Embed Size (px)
Citation preview

www.elsevier.com/locate/ybbrc
Biochemical and Biophysical Research Communications 332 (2005) 1012–1019
BBRC
A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation
Min Xia a,1, Qingfeng Jin a,b,1, Saıd Bendahhou c,1, Yusong He a,b,1,Marie-Madeleine Larroque c, Yiping Chen d, Qinshu Zhou a, Yiqing Yang b, Yi Liu a,
Ban Liu a, Qian Zhu b, Yanting Zhou d, Jie Lin b, Bo Liang a, Li Li a, Xiongjian Dong b,Zhiwen Pan a, Rongrong Wang b, Haiying Wan b, Weiqin Qiu a, Wenyuan Xu b,
Petra Eurlings a, Jacques Barhanin c, Yihan Chen a,b,*
a Institute of Medical Genetics, Tongji University, Shanghai, Chinab Department of Cardiology, Tongji Hospital, Tongji University, Shanghai, China
c Institut de Pharmacologie Moleculaire et Cellulaire, UMR 6097 CNRS, Valbonne, Franced Department of Cardiology, University Hospitals of Cleveland, Case Western Reserve University, Cleveland, OH, USA
Received 4 May 2005Available online 23 May 2005
Abstract
The inward rectifier K+ channel Kir2.1 mediates the potassium IK1 current in the heart. It is encoded by KCNJ2 gene that hasbeen linked to Andersen�s syndrome. Recently, strong evidences showed that Kir2.1 channels were associated with mouse atrialfibrillation (AF), therefore we hypothesized that KCNJ2 was associated with familial AF. Thirty Chinese AF kindreds were eval-uated for mutations in KCNJ2 gene. A valine-to-isoleucine mutation at position 93 (V93I) of Kir2.1 was found in all affected mem-bers in one kindred. This valine and its flanking sequence is highly conserved in Kir2.1 proteins among different species. Functionalanalysis of the V93I mutant demonstrated a gain-of-function consequence on the Kir2.1 current. This effect is opposed to the loss-of-function effect of previously reported mutations in Andersen�s syndrome. Kir2.1 V93I mutation may play a role in initiating and/or maintaining AF by increasing the activity of the inward rectifier K+ channel.� 2005 Elsevier Inc. All rights reserved.
Keywords: Kir2.1; KCNJ2; Ion channel; Atrial fibrillation; Molecular genetics
As the most common cardiac arrhythmia in clinicalpractice, atrial fibrillation (AF) is characterized by rapidand irregular activation of the atrium. AF prevalence in-creases progressively with age, with nearly 6% in thoseover 65 years of age. A recent Framingham Heart Studyshowed that lifetime risks for AF development wereabout 25% for men and women 40 years of age and old-er. AF may cause strokes and heart failures, and is asource of considerable morbidity and mortality [1–4].However, to date, a definitive treatment approach has
0006-291X/$ - see front matter � 2005 Elsevier Inc. All rights reserved.
doi:10.1016/j.bbrc.2005.05.054
* Corresponding author. Fax: +86 21 66371663.E-mail address: [email protected] (Y. Chen).
1 These authors contributed equally to this work.
not been established. According to a large survey per-formed in the arrhythmia clinic at Mayo Clinic inRochester, Minnesota, about 36% patients with AFhad no evident pathogeny, and 5% had a positive familyhistory [5]. This points to the fact that a subset of AFhas an important genetic background. Recently, wefound two causative genes (K+ channel genes KCNQ1and KCNE2) for familial AF [6,7]. However, moremolecular basis or causative genes remain to be identi-fied [5–10].
Understanding the electrophysiological process lead-ing to AF has been a subject of intensive study over lastcentury. Dobrev et al. [11,12] observed that the currentdensity of IK1 in human atrial myocytes was twofold

M. Xia et al. / Biochemical and Biophysical Research Communications 332 (2005) 1012–1019 1013
greater during AF than in sinus rhythm. Van Wagoneret al. [13,14] and Bosch et al. [15] also noted a significantincrease in IK1 in atrial myocytes of patients with AF.
Kir2.1 to Kir2.4 subfamily of inward rectifier K+
channel family underlies cardiac IK1 [16,17]. Kir2.1 isencoded by KCNJ2, a causative gene of Andersen syn-drome, which is characterized by a triad of periodicparalysis, cardiac arrhythmia, and skeletal developmen-tal abnormalities [18]. The role of Kir2.1 in forming car-diac IK1 was supported by the absence of IK1 current incardiac myocytes from Kir2.1�/� mice [19]. The overex-pression of Kir2.1 in the mouse heart upregulated IK1
and ultimately initiated multiple abnormalities of car-diac excitability including AF [20].
