Embed Size (px)
Citation preview

CRISPR-Cas9: A Potential Tool for Genome Editing
1. Introduction
1.1. Origins
In the year 1987, a team of Japanese scientists were the first to describe an unusual locus
found in the E. coli genome, adjacent to the iap gene, having short palindromic repeats interspersed
by similarly sized non-repetitive DNA spacers (Fig. 1) [1]. This clustered, regularly interspersed,
short palindromic repeat locus is therefore termed “CRISPR”. Nearly a decade later, up to forty
percent of sequenced bacteria and ninety percent of archaea were found to harbor this CRISPR locus
[2] [3].
In the year 2002, bacterial strains that survived bacteriophage infection were observed to
express the CRISPR loci, suggesting that the particular region may have a role in prokaryotic adaptive
immunity [4]. This hypothesis was subsequently confirmed when phage-resistant bacterial strains had
specific CRISPR loci spacers disrupted, they acquired susceptibility to phage infection, while
insertion of novel spacers into wild type stains demonstrated acquired resistance [5].
The proteins involved in the prokaryotic immune system were found to be conserved and
encoded in close proximity to the CRISPR locus (Fig. 1) [6]. As such, these proteins are known as
CRISPR-associated, or in short, “Cas”.
1.2. Mechanism of Action
The bacterial CRISPR-based acquired immunity (Fig. 1) encompasses three processes,
namely, spacer addition, CRISPR-RNA (crRNA) maturation and target elimination [7][8][9].
Upon initial exposure to foreign phage DNA, bacterial Cas1 locates the DNA’s unique
protospacer adjacent motif (PAM) and cuts a short DNA fragment (protospacer) that is directly beside
[7]. Integration of this protospacer into the CIRSPR locus is directed by the Cas1-Cas2 complex [7].
Successful integration of protospacers into the host genome are thereafter referred to as spacers.
A pre-crRNA is a long mRNA transcript of the CRISPR locus containing an array of spacer
and repeat sequences. It hybridizes with multiple trans-activating crRNAs (tracrRNA) to form RNA
duplex structures that are targeted for cleavage by RNase III [10]. The cleaved, mature crRNA,
encodes for a particular spacer and repeat sequence which remains hybridized to a tracrRNA, and this
short duplex is called the guide RNA (gRNA) [7][8].
Each gRNA has a unique spacer sequence that recognizes its complementary protospacer of
the phage DNA [7][8]. When an immunized bacterial cell reencounters the same phage DNA, the
appropriate gRNA guides Cas9, an endonuclease, to specifically target and eliminate the invading
DNA by inducing a site-specific double strand break (DSB) [7][8]. The PAM ensures that Cas9

complexes locate the correct protospacer sequence [7]. As the PAM is only found on invading phage
DNA, the DSB mechanism is able to discriminate self from non-self [7].
Figure 1: Bacterial CRISPR-based acquired immunity. Phase 1 (immunization): Initial injection of phage double strand DNA into a
bacterial cell followed by Cas1-Cas2 complex (large teal oval with associated grey ovals) excision of the protospacer (yellow rectangle)
from the phage DNA. The Cas1-Cas2 complex inserts the protospacer into the Type II CRISPR locus region containing short palindromic
repeats (black) and novel spacers derived from other foreign phage DNA (purple, green and red rectangles). Phase 2 (immunity): Pre-crRNA
(multi-colored linear mRNA) and tracrRNA (purple stem-loop mRNA) are transcribed and processed into guide RNA (mature crRNA-
tracrRNA duplex) which subsequently guides Cas9 (small teal oval) to cleave the invading DNA. (Figure adapted from [9])
1.3. Tool for Genome Editing
There are four major tools capable of inducing site-specific double strand breaks (DSB); zinc
finger nuclease (ZFN), transcription activator-like effector nuclease (TALEN), meganuclease and
CRISPR-Cas complex [11]. Any of these platforms may be programmed for DSB-induced genome
editing in prokaryotes and eukaryotes by exploiting their endogenous DNA repair mechanisms [11].
