Embed Size (px)
Citation preview

Feature review
A G protein-coupled receptor at work:the rhodopsin modelKlaus Peter Hofmann1,2, Patrick Scheerer1, Peter W. Hildebrand1, Hui-Woog Choe1,a,Jung Hee Park1, Martin Heck1 and Oliver P. Ernst1
1 Institut fur Medizinische Physik und Biophysik (CC2), Charite – Universitatsmedizin Berlin, Chariteplatz 1, D-10117 Berlin, Germany2 Zentrum fur Biophysik und Bioinformatik, Humboldt Universitat zu Berlin, Invalidenstrasse 42, D-10115 Berlin, Germany
Review
Glossary
a5 helix: C-terminal a-helix of the G protein a-subunit.
Agonist: ligand that fully activates a receptor by shifting an equilibrium
between inactive and active receptor conformations. Partial agonists induce
submaximal activation at saturating concentrations. Inverse agonists reduce
the basal activity of a receptor.
Antagonist: ligand that does not affect basal activity but instead competes
with other ligands for the binding site.
Ballesteros–Weinstein numbering: the most conserved residue in a transmem-
brane helix among rhodopsin-like GPCRs is designated x.50, where x is the
helix number and .50 is used for reference. All other residues on the same helix
are numbered relative to the x.50 reference residue [94].
E(D)R3.50Y: conserved motif located at the cytoplasmic end of TM3 in class A
GPCRs.
G protein: heterotrimeric guanine nucleotide-binding protein consisting of a, b
and g subunits. Heterotrimers are typically divided into four main classes
based on the primary sequence similarity of the Ga subunits Gas, Gai, Gaq and
Ga12 [38].
G protein-coupled receptor (GPCR) family: the human GPCR superfamily (�800
members) contains five main GPCR families rhodopsin, secretin, adhesion,
glutamate and frizzled/taste2. The rhodopsin family (or class A GPCRs) is by far
the largest (�670 members). For different nomenclature and classification
systems see Ref. [1], the rhodopsin family is further subdivided into four
groups, a, b, g and d [1].
Gt: heterotrimeric G protein transducin of rod cells. Gt enables vision by
G protein-coupled receptors (GPCRs) are ubiquitous sig-nal transducers in cell membranes, as well as importantdrug targets. Interaction with extracellular agoniststurns the seven transmembrane helix (7TM) scaffold ofa GPCR into a catalyst for GDP and GTP exchange inheterotrimeric Gabg proteins. Activation of the modelGPCR, rhodopsin, is triggered by photoisomerization ofits retinal ligand. From the augmentation of biochemicaland biophysical studies by recent high-resolution 3Dstructures, its activation intermediates can now be inter-preted as the stepwise engagement of protein domains.Rearrangement of TM5–TM6 opens a crevice at thecytoplasmic side of the receptor into which the C termi-nus of the Ga subunit can bind. The Ga C-terminal helixis used as a transmission rod to the nucleotide bindingsite. The mechanism relies on dynamic interactions be-tween conserved residues and could therefore be com-mon to other GPCRs.
Function of G protein-coupled receptorsTo transmit extracellular signals into living cells, naturehas evolved membrane-spanning receptor proteins thatconnect the extracellular environment to the cell interior.G protein-coupled-receptors (GPCRs) are the largestfamily of such receptors, with approximately 800 differentmembers in humans [1]. Environmental and physiologicalsignals such as hormones, neurotransmitters, odorants,gustatory substances and light are received by these recep-tors, which are also the targets for many drugs, including b
blockers and antihistamines. GPCRs are thought torespond to the binding of extracellular ligands with aconformational change in the ligand binding site [2], whichextends via their seven transmembrane helix (7TM) scaf-fold into the intracellular domain [3,4]. The active cyto-plasmic receptor surface enables binding of cognateheterotrimeric G proteins (Gabg) and catalysis ofGDP!GTP exchange in the Ga subunit. The GTP-boundG protein then decouples from the receptor and dissociatesinto Ga–GTP and Gbg subunits. Both Ga–GTP and Gbg
subunits can elicit cell-specific responses via particulareffector proteins and regulation of intracellular second
Corresponding authors: Hofmann, K.P. ([email protected]);Ernst, O.P. ([email protected])
a Present address: Department of Chemistry, College of Natural Science, ChonbukNational University, 561-756 Chonju, South Korea..
540 0968-0004/$ – see front matter � 2009 Elsevie
messenger levels. Hydrolysis of GTP to GDP within Ga
and subsequent reassociation of Ga–GDP with Gbg com-pletes the G protein cycle. In concert with shut-off of theactivated receptor by interactions with receptor kinase andarrestin, an enzymatic feedback sets the secondmessengerconcentration back to its original level. The catalyticnature of receptor–G protein interaction results in thegeneration of many copies of the GTP-bound activated Gprotein, establishing a first step in signal amplification andregulation. The three steps of reception, amplification andfeedback constitute a signaling module that might becommon to signal transduction systems in general [5].
This review focuses on the first key step in G protein-mediated signal transduction, in which the signal crossesthe membrane and the activated receptor couples to the Gprotein.New insight into thisprocess comes fromrhodopsin,the photoreceptor of the retinal rod cell and the eponym ofthe largest class of GPCRs, the rhodopsin-like GPCRs with�670 members in the human genome [1]. Rhodopsin con-sists of the opsin apoprotein and the covalently bound
activating a cGMP-specific phosphodiesterase.
NP7.50xxY(x)5,6F: conserved motif with Asn, Pro, and Tyr located in the
cytoplasmic end of TM7 and Phe in H8 of rhodopsin-like GPCRs.
Schiff base (imine): condensation product of an aldehyde group and a primary
amino group; in rhodopsin from retinal and the side chain of Lys296.
r Ltd. All rights reserved. doi:10.1016/j.tibs.2009.07.005 Available online 21 October 2009

Box 1. GPCR toolbox
A toolbox of functional elements utilized by GPCRs can be derived
from data available on their sequence, function and structure (Figure I).
The two highly conserved E(D)R3.50Y and NP7.50xxY(x)5,6F motifs [28]
are part of functional microdomains that constrain the 7TM helix
bundle in the compact inactive conformation and undergo structural
changes on receptor activation [6–8,32,87]. In rhodopsin, Arg1353.50 of
the E(D)R3.50Y motif on TM3 plays a key role in tethering TM3 to TM6.
Arg1353.50 forms a hydrogen bonding network with Glu1343.49 on TM3
and Glu2476.30 on TM6; in rhodopsin the network also includes
Thr2516.34. A functional network around Arg1353.50 – known as the
TM3–TM6 ionic lock – was proposed for class A GPCRs (rhodopsin
family) [25,26] and might also be utilized by class C GPCRs (glutamate
family) [95]. The NPxxY(x)5,6F motif on TM7–H8 is functionally bipartite
because it constrains TM7 with TM1 and TM2 and also constrains TM7
with H8. The latter is enabled by an electrostatic interaction between the
aromatic side chains of Tyr3067.53 on TM7 and Phe3137.60 on H8,
yielding the TM7–H8 microdomain. Asn3027.49 at the N-terminal part of
the NPxxY(x)5,6F motif and the two residues Ala2997.46 and Ser2987.45 in
the preceding helix turn are involved in a hydrogen bonding network
with Asn551.50 on TM1 and Asp832.50 on TM2 (TM1–TM2–TM7 network).
TM6 is interrupted due to a proline kink induced by Pro2676.50, which is
part of the conserved CWxP6.50 motif. The CWxP6.50 motif is the basis of
the rotamer toggle switch hypothesis [27]. In GPCR activation, the
rotamer states of Trp at position 6.48 and Phe at position 6.52 are
predicted to be coupled and to change during receptor activation, thus
providing a link between the CWxP6.50 motif and motion of the
cytoplasmic part of TM6 [27]. In the case of rhodopsin, the phenyl side
chain of Phe6.52 is missing but the b-ionone ring of retinal is in direct
contact with Trp2656.48. It is assumed that the b-ionone ring, together
with Ala2696.52, plays the role of Phe6.52 [75]. The Y5.58(x)7K(R)5.66 motif
on TM5 contains conserved residues (Tyr2235.58 and Lys2315.66) used as
microswitches in stabilizing the active receptor state (Box 2).
In rhodopsin, both ends of the retinal ligand are stabilized by nearby
residues. Glu1133.28 on TM3 and Glu181 in loop E2 form the complex
counterion of the protonated retinal Schiff base. The b-ionone ring of
retinal is surrounded by His2115.46, Trp1263.41 and Glu1223.37 (forming
the TM3–TM5 hydrogen bonding network) and Trp2656.48 of the
CWxP6.50 motif. On light-induced cis!trans isomerization of retinal,
the TM3–TM5 network is weakened and the dominant contribution to
the counterion shifts from Glu1133.28 on TM3 to Glu181.