In this study, we analyzed the Kir2.1 coding geneKCNJ2, the congenital long QT syndrome-associatedgenes (KCNQ1, HERG, SCN5A ANK-B, KCNE1, andKCNE2), and several other channel genes (KCNE3,KCNE4, and KCNE5) in 30 unrelated Chinese kindredswith AF. We found a missense mutation in KCNJ2 inone Chinese family. Heterologous expression in COSand HEK cells revealed a gain-of-function effect consis-tent with the AF clinical manifestations seen in thisfamily.
Materials and methods
Subjects.We identified 30 unrelated kindreds of familial AF amongChinese Han population. The diagnostic criteria for familial AF were apositive AF family history, electrocardiographic evidence of AF, andexclusion of organic heart diseases and other causative factors. Wescreened 10 ion channel or transporter related genes (KCNQ1, HERG,SCN5A, ANK-B, KCNJ2, KCNE1, KCNE2, KCNE3, KCNE4, andKCNE5) (GenBank Accession Nos. AJ006345, NT_007914,NT_022517, NW_105991, AP001720, NM_005136, NT_035430,XM_208561, NT_005403, and NM_012282. GenBank: http://www.ncbi.nlm.nih.gov/GenBank) in these 30 probands and in theirfamily members. The controls were 420 unrelated healthy Chinesesubjects of Han nationality. The study was approved by the EthicalCommittee of Chinese Human Genome Center at Shanghai. Writteninformed consent was obtained from all subjects.
Genetic analysis. DNA was extracted from whole venous bloodwith Wizard Genomic DNA Purification Kit (Promega). All codingsequences of the KCNQ1, HERG, SCN5A, ANK-B, KCNJ2, KCNE1,KCNE2, KCNE3, KCNE4, and KCNE5 were amplified using Hot-StarTaq DNA Polymerase (Qiagen). Products of PCR amplificationwere purified with MBI Fermentas DNA Extraction Kit (MBI). Bothstrands of each PCR product were sequenced with a DYEnamic ETdye terminator kit (Amersham Biosciences) under MegaBACE 500DNA Sequencing system (Amersham Biosciences).
RT-PCR. For RT-PCR, human atrial and ventricular tissue spec-imens were collected and preserved in RNAlater RNA stabilizationreagent (Qiagen). Total RNA was prepared using an RNeasy ProtectMini kit (Qiagen). An on-column DNase treatment was included in theRNA isolation step. Before RT-PCR, RNA samples were furthertreated with DNase (Invitrogen). RT was performed with KCNJ2 (5 0-GTGACACATCTGAAACCATAGCC-3 0) and control gene GAPDH
(5 0-CCACCACCCTGTTGCTGTAG-3 0) specific primers usingSuperScriptIIreverse transcriptase (Invitrogen). PCR was performed ina 25 ll reaction mixture with an annealing temperature at 62 �C, and
35 cycles. Q-solution was included in the reactions for the KCNJ2 andGAPDH transcripts. The primer pairs used for the concurrent ampli-fication of the two transcripts were for KCNJ2: forward—5 0-TCAGTAGACAGACCTTGGTAGAACC-3 0, reverse—50-GTGACACATCTGAAACCATAGCC-30, product size 621 bp; for GAPDH:forward—50-GCCAAGGTCATCCATGACA-30, reverse—50-CCACCACCCTGTTGCTGTAG-3 0, product size 496 bp. Genomic DNA-negative control for PCR was conducted by adding RNA templateinstead of RT product to the PCR mixture.
Mutagenesis. Forward (5 0-GCAGGATCCAAAGCAGAAGCACTGGAGTC-30) and reverse (5 0-GCAGCGGCCGCTGACCCATCTTGACCAGTACC-3 0) primers were used to amplify the codingregions and untranslated flanking regions of the human KCNJ2 geneusing pfuUltra high-fidelity DNA polymerase (Stratagene). Purified,BamHI and NotI digested PCR product was subcloned into the BamHIand NotI sites of the pXOOM expression vector (kindly provided byDr. T Jespersen, University of Copenhagen, Denmark).
Site-directed mutagenesis was performed using the QuickChange IIXL Site-Directed Mutageneses Kit. The forward and reverse primerswere 5 0-ATCTTCTGCCTGGCTTTCATCCTGTCATGGCTGTTTT-3 0 and 5 0-AAAACAGCCATGACAGGATGAAAGCCAGGCAGAAGAT-3 0, respectively. The entire coding region of the mutantclone was confirmed by sequencing.
Transfection and electrophysiology. Cells (COS-7 or HEK293) weremaintained as described [6]. Cells were transiently transfected byDEAE–dextran precipitate method with 2 lg wild-type (WT) KCNJ2-pXOOM DNA or 2 lg mutant-type (MT) KCNJ2-pXOOM DNA or1 lg WT and 1 lg MT KCNJ2-pXOOMDNA per 60-mm culture dish.Current recordings were performed 48 h after transfection in thewhole-cell configuration at room temperature (�22 �C), using EPC-10amplifier (HEKA Instruments), and as previously described [6].