Breaks in the genome are remediated via one of two main repair mechanisms; non-
homologous end-joining (NHEJ) or homology-directed repair (HDR) [11]. The NHEJ pathway does
not utilize a template to flank the break region, hence, there is random insertion and/or deletions
(indels) of nucleotides at the site of damage [11]. This error-prone repair mechanism results in
frameshifts at the break site (Fig. 2.), and if the region encodes for a gene, its function is knocked out
[11]. On the other hand, HDR employs a template sequence that is homologous to the break site [11].
This high fidelity repair mechanism is mainly active during the S and G2 phases of mammalian cell
cycle, and the sister chromatid is used as template for the break sites [11]. However, an exogenous
template may be introduced to flank these break sites to incorporate synthetic sequences (Fig. 2) [11].
The CRISPR-Cas system has been praised for its efficiency, precision, low cost and ease of
use [8]. Furthermore, for multiplexing purposes, this system has the ability to simultaneously edit
multiple target sites without the need of cumbersome protein engineering as required in ZFNs or
TALENs [7][8]. Three types of CRISPR systems have been identified, however, only the type II
CRISPR-Cas system is the least complex as it only requires three components to function; Cas

endonuclease, mature crRNA and auxiliary tracrRNA [2][7]. This system has been further simplified
by fusing the crRNA with the tracrRNA to form a single guide RNA (sgRNA) [7][11].
Figure 2: Genome double strand break (DSB) repair mechanism. Single endonuclease-induced DSB (left): Non-homologous end-joining
(NHEJ) knocks out gene function by altering the gene’s original stop codon location. Homology-directed repair (HDR) occurs via strand
invasion into complementary templates. HDR may knock-in point mutations when provided with a mutated exogenous flanking template
(light blue dsDNA with star). HDR may knock-in new gene function when the exogenous flanking template contains its coding sequence
(light blue dsDNA with orange regions). Dual endonuclease-induced DSB (right): NHEJ large sequence deletions. (Figure adapted from
[11])
The type II CRISPR-Cas9 system is derived from Streptococcus pyogenes, and this powerful
genome editing tool has already been employed to alter the genes of different organisms and cell
types ranging from microorganisms, insects, plants, fishes, to mammalian cell lines [12]. Here, I will
review a few CRISPR-Cas9 genome editing applications, limitations and optimizations.
2. Applications
2.1. Disease Modeling
Mice are routinely used to study mammalian genetics and associated diseases by knocking
out or knocking in specific genes through embryonic stem cell (ES) homologous DNA recombination
methods [13]. As these methods were first established in mouse ES cells, genome editing has been
limited to mice. This posed difficulties in editing other mammalian species that may be better suited
to model unique human diseases [13][14]. Furthermore, it may take up to a year or more to
successfully produce a genetically altered mouse [8].
In human cell lines, homologous recombination methods have had little rate of success such
that alternative approaches with short interfering RNA have become common [8]. These alternatives
also bring about their own set of limitations such as transient gene expression knock-downs and off
target effects contributing to experimental inaccuracies [8].

Recent developments in genome editing technologies have enabled researchers to circumvent
the limitations around animal and cell line based disease modeling. Advantages include the ability to
genetically tweak animals for which ES cell lines are unavailable, the potential to acquire
homozygous knock-outs within the first generation, and the opportunity to explore other types of
animal models [8].
With CRISPR-Cas9 multiplexed genome editing, multiple generations of interbreeding to
derive an animal model with many genetic modifications can be avoided. This multiplexing potential
has been demonstrated to knock-out two genes of a monkey in one step by co-injecting one-cell-stage
embryos with Cas9 mRNA and sgRNAs [15]. In mice, CRISPR-Cas9 multiplexed genome editing of
two genes produced biallelic mutations with eighty percent efficiency [16]. Also, the CRISPR-Cas9
system empowers researchers to study single nucleotide polymorphism-associated human diseases
efficiently in mice by introducing a donor template that encodes the mutated sequence (Fig. 2) [14].
These remarkable achievements suggest that there may be no technical barrier in using the CRISPR-
Cas9 system to model other animals or cell lines for the development of new pharmaceuticals.
