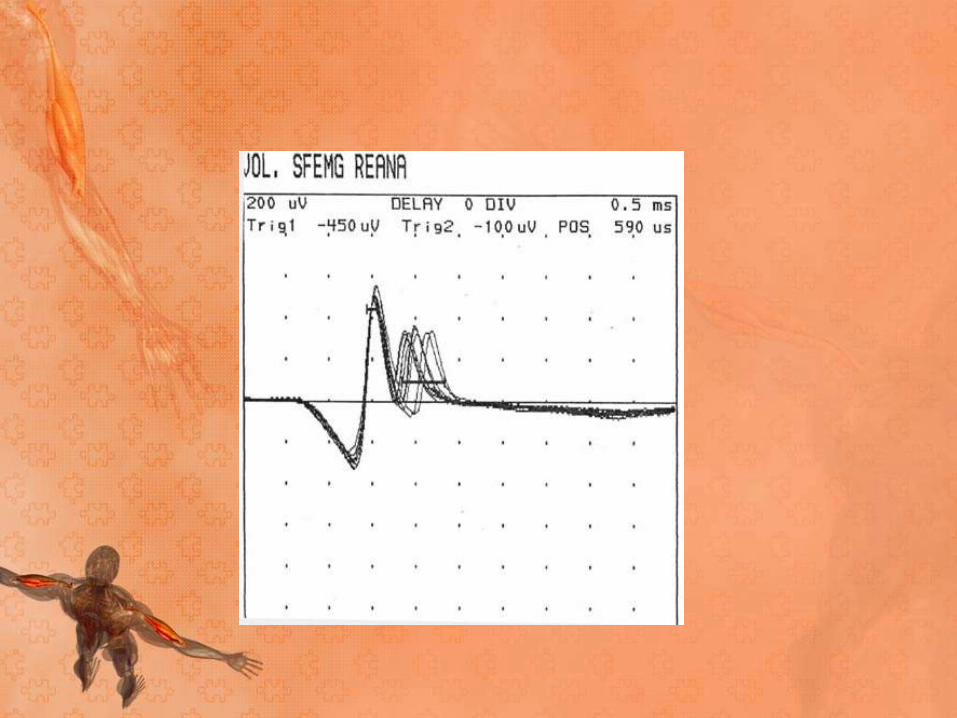

• SFEMG of the extensor digiti communis (EDC) yields
abnormal results in 87% of patients with generalized
MG.
• Examination of a second muscle raises the sensitivity
to 99%.
• In ocular MG, examination of the frontalis is more
useful than examination of the EDC.
• Frontalis findings are abnormal in almost 100% of
patients, but only about 60% of EDC findings are
abnormal

• Treatment with AChR inhibitors does not normalize
SFEMG results.
• SFEMG findings are abnormal in almost 100% of
patients, whereas RNS findings are abnormal in only 44-
65%.
• SFEMG is a good substitute for RNS in patients with
ocular MG

Anticholinesterase Test
• Edrophonium is used most commonly for diagnostic testing because of the rapid onset (30 s) and short duration (5 min) of its effect
• This test evaluates weakness (eg, ptosis, partial or complete ophthalmoplegia, and forced hand grip) in an involved group of muscles before and after intravenous (IV) administration of edrophonium
• 1 mg (0.1 mL) of edrophonium is given intravenously; if this dose is tolerated and no definite improvement in strength occurs after 45 s, another 3 to 6 mg is injected. If there is no response after another 45 s, an additional 3 to 5 mg may be given over approximately 1 min.
• A total dose of 10 mg is rarely necessary.
• Most patients who respond do so after 3 to 5 mg have been administered

• The use of neostigmine is sometimes preferable to edrophonium because the longer duration of its effect allows more deliberate and repeated testing of muscle function.
• Neostigmine methylsulfate is injected intramuscularly in a dose of 1.5 mg
• After intramuscular injection, objective and subjective improvement occurs within 10 to 15 min, reaches its peak at 20 min, and lasts up to 1 h, allowing for careful verification of the neurologic improvement
• the anticholinesterase-inhibiting drugs carry a rare risk of ventricular fibrillation and cardiac arrest, so that testing should be carried out preferably where emergency support is accessible.