Confocal microscopy. In order to monitor Kir2.1 subcellularlocalization, WT KCNJ2 and MT KCNJ2 were transferred into thepEYFP-N1 vector (enhanced yellow fluorescent protein, Clontech) andthe pECFP-N1 vector (enhanced cyan fluorescent protein, Clontech),respectively. The whole coding region of the two plasmid constructswas confirmed by sequencing. Fluorescence microscopy was performedwith a Leica confocal microscopy system.
Statistical analysis. Current changes were assessed by means ofANOVA and Dunnett�s t test. All reported p values are two-sided anda p value of 0.05 or less was considered to indicate statistical signifi-cance. Data are given as means ± SE.
Results
Identification of Kir2.1 V93I mutation
The probands in 30 kindreds of familial AF wereclinically and genetically evaluated. None of themhad evident etiologies for AF. We sequenced 10 ionchannel or transporter related genes (KCNQ1, HERG,SCN5A, ANK-B, KCNJ2, KCNE1, KCNE2, KCNE3,KCNE4, and KCNE5) after PCR amplification ofgenomic DNA. A novel missense mutation in KCNJ2
was found in one of the probands. With severalDNA samples unavailable and the phenotypes ofII:12, III:3, and III:6 uncertain, using the flankingmicrosatellite D17S949, a two-point linkage analysisbetween KCNJ2 and the familial AF gave a LOD scoreof 1.93 at recombination fraction 0 (detailed data notshown). The proband (II-3) who had paroxysmal AF

1014 M. Xia et al. / Biochemical and Biophysical Research Communications 332 (2005) 1012–1019
as frequently as two to three times a month was a het-erozygote for the 277G fi A transition, correspondingto a V93I substitution in KCNJ2. His father (I-1)and older sister (II-2) had permanent AF before death.V93I was also found in his younger sister (II-6), niece(III-2), and nephews (III-3 and III6). His younger sister(II-6) experienced paroxysmal AF as frequently as onceor twice a week. His niece (III-2) showed paroxysmalAF on 24-h electrocardiographic recordings. His neph-ews (III-3 and III-6 who was 42 and 33 years old,respectively) did not have AF on 24-h electrocardio-graphic monitoring. In view of the characteristic ofparoxysmal onset of AF, we did not exclude that III-3 and III-6 were AF patients. We did not obtain theblood sample of II-12 and did not know her pheno-type. Other unaffected family members did not carrythe V93I substitution. The substitution co-segregatedwith AF in the family with the exception of III-3 andIII-6 (Table 1 and Fig. 1). All affected members in thisfamily had normal QT interval on 12-lead ECG, expe-rienced no episodes of muscle weakness or syncope,and did not have frequent premature ventricular con-tractions or ventricular tachycardia on 24-h electrocar-diographic monitoring. Serum potassium levels werewithin normal limits. None of the patients exhibitedany developmental problems such as cleft palate, lowset ears, short stature, clinodactyly, syndactyly, orbrachydactyly, as reported for Andersen�s syndrome.
To further confirm that the V93I substitution ofKir2.1 was not a benign polymorphism, we checked thissubstitution in 420 healthy individuals. It was not foundin any of these healthy individuals.
Kir2.1 expression in human atrium
Although Kir2.1 is expressed in human heart, its dis-tribution in different heart chambers is not clear. Quali-tative RT-PCR was performed to determine the tissuedistribution of KCNJ2 mRNA in the human heart. Ourdata showed that Kir2.1 was strongly expressed in bothventricles and atria, with lower signals in atria (Fig. 1).
Table 1Clinical characteristics of subjects with Kir2.1 V93I mutation
Familymember
Sex Age(year)
Age at AFdiagnosis (year)
Recurrentpalpitation
Ac
I-1 M 82a 58 + PII-2 F 56a 50 + PII-3 M 59b 54 + PII-6 F 56b 50 + PIII-2 F 57b 57 � PIII-3 M 42b NA � NIII-6 M 33b NA � N
QTc, corrected QT interval; NA, not available or not applicable; LAD, lefta The age at death is shown.b Age is the current age.c The value in AF is shown.
Conservation of sequences flanking Kir2.1 V93I mutation
Kir2.1 subunits are made of two membrane-spanningsegments (M1 and M2) adjacent to a pore-formingloop, and cytoplasmic N- and C-termini. V93I substitu-tion is located in the outer helix of the M1 domain [21]which displays a highly conserved sequence among hu-man, domestic guinea pig, pig, dog, cow, Norway rat,rabbit, and house mouse (Table 2 and Fig. 2). V93I sub-stitution perturbs a highly conserved residue suggestingthat V93I may have important functional consequences.