2.2. Drug Development
Genome-wide association studies, a field of study in functional genomics, have provided us a
wealth of knowledge on polygenic diseases such as Alzheimer’s, schizophrenia, autism and diabetes
[7]. Drug development for these genetic diseases relies heavily on disease model studies in zebrafish,
drosophila, mice and even mammalian cell lines using RNA interference (RNAi) [17][18]. As
mentioned previously in the disease modeling section, RNAi techniques have the tendency for partial
knock-downs and unmeasurable phenotypes [17]. Despite these limitations, genome-wide screening
for novel therapeutic targets have been using subsets of the RNAi technology, specifically, short
hairpin RNA (shRNA) and small interfering RNA [17].
The advancement of programmable genome editing platforms has enabled the generation of
gene knock-outs and mutation knock-ins in clinically relevant animal and cell based models for target
validation and drug discovery [17]. Unlike RNAi, which silences a particular gene expression by
targeting its mRNA transcripts, the CRISPR-Cas system targets its DNA coding sequence directly,
leading to permanent and complete gene knock-out [18]. As amendments made to the genome via the
CRISPR-Cas9 system are irreversible, alternative methods have been explored to mimic the RNAi
gene silencing approach, but at the DNA level as it confers greater efficiency [18]. A team of
scientists managed to create a dead Cas9 (dCas9) by inactivating its DSB catalytic mechanism [19].
Therefore, when guided by an engineered sgRNA, the dCas9-sgRNA complex interferes with the
targeted gene’s transcription, strongly silencing its gene expression [19]. This CRISPR interference
(CRISPRi) method appears to be most analogous to the RNAi principle.
In summary, the CRISPR-Cas9 system has enabled the study of other clinically relevant
animal models. Also, the ability to perform multiplexed genome editing and interference has opened

up greater opportunities for novel therapeutic target screening. Overall, functional genomics has
uncharted territories for us to explore in terms of polygenic disease knowledge and novel drug
development.
2.3. Regenerative Medicine
Many genetic disorders require individuals to be placed under prolonged or lifelong
medication. Based on current developments in genetic engineering, gene therapy may soon become a
primary route of treatment for genetic diseases [7]. Several proof of concept studies have
demonstrated the potential of gene therapy to correct monogenic recessive disorders such as
hemophilia, cystic fibrosis and Duchenne muscular dystrophy (DMD) [7][11]. On the other hand,
non-genetic diseases may also be prevented by modifying one’s genome.
Knock-out gene therapy relies on the error-prone NHEJ repair mechanism to induce indels at
the targeted gene, causing a frameshift mutation (Fig. 2) [11]. Familial hypercholesterolemia is an
autosomal dominant genetic disease caused by a mutation in the PCSK9 gene [20]. This gene codes
for a proteinase that degrades low density lipoprotein receptors (LDLR) and therefore contributes to
decreased metabolism of LDL cholesterol by the liver, increasing the risk for cardiovascular diseases
[20]. Patients with this condition are continuously prescribed PCSK9 proteinase-antagonists, however,
a single co-injection of Cas9 together with PCSK9-targeted sgRNA into mice liver in vivo has shown
to knock-out the targeted gene and effectively lower cholesterol levels [21]. Another promising
application of knock-out gene therapy is by disrupting the CCR5 gene encoding a major co-receptor
essential for HIV-1 to infect CD4+ T-cells [7][11]. Successful clinical trials have been observed with
ZFN edited stem cells being reintroduced into patients [7]. Similarly, engrafting Cas9 edited human
hematopoietic stem and progenitor cells appears to be a powerful alternative to combat AIDS [22].
Knock-in gene therapy employs the high fidelity HDR mechanism of cells to rectify
mutations by providing an exogenous template encoding the correct gene sequence to flank the DSB
site (Fig. 2) [11]. In addition, an alternative error-free insertion of exogenous genetic elements up to
fifteen kilobases have been developed using NHEJ-mediated ligation [23]. This is achieved by using
nuclease-induced DSBs to generate compatible overhangs on both the exogenous template and the
endogenous target site [23]. Both methods are advantageous compared to conventional viral vector-
mediated gene therapy associated with random insertion mutagenesis [11].