• This test may give both false-negative results and false-
positive results.
• It has a low sensitivity in ocular MG; 50% of patients
presenting with eye symptoms will be missed.
• On the other hand, diseases other than MG, such as ALS
and cavernous sinus lesions can score positive on the test
• This test has been combined with EMG and ocular
tonography to increase its sensitivity in ocular MG;
however, it still produces false-negative and false-positive
results.
• The combination of edrophonium with electronystagmo-
graphic analysis of optokinetic nystagmus, seems
promising for the diagnosis of ocular MG
Ice Pack Test
• The ice pack test (ie, placing ice over the lid) has
gained interest among ophthalmologists for assessing
improvement in ptosis and diplopia in ocular MG.
• The rationale behind this test is that cooling might
improve neuromuscular transmission

Radiography
• On plain anteroposterior and lateral views, radiography
may identify a thymoma as an anterior mediastinal mass.
• A negative chest radiograph does not rule out a smaller
thymoma, in which case a chest computed tomography
(CT) scan is required.
• Chest CT scan is mandatory to identify or rule out
thymoma or thymic enlargement in all cases of MG
• It is essential to rule out mass lesions compressing the
cranial nerves in strictly ocular MG.
• CT or magnetic resonance imaging (MRI) of the brain and
orbit is indicated
• MRI can evaluate for intraorbital or intracranial lesions,
basal meningeal pathology, or multiple sclerosis.

Treatment
• Anticholinesterase Medications
• Immunosuppression
• Thymectomy

Anticholinesterase Drugs

• The usual dose of pyridostigmine is 30 to 90 mg given
every 6 h
• The maximum useful dose of pyridostigmine rarely
exceeds 120 mg every 4–6 h during daytime
• The oral dose of neostigmine ranges from 7.5 to 45 mg
given every 2 to 6 h.
• Neostigmine is poorly absorbed from the GI tract and
should be used only if pyridostigmine is unavailable
• For mild cases without thymic tumor, for patients in
partial remission after thymectomy, and for purely ocular
myasthenia, the use of anticholinesterase drugs may be
the only form of therapy necessary for some period of
time

Corticosteroids
• For the patient with myasthenia with moderate to severe
generalized weakness who is responding inadequately to
anticholinesterase drugs, the long-term administration of
corticosteroids is the most consistently effective form of
treatment
• The usual form of corticosteroid therapy is prednisone (or
corresponding doses of prednisolone), beginning with 15
to 20 mg/ day and increasing the dose gradually until a
satisfactory clinical response is obtained, or until a daily
dose of 50 to 60 mg is reached
• With higher doses or more rapid elevations of the doses,
worsening in the first week is common

• Once the maximal effect from prednisone has been
attained, the dosage can be reduced gradually over
months to the lowest point at which it is still
effective.
• The usual practice has been to then institute an
alternate-day schedule, which diminishes the side
effects
• At the outset of steroid therapy, anticholinesterase
drugs are given simultaneously; as the patient
improves, the dosage of the latter may be adjusted
downward

Immunosuppression
• Immunosuppression using glucocorticoids, azathioprine,
and other drugs is effective in nearly all patients with MG.
• The choice of drugs or other immunomodulatory
treatments should be guided by the relative benefits and
risks for the individual patient and the urgency of
treatment.
• It is helpful to develop a treatment plan based on short-
term, intermediate-term, and long-term objectives.
• If immediate improvement is essential either because of
the severity of weakness or because of the patient's need
to return to activity as soon as possible, IVIg should be
administered or plasmapheresis should be undertaken