Functional effect of V93I mutation on Kir2.1 channel
We expressed WT and MT KCNJ2 in COS-7 cells,and performed electrophysiological study using whole-cell patch-clamping technique. The data were as follows:(1) the currents for MT expression at potentials rangingfrom �140 to �80 mV or from �60 to �40 mV hadamplitudes significantly higher than those for WTexpression. For example, at �90 mV, the current forMT expression was �97 ± 7.8 pA/pF (n = 20) and thatfor WT expression �40 ± 4.9 pA/pF (n = 22)(p < 0.01); whereas at �50 mV, the former was97 ± 8.0 pA/pF (n = 20) and the latter 15 ± 3.0 pA/pF(n = 22) (p < 0.001) (Fig. 3); (2) at the same potentialrange, co-expression of MT and WT also significantlyenhanced the Kir2.1 current (Fig. 3); (3) compared withthe currents for co-expression of MT and WT, the cur-rents for MT expression at potentials ranging from�50 to �40 mV were significantly larger (Fig. 3). Similarresults were obtained in HEK293 cells (data not shown).Hence, it is concluded that the V93I mutation had again-of-function effect on Kir2.1 channels.
Subcellular trafficking of mutant Kir2.1 channel
To assess the subcellular trafficking of WT and MTKCNJ2 subunits, we performed confocal laser scanningmicroscopy inCOS-7 andHEK293 cells. Cells transfectedwith WT KCNJ2 showed the same plasma membrane
Flassification
Ventricularrate (bpm)
QTc (s) LAD(mm)
LVEF(%)
ermanent NA NA NA NAermanent 63/70c 0.42/0.43c 35 73aroxysmal 68/110c 0.41/0.44c 36 65aroxysmal 71/118c 0.43/0.43c 35 66aroxysmal 66/102c 0.42/0.43c 31 71A 83 0.41 33 68A 66 0.37 34 65
atrial dimension; LVEF, left ventricular ejection fraction.

Fig. 1. Familial AF associated with Kir2.1 V93I mutation. (A) Pedigree for a Chinese AF kindred. Asterisks indicate that the individual had theV93I mutation. (B) Normal KCNJ2 DNA sequence. (C) KCNJ2DNA sequence of the affected family members. Sequence analysis of DNA from theaffected family members after PCR amplification revealed a G-to-A substitution at nucleotide 277 in KCNJ2 causing a V93I mutation. (D) RT-PCRresults for transcripts of KCNJ2 and GAPDH genes concurrently expressed in human atrial and ventricular tissues.
M. Xia et al. / Biochemical and Biophysical Research Communications 332 (2005) 1012–1019 1015
fluorescence pattern as those transfected with MTKCNJ2. In cells co-transfected withWT andMTKCNJ2,both proteins also demonstrated a similar fluorescence
Table 2Conservation of sequences flanking Kir2.1 V93I mutation
Species Amino acids flanking residue 93
Human KGQRYLADIFTTCVDIRWRWMLVIFCLMutant V931 ........................................................................Domestic guinea pig ........................................................................Pig ........................................................................Dog ........................................................................Cow ........................................................................Norway rat ........................................................................Rabbit ........................................................................House mouse ........................................................................
a Identity is the percentage of the sequence that is identical to human sequeHuman Kir2.1, gi: 4504835; domestic guinea pig Kir2.1, gi: 1708549; pig KiNorway rat Kir2.1, gi: 2493597; rabbit Kir2.1, gi: 1352481; house mouse Ki
pattern (data not shown). Tagging with the fluorescentprotein did not affect the Kir2.1 channel function (datanot shown).
Identitiesa (%)
AFVLSWLFFGCVFWLIALLHGDLDASK.......I...................................................................................................................................... 99....................................................................... 99....................................................................... 99....................................................................... 99....................................................................... 99.....................................................................R 99..................................................................T... 98
nce as based on protein sequences from the following GenBank ID (gi):r2.1, gi: 3024031; dog Kir2.1, gi: 13878561; Cow Kir2.1, gi: 27805969;r2.1, gi: 547735.

Fig. 2. The proximity of residue valine 93 to the Kir2.1 channel pore.Residue V93 on the Kir2.1 sequence aligns to residue V72 on theKirBac1.1 crystal structure and is located in the outer helix.
1016 M. Xia et al. / Biochemical and Biophysical Research Communications 332 (2005) 1012–1019
Discussion
The availability of a large number of characterizedAF kindreds in our cardiology center allowed us to iden-tify KCNJ2 as a novel gene for familial AF. Interest-ingly, this gene has already been linked to Andersen�ssyndrome [22]. Unlike loss-of-function mutations inAndersen�s syndrome, the V93I substitution identifiedin our AF family had a Kir2.1 gain-of-function effect.This mutation was present in all affected family mem-bers and absent in 420 healthy subjects.
It was reported that overexpression of wild-typeKir2.1 in the mouse heart led to AF and other abnor-malities of cardiac excitability [20]. Thus, we hypothe-sized that Kir2.1 mutations might be associated withfamilial AF. Before the Kir2.1 coding gene KCNJ2
was sequenced, we performed two-point linkage analysison the four-generation family. Although several DNAsamples were unavailable and the phenotypes of II:12,III:3, and III:6 uncertain, a LOD score of 1.93 was ob-tained at recombination fraction zero at D17S949 (datanot shown).