In certain cases, where knock-out or knock-in gene therapies may be inappropriate to rectify a
particular genetic disease, deletion-based gene therapy may be explored. To delete large genetic
elements several megabases in size from the genome, two targeted DSBs flanking the region of
interest must be simultaneously administered (Fig. 2) [11]. Using the CRISPR-Cas9 platform, this
method has been proven to be helpful in treating hemoglobinopathis by removing an entire BCL11A
erythroid-specific enhancer region [24]. Additionally, in DMD, where internal gene deletions lead to

frameshifts and as a result causes protein dysfunction, deliberate targeted excision of its exons can
rectify the frameshifts to generate a truncated, partially functional protein [11].
Targeted gene therapy with the CRISPR-Cas9 system hold much promise in the upcoming
decade as numerous proof of concept studies have already been established [7][11], and it is only a
matter of time before a flood of new clinical trials using this system will be approved.
2.3. Medical Microbiology
The abuse of antibiotics in medicine and animal agriculture has led to great selection pressure
on bacteria such that multidrug-resistant strains, especially the human pathogens, are of growing
concern as our pre-existing arsenal of useful antibiotics is shrinking. Another issue is that these
antibiotics have broad-spectrum targets and does not discriminate pathogens from beneficial normal
flora in our systems. Alternative novel antimicrobials have been explored using the CRISPR-Cas9
system to circumvent these limitations [25].
The sgRNA in the CRISPR-Cas9 system can be engineered to target virtually any essential
genomic regions of the bacteria for killing. As for the mode of delivery, preliminary studies employed
bacteriophages with phagemids encoding the Streptococcus pyogenes Cas9, the engineered crRNA
and the auxiliary tracrRNA [26][27]. These studies demonstrated effective killing of pathogenic
Staphylococcus aureus, carbapenem-resistant Enterobacteriaceae and enterohemorrhagic Escherichia
coli by targeting their virulence genes [26][27]. Specificity was also observed in one of the study as
the sequence-specific Cas9 discriminatively targeted virulent from avirulent Staphylococcus aureus
[26].
The CRISPR-Cas9 system can also be re-programmed to target antibiotic resistance genes
residing within the bacterial genome or in plasmids [25]. This could possibly be a method to restrict
plasmid-borne antibiotic resistance between clinically relevant pathogenic strains. Also, re-sensitizing
pathogenic strains to previously obsolete antibiotics can re-expand our antimicrobial arsenal.
In summary, sequence-specific antimicrobials may be used to treat multidrug-resistant
infections without killing beneficial normal flora by either directly via genome disruption or indirectly
through re-sensitization to antibiotics. However, as with any antimicrobials, further studies have to be
conducted to evaluate the types of evolution this method could bring about due to selection pressure.
2.5. Industrial Microbiology
A number of industrially relevant fungi and yeast strains utilized for the production of biofuel
are tough to engineer because of their intricate genomes [28]. Saccharomyces cerevisiae, one of the
most commonly exploited microbial cell factories, have diploid or polyploidy stains that make
genome editing a tricky task. Conventional methods employ selection of markers that are co-
integrated into its genome for gene mutation, deletion and integration [28]. Metabolic engineering has

found the CRISPR-Cas9 system to be a versatile genome editing tool to overcome these types of
challenges [28].
Genome engineering of several different industrially relevant Saccharomyces cerevisiae
strains using the CRISPR-Cas9 platform has demonstrated successful biallelic disruption of a gene
with efficiency up to seventy-eight percent [29]. In addition, by harnessing the multiplex nature of the
CRISPR-Cas9 platform, genome engineering of up to five different genomic loci in Saccharomyces
cerevisiae has demonstrated to increase mevalonate titers up to forty-fold compared to wild-type
strains, although no overexpression of genes was done in the mevalonate pathway [30]. Therefore,
CRISPER-Cas9 can be used in strain optimization to yield greater amounts of important secondary
metabolites.
2.6. Agricultural Biotechnology
Genetically modified crops incorporated with transgene via the HDR mechanism undergoes
strict regulations, decreasing their commercial viability [7]. NHEJ knock-outs however, are said to be
non-transgenic and is beyond the United States Department of Agriculture (USDA) regulatory
authority [31]. Highlighted here are a few prominent CRISPR-Cas9 based crop and livestock genome
editing that have taken place over the past few years.