• For the intermediate term, glucocorticoids and
cyclosporine or tacrolimus generally produce clinical
improvement within a period of 1–3 months.
• The beneficial effects of azathioprine and mycophenolate
mofetil usually begin after many months (as long as a
year), but these drugs have advantages for the long-term
treatment of patients with MG.
• For the pts refractory to optimal treatment with
conventional immunosuppressive agents, a course of
high-dose cyclophosphamide may induce long-lasting
benefit by "rebooting" the immune system

Mycophenolate mofetil
• One of the most widely used drugs in the treatment of
MG because of its effectiveness and relative lack of side
effects.
• A dose of 1–1.5 g bid is recommended.
• Its mechanism of action involves inhibition of purine
synthesis by the de novo pathway.
• Since lymphocytes lack the alternative salvage pathway
that is present in all other cells, mycophenolate inhibits
proliferation of lymphocytes but not proliferation of other
cells.

• It does not kill or eliminate preexisting autoreactive
lymphocytes, and therefore clinical improvement may be
delayed for many months to a year, until the preexisting
autoreactive lymphocytes die spontaneously
• The advantage of mycophenolate lies in its relative lack
of adverse side effects, with only occasional production
of GI symptoms, rare development of leukopenia, and
very small risks of malignancy or PML inherent in all
immunosuppressive treatments

Azathioprine
• Until recently, it has been the m.c. used immuno-
suppressive agent for MG because of its relative safety in
most patients and long track record.
• Its therapeutic effect may add to that of glucocorticoids
and/or allow the glucocorticoid dose to be reduced.
• However, up to 10% of patients are unable to tolerate
azathioprine because of idiosyncratic reactions consisting
of flulike symptoms of fever and malaise, bone marrow
suppression, or abnormalities of liver function.

• An initial dose of 50 mg/d should be used for several
days to test for these side effects.
• If this dose is tolerated, it is increased gradually to
about 2–3 mg/kg of total body weight, or until the
white blood count falls to 3000 to 4000/L.
• The beneficial effect of azathioprine takes 3–6
months to begin and even longer to peak.

Calcineurin inhibitors
• Cyclosporine and tacrolimus (FK506) are approximately as
effective as azathioprine and are being used increasingly in
the management of MG.
• Their beneficial effect appears more rapidly than that of
azathioprine.
• Either drug may be used alone, but they are usually used
as an adjunct to glucocorticoids to permit reduction of the
glucocorticoid dose.
• The usual dose of cyclosporine is 4–5 mg/kg per d, and the
average dose of tacrolimus is 0.07–0.1 mg/kg per d, given
in two equally divided doses (to minimize side effects).
• Side effects of these drugs include hypertension and
nephrotoxicity, which must be closely monitored.

Rituximab
• Rituximab, a monoclonal antibody that depletes
CD20 B cells, has been used with variable—
sometimes dramatic—success in the treatment of
MG, especially in patients with anti-MuSK antibody.

Plasma Exchange and
Intravenous Immune Globulin
• For severe myasthenia that is refractory to treatment
with AChE drugs and prednisone, or during an acute
worsening
• This form of treatment may be lifesaving during a
myasthenic crisis
• To improve the patient's condition prior to surgery
(e.g., thymectomy)

Plasmapheresis
• It has been used therapeutically in MG.
• Plasma, which contains the pathogenic antibodies, is
mechanically separated from the blood cells, which are
returned to the patient.
• A course of five exchanges (3–4 L per exchange) is
generally administered over a 10- to 14-day period
• A 2-L exchange will remove 80 percent of circulating
antibodies
• Improvement is noted in a couple of days, but it does not
last for more than 2 months.