To date, over 20 Kir2.1 mutations have been found infamilies with Andersen�s syndrome, all of which resultedin loss-of-function with dominant-negative suppressionof inward currents, except for two, which displayed hap-lo-insufficiency [22–26]. In contrast, V93I had a gain-of-
function effect on Kir2.1 channels. The functionalexpression of the AF-associated V93I substitution dem-onstrated a significant gain-of-function in both inwardand outward Kir2.1 currents, without affecting thekinetics and rectification properties. Although not quan-tified, the results with the fluorescently labeled Kir2.1subunits are in favor of a normal trafficking of the mu-tated channels. A direct effect on channel conductance ismore likely, as suggested by the proximity of residue va-line 93 to the pore (Fig. 2).
Kir2.1 plays a major role in both the late phase ofrepolarization (phase 3) and the phase of resting mem-brane potential (phase 4), but almost no current is al-lowed to pass the channel during the plateau phase ofaction potential [16,27–31]. At potentials negative toresting membrane potential, inward rectifier channelspass a large inward current to bring the potential backto the resting status. In contrast, at potentials positiveto the resting membrane potential, inward rectifier chan-nels pass a much smaller outward current to repolarizethe cell. The activated inward rectifier channels may am-plify and accelerate the rate of return of the membraneto its resting membrane potential value, leading toshortening of action potential duration [30,32,33]. Asexpected, the Kir2.1 loss-of-function mutations led toQT interval prolongation in Andersen�s syndrome pa-tients. Kir2.1 V93I mutation increased inward potas-sium current at �90 to �80 mV and outwardpotassium current at �60 to �40 mV. These would sta-bilize resting membrane potential and shorten the repo-larization phase of the atrial action potential, resultingin a shortening of the atrial effective refractory period(ERP). Thus, the gain-of-function effect of V93I muta-tion may create a substrate favorable to a multiple wave-let re-entry, a dominant mechanism of AF [2].
An overexpression of Kir2.1 in mouse heart upregu-lated IK1 and initiated AF [20]. IK1 increase has alsobeen reported in human acquired AF [11–14]. Addition-ally, all the two human AF-associated mutations re-ported to date increased K+ channel current [6,7]. Thegain-of-function mutation of cardiac K+ channels couldbe a good trigger for AF. New evidence has been foundto support the speculation. A gain-of-function mutationof HERG, a K+ channel, caused not only short QT syn-drome but also AF [34].
According to the protein sequence alignment, the se-quences flanking the KCNJ2 V93I substitution are wellconserved in all mammals with KCNJ2 sequence known.However, the substitution can be found in birds (chick-ens and domestic pigeons) (GenBank Accession Nos.:chicken KCNJ2, gi: 45382445; domestic pigeon KCNJ2,gi: 6273353). This does not seem to support our hypoth-esis that KCNJ2 V93I is a novel mutation associatedwith familial AF. In view of this, we analyzed theexpression of KCNJ2 in the hearts of chicken anddomestic pigeon. We found that KCNJ2 was not
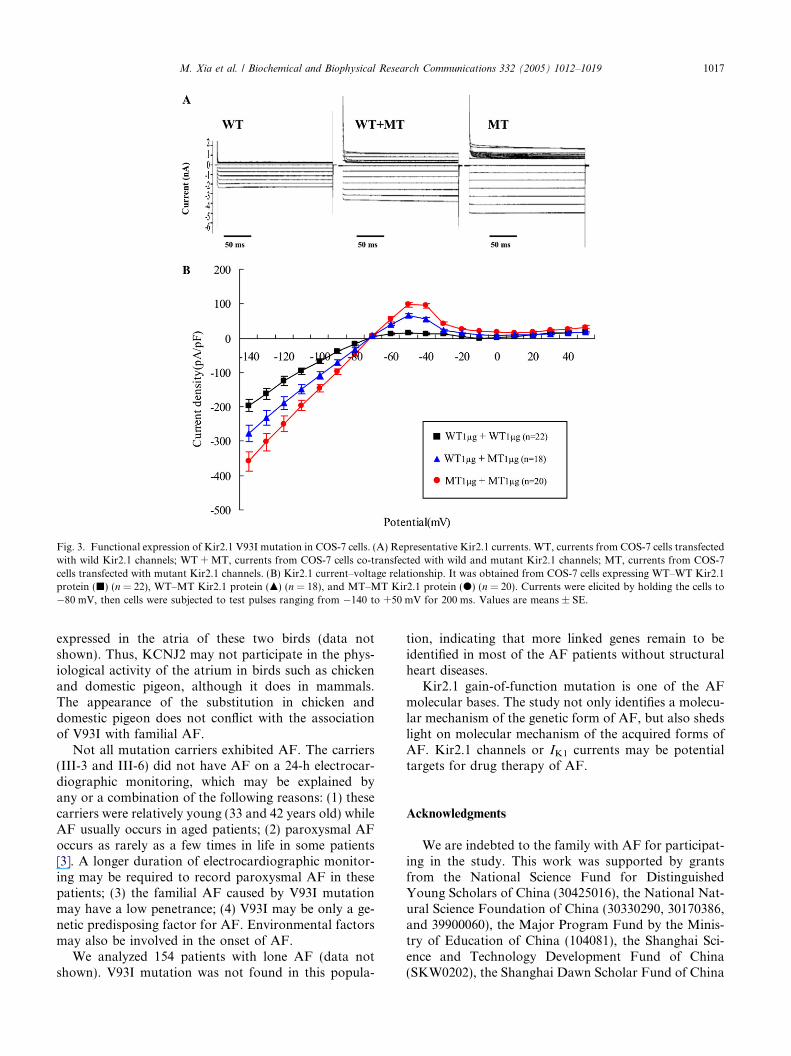