Figure I. Conserved residues and functional microdomains in GPCRs. (a) Rhodopsin (PDB accession 1GZM) with bound inverse agonist 11-cis-retinal is shown as a
representative GPCR. The basic GPCR architecture consists of 7TM helices, linked by cytoplasmic (C1–C3, top) and extracellular (E1–E3, bottom) loops. The cytoplasmic
helix H8 follows directly after TM7 and is frequently terminated by one or two palmitoylated Cys residues (Cys322 and Cys323 in rhodopsin). Oligosaccharide chains are
often attached at the N terminus (Asn2 and Asn15 in rhodopsin). A conserved disulfide bridge constrains the extracellular end of TM3 and the middle of loop E2 (Cys1103.25
and Cys187 in rhodopsin). The most conserved residue in each TM helix is shown in blue. According to Ballesteros–Weinstein numbering, these residues are designated
x.50, where x is the TM helix number, and other residues are designated relative to the reference residue on each helix [94]. Shaded areas and color-coded residues in (a)
correspond to the Logos color-coded sequence. For generation of the Logos sequence (http://weblogo.berkeley.edu), GPCR sequences covering the a-group of rhodopsin-
like GPCRs [1] for which crystal structures are available [19] were used. (b) Rhodopsin-specific microdomains (rhodopsin numbering). These include the TM3–TM5
network (blue shading) with Glu1223.37, Trp1263.41 on TM3 and His2115.46 on TM5, and the Schiff base network (purple shading) with the protonated Schiff base (PSB)
linkage between Lys2967.43 on TM7 and the inverse agonist 11-cis-retinal. All figures were prepared using PyMol software (www.pymol.org).
Review Trends in Biochemical Sciences Vol.34 No.11
541

Review Trends in Biochemical Sciences Vol.34 No.11
chromophore 11-cis-retinal, which acts in the dark as astrong inverse agonist and constrains rhodopsin in theinactive conformation (Box 1) [6–8]. Absorption of a photoncauses cis!trans isomerization of the retinal and generatesthe agonist all-trans-retinal in situ, which activates thereceptor. Although rhodopsin responds to light and not todiffusible ligands, it bears close similarities to ligand-acti-vated GPCRs. The photofunctional core, which alone dis-tinguishes rhodopsin from other GPCRs, is responsible forthe fast irreversible reactions linked to the photoisomeriza-tion event. However, the conversions that render the re-ceptor competent to interactwith its cognateGprotein occursubsequently on a slower time scale. These so-called Metastates are in G protein-dependent equilibrium and areanalogous to the high- and low-affinity ligand binding statesin other GPCRs [9,10]. Eventually, after the activationphase the photolyzed chromophore is released. In a phys-iological setting, 11-cis-retinal ismetabolically supplied andthe 11-cis-retinal-bound ground state is regenerated [11].The ligand-free apoprotein opsin, which remains whenregeneration does not occur, is much less active than theagonist-bound form [12,13]. In vitro, opsin can be stabilizedin an active conformation, thus serving as a template forstructural investigations.
Common structural features of GPCRsRecent advances in determining high-resolution crystalstructures of GPCRs include bovine rhodopsin [6–8], squidrhodopsin [14], the turkeyb1- and humanb2-adrenoceptors[15–17] and the human A2A adenosine receptor [18]. Allstructures confirmed the general 7TM architecture, with acytoplasmic eighth helix (H8) immediately following TM7and running parallel to the membrane surface. The fivevertebrate GPCR structures show high similarity of theTM regions, with a common structural core of 97 residueswith an average Ca root-mean-square deviation of 1.3 A[19], providing a good basis for homology modeling [20].Larger differences are observed in the ligand binding siteand the extracellular domain, which is very compact inrhodopsin to shield the hydrophobic retinal ligand from theaqueous phase. In the other GPCR structures revealed sofar, the extracellular domain shows a more open confor-mation to act as an entrance funnel for diffusible water-soluble ligands. In the cytoplasmic domain, small vari-ations in the length and secondary structure of the TM-connecting loops are thought to contribute to G proteinselectivity (for a review of GPCR structures see Refs[3,4,19,21,22]). The comparison of GPCR structures andsequences reveals a toolbox of functional microdomainsused in a typical GPCR setup (Box 1). From structural andfunctional studies it is now clear that many of the con-served residues have – often in concert with structurallyconserved water molecules – a dual role: they constrain the7TM bundle in its inactive conformation and are maindeterminants of the structural changes that occur on re-ceptor activation (Box 2) [3,21,23,24]. The most prominentresidues in this regard are Arg1353.50 and Tyr3067.53 of theE(D)R3.50Y and NP7.50xxY(x)5,6F motifs, respectively (thenumber after the residue refers to rhodopsin, the super-script refers to Ballesteros–Weinstein numbering). As out-lined below, both residues represent microswitches, which
542
in their off and on states stabilize the inactive and activeGPCR conformation, respectively. The E(D)R3.50Y motif ispart of a hydrogen bonding network linking TM3 and TM6,the so-called TM3–TM6 ionic lock, which is thought tostabilize the inactive state of rhodopsin, adrenoceptorsand perhaps other rhodopsin-like GPCRs [25,26]. TheNP7.50xxY(x)5,6F motif on TM7–H8 provides two con-straints. Asn3027.49 is part of a hydrogen bonding networklinking TM1, TM2 and TM7 (the TM1–TM2–TM7 net-work), whereas the Y(x)5,6F submotif constrains TM7 withH8 to form the TM7–H8 microdomain. Furthermore,GPCRs contain the conserved CWxP6.50 motif in TM6 witha proline kink due to Pro2676.50. According to the so-calledrotamer toggle switch hypothesis, the side chains ofTrp2656.48 in this motif and Phe6.52 (not present in rho-dopsin) undergo a rotamer change during GPCR activation[3,27]. Another important sequence is the Y5.58(x)7K(R)5.66
motif in TM5, featuring two residues (Tyr2235.58 andLys2315.66 in rhodopsin) that in the active receptor stateundergo specific interactions with residues released fromthe TM3–TM6 ionic lock (Box 2). Also highly conserved areproline residues Pro2155.50, Pro2676.50 and Pro3037.50,which induce kinks in TMs, as well as a disulfide bridgebetween Cys1103.25 on TM3 and Cys187 of the extracellu-lar loop E2 connecting TM4 and TM5 [6,28].
Rhodopsin-specific microdomainsIn rhodopsin, only one pair of ligands is natively present,11-cis-retinal and all-trans-retinal, which act as inverseagonist or agonist, respectively, with light energy trans-forming the inverse agonist into an agonist. As a light-operated switch for a single G protein pathway, rhodopsinlacks the complexity of diffusible ligand-activated GPCRsthat can couple to different pathways [29,30]. In concertwith the covalent Schiff base linkage between retinal andLys2967.43 on TM7, the inactive ground-state stability ofrhodopsin is ensured by two additional constraints. One isthe Schiff base network, a hydrogen bonding networkaround the chromophore. The network includes residuesin extracellular loop E2 andwater molecules, as well as theprotonated Schiff base, which forms a salt bridge withGlu1133.28 of its complex counterion [7,9,31]. The secondconstraint tethers TM3 and TM5 by a hydrogen bondingnetwork including Glu1223.37, Trp1263.41 and His2115.46
(Box 1) [8,32]. These residues can be expected to showsensitivity to movements of the b-ionone ring, with whichGlu1223.37 is in direct contact. The b-ionone ring is furtherrestricted by hydrophobic residues Phe2125.47, Phe2616.44
and Trp2656.48 and is thus in contact with TM6 [7].
Active conformation of the cytoplasmic domain in theopsin apoproteinA year ago, the structures of ligand-free opsin [33], aloneand in complex with a short G protein fragment [34], weresolved at 2.9 and 3.2 A, respectively (Box 2). The G proteinfragment was prepared as an 11-mer peptide derived fromthe key receptor binding site on the C terminus of the Ga
subunit, which is known to be mandatory for signal trans-fer from the receptor to the nucleotide binding site of Ga
[35–38]. The overall structures of opsin and opsin in com-plex with the Ga-derived peptide are similar to bovine

Review Trends in Biochemical Sciences Vol.34 No.11
rhodopsin in its 11-cis-retinal-bound ground state, the firstGPCR structure solved in 2000 [6]. The smallest differ-ences are observed in the extracellular domain. There theextracellular loops and the N terminus form the compactretinal plug structure [39,40], which remains intactalthough no ligand is present in the retinal binding pocketof opsin. A core structure composed of TM1–TM4 is alsovirtually unchanged. Relative to rhodopsin, prominentstructural changes are observed for TM5 and TM6. Thecytoplasmic part of TM6 is tilted outwards from the helixbundle by 6–7 A. TM5 is extended by 1.5–2.5 helix turns(depending on the reference structure and correspondingcrystal form) and moves 2–3 A towards TM6. Due to thishelix rearrangement a deep crevice is formed at the cyto-
Box 2. Crystal structures of inactive and active GPCR conformati
The structures of rhodopsin, opsin and opsin in complex with a Ga-
derived C-terminal peptide directly show that inactive and active
GPCR conformations differ at their cytoplasmic side by opening of the
binding crevice for the Ga C terminus [33,34]. This is made possible by
the movement of TM5 and TM6 and corresponding rearrangements of
residues of the TM3–TM6 ionic lock and the NP7.50xxY(x)5,6F micro-
domain (Figure I a,b). In the active opsin conformation Ops* (Figure I
b,c), Tyr2235.58 forms a hydrogen bond to Arg1353.50 and replaces
Glu1343.49, thereby stabilizing the cytoplasmic TM3–TM5 interface and
giving Arg1353.50 the role of a microswitch. Glu1343.49 faces towards a
more hydrophobic pocket between TM2 and TM4 and probably
interacts with a water molecule [34]. Glu2476.30, which loses its contact
with Arg1353.50, has a new interaction partner in Ops* and forms a salt
bridge with Lys2315.66 for which the cytoplasmic TM5–TM6 helix pair is
manifest. Parallel to changes in the TM3–TM6 ionic lock, changes in the
TM7–H8 constraint of the NPxxY(x)5,6F microdomain occur. Tyr3067.53
is released from Phe3137.60 and rotated into the helix bundle below
Arg1353.50 to keep TM6 in the outward position. Tyr3067.53, which acts
as a rotamer microswitch, was identified as an important element for
Figure I. Crystal structures of inactive rhodopsin and active opsin (Ops*) conformation
green, PDB accession 1U19) [7] and (b) ligand-free opsin (shown in orange, PDB acc
Glu2476.30), the Y5.58(x)7K(R)5.66 motif (Tyr2235.58, Lys2315.66) and the TM7–H8 microdo
on receptor activation are indicated by black arrows, whereas helix movement is indica
opsin in complex with a C-terminal peptide derived from the last 11 residues of the
residues are labeled in italics; PDB accession 3DQB) [34]. The peptide binds into the c
the peptide helical structure is involved in a hydrogen bonding network with Arg135
plasmic side of the receptor, which contains conservedArg1353.50 of the E(D)R3.50Ymotif at its floor. This opening,ormetaphorically speaking blossoming [41], of the receptorenables binding of the Ga C terminus, as observed in co-crystals of opsin and the corresponding Ga fragment. Thisprovides one of several lines of evidence for identification ofthe two available opsin structures as an active opsin state(denoted as Ops*), in which the cytoplasmic domain is inthe active G protein-binding conformation (Box 2) [34]. InOps*, the residues of the TM3–TM6 ionic lock andNPxxY(x)5,6F microdomains form new interactions. Inter-actions occur with residues of the Y5.58(x)7K(R)5.66 motif inTM5, where Tyr2235.58 and Lys2315.66 stabilize the Ops*conformation by tethering TM5–TM3 (linkage between
ons
the interaction between activated GPCR and G protein [87,96].