• In a crisis requiring plasma exchanges and mechanical
ventilation, anticholinesterase drugs should be
discontinued and resume them as the patient is being
weaned from the ventilator
• Complications are primarily limited to complications of
intravenous (IV) access (eg, central line placement) but
also may include hypotension and coagulation disorders
(though less commonly)

Intravenous immune globulin
• It is similarly useful in the short-term control of acutely
worsening myasthenia.
• The usual dose is 2 g/kg given in divided doses over 3
to 5 days
• The mechanism of action of IVIg is not known
• the treatment has no consistent effect on the
measurable amount of circulating AChR antibody
• Adverse reactions are headache, fluid overload, and
rarely aseptic meningitis or renal failure

Thymectomy
• Two separate issues should be distinguished: (1)
surgical removal of thymoma, and (2) thymectomy as a
treatment for MG.
• Surgical removal of a thymoma is necessary because
of the possibility of local tumor spread, although most
thymomas are histologically benign.
• In the absence of a tumor, the available evidence
suggests that up to 85% of patients experience
improvement after thymectomy; of these, 35% achieve
drug-free remission
• Complete removal of thymic tissue is widely considered
to be of the utmost importance, any small remnant
might lead to recurrence

• It is the consensus that thymectomy should be carried out
in all patients with generalized MG who are between the
ages of puberty and at least 55 years
• Remission occurs more frequently in young patients with a
short duration of disease, hyperplastic thymus, more
severe symptoms, and a high antibody titer
• There is also suggestive evidence that patients with MuSK
Ab–positive MG may respond less well to thymectomy
• In ocular MG, thymectomy should be delayed at least 2
years to allow for spontaneous remission or the
development of generalized MG

• The MGFA thymectomy classification is as follows:
T-1 transcervical thymectomy – Basic; extended
T-2 videoscopic thymectomy - Classic or VATS (video-
assisted thoracic surgery) thymectomy; VATET (video-
assisted thoracoscopic extended thymectomy)
T-3 transsternal thymectomy – Standard; extended
T-4 transcervical and transsternal thymectomy

Complications
• Systemically, myasthenic crisis is the most dreadful
complication.
• Aspiration pneumonia also may occur as a
consequence of poor oropharyngeal muscle function.
• Cholinergic crisis may follow excessive treatment with
cholinesterase inhibitors.
• The most common severe complication of MG is
respiratory failure, which often presents with the rapid
deterioration of respiratory effort that ultimately results
in apnea

Myasthenic crisis
• It is a life-threatening condition, which is defined as
weakness from acquired myasthenia gravis (MG) that is
severe enough to necessitate intubation or to delay
extubation following surgery
• The respiratory failure is due to weakness of respiratory
muscles.
• Severe bulbar (oropharyngeal) muscle weakness often
accompanies the respiratory muscle weakness, or may
be the predominant feature in some patients.
• When this results in upper airway obstruction or severe
dysphagia with aspiration, intubation and mechanical
ventilation are necessary

• Myasthenic crisis may be precipitated by a variety of
factors, most often a concurrent infection or some drugs.
• It can also follow a surgical intervention, pregnancy,
childbirth, or tapering of immunosuppressive medications.
• In addition, myasthenic crisis can occur spontaneously as
part of the natural history of myasthenia gravis (MG) itself
• Management of the crisis entails timely and careful
intubation followed by mechanical ventilation in a critical
care unit

• Anticholinergic drugs, which exaggerate secretions, are
best withdrawn at the time of intubation.
• The use of plasma exchange appears to hasten
improvement and weaning from the ventilator.
• When weaning from the ventilator is anticipated,
anticholinesterase agents are reintroduced slowly, and
treatment with corticosteroids can be instituted if
necessary

• Myasthenic crisis frequently occurs within the first 2
years after disease onset, and about one-fifth of the
patients develop crisis episodes within the first year
• Most often myasthenic crisis develops in patients with
generalized myasthenia
• A third of the patients who survive the first crisis
experience a second crisis.
• Myasthenic crisis is more common in patients of
myasthenia gravis associated with thymoma
• Infections are the most common precipitating factors, 30-
40% of cases