Engineering plants and crops to be resistant to diseases is one of the major goals for
agricultural biologists. Leveraging upon the superior gene silencing and multiplexing capabilities of
the CRISPR-Cas9 system over RNAi, a number of model plant and crop species have been further
studied [7]. Examples include Arabidopsis, tobacco, tomato, maize, rice and wheat [7]. The powdery
mildew disease is a deadly wheat infection caused by the fungus Blumeria graminis [7]. In bread
wheat, Triticum aestivum, a hexaploid crop, three mildew resistance locus (MLO) homoeoalleles were
mutated via CRISPR-Cas9 [32]. Subsequent self-fertilization yielded knock-outs for all six alleles,
conferring the bead wheat resistance to powdery mildew disease [32].
Biofortification is the process by which plants, crops and livestock are genetically engineered
to enhance their nutritional value. Improvements can be made directly by modulating the amount of
nutrient the foodstuff produces, or indirectly by eliminating anti-nutrients that decreases nutrient
bioavailability [7]. Goat, sheep, pig and cattle have already been successfully engineered using the
CRISPR-Cas9 system [7]. Other biofortification strategies using CRISPR-Cas9 are still under
development, and although ZNFs and TALENs have already been extensively used, the potential of
CRISPR technology to improve the nature of agricultural produce and livestock remains massive.
3. Limitations
3.1. Target Specificity
Off-targeting, whereby DSBs are made at sites other than the intended target region, has been
one of the greatest limitation of not only the CRISPR-Cas9 system, but also other genome editing
tools within the family of programmable nucleases [2][8]. Should off-target mutations occur
frequently using the CRISPR-Cas9 system, there may be a loss of genomic stability and functionality
of otherwise normal genes, reducing the reliability of the tool for biomedical and clinical applications.
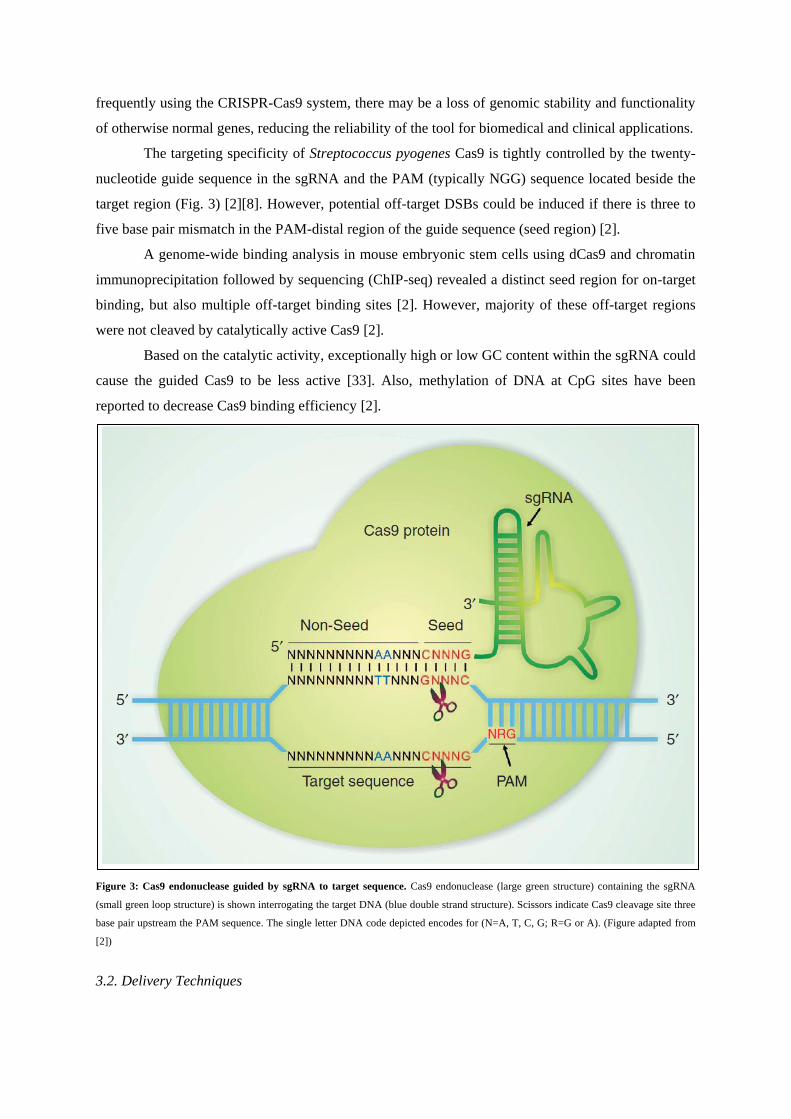
The targeting specificity of Streptococcus pyogenes Cas9 is tightly controlled by the twenty-
nucleotide guide sequence in the sgRNA and the PAM (typically NGG) sequence located beside the
target region (Fig. 3) [2][8]. However, potential off-target DSBs could be induced if there is three to
five base pair mismatch in the PAM-distal region of the guide sequence (seed region) [2].