Fig. 3. Functional expression of Kir2.1 V93I mutation in COS-7 cells. (A) Representative Kir2.1 currents. WT, currents from COS-7 cells transfectedwith wild Kir2.1 channels; WT +MT, currents from COS-7 cells co-transfected with wild and mutant Kir2.1 channels; MT, currents from COS-7cells transfected with mutant Kir2.1 channels. (B) Kir2.1 current–voltage relationship. It was obtained from COS-7 cells expressing WT–WT Kir2.1protein (j) (n = 22), WT–MT Kir2.1 protein (m) (n = 18), and MT–MT Kir2.1 protein (d) (n = 20). Currents were elicited by holding the cells to�80 mV, then cells were subjected to test pulses ranging from �140 to +50 mV for 200 ms. Values are means ± SE.
M. Xia et al. / Biochemical and Biophysical Research Communications 332 (2005) 1012–1019 1017
expressed in the atria of these two birds (data notshown). Thus, KCNJ2 may not participate in the phys-iological activity of the atrium in birds such as chickenand domestic pigeon, although it does in mammals.The appearance of the substitution in chicken anddomestic pigeon does not conflict with the associationof V93I with familial AF.
Not all mutation carriers exhibited AF. The carriers(III-3 and III-6) did not have AF on a 24-h electrocar-diographic monitoring, which may be explained byany or a combination of the following reasons: (1) thesecarriers were relatively young (33 and 42 years old) whileAF usually occurs in aged patients; (2) paroxysmal AFoccurs as rarely as a few times in life in some patients[3]. A longer duration of electrocardiographic monitor-ing may be required to record paroxysmal AF in thesepatients; (3) the familial AF caused by V93I mutationmay have a low penetrance; (4) V93I may be only a ge-netic predisposing factor for AF. Environmental factorsmay also be involved in the onset of AF.
We analyzed 154 patients with lone AF (data notshown). V93I mutation was not found in this popula-
tion, indicating that more linked genes remain to beidentified in most of the AF patients without structuralheart diseases.
Kir2.1 gain-of-function mutation is one of the AFmolecular bases. The study not only identifies a molecu-lar mechanism of the genetic form of AF, but also shedslight on molecular mechanism of the acquired forms ofAF. Kir2.1 channels or IK1 currents may be potentialtargets for drug therapy of AF.
Acknowledgments
We are indebted to the family with AF for participat-ing in the study. This work was supported by grantsfrom the National Science Fund for DistinguishedYoung Scholars of China (30425016), the National Nat-ural Science Foundation of China (30330290, 30170386,and 39900060), the Major Program Fund by the Minis-try of Education of China (104081), the Shanghai Sci-ence and Technology Development Fund of China(SKW0202), the Shanghai Dawn Scholar Fund of China

1018 M. Xia et al. / Biochemical and Biophysical Research Communications 332 (2005) 1012–1019
(SKW0103), and the Centre National de la RechercheScientifique Fund of France.
References
[1] D.M. Lloyd-Jones, T.J. Wang, E.P. Leip, M.G. Larson, D. Levy,R.S. Vasan, R.B. D�Agostino, J.M. Massaro, A. Beiser, P.A.Wolf, E.J. Benjamin, Lifetime risk for development of atrialfibrillation: the Framingham Heart Study, Circulation 110 (2004)1042–1046.
[2] S. Nattel, New ideas about atrial fibrillation 50 years on, Nature415 (2002) 219–226.
[3] S.S. Chugh, J.L. Blackshear, W.K. Shen, S.C. Hammill, B.J.Gersh, Epidemiology and natural history of atrial fibrillation:clinical implications, J. Am. Coll. Cardiol. 37 (2001) 371–378.