Cholinergic Crisis
• Patients taking an excess of acetylcholinesterase
inhibitors may precipitate a cholinergic crisis
characterized by both muscarinic and nicotinic toxicity
• Symptoms may include an increase in perspiration,
lacrimation, salivation and pulmonary secretions,
nausea, vomiting, diarrhea, bradycardia, and
fasciculations
• Although cholinergic crisis is an important consideration
in the evaluation of the patient in myasthenic crisis, it is
uncommon.
• Regardless of whether myasthenic or cholinergic crisis
is suspected, acetylcholinesterase inhibitors should be
significantly lowered or discontinued to avoid excessive
pulmonary secretions in the setting of respiratory
distress.

Drugs with Interactions in MG
• Antibiotics
• Aminoglycosides: e.g., streptomycin, tobramycin,
kanamycin
• Quinolones: e.g., ciprofloxacin, levofloxacin, ofloxacin,
gatifloxacin
• Macrolides: e.g., erythromycin, azithromycin,
• Nondepolarizing muscle relaxants for surgery
• D-Tubocurarine (curare), pancuronium, vecuronium,
atracurium
• Beta-blocking agents
• Propranolol, atenolol, metoprolol

• Local anesthetics and related agents
• Procaine, Xylocaine in large amounts
• Procainamide (for arrhythmias)
• Botulinum toxin
• Botox exacerbates weakness
• Quinine derivatives
• Quinine, quinidine, chloroquine, mefloquine (Lariam)
• Magnesium
• Decreases ACh release
• Penicillamine
• May cause MG

Lambert-Eaton Myasthenic
Syndrome (LEMS)
• Rare presynaptic disorder of neuromuscular
transmission in which quantal release of acetylcholine
(ACh) is impaired
• An autoimmune attack directed against the voltage-
gated calcium channels (VGCCs) on the presynaptic
motor nerve terminal results in a loss of functional
VGCCs at the motor nerve terminals
• VGCC antibody levels do not correlate with disease
severity among patients with LEMS.
• However, antibody levels do fall in individual patients
if the disease improves after cancer therapy or
immunosuppression

• In patients with LEMS who have SCLC or other cancer,
cancer cells presumably contain antigens that mimic
VGCCs and induce production of VGCC antibodies.
• In patients with LEMS but no cancer, VGCC antibodies
are probably produced as part of a more general
autoimmune state.
• In patients who have LEMS without cancer, an antibody
response to domain IV of the 1A subunit of P/Q-type
VGCCs is more common than in patients who have
LEMS with cancer.

• Symptoms of LEMS usually begin insidiously and
progress slowly.
• Many patients have symptoms for months or years
before the diagnosis is made.
• Weakness is the major symptom.
• Proximal muscles are more affected than distal
muscles; lower extremity muscles are affected
predominantly
• LEMS patients never present initially with ocular
weakness; 5% present with bulbar weakness, and 95%
present with limb weakness.

• Increased temperatures from fever or the environment
may worsen the weakness.
• Patients may experience transient worsening after hot
baths and showers or during systemic illnesses.
• The oropharyngeal and ocular muscles are mildly
affected in about one quarter of cases of LEMS, with
symptoms that may include ptosis, diplopia, and
dysarthria, but they are usually not affected to the same
extent or severity as in myasthenia gravis
• Respiratory muscles are not usually affected. When
respiratory muscle function often is involved, the
involvement is usually not as severe as with MG

• Most patients have a dry mouth, which frequently
precedes other symptoms of LEMS. (Many do not mention
this unless specifically questioned.)
• Many patients report an unpleasant metallic taste.
• Some patients have other manifestations of autonomic
dysfunction, including impotence in males and postural
hypotension.
• Exacerbation of weakness has been described after
administration of aminoglycoside or fluoroquinolone
antibiotics, magnesium, calcium channel blockers, and
iodinated intravenous contrast agents.