A genome-wide binding analysis in mouse embryonic stem cells using dCas9 and chromatin
immunoprecipitation followed by sequencing (ChIP-seq) revealed a distinct seed region for on-target
binding, but also multiple off-target binding sites [2]. However, majority of these off-target regions
were not cleaved by catalytically active Cas9 [2].
Based on the catalytic activity, exceptionally high or low GC content within the sgRNA could
cause the guided Cas9 to be less active [33]. Also, methylation of DNA at CpG sites have been
reported to decrease Cas9 binding efficiency [2].
Figure 3: Cas9 endonuclease guided by sgRNA to target sequence. Cas9 endonuclease (large green structure) containing the sgRNA
(small green loop structure) is shown interrogating the target DNA (blue double strand structure). Scissors indicate Cas9 cleavage site three
base pair upstream the PAM sequence. The single letter DNA code depicted encodes for (N=A, T, C, G; R=G or A). (Figure adapted from
[2])
3.2. Delivery Techniques

Lentiviral and Adeno-associated virus (AAV) vectors have been commonly used to transfer
genetic information into cells. However, they do not have sufficient capacity to shuttle an entire Cas9
genome editing infrastructure [7]. Although other viral vectors such as adenovirus possess greater
carrying-capacity, they are highly immunogenic and have limited cell-type infectivity [7].
4. Optimizations
4.1. System Modifications
Three different forms of modification have been made to the Cas9 system to increase
targeting fidelity. Firstly, inactivation of the RuvC nuclease domain generates a Cas9 with nickase
(nCas9) functionality, causing only targeted single strand breaks in DNA instead of DSBs [7].
Appropriately spaced nicks formed with two nCas9 can mimic the effects of DSB, reducing the off-
target frequency as compared to using only one endonuclease [7].
Secondly, as off-target DSBs are mainly due to the strong binding affinity between the
sgRNA and non-specific target site, a truncated guide sequence could reduce the binding affinity such
that mismatched base pairs are no longer well tolerated, decreasing off-target editing [7][8].
Lastly, dCas9 can be engineered with Fok I nuclease domain to generate fCas9 [7]. A paired
fCas9 functions analogously to the paired nCas9, and has up to 140-fold increase in specificity
compared to wild type Cas9 [7].
Earlier this year, researchers have reported the generation of a high-fidelity CRISPR-Cas9
system capable of precise genome edition with undetectable off-target genome disruption [36].
4.2. Alternative Delivery Techniques
To overcome the capacity limitations of viral vectors, the essential Cas9 genome editing
modules have been engineered into separate plasmids and co-transfected into cells [7]. For in vivo
disease modeling, mice have been engineered to express Cas9 in a Cre-driven manner, such that only
the sgRNA is required for efficient genome editing [7]. Another novel method to introduce Cas9
proteins into cells involve fusion with anionic supercharged GFP that are delivered to human cell
lines and mice using cationic liposomes [35].
Direct introduction of purified Cas9 proteins together with the engineered sgRNA into cells
have shown to minimize off-target DSBs compared to plasmid-based Cas9-sgRNA expression [34].
The increased fidelity was reported to be derived from the rapid degradation of these Cas9-sgRNA
complexes in cells [34].
5. Conclusion
In recent years, research and application of the CRISPR-Cas9 system have picked up pace.
The advantages associated with this system compared to other programmable nucleases are
efficiency, specificity, time and cost saving. Furthermore, its ability to conduct reliable multiplexed

applications have opened up the opportunity for us to explore the vast knowledge of functional
genomics unattainable previously with RNAi technology.