Glu1343.49 and Arg1353.50 of the E(D)R3.50Y motif also play a pivotal
role in this regard [97,98]. The bound Ga-derived peptide forms a near
ideal a-helix with a C-terminal open reverse turn (C cap) [34]. Although
the peptide mainly forms hydrophobic contacts to the inner surface of
TM5 and TM6, its recognition by the receptor seems to be more driven
by geometry and less by specific side chain contacts. Two backbone
carbonyl oxygens of the C cap of the Ga-derived peptide form a
hydrogen bonding network to Arg1353.50 and Gln3127.59, respectively
(Figure Ic). Importantly, the structure of the peptide in the crystal is
virtually identical to the structures of two homologous peptides as
determined by NMR spectroscopy in their conformation bound to light-
activated rhodopsin in native disk membranes [99,100]. In addition, X-
ray and NMR analysis yielded the same angle of the Ga-derived peptide
helix relative to the membrane normal [100]. EPR experiments also
indicated that the Ga-derived peptide binds to the open receptor [59].
The identical interaction of the Ga-derived peptide with opsin and light-
activated rhodopsin led us to identify both opsin structures as active
opsin (Ops*) conformations (Figure II).
s. Cytoplasmic and lateral views of (a) inactive rhodopsin ground state (shown in
ession 3CAP) [33]. Residues of the TM3–TM6 ionic lock (Glu1343.49, Arg1353.50,
main (Tyr3067.53, Phe3137.60) are shown as stick models. Side chain movements
ted by a yellow arrow. (c) Lateral view and close-up of the cytoplasmic domain of
transducin Ga subunit (shown in blue and magenta, respectively; Ga peptide
ytoplasmic crevice of opsin opened by movement of TM5 and TM6. The C cap of3.50 in the E(D)R3.50Y and Gln3127.59 in the NP7.50xxY(x)5,6F motifs, respectively.
543

Figure II. Structure of Ops* and model of channel for retinal through Ops*. (a) Lateral view of a surface model of Ops* (PDB accession 3CAP) showing two openings of
the retinal binding pocket (A,B) and a computationally docked retinal in its binding site (red). (b) A defined electron density for Lys2967.43 is missing in the crystal structure.
Depending on the rotamer state of Lys2967.43 (shown as orange or black stick model) and its interaction with Ser186 and Glu181 or Tyr2686.51, respectively, a channel for
retinal through the protein is open or closed [47]. (c,d) Coplanar cut through opsin revealing the channel with openings A and B. Electrostatic surface potentials are
contoured at �20 kT/e with negatively and positively charged surface areas in red and blue, respectively. Depending on the rotamer state of Lys2967.43 (indicated
schematically as an orange or black bar), the channel is closed (c) or open (d). In the model of the open channel, the e-amino group of Lys2967.43 is above the cutting plane,
explaining the apparent lack of positive surface charge (blue).
Review Trends in Biochemical Sciences Vol.34 No.11
Tyr2235.58 and Arg1353.50) and TM5–TM6 (linkage be-tween Lys2315.66 and Glu2476.30), respectively. Also inOps*, Tyr3067.53 shows a rotamer change and release fromPhe3137.60 on H8. Thus, Arg1353.50 and Tyr3067.53 aremicroswitches that have different roles and interactionsin the inactive and active state, respectively (Box 2) [33,34].
Conformation and gating of the retinal binding domainin opsinProfound structural rearrangements in Ops* compared to11-cis-retinal-bound rhodopsin are also observed in theretinal binding domain [33] (Box 2). Changes along theretinal binding pocket occur in regions adjacent to theretinal attachment site Lys2967.43, the C19 methyl groupin the middle and the b-ionone ring at the end of retinal.The side chain of Lys2967.43 seems to be flexible, assuggested by the lack of a defined electron density. A saltbridge between Lys2967.43 and Glu1133.28 as in rhodopsinis not evident. Instead, Glu181 in loop E2 is located closerto Lys2967.43, indicating a shift of the interaction fromGlu1133.28 to Glu181. This is reminiscent of the light-induced counterion shift in the metarhodopsin states[31,42,43].
The two bulky hydrophobic residues Phe2616.44 andTrp2656.48 on TM6 are moved due to the outward tilt ofTM6. The Trp2656.48 side chain is shifted towards theposition that is occupied by the retinal b-ionone ring inrhodopsin. Tentatively, it is possible that part of the spaceoccupied by retinal in rhodopsin might be used by water inOps*, as indicated by weak but at present uninterpretableelectron density in the retinal binding pocket. Interest-
544
ingly, in the Ops* structure with boundGa-derived peptide[34], the presence of the peptide seems to have a distanteffect on the retinal attachment site. In the complex be-tween Ops* and Ga-derived peptide, electron density isobserved for the Lys2967.43 side chain, indicating a poten-tial stabilizing network between Lys2967.43 and the resi-dues Ser186 and Glu181 from loop E2. Such long-rangeeffects are consistent with allostery between ligand and Gprotein binding domains, and thus with the postulate inclassical receptor theory that binding of G protein andligand are coupled in the active receptor conformation [44].
Thepositionandflexibility of Lys2967.43 are also relevantto its capacity to form the retinal Schiff base during rhodop-sin regeneration and for the entrance of 11-cis-retinal to andexit of all-trans-retinal from their common central bindingpocket. The retinal plug tightly shields the retinal bindingpocket fromtheextracellularenvironment inbothrhodopsinand Ops*, so the ligand binding site can only be accessedfrom the membrane phase, in agreement with the regener-ation observed for the N2C/D282C rhodopsin mutant inwhich the retinal plug is tightly fixed by an engineereddisulfide bond [45,46]. TheOps* structures reveal two open-ings of the retinal bindingpocket located in the extracellularhalf of the membrane spanning domain between TM1 andTM7 and TM5 and TM6, respectively (Box 2) [33,34]. Theopenings originate from structural rearrangements of theseTM pairs and from conformational changes of membrane-facing hydrophobic residues, giving the hydrophobic retinalaccess to its binding pocket. In recent modeling work usinggeometric mapping (i.e. skeleton search and retinal liganddocking), a channel was identified that meanders along the

Review Trends in Biochemical Sciences Vol.34 No.11
7TMbundleand links the two openings [47]. In the situationdiscussed above, where Lys2967.43 is part of a network withSer186andGlu181,a channel largeenough forretinalwouldnot continuously traverse the protein. This is only the casewhen the rotamer state of Lys2967.43 is such that it can behydrogen bonded to Tyr2686.51 [47] (Box 2, Figure II).Lys2967.43 might thus function as a gate allowing retinalto pass the channel [47].
Because the retinal channel is only found in Ops* andnot in all other known rhodopsin crystal structures, thequestion arises as to whether the Ops* conformation ismandatory for uptake or release of the retinal ligand. Forthe release reaction, this indeed seems to be the case,because the release rate of all-trans-retinal is maximum
Box 3. Photoactivation of rhodopsin and global changes for G pr
Light-induced activation of rhodopsin starts with cis!trans isomer-
ization of the retinal and energy storage in a twisted all-trans-
retinylidene-Lys2967.43 conformation (Figure I). Concomitant with the
release of strain in retinal, fast transformations in the protein occur.