• Reflexes usually are reduced or absent in LEMS.
• They can frequently be provoked or increased by having
the patient actively contract the muscle group in
question for 10 seconds prior to reflex testing or by
repeatedly tapping the muscles.
• An increase in reflex activity after contraction is a
hallmark of LEMS

Voltage-gated calcium channel antibodies
• Antibodies to voltage-gated calcium channels (VGCCs)
have been reported in 75-100% of LEMS patients who
have small cell lung cancer (SCLC) and in 50-90% of
LEMS patients who do not have underlying cancer.
• They are also found in fewer than 5% of patients with
myasthenia gravis (MG), in up to 25% of patients with
lung cancer without LEMS, and in some patients who do
not have LEMS but have high levels of circulating
immunoglobulins (eg, those with systemic lupus
erythematosus or rheumatoid arthritis).

Acetylcholine receptor antibodies
• ACh receptor (AChR) antibodies are most commonly
associated with myasthenia gravis (MG) and are
occasionally found in low titers in LEMS.
• The only true methods of differentiating MG from LEMS
are the detection of AChR antibodies and the presence
of underlying malignancy.

• In all adult patients with LEMS, diagnostic imaging (eg,
computed tomography [CT] or magnetic resonance
imaging [MRI]) of the chest for cancer detection should
be performed
• If imaging findings are negative in a patient with a
substantial risk of having lung cancer, bronchoscopy
should be performed.
• If both imaging and bronchoscopy results are initially
negative and risk factors for lung cancer are present,
positron emission tomography (PET) scanning should
be considered
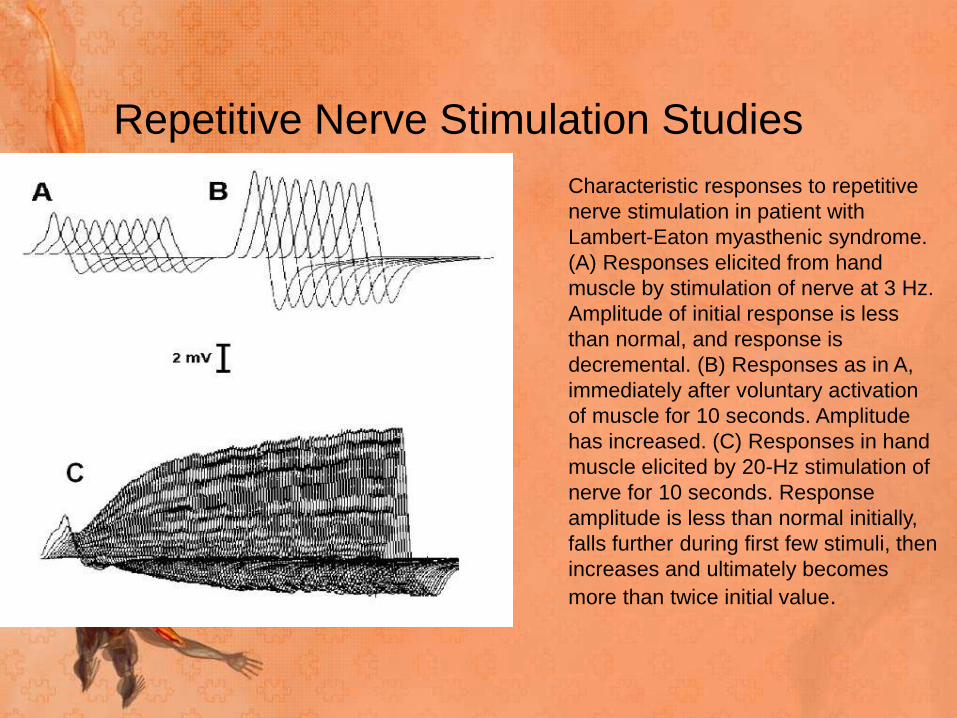
Repetitive Nerve Stimulation Studies
Characteristic responses to repetitive
nerve stimulation in patient with
Lambert-Eaton myasthenic syndrome.
(A) Responses elicited from hand
muscle by stimulation of nerve at 3 Hz.
Amplitude of initial response is less
than normal, and response is
decremental. (B) Responses as in A,
immediately after voluntary activation
of muscle for 10 seconds. Amplitude
has increased. (C) Responses in hand
muscle elicited by 20-Hz stimulation of
nerve for 10 seconds. Response
amplitude is less than normal initially,
falls further during first few stimuli, then
increases and ultimately becomes
more than twice initial value.