This genome editing tool is providing advancement in many scientific fields. Enhancing
agriculture, defending against pathogens and remediating human genetics are just few of the benefits
listed from the large pool of published studies. With the recent development of high-fidelity Cas9 that
generates no detectable genome-wide off targets, many human-based clinical trials harnessing this
powerful genome editing platform are expected to be approved sooner than later.
Many questions still remain about how the innate immune system of organisms targeted with
CRISPR-Cas9 will respond. As of now, little is known on the selection pressure or evolution these
types of genome editing can bring about. Nevertheless, as many genome editing preclinical studies
and clinical trials have been successful, there is much hope and optimism for the future of genome
editing with CRISPR-Cas9 technology.
6. References
1. Ishino, Y et al. (1987). Nucleotide Sequence of the iap Gene, Responsible for Alkaline
Phosphatase Isozyme Conversion in Escherichia coli, and Identification of the Gene Product.
J Bacteriol, 169, 5429-5433.
2. Zhang, XH et al. (2015). Off-target Effects in CRISPR/Cas9-mediated Genome Engineering.
Mol Ther Nucleic Acids, 4, e264. doi:10.1038/mtna.2015.37
3. Mojica, FJ et al. (2000). Biological significance of a family of regularly spaced repeats in the
genomes of Archaea, Bacteria and mitochondria. Mol Microbiol, 36, 244.
4. Tang, TH et al. (2002). Identification of 86 candidates for small non-messenger RNAs from
the archaeon Archaeoglobus fulgidus. Proc Natl Acad Sci USA, 99, 7536.
5. Barrangou, R et al. (2007). CRISPR provides acquired resistance against viruses in
prokaryotes. Science, 315, 1709.
6. Jansen, R et al. (2002). Identification of genes that are associated with DNA repeats in
prokaryotes. Mol Microbiol, 43, 1565.
7. Rajendran, SR et al. (2015). CRISPR-Cas9 Based Genome Engineering: Opportunities in
Agri-Food-Nutrition and Healthcare. OMICS, 19(5), 261-275. doi:10.1089/omi.2015.0023
8. Gupta, RM and Musunuru, K. (2014). Expanding the genetic editing tool kit: ZFNs,
TALENs, and CRISPR-Cas9. J Clin Invest, 124(10), 4154-4161. doi:10.1172/JCI72992

9. Wang, Y. (2014). Developing CRISPR/Cas9 Technologies for Research and Medicine. MOJ
Cell Science & Report, 1(1). doi:10.15406/mojcsr.2014.01.00006
10. Deltcheva, E et al. (2011). CRISPR RNA maturation by trans-encoded small RNA and host
factor RNase III. Nature, 471, 602. doi: 10.1038/nature09886
11. Maeder, ML and Gersbach, CA. (2016). Genome-editing Technologies for Gene and Cell
Therapy. Mol Ther. doi:10.1038/mt.2016.10
12. Sander, JD and Joung, JK. (2014). CRISPR-Cas systems for editing, regulating and targeting
genomes. Nat Biotechnol, 32, 347.
13. Fan, Z et al. (2014). Efficient gene targeting in golden Syrian hamsters by the CRISPR/Cas9
system. PLoS One, 9(10), e109755. doi:10.1371/journal.pone.0109755
14. Mashimo, T. (2014). Gene targeting technologies in rats: zinc finger nucleases, transcription
activator-like effector nucleases, and clustered regularly interspaced short palindromic
repeats. Dev Growth Differ, 56(1), 46-52. doi:10.1111/dgd.12110
15. Niu, Y et al. (2014). Generation of gene-modified cynomolgus monkey via Cas9/RNA-
mediated gene targeting in one-cell embryos. Cell, 156(4), 836-843.
doi:10.1016/j.cell.2014.01.027
16. Wang, H et al. (2013). One-step generation of mice carrying mutations in multiple genes by
CRISPR/Cas-mediated genome engineering. Cell, 153(4), 910-918.