The succession of corresponding spectrally distinguishable photo-
intermediates – i.e. Batho, Lumi and Meta I, absorbance maxima at
540, 497 and 478 nm, respectively – constitutes a fast photofunctional
core process. Up to Meta I, a state that is reached within a few
microseconds, the activation remains near the retinal binding pocket
and only minor structural changes occur in the more distant parts of
the protein [51,52,101]. With the formation of Meta I, the activation
path reaches the level of the Meta intermediates. The spectrally
identical Meta II substates Meta IIa, Meta IIb and Meta IIbH+
sequentially develop from one another until an equilibrium between
all the states is reached after a few milliseconds. At physiological pH
and temperature, Meta IIbH+ accumulates in the reaction sequence
[69]. In the course of the Meta conversions, deprotonation of the
retinal Schiff base and protonation of Glu1133.22 of its complex
counterion (step 1), motion of TM6 (step 2) and proton uptake to the
TM3–TM6 ionic lock microdomain (step 3) occur sequentially. Thus,
Figure I. Rhodopsin and light-activated photoproducts. The early photoproducts of rho
the late Meta intermediates in blue.
when the receptor is stabilized by the Ga-derived peptideand the amount of retinal released has the pH dependenceof the active conformation [48]. No clear answer yet existsfor retinal uptake. On the one hand, opsin recombinesreadily with 11-cis-retinal at intracellular pH values atwhich virtually no Ops* is present [11–13,49]. On the otherhand, intact rod cells show a short threshold elevationwithin the time frame of 11-cis-retinal uptake, which couldreflect transient formation of Ops* [50]. The informationrevealed by modeling work is that any function of thechannel in retinal passage through the receptor requiresconformational adjustments. A deeper understanding ofthe mechanism of 11-cis-retinal uptake will require a high-resolution structure of the inactive opsin conformation.
otein coupling
two of the main constraints of the inactive ground state, the retinal
Schiff base network and the TM3–TM6 ionic lock, are broken one after
another [59].
Opsin is reached with hydrolysis (in minutes) of the retinylidene
Schiff base and release of all-trans-retinal. In the living eye, fresh 11-
cis-retinal from a complex metabolism regenerates the light-sensitive
rhodopsin ground state [11]. Purified opsin, devoid of any retinoids
but in its native membrane host and at neutral pH, is several orders of
magnitude less active towards the G protein than Meta II [12,13], but
lower pH enhances its activity [62,102].
The residual activity of opsin can be explained by an active (Ops*)
conformation in pH-dependent equilibrium with inactive opsin
[49,62,103]. The free energy gap between Meta II and Ops* can be
estimated from the apparent pKa at which the respective species forms.
Meta II appears at an apparent pKa of >7.5, whereas the pKa for Ops*
formation is 4.1 [49,69]. The difference in pKa equates to a difference in
free energy of approximately 5 kcal/mol, which can be assigned to the
stabilizing effect of the all-trans-retinal agonist present in Meta II but not
in Ops*. Consistently, Meta II and Ops* exhibit a similar difference in
their pH-rate profiles for G protein activation [62,102–104].
dopsin related to the reactions of its photosensory core are underlined in red and
545

Review Trends in Biochemical Sciences Vol.34 No.11
The receptor activation path: unifying two paradigmsAs outlined above, the main difference between rhodopsinas a photoreceptor and other GPCRs is the photofunctionalcore with its fast light-induced transformations thatprecede G protein-dependent conformational conversionsin equilibrium. The ease of triggering the activation pro-cess by light facilitates the monitoring of fast transform-ations in the protein, which follow the cis!transisomerization of 11-cis-retinal (Box 3) [51–53]. Throughoutall the conversions from bathorhodopsin (Batho) via lumi-rhodopsin (Lumi) to the late metarhodopsin (Meta) photo-intermediates, retinal remains in the all-transconfiguration and its Schiff base bond remains in the anticonfiguration. The photointermediates reflect stages ofinteraction between the chromophoric ligand and itsimmediate protein environment, the ligand pocket. Aspecific structural change that occurs with the formationof the Meta I intermediate is a change in the complexcounterion of the protonated Schiff base, which shifts itscenter of interaction from Glu1133.28 on TM3 to Glu181 inloop E2 [31,42]. At this point in the photoexcitation path-way, we can assume that the interaction between thenewly formed agonist all-trans-retinal and the opsin apo-protein is firmly established. Meta I has limited signalingcapacity. An interaction with rhodopsin kinase has beenobserved [54] and there is also a non-productive interactionwith the G protein in which GDP is not released [55].
With the Meta I state, the activation path enters asystem of coupled equilibria between intermediates thatall bear all-trans-retinal bound by an intact but deproto-nated retinal Schiff base. This is the definition of thebleached intermediate Meta II [9]. Meta II formation isenhanced at the expense ofMeta I by interactionwith theGprotein (and also arrestin), which established Meta II asthe active G protein-binding species. Analogously, ligand-activated GPCRs show active (G protein binding) andinactive states characterized by high and low affinity foragonists, respectively [9,10,44]. Enhancement of Meta II atthe expense of Meta I is also observed when rhodopsin issolubilized in detergents such as dodecyl maltoside andoctyl glucoside [56,57]. The steps up to lumirhodopsinformation are detergent-insensitive [57]. Early on it wasnoted, paradoxically, that Meta II formation involves pro-ton uptake from solution, although the Schiff base becomesdeprotonated [9,56,58]. This is now explained by protontransfer in two different but thermodynamically coupledfunctional domains. These are the retinal Schiff base net-work and the TM3–TM6 ionic lock. In the course of rho-dopsin activation, proton transfer from the retinal Schiffbase to Glu1133.28 of its complex counterion and protonuptake to the TM3–TM6 ionic lock with Glu1343.49 asproton acceptor occur sequentially [9,59,60]. Proton trans-fer reactions per se are very fast, depending on the relativeorientation and distance of proton donor and acceptorgroups and on the electric field in their vicinity [61]. Theobserved reprotonation step is delayed because the systemwaits for the opportunity for proton transfer. Protonationchanges are thus both monitors and co-determinants ofconformational changes, in which donor and/or acceptorgroups are brought into a new environment, thereby gen-erating the opportunity for proton transfer.
546
It can be considered a paradigm of rhodopsin activationthat protonation changes and release of inactivating con-straints, occurring in the retinal Schiff base and in theTM3–TM6 ionic lock microdomains, are coupled [6,62,63].Protonation switches that govern the transition betweeninactive and active states of a GPCR have also beenpostulated for the b2-adrenoceptor, in which a confor-mational change is thought to influence the hydrophobicor hydrophilic character of Asp1423.49 and thus the proto-nation state of this residue [64]. A second paradigm ofrhodopsin activation was noted from observations of light-induced changes in the electron paramagnetic resonance(EPR) spectrum of site-specific spin-labeled rhodopsins,which occurred on the time scale of Meta II formationand reversed with its decay [65]. Later work revealed thatthe motion of TM6 was the dominant event and that bothhelix motion and activation of Gt could be prevented by TMcrosslinking using engineered disulfide bridges or metalion binding sites [66,67]. The effect was interpreted as asolid-state motion and outward tilt of TM6. Recent workhas confirmed the TM6 helix motion paradigm by distancemeasurements using double electron–electron resonance(DEER) spectroscopy on pairs of spin labels attached torhodopsin in a site-directed manner. The DEER measure-ments quantified a 5 A outward movement of TM6 andidentified smaller relative motions of TM1, TM7 and the C-terminal domain [68].
To unify the two paradigms, the temporal sequence ofevents was determined by measuring light-induced TM6motion and protonation changes of spin-labeled rhodopsinin parallel as a function of pH and temperature. Protonuptake in the TM3–TM6 ionic lockmicrodomain occurs as aconsequence of TM6 motion [59]. Both these events occurten times more slowly than retinal Schiff base deprotona-tion at 30 8C, but at the same rate at T < 10 8C. The datasuggest a temporal sequence of events (Box 3). Impor-tantly, TM6 motion is a thermally activated process inMeta II and occurs as a delayed consequence of Schiff basedeprotonation. Themotion of TM6most probably results ina change in the pKa of Glu1343.49 and subsequent protonuptake from solution [9]. The scheme in Box 3, which wasobtained from data on solubilized rhodopsin, has now beenconfirmed by two independent studies on native mem-branes [69,70]. Although the techniques applied cannotspecifically identify helix motion, a general activationscheme was deduced with the order (i) Schiff base depro-tonation, (ii) structural changes and (iii) proton uptake.