• RNS studies confirm the diagnosis of LEMS by
demonstrating characteristic findings
• Compound muscle action potentials (CMAPs) recorded
with surface electrodes are usually small, often less
than 10% of normal, and fall during 1- to 5-Hz RNS.
• During stimulation at 20-50 Hz, the CMAP increases in
size (ie, facilitation) and characteristically becomes at
least twice the size of the initial response.
• A similar increase in CMAP size is seen immediately
after the patient voluntarily contracts the muscle
maximally for several seconds

• Facilitation greater than 100% is seen in some but not all
muscles (or in all patients) with LEMS.
• Facilitation greater than 50% in any muscle suggests
LEMS.
• If facilitation is greater than 100% in most muscles tested
or greater than 400% in any muscle, the patient almost
certainly has LEMS.
• If facilitation is less than 50% in all muscles tested, the
patient still may have LEMS, especially if weakness has
been present for only a short time or the patient has been
partially treated.
• In LEMS, the CMAP amplitude is low in most muscles
tested

• When LEMS is mild, the electromyography (EMG)
findings may resemble those of MG, including normal
CMAP amplitudes, decremental response to RNS at low
rates, and little facilitation.
• One helpful feature is that in LEMS, the EMG findings
are usually more severe than the clinical findings would
suggest.
• The opposite is frequently true in MG

Single-fiber electromyography
• The jitter and blocking measured by single-fiber EMG is
increased markedly in LEMS, frequently out of
proportion to the severity of weakness.
• In many endplates, jitter and blocking decrease as the
firing rate increases. This pattern is not seen in all
endplates or in all patients with LEMS.
• Because jitter and blocking may also decrease at higher
firing rates in some endplates of patients with MG, this
pattern does not confirm an LEMS diagnosis unless it is
dramatic and seen in most muscles.

Management
• If an underlying neoplasm is present (eg, small cell lung
cancer [SCLC]), initial treatment should be aimed at the
neoplasm because weakness frequently improves with
effective cancer therapy.
• Typical treatments for patients with SCLC as the cause
of their LEMS would include combination therapy with
cisplatin and etoposide.
• Through both tumor modulation and its direct
immunosuppressive properties, chemotherapy does
seem to improve the symptoms of LEMS.
• In patients with LEMS who do not have cancer,
aggressive immunotherapy should be considered

• A Cochrane review identified only 4 controlled trials of
3,4-diaminopyridine (DAP) and a single cross-over study
that examined the use of IV immunoglobulin (IVIg) and
concluded that there was limited but moderate-to-high-
quality evidence to suggest improved muscle strength
with these interventions
• The initial pharmacotherapy for LEMS is with agents that
increase the transmission of acetylcholine (ACh) across
the neuromuscular junction, either by increasing the
release of ACh (eg,3,4 DAP) or by decreasing the action
of acetylcholinesterase (eg, pyridostigmine).

• If these treatments are not effective and the patient has
relatively mild weakness, aggressive immunotherapy may
be warranted. In such cases, plasma exchange (PEX) or
high-dose intravenous immunoglobulin (IVIg) may be
used initially to induce rapid, but transitory improvement.
• Prednisone and azathioprine, the most frequently used
immunosuppressants, can be used alone or in
combination.
• Cyclosporine may benefit patients with LEMS who are
candidates for immunosuppression but cannot take or do
not respond well to azathioprine