[4] K.M. Ryder, E.J. Benjamin, Epidemiology and significance ofatrial fibrillation, Am. J. Cardiol. 84 (1999) R131–R138.
[5] D. Darbar, K.J. Herron, J.D. Ballew, A. Jahangir, B.J. Gersh,W.K. Shen, S.C. Hammill, D.L. Packer, T.M. Olson, Familialatrial fibrillation is a genetically heterogeneous disorder, J. Am.Coll. Cardiol. 41 (2003) 2185–2192.
[6] Y.H. Chen, S.J. Xu, S. Bendahhou, X.L. Wang, Y. Wang, W.Y.Xu, H.W. Jin, H. Sun, X.Y. Su, Q.N. Zhuang, Y.Q. Yang, Y.B.Li, Y. Liu, H.J. Xu, X.F. Li, N. Ma, C.P. Mou, Z. Chen, J.Barhanin, W. Huang, KCNQ1 gain-of-function mutation infamilial atrial fibrillation, Science 299 (2003) 251–254.
[7] Y. Yang, M. Xia, Q. Jin, S. Bendahhou, J. Shi, Y. Chen, B.Liang, J. Lin, Y. Liu, B. Liu, Q. Zhou, D. Zhang, R. Wang, N.Ma, X. Su, K. Niu, Y. Pei, W. Xu, Z. Chen, H. Wan, J. Cui, J.Barhanin, Y. Chen, Identification of a KCNE2 gain-of-functionmutation in patients with familial atrial fibrillation, Am. J. Hum.Genet. 75 (2004) 899–905.
[8] R. Brugada, T. Tapscott, G.Z. Czernuszewicz, A.J. Marian, A.Iglesias, L. Mont, J. Brugada, J. Girona, A. Domingo, L.L.Bachinski, R. Roberts, Identification of a genetic locus forfamilial atrial fibrillation, N. Engl. J. Med. 336 (1997) 905–911.
[9] P.T. Ellinor, J.T. Shin, R.K. Moore, D.M. Yoerger, C.A.MacRae, Locus for atrial fibrillation maps to chromosome6q14-16, Circulation 107 (2003) 2880–2883.
[10] C. Oberti, L. Wang, L. Li, J. Dong, S. Rao, W. Du, Q. Wang,Genome-wide linkage scan identifies a novel genetic locus onchromosome 5p13 for neonatal atrial fibrillation associated withsudden death and variable cardiomyopathy, Circulation 110(2004) 3753–3759.
[11] D. Dobrev, E. Graf, E. Wettwer, H.M. Himmel, O. Hala, C.Doerfel, T. Christ, S. Schuler, U. Ravens, Molecular basis ofdownregulation of G-protein-coupled inward rectifying K(+)current Ik-ACh in chronic human atrial fibrillation: decrease inGIRK4 mRNA correlates with reduced Ik-ACh and muscarinicreceptor-mediated shortening of action potentials, Circulation 104(2001) 2551–2557.
[12] D. Dobrev, E. Wettwer, A. Kortner, M. Knaut, S. Schuler, U.Ravens, Human inward rectifier potassium channels in chronicand postoperative atrial fibrillation, Cardiovasc. Res. 54 (2002)397–404.
[13] D.R. Van Wagoner, A.L. Pond, P.M. McCarthy, J.S. Trimmer,J.M. Nerbonne, Outward K+ current densities and Kv1.5 expres-sion are reduced in chronic human atrial fibrillation, Circ. Res. 80(1997) 772–781.
[14] D.R. Van Wagoner, Electrophysiological remodeling in humanatrial fibrillation, Pacing Clin. Electrophysiol. 26 (2003) 1572–1575.
[15] R.F. Bosch, X. Zeng, J.B. Grammer, K. Popovic, C. Mewis, V.Kuhlkamp, Ionic mechanisms of electrical remodeling in humanatrial fibrillation, Cardiovasc. Res. 44 (1999) 121–131.
[16] A.N. Lopatin, C.G. Nichols, Inward rectifiers in the heart: anupdate on I(K1), J. Mol. Cell. Cardiol. 33 (2001) 625–638.
[17] C. Zobel, H.C. Cho, T.T. Nguyen, R. Pekhletski, R.J. Diaz, G.J.Wilson, P.H. Backx, Molecular dissection of the inward rectifierpotassium current (IK1) in rabbit cardiomyocytes: evidence forheteromeric coassembly of Kir2.1 and Kir2.2, J. Physiol. 550(2003) 365–372.
[18] E.D. Andersen, P.A. Krasilnikoff, H. Overvad, Intermittentmuscular weakness, extrasystoles, and multiple developmentalanomalies. A new syndrome? Acta Paediatr. Scand. 60 (1971) 559–564.
[19] J.J. Zaritsky, J.B. Redell, B.L. Tempel, T.L. Schwarz, Theconsequences of disrupting cardiac inwardly rectifying K+ current(IK1) as revealed by the targeted deletion of the murine Kir2.1 andKir2.2 genes, J. Physiol. 553 (2001) 697–710.