Structural correlation of activation stepsWe can now attempt to combine the above data on receptoractivation steps and receptor structure to derive a possiblerhodopsin activation scenario (Figure 1 and Table 1). Thiswill provide the basis for a description of the intermediatesas the stepwise engagement of structural changes in con-served regions (Box 1). Formation of the active state isinitiated by release of strain in the protein-bound retinalcaused by cis!trans isomerization-induced retinalelongation [53]. Both ends of retinal are fixed in the ligandbinding pocket, so the Schiff base and TM3–TM5 networksrespond first to retinal isomerization. The structuralelements that undergo changes in the transition to the
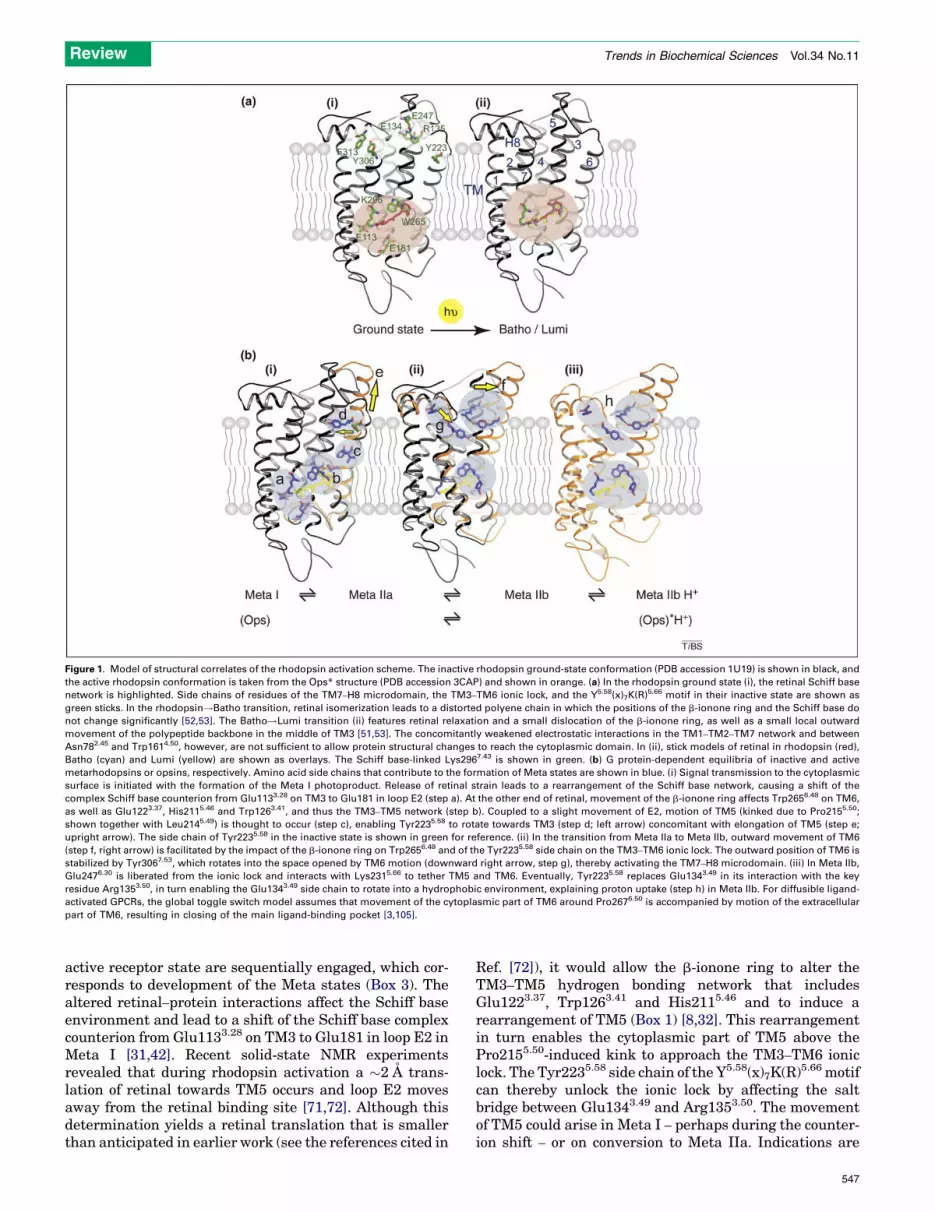
Figure 1. Model of structural correlates of the rhodopsin activation scheme. The inactive rhodopsin ground-state conformation (PDB accession 1U19) is shown in black, and
the active rhodopsin conformation is taken from the Ops* structure (PDB accession 3CAP) and shown in orange. (a) In the rhodopsin ground state (i), the retinal Schiff base
network is highlighted. Side chains of residues of the TM7–H8 microdomain, the TM3–TM6 ionic lock, and the Y5.58(x)7K(R)5.66 motif in their inactive state are shown as
green sticks. In the rhodopsin!Batho transition, retinal isomerization leads to a distorted polyene chain in which the positions of the b-ionone ring and the Schiff base do
not change significantly [52,53]. The Batho!Lumi transition (ii) features retinal relaxation and a small dislocation of the b-ionone ring, as well as a small local outward
movement of the polypeptide backbone in the middle of TM3 [51,53]. The concomitantly weakened electrostatic interactions in the TM1–TM2–TM7 network and between
Asn782.45 and Trp1614.50, however, are not sufficient to allow protein structural changes to reach the cytoplasmic domain. In (ii), stick models of retinal in rhodopsin (red),
Batho (cyan) and Lumi (yellow) are shown as overlays. The Schiff base-linked Lys2967.43 is shown in green. (b) G protein-dependent equilibria of inactive and active
metarhodopsins or opsins, respectively. Amino acid side chains that contribute to the formation of Meta states are shown in blue. (i) Signal transmission to the cytoplasmic
surface is initiated with the formation of the Meta I photoproduct. Release of retinal strain leads to a rearrangement of the Schiff base network, causing a shift of the
complex Schiff base counterion from Glu1133.28 on TM3 to Glu181 in loop E2 (step a). At the other end of retinal, movement of the b-ionone ring affects Trp2656.48 on TM6,
as well as Glu1223.37, His2115.46 and Trp1263.41, and thus the TM3–TM5 network (step b). Coupled to a slight movement of E2, motion of TM5 (kinked due to Pro2155.50;
shown together with Leu2145.49) is thought to occur (step c), enabling Tyr2235.58 to rotate towards TM3 (step d; left arrow) concomitant with elongation of TM5 (step e;
upright arrow). The side chain of Tyr2235.58 in the inactive state is shown in green for reference. (ii) In the transition from Meta IIa to Meta IIb, outward movement of TM6
(step f, right arrow) is facilitated by the impact of the b-ionone ring on Trp2656.48 and of the Tyr2235.58 side chain on the TM3–TM6 ionic lock. The outward position of TM6 is
stabilized by Tyr3067.53, which rotates into the space opened by TM6 motion (downward right arrow, step g), thereby activating the TM7–H8 microdomain. (iii) In Meta IIb,
Glu2476.30 is liberated from the ionic lock and interacts with Lys2315.66 to tether TM5 and TM6. Eventually, Tyr2235.58 replaces Glu1343.49 in its interaction with the key
residue Arg1353.50, in turn enabling the Glu1343.49 side chain to rotate into a hydrophobic environment, explaining proton uptake (step h) in Meta IIb. For diffusible ligand-
activated GPCRs, the global toggle switch model assumes that movement of the cytoplasmic part of TM6 around Pro2676.50 is accompanied by motion of the extracellular
part of TM6, resulting in closing of the main ligand-binding pocket [3,105].
Review Trends in Biochemical Sciences Vol.34 No.11
active receptor state are sequentially engaged, which cor-responds to development of the Meta states (Box 3). Thealtered retinal–protein interactions affect the Schiff baseenvironment and lead to a shift of the Schiff base complexcounterion from Glu1133.28 on TM3 to Glu181 in loop E2 inMeta I [31,42]. Recent solid-state NMR experimentsrevealed that during rhodopsin activation a �2 A trans-lation of retinal towards TM5 occurs and loop E2 movesaway from the retinal binding site [71,72]. Although thisdetermination yields a retinal translation that is smallerthan anticipated in earlier work (see the references cited in
Ref. [72]), it would allow the b-ionone ring to alter theTM3–TM5 hydrogen bonding network that includesGlu1223.37, Trp1263.41 and His2115.46 and to induce arearrangement of TM5 (Box 1) [8,32]. This rearrangementin turn enables the cytoplasmic part of TM5 above thePro2155.50-induced kink to approach the TM3–TM6 ioniclock. The Tyr2235.58 side chain of the Y5.58(x)7K(R)5.66 motifcan thereby unlock the ionic lock by affecting the saltbridge between Glu1343.49 and Arg1353.50. The movementof TM5 could arise in Meta I – perhaps during the counter-ion shift – or on conversion to Meta IIa. Indications are
547

Table 1. GPCR signaling: what can be learned from rhodopsin?
Review Trends in Biochemical Sciences Vol.34 No.11
given by the crystal structures of the b-adrenoceptors andof the A2A adenosine receptor [15–18], which in theirpartial inverse agonist or antagonist bound state – relativeto inactive rhodopsin – exhibit a clearmovement of TM5anda flip of Tyr5.58 (or Tyr2275.58 Ala [15]) into the direction ofthe TM3–TM6 ionic lock, whereas a large TM6movement islacking. It is interesting to note that the structures of boththe b-adrenoceptors and of theA2A adenosine receptor showstabilization of the cytoplasmic loop C2 by interaction withArg1353.50. The crystal structure of a photoactivatedintermediate of rhodopsin, which, according to its 380-nmabsorbance maximum, can be either Meta IIa or Meta IIb,lacks TM5 movement and shows little if any movement ofTM6 [73]. Together with the information from the EPRwork, we can assign this photoactivated form to the MetaIIa state.