doi:10.1016/j.cell.2013.04.025
17. Moore, JD. (2015). The impact of CRISPR-Cas9 on target identification and validation. Drug
Discov Today, 20(4), 450-457. doi:10.1016/j.drudis.2014.12.016
18. Taylor, J and Woodcock, S. (2015). A Perspective on the Future of High-Throughput RNAi
Screening: Will CRISPR Cut Out the Competition or Can RNAi Help Guide the Way? J
Biomol Screen, 20(8), 1040-1051. doi:10.1177/1087057115590069
19. Qi, L et al. (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific
control of gene expression. Cell, 152(5), 1173-1183. doi:10.1016/j.cell.2013.02.022

20. Abifadel, M et al. (2003). Mutations in PCSK9 cause autosomal dominant
hypercholesterolemia. Nat Genet, 34(2), 154-156. doi:10.1038/ng1161
21. Ran, FA et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature,
520(7546), 186-191. doi:10.1038/nature14299
22. Mandal, PK et al. (2014). Efficient ablation of genes in human hematopoietic stem and
effector cells using CRISPR/Cas9. Cell Stem Cell, 15(5), 643-652.
doi:10.1016/j.stem.2014.10.004
23. Maresca, M et al. (2013). Obligate ligation-gated recombination (ObLiGaRe): custom-
designed nuclease-mediated targeted integration through nonhomologous end joining.
Genome Res, 23(3), 539-546. doi:10.1101/gr.145441.112
24. Canver, MC et al. (2015). BCL11A enhancer dissection by Cas9-mediated in situ saturating
mutagenesis. Nature, 527(7577), 192-197. doi:10.1038/nature15521
25. Beisel, CL et al. (2014). A CRISPR design for next-generation antimicrobials. Genome
Biology, 15(11), 1-4. doi:10.1186/s13059-014-0516-x
26. Bikard, D et al. (2014). Exploiting CRISPR-Cas nucleases to produce sequence-specific
antimicrobials. Nat Biotechnol, 32(11), 1146-1150. doi:10.1038/nbt.3043
27. Citorik, RJ et al. (2014). Sequence-specific antimicrobials using efficiently delivered RNA-
guided nucleases. Nat Biotechnol, 32(11), 1141-1145. doi:10.1038/nbt.3011
28. Estrela, R and Cate, JH. (2016). Energybiotechnology in the CRISPR-Cas9 era. Curr Opin
Biotechnol, 38, 79-84. doi:10.1016/j.copbio.2016.01.005
29. Stovicek, V et al. (2015). CRISPR–Cas system enables fast and simple genome editing of
industrial Saccharomyces cerevisiae strains. Metabolic Engineering Communications, 2, 13-
22. doi:10.1016/j.meteno.2015.03.001
30. Jakociunas, T et al. (2015). Multiplex metabolic pathway engineering using CRISPR/Cas9 in

Saccharomyces cerevisiae. Metab Eng, 28, 213-222. doi:10.1016/j.ymben.2015.01.008
31. Waltz, E. (2012). Tiptoeing around transgenics. Nat Biotechnol, 30(3), 215-217.
doi:10.1038/nbt.2143
32. Wang, Y et al. (2014). Simultaneous editing of three homoeoalleles in hexaploid bread wheat
confers heritable resistance to powdery mildew. Nat Biotechnol, 32(9), 947-951.
doi:10.1038/nbt.2969
33. Doench, JG et al. (2014). Rational design of highly active sgRNAs for CRISPR-Cas9-
mediated gene inactivation. Nat Biotechnol, 32(12), 1262-1267. doi:10.1038/nbt.3026
34. Kim, S et al. (2014). Highly efficient RNA-guided genome editing in human cells via
delivery of purified Cas9 ribonucleoproteins. Genome Res, 24(6), 1012-1019.
doi:10.1101/gr.171322.113
35. Zuris, JA et al. (2015). Cationic lipid-mediated delivery of proteins enables efficient protein-
based genome editing in vitro and in vivo. Nat Biotechnol, 33(1), 73-80.
doi:10.1038/nbt.3081
36. Kleinstiver, BP et al. (2016). High-fidelity CRISPR-Cas9 nucleases with no detectable
genome-wide off-target effects. Nature, 529(7587), 490-495. doi:10.1038/nature16526





