[20] J. Li, M. McLerie, A.N. Lopatin, Transgenic upregulation of IK1in the mouse heart leads to multiple abnormalities of cardiacexcitability, Am. J. Physiol. Heart Circ. Physiol. 287 (2004)H2790–H2802.
[21] Y. Guo, G.J. Waldron, R. Murrell-Lagnado, A role for themiddle C terminus of G-protein-activated inward rectifier potas-sium channels in regulating gating, J. Biol. Chem. 277 (2002)48289–48294.
[22] N.M. Plaster, R. Tawil, M. Tristani-Firouzi, S. Canun, S.Bendahhou, A. Tsunoda, M.R. Donaldson, S.T. Iannaccone, E.Brunt, R. Barohn, J. Clark, F. Deymeer, A.L. George Jr., F.A.Fish, A. Hahn, A. Nitu, C. Ozdemir, P. Serdaroglu, S.H.Subramony, G. Wolfe, Y.H. Fu, L.J. Ptacek, Mutations inKir2.1 cause the developmental and episodic electrical phenotypesof Andersen�s syndrome, Cell 105 (2001) 511–519.
[23] S. Bendahhou, M.R. Donaldson, N.M. Plaster, M. Tristani-Firouzi, Y.H. Fu, L.J. Ptacek, Defective potassium channelKir2.1 trafficking underlies Andersen–Tawil syndrome, J. Biol.Chem. 278 (2003) 51779–51785.
[24] M. Tristani-Firouzi, J.L. Jensen, M.R. Donaldson, V. Sansone,G. Meola, A. Hahn, S. Bendahhou, H. Kwiecinski, A. Fidzianska,N. Plaster, Y.H. Fu, L.J. Ptacek, R. Tawil, Functional andclinical characterization of KCNJ2 mutations associated withLQT7 (Andersen syndrome), J. Clin. Invest. 110 (2002) 381–388.
[25] K. Ho, C.G. Nichols, W.J. Lederer, J. Lytton, P.M. Vassilev,M.V. Kanazirska, S.C. Hebert, Cloning and expression of aninwardly rectifying ATP-regulated potassium channel, Nature 362(1993) 31–38.
[26] H. Fodstad, H. Swan, M. Auberson, I. Gautschi, J. Loffing, L.Schild, K. Kontula, Loss-of-function mutations of the K(+)channel gene KCNJ2 constitute a rare cause of long QTsyndrome, J. Mol. Cell. Cardiol. 37 (2004) 593–602.
[27] Y. Shimoni, R.B. Clark, W.R. Giles, Role of an inwardlyrectifying potassium current in rabbit ventricular action potential,J. Physiol. 448 (1992) 709–727.
[28] Y. Kubo, T.J. Baldwin, Y.N. Jan, L.Y. Jan, Primary structureand functional expression of a mouse inward rectifier potassiumchannel, Nature 362 (1993) 127–133.
[29] H.J. Jongsma, R. Wilders, Channelopathies: Kir2.1 mutationsjeopardize many cell functions, Curr. Biol. 11 (2001) R747–R750.
[30] J.M.B. Anumonwo, Biophysic properties of inward rectifierpotassium channels, in: D.P. Zipes, J. Jalife (Eds.), CardiacElectrophysiology: from Cell to Bedsides, first ed., Saunders,Philadelphia, 2004, pp. 112–119.
[31] A.S. Dhamoon, S.V. Pandit, F. Sarmast, K.R. Parisian, P. Guha,Y. Li, S. Bagwe, S.M. Taffet, J.M. Anumonwo, Unique Kir2.xproperties determine regional and species differences in the cardiacinward rectifier K+ current, Circ. Res. 94 (2004) 1332–1339.
[32] B.A. Wible, M. De Biasi, K. Majumder, M. Taglialatela, A.M.Brown, Cloning and functional expression of an inwardly recti-fying K+ channel from human atrium, Circ. Res. 76 (1995) 343–350.

M. Xia et al. / Biochemical and Biophysical Research Communications 332 (2005) 1012–1019 1019
[33] C. Cabo, Computation of the action potential of a cardiac cell, in:C. Cabo, D.S. Rosenbaum (Eds.), Quantitative Cardiac Electro-physiology, Marcel Dekker, New York, 2002, pp. 61–104.
[34] K. Hong, P. Bjerregaard, I. Gussak, R. Brugada, Short QTsyndrome and atrial fibrillation caused by mutation in KCNH2, J.Cardiovasc. Electrophysiol. 16 (2005) 394–396.

















![[Guideline of atrial fibrillation]](https://img.pdfslide.net/doc/110x75/634997ca8e60111deb0a49ae/guideline-of-atrial-fibrillation.jpg)