In the subsequent Meta IIa!Meta IIb transition ofrhodopsin, TM6 moves outward in concert with disruptionof the TM3–TM6 ionic lock network [59,60,74] and sup-ported by the b-ionone ring interaction with Trp2656.48 ofthe CWxPmotif. According to the activating Trp6.48–Phe6.52
rotamer toggle switch hypothesis [27], the retinal b-iononering acts in place of the phenyl ring, which is not present inrhodopsin due to substitution of Phewith Ala2696.52 [75]. InMeta IIb, TM6 is fixed in the outward position due torearrangement of the NPxxY(x)5,6F microdomain and theTM3–TM6 ionic lock involving a rotamer change of theTyr3067.53 side chain and an interaction of Glu2476.30 withLys2315.66 of the Y5.58(x)7K(R)5.66 motif. Thus, in rhodopsintheNPxxY(x)5,6Fmicrodomainand theTM3–TM6 ionic lockexist in two well-defined on and off states. In the GPCRstructuresof theb1,b2 andA2A receptors, partial steps of therearrangement of these two microdomains are observed,whichmight be explained by the lack of full inverse agonismof the bound ligands. However, all-trans-retinal is, as a fullagonist, expected to drive the complete inactive!activeconversion of the TM3–TM6 ionic lock and NPxxY(x)5,6Fmicrodomain. Complete structural conversion to the activeGPCR state is manifest in the new stabilization ofArg1353.50 by Tyr2235.58 and protonation of Glu1343.49
(which is released from Arg1353.50), forming Meta IIbH+.In the active opsin structure in which an agonist is lacking,
548
pH and crystallization conditions act as stabilizing forces.Detergents shift the metarhodopsin equilibrium to theactive conformation, so it is conceivable that octyl glucosideused in crystallization favors the Ops* conformation.
In this regard the largely different conformations ofrhodopsin and Ops* can be viewed as the extremes necess-ary for the switch-like operation of rhodopsin. However,partial agonism does exist in rhodopsin. Pigments withartificial retinals lacking either the ring or the C19 methylgroup [74,76–78] are impaired in their capacity to activatethe G protein, but also show an equilibrium of Meta states.One known case of a native partial agonist is all-trans-retinal bound by a reprotonated and distorted Schiff basebond (syn instead of anti configuration [79]). This so-calledMeta III intermediate [48] is in equilibrium with an activeform [80], similar to the equilibrium between Meta I andMeta II or Ops and Ops*.
Signal transfer to the G protein: role of the C-terminala5 helix in GaInformation on the active state of rhodopsin can now beused to obtain insights into the mechanism of signaltransfer to the G protein (Box 4). Available evidencesuggests that GPCRs can exist as monomers and dimersand even higher oligomers [81]. Recent work has shownthat active rhodopsin forms a 1:1 complex with transducin[82] and that rhodopsin and the b2-adrenoceptor as mono-mers can activate G proteins efficiently [82–85]. This hasprovided the basis for the following discussion of signaltransfer in a 1:1 rhodopsin–transducin complex.
It has been firmly established that the C terminus of theGa subunit is a key receptor interaction site [35] thatundergoes a conformational change on Gbg and receptorbinding [86] and several studies have identified the C-terminal a5 helix of Ga as an indispensable part of thesignal transmission machinery [36–38]. We can now alsounderstand why cytoplasmic loops C2, C3 and C4 are allimplicated in signal transmission and why impairments inthe TM7–H8 microdomain lead to severe defects in Gt
activation [34,87,88].To further explore the role of the a5 helix as a signal
transmission element, Oldham and co-workers used EPR

Box 5. Outstanding questions
� In rhodopsin, presentation of a crevice-like binding site for the G
protein requires motion and rearrangement of transmembrane
helices. Is this a general principle in GPCRs?
� What is the role of the cytoplasmic loops of GPCRs in the
rearrangement of transmembrane helices and how do they
contribute to G protein specificity?
� Do the two elements TM5 and TM6 move simultaneously or
sequentially? Can the two motions be assigned to specific
intermediates, as outlined in Box 3 and Figure 1?
� The Ops* conformation has a ligand channel with two openings
into the hydrophobic lipid environment. Is this the channel for
retinal uptake and release and is the channeling unidirectional? Is
such a channel common to all GPCRs that bind hydrophobic
ligands?
� GPCRs bind the a5 helix of the Ga subunit to trigger GDP release
from the nucleotide binding pocket. How are other structural
elements in the G protein involved?
� The C terminus of the a5 helix is flexible before it binds into the
receptor crevice, but is bound as an a helix with a C cap structure.
What is the nature of this conversion (induced fit or conforma-
tional selection) and is it a general principle of signal transfer
between receptors and cognate proteins?
Box 4. A hypothesis of receptor–G protein coupling
Signal transfer over a cell membrane by a GPCR is based on the
presentation of an active receptor. Driven by the binding of an
agonist, the active conformation is selected from an ensemble of
receptor conformations. A crevice within the cytoplasmic receptor
surface is the main binding domain for the G protein. The activating
latch of the G protein is the C terminus of the a subunit. This
element is flexible before it binds, but adopts a helical conformation
with a C cap structure when the G protein recognizes the active
receptor. Signal transmission over a distance of >40 A, from the
agonist binding site to the nucleotide binding site of the G protein,
proceeds through a well-defined solid-state rearrangement of a
helices. Helix motion is supported by proton transfer reactions and
fast backbone and side chain fluctuations. First, TM5 and TM6
engage in new interactions to form a crevice into which the G
protein a5 helix can bind. Second, the bound a5 helix switches into
a new position, thereby acting as a transmission rod to the
nucleotide binding site (Figure I).
Figure I. The rhodopsin model: Signal transmission from the retinal binding
site to the nucleotide binding site in the G protein.
Review Trends in Biochemical Sciences Vol.34 No.11
techniques to investigate the dynamicmotion of thea5helixwithin the G protein, which occur on interaction with light-activated rhodopsin. They found a distance change betweenthe a5 helix and the b2 strand of Ga consistent with arotation and translation of the a5 helix [37]. In recent work,a disulfide bond was engineered to constrain the a5 helixin its receptor-associated conformation [89]. The levelof basal nucleotide exchange was strongly enhanced,supporting the role of the a5 helix as a transmission rodfrom the receptor–G protein interface to the nucleotidebinding site.
A complementary molecular modeling approachrevealed two modes of interaction of the a5 helix withOps* [88]. One of them closely matches the position of theGa-derived peptide in the crystal structure and repro-duces the hydrogen bonding network at the C cap of thepeptide (Box 2). In the alternative fit, the a5 helix binds tothe inner surface of the TM5–TM6 helix pair and runs
parallel to themembrane. Thiswas interpreted as a switchfrom an initial transient interaction to the final stableinteraction within the open crevice of the active receptor(helix switch [88]). Additional conformational changes inthe G protein accompany the motion of a5 and additionalinteractions between the receptor and G protein arerequired to provide a firm basis for the a5 switch [38,88].
Switches and lubricants behind signal transmissionAs soon as the agonist all-trans-retinal is fitted in itsbinding pocket (Box 3 and Figure 1), the rhodopsin acti-vation process is initiated and proceeds through changes inmicrodomains using highly conserved microswitches. Howdoes the signaling free energy flow from the ligand bindingpocket to the binding domain for the G protein? Nygaardand co-workers [3] have discussed alternative mechanismsfor the propagation of structural changes, namely: (i) adomino effect from residue to residue and (ii) an allostericeffect thatmobilizes larger domains in a concerted action oftheMonod–Wyman–Changeux type, termed a global toggleswitch. A continuous domino effect through the wholeprotein is unlikely because of the variable thermodynamiccoupling between the two protonation switches in theretinal Schiff base network and at the TM3–TM6 ioniclock (transitions between metarhodopsin states; Box 3).The data available for rhodopsin and opsin consistentlysupport a mechanism of the solid state and thus of theglobal toggle switch type. This does, however, not exclude –
but rather requires – microscopic rearrangement of aminoacid side chains, which has a role in both switching andsliding of helices or domains. The large conformationalswitches observed in helix motion alternate with localchanges in protonation in a cause–consequence relation-ship (Box 3). The motion of TM6 at 30 8C occurs ten timesmore slowly than the preceding reaction, deprotonation ofthe retinal Schiff base. During this time TM6 remainslocked in its inward position (corresponding to MetaIIa), presumably stabilized by hydrophobic and/or van
549

Review Trends in Biochemical Sciences Vol.34 No.11
der Waals interactions (Box 3). Interactions relevant toswitching occur not only between residues of the protein,but also between water molecules in strongly hydrogen-bonded water networks or in protonated water clusters[90]. Any such process is based on the precise and evolu-tionarily conserved [23,24] arrangement of water mol-ecules in the protein matrix.
Water is likely to have a role not only in the protonationswitches between the global solid-state motions, but also insupporting the sliding motion of helices by solvating helix–
helix contacts [91]. Generally, hydrogen bonding networksmight open or close in a coordinated manner and define thesolid-state motion or sliding of helices [91]. Within thehydrophobic milieu of the membrane, the strength ofhydrogen bonds will be diminished by the high dielectriceffect of buried water molecules and the competition fromwater for hydrogen bonds. Moreover, surfaces of solvatedcontacts are more easily separated [92]. This could play arole in the interaction of the Ga C-terminal a5 helix withopsin, in which hydrogen bonding networks between the Ccap and the opsin binding pocket likely include interfacialwater molecules to guide the a5 helix through the helixswitch [88].
These considerations teach us that understanding sig-nal transduction in GPCRs will not only require knowledgeof the frozen conformations in active and inactive states,the function of the intermediates, and the thermodynamicand kinetic properties of their interconversion. It will alsobe necessary to understand the underlying proteindynamics and the specific conformational space that thesignaling proteins can explore [93]. Questions that need tobe addressed are listed in Box 5.
AcknowledgementsWe thank Martha Sommer for critical reading of the manuscript. Thiswork was supported by the Deutsche Forschungsgemeinschaft (Sfb449and Sfb740).
References1 Lagerstrom, M.C. and Schioth, H.B. (2008) Structural diversity of G
protein-coupled receptors and significance for drug discovery. Nat.Rev. Drug Discov. 7, 339–357
2 Swaminath, G. et al. (2004) Sequential binding of agonists to the b2adrenoceptor. Kinetic evidence for intermediate conformationalstates. J. Biol. Chem. 279, 686–691
3 Nygaard, R. et al. (2009) Ligand binding and micro-switches in 7TMreceptor structures. Trends Pharmacol. Sci. 30, 249–259
4 Rosenbaum, D.M. et al. (2009) The structure and function of G-protein-coupled receptors. Nature 459, 356–363
5 Hofmann, K.P. et al. (2006) Building functional modules frommolecular interactions. Trends Biochem. Sci. 31, 497–508
6 Palczewski, K. et al. (2000) Crystal structure of rhodopsin: aG protein-coupled receptor. Science 289, 739–745
7 Okada, T. et al. (2004) The retinal conformation and its environmentin rhodopsin in light of a new 2.2 A crystal structure. J. Mol. Biol. 342,571–583
8 Li, J. et al. (2004) Structure of bovine rhodopsin in a trigonal crystalform. J. Mol. Biol. 343, 1409–1438
9 Okada, T. et al. (2001) Activation of rhodopsin: new insightsfrom structural and biochemical studies. Trends Biochem. Sci. 26,318–324
10 Yao, X.J. et al. (2009) The effect of ligand efficacy on the formation andstability of a GPCR–G protein complex. Proc. Natl. Acad. Sci. U. S. A.106, 9501–9506
11 Lamb, T.D. and Pugh, E.N., Jr (2004) Dark adaptation and theretinoid cycle of vision. Prog. Retin. Eye Res. 23, 307–380
550
12 Melia, T.J., Jr et al. (1997) A comparison of the efficiency of G proteinactivation by ligand-free and light-activated forms of rhodopsin.Biophys. J. 73, 3182–3191
13 Cornwall, M.C. and Fain, G.L. (1994) Bleached pigment activatestransduction in isolated rods of the salamander retina. J. Physiol. 480,261–279
14 Murakami, M. and Kouyama, T. (2008) Crystal structure of squidrhodopsin. Nature 453, 363–367
15 Warne, T. et al. (2008) Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 454, 486–491
16 Cherezov, V. et al. (2007) High-resolution crystal structure of anengineered human beta2-adrenergic G protein-coupled receptor.Science 318, 1258–1265
17 Rasmussen, S.G. et al. (2007) Crystal structure of the human b2adrenergic G-protein-coupled receptor. Nature 450, 383–387
18 Jaakola, V.P. et al. (2008) The 2.6 A crystal structure of a human A2Aadenosine receptor bound to an antagonist. Science 322, 1211–1217
19 Hanson, M.A. and Stevens, R.C. (2009) Discovery of new GPCRbiology: one receptor structure at a time. Structure 17, 8–14
20 Vaidehi, N. et al. (2009) Modeling small molecule-compound bindingto G-protein-coupled receptors. Methods Enzymol. 460, 263–288
21 Weis, W.I. and Kobilka, B.K. (2008) Structural insights into G-protein-coupled receptor activation. Curr. Opin. Struct. Biol. 18, 734–740
22 Mustafi, D. and Palczewski, K. (2009) Topology of class A G protein-coupled receptors: insights gained from crystal structures ofrhodopsins, adrenergic and adenosine receptors. Mol. Pharmacol.75, 1–12
23 Pardo, L. et al. (2007) The role of internal water molecules in thestructure and function of the rhodopsin family of G protein-coupledreceptors. ChemBiochem 8, 19–24
24 Angel, T.E. et al. (2009) Conserved waters mediate structural andfunctional activation of family A (rhodopsin-like) G protein-coupledreceptors. Proc. Natl. Acad. Sci. U. S. A. 106, 8555–8560
25 Ballesteros, J.A. et al. (2001) Activation of the b2-adrenergic receptorinvolves disruption of an ionic lock between the cytoplasmic ends oftransmembrane segments 3 and 6. J. Biol. Chem. 276, 29171–29177
26 Dror, R.O. et al. (2009) Identification of two distinct inactiveconformations of the b2-adrenergic receptor reconciles structural andbiochemical observations.Proc.Natl. Acad.Sci.U.S.A.106, 4689–4694
27 Shi, L. et al. (2002) b2 adrenergic receptor activation. Modulation ofthe proline kink in transmembrane 6 by a rotamer toggle switch. J.Biol. Chem. 277, 40989–40996
28 Mirzadegan, T. et al. (2003) Sequence analyses of G-protein-coupledreceptors: similarities to rhodopsin. Biochemistry 42, 2759–2767
29 Lefkowitz, R.J. and Shenoy, S.K. (2005) Transduction of receptorsignals by b-arrestins. Science 308, 512–517
30 Kobilka, B.K. and Deupi, X. (2007) Conformational complexity of G-protein-coupled receptors. Trends Pharmacol. Sci. 28, 397–406
31 Ludeke, S. et al. (2005) The role of Glu181 in the photoactivation ofrhodopsin. J. Mol. Biol. 353, 345–356
32 Vogel, R. et al. (2007) Coupling of protonation switches duringrhodopsin activation. Photochem. Photobiol. 83, 286–292
33 Park, J.H. et al. (2008) Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454, 183–187
34 Scheerer, P. et al. (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455, 497–502
35 Hamm, H.E. et al. (1988) Site of G protein binding to rhodopsinmapped with synthetic peptides from the a subunit. Science 241,832–835
36 Nanoff, C. et al. (2006) The carboxyl terminus of the Ga-subunit is thelatch for triggered activation of heterotrimeric G proteins. Mol.Pharmacol. 69, 397–405
37 Oldham, W.M. et al. (2006) Mechanism of the receptor-catalyzedactivation of heterotrimeric G proteins. Nat. Struct. Mol. Biol. 13,772–777
38 Oldham, W.M. and Hamm, H.E. (2008) Heterotrimeric G proteinactivation by G-protein-coupled receptors. Nat. Rev. Mol. Cell. Biol.9, 60–71
39 Filipek, S. et al. (2003) G protein-coupled receptor rhodopsin: aprospectus. Annu. Rev. Physiol. 65, 851–879
40 Janz, J.M. et al. (2003) Stability of dark state rhodopsin is mediatedby a conserved ion pair in intradiscal loop E-2. J. Biol. Chem. 278,16982–16991

Review Trends in Biochemical Sciences Vol.34 No.11
41 Meng, E.C. and Bourne, H.R. (2001) Receptor activation: whatdoes the rhodopsin structure tell us? Trends Pharmacol. Sci. 22,587–593
42 Yan, E.C. et al. (2003) Retinal counterion switch in thephotoactivation of the G protein-coupled receptor rhodopsin. Proc.Natl. Acad. Sci. U. S. A. 100, 9262–9267
43 Standfuss, J. et al. (2008) Structural impact of the E113Q counterionmutation on the activation and deactivation pathways of the Gprotein-coupled receptor rhodopsin. J. Mol. Biol. 380, 145–157
44 De Lean, A. et al. (1980) A ternary complex model explains theagonist-specific binding properties of the adenylate cyclase-coupledb-adrenergic receptor. J. Biol. Chem. 255, 7108–7117
45 Xie, G. et al. (2003) An opsin mutant with increased thermal stability.Biochemistry 42, 1995–2001
46 Standfuss, J. et al. (2007) Crystal structure of a thermally stablerhodopsin mutant. J. Mol. Biol. 372, 1179–1188
47 Hildebrand, P.W. et al. (2009) A ligand channel through the G proteincoupled receptor opsin. PLoS ONE 4, e4382
48 Heck, M. et al. (2003) Signaling states of rhodopsin. Formation of thestorage form, metarhodopsin III, from active metarhodopsin II. J.Biol. Chem. 278, 3162–3169
49 Vogel, R. and Siebert, F. (2001) Conformations of the active andinactive states of opsin. J. Biol. Chem. 276, 38487–38493
50 Kefalov, V.J. et al. (2001) Role of noncovalent binding of 11-cis-retinalto opsin in dark adaptation of rod and cone photoreceptors.Neuron 29,749–755
51 Nakamichi, H. and Okada, T. (2006) Local peptide movement in thephotoreaction intermediate of rhodopsin. Proc. Natl. Acad. Sci. U. S.A. 103, 12729–12734
52 Nakamichi, H. and Okada, T. (2006) Crystallographic analysis ofprimary visual photochemistry. Angew. Chem. Int. Ed. 45, 4270–4273
53 Ritter, E. et al. (2008) Activity switches of rhodopsin. Photochem.Photobiol. 84, 911–920
54 Pulvermuller, A. et al. (1993) Interaction between photoactivatedrhodopsin and its kinase: stability and kinetics of complexformation. Biochemistry 32, 14082–14088
55 Morizumi, T. et al. (2005) Direct observation of the complex formationof GDP-bound transducin with the rhodopsin intermediate having avisible absorption maximum in rod outer segment membranes.Biochemistry 44, 9936–9943
56 Arnis, S. and Hofmann, K.P. (1993) Two different forms ofmetarhodopsin II: Schiff base deprotonation precedes proton uptakeand signaling state. Proc. Natl. Acad. Sci. U. S. A. 90, 7849–7853
57 Epps, J. et al. (2006) Lumi I!Lumi II: the last detergent independentprocess in rhodopsin photoexcitation. Photochem. Photobiol. 82, 1436–
144158 Arnis, S. et al. (1994) A conserved carboxylic acid group mediates
light-dependent proton uptake and signaling by rhodopsin. J. Biol.Chem. 269, 23879–23881
59 Knierim, B. et al. (2007) Sequence of late molecular events in theactivation of rhodopsin.Proc.Natl. Acad.Sci.U.S.A.104, 20290–20295
60 Vogel, R. et al. (2008) Functional role of the ‘‘ionic lock’’ – aninterhelical hydrogen-bond network in family A heptahelicalreceptors. J. Mol. Biol. 380, 648–655
61 Gutman, M. and Nachliel, E. (1990) The dynamic aspects of proton-transfer processes. Biochim. Biophys. Acta 1015, 391–414
62 Cohen, G.B. et al. (1992) Mechanism of activation and inactivation ofopsin: role of Glu113 and Lys296. Biochemistry 31, 12592–12601
63 Cohen, G.B. et al. (1993) Constitutive activation of opsin: influence ofcharge at position 134 and size at position 296. Biochemistry 32,6111–6115
64 Scheer, A. et al. (1997) The activation process of the a1B-adrenergicreceptor: potential role of protonation and hydrophobicity of a highlyconserved aspartate. Proc. Natl. Acad. Sci. U. S. A. 94, 808–813
65 Farahbakhsh, Z.T. et al. (1993) Photoactivated conformationalchanges in rhodopsin: a time-resolved spin label study. Science262, 1416–1419
66 Farrens, D.L. et al. (1996) Requirement of rigid-body motion oftransmembrane helices for light activation of rhodopsin. Science274, 768–770
67 Sheikh, S.P. et al. (1996) Rhodopsin activation blocked by metal-ion-binding sites linking transmembrane helices C and F. Nature 383,347–350
68 Altenbach, C. et al. (2008) High-resolution distance mapping inrhodopsin reveals the pattern of helix movement due to activation.Proc. Natl. Acad. Sci. U. S. A. 105, 7439–7444
69 Mahalingam, M. et al. (2008) Two protonation switches controlrhodopsin activation in membranes. Proc. Natl. Acad. Sci. U. S. A.105, 17795–17800
70 Hoersch, D. et al. (2008) Monitoring the conformational changes ofphotoactivated rhodopsin from microseconds to seconds by transientfluorescence spectroscopy. Biochemistry 47, 11518–11527
71 Ahuja, S. et al. (2009) Helix movement is coupled to displacement ofthe second extracellular loop in rhodopsin activation.Nat. Struct. Mol.Biol. 16, 168–175
72 Ahuja, S. et al. (2009) Location of the retinal chromophore in theactivated state of rhodopsin*. J. Biol. Chem. 284, 10190–10201
73 Salom, D. et al. (2006) Crystal structure of a photoactivateddeprotonated intermediate of rhodopsin. Proc. Natl. Acad. Sci. U.S. A. 103, 16123–16128
74 Knierim, B. et al. (2008) Rhodopsin and 9-demethyl-retinal analog:effect of a partial agonist on displacement of transmembrane helix 6 inclass A G protein-coupled receptors. J. Biol. Chem. 283, 4967–4974
75 Crocker, E. et al. (2006) Location of Trp265 in metarhodopsin II:implications for the activation mechanism of the visual receptorrhodopsin. J. Mol. Biol. 357, 163–172
76 Bartl, F.J. et al. (2005) Partial agonism in a G protein-coupledreceptor: role of the retinal ring structure in rhodopsin activation.J. Biol. Chem. 280, 34259–34267
77 Meyer, C.K. et al. (2000) Signaling states of rhodopsin. Retinalprovides a scaffold for activating proton transfer switches. J. Biol.Chem. 275, 19713–19718
78 Vogel, R. et al. (2006) Agonists and partial agonists of rhodopsin:retinal polyene methylation affects receptor activation. Biochemistry45, 1640–1652
79 Vogel, R. et al. (2003) Deactivation of rhodopsin in the transition fromthe signaling state meta II to meta III involves a thermalisomerization of the retinal chromophore C D. Biochemistry 42,9863–9874
80 Ritter, E. et al. (2004) Transition of rhodopsin into the activemetarhodopsin II state opens a new light-induced pathway linkedto Schiff base isomerization. J. Biol. Chem. 279, 48102–48111
81 Palczewski, K. (2006) G protein-coupled receptor rhodopsin. Annu.Rev. Biochem. 75, 743–767
82 Ernst, O.P. et al. (2007) Monomeric G protein-coupled receptorrhodopsin in solution activates its G protein transducin at thediffusion limit. Proc. Natl. Acad. Sci. U. S. A. 104, 10859–10864
83 Bayburt, T.H. et al. (2007) Transducin activation by nanoscale lipidbilayers containing one and two rhodopsins. J. Biol. Chem. 282,14875–14881
84 Whorton, M.R. et al. (2008) Efficient coupling of transducin tomonomeric rhodopsin in a phospholipid bilayer. J. Biol. Chem. 283,4387–4394
85 Banerjee, S. et al. (2008) Rapid incorporation of functional rhodopsininto nanoscale apolipoprotein bound bilayer (NABB) particles. J. Mol.Biol. 377, 1067–1081
86 Ridge, K.D. et al. (2006) NMR analysis of rhodopsin-transducininteractions. Vision Res. 46, 4482–4492
87 Fritze, O. et al. (2003) Role of the conserved NPxxY(x)5,6F motif in therhodopsin ground state and during activation. Proc. Natl. Acad. Sci.U. S. A. 100, 2290–2295
88 Scheerer, P. et al. (2009) Structural and kinetic modeling of anactivating helix switch in the rhodopsin-transducin interface. Proc.Natl. Acad. Sci. U. S. A. 106, 10660–10665
89 Preininger, A. et al. (2009) Helix dipole movement and conformationalvariability contribute to allosteric GDP release in Gai subunits.Biochemistry 48, 2630–2642
90 Garczarek, F. and Gerwert, K. (2006) Functional waters inintraprotein proton transfer monitored by FTIR differencespectroscopy. Nature 439, 109–112
91 Hildebrand, P.W. et al. (2008) Hydrogen-bonding and packingfeatures of membrane proteins: functional implications. Biophys. J.94, 1945–1953
92 MacCallum, J.L. et al. (2007) Hydrophobic association of a-helices,steric dewetting, and enthalpic barriers to protein folding. Proc. Natl.Acad. Sci. U. S. A. 104, 6206–6210
551

Review Trends in Biochemical Sciences Vol.34 No.11
93 Smock, R.G. and Gierasch, L.M. (2009) Sending signals dynamically.Science 324, 198–203
94 Ballesteros, J.A. and Weinstein, H. (1995) Integrated methods for theconstruction of three-dimensional models and computational probingof structure-function relations in G protein-coupled receptors.Methods Neurosci. 25, 366–428
95 Binet, V. et al. (2007) Common structural requirements forheptahelical domain function in class A and class C G protein-coupled receptors. J. Biol. Chem. 282, 12154–12163
96 Prioleau, C. et al. (2002) Conserved helix 7 tyrosine acts as amultistate conformational switch in the 5HT2C receptor.Identification of a novel ‘‘locked-on’’ phenotype and doublerevertant mutations. J. Biol. Chem. 277, 36577–36584
97 Franke, R.R. et al. (1990) Rhodopsin mutants that bind but fail toactivate transducin. Science 250, 123–125
98 Ernst, O.P. et al. (1995) Characterization of rhodopsin mutants thatbind transducinbut fail to induceGTPnucleotideuptake.Classificationof mutant pigments by fluorescence, nucleotide release, and flash-induced light-scattering assays. J. Biol. Chem. 270, 10580–10586
552
99 Kisselev, O.G. et al. (1998) Light-activated rhodopsin inducesstructural binding motif in G protein alpha subunit. Proc. Natl.Acad. Sci. U. S. A. 95, 4270–4275
100 Koenig, B.W. et al. (2002) Structure and orientation of a G proteinfragment in the receptor bound state from residual dipolar couplings.J. Mol. Biol. 322, 441–461
101 Ruprecht, J.J. et al. (2004) Electron crystallography reveals thestructure of metarhodopsin I. EMBO J. 23, 3609–3620
102 Kono, M. et al. (2008) 11-cis- and all-trans-retinols can activaterod opsin: rational design of the visual cycle. Biochemistry 47,7567–7571
103 Buczylko, J. et al. (1996) Mechanisms of opsin activation. J. Biol.Chem. 271, 20621–20630
104 Kisselev, O.G. et al. (1999) Signal transfer from rhodopsin to the G-protein: evidence for a two-site sequential fit mechanism. Proc. Natl.Acad. Sci. U. S. A. 96, 4898–4903
105 Schwartz, T.W. et al. (2006) Molecular mechanism of 7TM receptoractivation – a global toggle switch model. Annu. Rev. Pharmacol.Toxicol. 46, 481–519





























